 Melika Hajimohammadebrahim-Ketabforoush
Melika Hajimohammadebrahim-Ketabforoush Alireza Zali
Alireza Zali Mohammadreza Shahmohammadi2
Mohammadreza Shahmohammadi2 Amir Ali Hamidieh
Amir Ali Hamidieh- 1Student Research Committee, Department of Clinical Nutrition and Dietetics, Faculty of Nutrition Sciences and Food Technology, National Nutrition and Food Technology Research Institute, Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 2Functional Neurosurgery Research Center, Shohada Tajrish Comprehensive Neurosurgical Center of Excellence, Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 3Pediatric Cell and Gene Therapy Research Center, Gene, Cell & Tissue Research Institute, Tehran University of Medical Sciences, Tehran, Iran
Despite reaching enormous achievements in therapeutic approaches worldwide, GBM still remains the most incurable malignancy among various cancers. It emphasizes the necessity of adjuvant therapies from the perspectives of both patients and healthcare providers. Therefore, most emerging studies have focused on various complementary and adjuvant therapies. Among them, metabolic therapy has received special attention, and metformin has been considered as a treatment in various types of cancer, including GBM. It is clearly evident that reaching efficient approaches without a comprehensive evaluation of the key mechanisms is not possible. Among the studied mechanisms, one of the more challenging ones is the effect of metformin on apoptosis and senescence. Moreover, metformin is well known as an insulin sensitizer. However, if insulin signaling is facilitated in the tumor microenvironment, it may result in tumor growth. Therefore, to partially resolve some paradoxical issues, we conducted a narrative review of related studies to address the following questions as comprehensively as possible: 1) Does the improvement of cellular insulin function resulting from metformin have detrimental or beneficial effects on GBM cells? 2) If these effects are detrimental to GBM cells, which is more important: apoptosis or senescence? 3) What determines the cellular decision between apoptosis and senescence?
1 Introduction
Glioblastoma multiforme (GBM) is known as the most devastating and incurable primary brain tumor. Patients with GBM experience a wide range of side effects and discomforts, resulting in an average life expectancy of approximately 14 months (1–4). Therefore, more of them are seeking additional remedies. Current treatments for GBM include surgery to remove as much of the tumor as possible, followed by radiation and temozolomide (TMZ) (5–7). Moreover, the use of nitrosourea-based regimens, especially lomustine (oral form) and carmustine (infusion form), has been evidenced as efficient adjuvant therapy in GBM (8–10). However, existing therapeutic strategies are still controversial, and it has remained an incurable disease in health services so far (4, 11). Therefore, even the smallest effort to find new insights into GBM therapy may be extremely valuable. According to it, complementary medicines and diets have already been imported and studied in the literature (12–17). Among these studies, the results are still very controversial. For example, recent studies have discussed how a ketogenic diet could improve GBM prognosis by reducing tumor size, tumor progression, and enhancing survival and quality of life. These impacts may be attributed to the anti-tumor properties of ketone bodies (18, 19), However, studies from a mechanistic perspective suggest that increased activation of fatty acid synthase has progressive effects on GBM cells (20, 21). Studies on the benefits of statin drugs in GBM patients have confirmed these results (4). On the other hand, blood glucose and serum insulin levels are significant metabolic culprits in the extensive body of cancer studies. An increasing number of recent studies have confirmed the benefits of metformin for cancer patients, including those with GBM (4, 22–27). However, it is still not well known whether these benefits truly result from reducing glucose and systemic insulin, or if other mechanisms are involved. It is noteworthy that metformin is recognized as an insulin-sensitizing drug that enhances insulin function at the cellular level, leading to reduced systemic insulin levels. With careful focus on literature, it is well established that metformin exerts anti-tumor effects on various cancer types through various possible routes, including cancer metabolism, epigenetics, cell cycle arrest, cancer invasion, migration, and metastasis, cell death, senescence, cancer stem cells, cancer immunity, and gut microbes, as demonstrated in in vitro and in vivo studies (28–41). The activation of adenosine monophosphate-activated protein kinase (AMPK), phosphorylation, and activation of P53, leading to the apoptotic state, is the most frequently mentioned mechanism of metformin function in many studies on various types of cancer (23, 42, 43) and GBM (44–50). In this regard, some studies have discussed both apoptosis and senescence as forms of cell death. However, it is well known that senescence is a state in which a cell no longer divides but has not yet undergone cell death. Moreover, unlike apoptosis, senescence is associated with the senescence-associated secretory phenotype (SASP) and resistance to apoptosis (51–54). Among the studies, most have focused on insulin, metformin, and apoptosis in GBM, a recent high-quality study suggested that metformin and simvastatin alone, but especially their combination, result in decreased cell proliferation and VEGF, increased apoptosis, and senescence in GBM cells (4). However, because senescence is associated with SASP and could induce immunosenescence, recent studies have shown that metformin can delay aging by reducing cellular senescence (29, 55, 56). On the other hand, taken together, to the best of our knowledge, insulin has a growing effect on cancer cells (57–59), therefore, the study mentioned above (4) may raise questions about how metformin’s improvement of insulin function at the cellular level could lead to anti-tumor effects on GBM cells. Among these enormous controversies, it seems that finding the exact mechanisms could partially help to solve these issues and to use supplementary drugs and diets to enhance the recovery of GBM patients. The present study aimed to conduct a narrative review of related studies to address the following questions as comprehensively as possible: 1) Does the improvement of cellular insulin function resulting from metformin have detrimental or beneficial effects on GBM cells? 2) If these effects are detrimental to GBM cells, which is more important: apoptosis or senescence? 3) What determines the cellular decision between apoptosis and senescence?
2 The studied mechanisms of the antitumor effects of metformin, especially in GBM
Mechanistically, studies have discussed multiple major aspects of the antitumor efficacy of metformin. These aspects are almost always related to systemic, cell-autonomous pathways, which can be AMPK-dependent or independent (60–66). Although there are innumerable underlying mechanisms, and their comprehensive explanations may not be entirely satisfying, we have made our best effort to review the most important ones as thoroughly as possible. The anticancer mechanisms of action for metformin, along with the possible responses to the rest of our research queries, have been summarized in a graphical abstract in Figure 1.
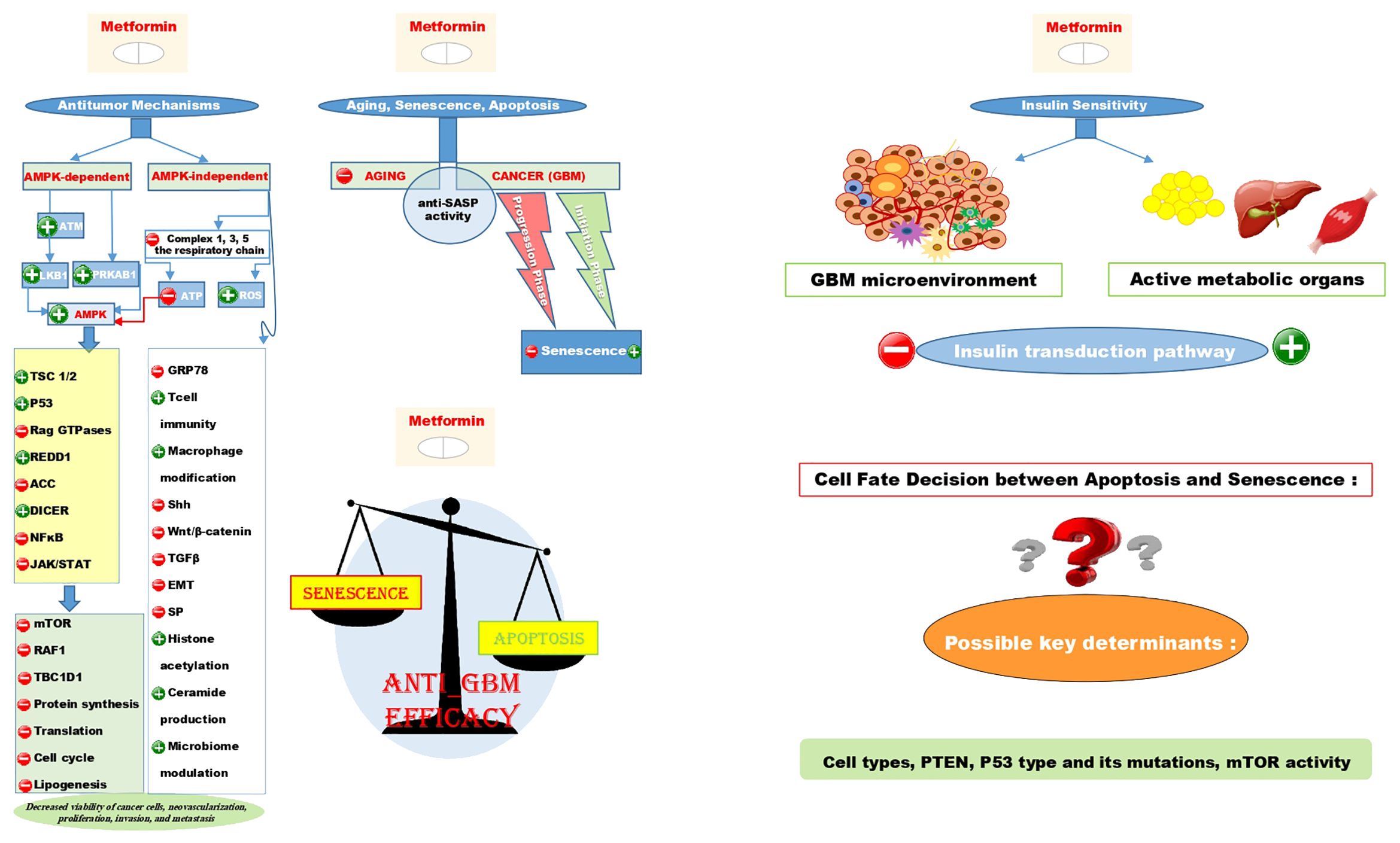
Figure 1. Illustrates the AMPK-dependent and AMPK-independent antitumor mechanisms of metformin using + signs for induction and - signs for repression on the left side of the figure. It also shows that the anti-SASP activity contributes to both the anti-aging and anti-cancer effects of metformin. Interestingly, metformin can induce senescence in the initiation phase of GBM while repressing it in the progression phase. The scale schematic suggests that the primary anti-tumor effect of metformin is linked to apoptosis rather than senescence, with cell types, PTEN, P53 type and mutations, and mTOR activity as key factors in determining cell fate decision between these two situations. Additionally, despite metformin being an insulin sensitizer metabolically, it could induce insulin resistance in the GBM microenvironment.
2.1 AMPK-dependent mechanisms
AMPK-dependent pathways are most frequently mentioned, identifying AMPK as a master regulator in this regard. Genome-wide association studies have confirmed that metformin can induce the Ataxia-telangiectasia mutated kinase (ATM) in cancer cell lines (67–71). The activated ATM phosphorylates Liver kinase B1 (LKB1) at Thr366, and subsequently, the activated LKB1 phosphorylates and activates AMPK at Thr172 (60). Notably, some studies have suggested that metformin also significantly increases the concentration of the regulatory subunit of AMPK (PRKAB1), which interacts with the catalytic subunit (61). Ultimately, metformin activates AMPK, exerting anti-tumorigenic properties in cancers by phosphorylating and activating two crucial tumor suppressors, TSC 1/2 and P53 (28, 65, 72, 73). The activation of TSC 1/2 could inhibit the mammalian target of rapamycin (mTOR) signaling, thereby downregulating proliferation-related proteins such as RAF1 and TBC1D1, which in turn inhibits protein synthesis, translation, and cell cycle progression (61). Furthermore, metformin inhibits Rag GTPases and activates REDD1 (regulated in development and DNA damage responses 1), a negative regulator of mTOR, resulting in the blockage of mTOR signaling. These results indicate the inhibition of both protein synthesis and gluconeogenesis (61, 65, 74). At the same time, activation of AMPK by metformin puts the body into a state of starvation and catabolism, leading to the repression of protein synthesis and lipogenesis. This repression occurs through the inhibition of Acetyl CoA Carboxylase (ACC) and fatty acid synthetase (FASN), ultimately resulting in decreased viability of cancer cells, neovascularization, proliferation, invasion, and metastasis in both in vitro and in vivo studies (75–79).
2.1.1 Mitochondrial respiratory chain complexes
On the other hand, mitochondria have always been the best therapeutic target in cancer because mitochondrial activity and biogenesis are vital for tumor cell activity. Interestingly, studies have suggested that metformin can enter cancer cells via the organic cationic transporter (OCT) and directly affect mitochondria (66, 80). In this way, metformin inhibits complex 1 of the respiratory chain, alongside decreased protein subunit complex 1 and fifth components of complex 3. Subsequently, increased reactive oxygen species (ROS) resulting from respiratory chain disruption leads to DNA damage and apoptosis (61, 81, 82). The inhibitory effects of metformin on the mitochondrial respiratory chain led to decreased ATP production, causing an energetic stress state and activating AMPK and its subsequent hierarchy as mentioned in the previous section.
2.1.2 Cell cycle
Most authors believe that the antitumor effects of metformin are primarily mediated by a disruption in the cell cycle, which is caused by the activation of P53 induced by AMPK. Exposure to metformin further activated P53, leading to apoptosis and autophagy. It induced cell cycle arrest at the G0/G1 and G2/M phases by activating P21, P27, Bax, cyclin E, and decreasing cyclin D1 consequently downregulating cyclin-dependent kinases such as CDK1,CDK2,CDK4, and CDK6 in various cancer cell lines (83–86). Moreover, it has been suggested that a decrease in ATP production in mitochondria can lead to a pro-apoptotic (Bax)/anti-apoptotic (Bcl2) imbalance, followed by the entry of Bax into the mitochondria and the release of cytochrome C, which is a signal for cell death (87, 88).
2.1.3 MicroRNAs
The AMPK-dependent antitumor effects of metformin extend beyond this. Studies have shown that AMPK is a positive regulator of DICER, which in turn leads to alterations and modifications in various subpopulations of microRNAs. Both basic and clinical studies have shown the importance of inhibiting microRNA expression in cancer pathogenesis (89–92). DICER, a helicase with an RNase motif, can selectively activate the RNA-induced silencing complex (RISC), resulting in the inhibition of translation or fragmentation of the oncogenic target mRNA. This novel discusses the antitumor mechanism recently attributed to metformin in studies.
2.1.4 Inflammation
AMPK can also disrupt inflammatory pathways, thereby exerting metformin-dependent antitumor effects. In this way, by blocking the NFκB signaling, which is an important factor for tumor progression in inflammation-related cancers, and by decreasing TNF-α and some inflammatory cytokines such as IL-6, tumor progression can be alleviated (93–95). Furthermore, evidence has shown that metformin can stimulate CD8 cells and induce T-cell immunity, thereby attenuating cancer and inhibiting cancer occurrence in various cancer studies, respectively (96–98). Interestingly, metformin can suppress inflammation through NFκB signaling inhibition mediated by AMPK, or through AMPK-independent pathways as suggested in studies (65, 99, 100). This suppression can lead to a transition of the tumor-associated macrophage M2 phenotype to the M1 phenotype, causing macrophages and their secretions to attack the tumor itself rather than promoting its progression. Recent studies have mentioned these mechanisms, along with those related to the regulation of DICER, as potential anticancer stem cell (Anti-CSC) mechanisms (65).
2.2 Possible AMPK-independent mechanisms
Some other metformin anti-CSC mechanisms, which are completely unrelated to the AMPK signaling, involve the direct inhibition of self-renewal and metastatic pathways, including Sonic Hedgehog (Shh), Wnt/β-catenin, and TGFβ signaling. Moreover, metformin directly inhibited epithelial-to-mesenchymal transition (EMT), a dedifferentiation pathway that gives rise to a tendency for tumorigenesis and invasiveness (101–103). Another suggested mechanism is related to the unfolded protein response (UPR). When proteins are misfolded pathologically or unfolded, there are three receptors on the endoplasmic reticulum (ER) membrane that sense them and dislocate from the membrane, triggering a signal transduction for protein degradation and apoptosis. It is worth noting that metformin has been suggested as a downregulator of GRP78, a heat shock protein that regulates UPR. This can lead to an increase in unfolded protein levels in the ER lumen, causing ER stress and inducing cell death (61). It is interesting that even in this mechanism, some studies found a clue related to AMPK and attributed it to the mechanism. In this regard, metformin induces CHOP-related apoptosis in some cancer cell lines, and the regulation of certain microRNA expressions is also involved in this effect of metformin (61, 87, 104–106). Some discussions have focused on the JAK/STAT pathway, with studies indicating that STAT signaling is abnormally activated in most solid tumors and is associated with increased proliferation and invasion properties. Recent evidence has shown an AMPK-dependent effect of metformin that leads to limited STAT signaling in cancers (66). As we have reviewed so far, the antitumor effects of metformin are often directly or indirectly mediated by AMPK. However, some of the mentioned mechanisms appear to be free from any clues of the AMPK. For instance, previous studies have suggested that metformin may induce epigenetic modifications. Some authors have reported that metformin can inhibit members of the transcription factor family SP, especially SP1 (107, 108). This inhibition has been linked to the downregulation of several pro-oncogenic genes, including Bcl2, survivin, cyclin D1, VEGF, and FASN, in various in vitro and in vivo cancer studies. Furthermore, metformin-mediated SP1 degradation led to the downregulation of the oncogenic isoform of pyruvate kinase M2. Overall, inhibition and degradation of SP1 have been reported to be associated with the control of most SP1- and metabolic pathways-related cancers (61). Moreover, the inhibition of the tricarboxylic acid cycle, leading to the accumulation and forwarding of acetyl CoA for histone and non-histone acetylation, is reported as another epigenetic antitumor mechanism of metformin (109). On the other hand, metformin disrupts sphingolipid metabolism, thereby interfering with onco-promoter signaling and increasing ceramide production, a pro-apoptotic agent (110). It has also been suggested that metformin directly disrupts the integrity of the mitochondrial membrane and induces apoptosis in cancer cell lines by disrupting calcium flow (111). Recent and novel evidence has discussed the gut microbiota-related antitumor effects of metformin in both in vitro and in vivo studies. In these studies, metformin was found to modulate the gut microbiome, leading to decreased carcinogenesis and tumor progression, particularly in colorectal cancers (28, 112).
2.3 Focusing on GBM
To the best of our knowledge, the underlying antitumor mechanisms of metformin that have been mentioned so far are reported as the most frequent and important in various cancers, including breast (22), pancreas (78), ovary (113), lung (114), liver (77), esophagus (83), gastric (33), colorectal (115), thyroid (116), endometrial (85), prostate (106), melanoma (117), leukemia (118), and myeloma (86). However, there are still limited studies related to brain tumors. Some studies have shown similar mechanisms to those mentioned in previous sections also in cases of glioma and glioblastoma. For instance, Moretti et al. (119) evaluated the effects of metformin on two GBM cell lines that were stimulated with lipopolysaccharide (LPS) as an agonist for the TLR4 pathway. Metformin, when used alongside TMZ in both GBM cell lines, leads to the disruption of mitochondrial respiration, resulting in oxidative stress. In the mentioned study, decreased cell viability, increased apoptosis due to ER stress, and downregulation of BCL2 were observed after these treatments. Moreover, they have demonstrated through in silico analysis of TCGA-GBM-RNASeq that GPM-GBM cases with an activated TLR4 pathway may benefit from metformin treatment, but the simultaneous upregulation of CXCL8/IL8 may require a combination therapy with an IL8 inhibitor. Metformin combined with an antioxidant inhibitor, such as anti-SOD1, may be recommended for cases of mitochondrial-GBM. In another study, Song et al. (64) have shown that metformin inhibits the invasive properties of GBM cells by blocking the epithelial-to-mesenchymal transition-like process and consequently inhibiting the related TGF-β1 pathway. As a whole, these studies have illustrated that metformin plays an anti-cancer stem-like role in GBM cell lines via the AKT/mTOR/ZEB1 pathway. This provides evidence of metformin for further clinical investigation targeting GBM. As highlighted previously in this review, metformin can enter mitochondrial or other tissue and cell membranes, as well as cancer cells, via the OCT transporter. Furthermore, studies have shown that many transporters are identified for facilitating the entrance of metformin into cells based on the type of organs. For example, the carnitine/organic cation transporter 2 (OCTN2), plasma membrane monoamine transporter (PMAT), thiamine transporter 2, and serotonin transporter are attributed to intestinal cells through which metformin enters the cells and then shifts to the blood. On the other hand, multidrug and toxin extruder (MATE) 1 and 2 are responsible for metformin entering renal tubes and then shifting to urine (120). Although the blood-brain barrier (BBB) is tightly conserved, animal studies have shown that after a few hours of metformin administration, there is an equal concentration of metformin in brain tissue and plasma (121). This interesting fact indicates that metformin can somewhat penetrate the BBB. The exact mechanism is not clearly defined yet, but some studies suggest that four types of organic cationic transporters - OCT1, OCT2, OCT3, and PMAT, as well as plasma glycoprotein and MATE-1, are responsible for metformin penetration into the brain through the BBB (120, 122–124). Also, recent studies suggest that the CLIC1 protein, which is sensitive to metformin, accumulates in the transmembrane of GBM stem cells and can lose its function due to metformin, demonstrating the antitumoral action of metformin in the brain (125, 126). The exact effective concentration of metformin in the brain still remains to be well understood in the literature. However, some in vitro studies have reported antitumor effects of metformin with increasing concentrations in a dose-response manner in GBM cell lines (4, 127–130). One study showed that low metformin concentrations are related to cytostatic effects, while higher concentrations (about 10 mM) had cytotoxic effects on GBM-initiating cells (131). Another study demonstrated that metformin concentrations beyond 2.5 mM reached a plateau in terms of anti-GBM activity (127). Moreover, in terms of the response of GBM to metformin, the following theme could be worth noting. O6-methylguanine (O6-MeG)-DNA methyltransferase (MGMT) is considered a clinical biomarker in GBM based on numerous previous studies (132). The primary reason for this is the frequent occurrence of drug resistance issues in these patients. The key mechanism of the cytotoxic efficacy of TMZ, the first-line chemotherapy in GBM, is the alkylation of DNA, which subsequently hinders replication. In this process, a methyl group is added to the O6 N7 position of guanine and the N3 position of adenine. This methylation leads to mispairing of bases and breaks in DNA strands. MGMT, through the transfer of the methyl group, disrupts this process and hinders the anti-GBM effect of TMZ (132–134). Studies have indicated that MGMT promoter status is a crucial factor in determining TMZ sensitivity (132, 135). A methylated promoter results in MGMT gene silencing, thereby enhancing TMZ efficacy (132). However, evidence shows that 30-60% of patients with a methylated MGMT (mMGMT) promoter still exhibit TMZ resistance due to MGMT expression (132). Interestingly, one study suggested that combining metformin with TMZ and/or radiotherapy enhances the efficacy of these treatments, even in resistant GBM cells with unmethylated MGMT promoters (133). On the other side, studies have discussed that metformin alone or in combination with TMZ can significantly suppress the induction of MGMT proteins in a dose-response manner (134). Furthermore, some studies have suggested that metformin can sensitize TMZ-resistant GBM cells through the suppression of MGMT expression and more detailed mechanisms. Therefore, it seems that metformin could be a suitable adjuvant therapy in the current situation where there is no substitute for TMZ. In practice, scientific associations still believe in the standard of care based on TMZ even in resistant GBM cells with unmethylated MGMT promoter due to the indefinite predictive value of MGMT promoter status. However, a recent cohort study belonging to the JAMA network (136) has shown that glioma patients with mMGMT promoter had experienced longer survival than those with unmethylated MGMT promoter in terms of both progression-free survival and overall survival. Interestingly, some studies have shown that metformin use had a beneficial effect on survival in GBM patients with mMGMT promoter (137). Altogether, it seems that there is a need for more rigorous trials to evaluate these effects, considering pivotal factors such as MGMT promoter status and excluding patients with diabetes.
3 The effects of metformin on cellular insulin function in healthy cells compared to GBM cells
It is evident that after insulin interacts with its receptor and undergoes autophosphorylation of the insulin receptor substrate (IRS) cytoplasmic domain, it triggers a crucial signal transduction pathway mediated by signaling molecules such as Shc, Grb2, SoS, Ras, Raf-1, MEK. This ultimately activates ERK1/2, leading to cell proliferation and growth. Moreover, insulin activates PI3K and Akt, leading to enhanced cell glucose uptake, as well as promoting glycogen, lipid, and protein synthesis (138–141). In healthy cells, particularly in active metabolic organs, metformin, a well-known insulin sensitizer drug, enhances the effects of insulin on growth by accelerating the IRS signaling, leading to increased glucose uptake and reduced systemic circulating glucose and insulin. This, in turn, mediates the definition of metformin’s antitumor efficacy, due to the well-established tumorigenesis and tumor growth effects of high glucose levels and hyperinsulinemia in various cancer studies (142–144). Interestingly, evidence from studies on cancers and GBM cell lines discusses the fact that metformin could disrupt the insulin signaling pathway in cancerous cells by downregulating key signaling molecules in this pathway, such as PI3K, Akt, and ERK1/2. Therefore, this suggests that the impact of metformin as an insulin sensitizer may be reversed in cancer cell microenvironments, inhibiting the growth-promoting efficacy of insulin in this context (26, 27, 131, 145–148). One of the astonishing challenges in biology is the recognition that the effect of metformin varies greatly depending on the context or type of cell. A study illustrating potential molecular targets in glioblastoma has reported that the PI3K/AKT/mTOR pathway is the most significant target compared to other culprits even mutated P53 so that this pathway is overexpressed in 90% of GBM cases (27). Therefore, it seems that metformin has potential therapeutic effects on GBM by disrupting this pathway. On the other hand, there is an interesting controversy in some studies claiming that ERK1/2, a key factor in cell proliferation and growth activated by the insulin signaling cascade, could lead to cell death. These studies have indicated that ERK1/2 mediates cell death based on stimuli and activated cell types. The intensity, duration, and balance between pro-versus anti-apoptotic signals determine whether a cell duplicates or undergoes an apoptotic state (149–152). Although the exact mechanisms remain to be understood, some studies have suggested ERK-associated DNA damage (151) or IFNγ-induced cell death (153). Furthermore, the involvement of ERK1/2 in inhibiting survival signaling and Fas-mediated cell death are other proposed mechanisms in this context (150). This evidence suggests that Akt is downstream of ERK1/2 activation in the cytosol, so it does not phosphorylate ERK1/2. Instead, it phosphorylates and stabilizes PEA-15, acting as a confiscator of ERK1/2 in the cytosol. As a result, the translocation of ERK to the nuclei does not occur, and ELK-1-associated transcription is not activated (150, 152). Therefore, it seems that PI3K inhibitors, such as metformin in the context of cancer, that block Akt phosphorylation and activation, may conversely lead to the restoration of ERK1/2 translocation to the nucleus and cell growth effects. So it is noteworthy that further future studies examine the antitumor efficacy of metformin from the perspective of its effect on ERK1/2 and its translocation to the nucleus. In their valuable reports from both the orthotopic GBM mouse model and patient samples, Noch et al. (154) focused on the issue of PI3K inhibitors for GBM treatment. They highlighted that these inhibitors have failed due to their potential to elevate blood glucose, induce insulin receptor hyperactivity, trigger insulin feedback, and cause hyperinsulinemia. In this study, it is interesting to note that metformin could enhance the effectiveness of PI3K inhibition in the GBM microenvironment, while also reducing systemic glucose and insulin levels. Furthermore, PI3K inhibition led to increased T-cell and microglia presence in GBM samples from patients (154). Numerous other studies have even mentioned metformin as an independent and efficient inhibitor of the tumor niche-associated PI3K in various cancers, especially GBM. For example, Al Hassan et al. (127) demonstrated that metformin inhibits AKT, a key molecule in the PI3K signaling pathway. By blocking this pathway, metformin exerts its anticancer, anti-invasive, and anti-migratory effects on GBM cell lines. The study highlighted the significance of the PI3K/Akt pathway in glioblastoma by demonstrating reduced motility and EGF stimulating properties in GBM cells treated with wortmannin, a PI3K inhibitor. These observations were correlated with the effects observed after the use of metformin in their study. Another study by Lo Dico et al. (155) reported that they treated both GBM cell lines, including those susceptible and resistant to TMZ with metformin in combination with TMZ. During hypoxia, metformin in combination with TMZ, could decrease cell viability. This phenomenon was partly associated with the inhibition of the PI3K/mTOR axis. However, this effect on TMZ-resistant GBM cell line is facilitated by adding BEZ235, a PI3K/mTOR inhibitor. This study concluded that TMZ + metformin could reverse the resistance of GBM cells to treatment, and this effect was potentiated by a disruption in the PI3K/mTOR axis. Furthermore, Würth et al. (131) have also demonstrated that different doses of metformin treatment (4.9 - 9.4 mM) administered to four types of GBM cell lines for 48 hours resulted in decreased Akt phosphorylation, cell survival, and proliferation.
4 Dietary patterns that improve cellular insulin function and their effect on GBM
As frequently mentioned, metformin improves cellular insulin function in metabolic organs, leading to enhanced cellular glucose uptake and decreased circulating glucose and insulin. Furthermore, metformin activates AMPK, shifting the body into a calorie-restricted state. The well-known antitumor efficacy of metformin appears to mimic dietary patterns such as intermittent fasting, hypocaloric and calorie restriction, as well as exercise, both reasonably and mechanistically (156, 157). In this regard, some preclinical and clinical studies have shown promising results so far (158, 159). However, clinical studies examining the impact of intermittent fasting, hypocaloric diets, and calorie restriction on tumor characteristics and clinical outcomes in GBM patients have not yet produced sufficient evidence. Most studies have examined the feasibility of these treatment plans in GBM (18, 160, 161), with the majority of the literature focusing on animal studies in this area (162–164). Safdie et al. (165) demonstrated that fasting for 48 hours before radiotherapy or chemotherapy could sensitize GBM cell lines from mice, rats, and humans to the therapy. Furthermore, in live organisms, this fasting regimen resulted in improved survival and a significant decrease in circulating glucose and IGF1 levels. Another study by Duffy et al. (166) interestingly demonstrated that fasting selectively enhanced the cytotoxicity of TMZ in human GBM cell lines, while having no effect on normal astroglial cells. Moreover, Schreck et al. (160) investigated a fasting schedule for astrocytoma patients with grade II to IV. In this regimen, patients followed the glioma Atkins diet and fasted for 2 days during an 8-week period. The schedule was accepted by 48% of the participants, with reasonable safety and tolerability. This regimen resulted in increased levels of β-hydroxybutyrate and acetone in the brain, and decreased levels of HbA1C and insulin. It is noteworthy that altered carbohydrate metabolism is a common and important trait of cancer, well known as the “Warburg effect.” Therefore, cancer cells preferentially utilize anaerobic glycolysis, which reduces ATP production and causes cancer cells to require excessive glucose to survive. Fasting leads to decreased systemic glucose and anabolic hormones, as well as increased ketone bodies such as β-hydroxybutyrate, which results in the regression of tumors. It seems that various types of fasting are more effective, while traditionally any type of ketogenic diet has been used for this purpose. In addition to glucose, glutamine is also considered to be an important fuel for GBM cells, powering their growth. Mukherjee et al. (162) utilized a glutamine antagonist (6-diazo-5-oxo-L-norleucine) in combination with a calorie-restricted ketogenic diet as a diet/drug strategy. They demonstrated the ability to kill GBM cells, reverse symptoms, reduce edema, hemorrhage, inflammation, and enhance survival in mice with late-stage GBM. Most of the literature focuses on ketogenic metabolic therapy as an adjuvant treatment in GBM. However, its efficacy remains to be determined due to the limited nature of these studies, with most being case reports (167, 168) or animal/in vitro studies. However, some studies have shown that the ketogenic diet, by increasing fatty acids and ketone bodies, can lead to aggressive tumor growth through increased utilization of fats and ketones in GBM cells, and reduced survival in mouse models (20). Therefore, applying a ketogenic diet in GBM from the cellular level to clinical practice remains very challenging, and further studies are necessary for the future.
5 Metformin, senescence, and aging
According to the literature, various types of stress on the body, such as oxidative, psychological, or genotoxic, DNA damage, epigenetic changes, metabolic function disorders, oncogene overexpression, genetic mutations, and mitochondrial disorders, ultimately resulting in the senescence state. Senescence is a state in which cells irreversibly enter a cell cycle arrest state. A senescent cell undergoes increased volume, secretes inflammatory factors, and contributes to the creation of the SASP. Studies have shown that the accumulation of senescent cells, which commonly occurs with aging, is associated with inflammation and chronic age-related diseases (53, 169, 170). Recent studies suggest that metformin can induce anti-aging transcriptional changes (29, 158, 171). However, it is still debatable whether this effect of metformin is also true for healthy individuals, and the exact mechanisms are unknown. The potential benefit of metformin lies in reducing mortality associated with the disease by lowering glucose levels. Also, studies have shown that metformin can reverse telomere attrition, which is the most prominent characteristic of senescent cells (29, 171–174). Moreover, some top mentioned mechanisms in literature, which have been attributed to the anti-aging efficacy of metformin, consist of intervening in nutrient sensing, DNA damage, the accumulation of ROS, telomere attrition, inflammation, cellular senescence, stem cell depletion, and autophagy. These mechanisms can then be attributed to prevent aging-related diseases (171).
6 Metformin, senescence, and cancer, especially GBM
As described in the previous section, one of the important anti-aging properties of metformin is its anti-senescence effect. However, when focusing on cancer studies, it is evident that metformin can exert anticancer properties through various mechanisms and traits, such as inducing cell senescence, which is one of the most important mechanisms (36, 175, 176). As a result, there is a paradoxical interpretation of the effect of metformin on cell senescence. It seems that this is related to the paradigm in which the effects of metformin could be highly tissue-specific and context-dependent. In this study, we discuss the context-dependent effects of metformin for the second time. On the other hand, metformin-induced cell senescence in cancer cells, which is considered a beneficial effect of this drug on tumor initiation, may have a detrimental impact on tumor progression. This is due to the development of SASP phenotype and subsequent immunosenescence (29, 177, 178). Studying in this field is fascinating, and the exact interrelated concept is still not entirely clear. Taken together, the prevailing observation is that most diseases, particularly cancers, are age-related and tend to occur with aging. Therefore, metformin, through its established effects on delaying aging, primarily by interfering with SASP, can reduce the incidence of age-related cancers (37, 179, 180). According to available data, a plausible explanation for the bidirectional effects of metformin on senescence could be that metformin decreases the cell senescence threshold, leading to an accelerated initiation of cell senescence in response to oncogenes (36, 176, 179). Metformin induces SASP during the early stages of tumorigenesis to restore immune surveillance, promoting immune-mediated clearance of senescent cells that may act as potential premalignant agents, as well as existing malignant lesions, and to impede the evasion of cancer cells from antitumor defenses (176). Therefore, it seems that the net effect of metformin should be a significant reduction in dysfunction and malignant cells. Some studies suggest that metformin acts as a radiosensitizer in cancer therapy, enhancing radiation-induced senescence and increasing the effectiveness of radiotherapy (181–183). However, from another perspective, studies on cancer have shown that metformin exhibits anti-SASP activity (4, 29, 184, 185). It has been reported that senescent cells have the ability to undergo genomic reprogramming, allowing them to re-enter the cell cycle and regain their stemness phenotype. Furthermore, senescent cells with aggressive SASP activity could induce immunosenescence over time, leading to immune dysfunction, particularly in relation to antitumor immune responses (186). Additionally, there is evidence indicating that the accumulation of senescent cells can lead to mTOR activation and create a cancerophil microenvironment (187, 188). Based on the literature, although metformin is not an immunosuppressive agent, it appears that in a proinflammatory context such as senescence, it could inhibit the NFKB pathway, inflammation, and most cytokines related to SASP (37). Therefore, metformin has been identified as a senostatic agent due to its anti-SASP activity, which can inhibit tumor progression. Some studies have shown that the remnant of senescent cells after chemotherapy and radiotherapy is associated with cancer relapse. Metformin, as an adjuvant and effective senotherapy drug, has been found to decrease the presence of these cells (189). This belief still remains that the bidirectional effects of metformin mentioned above warrant further preclinical and clinical studies in the future. So far, the effects of metformin have been discussed in various types of cancer, including breast (94), prostate (109), ovary (95), pancreas (78), and hepatocarcinoma (25). However, research on brain cancers is relatively scarce. After conducting a comprehensive search on PubMed based on following terms and search strategy: GBM OR glioblastoma OR “glioblastoma multiform” AND senescence OR SASP OR senesc* AND metformin OR glucophage, just was funded 2 articles. Then, when (head OR neck OR cerebral OR cranial OR brain AND tumor OR carcin* OR malignan* OR neoplasm*) were substituted for GBM, the search yielded 9 results. Among them, an important recent study worth mentioning is the work of Fuentes-Fayos et al. (4) The results of their study showed that treatment with metformin and simvastatin alone, and with a stronger effect in combination, could induce a senescent state and increase the number of senescent cells. These observations were confirmed via β-galactosidase assay, along with effective alterations in key genes related to SASP in GBM cell lines. Moreover, according to their report, this treatment resulted in telomere lengthening, which is an outcome of the senescence state, as well as phosphorylation of ERK1/ERK2 and P53, and inhibition of some oncogenic factors. This led to the blocking of proliferation capacity in GBM cell lines and a decrease in aggressive features in the most aggressive form of cancer (4). However, some other studies on head and neck squamous cell carcinoma (HNSCC) have reported that an early senescence state, numerous senescent cells, and consequently SASP lead to resistance against radio (chemo) therapy. Metformin, with a decrease in senescence state, could alleviate this resistance (185). Similarly, Skinner et al. (183) showed that metformin was a radiosensitizer in HNSCC with a disruptive P53. On the other hand, Woo et al. (190) reported that metformin inhibits malic enzyme 2, leading to increased senescence mediated by ROS in HNSCC. Curry et al. (191) demonstrated that administering a diabetic dose of metformin (2000 mg/day) for 9 days or more before surgery could increase senescence in cell lines derived from HNSCC patients. Moreover, Hu et al. (185) demonstrated that metformin trough induces cell cycle arrest in vitro and in vivo, alongside modulation of SASP via inhibition of mTOR and Stat3 pathways, and has antitumor activity in HNSCC. Taken together, it is concluded that metformin should be used as a senescence inducer along with anti-SASP activity, which together account for an important part of metformin’s antitumor efficacy and may be used as a promising therapy in these patients.
7 Are the antitumor effects of metformin more attributed to apoptosis or senescence?
An intriguing recent study of high quality discusses how senescent cells contribute to the aggressiveness of malignant cells in mouse and human glioblastoma (192). Senescent cells accounted for approximately 7% of tumors, and their removal was associated with improving the tumor microenvironment in the study mentioned (192). This result supports our interpretation as outlined in the previous sections. According to the literature, senescence may be a beneficial phenomenon only in the initial stage of GBM, but in the progression phase, it could actually be detrimental (4, 53, 189). Therefore, it seems that apoptosis or autophagy play a fundamental role in eventually clearing senescent cells and have a greater impact on antitumor efficacy than senescence. In line with previous findings, Guarnaccia et al. (128) have demonstrated that metformin interferes with sphingolipid metabolism, leading to the production of pro-apoptotic ceramide and ultimately facilitating apoptosis in GBM cells. Furthermore, Xiong et al. (47) concluded that metformin could impact the rise in caspase 3 activity in human GBM cells. Sesen et al. (133) demonstrated that metformin could induce apoptosis by increasing the expression of the pro-apoptotic protein Bax and decreasing the expression of the anti-apoptotic protein Bcl2 in GBM cells. Furthermore, some studies have reported that metformin enhances the sensitivity of GBM cells to Temozolomide, resulting in increased apoptosis due to DNA damage (193, 194). Other studies have also reported that metformin induces apoptosis or autophagy in animal and human GBM (49, 121, 128, 133). Hence, it appears that the antitumor effects of metformin in GBM cells can be attributed to apoptosis rather than senescence.
8 What influential factor determines the cellular decision between apoptosis and senescence?
The cellular decision between apoptosis and senescence is influenced by complex and diverse factors, including the type and intensity of the cancer cell’s DNA damage, the tumor microenvironment, the presence of immune cells, and the overall physiological status (195–198). Typically, studies have indicated that apoptosis occurs after more extend cellular stress than senescence. In this regard, some studies have demonstrated that the dosage of anticancer drugs is an important factor. Song et al. (199) reported that lower doses of doxorubicin induce senescence, while higher doses lead to apoptosis in breast cancer cells. Yi et al. (35) reported the same result regarding metformin in hepatoma cells. Moreover, cell type is a crucial determinant, as illustrated by the study of Curry et al. (191), which showed that metformin induces senescence in stromal cells whereas exert apoptosis in carcinoma cells in head and neck cancer samples. On the other hand, some cancer studies have highlighted the role of PTEN as a key determinant in the decision-making process of cancer cells. PTEN-deficient cells enter a state of senescence, while cells with adequate PTEN levels undergo apoptosis (196, 200, 201). Some interesting results suggest a potential role for P53 in radiation-induced senescence, as one study has shown a correlation between the two. The results of this study show that breast cancer cells with a missense mutation in the DNA-binding domain of P53 are susceptible to apoptosis rather than senescence (202), while the opposite was observed in GBM cells (203). This missense mutation resulted in escape from radiation in GBM cells. In line with this, some mechanistic studies, which identified P53 as wild type, have shown that adjuvant anticancer therapies such as metformin dramatically induce SASP. This, in turn, leads to inappropriate activation of AKT/ERK-mTORC1-4EBP1-MCL1/SURVIVIN in P53-deficient cancer samples with wild type (204). However, this result has not been supported in P53 deficient cancer cells without wild type. In GBM, temozolomide, which is the first-line therapy, has interestingly been shown to induce senescence more than apoptosis. This, in turn, has been mentioned as the cause of resistance to the therapy and tumor relapse (205–207). Taken together, it seems that more future studies should evaluate the effect of metformin as a senotherapy on GBM in vitro and in vivo, taking into consideration the type of P53.
9 Conclusion
In the current review, we carefully evaluated the exact mechanisms that account for the therapeutic modalities of metformin in cancer settings, with specific emphasis on GBM. Our findings encourage scientists to conduct more well-designed studies that take into account crucial factors concerning metformin and GBM research, including MGMT promoter status and GBM patients without diabetes. Several studies have illustrated that metformin could induce senescence in GBM cells. However, most other studies have stated that metformin decreases some key component of SASP and plays a role in alleviating GBM. In the current review, we conclude that metformin has anti-tumor efficacy as a senescence inducer in the initiation phase of cancer. It is worth noting that senescence, along with SASP production, is not a beneficial phenomenon in the progression phase of cancers. Metformin has been shown to act as an anti-senescent agent during this stage, as indicated by related studies. Actually, metformin has anti-SASP activity, which is a common factor explaining its anti-aging and anti-cancer effects. Moreover, the effects of metformin are highly context-dependent. As a good example of this, metformin leads to increased insulin sensitivity in active metabolic organs, while it inhibits insulin signaling in the GBM microenvironment. This effect has also been verified in other types of cancer. On the other hand, cells in which mTOR signaling is more active undergo a senescent state, while those with inhibited signaling enter a quiescent state that eventually results in apoptosis. Senescent cells, however, are resistant to apoptosis. Studies have indicated that the primary anti-tumor efficacy of metformin is associated with apoptosis rather than senescence. The key determinants for cancer cells to choose between apoptosis and senescence include cell types, PTEN, P53 type and its mutations, and the mTOR activity of cells. Finally, although the vast mechanisms have been discussed and metformin effects have often been reported as effective, some clinical trials have shown that metformin has no effect on clinical outcomes such as survival and tumor recurrence. Therefore, it seems that there is still a need for studies with a more meticulous perspective on related mechanisms to clarify existing controversies, especially in cases of GBM. In the next step, it is recommended that more extensive and rigorous clinical trials be conducted to either approve or reject the clinical efficacy of metformin as a senotherapeutic agent in the GBM treatment protocol.
Author contributions
MH-K: Conceptualization, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. AZ: Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. MS: Methodology, Supervision, Writing – original draft, Writing – review & editing. AH: Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro-oncology. (2020) 22:iv1–iv96. doi: 10.1093/neuonc/noaa200
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-oncology. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
3. Philips A, Henshaw DL, Lamburn G, O’Carroll MJ. Brain tumours: rise in glioblastoma multiforme incidence in England 1995–2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health. (2018) 2018:1–10. doi: 10.1155/2018/7910754
4. Fuentes-Fayos AC, G-García ME, Pérez-Gómez JM, Montero-Hidalgo AJ, Martín-Colom J, Doval-Rosa C, et al. Metformin and simvastatin exert additive antitumour effects in glioblastoma via senescence-state: clinical and translational evidence. EBioMedicine. (2023) 90:1–21. doi: 10.1016/j.ebiom.2023.104484
5. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. (2021) 18:170–86. doi: 10.1038/s41571-020-00447-z
6. Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-oncology. (2020) 22:1073–113. doi: 10.1093/neuonc/noaa106
7. Simon J, Toubiana T, Lang P, Taillibert S, Mazeron J. Radiotherapy for glioblastomas: from radiobiology to concomitant chemotherapy. Cancer Radiotherapie: J la Societe Francaise Radiotherapie Oncologique. (2005) 9:322–31. doi: 10.1016/j.canrad.2005.07.001
8. Weller M, Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev. (2020) 87:102029. doi: 10.1016/j.ctrv.2020.102029
9. Kotecha R, Odia Y, Khosla AA, Ahluwalia MS. Key clinical principles in the management of glioblastoma. JCO Oncol practice. (2023) 19:180–9. doi: 10.1200/OP.22.00476
10. Xiao Z-Z, Wang Z-F, Lan T, Huang W-H, Zhao Y-H, Ma C, et al. Carmustine as a supplementary therapeutic option for glioblastoma: a systematic review and meta-analysis. Front Neurology. (2020) 11:1036. doi: 10.3389/fneur.2020.01036
11. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA: Cancer J Clin. (2020) 70:299–312. doi: 10.3322/caac.21613
12. Mulpur BH, Nabors LB, Thompson RC, Olson JJ, LaRocca RV, Thompson Z, et al. Complementary therapy and survival in glioblastoma. Neuro-Oncology Practice. (2015) 2:122–6. doi: 10.1093/nop/npv008
13. Tagde P, Tagde P, Tagde S, Bhattacharya T, Garg V, Akter R, et al. Natural bioactive molecules: An alternative approach to the treatment and control of glioblastoma multiforme. Biomedicine Pharmacotherapy. (2021) 141:111928. doi: 10.1016/j.biopha.2021.111928
14. Shakya S, Gromovsky AD, Hale JS, Knudsen AM, Prager B, Wallace LC, et al. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta neuropathologica Commun. (2021) 9:1–18. doi: 10.1186/s40478-021-01205-7
15. Guyon J, Fernandez-Moncada I, Larrieu CM, Bouchez CL, Pagano Zottola AC, Galvis J, et al. Lactate dehydrogenases promote glioblastoma growth and invasion via a metabolic symbiosis. EMBO Mol Med. (2022) 14:e15343. doi: 10.15252/emmm.202115343
16. Kwak S, Park S-H, Kim S-H, Sung G-J, Song J-H, Jeong J-H, et al. miR-3189-targeted GLUT3 repression by HDAC2 knockdown inhibits glioblastoma tumorigenesis through regulating glucose metabolism and proliferation. J Exp Clin Cancer Res. (2022) 41:87. doi: 10.1186/s13046-022-02305-5
17. Liu R, Liang W, Hua Q, Wu L, Wang X, Li Q, et al. Fatty acid metabolic signaling pathway alternation predict prognosis of immune checkpoint inhibitors in glioblastoma. Front Immunol. (2022) 13:819515. doi: 10.3389/fimmu.2022.819515
18. Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ, et al. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J neuro-oncology. (2014) 117:125–31. doi: 10.1007/s11060-014-1362-0
19. Sargaço B, Oliveira PA, Antunes ML, Moreira AC. Effects of the ketogenic diet in the treatment of gliomas: a systematic review. Nutrients. (2022) 14:1007. doi: 10.3390/nu14051007
20. Sperry J, Condro MC, Guo L, Braas D, Vanderveer-Harris N, Kim KK, et al. Glioblastoma utilizes fatty acids and ketone bodies for growth allowing progression during ketogenic diet therapy. Iscience. (2020) 23(9):1–17. doi: 10.1016/j.isci.2020.101453
21. Klement RJ, Sweeney RA, Gross EC, Champ CE. Problems associated with a highly artificial ketogenic diet: Letter to the Editor Re: van der Louw EJTM, Olieman JF, van den Bemt PMLA, et al.‘Ketogenic diet treatment as adjuvant to standard treatment of glioblastoma multiforme: a feasibility and safety study’. Ther Adv Med Oncol. (2019) 11:1758835919879268. doi: 10.1177/1758835919879268
22. Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PloS One. (2014) 9:e98207. doi: 10.1371/journal.pone.0098207
23. Li B, Zhou P, Xu K, Chen T, Jiao J, Wei H, et al. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int J Biol Sci. (2020) 16:74. doi: 10.7150/ijbs.33787
24. De Santi M, Baldelli G, Diotallevi A, Galluzzi L, Schiavano GF, Brandi G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Sci Rep. (2019) 9:66. doi: 10.1038/s41598-018-37247-6
25. Gao C, Fang L, Zhang H, Zhang W-S, Li X-O, Du S-Y. Metformin induces autophagy via the AMPK-mTOR signaling pathway in human hepatocellular carcinoma cells. Cancer Manage Res. (2020) 12:5803–11. doi: 10.2147/CMAR.S257966
26. Barzegar Behrooz A, Talaie Z, Jusheghani F, Łos MJ, Klonisch T, Ghavami S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int J Mol Sci. (2022) 23:1353. doi: 10.3390/ijms23031353
27. Khabibov M, Garifullin A, Boumber Y, Khaddour K, Fernandez M, Khamitov F, et al. Signaling pathways and therapeutic approaches in glioblastoma multiforme. Int J Oncol. (2022) 60:1–18. doi: 10.3892/ijo
28. Hua Y, Zheng Y, Yao Y, Jia R, Ge S, Zhuang A. Metformin and cancer hallmarks: shedding new lights on therapeutic repurposing. J Trans Med. (2023) 21:403. doi: 10.1186/s12967-023-04263-8
29. Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
30. Huo M, Zhang J, Huang W, Wang Y. Interplay among metabolism, epigenetic modifications, and gene expression in cancer. Front Cell Dev Biol. (2021) 9:793428. doi: 10.3389/fcell.2021.793428
31. Cuyàs E, Fernández-Arroyo S, Verdura S, García RÁ-F, Stursa J, Werner L, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. (2018) 37:963–70. doi: 10.1038/onc.2017.367
32. Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, et al. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PloS Biol. (2015) 13:e1002309. doi: 10.1371/journal.pbio.1002309
33. Tseng H-H, Chen Y-Z, Chou N-H, Chen Y-C, Wu C-C, Liu L-F, et al. Metformin inhibits gastric cancer cell proliferation by regulation of a novel Loc100506691-CHAC1 axis. Mol Therapy-Oncolytics. (2021) 22:180–94. doi: 10.1016/j.omto.2021.08.006
34. Yan L, Zhou J, Gao Y, Ghazal S, Lu L, Bellone S, et al. Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene. (2015) 34:3076–84. doi: 10.1038/onc.2014.236
35. Yi G, He Z, Zhou X, Xian L, Yuan T, Jia X, et al. Low concentration of metformin induces a p53-dependent senescence in hepatoma cells via activation of the AMPK pathway. Int J Oncol. (2013) 43:1503–10. doi: 10.3892/ijo.2013.2077
36. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Quirantes R, Segura-Carretero A, Micol V, et al. Metformin lowers the threshold for stress-induced senescence: a role for the microRNA-200 family and miR-205. Cell Cycle. (2012) 11:1235–46. doi: 10.4161/cc.11.6.19665
37. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κ B activation. Aging Cell. (2013) 12:489–98. doi: 10.1111/acel.12075
38. Yang J, Liu H-C, Zhang J-Q, Zou J-Y, Zhang X, Chen W-M, et al. The effect of metformin on senescence of T lymphocytes. Immun Ageing. (2023) 20:73. doi: 10.1186/s12979-023-00394-0
39. Kim JH, Lee KJ, Seo Y, Kwon J-H, Yoon JP, Kang JY, et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci Rep. (2018) 8:409. doi: 10.1038/s41598-017-18762-4
40. Ma R, Yi B, Riker AI, Xi Y. Metformin and cancer immunity. Acta Pharmacologica Sinica. (2020) 41:1403–9. doi: 10.1038/s41401-020-00508-0
41. Zhao L-Y, Mei J-X, Yu G, Lei L, Zhang W-H, Liu K, et al. Role of the gut microbiota in anticancer therapy: from molecular mechanisms to clinical applications. Signal Transduction Targeted Ther. (2023) 8:201. doi: 10.1038/s41392-023-01406-7
42. Park D-B. Metformin promotes apoptosis but suppresses autophagy in glucose-deprived H4IIE hepatocellular carcinoma cells. Diabetes Metab J. (2015) 39:518–27. doi: 10.4093/dmj.2015.39.6.518
43. Li P, Zhao M, Parris AB, Feng X, Yang X. p53 is required for metformin-induced growth inhibition, senescence and apoptosis in breast cancer cells. Biochem Biophys Res Commun. (2015) 464:1267–74. doi: 10.1016/j.bbrc.2015.07.117
44. Lee JE, Lim JH, Hong YK, Yang SH. High-dose metformin plus temozolomide shows increased anti-tumor effects in glioblastoma in vitro and in vivo compared with monotherapy. Cancer Res treatment: Off J Korean Cancer Assoc. (2018) 50:1331–42. doi: 10.4143/crt.2017.466
45. Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, et al. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci. (2007) 64:1290–302. doi: 10.1007/s00018-007-7080-4
46. Songthaveesin C, Sa-Nongdej W, Limboonreung T, Chongthammakun S. Combination of metformin and 9-cis retinoic acid increases apoptosis in C6 glioma stem-like cells. Heliyon. (2018) 4(5):1–16. doi: 10.1016/j.heliyon.2018.e00638
47. Xiong ZS, Gong SF, Si W, Jiang T, Li QL, Wang TJ, et al. Effect of metformin on cell proliferation, apoptosis, migration and invasion in A172 glioma cells and its mechanisms. Mol Med Rep. (2019) 20:887–94. doi: 10.3892/mmr
48. Wang Y, Meng Y, Zhang S, Wu H, Yang D, Nie C, et al. Phenformin and metformin inhibit growth and migration of LN229 glioma cells in vitro and in vivo. OncoTargets Ther. (2018) 11:6039–48. doi: 10.2147/OTT
49. Ding W, Liao L, Liu J, Zhao J, Tang Q, Liao Y. Lower dose of metformin combined with artesunate induced autophagy-dependent apoptosis of glioblastoma by activating ROS-AMPK-mTOR axis. Exp Cell Res. (2023) 430:113691. doi: 10.1016/j.yexcr.2023.113691
50. Sanati M, Aminyavari S, Mollazadeh H, Motamed-Sanaye A, Bibak B, Mohtashami E, et al. The potential therapeutic impact of metformin in glioblastoma multiforme. Curr Medicinal Chem. (2023) 30:857–77. doi: 10.2174/0929867329666220707103525
51. Langhi Prata LG, Tchkonia T, Kirkland JL. Cell senescence, the senescence-associated secretory phenotype, and cancers. PloS Biol. (2023) 21:e3002326. doi: 10.1371/journal.pbio.3002326
52. Galluzzi L, Myint M. Cell death and senescence. J Trans Med. (2023) 21:425. doi: 10.1186/s12967-023-04297-y
53. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrology. (2022) 18:611–27. doi: 10.1038/s41581-022-00601-z
54. Raffetto JD, Leverkus M, Park H-Y, Menzoian JO. Synopsis on cellular senescence and apoptosis. J Vasc surgery. (2001) 34:173–7. doi: 10.1067/mva.2001.115964
55. Glossmann HH, Lutz O. Metformin and aging: a review. Gerontology. (2019) 65:581–90. doi: 10.1159/000502257
56. Soukas AA, Hao H, Wu L. Metformin as anti-aging therapy: is it for everyone? Trends Endocrinol Metab. (2019) 30:745–55. doi: 10.1016/j.tem.2019.07.015
57. Vigneri R, Sciacca L, Vigneri P. Rethinking the relationship between insulin and cancer. Trends Endocrinol Metab. (2020) 31:551–60. doi: 10.1016/j.tem.2020.05.004
58. Chiefari E, Mirabelli M, La Vignera S, Tanyolaç S, Foti DP, Aversa A, et al. Insulin resistance and cancer: In search for a causal link. Int J Mol Sci. (2021) 22:11137. doi: 10.3390/ijms222011137
59. Poloz Y, Stambolic V. Obesity and cancer, a case for insulin signaling. Cell Death disease. (2015) 6:e2037–e. doi: 10.1038/cddis.2015.381
60. Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood J Am Soc Hematology. (2013) 121:4036–45. doi: 10.1182/blood-2012-09-456897
61. Sacco F, Calderone A, Castagnoli L, Cesareni G. The cell-autonomous mechanisms underlying the activity of metformin as an anticancer drug. Br J Cancer. (2016) 115:1451–6. doi: 10.1038/bjc.2016.385
62. Chan AT. Metformin for cancer prevention: a reason for optimism. Lancet Oncol. (2016) 17:407–9. doi: 10.1016/S1470-2045(16)00006-1
63. Chang S-H, Luo S, O'Brian KK, Thomas TS, Colditz GA, Carlsson NP, et al. Association between metformin use and progression of monoclonal gammopathy of undetermined significance to multiple myeloma in US veterans with diabetes mellitus: a population-based retrospective cohort study. Lancet Haematology. (2015) 2:e30–e6. doi: 10.1016/S2352-3026(14)00037-4
64. Song Y, Chen Y, Li Y, Lyu X, Cui J, Cheng Y, et al. Metformin inhibits TGF-β1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget. (2018) 9:7023. doi: 10.18632/oncotarget.v9i6
65. Saini N, Yang X. Metformin as an anti-cancer agent: actions and mechanisms targeting cancer stem cells. Acta Biochim Biophys Sinica. (2018) 50:133–43. doi: 10.1093/abbs/gmx106
66. Zhao B, Luo J, Yu T, Zhou L, Lv H, Shang P. Anticancer mechanisms of metformin: A review of the current evidence. Life Sci. (2020) 254:117717. doi: 10.1016/j.lfs.2020.117717
67. Harries LW, Hattersley AT, Doney AS, Colhoun H, Morris AD, Sutherland C, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. (2011) 43:117–20. doi: 10.1038/ng.735
68. Dawed AY, Mari A, Brown A, McDonald TJ, Li L, Wang S, et al. Pharmacogenomics of GLP-1 receptor agonists: a genome-wide analysis of observational data and large randomised controlled trials. Lancet Diabetes Endocrinology. (2023) 11:33–41. doi: 10.1016/S2213-8587(22)00340-0
69. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. (2010) 11:554–65. doi: 10.1016/j.cmet.2010.04.001
70. Cuyàs E, Buxó M, Ferri Iglesias MJ, Verdura S, Pernas S, Dorca J, et al. The C allele of ATM rs11212617 associates with higher pathological complete remission rate in breast cancer patients treated with neoadjuvant metformin. Front Oncol. (2019) 9:193. doi: 10.3389/fonc.2019.00193
71. Sacco F, Silvestri A, Posca D, Pirrò S, Gherardini PF, Castagnoli L, et al. Deep proteomics of breast cancer cells reveals that metformin rewires signaling networks away from a pro-growth state. Cell Systems. (2016) 2:159–71. doi: 10.1016/j.cels.2016.02.005
72. Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans cancer Cell. (2016) 167:1705–18. e13. doi: 10.1016/j.cell.2016.11.055
73. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. elife. (2014) 3:e02242. doi: 10.7554/eLife.02242
74. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, Independent of AMPK, Induces mTOR Inhibition and cell-cycle arrest through REDD C-CA. Cancer Res. (2011) 71:4366. doi: 10.1158/0008-5472.CAN-10-1769
75. Viollet B, Guigas B, Garcia NS, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin science. (2012) 122:253–70. doi: 10.1042/CS20110386
76. Saraei P, Asadi I, Kakar MA, Moradi-Kor N. The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manage Res. (2019) 11:3295–313. doi: 10.2147/CMAR
77. Yan J-B, Lai C-C, Jhu J-W, Gongol B, Marin TL, Lin S-C, et al. Insulin and metformin control cell proliferation by regulating TDG-mediated DNA demethylation in liver and breast cancer cells. Mol Therapy-Oncolytics. (2020) 18:282–94. doi: 10.1016/j.omto.2020.06.010
78. Chang H-H, Moro A, Chou CEN, Dawson DW, French S, Schmidt AI, et al. Metformin decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity in the conditional KrasG12D mouse model. Sci Rep. (2018) 8:5899. doi: 10.1038/s41598-018-24337-8
79. Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discovery. (2022) 21:283–305. doi: 10.1038/s41573-021-00367-2
80. Chan P, Shao L, Tomlinson B, Zhang Y, Liu Z-M. Metformin transporter pharmacogenomics: insights into drug disposition—where are we now? Expert Opin Drug Metab Toxicol. (2018) 14:1149–59. doi: 10.1080/17425255.2018.1541981
81. Feng J, Wang X, Ye X, Ares I, Lopez-Torres B, Martínez M, et al. Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol Res. (2022) 177:106114. doi: 10.1016/j.phrs.2022.106114
82. Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism (s) of action of metformin. Front endocrinology. (2019) 10:294. doi: 10.3389/fendo.2019.00294
83. Cai X, Hu X, Tan X, Cheng W, Wang Q, Chen X, et al. Metformin induced AMPK activation, G0/G1 phase cell cycle arrest and the inhibition of growth of esophageal squamous cell carcinomas in vitro and in vivo. PloS One. (2015) 10:e0133349. doi: 10.1371/journal.pone.0133349
84. Sahra IB, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. (2008) 27:3576–86. doi: 10.1038/sj.onc.1211024
85. Takahashi A, Kimura F, Yamanaka A, Takebayashi A, Kita N, Takahashi K, et al. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. (2014) 14:1–12. doi: 10.1186/1475-2867-14-53
86. Wang Y, Xu W, Yan Z, Zhao W, Mi J, Li J, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res. (2018) 37:1–12. doi: 10.1186/s13046-018-0731-5
87. Lee DE, Lee GY, Lee HM, Choi SY, Lee SJ, Kwon O-S. Synergistic apoptosis by combination of metformin and an O-GlcNAcylation inhibitor in colon cancer cells. Cancer Cell Int. (2023) 23:108. doi: 10.1186/s12935-023-02954-2
88. Ma L, Wei J, Wan J, Wang W, Wang L, Yuan Y, et al. Low glucose and metformin-induced apoptosis of human ovarian cancer cells is connected to ASK1 via mitochondrial and endoplasmic reticulum stress-associated pathways. J Exp Clin Cancer Res. (2019) 38:1–14. doi: 10.1186/s13046-019-1090-6
89. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. (2012) 3:865. doi: 10.1038/ncomms1859
90. Pulito C, Mori F, Sacconi A, Goeman F, Ferraiuolo M, Pasanisi P, et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell discovery. (2017) 3:1–19. doi: 10.1038/celldisc.2017.22
91. Jiang W, Finniss S, Cazacu S, Xiang C, Brodie Z, Mikkelsen T, et al. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget. (2016) 7:56456. doi: 10.18632/oncotarget.v7i35
92. Alimoradi N, Firouzabadi N, Fatehi R. How metformin affects various Malignancies by means of microRNAs: a brief review. Cancer Cell Int. (2021) 21:1–13. doi: 10.1186/s12935-021-01921-z
93. Nguyen TT, Ung TT, Li S, Lian S, Xia Y, Park SY, et al. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci Rep. (2019) 9:2003. doi: 10.1038/s41598-019-38778-2
94. Yenmis G, Sarac EY, Besli N, Soydas T, Tastan C, Kancagi DD, et al. Anti-cancer effect of metformin on the metastasis and invasion of primary breast cancer cells through mediating NF-kB activity. Acta Histochemica. (2021) 123:151709. doi: 10.1016/j.acthis.2021.151709
95. Xu S, Yang Z, Jin P, Yang X, Li X, Wei X, et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol Cancer Ther. (2018) 17:1291–302. doi: 10.1158/1535-7163.MCT-17-0927
96. Abdelmoneim M, Aboalela MA, Naoe Y, Matsumura S, Eissa IR, Bustos-Villalobos I, et al. The impact of metformin on tumor-infiltrated immune cells: preclinical and clinical studies. Int J Mol Sci. (2023) 24:13353. doi: 10.3390/ijms241713353
97. Wu Z, Zhang C, Najafi M. Targeting of the tumor immune microenvironment by metformin. J Cell communication Signaling. (2022) 16:333–48. doi: 10.1007/s12079-021-00648-w
98. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4+ CD25+ regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine. (2017) 25:154–64. doi: 10.1016/j.ebiom.2017.10.009
99. Chiang C-F, Chao T-T, Su Y-F, Hsu C-C, Chien C-Y, Chiu K-C, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget. (2017) 8:20706. doi: 10.18632/oncotarget.v8i13
100. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. (2011) 12:715–23. doi: 10.1038/ni.2060
101. Zhang C, Wang Y. Metformin attenuates cells stemness and epithelial−mesenchymal transition in colorectal cancer cells by inhibiting the Wnt3a/β−catenin pathway. Mol Med Rep. (2019) 19:1203–9. doi: 10.3892/mmr.2018.9765
102. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal transduction targeted Ther. (2020) 5:8. doi: 10.1038/s41392-020-0110-5
103. Samuel SM, Varghese E, Koklesová L, Líšková A, Kubatka P, Büsselberg D. Counteracting chemoresistance with metformin in breast cancers: targeting cancer stem cells. Cancers. (2020) 12:2482. doi: 10.3390/cancers12092482
104. Hu M, Chen Y, Ma T, Jing L. Repurposing Metformin in hematologic tumor: State of art. Curr Problems Cancer. (2023) 47(4):100972. doi: 10.1016/j.currproblcancer.2023.100972
105. Li Y, Zhang Q, Yang J, He W, Jiang Y, Chen Y, et al. Metformin combined with glucose starvation synergistically suppress triple-negative breast cancer by enhanced unfolded protein response. Biochem Biophys Res Commun. (2023) 675:146–54. doi: 10.1016/j.bbrc.2023.07.029
106. Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P, et al. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis. (2015) 4:e158–e. doi: 10.1038/oncsis.2015.18
107. Vizcaíno C, Mansilla S, Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol Ther. (2015) 152:111–24. doi: 10.1016/j.pharmthera.2015.05.008
108. Wei L, Deng C, Zhang B, Wang G, Meng Y, Qin H. SP4 facilitates esophageal squamous cell carcinoma progression by activating PHF14 transcription and Wnt/β-Catenin signaling. Mol Cancer Res. (2024) 22:55–69. doi: 10.1158/1541-7786.MCR-22-0835
109. Galdieri L, Gatla H, Vancurova I, Vancura A. Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J Biol Chem. (2016) 291:25154–66. doi: 10.1074/jbc.M116.742247
110. Hart PC, Chiyoda T, Liu X, Weigert M, Curtis M, Chiang C-Y, et al. SPHK1 is a novel target of metformin in ovarian cancer. Mol Cancer Res. (2019) 17:870–81. doi: 10.1158/1541-7786.MCR-18-0409
111. Loubiere C, Clavel S, Gilleron J, Harisseh R, Fauconnier J, Ben-Sahra I, et al. The energy disruptor metformin targets mitochondrial integrity via modification of calcium flux in cancer cells. Sci Rep. (2017) 7:5040. doi: 10.1038/s41598-017-05052-2
112. Huang X, Hong X, Wang J, Sun T, Yu T, Yu Y, et al. Metformin elicits antitumour effect by modulation of the gut microbiota and rescues Fusobacterium nucleatum-induced colorectal tumourigenesis. EBioMedicine. (2020) 61:1–16. doi: 10.1016/j.ebiom.2020.103037
113. Brown JR, Chan DK, Shank JJ, Griffith KA, Fan H, Szulawski R, et al. Phase II clinical trial of metformin as a cancer stem cell–targeting agent in ovarian cancer. JCI Insight. (2020) 5(11):1–12. doi: 10.1172/jci.insight.133247
114. Stirrups R. Metformin plus EGFR TKIs for lung adenocarcinoma. Lancet Oncol. (2019) 20:e563. doi: 10.1016/S1470-2045(19)30571-6
115. Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. (2016) 17:475–83. doi: 10.1016/S1470-2045(15)00565-3
116. Greco A, Coperchini F, Croce L, Magri F, Teliti M, Rotondi M. Drug repositioning in thyroid cancer treatment: the intriguing case of anti-diabetic drugs. Front Pharmacol. (2023) 14. doi: 10.3389/fphar.2023.1303844
117. Augustin RC, Huang Z, Ding F, Zhai S, McArdle J, Santisi A, et al. Metformin is associated with improved clinical outcomes in patients with melanoma: a retrospective, multi-institutional study. Front Oncol. (2023) 13:1075823. doi: 10.3389/fonc.2023.1075823
118. Hua L, Yang N, Li Y, Huang K, Jiang X, Liu F, et al. Metformin sensitizes AML cells to venetoclax through endoplasmic reticulum stress—CHOP pathway. Br J Haematology. (2023) 202:971–84. doi: 10.1111/bjh.18968
119. Moretti IF, Lerario AM, Sola PR, Macedo-da-Silva J, Baptista M, Palmisano G, et al. GBM cells exhibit susceptibility to metformin treatment according to TLR4 pathway activation and metabolic and antioxidant status. Cancers. (2023) 15:587. doi: 10.3390/cancers15030587
120. Isop LM, Neculau AE, Necula RD, Kakucs C, Moga MA, Dima L. Metformin: the winding path from understanding its molecular mechanisms to proving therapeutic benefits in neurodegenerative disorders. Pharmaceuticals. (2023) 16:1714. doi: 10.3390/ph16121714
121. Ibrahim RS, Ibrahim SS, El-Naas A, Koklesová L, Kubatka P, Büsselberg D. Could metformin and resveratrol support glioblastoma treatment? A mechanistic view at the cellular level. Cancers. (2023) 15:3368. doi: 10.3390/cancers15133368
122. Sharma S, Zhang Y, Sifat AE, Akter KA, Archie SR, Nozohouri S, et al. Evaluation of influx and efflux transporters of metformin in an In vitro blood-brain barrier model during normoxic and ischemic conditions. ASPET;. (2023) 385(S3):563. doi: 10.1124/jpet.122.156090
123. Sharma S, Zhang Y, Akter KA, Nozohouri S, Archie SR, Patel D, et al. Permeability of metformin across an in vitro blood–brain barrier model during normoxia and oxygen-glucose deprivation conditions: role of organic cation transporters (octs). Pharmaceutics. (2023) 15:1357. doi: 10.3390/pharmaceutics15051357
124. Ailuno G, Baldassari S, Balboni A, Drava G, Spalletti C, Tantillo E, et al. Development and validation of a GC-MS method for determination of metformin in normal brain and in glioblastoma tissues. J Pharm Biomed Analysis. (2023) 234:115503. doi: 10.1016/j.jpba.2023.115503
125. Verduci I, Cannavale G, Rey G, Cianci F, Fantin A, Mazzanti M. Repetitive membrane potential oscillations enhance metformin's antiproliferative effect in glioblastoma stem cells. Cancer Res. (2024) 84:4705–. doi: 10.1158/1538-7445.AM2024-4705
126. Cannavale G. Metformin, clic1 and transcranial stimulation: the basis of a new targeted therapy against glioblastoma relapse. IRIS Institutional Research Information System - AIR Archivio Istituzionale della Ricerca. Neuro Oncol (2022) 24(Issue Supplement_2):ii93. doi: 10.1093/neuonc/noac174.325
127. Al Hassan M, Fakhoury I, El Masri Z, Ghazale N, Dennaoui R, El Atat O, et al. Metformin treatment inhibits motility and invasion of glioblastoma cancer cells. Analytical Cell Pathol. (2018) 2018:1–9. doi: 10.1155/2018/5917470
128. Guarnaccia L, Navone SE, Masseroli MM, Balsamo M, Caroli M, Valtorta S, et al. Effects of metformin as add-on therapy against glioblastoma: an old medicine for novel oncology therapeutics. Cancers. (2022) 14:1412. doi: 10.3390/cancers14061412
129. Ucbek A, Özünal ZG, Uzun Ö, Gepdİremen A. Effect of metformin on the human T98G glioblastoma multiforme cell line. Exp Ther Med. (2014) 7:1285–90. doi: 10.3892/etm.2014.1597
130. Mouhieddine TH, Nokkari A, Itani MM, Chamaa F, Bahmad H, Monzer A, et al. Metformin and ara-a effectively suppress brain cancer by targeting cancer stem/progenitor cells. Front Neurosci. (2015) 9:442. doi: 10.3389/fnins.2015.00442
131. Würth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. (2013) 12:145–56. doi: 10.4161/cc.23050
132. Butler M, Pongor L, Su Y-T, Xi L, Raffeld M, Quezado M, et al. MGMT status as a clinical biomarker in glioblastoma. Trends cancer. (2020) 6:380–91. doi: 10.1016/j.trecan.2020.02.010
133. Sesen J, Dahan P, Scotland SJ, Saland E, Dang V-T, Lemarié A, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PloS One. (2015) 10:e0123721. doi: 10.1371/journal.pone.0123721
134. Feng S-W, Chang P-C, Chen H-Y, Hueng D-Y, Li Y-F, Huang S-M. Exploring the mechanism of adjuvant treatment of glioblastoma using temozolomide and metformin. Int J Mol Sci. (2022) 23:8171. doi: 10.3390/ijms23158171
135. Takhwifa F, Aninditha T, Setiawan H, Sauriasari R. The potential of metformin as an antineoplastic in brain tumors: A systematic review. Heliyon. (2021) 7(4):1–7. doi: 10.1016/j.heliyon.2021.e06558
136. Kinslow CJ, Mercurio A, Kumar P, Rae AI, Siegelin MD, Grinband J, et al. Association of MGMT promoter methylation with survival in low-grade and anaplastic gliomas after alkylating chemotherapy. JAMA Oncol. (2023) 9:919–27. doi: 10.1001/jamaoncol.2023.0990
137. Mohammad AH, Jatana S, Ruiz-Barerra MA, Khalaf R, Al-Saadi T, Diaz RJ. Metformin use is associated with longer survival in glioblastoma patients with MGMT gene silencing. J Neuro-Oncology. (2023) 165:209–18. doi: 10.1007/s11060-023-04485-2
138. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harbor Perspect Biol. (2014) 6:a009191. doi: 10.1101/cshperspect.a009191
139. De Meyts P. The insulin receptor and its signal transduction network. Endotext. (2016) NBK378978.
140. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. (2006) 7:85–96. doi: 10.1038/nrm1837
141. Saltiel AR. Insulin signaling in health and disease. J Clin Invest. (2021) 131(1):1–12. doi: 10.1172/JCI142241
142. Foretz M, Guigas B, Viollet B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinology. (2023) 19(8):460–76. doi: 10.1038/s41574-023-00833-4
143. LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocrine Rev. (2021) 42:77–96. doi: 10.1210/endrev/bnaa023
144. Lv Z, Guo Y. Metformin and its benefits for various diseases. Front endocrinology. (2020) 11:191. doi: 10.3389/fendo.2020.00191
145. Bi T, Zhu A, Yang X, Qiao H, Tang J, Liu Y, et al. Metformin synergistically enhances antitumor activity of cisplatin in gallbladder cancer via the PI3K/AKT/ERK pathway. Cytotechnology. (2018) 70:439–48. doi: 10.1007/s10616-017-0160-x
146. Mazurek M, Litak J, Kamieniak P, Kulesza B, Jonak K, Baj J, et al. Metformin as potential therapy for high-grade glioma. Cancers. (2020) 12:210. doi: 10.3390/cancers12010210
147. Ferla R, Haspinger E, Surmacz E. Metformin inhibits leptin-induced growth and migration of glioblastoma cells. Oncol letters. (2012) 4:1077–81. doi: 10.3892/ol.2012.843
148. Xiao Q, Xiao J, Liu J, Liu J, Shu G, Yin G. Metformin suppresses the growth of colorectal cancer by targeting INHBA to inhibit TGF-β/PI3K/AKT signaling transduction. Cell Death Disease. (2022) 13:202. doi: 10.1038/s41419-022-04649-4
149. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. (2010) 277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x
150. Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. (2009) 8:1168–75. doi: 10.4161/cc.8.8.8147
151. Shin S, Buel GR, Wolgamott L, Plas DR, Asara JM, Blenis J, et al. ERK2 mediates metabolic stress response to regulate cell fate. Mol Cell. (2015) 59:382–98. doi: 10.1016/j.molcel.2015.06.020
152. Buscà R, Pouysségur J, Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol. (2016) 4:53. doi: 10.3389/fcell.2016.00053
153. Champhekar A, Heymans R, Saco J, Turon Font G, Gonzalez C, Gao A, et al. ERK mediates interferon gamma-induced melanoma cell death. Mol Cancer. (2023) 22:165. doi: 10.1186/s12943-023-01868-x
154. Noch EK, Palma LN, Yim I, Bullen N, Qiu Y, Ravichandran H, et al. Insulin feedback is a targetable resistance mechanism of PI3K inhibition in glioblastoma. Neuro-oncology. (2023) 25:2165–76. doi: 10.1093/neuonc/noad117
155. Lo Dico A, Valtorta S, Ottobrini L, Moresco RM. Role of metformin and AKT axis modulation in the reversion of hypoxia induced TMZ-resistance in glioma cells. Front Oncol. (2019) 9:463. doi: 10.3389/fonc.2019.00463
156. De Cabo R, Mattson MP. Effects of intermittent fasting on health, aging, and disease. New Engl J Med. (2019) 381:2541–51. doi: 10.1056/NEJMra1905136
157. Onken B, Driscoll M. Metformin induces a dietary restriction–like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PloS One. (2010) 5:e8758. doi: 10.1371/journal.pone.0008758
158. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front endocrinology. (2021) 12:933. doi: 10.3389/fendo.2021.718942
159. Teong XT, Liu K, Vincent AD, Bensalem J, Liu B, Hattersley KJ, et al. Intermittent fasting plus early time-restricted eating versus calorie restriction and standard care in adults at risk of type 2 diabetes: a randomized controlled trial. Nat Med. (2023) 29:963–72. doi: 10.1038/s41591-023-02287-7
160. Schreck KC, Hsu F-C, Berrington A, Henry-Barron B, Vizthum D, Blair L, et al. Feasibility and biological activity of a ketogenic/intermittent-fasting diet in patients with glioma. Neurology. (2021) 97:e953–e63. doi: 10.1212/WNL.0000000000012386
161. Voss M, Wagner M, von Mettenheim N, Harter PN, Wenger KJ, Franz K, et al. ERGO2: a prospective, randomized trial of calorie-restricted ketogenic diet and fasting in addition to reirradiation for Malignant glioma. Int J Radiat Oncol Biol Physics. (2020) 108:987–95. doi: 10.1016/j.ijrobp.2020.06.021
162. Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol. (2019) 2:200. doi: 10.1038/s42003-019-0455-x
163. Guadilla I, González S, Cerdán S, Lizarbe B, López-Larrubia P. Magnetic resonance imaging to assess the brain response to fasting in glioblastoma-bearing rats as a model of cancer anorexia. Cancer Imaging. (2023) 23:1–12. doi: 10.1186/s40644-023-00553-y
164. Clontz AD. Ketogenic therapies for glioblastoma: Understanding the limitations in transitioning from mice to patients. Front Nutr. (2023) 10:1110291. doi: 10.3389/fnut.2023.1110291
165. Safdie F, Brandhorst S, Wei M, Wang W, Lee C, Hwang S, et al. Fasting enhances the response of glioma to chemo-and radiotherapy. PLoS One. (2012) 7(9):1–9. doi: 10.1371/journal.pone.0044603
166. Duffy J. Evaluating the effects of fasting conditions in combination with temozolomide for the treatment of glioblastoma multiforme. Northern Michigan University ProQuest Dissertations & Theses. (2020) 638. Available at: https://commons.nmu.edu/theses/638.
167. Elsakka AM, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P, et al. Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: a 24-month follow-up. Front Nutr. (2018) 5:20. doi: 10.3389/fnut.2018.00020
168. Seyfried TN, Shivane AG, Kalamian M, Maroon JC, Mukherjee P, Zuccoli G. Ketogenic metabolic therapy, without chemo or radiation, for the long-term management of IDH1-mutant glioblastoma: an 80-month follow-up case report. Front Nutr. (2021) 281. doi: 10.3389/fnut.2021.682243
169. Lee Y, Kim J, Kim M-S, Kwon Y, Shin S, Yi H, et al. Coordinate regulation of the senescent state by selective autophagy. Dev Cell. (2021) 56:1512–25. e7. doi: 10.1016/j.devcel.2021.04.008
170. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
171. Cheng F-F, Liu Y-L, Du J, Lin J-T. Metformin's mechanisms in attenuating hallmarks of aging and age-related disease. Aging disease. (2022) 13:970. doi: 10.14336/AD.2021.1213
172. Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. (2017) 40:31–44. doi: 10.1016/j.arr.2017.08.003
173. Zhu Z, Zeng Q, Liu Q, Wen J, Chen G. Association of glucose-lowering drugs with outcomes in patients with diabetes before hospitalization for COVID-19: A systematic review and network meta-analysis. JAMA Network Open. (2022) 5:e2244652–e. doi: 10.1001/jamanetworkopen.2022.44652
174. Luo S, Wong ICK, Chui CSL, Zheng J, Huang Y, Schooling CM, et al. Effects of putative metformin targets on phenotypic age and leukocyte telomere length: a mendelian randomisation study using data from the UK Biobank. Lancet Healthy Longevity. (2023) 4:e337–e44. doi: 10.1016/S2666-7568(23)00085-5
175. Serrano M. Cancer regression by senescence. New Engl J Med. (2007) 356:1996–7. doi: 10.1056/NEJMcibr071461
176. Menendez JA, Cufí S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, et al. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY). (2011) 3:1063. doi: 10.18632/aging.v3i11
177. Schmitt CA, Wang B, Demaria M. Senescence and cancer—role and therapeutic opportunities. Nat Rev Clin Oncol. (2022) 19:619–36. doi: 10.1038/s41571-022-00668-4
178. Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab. (2021) 3:1290–301. doi: 10.1038/s42255-021-00483-8
179. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. (2020) 34:1565–76. doi: 10.1101/gad.343129.120
180. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. (2022) 28:1556–68. doi: 10.1038/s41591-022-01923-y
181. Schoetz U, Klein D, Hess J, Shnayien S, Spoerl S, Orth M, et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death disease. (2021) 12:1162. doi: 10.1038/s41419-021-04454-5
182. Jeong YK, Kim M-S, Lee JY, Kim EH, Ha H. Metformin radiosensitizes p53-deficient colorectal cancer cells through induction of G2/M arrest and inhibition of DNA repair proteins. PloS One. (2015) 10:e0143596. doi: 10.1371/journal.pone.0143596
183. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. (2012) 18:290–300. doi: 10.1158/1078-0432.CCR-11-2260
184. Abdelgawad IY, Agostinucci K, Sadaf B, Grant MK, Zordoky BN. Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front Aging. (2023) 4:1170434. doi: 10.3389/fragi.2023.1170434
185. Hu Q, Peng J, Jiang L, Li W, Su Q, Zhang J, et al. Metformin as a senostatic drug enhances the anticancer efficacy of CDK4/6 inhibitor in head and neck squamous cell carcinoma. Cell Death disease. (2020) 11:925. doi: 10.1038/s41419-020-03126-0
186. Milanovic M, Fan DN, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
187. Carroll B, Nelson G, Rabanal-Ruiz Y, Kucheryavenko O, Dunhill-Turner NA, Chesterman CC, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol. (2017) 216:1949–57. doi: 10.1083/jcb.201610113
188. Weichhart T. mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Gerontology. (2018) 64:127–34. doi: 10.1159/000484629
189. Wang B, Kohli J, Demaria M. Senescent cells in cancer therapy: friends or foes? Trends Cancer. (2020) 6:838–57. doi: 10.1016/j.trecan.2020.05.004
190. Woo SH, Yang LP, Chuang HC, Fitzgerald A, Lee HY, Pickering C, et al. Down-regulation of Malic enzyme 1 and 2: sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. Head neck. (2016) 38:E934–E40. doi: 10.1002/hed.24129
191. Curry J, Johnson J, Tassone P, Vidal MD, Menezes DW, Sprandio J, et al. Metformin effects on head and neck squamous carcinoma microenvironment: window of opportunity trial. Laryngoscope. (2017) 127:1808–15. doi: 10.1002/lary.26489
192. Salam R, Saliou A, Bielle F, Bertrand M, Antoniewski C, Carpentier C, et al. Cellular senescence in Malignant cells promotes tumor progression in mouse and patient Glioblastoma. Nat Commun. (2023) 14:441. doi: 10.1038/s41467-023-36124-9
193. Valtorta S, Dico AL, Raccagni I, Gaglio D, Belloli S, Politi LS, et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget. (2017) 8:113090. doi: 10.18632/oncotarget.v8i68
194. Yu Z, Zhao G, Li P, Li Y, Zhou G, Chen Y, et al. Temozolomide in combination with metformin act synergistically to inhibit proliferation and expansion of glioma stem-like cells. Oncol Letters. (2016) 11:2792–800. doi: 10.3892/ol.2016.4315
195. Gupta S, Silveira DA, Mombach JCM. Towards DNA-damage induced autophagy: A Boolean model of p53-induced cell fate mechanisms. DNA repair. (2020) 96:102971. doi: 10.1016/j.dnarep.2020.102971
196. Childs BG, Baker DJ, Kirkland JL, Campisi J, Van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. (2014) 15:1139–53. doi: 10.15252/embr.201439245
197. Dhokia V, Moss JA, Macip S, Fox JL. At the crossroads of life and death: the proteins that influence cell fate decisions. Cancers. (2022) 14:2745. doi: 10.3390/cancers14112745
198. Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, Bourgeois BR, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. (2017) 169:132–47. e16. doi: 10.1016/j.cell.2017.02.031
199. Song YS, Lee BY, Hwang ES. Dinstinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech Ageing Dev. (2005) 126:580–90. doi: 10.1016/j.mad.2004.11.008
200. Brandmaier A, Hou S-Q, Shen WH. Cell cycle control by PTEN. J Mol Biol. (2017) 429:2265–77. doi: 10.1016/j.jmb.2017.06.004
201. Vidotto T, Melo CM, Castelli E, Koti M, Dos Reis RB, Squire JA. Emerging role of PTEN loss in evasion of the immune response to tumours. Br J cancer. (2020) 122:1732–43. doi: 10.1038/s41416-020-0834-6
202. Jones K, Elmore L, Jackson-Cook C, Demasters G, Povirk L, Holt S, et al. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol. (2005) 81:445–58. doi: 10.1080/09553000500168549
203. Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J neurosurgery. (2006) 105:111–8. doi: 10.3171/jns.2006.105.1.111
204. Li X, Li B, Ni Z, Zhou P, Wang B, He J, et al. Metformin synergizes with BCL-XL/BCL-2 inhibitor ABT-263 to induce apoptosis specifically in p53-defective cancer cells. Mol Cancer Ther. (2017) 16:1806–18. doi: 10.1158/1535-7163.MCT-16-0763
205. Beltzig L, Schwarzenbach C, Leukel P, Frauenknecht KB, Sommer C, Tancredi A, et al. Senescence is the main trait induced by temozolomide in glioblastoma cells. Cancers. (2022) 14:2233. doi: 10.3390/cancers14092233
206. Kaina B. Temozolomide in glioblastoma therapy: role of apoptosis, senescence and autophagy. comment on strobel, temozolomide and other alkylating agents in glioblastoma therapy. biomedicines. (2019) 7:69. doi: 10.3390/biomedicines7040090
207. Reich TR, Schwarzenbach C, Vilar JB, Unger S, Mühlhäusler F, Nikolova T, et al. Localization matters: nuclear-trapped Survivin sensitizes glioblastoma cells to temozolomide by elevating cellular senescence and impairing homologous recombination. Cell Mol Life Sci. (2021) 78:5587–604. doi: 10.1007/s00018-021-03864-0
Keywords: metformin, GBM, apoptosis, senescence, calorie restriction, AMPK, mTOR, P53
Citation: Hajimohammadebrahim-Ketabforoush M, Zali A, Shahmohammadi M and Hamidieh AA (2024) Metformin and its potential influence on cell fate decision between apoptosis and senescence in cancer, with a special emphasis on glioblastoma. Front. Oncol. 14:1455492. doi: 10.3389/fonc.2024.1455492
Received: 26 June 2024; Accepted: 12 August 2024;
Published: 29 August 2024.
Edited by:
Mario Cioce, Campus Bio-Medico University, ItalyReviewed by:
Meike Kaehler, University Hospital Schleswig-Holstein, GermanyAhmed Sohaib, University of Menoufia, Egypt
Copyright © 2024 Hajimohammadebrahim-Ketabforoush, Zali, Shahmohammadi and Hamidieh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melika Hajimohammadebrahim-Ketabforoush, a2V0YWJmb3Jvc2htQGdtYWlsLmNvbQ==; a2V0YWJmb3JvdXNoQHNibXUuYWMuaXI=