 Le Yu
Le Yu Ruoyi Yang
Ruoyi Yang Zeng Long2
Zeng Long2 Bin Liu
Bin Liu- 1Sichuan Cancer Hospital, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 2School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan, China
Lung cancer is a leading cause of cancer-related deaths globally, and traditional chemotherapy has limited efficacy in treating advanced non-small cell lung cancer (NSCLC). In recent years, the prognosis for patients with NSCLC has significantly improved due to the development of new treatment modalities, including targeted therapies. Targeted therapies utilize monoclonal antibodies (mAbs), antibody-drug conjugates (ADCs), or small molecule tyrosine kinase inhibitors (TKIs) directed against specific mutated genes such as EGFR and ALK. The development of these drugs has deepened our understanding of NSCLC and improved treatment outcomes for patients. This review aims to summarize the mechanisms and current status of targeted therapy for NSCLC, discuss strategies to overcome acquired resistance, and address current challenges in the field.
1 Introduction
Lung cancer is a leading cause of cancer deaths globally, accounting for 18% of cancer-related deaths (1). In China, it is the most common type of cancer, with approximately 630,500 deaths annually, representing 27% of all cancer deaths (2). Lung cancer is classified into multiple subtypes based on histology, with non-small cell lung cancer (NSCLC) comprising 85%, including adenocarcinoma (40%), squamous cell carcinoma (30%), large cell (undifferentiated) carcinoma (15%), and other rare types (3). Due to the lack of specific early screening methods, most NSCLC patients are diagnosed at advanced stages or with widespread metastasis, resulting in poor prognosis.
For many years, chemotherapy, radiation therapy, and surgical tumor resection have been the mainstays of NSCLC treatment, but advancements in chemotherapy have not significantly improved patient survival rates. Over the past decade, significant improvements in prognosis for lung cancer patients have been achieved due to research and application of new treatment modalities such as targeted therapy and immunotherapy. Targeted therapies, utilizing monoclonal antibodies (mAbs) or tyrosine kinase inhibitors (TKIs) against specific mutated genes such as EGFR and ALK, have greatly enhanced outcomes for lung cancer patients (4, 5). On this basis, the Antibody-Drug Conjugate (ADC) drugs have shown exciting results in the treatment of NSCLC (6). In addition, patients with advanced NSCLC, immunotherapy is an important treatment strategy, and its combination with other targeted therapies helps further improve patient prognosis (7). In this review, we aim to summarize the mechanisms and current status of targeted therapy for NSCLC, discuss strategies to overcome acquired resistance, and address current challenges in the field.
2 TKIs therapy
Tyrosine kinases (TKs) are crucial cell signaling enzymes that regulate pathways related to cell growth, differentiation, and apoptosis (8). Their oncogenic mutations or overexpression are markers of cell cycle dysregulation and play a role in the development and progression of various cancers, including NSCLC. TKIs selectively inhibit TK proteins, preventing tumor cell proliferation and growth, and play a significant role in targeted therapy for NSCLC (9). Due to this highly specific mechanism, TKIs often have minimal impact on normal cells, resulting in relatively fewer side effects, however, resistance remains a challenge in clinical cancer treatment (10). Some acquired resistance mechanisms have been identified, and further research into these targets and resistance mechanisms is essential for improving the prognosis of NSCLC (Figure 1).
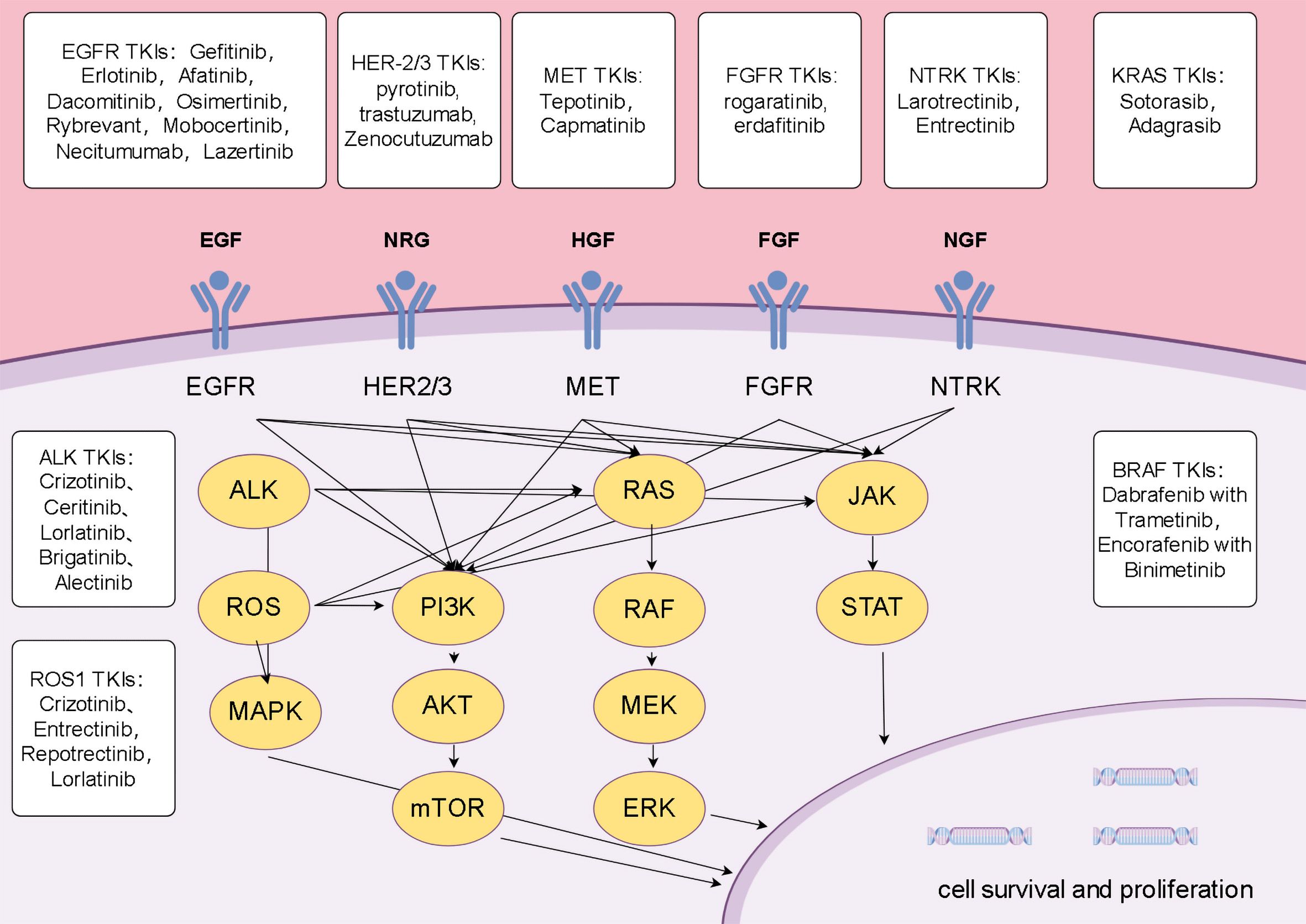
Figure 1. Mechanisms of lung cancer progression and potential therapeutic targets.
2.1 EGFR
Epidermal growth factor receptor(EGFR)is a receptor tyrosine kinase, also known as ErbB1/human epidermal growth factor receptor 1 (HER1), belonging to the tyrosine kinase receptor ErbB family. This family also includes ErbB2/human epidermal growth factor receptor 2 (HER-2), ErbB3/human epidermal growth factor receptor 3 (HER-3), and ErbB4/human epidermal growth factor receptor 4 (HER4). Upon binding with growth factors, the domains of EGFR and other ErbB family members undergo phosphorylation, activating their cytoplasmic tyrosine kinase domains, thereby initiating intracellular signal transduction that affects cell proliferation and differentiation. Overexpression or mutation activation of EGFR is closely associated with the development and progression of many human malignancies (8, 11).
In lung cancer patients, EGFR mutation is one of the most common variants in NSCLC. In Asian populations, at least 50% of patients have EGFR gene mutations, with exon 19 deletions (Ex 19Del) and exon 21 single amino acid substitutions (L858R) being the most common. These mutations alter the receptor’s conformation, leading to dimerization and increased activity, while reducing affinity for ATP (12–14).This allows first-generation EGFR TKIs such as gefitinib, erlotinib, and afatinib to competitively and reversibly inhibit downstream signaling, significantly improving response and survival rates in untreated EGFR mutation-positive patients. Clinical results show erlotinib achieves a response rate (RR) of 72% and a median progression-free survival (PFS) of nearly 10 months in patients with EGFR mutations (Ex 19Del or L858R), while maintaining good safety profiles (15).
However, a considerable proportion of patients develop resistance to first-generation EGFR therapy, with 60% of resistant cases associated with the T790M gatekeeper mutation. This mutation increases receptor affinity for ATP, hindering competitive inhibition by first-generation EGFR TKIs (16, 17). To overcome this resistance mechanism, second-generation EGFR TKIs, such as afatinib and dacomitinib, were developed as irreversible inhibitors. They form irreversible covalent bonds with the C797 residue of EGFR, blocking ATP binding and controlling disease progression. Despite demonstrating significant PFS and overall survival (OS) benefits in first-line therapy, second-generation EGFR TKIs exhibit poor efficacy against T790M-mediated resistance to first-generation EGFR therapy due to their lower selectivity for the T790M EGFR mutation compared to wild-type (WT) EGFR. Additionally, their clinical use is associated with higher rates of adverse events compared to first-generation drugs (18).
Third-generation EGFR-TKIs, such as osimertinib, have shown significant efficacy in overcoming acquired resistance mediated by the T790M mutation from first- and second-generation EGFR-TKIs. These drugs also form irreversible covalent complexes with EGFR, not only overcoming enhanced ATP affinity conferred by the T790M mutation but also showing higher selectivity for L858R/T790M mutant EGFR, with 200 times the potency against WT EGFR, thus significantly improving treatment outcomes (19). For instance, results from the AURA3 clinical trial demonstrate that osimertinib is superior to platinum-based chemotherapy plus pemetrexed in terms of efficacy in T790M-positive advanced NSCLC patients who progressed during first-line EGFR-TKI therapy, achieving a 71% objective RR and a median PFS of 10.1 months in patients with advanced disease, including central nervous system metastases (20). The FLAURA trial further confirms osimertinib’s superiority over first-generation EGFRis in first-line treatment of EGFR mutation-positive advanced NSCLC, showing longer median PFS (18.9 months vs. 10.2 months), similar objective RR (80% vs. 76%), and lower rates of severe adverse events (34% vs. 45%) (21). However, even third-generation EGFR-TKIs cannot avoid the development of resistance. Resistance mechanisms to third-generation EGFR-TKIs in advanced NSCLC are highly complex and vary between patients, highlighting the critical importance of next-generation sequencing (NGS) testing for evaluating resistance mechanisms and identifying subsequent treatment targets. Currently, novel fourth-generation EGFR-TKIs are under preclinical investigation, aiming to overcome existing EGFR-mediated acquired resistance and provide new perspectives for subsequent treatment of EGFR mutation-positive patients (22, 23).
2.2 ALK
The ALK gene encodes a tyrosine kinase receptor belonging to the insulin receptor superfamily. Its activation leads to receptor dimerization and autophosphorylation, subsequently activating downstream signaling pathways crucial for cell proliferation, migration, and differentiation. In malignant tumors, most ALK gene mutations occur in the form of translocations with another partner gene, leading to aberrant activation of ALK and its downstream signaling pathways, thereby promoting tumor progression (24, 25).In NSCLC, ALK fusion genes are present in approximately 3%-5% of patients, more commonly observed in younger individuals (26). Among these, the EML4-ALK fusion, involving the echinoderm microtubule-associated protein-like 4 (EML4) gene, is the most frequent, found in about 85% of all ALK fusion cases. Additionally, ALK gene rearrangements with partner genes such as KIF5B, TFG, TPR, BCL11A, though the choice of ALK inhibitors currently does not depend on the specific fusion type (27–30).
Crizotinib is a first-generation multi-targeted ALK inhibitor that also exhibits activity against MET and ROS1 (31). Clinical studies have shown a RR of 65% with crizotinib, and a median PFS of 7.7 months, which is superior to standard chemotherapy, though it has not significantly improved OS compared to chemotherapy (32). Second-generation ALK inhibitors include ceritinib and alectinib, which have higher affinity for ALK and better penetration through the blood-brain barrier. Alectinib has demonstrated higher PFS rates compared to crizotinib, with a 12-month event-free survival rate of 68.4% (33). Similar to alectinib, the brigatinib group shows a higher PFS compared to the crizotinib group (34)..
Most patients inevitably develop treatment resistance after receiving second-generation ALK TKI therapy. The G1202R mutation is the most common acquired resistance mutation among patients treated with second-generation ALK inhibitors. Its occurrence rates in patients treated with ceritinib, alectinib, and brigatinib are 21%, 29%, and 43%, respectively (35, 36). Lorlatinib, a reversible third-generation ALK and ROS1 inhibitor, overcomes multiple ALK resistance mutations including G1202R, and exhibits good blood-brain barrier penetration (37). Clinical studies indicate that in ALK-positive patients who have previously received at least one ALK TKI, the objective RR of lorlatinib is 47.0%. Among patients who have previously received two or more ALK TKIs, the objective RR is 38.7% (38). CROWN trial assessed the efficacy of lorlatinib in treatment-naive, ALK-positive NSCLC patients. The results showed that the 3-year PFS rate was 64% in the lorlatinib group compared to 19% in the crizotinib group. Compared to crizotinib, lorlatinib demonstrated improvements in PFS, objective RR, intracranial objective RR, intracranial progression time, and duration of response, indicating durable benefits with lorlatinib (39).Apart from further refining treatment targets, immunotherapy may represent a new strategy for the treatment of ALK-positive NSCLC after development of resistance. The complex interaction between ALK rearrangements and immune cells suggests that high PD-L1 expression could serve as a poor prognostic biomarker in ALK-positive NSCLC, indicating that immune checkpoint inhibitors combined with targeted therapy may potentially improve patient outcomes (40).
2.3 ROS1
ROS1 is an oncogene located on chromosome 6Q22.1 that encodes a member of the insulin receptor subfamily. ROS1 fusion genes have been identified in various types of tumors, with the most prevalent fusion being CD74-ROS1, found in 44% of cases, followed by EZRs-ROS1, SDC4-ROS1, and SLC34A2 (41–43). The ROS1 fusion protein activates downstream signaling pathways to promote cell proliferation, activation, and cell cycle progression, accelerating the development and progression of NSCLC. ROS1 rearrangements are detected in 1%-2% of NSCLC patients, and 36% of ROS1-positive NSCLC patients harbor concurrent oncogenic mutations such as EGFR or KRAS mutations, MET amplification, or ALK translocations (44). The central nervous system is a common site of metastasis in ROS1 fusion-positive NSCLC patients, with up to 36% diagnosed with brain metastases, and many others potentially developing intracranial metastases subsequently. Therefore, treatment strategies for ROS1-positive NSCLC patients should encompass the central nervous system, and ROS1 testing is recommended for all NSCLC patients with brain metastases (45).
ROS1 belongs to the insulin receptor family. While not all ALK TKIs exhibit dual inhibitory activity against both ALK and ROS1, several TKI targeting multiple ALK targets have shown efficacy in treating ROS1. Due to its unique structure, crizotinib inhibits ROS1 with five times the potency compared to ALK, showing significant efficacy in ROS1-mutant patients. Results from the PROFILE 1001 study demonstrate an objective RR of 72% with crizotinib in advanced ROS1-rearranged NSCLC patients, with a median duration of response of 24.7 months. Consistent objective RRs were observed across different subgroups, with a median PFS of 19.3 months and a median OS of 51.4 months (46). The EUCROSS study evaluated crizotinib in ROS1-positive NSCLC, reporting an objective RR of 70% and a median PFS of 20.0 months. These studies underscore the therapeutic efficacy of crizotinib while indicating a poorer prognosis for patients with brain metastases, likely due to its limited ability to penetrate the blood-brain barrier. In contrast, entrectinib effectively crosses the blood-brain barrier, offering an advantage in treating patients with central nervous system metastases (47). Multiple studies confirm entrectinib’s role in ROS1-mutant NSCLC, demonstrating an objective RR of 77% and a comprehensive RR of 67.1%, with 12-month PFS and OS rates of 55% and 81%, respectively. In patients with central nervous system metastases, entrectinib achieves an intracranial objective RR of 79.2% and a median intracranial PFS of 12.0 months (48).
Lorlatinib, a third-generation TKI targeting the kinase domains of ALK and ROS1, has shown efficacy in NSCLC patients resistant to first and second-generation ALK inhibitors. Studies in advanced ROS1-positive NSCLC demonstrate an objective RR of 62% in TKI-naïve patients and 35% in patients previously treated with crizotinib. Lorlatinib achieves a 64% intracranial RR in TKI-naïve patients and 50% in crizotinib-resistant patients (49). However, the PFROST study indicates limited efficacy of lorlatinib in treating secondary ROS1 resistance mutations induced by crizotinib (50). Mechanisms of acquired resistance to ROS1 inhibitors are still under investigation, and several potential therapeutic agents have been developed to further enhance treatment outcomes for ROS1-mutant patients.
2.4 BRAF
BRAF is a serine/threonine protein kinase belonging to the RAF kinase family. While all RAF proteins can phosphorylate MEK (MEK1 and MEK2), BRAF exhibits the strongest activation capability. Upon activation by RAS, BRAF further activates MEK to phosphorylate ERK (ERK1 and ERK2) in the cytoplasm, which subsequently translocates to the nucleus. In the nucleus, ERK1/2 phosphorylates and activates various transcription factors, thereby participating in the regulation of apoptosis, proliferation, migration, and enhancing the expression of genes involved in many oncogenic processes (51–54).BRAF mutations occur in 3-5% of NSCLCs, predominantly in adenocarcinoma histology. Based on their dimerization status, kinase activity, and RAS dependency, BRAF mutations are classified into three types. Type 1 mutations, characterized by a monomeric state and high BRAF kinase activity independent of RAS, include the most common V600E point mutation found in 90% of BRAF-mutant tumors on exon 15. Compared to type 1 alterations, type 2 and type 3 mutations are associated with a higher risk of brain metastases at diagnosis (55–58).
The role and clinical significance of BRAF mutations in solid tumors have been extensively studied, leading to the development of various BRAF TKIs and therapeutic strategies targeting BRAF mutations. Following positive results in metastatic melanoma patients with BRAF V600E mutations, anti-BRAF therapies have been extended to other tumor types harboring the same mutation. The EURAF cohort compared dabrafenib monotherapy with dabrafenib in combination with trametinib, showing an overall RR of 53% and a disease control rate of 85% for BRAF-targeted therapy. Median PFS with BRAF-targeted therapy was 5.0 months, and OS was 10.8 months (59).
Results from phase II clinical trials of dabrafenib-trametinib therapy in previously treated metastatic BRAF V600E mutation NSCLC patients showed an overall RR of 63.2%. In treatment-naïve metastatic BRAF V600E mutation NSCLC, first-line dabrafenib combined with trametinib achieved an overall RR of 64% (60, 61). Based on these findings, dabrafenib-trametinib combination therapy has been FDA-approved as first-line treatment for BRAF V600 mutant lung cancer (62). Apart from dabrafenib-trametinib, FDA approvals also include vemurafenib/cobimetinib and encorafenib/binimetinib for advanced or metastatic NSCLC with BRAF (V600E/K) mutations.
2.5 MET
The MET gene, located on chromosome 7q21-q31, encodes the protein tyrosine kinase MET, a transmembrane receptor in the RTK family. MET is activated by HGF, regulating cell growth and development by promoting mitosis, movement, and invasion. Pathological MET activation drives tumorigenesis in various cancers by enhancing proliferation, invasive growth, and angiogenesis (63, 64). Oncogenic MET alterations include mutations, amplifications, overexpression, chromosomal rearrangements, and fusions, disrupting the HGF/MET axis and contributing to cancers like NSCLC. METex14 skipping mutations, found in 3%-4% of NSCLC patients, are linked to poor prognosis and responsiveness to MET-targeted therapy, serving as a predictive biomarker for MET TKI sensitivity (65–67). The PROFILE 1001 study showed that crizotinib monotherapy was effective for advanced NSCLC patients with METex14 mutations (38% treatment-naive): objective response rate was 32%, median duration of response was 9.1 months, and median progression-free survival was 7.3 months. Crizotinib is approved for previously platinum-treated, metastatic NSCLC patients with METex14 mutations.Tepotinib is a selective MET inhibitor that suppresses tumor growth by disrupting MET signaling pathways. Results from the VISION study shows that in NSCLC patients with MET exon 14 skipping mutations, the ORR was 56%. The median PFS was 8.9 months. There was no statistically significant difference in outcomes between treatment-naïve and previously treated patients (68).
In addition to influencing tumor progression through various mechanisms, MET amplification is recognized as a distinct mechanism of acquired resistance to EGFR TKIs (EGFR-TKIs) in advanced EGFR-mutant NSCLC patients. MET amplification is detected in 5% to 22% of NSCLC patients resistant to first-generation EGFR TKIs, representing the second most common acquired resistance mechanism after the EGFR T790M mutation (69).Combination therapy with MET TKIs and EGFR-TKIs has shown significant clinical benefits for this patient subset, demonstrating preliminary clinical activity with a Phase Ib/II ORR of 27%. Increased activity has been observed particularly in patients with high MET amplification levels; in patients with MET gene copy number ≥6, the Phase II ORR was 47% (70).Thus, MET TKIs have become a potential effective option for patients who have developed resistance to EGFR-TKIs. Several new MET TKIs are currently under development and in clinical trials, aiming to provide new opportunities for patients with MET mutations (71).
2.6 KRAS
The Kirsten rat sarcoma viral oncogene homolog (KRAS), along with its homologs NRAS and HRAS, belongs to the RAS GTPase protein family, situated on the inner surface of the cell membrane. It dynamically regulates between an inactive GDP-bound state (KRAS-GDP) and an active GTP-bound state (KRAS-GTP), acting as a molecular switch in signaling cascades that govern cell proliferation, survival, and differentiation (72).KRAS gene mutations lead to an increase in KRAS-GTP state, thereby triggering downstream oncogenic pathways. KRAS mutations are among the most common genomic alterations in solid tumors, accounting for 85% of observed RAS mutations in human cancers. Among various KRAS mutation variants, KRAS G12C is the most prevalent, and occurs in 13% of NSCLC cases (73). KRAS-driven cancers depend on sustained activation and signaling of KRAS, making it an ideal therapeutic target. However, due to the lack of deep binding pockets for specific small molecule inhibitors, it has historically been considered an “undruggable” target (74).
In clinical practice, first-line platinum-based chemotherapy ± immunotherapy is the recommended choice for KRAS-mutant patients. Attempts targeting KRAS have not been particularly successful. Research results of sotorasib in patients with advanced NSCLC harboring KRAS p.G12C mutation who have previously received standard therapy show an objective RR of 37.1%. The median duration of response is 11.1 months. The median PFS is 6.8 months, and the median OS is 12.5 months (75). In May 2021, the FDA accelerated the approval of sotorasib for the treatment of locally advanced or metastatic NSCLC in adults with KRAS (G12C) mutation who have received at least one prior FDA-approved systemic therapy. Further clinical trials are ongoing to bring new strategies for NSCLC treatment (71).
Based on clinical trial results, the FDA has approved various TKI drugs for the treatment of NSCLC (Table 1). With the deepening of fundamental research and the continuous improvement of genetic testing methods, understanding of NSCLC has advanced further. More potential therapeutic targets for NSCLC, such as PIK3CA and HER-2, are being explored (76–78). These ongoing studies and the development of next-generation inhibitors aim to enhance efficacy, reduce side effects, and address current drug resistance, potentially leading to better outcomes for NSCLC patients in the future (79). Besides new drug development, novel treatment strategies, such as combining targeted therapy with chemotherapy or immunotherapy, are being developed to further improve patient prognosis (80). With the advancement of new-generation testing technologies like NGS, we will be able to obtain more comprehensive and convenient molecular profiles of NSCLC tumors, enabling personalized drug selection and the formulation of optimal treatment strategies.
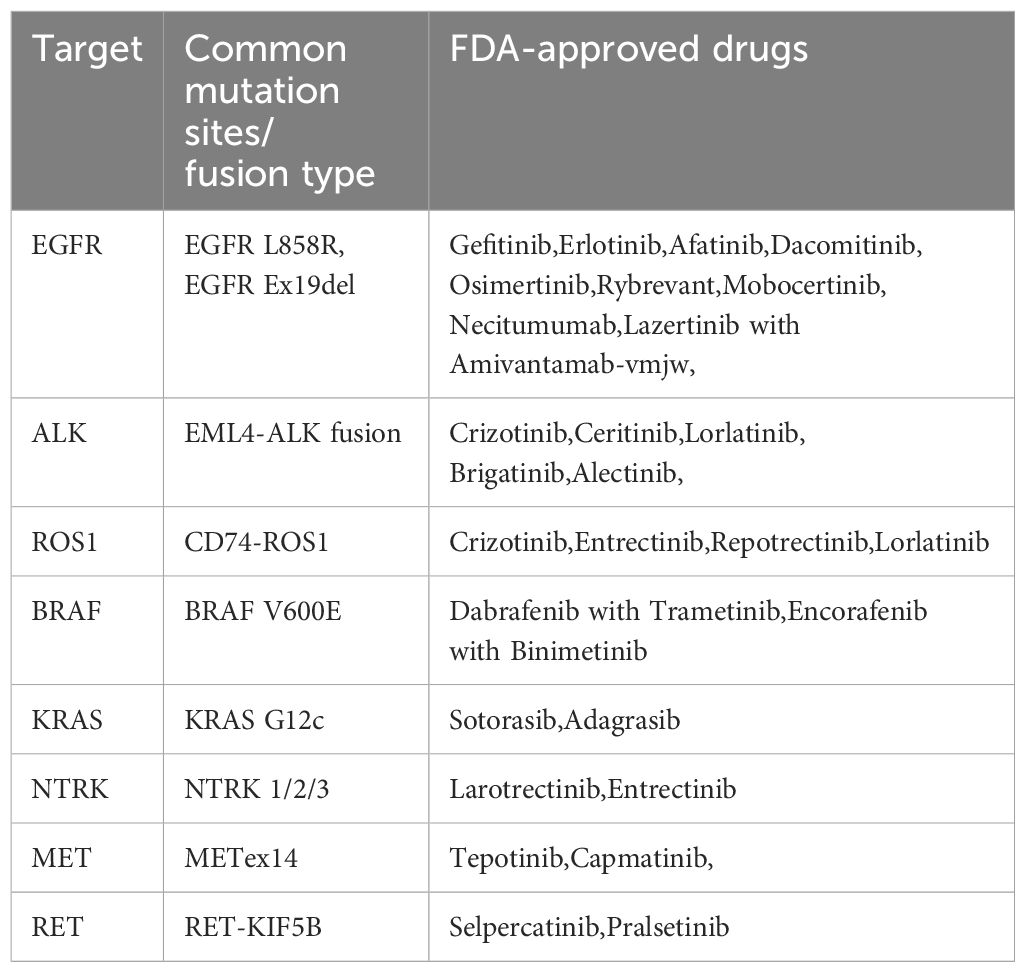
Table 1. FDA-approved TKI drugs for NSCLC treatment.
3 Monoclonal antibody therapy
Conventional chemotherapy and/or radiotherapy have shown limited efficacy in treating NSCLC patients, partly due to the high doses required for tumor eradication, which often lead to irreversible damage to normal tissues (81). As a more precise treatment, monoclonal antibody therapy not only enhances therapeutic efficacy for patients but also reduces side effects, thereby improving patient tolerance and compliance with treatment. Approved biologics for treating NSCLC include cetuximab, bevacizumab, nivolumab, and pembrolizumab. Among them, nivolumab, pembrolizumab, and immunotherapy are highly correlated.
3.1 EGFR monoclonal antibodies
Cetuximab is a recombinant chimeric human/mouse IgG1 monoclonal antibody that binds to the EGFR and competitively inhibits the binding of epidermal growth factor (EGF) and other ligands, thereby blocking ligand-induced EGFR phosphorylation and downstream signaling pathways (82). In advanced lung cancer patients, the role of cetuximab in combination with conventional chemotherapy remains unclear. Clinical study results of cetuximab combined with paclitaxel/carboplatin as first-line therapy for advanced NSCLC show: median PFS was 4.40 months in the cetuximab group and 4.24 months in the TC (Taxane/Carboplatin) group. Median OS was 9.69 months in the cetuximab group and 8.38 months in the TC group. ORR was 25.7% in the cetuximab group and 17.2% in the TC group. No significant benefit was observed in advanced NSCLC patients (83).
3.2 VEGF monoclonal antibodies
VEGF monoclonal antibodies promote tumor growth by enhancing endothelial cell proliferation and survival, increasing endothelial cell migration and invasion, increasing vascular permeability, and enhancing chemotaxis and homing of bone marrow-derived precursor cells (84). Bevacizumab is a humanized monoclonal antibody against VEGF that reduces tumor expansion by controlling abnormal vascular growth around tumors. It was the first anti-tumor angiogenesis drug approved for first-line treatment of metastatic colorectal cancer by the FDA in 2004. The expression of HER-1/EGFR and VEGF molecules in NSCLC is associated with poor prognosis. The AVAil trial evaluated cisplatin/gemcitabine (CG) plus bevacizumab in advanced non-squamous NSCLC, with a high-dose PFS of 6.5 months, a low-dose PFS of 6.7 months, and a control group of 6.1 months, with ORRs of 20.1%, 34.1%, and 30.4%, respectively, for placebo, low-dose bevacizumab, and high-dose bevacizumab plus CG, suggesting that bevacizumab (7.5 or 15 mg/kg) in combination with CG significantly improves PFS and objective RR (85). The role of bevacizumab combined with chemotherapy (docetaxel or pemetrexed) or erlotinib in refractory non-squamous NSCLC suggests a one-year survival rate of 57.4% for bevacizumab-erlotinib, 53.8% for bevacizumab-chemotherapy, and 33.1% for chemotherapy alone. The results of PFS and OS favor bevacizumab combined with chemotherapy or erlotinib over monotherapy (86).
4 Antibody-drug conjugate
ADC drugs are a novel class of anti-tumor medications that link small molecule cytotoxic drugs with monoclonal antibodies through linkers. These drugs can specifically target tumor cells, combining the potent cytotoxic effects of traditional small molecule chemotherapy with the tumor-targeting properties of antibody drugs (6, 87). Currently, several ADC drugs are under research in the field of lung cancer and have shown significant clinical efficacy (Figure 2).
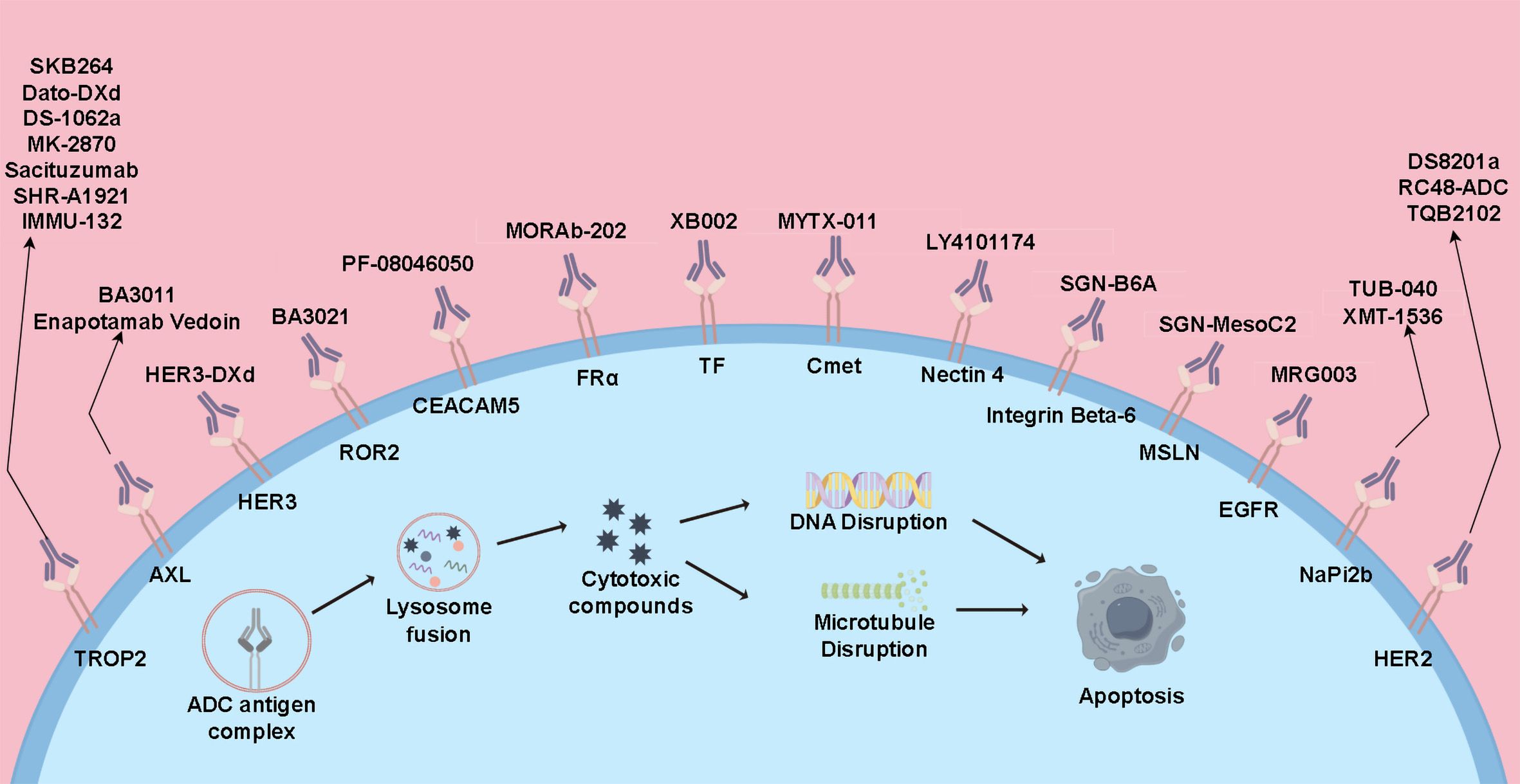
Figure 2. Mechanisms of action of ADCs and current therapeutic drugs.
4.1 HER-2 ADC therapy
Trastuzumab deruxtecan (T-DXd; DS-8201) consists of a humanized anti-HER-2 monoclonal antibody (trastuzumab) linked to a topoisomerase I inhibitor. Previous clinical trials have validated its efficacy in HER-2-positive breast and gastric cancers, leading to its approval for metastatic HER-2-positive breast cancer and gastric cancer (88, 89). DESTINY-Lung01 evaluated T-DXd in refractory HER-2-overexpressing or HER-2-mutant NSCLC, showing a median PFS of 8.2 months, median OS of 18.6 months, median DOR of 10.6 months, and DCR of 92.3%, demonstrating its efficacy in treating HER-2-positive NSCLC patients (90). DESTINY-Lung02 studied the efficacy and safety of trastuzumab deruxtecan (DS-8201) in previously treated HER-2-mutant advanced NSCLC. In patients receiving 5.4 mg/kg dose of trastuzumab deruxtecan, the ORR was 49%, with median PFS of 9.9 months. In the 6.4 mg/kg dose group, the ORR was 56.0%, and median PFS reached 15.4 months (91). Based on these trials, T-DXd has been recommended for treating previously treated HER-2-mutant advanced NSCLC patients.
T-DM1 is an ADC drug that consists of an anti-HER-2 antibody (trastuzumab) linked to the microtubule inhibitor emtansine (DM1) via a non-cleavable thioether linker. In single-agent treatment, it achieved an ORR of 6.7% in recurrent HER-2-positive NSCLC. The median follow-up time was 9.2 months, with median PFS and median OS of 2.0 months and 10.9 months, respectively. Due to limited efficacy, the study was terminated early (92). This treatment approach did not achieve satisfactory results in other studies involving HER-2-positive NSCLC, warranting further investigation into its efficacy (93, 94).
4.2 HER-3 ADC therapy
HER-3-DXd is an antibody-drug conjugate composed of a HER-3 antibody linked to a topoisomerase I inhibitor. HER-3 receptor tyrosine kinase protein is expressed in most EGFR-mutant lung cancers, although it is not a known mechanism of resistance to EGFR inhibitors. The HETHENA-Lung01 trial evaluated the efficacy and safety of patritumab deruxtecan in locally advanced or metastatic NSCLC patients with EGFR mutations who progressed on EGFR-TKI therapy. It reported an ORR of 29.8%, median PFS of 5.5 months, and median OS of 11.9 months. The study observed efficacy across a broad range of tumor HER-3 membrane expression levels and different mechanisms of EGFR TKI resistance (95).
4.3 TROP-2 ADC therapy
Datopotamab deruxtecan (Dato-DXd) is an ADC drug targeting the transmembrane protein TROP-2, consisting of a humanized anti-TROP-2 IgG1 monoclonal antibody conjugated with a topoisomerase I inhibitor. Currently, Dato-DXd is undergoing multiple studies in the field of lung cancer. The TROPION-Lung01 trial aimed to evaluate the efficacy and safety of Dato-DXd monotherapy compared to docetaxel in patients with advanced NSCLC who had received at least one prior treatment, with or without driver gene mutations. The Dato-DXd group demonstrated a significant PFS (PFS) benefit compared to the docetaxel group (4.4 months vs. 3.7 months). It is noteworthy that in the non-squamous subgroup, patients showed better PFS with Dato-DXd compared to docetaxel, with PFS of 5.6 months and 3.7 months, respectively (96).
TROPION-Lung02 evaluated the safety and efficacy of Dato-DXd (at 4 or 6 mg/kg) plus pembrolizumab ± platinum-based chemotherapy in patients with advanced NSCLC in the first-line or previously treated setting. The results showed an ORR of 38% in the doublet group and 49% in the triplet group. In the first-line treatment population, the ORR was 50% in the doublet group and 57% in the triplet group (97).
Sacituzumab govitecan is another ADC targeting TROP-2. The EVOKE-02 trial evaluated sacituzumab govitecan in combination with pembrolizumab as first-line treatment for patients with advanced or metastatic NSCLC negative for driver gene mutations. The results showed an ORR of 55.5% and a disease control rate (DCR) of 82% in the patient population. In the subgroup of patients with PD-L1 tumor proportion score (TPS) ≥50%, the ORR was notably higher at 69%, with a DCR of 86%, demonstrating promising early efficacy and manageable safety (98).
ADCs have received extensive attention in the treatment of NSCLC and have shown promising results in clinical trials. However, the complex combination of antibody and drug brings not only strong efficacy but also a high incidence of toxic side effects. A large-scale meta-analysis of existing ADC clinical trials indicates that the overall incidence of any-grade adverse events is 100.0%, and the incidence of ≥3 grade adverse events is 6.2%, limiting their further clinical use (99). The toxicity of ADCs mainly arises from two sources: the toxicity of the antibody molecule and the toxicity of the cytotoxic compounds. The antibody-induced toxicity mainly stems from damage to normal tissues expressing the target antigen, varying greatly based on target and patient expression. The cytotoxic compounds toxicity is primarily due to the toxin molecules’ release into the bloodstream, causing damage to normal cells, or non-specific internalization of ADCs by normal cells, often resulting in adverse effects similar to those of chemotherapeutic agents, such as hematologic toxicity, alopecia, and gastrointestinal reactions (6, 87).
In the DESTINY-Lung02 study, NSCLC patients were treated with Trastuzumab Deruxtecan at doses of 5.4 mg/kg and 6.4 mg/kg every 3 weeks. The results showed that 96% of patients in the low-dose group reported any-grade adverse events. Among them, 38.6% in the low-dose group experienced drug-related ≥3 grade AEs (91).In the TROPION-Lung01 study, NSCLC patients were treated with docetaxel or datopotamab deruxtecan. Results showed 25% and 41% of patients had grade 3 or higher TRAEs in the datopotamab deruxtecan and docetaxel groups, respectively. The Dato-DXd group had a higher incidence of grade 3 or higher drug-related ILD (3% vs. 1%) (100).
The high clinical efficacy of ADCs, coupled with their high incidence of adverse events, limits their further clinical application. Research on ADCs aims to develop safer and more effective drugs through mechanism exploration and to identify better dosing strategies through clinical trials to benefit patients. In new drug development, peptide-drug conjugates (PDCs) are a potential option, where antibody-binding regions are masked by unique peptides. Protease activity in the tumor microenvironment can cut these masking peptides, allowing PDCs to bind to target antigens and reduce toxicity (101, 102). Additionally, developing bispecific antibodies targeting two tumor-associated antigens, which strongly bind only to cells co-expressing both antigens, could further enhance drug selectivity. Improving the cytotoxicity of the toxin and the stability of the linker can also enhance drug efficacy (103). Clinical trials are exploring optimal drug dosing and combination strategies, such as ADCs with chemotherapy or immunotherapy, to benefit patients. Several drugs targeting NSCLC are currently under development and undergoing related clinical trials, with the hope that these studies will provide new treatment options for NSCLC patients (Table 2).
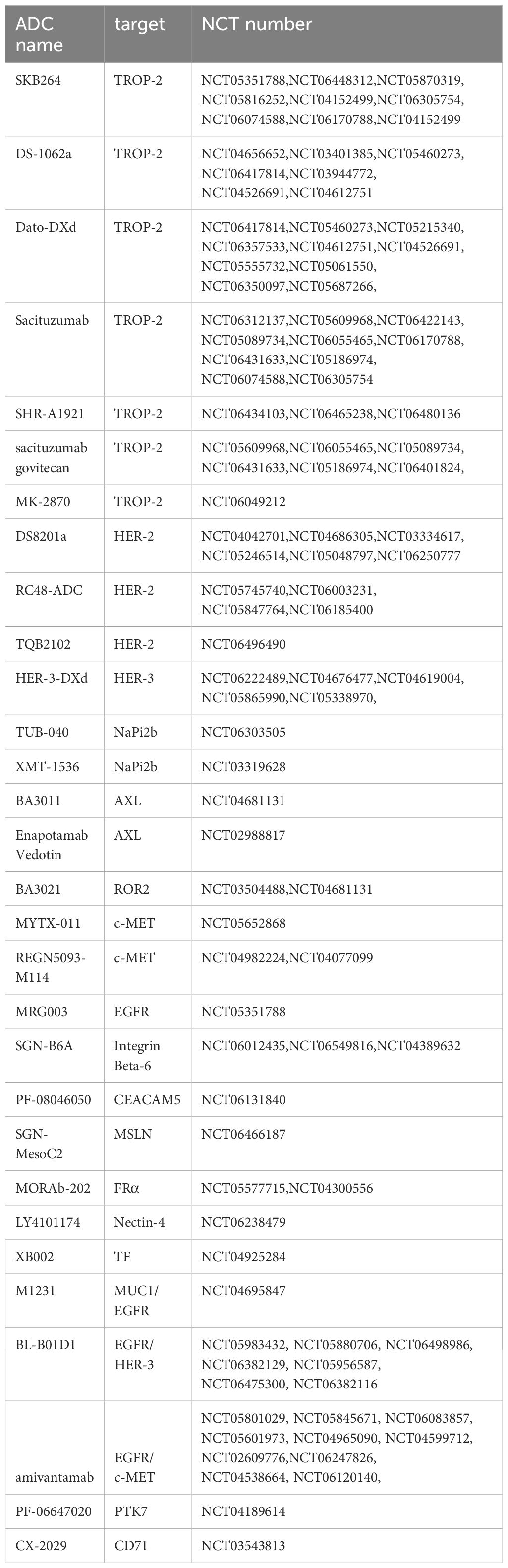
Table 2. Clinical trials of ADC therapies for NSCLC currently underway.
Despite years of progress in basic research and clinical trials, lung cancer remains one of the deadliest diseases globally, attracting extensive research attention. we summarized several current major targets used in treatment and clinical trials, and provides an initial overview of the resistance mechanisms associated with these targets and current resistance-related treatment approaches. In addition to the main targets mentioned in this article, numerous other targets are under investigation and clinical trials. New treatment modalities, including immunotherapy combinations and multi-targeted TKIs, are expected to further optimize targeted treatment outcomes.
Author contributions
LY: Data curation, Investigation, Writing – original draft. RY: Data curation, Investigation, Writing – original draft. ZL: Data curation, Formal Analysis, Investigation, Writing – original draft. T: Data curation, Writing – review & editing, Investigation. BL: Data curation, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: Cancer J Clin. (2022) 72:7–33. doi: 10.3322/caac.21708
2. Gao S, Li N, Wang S, Zhang F, Wei W, Li N, et al. Lung cancer in people’s republic of China. J Thorac Oncol. (2020) 15:1567–76. doi: 10.1016/j.jtho.2020.04.028
3. Broderick SR. Adjuvant and neoadjuvant immunotherapy in non–small cell lung cancer. Thorac Surg clinics. (2020) 30:215–20. doi: 10.1016/j.thorsurg.2020.01.001
4. Reck M, Remon J, Hellmann MD. First-line immunotherapy for non–small-cell lung cancer. J Clin Oncol. (2022) 40:586–97. doi: 10.1200/JCO.21.01497
5. La Montagna M, Ginn L, Garofalo M. Mechanisms of drug resistance mediated by long non-coding RNAs in non-small-cell lung cancer. Cancer Gene Ther. (2021) 28:175–87. doi: 10.1038/s41417-020-00214-3
6. Desai A, Abdayem P, Adjei AA, Planchard D. Antibody-drug conjugates: A promising novel therapeutic approach in lung cancer. Lung Cancer. (2022) 163:96–106. doi: 10.1016/j.lungcan.2021.12.002
7. Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, Bisht B, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol cancer. (2023) 22:40. doi: 10.1186/s12943-023-01740-y
8. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathology: Mech Disease. (2011) 6:49–69. doi: 10.1146/annurev-pathol-011110-130206
9. Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer—is it becoming a reality? Nat Rev Clin Oncol. (2010) 7:401–14. doi: 10.1038/nrclinonc.2010.64
10. Rothschild SI. Targeted therapies in non-small cell lung cancer—beyond EGFR and ALK. Cancers. (2015) 7:930–49. doi: 10.3390/cancers7020816
11. Gazdar A. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. (2009) 28:S24–31. doi: 10.1038/onc.2009.198
12. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. New Engl J Med. (2009) 361:958–67. doi: 10.1056/NEJMoa0904554
13. D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. (2011) 29:2066. doi: 10.1200/JCO.2010.32.6181
14. Harrison PT, Vyse S, Huang PH eds. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. In: Seminars in cancer biology ( Semin Cancer Biol). Elsevier.
15. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2012) 13:239–46. doi: 10.1016/S1470-2045(11)70393-X
16. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. (2013) 19:2240–7. doi: 10.1158/1078-0432.CCR-12-2246
17. Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci. (2008) 105:2070–5. doi: 10.1073/pnas.0709662105
18. Sos ML, Rode HB, Heynck S, Peifer M, Fischer F, Klüter S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res. (2010) 70:868–74. doi: 10.1158/0008-5472.CAN-09-3106
19. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer discovery. (2014) 4:1046–61. doi: 10.1158/2159-8290.CD-14-0337
20. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. New Engl J Med. (2017) 376:629–40. doi: 10.1056/NEJMoa1612674
21. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. New Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
22. Liu X, Zhang X, Yang L, Tian X, Dong T, Ding CZ, et al. Preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Cancer Res. (2019) 79:1320–. doi: 10.1158/1538-7445.AM2019-1320
23. Schalm S, Dineen T, Lim SM, Park C-W, Hsieh J, Woessner R, et al. 1296P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann Oncol. (2020) 31:S839. doi: 10.1016/j.annonc.2020.08.1610
24. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. (2008) 8:11–23. doi: 10.1038/nrc2291
25. Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol cancer. (2018) 17:1–15. doi: 10.1186/s12943-018-0810-4
26. Kohno T, Nakaoku T, Tsuta K, Tsuchihara K, Matsumoto S, Yoh K, et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Trans Lung Cancer Res. (2015) 4:156. doi: 10.3978/j.issn.2218-6751.2014.11.11
27. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. (2007) 131:1190–203. doi: 10.1016/j.cell.2007.11.025
28. Du X, Shao Y, Qin HF, Tai YH, Gao HJ. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac cancer. (2018) 9:423–30. doi: 10.1111/tca.2018.9.issue-4
29. Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. (2009) 15:3143–9. doi: 10.1158/1078-0432.CCR-08-3248
30. Choi YL, Lira ME, Hong M, Kim RN, Choi SJ, Song JY, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol. (2014) 9:563–6. doi: 10.1097/JTO.0000000000000093
31. Hiroyuki Y, de Figueiredo-Pontes LL, Susumu K, Daniel BC. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer - scienceDirect. J Thorac Oncol. (2012) 7:1086–90. doi: 10.1097/JTO.0b013e3182570919
32. Shaw AT, Kim DW, Nakagawa K, Seto T, Jänne PA. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. New Engl J Med. (2013) 368:2385–94. doi: 10.1056/NEJMoa1214886
33. Peters S, Camidge DR, Shaw AT, Gadgeel S, Mok T. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med. (2017) 377:829. doi: 10.1056/NEJMoa1704795
34. Camidge DR, Kim HR, Ahn MJ, Yang CH, Popat S. Brigatinib versus crizotinib in ALK-positive non–small-cell lung cancer. New Engl J Med. (2018) 379:829–38. doi: 10.1056/NEJMoa1810171
35. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery. (2016) 6(10):1118–33. doi: 10.1158/2159-8290.CD-16-0596
36. Pan Y, Deng C, Qiu Z, Wu F. The resistance mechanisms and treatment strategies for ALK-rearranged non-small cell lung cancer. Front Oncol. (2021) 11. doi: 10.3389/fonc.2021.713530
37. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. (2015) 28:70–81. doi: 10.1016/j.ccell.2015.05.010
38. Solomon BJ, Besse B, Bauer TM, Ou SHI. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study (vol 19, pg 1654, 2018). Lancet Oncol. (2018) 2019:20. doi: 10.1016/S1470-2045(18)30649-1
39. Solomon BJ, Bauer TM, Mok TS, Liu G, Mazieres J, de Marinis F, et al. Efficacy and safety of first-line lorlatinib versus crizotinib in patients with advanced, ALK-positive non-small-cell lung cancer: updated analysis of data from the phase 3, randomised, open-label CROWN study. Lancet Respir Med. (2023) 11:354–66. doi: 10.1016/S2213-2600(22)00437-4
40. Tian X, Li Y, Huang Q, Zeng H, Wei Q, Tian P. High PD-L1 expression correlates with an immunosuppressive tumour immune microenvironment and worse prognosis in ALK-rearranged non-small cell lung cancer. Biomolecules. (2023) 13:991. doi: 10.3390/biom13060991
41. D'Angelo A, Sobhani N, Chapman R, Bagby S, Roviello G. Focus on ROS1-positive non-small cell lung cancer (NSCLC): crizotinib, resistance mechanisms and the newer generation of targeted therapies. Cancers. (2020) 3293:3293. doi: 10.3390/cancers12113293
42. Jun HJ, Johnson H, Bronson RT, De Feraudy S, White F, Charest A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-syt1 phosphorylation. Cancer Res. (2012) 72:3764–74. doi: 10.1158/0008-5472.CAN-11-3990
43. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial Malignancies. Nat Rev Cancer. (2013) 13(11):772–87. doi: 10.1038/nrc3612
44. Wiesweg M, Eberhardt WEE, Reis H, Ting S, Schuler M. High prevalence of concomitant oncogene mutations in prospectively identified patients with ROS1-positive metastatic lung cancer. J Thorac Oncol. (2017) 12:54–64. doi: 10.1016/j.jtho.2016.08.137
45. Tejas P, Smith DE, Bunn PA, Aisner DL, Le AT, Mark H, et al. The incidence of brain metastases in stage IV ROS1-rearranged non-small cell lung cancer and rate of central nervous system progression on crizotinib. J Thorac Oncol. (2018) 13(11):S155608641830772X–. doi: 10.1093/annonc/mdz131
46. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Varella-Garcia M, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. (2019) 30(7):1121–6. doi: 10.1093/annonc/mdz063.005
47. Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a pan–TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. (2016) 628:628–39. doi: 10.1158/1535-7163.MCT-15-0758
48. Dziadziuszko R, Krebs MG, Braud FD, Siena S, Drilon A, Doebele RC, et al. Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion–positive non–small-cell lung cancer. J Clin Oncol. (2021) 39:1253–63. doi: 10.1200/JCO.20.03025
49. Shaw AT, Solomon BJ, Chiari R, Riely GJ, Besse B, Soo RA, et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. (2019) 20:1691–701. doi: 10.1016/S1470-2045(19)30655-2
50. Landi L, Tiseo M, Heukamp L, Menon R, Spitaleri G, Cortinovis D, et al. Secondary ROS1 mutations and lorlatinib sensitivity in crizotinib-refractory ROS1 positive NSCLC: Results of the prospective PFROST trial. Ann Oncol. (2019) 30:v609–v10. doi: 10.1093/annonc/mdz260.011
51. Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. (2006) 31:268–75. doi: 10.1016/j.tibs.2006.03.009
52. Roskoski J. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol Res. (2018) 135:239–58. doi: 10.1016/j.phrs.2018.08.013
53. Śmiech M, Leszczyński P, Kono H, Wardell C, Taniguchi H. Emerging BRAF mutations in cancer progression and their possible effects on transcriptional networks. Genes. (2020) 11:1342. doi: 10.3390/genes11111342
54. Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. (2004) 6:313–9. doi: 10.1016/j.ccr.2004.09.022
55. Guaitoli G, Zullo L, Tiseo M, Dankner M, Rose AA, Facchinetti F. Non-small-cell lung cancer: how to manage BRAF-mutated disease. Drugs Context. (2023) 12:2022–11–3. doi: 10.7573/dic.2022-11-3
56. Dagogo-Jack I, Martinez P, Yeap BY, Ambrogio C, Ferris LA, Lydon C, et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin Cancer Res. (2019) 25:158–65. doi: 10.1158/1078-0432.CCR-18-2062
57. Dankner M, Rose AA, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. (2018) 37:3183–99. doi: 10.1038/s41388-018-0171-x
58. Perrone F, Mazzaschi G, Minari R, Verzè M, Azzoni C, Bottarelli L, et al. Multicenter observational study on metastatic non-small cell lung cancer harboring BRAF mutations: focus on clinical characteristics and treatment outcome of V600E and non-V600E subgroups. Cancers. (2022) 14:2019. doi: 10.3390/cancers14082019
59. Gautschi O, Milia J, Cabarrou B, Bluthgen M-V, Besse B, Smit EF, et al. Targeted therapy for patients with BRAF-mutant lung cancer results from the european EURAF cohort. J Thorac Oncol. (2015) 10:1451–7. doi: 10.1097/JTO.0000000000000625
60. Planchard D, Besse B, Groen HJ, Souquet P-J, Quoix E, Baik CS, et al. An open-label phase 2 trial of dabrafenib plus trametinib in patients with previously treated BRAF V600E–mutant metastatic non-small cell lung cancer. Lancet Oncol. (2016) 17:984. doi: 10.1016/S1470-2045(16)30146-2
61. Planchard D, Smit EF, Groen HJ, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. (2017) 18:1307–16. doi: 10.1016/S1470-2045(17)30679-4
62. Odogwu L, Mathieu L, Blumenthal G, Larkins E, Goldberg KB, Griffin N, et al. FDA approval summary: dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. oncologist. (2018) 23:740–5. doi: 10.1634/theoncologist.2017-0642
63. Spagnolo CC, Ciappina G, Giovannetti E, Squeri A, Granata B, Lazzari C, et al. Targeting MET in non-small cell lung cancer (NSCLC): A new old story? Int J Mol Sci. (2023) 24:10119. doi: 10.3390/ijms241210119
64. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer discovery. (2015) 5:850–9. doi: 10.1158/2159-8290.CD-15-0285
65. Koch JP, Aebersold DM, Zimmer Y, Medová M. MET targeting: time for a rematch. Oncogene. (2020) 39:2845–62. doi: 10.1038/s41388-020-1193-8
66. Digumarthy SR, Mendoza DP, Zhang EW, Lennerz JK, Heist RS. Clinicopathologic and imaging features of non-small-cell lung cancer with MET exon 14 skipping mutations. Cancers. (2019) 11:2033. doi: 10.3390/cancers11122033
67. Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543. doi: 10.1038/nature13385
68. Paik PK, Felip E, Veillon R, Sakai H, Le X. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. New Engl J Med. (2020) 383:931–43. doi: 10.1056/NEJMoa2004407
69. Westover D, Zugazagoitia J, Cho B, Lovly C, Paz-Ares L. Mechanisms of acquired resistance to first-and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. (2018) 29:i10–i9. doi: 10.1093/annonc/mdx703
70. Wu Y-L, Zhang L, Kim D-W, Liu X, Lee DH, Yang JC-H, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. (2018) 36:3101–+. doi: 10.1200/JCO.2018.77.7326
71. Wolf J, Seto T, Han J-Y, Reguart N, Garon EB, Groen HJ, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. Am Soc Clin Oncol. (2019) 43(6):1092–111. doi: 10.1200/JCO.2019.37.15_suppl.9004
72. Lindsay CR, Garassino MC, Nadal E, Öhrling K, Scheffler M, Mazieres J. On target: Rational approaches to KRAS inhibition for treatment of non-small cell lung carcinoma. Lung Cancer. (2021) 160:152–65. doi: 10.1016/j.lungcan.2021.07.005
73. Lim TKH, Skoulidis F, Kerr KM, Ahn M-J, Kapp JR, Soares FA, et al. KRAS G12C in advanced NSCLC: prevalence, co-mutations, and testing. Lung Cancer. (2023) 184:107293. doi: 10.1016/j.lungcan.2023.107293
74. Salgia R, Pharaon R, Mambetsariev I, Nam A, Sattler M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep Med. (2021) 2:107293. doi: 10.1016/j.xcrm.2020.100186
75. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p. G12C mutation. New Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
76. Wang Y, Wang Y, Li J, Li J, Che G. Clinical significance of PIK3CA gene in non-small-cell lung cancer: a systematic review and meta-analysis. BioMed Res Int. (2020) 2020:3608241. doi: 10.1155/2020/3608241
77. Sun R, Hou Z, Zhang Y, Jiang B. Drug resistance mechanisms and progress in the treatment of EGFR−mutated lung adenocarcinoma. Oncol Letters. (2022) 24:1–16. doi: 10.3892/ol.2022.13528
78. Liu F, Wei Y, Zhang H, Jiang J, Zhang P, Chu Q. NTRK fusion in non-small cell lung cancer: diagnosis, therapy, and TRK inhibitor resistance. Front Oncol. (2022) 12:864666. doi: 10.3389/fonc.2022.864666
79. Garg P, Singhal S, Kulkarni P, Horne D, Malhotra J, Salgia R, et al. Advances in non-small cell lung cancer: current insights and future directions. J Clin Med. (2024) 13:4189. doi: 10.3390/jcm13144189
80. Salmani-Javan E, Jadid MFS, Zarghami N. Recent advances in molecular targeted therapy of lung cancer: Possible application in translation medicine. Iranian J Basic Med Sci. (2024) 27:122. doi: 10.22038/IJBMS.2023.72407.15749
81. Silva AP, Coelho PV, Anazetti M, Simioni PU. Targeted therapies for the treatment of non-small-cell lung cancer: Monoclonal antibodies and biological inhibitors. Hum Vaccines immunotherapeutics. (2017) 13:843–53. doi: 10.1080/21645515.2016.1249551
82. Mazzarella L, Guida A, Curigliano G. Cetuximab for treating non-small cell lung cancer. Expert Opin Biol Ther. (2018) 18:483–93. doi: 10.1080/14712598.2018.1452906
83. Lynch TJ, Patel T, Dreisbach L, McCleod M, Heim WJ, Hermann RC, et al. Cetuximab and first-line taxane/carboplatin chemotherapy in advanced non–small-cell lung cancer: Results of the randomized multicenter phase III trial BMS099. J Clin Oncol. (2010) 28:911–7. doi: 10.1200/JCO.2009.21.9618
84. Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. (2002) 2:826–35. doi: 10.1038/nrc925
85. Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non–small-cell lung cancer: AVAiL. J Clin Oncol. (2009) 27:1227–34. doi: 10.1200/JCO.2007.14.5466
86. Herbst RS, O'Neill VJ, Fehrenbacher L, Belani CP, Bonomi PD, Hart L, et al. Phase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non-small-cell lung cancer. J Clin Oncol. (2007) 25:4743–50. doi: 10.1200/JCO.2007.12.3026
87. Passaro A, Jänne PA, Peters S. Antibody-drug conjugates in lung cancer: recent advances and implementing strategies. J Clin Oncol. (2023) 41:3747–61. doi: 10.1200/JCO.23.00013
88. Shitara K, Bang Y-J, Iwasa S, Sugimoto N, Ryu M-H, Sakai D, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. New Engl J Med. (2020) 382:2419–30. doi: 10.1056/NEJMoa2004413
89. Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. New Engl J Med. (2020) 382:610–21. doi: 10.1056/NEJMoa1914510
90. Li B, Smit E, Goto Y, Nakagawa K, Goto K, Mazieres J, et al. 976P Phase II trial of trastuzumab deruxtecan (T-DXd) in patients (Pts) with HER2-mutated (HER2m) metastatic non-small cell lung cancer (NSCLC): Registrational data from DESTINY-Lung01. Ann Oncol. (2022) 33:S995–S6. doi: 10.1016/j.annonc.2022.07.1104
91. Goto K, Goto Y, Kubo T, Ninomiya K, Kim S-W, Planchard D, et al. Trastuzumab deruxtecan in patients with HER2-mutant metastatic non–small-cell lung cancer: Primary results from the randomized, phase II DESTINY-Lung02 trial. J Clin Oncol. (2023) 41:4852–63. doi: 10.1200/JCO.23.01361
92. Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, et al. A phase II study of trastuzumab emtansine in HER2-positive non–small cell lung cancer. J Thorac Oncol. (2018) 13:273–9. doi: 10.1016/j.jtho.2017.10.032
93. Iwama E, Zenke Y, Sugawara S, Daga H, Morise M, Yanagitani N, et al. Trastuzumab emtansine for patients with non–small cell lung cancer positive for human epidermal growth factor receptor 2 exon-20 insertion mutations. Eur J Cancer. (2022) 162:99–106. doi: 10.1016/j.ejca.2021.11.021
94. Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: results from a phase II basket trial. J Clin Oncol. (2018) 36:2532. doi: 10.1200/JCO.2018.77.9777
95. Yu HA, Goto Y, Hayashi H, Felip E, Chih-Hsin Yang J, Reck M, et al. HERTHENA-Lung01, a phase II trial of patritumab deruxtecan (HER3-DXd) in epidermal growth factor receptor–mutated non–small-cell lung cancer after epidermal growth factor receptor tyrosine kinase inhibitor therapy and platinum-based chemotherapy. J Clin Oncol. (2023) 41:5363–75. doi: 10.1200/JCO.23.01476
96. Atmaca A, Ahn M, Lisberg A, Paz-Ares L, Cornelissen R, Girard N, et al. Datopotamab deruxtecan (Dato-DXd) vs docetaxel in previously treated advanced/metastatic (adv/met) non-small cell lung cancer (NSCLC): Results of the randomized phase 3 study TROPION-Lung01. Pneumologie. (2024) 78:Po 108. doi: 10.1055/s-0044-1781395
97. Goto Y, Su W-C, Levy BP, Rixe O, Yang T-Y, Tolcher AW, et al. TROPION-Lung02: Datopotamab deruxtecan (Dato-DXd) plus pembrolizumab (pembro) with or without platinum chemotherapy (Pt-CT) in advanced non-small cell lung cancer (aNSCLC). J Clin Oncol. (2023) 41:9004–. doi: 10.1200/JCO.2023.41.16_suppl.9004
98. Cho BC, Dols MC, Reyes Cabanillas R, Vicente Baz D, Fuentes Pradera J, Grisanti S, et al. OA05.04 sacituzumab govitecan + Pembrolizumab in 1L metastatic non-small cell lung cancer: preliminary results of the EVOKE-02 study. J Thorac Oncol. (2023) 18:S54. doi: 10.1016/j.jtho.2023.09.041
99. Zhu Y, Liu K, Wang K, Zhu H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: A systematic review and meta-analysis. Cancer. (2023) 129:283–95. doi: 10.1002/cncr.v129.2
100. Girard N, Okamoto I, Lisberg A, Pons-Tostivint E, Cornelissen R, Hong M, et al. 59P Datopotamab deruxtecan (Dato-DXd) in patients with previously treated advanced non-small cell lung cancer (NSCLC): Nonsquamous (NSQ) histology in the phase III TROPION-Lung01 trial. ESMO Open. (2024) 9:102638. doi: 10.1016/j.esmoop.2024.102638
101. Ma L, Wang C, He Z, Cheng B, Zheng L, Huang K. Peptide-drug conjugate: a novel drug design approach. Curr medicinal Chem. (2017) 24:3373–96. doi: 10.2174/0929867324666170404142840
102. Yang S-B, Banik N, Han B, Lee D-N, Park J. Peptide-based bioconjugates and therapeutics for targeted anticancer therapy. Pharmaceutics. (2022) 14:1378. doi: 10.3390/pharmaceutics14071378
Keywords: NSCLC, target therapy, TKIs, monoclonal antibodies, antibody-drug conjugate
Citation: Yu L, Yang R, Long Z, Tao Q and Liu B (2024) Targeted therapy of non-small cell lung cancer: mechanisms and clinical trials. Front. Oncol. 14:1451230. doi: 10.3389/fonc.2024.1451230
Received: 18 June 2024; Accepted: 09 September 2024;
Published: 26 September 2024.
Edited by:
Shiyou Wei, Sichuan University, ChinaReviewed by:
Jinwei Zhang, Chinese Academy of Sciences (CAS), ChinaWenyu Zhai, Sun Yat-sen University Cancer Center, China
Copyright © 2024 Yu, Yang, Long, Tao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Liu, YmlubGl1MjAyMDAzQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship