 William Han Bae
William Han Bae Stefania Maraka
Stefania Maraka Ahmad Daher
Ahmad Daher- 1Division of Hematology/Oncology, Department of Internal Medicine, University of Illinois Chicago, Chicago, IL, United States
- 2Department of Neurology and Rehabilitation, University of Illinois Chicago, Chicago, IL, United States
Glioblastoma remains the most prevalent and aggressive primary malignant brain tumor in adults, characterized by limited treatment options and a poor prognosis. Previous drug repurposing efforts have yielded only marginal survival benefits, particularly those involving inhibitors targeting receptor tyrosine kinase and cyclin-dependent kinase-retinoblastoma pathways. This limited efficacy is likely due to several critical challenges, including the tumor’s molecular heterogeneity, the dynamic evolution of its genetic profile, and the restrictive nature of the blood-brain barrier that impedes effective drug delivery. Emerging diagnostic tools, such as circulating tumor DNA and extracellular vesicles, offer promising non-invasive methods for real-time tumor monitoring, potentially enabling the application of targeted therapies to more selected patient populations. Moreover, innovative drug delivery strategies, including focused ultrasound, implantable drug-delivery systems, and engineered nanoparticles, hold potential for enhancing the bioavailability and therapeutic efficacy of treatments.
1 Introduction
Glioblastoma (GB) is the most common and aggressive adult primary malignant brain tumor. First-line FDA-approved treatment relies on a multimodal approach of surgery, radiation, temozolomide (TMZ) chemotherapy, and tumor-treating fields. Unfortunately, the prognosis remains poor, with a median overall survival (mOS) of 20.9 months and a median progression-free survival (mPFS) of 6.7 months (1). The therapeutic outcomes are even less favorable in recurrent disease, where there is no standard of care, and the mOS from the first recurrence is around 6.5 months (2). Currently, there are only limited FDA-approved drugs for GB, such as TMZ, Bevacizumab, CCNU, and BiCNU, highlighting the urgent need for new treatment options for GB. One of the most promising approaches in cancer therapy is implementing next-generation sequencing (NGS) techniques to uncover actionable mutations that can be targeted in a tissue-agnostic fashion. In that regard, repurposing existing anticancer medications offers a potentially efficient and effective tool to discover new therapeutic agents for GB.
Here, we will discuss the previously explored repurposing efforts in the treatment of GB based on targeting two of the three most altered signal transduction pathways in GB: Receptor tyrosine kinase (RTK) and cyclin-dependent kinase (CDK)-retinoblastoma (Rb) (3). Approximately 57-60% of glioblastomas exhibit alterations in the RTK/PI3K pathway, with EGFR amplification or mutation being the most common, occurring in about 40-50% of cases (3). The CDK-Rb pathway is also frequently altered, with aberrations occurring in about 78-80% of cases, often involving the loss of CDKN2A, amplification of CDK4 or CDK6, and/or inactivation of the Rb1 protein. There are no validated targeted therapeutics for the murine double minute 2 (MDM2)-p53 pathway, the third commonly altered pathway in GB or other malignancies, primarily due to its crucial role in normal cell functions (4).
Despite an increased understanding of GB tumor biology with discoveries of potential targets, the unique challenges posed by this aggressive tumor continue to thwart treatment benefits. The tumor microenvironment (TME) of GB is highly heterogeneous and exceptionally dynamic, creating a landscape where cancer cells can evade therapies and rapidly adapt to treatment pressures. Compounding this complexity is the formidable blood-brain barrier (BBB), which acts as a gatekeeper, severely limiting the delivery of therapeutic agents to the tumor bed. Moreover, the inherent risk and challenges with accessing GB tissue at recurrence further hamper the ability to tailor treatments to individual patients, making it difficult to combat this relentless disease.
However, hope lies in innovative approaches designed to overcome these barriers. Cutting-edge techniques aimed at enhancing the detection of cancer biomarkers are on the horizon, offering the potential for more precise targeting of GB. Additionally, novel drug delivery vehicles, such as nanoparticles (NPs), are being developed to penetrate the BBB and deliver therapies directly to the tumor site. These advancements not only hold promise for improving treatment outcomes but also represent a bold step forward in the fight against GB.
2 RTK pathway inhibitors
2.1 Epidermal growth factor receptor-targeted agents
Anti-EGFR tyrosine kinase inhibitors (TKIs) have been a focal point in GB treatment trials due to the high rates of EGFR alterations, reaching ~60% in GB (5). Gefitinib was the first anti-EGFR TKI tested in GB. Various trials evaluated the role of gefitinib monotherapy in recurrent glioblastoma (rGB) treatment, showing good safety data but without promising efficacy (6, 7). The use of gefitinib with other RTK pathway inhibitors (8) and in newly diagnosed GB (nGB) treatment (9) was similarly inefficacious.
Erlotinib demonstrated limited efficacy in the treatment of GB. Early clinical trials indicated minimal activity, primarily because the drug couldn’t penetrate the blood-brain barrier (BBB) effectively and because the tumor growth wasn’t largely dependent on the targeted pathway (10). Subsequent trials exploring combinations of erlotinib with various chemotherapeutic agents, such as carboplatin, failed to yield significant improvements in PFS or OS in rGB (11). Additionally, while the addition of receptor tyrosine kinase (RTK) pathway inhibitor sirolimus, the mammalian target of rapamycin (mTOR) inhibitor temsirolimus, Ras/Raf/mitogen-activated protein kinase (MAPK) inhibitor sorafenib, and the anti-angiogenic agent bevacizumab was well tolerated, these combinations did not translate into clinically meaningful survival benefits (12–15). Furthermore, erlotinib demonstrated limited efficacy in treating newly diagnosed glioblastoma (nGB) (16).
Lapatinib, a dual inhibitor of EGFR and human epidermal growth factor receptor 2 (HER2), is the most frequently tested EGFR inhibitor in GB trials. However, its activity has been minimal when used as alone (17) and in combination with TMZ (18) or with pazopanib in rGB (19). In the nGB setting, pulsatile dosing of lapatinib in conjunction with TMZ chemoradiation was well-tolerated and showed promise in a phase 2 study, although further clinical data is currently lacking to establish its efficacy (20). The anti-EGFR monoclonal antibody cetuximab has not consistently outperformed existing GB therapies (21, 22). Nevertheless, a subgroup analysis from a phase 2 trial involving rGB patients with EGFR mutations revealed that tumors with EGFR amplification but without the EGFR variant III mutation experienced a statistically significant increase in progression-free survival (PFS) of 3.03 months compared to 1.63 months (p = 0.006), with a trend toward improved overall survival (OS) of 5.56 months versus 3.97 months (p = 0.12) (22). This suggests that certain EGFR mutations may confer a selective advantage in anti-EGFR treatment for GB.
Nimotuzumab is another anti-EGFR monoclonal antibody that has shown some promise, particularly when combined with radiation or TMZ chemoradiation. A phase 2 study reported an improved median overall survival (mOS) when nimotuzumab was combined with radiation in patients with high-grade glioma, showing a mOS of 17.76 months compared to 12.63 months in the placebo plus radiation group (23). Another phase 2 trial yielded similar positive outcomes with nimotuzumab (24). However, a subsequent phase 3 trial that combined nimotuzumab with TMZ chemoradiation did not replicate these survival benefits (25), suggesting a nuanced potential for monoclonal antibodies in targeted GB therapy, dependent on specific patient genetic profiles.
Recent preclinical studies utilizing in vitro glioblastoma stem cells (GCS) models and GB orthotopic xenograft model with EGFR variant III showed antitumor activity along with inhibition of EGFR downstream signaling pathway for the third-generation EGFR inhibitor osimertinib (26). Initial clinical studies of osimertinib in GB treatment are promising. Two small retrospective studies on rGB patients with EGFR mutations showed some benefit when used as monotherapy (27) and in combination with bevacizumab (28). These promising findings indicate that osimertinib’s primary advantage over earlier generations of EGFR inhibitors in GB therapy lies in its superior ability to penetrate BBB.
2.2 Platelet-derived growth factor receptor-targeted agents
Imatinib, a drug primarily approved for leukemia, has been explored for its potential in treating rGB with mixed outcomes. An initial phase 2 study combining imatinib with hydroxyurea demonstrated some antitumor activity, with a median PFS of 14.4 weeks and 9% of patients achieving radiographic responses (29). However, subsequent trials found no significant clinical benefit with imatinib, either as a monotherapy or in combination with hydroxyurea (30–32). Similarly, other early-phase trials that combined imatinib with TMZ (33) or the vascular endothelial growth factor receptor (VEGFR) inhibitor vatalanib (34) reported minimal clinical activity. While neoadjuvant administration of imatinib resulted in detectable drug levels in brain tissue, it had a limited impact on tumor proliferation and patient survival (35, 36), highlighting the drug’s limited efficacy in this context.
Dasatinib, another leukemia-approved drug, was tested in rGB with minimal success. A retrospective study combining dasatinib with bevacizumab showed little activity (37), and further trials involving dasatinib with CCNU highlighted significant hematologic toxicities (38), curtailing additional studies with this combination. More focused clinical trials on rGB harboring activation or overexpression mutations of dasatinib targets, such as SRC, c-kit, EPHA, and PDGFR, also indicated insufficient clinical benefits, even with pulse-dosing strategies combined with bevacizumab (39).
Ripretinib (DCC-2618), an innovative type 2 tyrosine switch control inhibitor of the KIT and PDGFRA activating mutations, showed some potential in an early-phase study in which five high-grade glioma patients were enrolled. One of the two GB patients carrying triple amplification of PDGFRA, KIT, and KDR (4q12 amplicon) showed a remarkable 94% tumor reduction and survived through over 20 cycles. However, larger-scale evidence is still lacking (40).
Despite the theoretical promise based on their successful oncological applications elsewhere, none of those above PDGFR-targeted agents demonstrated substantial benefits in GB, particularly in recurrent settings.
2.3 VEGFR-targeted agents
Cabozantinib, a multikinase inhibitor targeting targets Met, VEGFR, and Axl, has been approved for different cancers. It has been tested in rGBs with mixed results. In large phase 2 trials, cabzantinib demonstrated reasonable tolerance and some clinical activity in rGB patients who were naïve to antiangiogenic therapy (41) or those previously exposed to an antiangiogenic agent (42).
Another multi-kinase inhibitor, Sunitinib, showed limited antitumor activity across several GB trials. Its use with irinotecan resulted in moderate toxicities and insufficient clinical effectiveness (43, 44), leading to the early termination of a subsequent phase 2 trial (45). Another phase 2 trial using sunitinib monotherapy in non-resectable nGB also failed to show antitumor activity (46). Despite these setbacks, interest in sunitinib continues with ongoing trials exploring different dosing strategies in the rGB setting.
Pazopanib, approved for sarcoma, is also a multikinase inhibitor targeting VEGFR and PDGFR. It has similarly struggled to demonstrate efficacy in GB. An initial trial using pazopanib monotherapy in rGB failed to prolong PFS, and a subsequent trial combining pazopanib with lapatinib in rGB yielded questionable antitumor activity with 0% and 15% PFS rates in PTEN/EGFRvIII-positive and PTEN/EGFRvIII- negative cohorts respectively (19). A complex combination therapy trial combining pazopanib and four other drugs showed promising clinical response rates: complete response (CR) in 18.2.%, partial response (PR) in 36.3%, and stable disease (SD) in 27.3% of patients. However, issues with patient compliance halted further exploration of this regimen (47). Additional studies pairing pazopanib with topotecan and bevacizumab also did not meet the anticipated outcomes, recording poor mPFS and mOS rates compared to historical controls, as reported in preliminary results on clinicaltrials.gov (48).
2.4 Fibroblast growth factor receptor-targeted agents
The frequency of FGFR mutations in GB is relatively low, resulting in limited use of FGFR inhibitors in this disease. However, various preclinical studies showed that FGFR signaling has a significant impact on GB progression (49).
Infigratinib monotherapy was tested on twenty-six rGB patients in a phase 2 trial, and it showed limited efficacy overall. However, durable disease control was observed in subgroups of patients harboring FGFR1 or FGFR3 point mutations or with FGFR3-TACC fusion mutation (50).
Two separate FGFR-mutated solid malignancy basket trials with erdafitnib (51) and pemigitinib (52), including thirty-two and twelve glioma patients, respectively, showed promising clinical benefits that have not been verified in subsequent trials yet.
2.5 PIK/Akt/mTOR pathway inhibitors
The PI3K/Akt/mTOR pathway, mutated in approximately 45.6% of GB cases, has been one of the key pathways implicated in tumorigenesis.
Paxslisib is a selective small-molecule PI3K inhibitor used in two glioma trials. A phase 1 study on forty-seven recurrent high-grade glioma patients showed reasonable safety and promising efficacy, with 40% having SD (53). A subsequent multi-center phase 2 study on thirty patients with O6-methylguanine-DNA methyltransferase (MGMT) promoter unmethylated nGB showed encouraging survival data with median PFS and OS of 8.6 months and 15.7 months, respectively (54).
Buparlisib, a pan-PI3K inhibitor, has been the focus of several clinical trials in GB. Although it showed good brain penetrance and tolerability in a phase 2 trial for rGB with PI3K pathway activation mutations, its efficacy was limited, with only a small fraction of patients achieving PFS at six months (55). Subsequent trials combining buparlisib with other therapies like bevacizumab, carboplatin (56), or the c-met inhibitor capmatinib (57) did not demonstrate superior efficacy over monotherapy or existing treatments.
Another therapeutic approach involved the mTOR inhibitors temsirolimus and everolimus, both of which have been extensively evaluated in GB in various clinical settings. An early phase trial using temsirolimus in rGB showed promising results, with 36% of participants showing radiographic responses and significantly longer time to progression than non-responders (58). However, the drug failed to meet efficacy endpoints in subsequent studies, including combinations with TMZ chemoradiation (59) and sorafenib (60). These combinations often resulted in increased toxicity, most notably severe hematologic toxicity, and increased infection risk (61).
Everolimus use with TMZ and radiation in nGB has shown reasonable tolerability in phase 1 trials (62–64). However, subsequent phase 2 trials did not demonstrate a significant survival benefit over historical controls (65) or over TMZ arm in a randomized trial (66). A phase 2 study for nGB treatment with concurrent TMZ, bevacizumab and radiation therapy followed by adjuvant treatment with bevacizumab/everolimus showed a favorable response with median PFS of 11.3 months but not mOS benefit compared to historical control. Additionally, the radiographic objective response rate (ORR) of 61% could have been influenced by the use of bevacizumab, considering its known radiographic effects (67).
2.6 Pan-kinase inhibitors
Anlotinib is a multi-kinase inhibitor targeting VEGFR, PDGFR, and FGFR. In a phase 2 study involving 21 patients with recurrent rGB, anlotinib combined with temozolomide demonstrated efficacy, achieving a median progression-free survival (PFS) of 7.3 months and a median overall survival (OS) of 16.9 months (68). Additionally, anlotinib showed promising activity in patients with MGMT promoter-unmethylated nGB when used in place of temozolomide in a phase 2 study of 32 patients (69).
Regorafinib, a pankinase inhibitor, was initially tested as monotherapy on rGB patients in a phase 2 trial, which showed a survival benefit when compared to CCNU (70). Recently, a large multi-center prospective observational trial on 190 rGB patients showed similar promising mOS and better drug tolerability compared to that seen by Lombardi et al. (71).
3 CDK-Rb targeting agents
Targeting this pathway in oncology has been primarily limited to CDK4/6 inhibitors.
Abemaciclib activity in rGB was assessed in a basket trial involving seventeen GB patients and showed limited effectiveness. A subsequent phase 2 trial on thirty-two rGB with documented CDK mutations showed SD in 35.5% and PR in 3.2% of the patients (72). More recently, abemaciclib was tested on seventy-three nGB patients in a phase 2 study, resulting in improved PFS compared to standard of care (HR 0.72; one-sided p =0.046), suggesting some potential for this drug in specific GB populations, but it failed to demonstrate significant overall survival (OS) benefit (73).
Palbociclib role in GB has been less encouraging. A phase 2 trial on heavily pretreated rGB patients noted adequate pharmacokinetics but ultimately showed limited efficacy, leading to the trial’s termination (74). No further studies are currently investigating its role in GB.
Ribociclib, another CDK4/6 inhibitor, also displayed minimal clinical activity in a phase 0/2 surgical trial on rGB patients (75). The trial identified upregulation of the mTOR pathway as a potential resistance mechanism, suggesting the addition of mTOR inhibitor as a potential strategy to enhance ribociclib effectiveness in GB treatment.
4 Repurposing targeted drugs in GB: challenges and solutions
Below described are the main barriers to GB targeted therapy, which is a field largely dependent on repurposed drugs, with a few exceptions outside the scope of this review.
4.1 Complexity of GB biology
Intra-tumoral heterogeneity, which has also been previously described in GB using single-cell RNA-seq profiling (76), complicates accurate targeted therapy in several ways. Different regions within the tumor exhibit distinct genetic, epigenetic, and transcriptional profiles. As a result, tissue samples, particularly those obtained through biopsy, may not capture the full spectrum of mutations within the tumor, leading to suboptimal treatment strategies that fail to target all tumor cell populations. Furthermore, this variability complicates the identification of reliable therapeutic targets, as a target found in one tumor region may be absent in another, increasing the likelihood of treatment failure. Resistance mechanisms also play a critical role, with certain cell populations potentially being resistant to specific therapies due to their unique genetic or epigenetic characteristics. This resistance can lead to disease progression and recurrence, especially as therapy may select for these resistant clones over time. While single-cell RNA-seq provides valuable insights into tumor heterogeneity, its application in clinical practice is limited by the complexity and resources required for its clinical use.
Redundant signaling pathways in gliomagenesis limit the effectiveness of targeted therapies that focus on a single gene or pathway, even when the tumor’s complete molecular profile is known. Combination strategies that target multiple actionable mutations within a tumor could potentially enhance the efficacy of targeted therapies for GB. However, the potential benefits of such approaches must be carefully weighed against the risk of cumulative toxicities associated with combination treatments. Additionally, our understanding of the role of downstream mutations within gliomagenesis signaling pathways remains incomplete. For example, PTEN mutations, which occur downstream of EGFR signaling, have been identified as a resistance factor in anti-EGFR therapy for GB, highlighting the complexity of these pathways and the challenges in developing effective targeted treatments (77). Figure 1 further illustrates the intricate and overlapping signaling cascades within the RTK pathway, emphasizing the complexity involved in the context of the various targeted therapeutics discussed above.
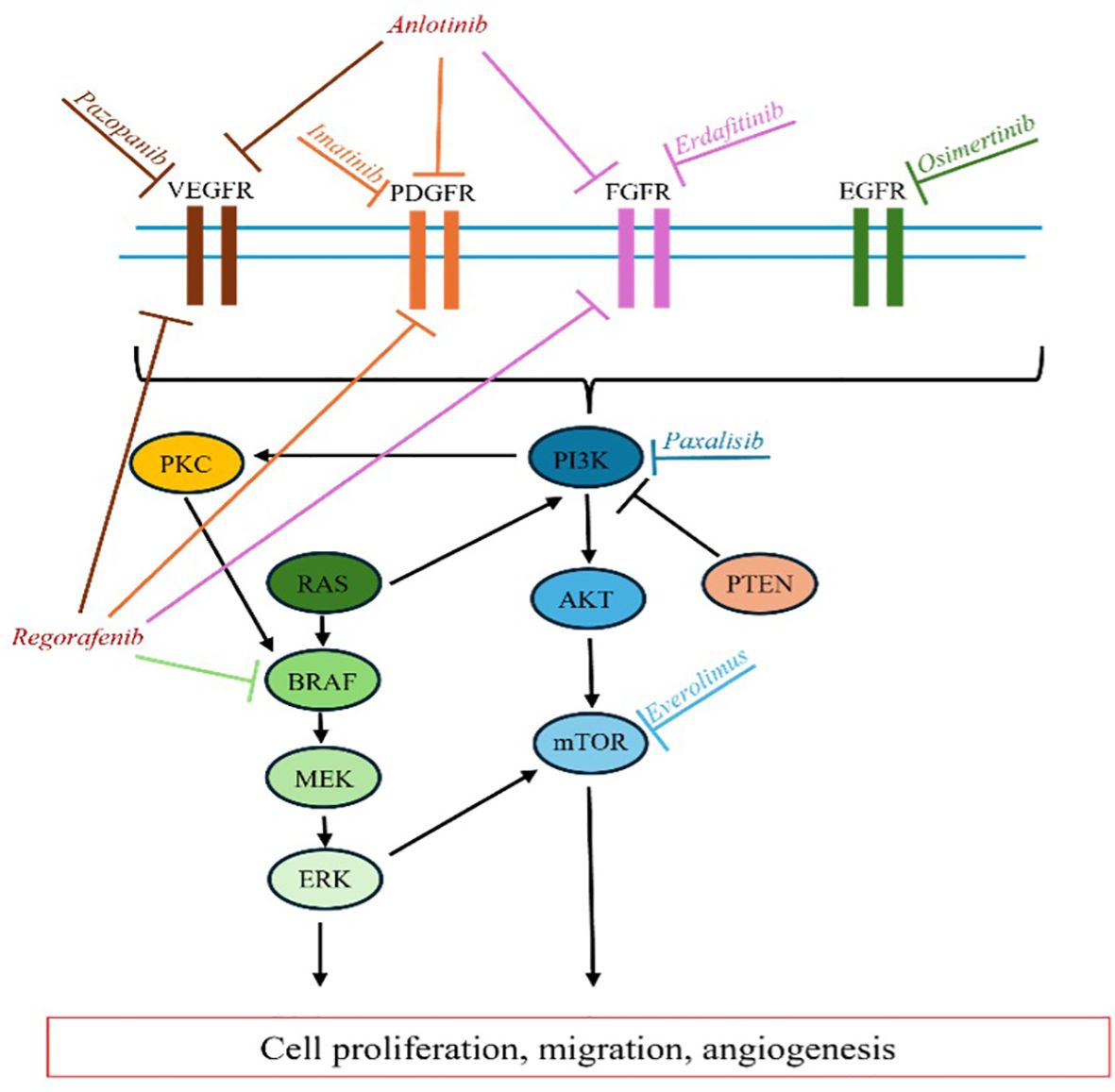
Figure 1. RTK Pathway. Overview of targeted inhibition of various RTK signaling enzymes.
Another challenge is the lack of a consensus on what level of increased expression is considered significant enough to influence clinical decisions. NGS platforms routinely provide the percentage of overexpression of an amplified gene, but there are no studies that have stratified clinical response to a drug by the fold-increase of its target. Retrospective analysis of clinical trial data using targeted therapeutics based on survival by target fold-increase may help refine personalized therapies.
Reliance on a single molecular profiling platform, such as NGS, can diminish the importance of other platforms in identifying personalized treatment response signatures. For instance, methylation profiling of GB specimens is currently the only sequencing method that can identify a subset of IDH-wild type gliomas. This subset represents a negative prognostic marker that is sufficient to diagnose GB in a glioma, regardless of its histopathological grade (78). On the other hand, methylation of MGMT gene promoter is associated with a more favorable prognosis (79). High-throughput drug screening combined with pan-omic molecular profiling of response can help generate relevant predictive biomarker libraries (80), resulting in a more nuanced approach to targeted therapy, one that is not single actionable mutation-based. Such efforts can also help inform future clinical trial design.
4.2 Dynamic evolution of the tumor
GB is a dynamic tumor that continuously evolves in response to therapeutic interventions like radiation and chemotherapy. This evolution is driven by clonal diversity within the tumor, where different cell populations harbor distinct genetic mutations. As treatments impose selective pressures, sensitive clones are eliminated, while resistant clones survive and proliferate. For example, radiation and chemotherapy, such as alkylating agents, work by imposing lethal DNA damage. However, clones with efficient DNA repair mechanisms can selectively survive through radiochemical stress and flourish in less competitive environments. On the other hand, the increased mutation burden from radiation or chemotherapy can potentially lead to the emergence of new mutations that confer resistance to the therapy (81). This dynamic nature means that the tumor’s mutational profile at recurrence differs from its profile at diagnosis, and the problem is exacerbated by limited access to GB tissue at recurrence, as re-resection is not always safe or preferred. The failure of most targeted therapy trials for rGB can be attributed to their focus on the tumor’s initial mutational profile, which may not reflect the tumor’s current genetic state due to its evolution over time. Advances in liquid biopsy techniques for non-invasive profiling of GB can help to overcome this challenge and will be discussed in more detail in the next section.
For nGB, the presence of FDA-approved standard chemotherapy (TMZ) limits the application of new therapies and delays the approval of new drugs for nGB. Advances in clinical trial design for these patients fall under one of three categories:
1. Combining the experimental drug with TMZ so as not to deprive patients of standard of care treatment.
2. Using an experimental drug instead of TMZ in the setting of unmethylated MGMT promoter which results in high expression of the DNA-repair enzyme MGMT. Overexpression of MGMT results in decreased response to alkylating agents such as TMZ, as evidenced by the significantly improved survival outcomes in the MGMT promotor methylated group (Median PFS 19 mo vs. 6 months; p=.014) when adjuvant TMZ, lomustine, and radiation therapy was used (82).
3. Utilizing adaptive trial platforms to accelerate drug discovery in an efficient and cost-effective manner. This approach involves continuously adjusting multiple treatment arms, including adding new ones and terminating others early based on emerging data, all within a single master protocol. Response-adaptive randomization allows trials to progress through different phases more quickly and facilitates the rapid identification of promising therapies and response biomarkers. Currently, two major adaptive trials in GB, AGILE and INSIGhT, have successfully increased enrollment rates by leveraging this methodology (83)
Lastly, the frequency of an actionable mutation within a tumor, as indicated by variant allele frequency (VAF) in NGS reports, varies between different GB specimens. However, targeted therapy trials almost never include a VAF cutoff as an eligibility criterion. Consequently, these trials’ perceived low response rate may be diluted by a subset of patients with low VAF, highlighting the need for detailed subgroup analysis upon trial completion.
4.3 GB microenvironment
4.3.1 BBB
BBB is a critical and complex structure that serves as a dynamic interface between the bloodstream and the CNS. It is primarily composed of endothelial cells, basement membrane, pericyte, and astrocyte. The endothelial cells are tightly connected by intracellular junctions that play a crucial role in maintaining the highly selective permeability of the BBB, allowing it to regulate the movement of ions, molecules, and cells between the blood and the neural tissue (84, 85). This selective permeability is essential for maintaining CNS homeostasis, protecting the brain from toxins, pathogens, and immune cells, and regulating the influx and efflux of nutrients and other compounds necessary for brain function. The BBB’s integrity is hallmarked by its high transendothelial electrical resistance (TEER), which restricts the passage of most water-soluble compounds, including polar drugs (86). Only small, lipophilic molecules, such as oxygen and carbon dioxide, can passively diffuse across the BBB. Additionally, specific transporters and receptors on the endothelial cells facilitate the selective transport of essential molecules like glucose, amino acids, and nucleosides, while actively effluxing potentially harmful substances, including many therapeutic drugs, back into the bloodstream.
This highly selective and regulated nature of the BBB presents a significant challenge in the treatment of GB. The barrier limits the delivery of therapeutic agents, particularly large or hydrophilic molecules, into the brain. Most anti-neoplastic drugs, which are often hydrophilic and large, cannot cross the BBB efficiently due to their size and polarity (87, 88). Furthermore, the presence of active efflux transporters like P-glycoproteins exacerbates this challenge by pumping out drugs that manage to penetrate the endothelial cells (89). Moreover, the BBB’s integrity is not uniform throughout the GB tumor (90). While the BBB may be compromised in some regions of the tumor, allowing partial drug penetration, other areas may still have an intact barrier, further complicating effective drug delivery. This heterogeneity in BBB disruption within GB tumors means that even if a therapeutic agent reaches some parts of the tumor, it may not reach all areas, leading to incomplete treatment and potential recurrence. Consequently, innovative strategies are required to either bypass or transiently disrupt the BBB to improve drug delivery and therapeutic efficacy in GB treatment. One strategy involves incorporating a window-of-opportunity component into surgical clinical trials. In this approach, patients receive the experimental drug before the tumor resection. Pharmacodynamic studies on the resected tumor can then determine whether the drug crossed the blood-brain barrier (BBB) and performed its expected function. This helps differentiate between a mechanistic failure and a delivery failure of the experimental drug (91). BBB disruption is another strategy that will be discussed in more detail below.
4.3.2 Hypoxia
GB TME features a necrotic core primarily formed due to high cell density or vaso-occlusive events leading to hypoxia, a pervasive feature in GB. The hypoxic niches contribute to the development of therapy resistance to conventional chemotherapy, which often relies on oxygen to generate reactive oxygen species (ROS) that damage cancer cells. This results in increased expression of hypoxia-inducible factors (HIFs), which promote angiogenesis (via VEGF upregulation) and invasion (92, 93). In the context of radiotherapy, hypoxic tumor cells exhibit increased resistance due to their impaired ability to produce damaging oxygen radicals upon irradiation (94). Additionally, HIFs help mitigate the DNA damage caused by radiotherapy, further reducing its effectiveness (95).
4.3.3 Acidosis
Tumor acidosis crucially impacts the effectiveness of various therapeutic interventions by modulating the TME and promoting oncogenesis. It enhances the expression of glioma stem cell (GSC) markers, fostering tumor growth through paracrine actions that involve angiogenic factors controlled by HIFs, particularly HIF-2α (96). Acidosis also heightens autophagic activity linked to the maintenance and aggressiveness of GSCs (97). Additionally, it supports tumor invasion by activating cathepsin L, which converts plasminogen into plasmin, leading to the degradation of key extracellular matrix proteins and activation of latent matrix metalloproteinases (98). It also compromises the efficacy of chemotherapeutics, particularly weak base drugs like doxorubicin and vincristine, through ion trapping that reduces their intracellular concentration and by increasing the efflux activity of the p-glycoprotein (99–101). Moreover, acidosis facilitates tumor immune escape and resistance to immunotherapy by impairing CD8+ T lymphocytes, reducing their cytokine secretion and expression of critical receptors, thus dampening key immune signaling pathways (102, 103). It also lowers the production of vital immune effectors by T cells and monocytes, enhances the number of myeloid-derived suppressor cells (MDSCs), and inhibits the cytotoxic functions of NK and NKT cells (104).
4.3.4 Glutathione
Elevated glutathione levels in GB lead to reduced oxidative stress which is crucial for disease progression (105). Although increased glutathione protects healthy cells from oxidative stress, it concurrently promotes resistance to many chemotherapeutics which exert their cancer killing properties by ROS production. This is further highlighted by studies showing that resistant cells had higher levels of glutathione and lower levels of ROS than TMZ-sensitive cells (106, 107).
4.3.5 Altered drug mechanism of action
Repurposed drugs may exert their effect on the CNS tumor differently from their actions in the originally FDA-approved tumor type (108, 109). Many drugs repurposed for GB treatment, like cabozantinib, inhibit multiple pathways. While they are primarily known as VEGF inhibitors, their role as multi-targeted tyrosine kinase inhibitors can lead to a range of effects, resulting in a broader range of biological effects. This variability in drug action within the unique CNS tumor microenvironment further complicates the therapeutic efficacy and predictability of these agents (110).
The cumulative effect of all these features is a selective pressure favoring the growth of resistant clones, complicating future therapies, whether targeted or otherwise.
5 Emerging diagnostic tools: liquid biopsy
As previously mentioned, the failure of drug repurposing efforts in GB can be attributed to several factors. Tumor heterogeneity, characterized by the presence of multiple clones, increases the likelihood of drug-resistant clones, resulting in treatment failure or recurrence. Even if the tumor initially responds to treatment, tumor cells may evolve into different phenotypes with new mechanisms of therapeutic resistance. In clinical practice, disease monitoring predominantly relies on imaging modalities, limiting the ability to observe real-time tumor dynamics and variations in biomarkers. While tumor biopsy at recurrence is an option, it is infrequently performed due to its morbidity risks and the lack of clear benefit. It remains uncertain whether molecular characterization of recurrent tumors provides a definitive survival advantage in patients treated with targeted therapies. In that regard, liquid biopsy rapidly emerges as a promising non-invasive diagnostic tool, showing encouraging results.
By analyzing molecular biomarkers in bodily fluids, liquid biopsies provide a real-time snapshot of the tumor’s genetic landscape without the need for invasive surgical procedures. This approach can detect changes in the tumor’s mutational profile over time, allowing for more precise and adaptive treatment strategies that respond to the tumor’s evolving nature. Liquid biopsies offer a promising tool to enhance the effectiveness of targeted therapies and improve the prognosis for GB patients by enabling continuous monitoring and timely adjustments to therapeutic interventions.
While the field of liquid biopsy in GB began with studies on circulating tumor cells, there are no validated or reproducible studies on isolating them. Therefore, we will focus here on the three main molecular markers studied in liquid biopsy (Figure 2).
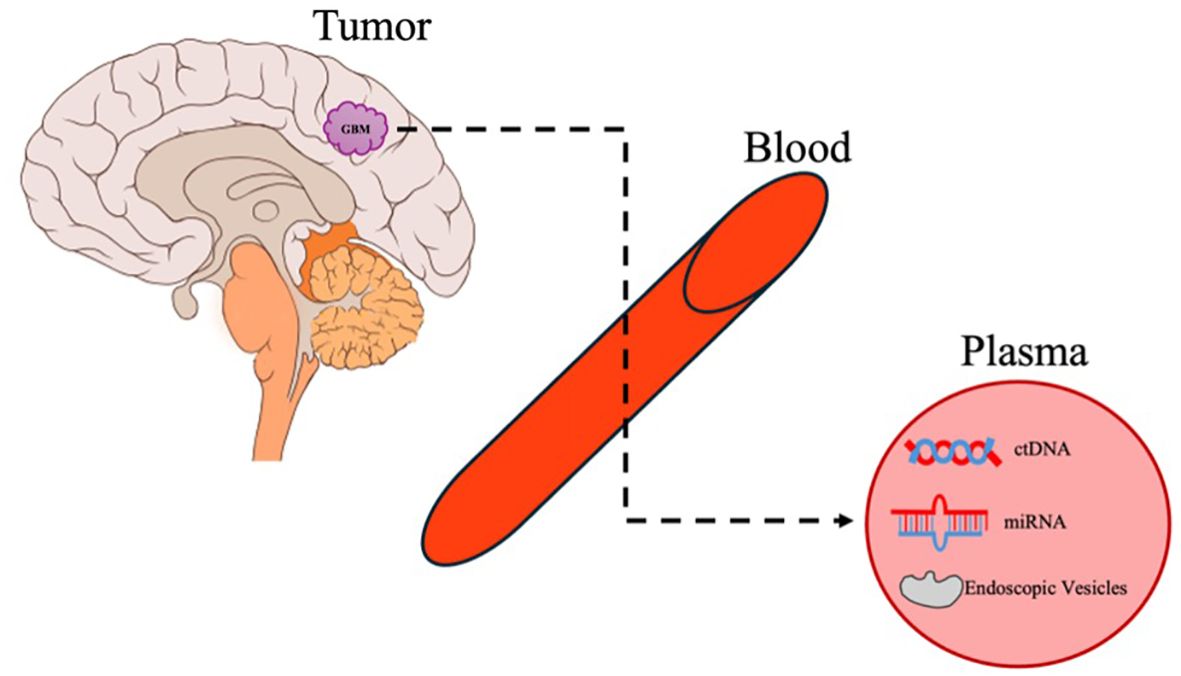
Figure 2. Liquid biopsy. An overview of the release of GB molecular biomarkers and cargo into the systemic circulation and its detection in plasma.
5.1 ctDNA
The detection of circulating tumor DNA (ctDNA) in glioma patients varies significantly between blood and cerebrospinal fluid (CSF). The BBB limits ctDNA detection in blood to less than 10% of glioma patients, primarily due to ctDNA’s inability to effectively penetrate the BBB (111). In contrast, ctDNA levels in CSF are generally higher (112, 113), as it is directly shed into the CSF from the tumor. However, monitoring treatment response by serial CSF ctDNA measurements requires repeated lumbar punctures and exposes patients to potentially unjustified morbidities.
Peripheral blood is a less invasive option for serial monitoring, facilitating longitudinal monitoring studies, but it presents several challenges. The rapid clearance of ctDNA with a half-life of about 1.5h (114) and the small size of ctDNA fragments, which may not include relevant genetic alterations, contribute to plasma ctDNA’s low sensitivity. However, recent advances in NGS have increased the sensitivity of their detection. According to Piccioni et al., the NGS panel could detect ctDNA mutation in blood from 50% of all brain tumor patients and 55% of GB patients (115), allowing for measurement of GB’s mutational profile evolution during treatment. A more recent pilot study enrolling ten glioma patients utilizing the CAPP-seq-based NGS technique reported a detection rate of up to 93.8% plasma samples with successful tracking of change in mutation profiles (116). The serial ctDNA analyses detected an emergence of mismatch repair gene MSH2 and MSH6 gene mutations, which is associated with hypermutation and potential development of TMZ resistance during treatment with TMZ. Another study utilizing a droplet digital PCR technique reliably detected the IDH1 mutation with 84% sensitivity in cross-comparison with tissue mutations (117). TERTp C228T mutation was detected in 88% of patients, and EGFRvIII mutation was detected in 71% of patients.
5.2 miRNA
There have been several studies exploring the role of circulating miRNA in GB. Differentially expressed miRNAs were identified in several studies comparing the plasma of GB patients to that of healthy controls (118–120). Circulating miRNAs can also serve as biomarkers correlating with OS and PFS in GB patients (121, 122). However, the clinical implications of these findings in relation to treatment strategies warrant further exploration.
5.3 Extracellular vesicles
Extracellular vesicles (EVs) are small membrane-bound vesicles released by cells under both normal and abnormal conditions, playing a crucial role in cell-to-cell communication. These vesicles transport genetic materials like DNA, mRNA, and miRNA across the body, influencing the behavior and phenotype of distant cells, including endothelial cells (123, 124).
EVs originating from gliomas or other cells in the tumor microenvironment appear to play crucial roles in tumor cell proliferation, invasion, malignancy, and drug resistance (125). Cancer cells release a greater number of EVs with differing protein and RNA contents, compared to non-malignant cells (126). These EVs facilitate communication with surrounding cells to alter the TME by influencing the behavior of local and recruited stromal cells, contributing to the creation of a tumor-supportive environment that enhances angiogenesis, immunosuppression, and the malignant transformation of cancer cells (127). Tumor-derived EVs can also enter into the circulation and prepare distant organs for metastasis by creating favorable conditions for tumor cell growth (128). This process, known as pre-metastatic niche formation, involves steps such as inducing vascular leakiness, altering stromal components, and suppressing the immune system. These interactions underscore the significance of EVs in the progression and maintenance of cancer.
From a diagnostic standpoint, EVs are increasingly recognized for their potential in identifying tumor molecular signatures, particularly due to their ability to traverse an intact BBB (129). Manda et al. reported that variant EGFR RNA transcripts were detected with similar frequency in GB tissue (39.5%) and their matched serum exosomes (44.7%). The presence of circulating exosomal EGFRvIII variants correlated with poor outcomes (130). Studies on miRNA contents of EVs revealed that the exosomal levels and types of miRNA within the EVs were associated with the aggressive potentials of the GB (131). For example, an in vitro functional study using glioma cell lines has indicated that miR-221 silencing can reduce cell proliferation, migratory potential, and resistance to temozolomide (132). In addition, exosomal levels of miR-221 were increased in parallel with glioma grades. Another study reported that syndecan-1 plasma EVs could distinguish between low-grade and high-grade gliomas with a sensitivity and specificity greater than 70% (133).
However, challenges remain. We still lack an understanding of the mechanisms that drive the RNA incorporation into EVs of various sizes and types in different RNA concentrations. The technical challenges of isolating purified tumor-specific EVs of different sizes in high-yield continue to complicate the interpretation and utility of these biomarkers in clinical settings.
Liquid biopsy offers significant potential in enhancing the management of GB by providing a non-invasive method to monitor treatment response in real-time, tracking changes in tumor-derived biomarkers. By identifying specific genetic mutations and alterations, liquid biopsies can guide targeted therapies, facilitating more personalized and effective treatment strategies. However, the clinical implementation of liquid biopsy in GB is still in its early stages. There is a need for standardized methodologies across laboratories to ensure consistent and reliable results. Larger prospective studies are required to validate the clinical utility of liquid biopsy biomarkers in GB.
6 Strategies to improve drug delivery to GB
Effective drug delivery to the CNS is significantly hindered by the BBB, posing a substantial challenge in GB treatment. The BBB is a multi-layered cellular physical barrier composed of endothelial cells, astrocytes, and pericytes, which effectively prevent the diffusion of small molecules. It facilitates the exchange of essential nutrients and metabolites through a selective transport system. Even if small molecules manage to penetrate the BBB, they are often actively transported back out via efflux pumps (134). Overcoming this barrier to enhance bioavailability is a critical area of ongoing research.
6.1 Focused ultra-sound
FUS has been investigated as a method to transiently and non-invasively increase the permeability of the BBB, thereby enhancing the delivery of therapeutic agents to GB tissue. This technique involves the use of pulsed ultrasound waves in conjunction with microbubbles, which oscillate and cause transient disruption of the BBB. Preclinical studies have demonstrated that FUS can improve the concentration of chemotherapeutic agents such as TMZ in brain tissue, leading to prolonged survival in animal models (135, 136). Adding MRI to FUS (MR-guided FUS) further improves the precision of the drug delivery, thus minimizing damage to healthy tissue. Clinical trials have begun to explore the safety and efficacy of FUS in humans, with early results indicating that FUS can be safely performed and may improve drug delivery and patient survival (137, 138).
Animal studies have shown that FUS can improve the penetration of immunotherapy into brain tumors, the immune response against the cancer cells, and survival outcomes (139, 140). The clinical use of immunotherapy in GB treatment is currently not validated with debatable early study outcomes.
FUS is actively investigated for its use in direct tumor ablation by delivering high-energy ultrasound waves. Initial clinical studies using MR-guided FUS demonstrated precise ablation of brain tumors after craniotomy (141). However, significant attenuation of ultrasound waves by the skull, significant damage to healthy brain tissue, and lack of clinical validity currently limit its use.
6.2 Implantable drug-delivery systems
The development of new extended-release drug-delivery vehicles led to several promising strategies for improving patient outcomes after tumor resection.
The pioneering work of Langer’s group in the 1980s led to the development of localized controlled-release therapies for GB, culminating in the FDA approval of the first implantable intra-cavity wafer, Gliadel, in 1996 (142). Gliadel, a polyanhydride-based wafer containing carmustine (BCNU), is designed for optimal drug release, achieving substantial polymer degradation within three weeks of implantation. Clinical trials demonstrated survival benefits for patients receiving Gliadel compared to placebo (13.8 months vs 11.6 months; p=0.018) (25). On the other hand, implantation is associated with several negative side effects, including seizures, vasogenic edema, meningitis, and impaired wound healing (143). It can also dislodge and cause micro-tears (144). Also, its content (BCNU) has a low diffusion rate (145). Therefore, it is no longer commonly used in clinical practice.
Following Gliadel’s approval, there has been a surge in the development of locally administered chemotherapeutic devices. Sheleg et al. explored a biodegradable polymer device loaded with cisplatin (146). Twenty 1.5 x 1.5cm polymer plates loaded with cisplatin with a drug density of 1mg/cm2 were implanted in the surgical bed after subtotal removal of GB. This strategy resulted in extended OS when administered with radiation therapy compared to radiation alone (427.5 vs 211.0 days; p = 0.00001) (146). Similarly, Di Mascolo et al. developed microfabricated PLGA meshes loaded with diclofenac and docetaxel, which effectively prevented tumor recurrence and significantly extended survival in orthotopic brain tumor mouse models, emphasizing the advantage of the meshes’ flexibility over solid films (147).
Technological advancement led to different designs of implantable drug-delivery systems, including hydrogel and microparticles. Hydrogel is a hydrophilic polymer network with high water contents that can be loaded with water-soluble biomacromolecules such as small molecules and NPs. For example, OncoGel, a type of thermoresponsive PLGA-PEG matrix hydrogel loaded with paclitaxel, transitions to a semisolid state at the body temperature once applied to the tumor bed and maintains drug release over six weeks (148). PLGA microparticles have been studied extensively, offering controlled release of anticancer drugs like 5-fluorouracil (149), which has shown to work synergistically with radiotherapy in enhancing survival in animal models (150).
There are no implantable targeted drug-delivery systems, likely due to cost considerations. However, as this technology advances and shows more promising data, it can potentially be applied to targeted therapeutics.
In the context of targeted therapy, only EGFR-targeting nanoparticles have been investigated in human studies. The first was a phase 1/2 trial involving fourteen patients with rGB. In this study, doxorubicin-loaded nanoparticles targeting EGFR were used to deliver the chemotherapeutic agent into GB tumor cells (155). While the maximum tolerated dose (MTD) was determined, the trial was prematurely terminated by the sponsor, leaving efficacy data unavailable. Similarly, a phase 1 study utilizing anti-EGFR doxorubicin-loaded immunoliposomes was conducted in nine rGB patients with EGFR amplification (155). This study confirmed the successful delivery of nanoliposomes to GB tissue; however, the small sample size and absence of a control group limited the assessment of therapeutic efficacy.
6.3 Nanoparticles
NPs can be engineered specifically to cross the BBB and target tumor cells directly, minimizing side effects. NPs, with their tunable physicochemical properties, can be loaded with various therapeutic agents to deliver the intended targets. The NPs can be tailored to have specific intrinsic (electronic, optical, and magnetic) and extrinsic (size, surface-to-volume ratio, or surface energy) characteristics to increase delivery efficiency, decrease off-target effects, and improve drug kinetics. These NPs act as “Trojan horses,” facilitating the delivery of drugs like doxorubicin and paclitaxel and biological molecules such as antibodies, DNA, and peptides directly to GB cells.
The surface of NPs can be modified with specific ligands that recognize and bind to receptors on endothelial cells lining the brain, facilitating their entry into the brain through mechanisms like receptor-mediated transcytosis. For example, the transferrin receptor (TfR) and low-density lipoprotein receptor (LRP1) are common targets on brain endothelial cells that NPs exploit to achieve transcytosis (151, 152). These receptors allow NPs to bypass the typical barriers posed by the BBB, improving the delivery efficiency of chemotherapeutics into the brain. Since the brain uptakes glucose via like the glucose transporter-1 (GLUT1), Anraku et al. utilized glycosylated micelles to transport bioactive substances via the GLUT1 transporter. The precisely calculated glucose density on the surface of the NP allowed the regulation of its distribution within the brain, thus successfully increasing the number of nanocarriers within the brain (153). Further, PEGylation of these NPs further prolongs their circulation time in the bloodstream, reducing protein interactions and enhancing their therapeutic efficacy (154).
In the context of targeted therapy, only EGFR-targeting nanoparticles have been investigated in human studies. The first was a phase 1/2 trial involving fourteen patients with rGB. In this study, doxorubicin-loaded nanoparticles targeting EGFR were used to deliver the chemotherapeutic agent into GB tumor cells (155). While the maximum tolerated dose (MTD) was determined, the trial was prematurely terminated by the sponsor, leaving efficacy data unavailable. Similarly, a phase 1 study utilizing anti-EGFR doxorubicin-loaded immunoliposomes was conducted in nine rGB patients with EGFR amplification (156). This study confirmed the successful delivery of nanoliposomes to GB tissue; however, the small sample size and absence of a control group limited the assessment of therapeutic efficacy.
7 Conclusions/future directions
Treatment of GB remains a formidable challenge with limited treatment options, particularly in recurrent settings. Many past attempts to employ targeted therapy, including clinical trials selecting patients whose tumors possess the actionable mutation of interest, have not resulted in substantial clinical benefits. This is largely due to the unique biological and clinical characteristics of GB, including dynamic evaluation of the tumor, its highly heterogeneous tumor microenvironment, and poor drug penetration through BBB, as illustrated in Figure 3. These obstacles underscore the need for innovative approaches to improve the understanding and monitoring of tumor biology.
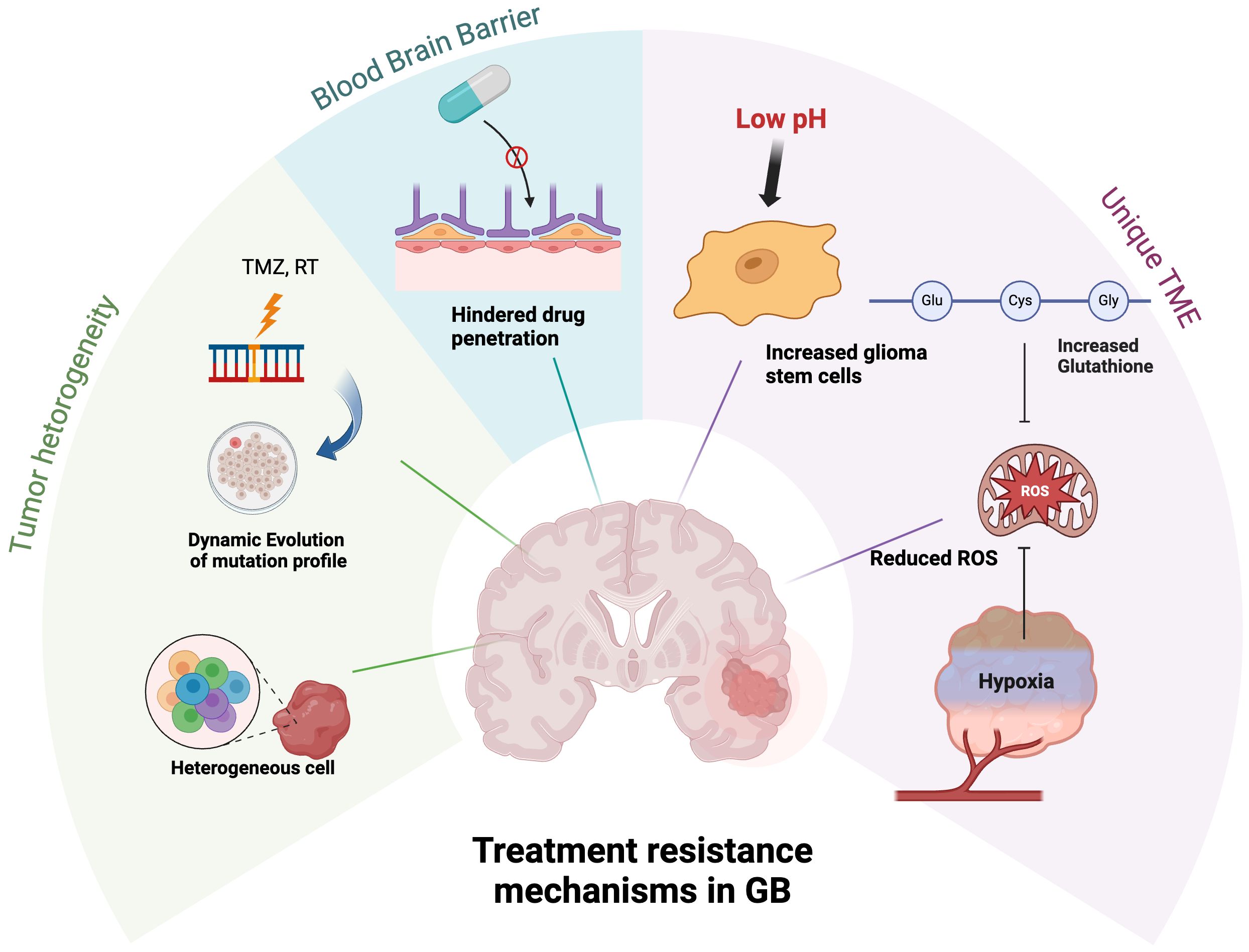
Figure 3. Treatment resistance mechanisms of GB. The tumor cells continuously evolve and diversify the tumor’s genetic profile. Targeted treatment strategies face additional challenges due to poor drug penetration across the BBB. The unique biochemical environment of GB TME, characterized by low pH, hypoxia, and elevated glutathione levels, promotes the proliferation of glioma stem cells and reduces ROS, thereby diminishing the efficacy of anti-neoplastic therapies.
Emerging diagnostic tools such as liquid biopsy offer promising, non-invasive methods to monitor GB. Liquid biopsies using ctDNA provide real-time snapshots of the tumor’s genetic landscape, allowing for adaptive treatment strategies that respond to the tumor’s evolving nature. Although ctDNA detection in blood is limited by the BBB, advances in NGS have improved sensitivity. The regulatory role of miRNA sets them apart as particularly promising biomarkers in liquid biopsy. Further validation of the role of plasma miRNA in GB may result in the identification of GB-specific miRNAs that can be used in lieu of surgery for some patients. EVs can traverse an intact BBB and are valuable in identifying tumor molecular signatures and monitoring treatment response with increased sensitivity. There are other uses of plasma from GB patients. A recent study identified unique metabolomic signatures in the plasma of GB patients at diagnosis and recurrence (157). We believe that serial plasma collection from GB patients during their treatment, along with multi-platform profiling at clinically relevant endpoints (e.g., pre-surgery, post-surgery, recurrence, tissue-proven radionecrosis), can further revolutionize this field and be an extremely valuable diagnostic tool for GB patients.
Recent innovative strategies have shown potential in overcoming GB treatment challenges and enhancing therapeutic agents’ delivery and efficacy. NPs can be engineered to cross the BBB and deliver drugs directly to the tumor site, leveraging mechanisms like receptor-mediated transcytosis. Additionally, MRgFUS can transiently disrupt the BBB, allowing therapeutic agents to penetrate the brain more effectively. Combining this with implantable drug delivery systems, which provide sustained and localized release of therapeutic agents directly to the tumor bed following surgical resection, shows promise. Innovations such as hydrogel-based delivery systems and biodegradable polymer devices have demonstrated efficacy in preclinical models, offering prolonged drug release and improved survival outcomes. These technologies collectively enhance drug delivery efficiency, reduce off-target effects, and improve therapeutic efficacy.
In summary, while the treatment of GB has faced significant difficulties, new strategies such as NPs, FUS, implantable drug delivery systems, liquid biopsy, and adaptive trial platforms offer promising solutions. Continued research and clinical trials are essential to fully realize the potential of these innovative therapies, ultimately improving the prognosis for patients with this aggressive and devastating disease.
Author contributions
WB: Writing – original draft, Writing – review & editing. SM: Supervision, Writing – review & editing. AD: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
2. van Linde ME, Brahm CG, de Witt Hamer PC, Reijneveld JC, Bruynzeel AME, Vandertop WP, et al. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. J Neurooncol. (2017) 135:183–92. doi: 10.1007/s11060-017-2564-z
3. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. (2008) 455:1061–8. doi: 10.1038/nature07385
4. Alaseem AM. Advancements in MDM2 inhibition: clinical and pre-clinical investigations of combination therapeutic regimens. Saudi Pharm J. (2023) 31:101790. doi: 10.1016/j.jsps.2023.101790
5. Xu H, Zong H, Ma C, Ming X, Shang M, Li K, et al. Epidermal growth factor receptor in glioblastoma. Oncol Lett. (2017) 14:512–6. doi: 10.3892/ol.2017.6221
6. Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. (2004) 22:133–42. doi: 10.1200/JCO.2004.08.110
7. Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther. (2011) 10:1102–12. doi: 10.1158/1535-7163.MCT-11-0048
8. Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (Gbm). J Neurooncol. (2009) 92:99–105. doi: 10.1007/s11060-008-9741-z
9. Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. (2013) 85:1206–11. doi: 10.1016/j.ijrobp.2012.10.008
10. van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: eortc brain tumor group study 26034. J Clin Oncol. (2009) 27:1268–74. doi: 10.1200/JCO.2008.17.5984
11. de Groot JF, Gilbert MR, Aldape K, Hess KR, Hanna TA, Ictech S, et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neurooncol. (2008) 90:89–97. doi: 10.1007/s11060-008-9637-y
12. Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE 2nd, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. (2010) 96:219–30. doi: 10.1007/s11060-009-9950-0
13. Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent Malignant gliomas: north American brain tumor consortium trial 04-02. Neuro Oncol. (2014) 16:567–78. doi: 10.1093/neuonc/not247
14. Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, et al. Nabtt 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. (2013) 15:490–6. doi: 10.1093/neuonc/nos322
15. Sathornsumetee S, Desjardins A, Vredenburgh JJ, McLendon RE, Marcello J, Herndon JE, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent Malignant glioma. Neuro Oncol. (2010) 12:1300–10. doi: 10.1093/neuonc/noq099
16. Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. (2010) 98:93–9. doi: 10.1007/s11060-009-0067-2
17. Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A phase I/II trial of GW572016 (Lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. (2010) 65:353–61. doi: 10.1007/s00280-009-1041-6
18. Karavasilis V, Kotoula V, Pentheroudakis G, Televantou D, Lambaki S, Chrisafi S, et al. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. J Neurol. (2013) 260:1469–80. doi: 10.1007/s00415-012-6812-z
19. Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed Malignant glioma. Clin Cancer Res. (2013) 19:900–8. doi: 10.1158/1078-0432.CCR-12-1707
20. Yu A, Faiq N, Green S, Lai A, Green R, Hu J, et al. Report of safety of pulse dosing of lapatinib with temozolomide and radiation therapy for newly-diagnosed glioblastoma in a pilot phase II study. J Neurooncol. (2017) 134:357–62. doi: 10.1007/s11060-017-2533-6
21. Hasselbalch B, Eriksen JG, Broholm H, Christensen IJ, Grunnet K, Horsman MR, et al. Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS. (2010) 118:585–94. doi: 10.1111/j.1600-0463.2010.02631.x
22. Lv S, Teugels E, Sadones J, De Brakeleer S, Duerinck J, Du Four S, et al. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int J Oncol. (2012) 41:1029–35. doi: 10.3892/ijo.2012.1539
23. Solomon MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer. (2013) 13:299. doi: 10.1186/1471-2407-13-299
24. Du XJ, Li XM, Cai LB, Sun JC, Wang SY, Wang XC, et al. Efficacy and safety of nimotuzumab in addition to radiotherapy and temozolomide for cerebral glioblastoma: A phase II multicenter clinical trial. J Cancer. (2019) 10:3214–23. doi: 10.7150/jca.30123
25. Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer. (2015) 51:522–32. doi: 10.1016/j.ejca.2014.12.019
26. Chagoya G, Kwatra SG, Nanni CW, Roberts CM, Phillips SM, Nullmeyergh S, et al. Efficacy of osimertinib against EGFRvIII+ Glioblastoma. Oncotarget. (2020) 11:2074–82. doi: 10.18632/oncotarget.27599
27. Abousaud M, Faroqui NM, Lesser G, Strowd RE, Ramkissoon SH, Kwatra M, et al. Clinical experience using osimertinib in patients with recurrent Malignant gliomas containing EGFR alterations. J Cancer Sci Clin Ther. (2021) 5:210–20. doi: 10.26502/jcsct.5079114
28. Cardona AF, Jaramillo-Velasquez D, Ruiz-Patino A, Polo C, Jimenez E, Hakim F, et al. Efficacy of osimertinib plus bevacizumab in glioblastoma patients with simultaneous EGFR amplification and EGFRvIII mutation. J Neurooncol. (2021) 154:353–64. doi: 10.1007/s11060-021-03834-3
29. Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. (2005) 23:9359–68. doi: 10.1200/JCO.2005.03.2185
30. Hassler MR, Vedadinejad M, Flechl B, Haberler C, Preusser M, Hainfellner JA, et al. Response to imatinib as a function of target kinase expression in recurrent glioblastoma. Springerplus. (2014) 3:111. doi: 10.1186/2193-1801-3-111
31. Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. (2010) 96:393–402. doi: 10.1007/s11060-009-9976-3
32. Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A European organisation for research and treatment of cancer brain tumor group study. J Clin Oncol. (2008) 26:4659–65. doi: 10.1200/JCO.2008.16.9235
33. Reardon DA, Desjardins A, Vredenburgh JJ, Sathornsumetee S, Rich JN, Quinn JA, et al. Safety and pharmacokinetics of dose-intensive imatinib mesylate plus temozolomide: phase 1 trial in adults with Malignant glioma. Neuro Oncol. (2008) 10:330–40. doi: 10.1215/15228517-2008-003
34. Reardon DA, Egorin MJ, Desjardins A, Vredenburgh JJ, Beumer JH, Lagattuta TF, et al. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (Ptk787) plus imatinib and hydroxyurea for Malignant glioma. Cancer. (2009) 115:2188–98. doi: 10.1002/cncr.24213
35. Holdhoff M, Supko JG, Gallia GL, Hann CL, Bonekamp D, Ye X, et al. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme. J Neurooncol. (2010) 97:241–5. doi: 10.1007/s11060-009-0008-0
36. Razis E, Selviaridis P, Labropoulos S, Norris JL, Zhu MJ, Song DD, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. (2009) 15:6258–66. doi: 10.1158/1078-0432.CCR-08-1867
37. Lu-Emerson C, Norden AD, Drappatz J, Quant EC, Beroukhim R, Ciampa AS, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol. (2011) 104:287–91. doi: 10.1007/s11060-010-0489-x
38. Franceschi E, Stupp R, van den Bent MJ, van Herpen C, Laigle Donadey F, Gorlia T, et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol. (2012) 14:1503–10. doi: 10.1093/neuonc/nos256
39. Lassman AB, Pugh SL, Gilbert MR, Aldape KD, Geinoz S, Beumer JH, et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (Rtog 0627). Neuro Oncol. (2015) 17:992–8. doi: 10.1093/neuonc/nov011
40. de Groot J, George S, Razak A, Gordon M, Janku F, Ligon K, et al. ACTR-02. DCC-2618, a novel pan-KIT and PDGFRa kinase switch control inhibitor, shows encouraging signal in a patient (PT) with glioblastoma (GBM). Neuro-Oncology. (2017) 19:vi1. doi: 10.1093/neuonc/nox168.002
41. Wen PY, Drappatz J, de Groot J, Prados MD, Reardon DA, Schiff D, et al. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. (2018) 20:249–58. doi: 10.1093/neuonc/nox154
42. Cloughesy TF, Drappatz J, de Groot J, Prados MD, Reardon DA, Schiff D, et al. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients with prior antiangiogenic therapy. Neuro Oncol. (2018) 20:259–67. doi: 10.1093/neuonc/nox151
43. Hutterer M, Nowosielski M, Haybaeck J, Embacher S, Stockhammer F, Gotwald T, et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (Surge 01-07). Neuro Oncol. (2014) 16:92–102. doi: 10.1093/neuonc/not161
44. Reardon DA, Vredenburgh JJ, Coan A, Desjardins A, Peters KB, Gururangan S, et al. Phase I study of sunitinib and irinotecan for patients with recurrent Malignant glioma. J Neurooncol. (2011) 105:621–7. doi: 10.1007/s11060-011-0631-4
45. Grisanti S, Ferrari VD, Buglione M, Agazzi GM, Liserre R, Poliani L, et al. Second line treatment of recurrent glioblastoma with sunitinib: results of a phase II study and systematic review of literature. J Neurosurg Sci. (2019) 63:458–67. doi: 10.23736/S0390-5616.16.03874-1
46. Balana C, Gil MJ, Perez P, Reynes G, Gallego O, Ribalta T, et al. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Target Oncol. (2014) 9:321–9. doi: 10.1007/s11523-014-0305-1
47. Stanislaw R, Burzynski TJ, Burzynski GS, Brookman S. Preliminary findings on the use of targeted therapy with pazopanib and other agents in combination with sodium phenylbutyrate in the treatment of glioblastoma multiforme. J Cancer Ther. (2014) 5:1423–37. doi: 10.4236/jct.2014.514144
48. Gilbert M. Oral Pazopanib Plus Oral Topotecan Metronomic Antiangiogenic Therapy for Recurrent Glioblastoma Multiforme (a) without Prior Bevacizumab Exposure and (B) after Failing Prior Bevacizumab. National Cancer Institute (NCI), Bethesda, USA: ClinicalTrials.gov (2020). Available at: https://clinicaltrials.gov/study/NCT01931098?tab=results#outcome-measures.
49. Jimenez-Pascual A, Siebzehnrubl FA. Fibroblast growth factor receptor functions in glioblastoma. Cells. (2019) 8. doi: 10.3390/cells8070715
50. Lassman AB, Sepulveda-Sanchez JM, Cloughesy TF, Gil-Gil MJ, Puduvalli VK, Raizer JJ, et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: A multicenter phase II study. Clin Cancer Res. (2022) 28:2270–7. doi: 10.1158/1078-0432.CCR-21-2664
51. Pant S, Schuler M, Iyer G, Witt O, Doi T, Qin S, et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. (2023) 24:925–35. doi: 10.1016/S1470-2045(23)00275-9
52. Rodon J, Damian S, Furqan M, Garcia-Donas J, Imai H, Italiano A, et al. Pemigatinib in previously treated solid tumors with activating FGFR1-FGFR3 alterations: phase 2 fight-207 basket trial. Nat Med. (2024) 30:1645–54. doi: 10.1038/s41591-024-02934-7
53. Wen PY, Cloughesy TF, Olivero AG, Morrissey KM, Wilson TR, Lu X, et al. First-in-human phase I study to evaluate the brain-penetrant PI3K/mtor inhibitor GDC-0084 in patients with progressive or recurrent high-grade glioma. Clin Cancer Res. (2020) 26:1820–8. doi: 10.1158/1078-0432.CCR-19-2808
54. Wen PY, de Groot J, Battiste J, Goldlust S, Damek D, Ellingson B, et al. CTNI-27. Multi-center, phase 2 study evaluating the pharmacokinetics, safety and preliminary efficacy of paxalisib in newly diagnosed adult patients with unmethylated glioblastoma (GBM). Neuro-Oncology. (2022) 24:vii76–vii7. doi: 10.1093/neuonc/noac209.292
55. Wen PY, Touat M, Alexander BM, Mellinghoff IK, Ramkissoon S, McCluskey CS, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J Clin Oncol. (2019) 37:741–50. doi: 10.1200/JCO.18.01207
56. Rosenthal M, Clement PM, Campone M, Gil-Gil MJ, DeGroot J, Chinot O, et al. Buparlisib plus carboplatin or lomustine in patients with recurrent glioblastoma: A phase Ib/II, open-label, multicentre, randomised study. ESMO Open. (2020) 5. doi: 10.1136/esmoopen-2020-000672
57. van den Bent M, Azaro A, De Vos F, Sepulveda J, Yung WKA, Wen PY, et al. A phase Ib/II, open-label, multicenter study of INC280 (Capmatinib) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. J Neurooncol. (2020) 146:79–89. doi: 10.1007/s11060-019-03337-2
58. Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A north central cancer treatment group study. J Clin Oncol. (2005) 23:5294–304. doi: 10.1200/JCO.2005.23.622
59. Wick W, Gorlia T, Bady P, Platten M, van den Bent MJ, Taphoorn MJ, et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clin Cancer Res. (2016) 22:4797–806. doi: 10.1158/1078-0432.CCR-15-3153
60. Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: north American brain tumor consortium study 05-02. Neuro Oncol. (2012) 14:1511–8. doi: 10.1093/neuonc/nos264
61. Sarkaria JN, Galanis E, Wu W, Dietz AB, Kaufmann TJ, Gustafson MP, et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin Cancer Res. (2010) 16:5573–80. doi: 10.1158/1078-0432.CCR-10-1453
62. Sarkaria JN, Galanis E, Wu W, Peller PJ, Giannini C, Brown PD, et al. North central cancer treatment group phase I trial N057k of everolimus (Rad001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. (2011) 81:468–75. doi: 10.1016/j.ijrobp.2010.05.064
63. Chinnaiyan P, Won M, Wen PY, Rojiani AM, Wendland M, Dipetrillo TA, et al. RTOG 0913: A phase 1 study of daily everolimus (RAD001) in combination with radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. (2013) 86:880–4. doi: 10.1016/j.ijrobp.2013.04.036
64. Mason WP, Macneil M, Kavan P, Easaw J, Macdonald D, Thiessen B, et al. A phase I study of temozolomide and everolimus (RAD001) in patients with newly diagnosed and progressive glioblastoma either receiving or not receiving enzyme-inducing anticonvulsants: an NCIC CTG study. Invest New Drugs. (2012) 30:2344–51. doi: 10.1007/s10637-011-9775-5
65. Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, Peller PJ, et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057k. Neuro Oncol. (2015) 17:1261–9. doi: 10.1093/neuonc/nou328
66. Chinnaiyan P, Won M, Wen PY, Rojiani AM, Werner-Wasik M, Shih HA, et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: results of NRG oncology RTOG 0913. Neuro Oncol. (2018) 20:666–73. doi: 10.1093/neuonc/nox209
67. Hainsworth JD, Shih KC, Shepard GC, Tillinghast GW, Brinker BT, Spigel DR. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clin Adv Hematol Oncol. (2012) 10:240–6.
68. Xu Q, Huang K, Meng X, Weng Y, Zhang L, Bu L, et al. Safety and efficacy of anlotinib hydrochloride plus temozolomide in patients with recurrent glioblastoma. Clin Cancer Res. (2023) 29:3859–66. doi: 10.1158/1078-0432.CCR-23-0388
69. Yang K, Wu B, Zhang Z, Peng G, Huang J, Hong X, et al. Anlotinib for patients with newly diagnosed glioblastoma with unmethylated mgmt promoter: an open-label, single-center, phase 2 clinical trial. J Clin Oncol. (2023) 41:e14039–e. doi: 10.1200/JCO.2023.41.16_suppl.e14039
70. Lombardi G, De Salvo GL, Brandes AA, Eoli M, Ruda R, Faedi M, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (Regoma): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. (2019) 20:110–9. doi: 10.1016/S1470-2045(18)30675-2
71. Caccese M, Desideri I, Villani V, Simonelli M, Buglione M, Chiesa S, et al. Regoma-oss: A large, italian, multicenter, prospective, observational study evaluating the efficacy and safety of regorafenib in patients with recurrent glioblastoma. ESMO Open. (2024) 9:102943. doi: 10.1016/j.esmoop.2024.102943
72. Lee E, Muzikansky A, Arrillaga-Romany I, Chukwueke U, Cloughesy T, Colman H, et al. CTNI-47. Phase II study of abemaciclib in recurrent GBM patients with CDKN2A/B loss and intact RB. Neuro-Oncology. (2020) 22:ii53. doi: 10.1093/neuonc/noaa215.213
73. Rahman R, Trippa L, Lee EQ, Arrillaga-Romany I, Fell G, Touat M, et al. Inaugural results of the individualized screening trial of innovative glioblastoma therapy: A phase II platform trial for newly diagnosed glioblastoma using bayesian adaptive randomization. J Clin Oncol. (2023) 41:5524–35. doi: 10.1200/JCO.23.00493
74. Taylor JW, Parikh M, Phillips JJ, James CD, Molinaro AM, Butowski NA, et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J Neurooncol. (2018) 140:477–83. doi: 10.1007/s11060-018-2977-3
75. Tien AC, Bao X, Derogatis A, Kim S, Mehta S, Li J, et al. ACTR-45. Phase 0/2 study of ribociclib in patients with recurrent glioblastoma. Neuro-Oncology. (2018) 20:vi21. doi: 10.1093/neuonc/noy148.077
76. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
77. Mellinghoff IK, Cloughesy TF, Mischel PS. Pten-mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. (2007) 13:378–81. doi: 10.1158/1078-0432.CCR-06-1992
78. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: A summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
79. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. (2016) 164:550–63. doi: 10.1016/j.cell.2015.12.028
80. Daher A, de Groot J. Rapid identification and validation of novel targeted approaches for glioblastoma: A combined ex vivo-in vivo pharmaco-omic model. Exp Neurol. (2018) 299:281–8. doi: 10.1016/j.expneurol.2017.09.006
81. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
82. Herrlinger U, Rieger J, Koch D, Loeser S, Blaschke B, Kortmann RD, et al. Phase II trial of lomustine plus temozolomide chemotherapy in addition to radiotherapy in newly diagnosed glioblastoma: UKT-03. J Clin Oncol. (2006) 24:4412–7. doi: 10.1200/JCO.2006.06.9104
83. Cho NS, Wong WK, Nghiemphu PL, Cloughesy TF, Ellingson BM. The future glioblastoma clinical trials landscape: early phase 0, window of opportunity, and adaptive phase I-III studies. Curr Oncol Rep. (2023) 25:1047–55. doi: 10.1007/s11912-023-01433-1
84. Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol. (2015) 209:493–506. doi: 10.1083/jcb.201412147
85. Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist's view. Brain Res Brain Res Rev. (2003) 42:221–42. doi: 10.1016/s0165-0173(03)00177-2
86. Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J Physiol. (1990) 429:47–62. doi: 10.1113/jphysiol.1990.sp018243
87. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
88. Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to Malignant glioma, a disease of the whole brain. Expert Rev Mol Med. (2011) 13:e17. doi: 10.1017/S1462399411001888
89. Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. (2020) 20:26–41. doi: 10.1038/s41568-019-0205-x
90. Zhang F, Xu CL, Liu CM. Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Des Devel Ther. (2015) 9:2089–100. doi: 10.2147/DDDT.S79592
91. Srinivasan VM, Ene C, Kerrigan BP, Lang FF. Window of opportunity clinical trials to evaluate novel therapies for brain tumors. Neurosurg Clin N Am. (2021) 32:93–104. doi: 10.1016/j.nec.2020.09.002
92. Jensen RL, Ragel BT, Whang K, Gillespie D. Inhibition of hypoxia inducible factor-1alpha (HIF-1alpha) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in Malignant gliomas. J Neurooncol. (2006) 78:233–47. doi: 10.1007/s11060-005-9103-z
93. Cassavaugh J, Lounsbury KM. Hypoxia-mediated biological control. J Cell Biochem. (2011) 112:735–44. doi: 10.1002/jcb.22956
94. Burroughs SK, Kaluz S, Wang D, Wang K, Van Meir EG, Wang B. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med Chem. (2013) 5:553–72. doi: 10.4155/fmc.13.17
95. Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. (2017) 127:415–26. doi: 10.1172/JCI89587
96. Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. (2011) 18:829–40. doi: 10.1038/cdd.2010.150
97. Peppicelli S, Andreucci E, Ruzzolini J, Laurenzana A, Margheri F, Fibbi G, et al. The acidic microenvironment as a possible niche of dormant tumor cells. Cell Mol Life Sci. (2017) 74:2761–71. doi: 10.1007/s00018-017-2496-y
98. Mignatti P, Rifkin DB. Plasminogen activators and matrix metalloproteinases in angiogenesis. Enzyme Protein. (1996) 49:117–37. doi: 10.1159/000468621
99. Gerweck LE, Vijayappa S, Kozin S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol Cancer Ther. (2006) 5:1275–9. doi: 10.1158/1535-7163.MCT-06-0024
100. Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta. (2013) 1832:606–17. doi: 10.1016/j.bbadis.2013.01.020
101. Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M. Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia. (2006) 8:143–52. doi: 10.1593/neo.05697
102. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
103. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. (2010) 184:1200–9. doi: 10.4049/jimmunol.0902584
104. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol. (2017) 8:1887. doi: 10.3389/fimmu.2017.01887
105. Ogunrinu TA, Sontheimer H. Hypoxia increases the dependence of glioma cells on glutathione. J Biol Chem. (2010) 285:37716–24. doi: 10.1074/jbc.M110.161190
106. Rocha CR, Garcia CC, Vieira DB, Quinet A, de Andrade-Lima LC, Munford V, et al. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. (2014) 5:e1505. doi: 10.1038/cddis.2014.465
107. Zhu Z, Du S, Du Y, Ren J, Ying G, Yan Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J Neurochem. (2018) 144:93–104. doi: 10.1111/jnc.14250
108. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. (2012) 32:1959–72. doi: 10.1038/jcbfm.2012.126
109. Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discovery. (2016) 15:275–92. doi: 10.1038/nrd.2015.21
110. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. (2008) 359:492–507. doi: 10.1056/NEJMra0708126
111. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human Malignancies. Sci Transl Med. (2014) 6:224ra24. doi: 10.1126/scitranslmed.3007094
112. Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U.S.A. (2015) 112:9704–9. doi: 10.1073/pnas.1511694112
113. Wang Z, Jiang W, Wang Y, Guo Y, Cong Z, Du F, et al. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. BioMed Rep. (2015) 3:543–8. doi: 10.3892/br.2015.462
114. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. (2008) 14:985–90. doi: 10.1038/nm.1789
115. Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, Barkhoudarian G, et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. (2019) 8:CNS34. doi: 10.2217/cns-2018-0015
116. Jones JJ, Jones KL, Wong SQ, Whittle J, Goode D, Nguyen H, et al. Plasma ctdna enables early detection of temozolomide resistance mutations in glioma. Neurooncol Adv. (2024) 6:vdae041. doi: 10.1093/noajnl/vdae041
117. Jones JJ, Nguyen H, Wong SQ, Whittle J, Iaria J, Stylli S, et al. Plasma ctdna liquid biopsy of IDH1, TERTp, and EGFRvIII mutations in glioma. Neurooncol Adv. (2024) 6:vdae027. doi: 10.1093/noajnl/vdae027
118. Roth P, Wischhusen J, Happold C, Chandran PA, Hofer S, Eisele G, et al. A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem. (2011) 118:449–57. doi: 10.1111/j.1471-4159.2011.07307.x
119. Wang Q, Li P, Li A, Jiang W, Wang H, Wang J, et al. Plasma specific mirnas as predictive biomarkers for diagnosis and prognosis of glioma. J Exp Clin Cancer Res. (2012) 31:97. doi: 10.1186/1756-9966-31-97
120. Geczi D, Nagy B, Szilagyi M, Penyige A, Klekner A, Jenei A, et al. Analysis of circulating miRNA profile in plasma samples of glioblastoma patients. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22105058
121. Swellam M, Ezz El Arab L, Al-Posttany AS, Said SB. Clinical impact of circulating oncogenic miRNA-221 and miRNA-222 in glioblastoma multiform. J Neurooncol. (2019) 144:545–51. doi: 10.1007/s11060-019-03256-2
122. Zhao H, Shen J, Hodges TR, Song R, Fuller GN, Heimberger AB. Serum microrna profiling in patients with glioblastoma: A survival analysis. Mol Cancer. (2017) 16:59. doi: 10.1186/s12943-017-0628-5
123. Shankar GM, Balaj L, Stott SL, Nahed B, Carter BS. Liquid biopsy for brain tumors. Expert Rev Mol Diagn. (2017) 17:943–7. doi: 10.1080/14737159.2017.1374854
124. Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). (2013) 91:431–7. doi: 10.1007/s00109-013-1020-6
125. Balakrishnan A, Roy S, Fleming T, Leong HS, Schuurmans C. The emerging role of extracellular vesicles in the glioma microenvironment: biogenesis and clinical relevance. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12071964
126. Matarredona ER, Pastor AM. Extracellular vesicle-mediated communication between the glioblastoma and its microenvironment. Cells. (2019) 9. doi: 10.3390/cells9010096
127. Bebelman MP, Smit MJ, Pegtel DM, Baglio SR. Biogenesis and function of extracellular vesicles in cancer. Pharmacol Ther. (2018) 188:1–11. doi: 10.1016/j.pharmthera.2018.02.013
128. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells toward a Pro-Metastatic Phenotype through Met. Nat Med. (2012) 18:883–91. doi: 10.1038/nm.2753
129. Garcia-Romero N, Carrion-Navarro J, Esteban-Rubio S, Lazaro-Ibanez E, Peris-Celda M, Alonso MM, et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. (2017) 8:1416–28. doi: 10.18632/oncotarget.13635
130. Manda SV, Kataria Y, Tatireddy BR, Ramakrishnan B, Ratnam BG, Lath R, et al. Exosomes as a biomarker platform for detecting epidermal growth factor receptor-positive high-grade gliomas. J Neurosurg. (2018) 128:1091–101. doi: 10.3171/2016.11.JNS161187
131. Khayamzadeh M, Niazi V, Hussen BM, Taheri M, Ghafouri-Fard S, Samadian M. Emerging role of extracellular vesicles in the pathogenesis of glioblastoma. Metab Brain Dis. (2023) 38:177–84. doi: 10.1007/s11011-022-01074-6
132. Yang JK, Yang JP, Tong J, Jing SY, Fan B, Wang F, et al. Exosomal miR-221 targets DNM3 to induce tumor progression and temozolomide resistance in glioma. J Neurooncol. (2017) 131:255–65. doi: 10.1007/s11060-016-2308-5
133. Indira Chandran V, Welinder C, Mansson AS, Offer S, Freyhult E, Pernemalm M, et al. Ultrasensitive immunoprofiling of plasma extracellular vesicles identifies syndecan-1 as a potential tool for minimally invasive diagnosis of glioma. Clin Cancer Res. (2019) 25:3115–27. doi: 10.1158/1078-0432.CCR-18-2946
134. Mason WP. Blood-brain barrier-associated efflux transporters: A significant but underappreciated obstacle to drug development in glioblastoma. Neuro Oncol. (2015) 17:1181–2. doi: 10.1093/neuonc/nov122
135. Liu HL, Huang CY, Chen JY, Wang HY, Chen PY, Wei KC. Pharmacodynamic and therapeutic investigation of focused ultrasound-induced blood-brain barrier opening for enhanced temozolomide delivery in glioma treatment. PloS One. (2014) 9:e114311. doi: 10.1371/journal.pone.0114311
136. Wei HJ, Upadhyayula PS, Pouliopoulos AN, Englander ZK, Zhang X, Jan CI, et al. Focused ultrasound-mediated blood-brain barrier opening increases delivery and efficacy of etoposide for glioblastoma treatment. Int J Radiat Oncol Biol Phys. (2021) 110:539–50. doi: 10.1016/j.ijrobp.2020.12.019
137. Park SH, Kim MJ, Jung HH, Chang WS, Choi HS, Rachmilevitch I, et al. One-year outcome of multiple blood-brain barrier disruptions with temozolomide for the treatment of glioblastoma. Front Oncol. (2020) 10:1663. doi: 10.3389/fonc.2020.01663
138. Carpentier A, Stupp R, Sonabend AM, Dufour H, Chinot O, Mathon B, et al. Repeated blood-brain barrier opening with a nine-emitter implantable ultrasound device in combination with carboplatin in recurrent glioblastoma: A phase I/II clinical trial. Nat Commun. (2024) 15:1650. doi: 10.1038/s41467-024-45818-7
139. Hersh AM, Bhimreddy M, Weber-Levine C, Jiang K, Alomari S, Theodore N, et al. Applications of focused ultrasound for the treatment of glioblastoma: A new frontier. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14194920
140. Ye D, Yuan J, Yue Y, Rubin JB, Chen H. Focused ultrasound-enhanced delivery of intranasally administered anti-programmed cell death-ligand 1 antibody to an intracranial murine glioma model. Pharmaceutics. (2021) 13. doi: 10.3390/pharmaceutics13020190
141. Ram Z, Cohen ZR, Harnof S, Tal S, Faibel M, Nass D, et al. Magnetic resonance imaging-guided, high-intensity focused ultrasound for brain tumor therapy. Neurosurgery. (2006) 59:949–55. doi: 10.1227/01.NEU.0000254439.02736.D8
142. Tamada J, Langer R. The development of polyanhydrides for drug delivery applications. J Biomater Sci Polym Ed. (1992) 3:315–53. doi: 10.1163/156856292x00402
143. Perry J, Chambers A, Spithoff K, Laperriere N. Gliadel wafers in the treatment of Malignant glioma: A systematic review. Curr Oncol. (2007) 14:189–94. doi: 10.3747/co.2007.147
144. Xing WK, Shao C, Qi ZY, Yang C, Wang Z. The role of gliadel wafers in the treatment of newly diagnosed GBM: A meta-analysis. Drug Des Devel Ther. (2015) 9:3341–8. doi: 10.2147/DDDT.S85943
145. Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. (2002) 41:403–19. doi: 10.2165/00003088-200241060-00002
146. Sheleg SV, Korotkevich EA, Zhavrid EA, Muravskaya GV, Smeyanovich AF, Shanko YG, et al. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J Neurooncol. (2002) 60:53–9. doi: 10.1023/a:1020288015457
147. Di Mascolo D, Palange AL, Primavera R, Macchi F, Catelani T, Piccardi F, et al. Conformable hierarchically engineered polymeric micromeshes enabling combinatorial therapies in brain tumours. Nat Nanotechnol. (2021) 16:820–9. doi: 10.1038/s41565-021-00879-3
148. Elstad NL, Fowers KD. Oncogel (Regel/paclitaxel)–clinical applications for a novel paclitaxel delivery system. Adv Drug Delivery Rev. (2009) 61:785–94. doi: 10.1016/j.addr.2009.04.010
149. Boisdron-Celle M, Menei P, Benoit JP. Preparation and characterization of 5-fluorouracil-loaded microparticles as biodegradable anticancer drug carriers. J Pharm Pharmacol. (1995) 47:108–14. doi: 10.1111/j.2042-7158.1995.tb05760.x
150. Roullin VG, Mege M, Lemaire L, Cueyssac JP, Venier-Julienne MC, Menei P, et al. Influence of 5-fluorouracil-loaded microsphere formulation on efficient rat glioma radiosensitization. Pharm Res. (2004) 21:1558–63. doi: 10.1023/b:pham.0000041448.22771.48
151. Wiley DT, Webster P, Gale A, Davis ME. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci U.S.A. (2013) 110:8662–7. doi: 10.1073/pnas.1307152110
152. Tian X, Leite DM, Scarpa E, Nyberg S, Fullstone G, Forth J, et al. On the shuttling across the blood-brain barrier via tubule formation: mechanism and cargo avidity bias. Sci Adv. (2020) 6. doi: 10.1126/sciadv.abc4397
153. Anraku Y, Kuwahara H, Fukusato Y, Mizoguchi A, Ishii T, Nitta K, et al. Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain. Nat Commun. (2017) 8:1001. doi: 10.1038/s41467-017-00952-3
154. Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein pegylation. Adv Drug Delivery Rev. (2002) 54:459–76. doi: 10.1016/s0169-409x(02)00022-4
155. Whittle JR, Lickliter JD, Gan HK, Scott AM, Simes J, Solomon BJ, et al. First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. J Clin Neurosci. (2015) 22:1889–94. doi: 10.1016/j.jocn.2015.06.005
156. Kasenda B, Konig D, Manni M, Ritschard R, Duthaler U, Bartoszek E, et al. Targeting immunoliposomes to EGFR-positive glioblastoma. ESMO Open. (2022) 7:100365. doi: 10.1016/j.esmoop.2021.100365
Keywords: glioblastoma, targeted therapy, drug repurposing, liquid biopsy, extracellular vesicle (EV), ctDNA
Citation: Bae WH, Maraka S and Daher A (2024) Challenges and advances in glioblastoma targeted therapy: the promise of drug repurposing and biomarker exploration. Front. Oncol. 14:1441460. doi: 10.3389/fonc.2024.1441460
Received: 31 May 2024; Accepted: 23 September 2024;
Published: 08 October 2024.
Edited by:
Teresita Padilla-Benavides, Wesleyan University, United StatesReviewed by:
Mario Chiariello, Italian National Research Council, ItalyLisa Gherardini, National Research Council (CNR), Italy
Copyright © 2024 Bae, Maraka and Daher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmad Daher, YWRhaGVyQHVpYy5lZHU=