
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 17 July 2024
Sec. Cancer Molecular Targets and Therapeutics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1427802
 Mohd Mustafa1
Mohd Mustafa1 Kashif Abbas2
Kashif Abbas2 Mudassir Alam2
Mudassir Alam2 Safia Habib1
Safia Habib1 Zulfareen3
Zulfareen3 Gulam Mustafa Hasan4
Gulam Mustafa Hasan4 Sidra Islam5
Sidra Islam5 Anas Shamsi6*
Anas Shamsi6* Imtaiyaz Hassan3*
Imtaiyaz Hassan3*Pancreatic adenocarcinoma, a clinically challenging malignancy constitutes a significant contributor to cancer-related mortality, characterized by an inherently poor prognosis. This review aims to provide a comprehensive understanding of pancreatic adenocarcinoma by examining its multifaceted etiologies, including genetic mutations and environmental factors. The review explains the complex molecular mechanisms underlying its pathogenesis and summarizes current therapeutic strategies, including surgery, chemotherapy, and emerging modalities such as immunotherapy. Critical molecular pathways driving pancreatic cancer development, including KRAS, Notch, and Hedgehog, are discussed. Current therapeutic strategies, including surgery, chemotherapy, and radiation, are discussed, with an emphasis on their limitations, particularly in terms of postoperative relapse. Promising research areas, including liquid biopsies, personalized medicine, and gene editing, are explored, demonstrating the significant potential for enhancing diagnosis and treatment. While immunotherapy presents promising prospects, it faces challenges related to immune evasion mechanisms. Emerging research directions, encompassing liquid biopsies, personalized medicine, CRISPR/Cas9 genome editing, and computational intelligence applications, hold promise for refining diagnostic approaches and therapeutic interventions. By integrating insights from genetic, molecular, and clinical research, innovative strategies that improve patient outcomes can be developed. Ongoing research in these emerging fields holds significant promise for advancing the diagnosis and treatment of this formidable malignancy.
Pancreatic cancer, characterized by its aggressive behavior, a tendency for late-stage identification, and limited therapeutic options, poses a significant challenge in the advancing field of oncology (1). The tumor microenvironment (TME) comprises a dynamic amalgamation of immune cells, extracellular matrix, and stromal cells, significantly influencing the disease trajectory and complicating treatment resistance (2). Epithelial-mesenchymal transition (EMT) promotes cancer cell invasion and migratory capabilities, intensifying cancer cell complexity. The immune evasion mechanisms employed by pancreatic cancer cells pose a formidable barrier to effectively utilizing immunotherapy, necessitating innovative solutions (3).
Critical signaling pathways govern the crucible of cellular life. Furthermore, persistent activation of the KRAS pathway is a hallmark feature of uncontrolled cell survival and proliferation (4). The Hedgehog and Notch pathways contribute to the resilience of cancer stem cells, increasing their resistance to treatment (5). Dysregulation of the PI3K/AKT/mTOR pathway promotes increased cellular growth and survival (6). The Wnt/β-catenin signaling pathway activates tumor growth, further complicating the battle against pancreatic cancer (7). Surgical excision remains the primary curative option for early-stage patients (8). In more advanced stages, accepted standard-of-care options include chemotherapy regimens such as gemcitabine, FOLFIRINOX, and nab-paclitaxel (9). Localized tumors may undergo radiation treatment to eliminate or reduce their presence. Several targeted therapies, particularly PARP inhibitors, are currently under rigorous investigation to treat pancreatic cancer (10). Immunotherapy involving checkpoint inhibitors and vaccines holds promise for enhancing the immune system’s response to pancreatic cancer (11).
Liquid biopsies are being explored as noninvasive diagnostic tools for the primary detection of pancreatic cancer, potentially enabling intervention at a more treatable stage (12). Personalized medicine approaches, tailored to individuals’ genetic and molecular profiles, are poised to optimize therapeutic strategies, providing a specialized toolkit against this resilient adversary. The application of CRISPR/Cas9 genome editing tools for the exploration and potential correction of genetic mutations is actively being explored, revealing the possibility of addressing the illness at its molecular origins. Artificial intelligence has been harnessed to expedite the early identification and prediction of therapeutic responses in pancreatic cancer patients, demonstrating the power of technology to treat pancreatic carcinoma (13).
The primary objectives of this review are to provide a comprehensive overview of the current molecular and genetic landscape of PDAC, including an in-depth examination of key molecular pathways such as KRAS, Notch, and Hedgehog, and their roles in the pathogenesis and progression of the disease. Additionally, this review aims to critically analyse existing therapeutic strategies and their limitations, offering a thorough evaluation of conventional treatments like surgery, chemotherapy, and radiation, as well as emerging therapies such as immunotherapy and targeted molecular treatments. The challenges associated with these treatments, particularly issues related to drug resistance and the tumor microenvironment, will be highlighted. Furthermore, the review seeks to highlight the potential of emerging diagnostic and therapeutic technologies. This involves exploring novel approaches such as liquid biopsies for early detection, personalized medicine based on genomic and transcriptomic profiling, and the application of CRISPR/Cas9 gene editing technology. The review aims to identify key challenges and propose future research directions. This includes recognizing major obstacles in the treatment and management of PDAC, such as the tumor micro-environment and immune evasion mechanisms. The review will propose future research directions aimed at overcoming these challenges, thereby facilitating the way for more effective diagnostic and therapeutic strategies.
Pancreatic intraepithelial neoplasia (PanIN), a frequently encountered preneoplastic lesion, serves as the primary instigator of pancreatic cancer (14). Furthermore, more sophisticated precursor abnormalities, such as mucinous cystic neoplasms and intraductal papillary mucinous neoplasms (IPMNs), actively contribute to the development of this condition (15). The progression of pancreatic cancer involves complex molecular and cellular processes, with distorted autocrine and paracrine signaling pathways playing crucial roles in fostering the growth, migration, invasion, and metastasis of cancer cells. Critical factors, including transforming growth factor-α (TGFα) (16), insulin-like growth factor 1 (IGF1) (17), fibroblast growth factors (FGFs) (18), and hepatocyte growth factor (HGF) (19), along with their corresponding tyrosine kinase receptors, such as epidermal growth factor receptor (EGFR) (20), receptor tyrosine-protein kinase erbB-2 (ERBB2/HER2) (21), HER3 (22), the IGF1 receptor (IGF1R) (23), FGF receptors (FGFRs) (24), and the HGF receptor (HGFR/MET) (25), trigger several pathways contributing to cell growth (Figure 1).
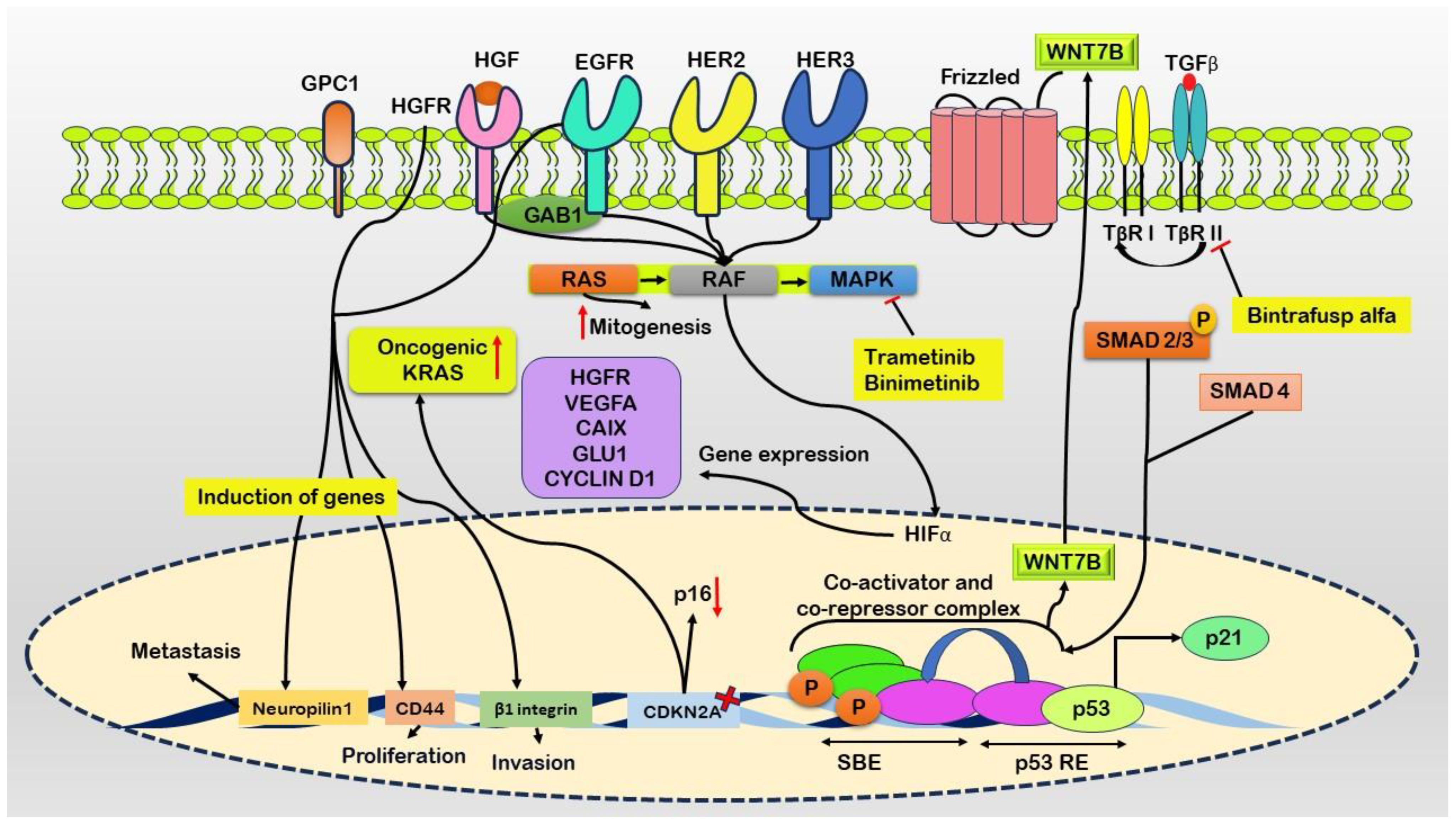
Figure 1 Schematic representation of molecular events driving pancreatic cancer progression. Ligand binding to the Epidermal Growth Factor Receptor (EGFR) initiates heterodimerization with ERRB2/HER2 and HER3. Co-occurrence of oncogenic KRAS mutations and elevated ligand expression enhances downstream signaling. Growth factor receptor-bound protein 1 (GAB1) augments activation of EGFR and Hepatocyte Growth Factor Receptor (HGFR). Glypican-1 (GPC1) maintains signaling pathways promoting mitogenesis, invasion, and metastasis via canonical RAS, RAF, MAPK, STAT3, PI3K, and AKT pathways. Fibroblast growth factor receptor substrate 2 (FRS2) is crucial for downstream signaling from FGFR1, triggering the Ras cascade. MAPK translocates to the nucleus to regulate transcription, including Hypoxia-Inducible Factor 1 (HIF1) induction. Dysfunctional retinoblastoma protein 1 (RB1) exacerbates mitogenic signaling and may convert Transforming Growth Factor-beta (TGFβ) into a direct mitogen through non-canonical MAPK and PI3K pathways. TGFβ-mediated activation of WNT7B occurs via a SMAD4-dependent mechanism. Elevated expression of growth factor receptors, such as HGFR and EGFR, induces genes like Neuropilin1, CD44, and β1 integrin, contributing to metastasis, proliferation, and invasion. Inhibitors such as Trametinib and Binimetinib target the MAPK pathway, while Bintrafusp alfa binds to TGFβ, leading to its blockade.
Initial activation ensues upon ligand binding, activating EGFR and forming heterodimers with the receptor tyrosine-protein kinase erbB-2 (ERRB2/HER2) and HER3 (26). The coexistence of oncogenic KRAS and heightened ligand expression synergistically amplifies downstream signaling cascades (27). Docking protein growth factor receptor-bound protein 2 (GRB2)-associated binding protein 1 (GAB1) further enhances the activation of both EGFR and the hepatocyte growth factor (HGF) receptor (HGFR) (28). Prolonged signaling is sustained through the overexpression of heparan sulfate proteoglycan glypican 1 (GPC1), which promotes mitogenesis, invasion, and metastasis via canonical RAS, RAF, mitogen-activated protein kinase (MAPK), and other pathways, including signal transducer and activator of transcription 3 (STAT3), phosphatidylinositol 3-kinase (PI3K), and AKT pro-survival signaling (29).
The crucial adaptor protein fibroblast growth factor receptor substrate 2 (FRS2) is indispensable for downstream signaling from fibroblast growth factor receptor 1 (FGFR1), thereby activating the Ras signaling cascade (30). Subsequently, MAPK translocate to the nucleus, where it coordinates transcriptional activities, including the induction of hypoxia-inducible transcription factor 1 (HIF1) (31). Concurrently, dysfunctional retinoblastoma-associated protein (RB1) intensifies mitogenic signaling, potentially converting transforming growth factor-beta (TGFβ) into a direct mitogen through noncanonical pathways (MAPK and PI3K). Additionally, TGFβ-mediated activation of WNT7B is facilitated via a SMAD4-dependent mechanism (32).
Pancreatic cancer involves pathways that promote cell survival and inhibit apoptosis, particularly pathways involving AKT, NF-κB, and STAT3 (33). The reactivation of developmental genes such as WNT, SHH, and NOTCH occurs in certain pancreatic tumors (34). Aberrant crosstalk pathways and multiple nodes further compound the complicated signaling network of pancreatic cancer. For instance, heightened action of HGFR and EGFR leads to the induction of neuropilin1, CD44, and β1 integrin, contributing to an abnormal signaling node (35). The formation of heterodimers between HGFR and EGFR aggravates this complexity (36).
Simultaneously, these molecular changes occur with the deletion of CDKN2A, which is responsible for encoding the tumor suppressor p16, and the activation of oncogenic KRAS (37). Metabolic irregularities and a diminished response to growth-inhibitory pathways mark pancreatic cancer. One example of a lack of negative growth limitations is dysregulated TGFβ signaling, which is usually a tumor suppressor but paradoxically promotes tumor development in pancreatic cancer. TGFβ exerts paracrine effects within the tumor microenvironment, augmenting growth and metastatic processes (38).
Moreover, pancreatic cancer cell proliferation is directly stimulated by TGFβ through noncanonical signaling pathways. These pathways involve the phosphorylation of MAPK, the proto-oncogene tyrosine-protein kinase Src (SRC), AKT phosphorylation, and canonical SMAD4-dependent mechanisms that lead to the upregulation of WNT7B expression (39). Trametinib and binimetinib function as inhibitors of the MAPK pathway.
K-Ras plays a vital role in pancreatic ductal adenocarcinoma (PDAC), and K-Ras point mutations are highly prevalent among most PDAC patients. These mutations underscore fundamental genetic modifications originating in early pancreatic lesions, especially in low-grade PanIN (40). The persistent proliferation and survival of pancreatic cancer cells rely on the signaling activity of K-Ras (41). The initiation of the KRAS protein triggers its downstream intracellular pathways (Figure 2). Following the activation of growth factor receptors, such as tyrosine kinase or G-coupled receptors, growth factor receptor-bound protein 2 (GRB2) associates with the guanine nucleotide exchange factor son of sevenless (SOS) and engages with the KRAS protein (42). To be active, KRAS must be anchored to the cell membrane, where influential membrane association occurs. Once this association is established, KRAS becomes activated when it is bound to GTP (43).
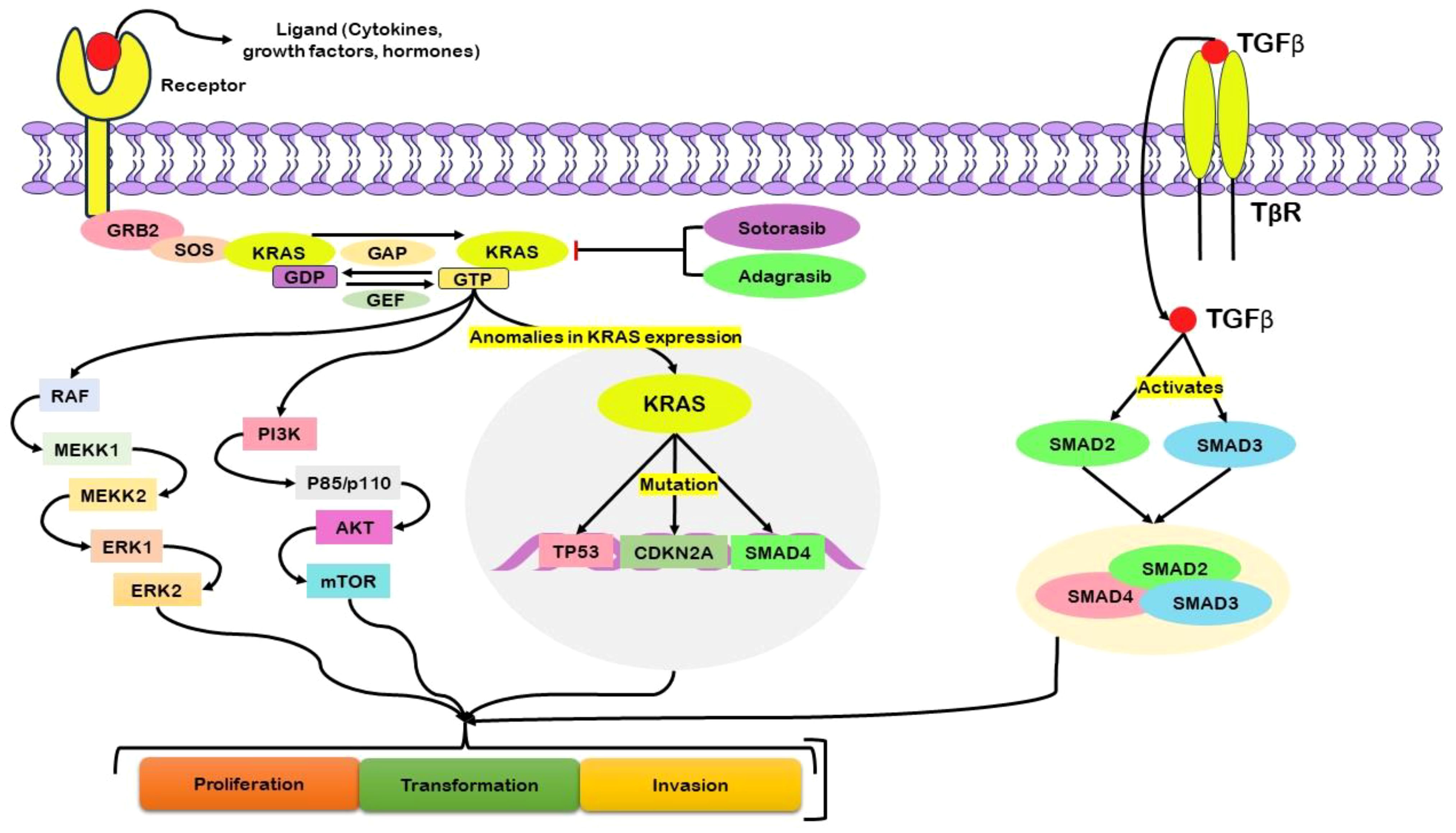
Figure 2 Schematic illustration of the central role of KRAS, particularly in its mutated form, in driving abnormal cellular activities associated with pancreatic cancer. Activation of growth factor receptors engages critical mediators, including growth factor receptor-bound protein 2 (GRB2), the guanine nucleotide exchange factor son of sevenless (SOS), and KRAS. Activation of KRAS, reliant on its membrane association and binding to guanosine triphosphate (GTP), initiates downstream signaling pathways. The complex regulation of KRAS GTP–guanosine diphosphate (GDP) cycling is governed by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). Mutations in KRAS disrupt this regulatory mechanism, resulting in persistent GTP binding and continuous downstream signaling. This dysregulation impacts nuclear transcription factors, influencing cellular proliferation, survival, and transformation. Abnormal expression of KRAS is associated with mutations in key genes such as TP53, CDKN2A, and SMAD4, further promoting oncogenesis. KRAS activity also relates to the activation of Transforming Growth Factor-beta (TGF-β) and subsequent downstream signaling. Therapeutic interventions targeting KRAS, such as Sotorasib and Adagrasib, are commonly used in the treatment of pancreatic cancer.
Inherent KRAS GTP–GDP cycling is regulated by guanine nucleotide exchange factors (GEFs), which facilitate nucleotide exchange, and by GTPase-activating proteins (GAPs), which accelerate the intrinsic GTP hydrolysis activity of KRAS (44). In cases of KRAS mutation, the intrinsic GTPase activity is compromised, impeding the role of GAPs in facilitating the conversion of GTP to GDP (45). Consequently, KRAS remains persistently bound to GTP, initiating downstream signaling pathways. This, in turn, activates nuclear transcription factors, ultimately leading to cellular processes such as proliferation, survival, and transformation (46). The dysregulation of KRAS function due to mutation underscores its pivotal role in driving aberrant cellular activities associated with pancreatic cancer. Mutations in codons G12D or G12V cause acinar to ductal metaplasia and PanIN, which advances PDAC (47). Mutations in tumor suppressor genes, viz. P16/CDKN2A, SMAD4, and p53, combined with a positive K-Ras mutation, enhance cancer development in mouse models (48).
Various downstream effectors, including classical Raf/MAPK/extracellular signal-regulated kinase (Erk) (49), PI3Ks/(PDK-1)/Akt, RalGEFs, and phospholipase Cϵ, play crucial roles in the signaling cascade of K-Ras. Disruptions or mutations within these downstream cascades introduce complexities in K-RAS-driven PDAC (50). The presence of a persistently active oncogenic class 1A PI3K, such as PI3CA H1047R, hinders K-RasG12D-driven PDAC, triggers acinar to ductal metaplasia, and initiates precancerous PanIN while also precluding the involvement of PDK-1 (51). The most commonly used active suppressor in pancreatic cancer is P16/CDKN2A (52). It prevents retinoblastoma from being phosphorylated by CDK4/6, preventing cells from entering the S phase of the cell cycle (53).
Various factors contribute to P16/CDKN2A inhibition, such as epigenetic suppression and homozygous deletion, highlighting the critical role of this tumor suppressor gene in this disease (54). Moreover, the haploinsufficiency of P16/CDKN2A, especially in K-Ras-mutant mice, significantly advances the development of PanIN lesions and PDAC (55). SMAD4, another notable tumor suppressor gene, functions downstream of TGF-β signaling, exerting control over cell cycle progression and promoting apoptosis. TGF-β triggers the activation of Smad2 and Smad3, resulting in their binding with Smad4. Subsequently, this complex relocates to the nucleus, influencing gene expression (56).
In PDAC, SMAD4 loss promotes carcinogenesis and potentiates K-RasG12D-driven acinar to ductal metaplasia, PanIN, and PDAC (57). SMAD4 inactivation often occurs through homozygous deletion, highlighting its crucial role as a gatekeeper in pancreatic cancer (58). The tumor suppressor p53, encoded by TP53, is mutated in most pancreatic cancer patients (59). Interestingly, a heterozygous inactivating mutation (p53R172H/+) in combination with K-RasG12D amplifies PanIN and PDAC development in mouse models (60). Thus, p53 acts as a crucial barrier against K-Ras-driven pancreatic carcinogenesis. P53 regulates various cellular functions, including halting the cell cycle, facilitating DNA repair, inducing senescence, and promoting apoptosis (61). The deviant activation of K-Ras leads to mutations in TP53, CDKN2A, and SMAD4, which propels the development and progression of pancreatic cancer (62). These molecular mechanisms highlight the challenges in targeting K-Ras directly, as its mutational landscape and downstream signaling pathways are highly complex and context dependent. Drugs such as sotorasib (FDA-approved) and adagrasib target KRAS as therapeutic interventions for various types of cancer, including pancreatic cancer (63).
The Notch signaling pathway significantly contributes to the pathogenesis of pancreatic cancer by precisely governing cellular processes, including proliferation, differentiation, and apoptosis, and plays a vital role in growth and tissue homeostasis (64). Dysregulation of Notch signaling promotes carcinoma initiation and onset (Figure 3). The human Notch family comprises five ligands (Delta-like 1, 3, 4, and Jagged 1, 2) and four receptors (Notch1-4) (65). The initiation of Notch signaling involves ligand-;receptor interactions, leading to the proteolytic cleavage of the Notch receptor by γ-secretase (66). During this stage, the Notch intracellular domain (NICD) is liberated, moves to the nucleus, and associates with Mastermind-like (MAML), CSL (CBF1/RBPJκ in mammals), and other coactivators. This associated assembly then stimulates the transcription of target genes, including those belonging to the Hes and Hey families (67).
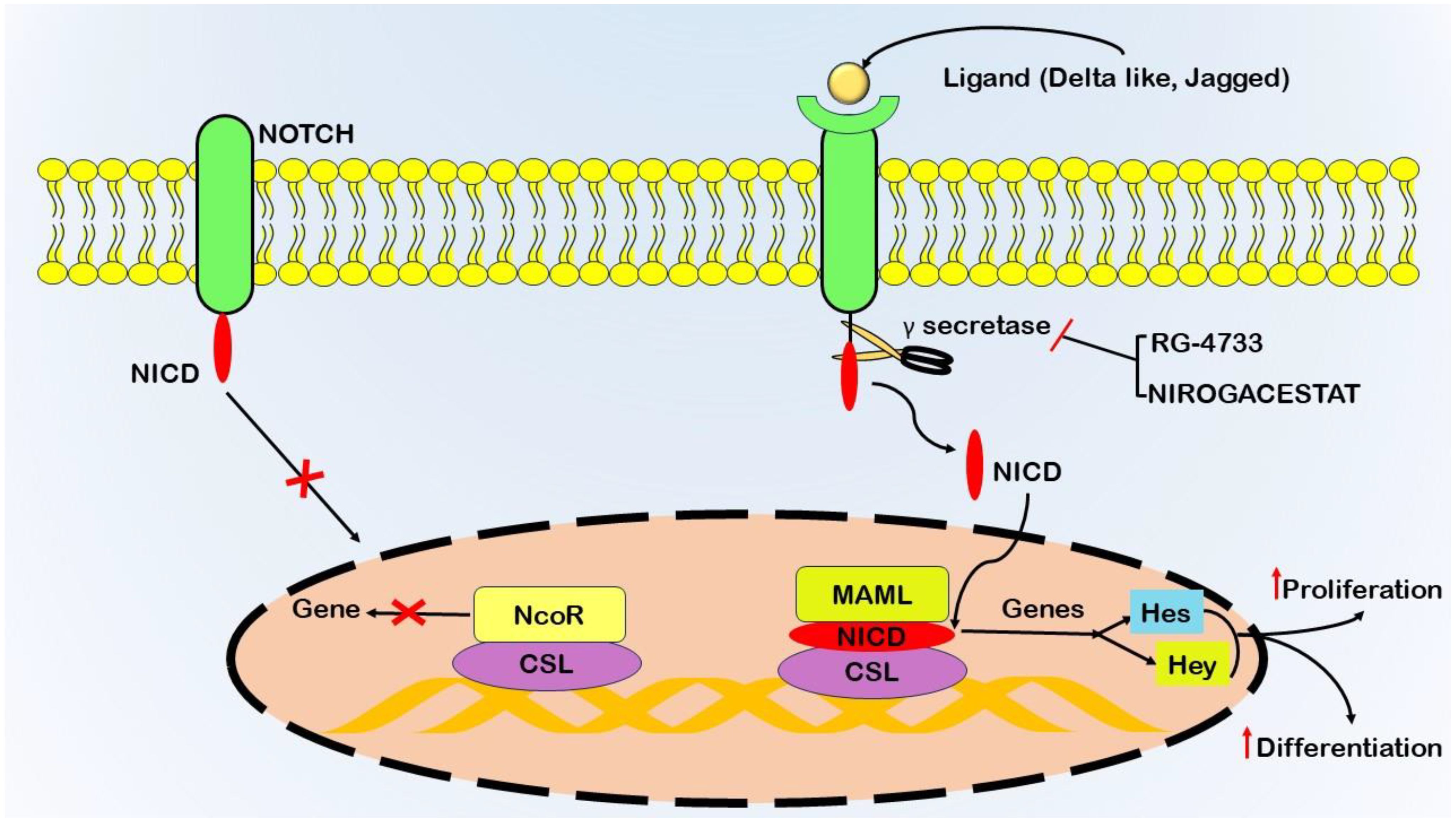
Figure 3 Schematic representation of gamma-secretase activity in initiating the Notch signaling pathway, a critical system involving transmembrane receptors (Notch1–4) and ligands (Delta-like 1, 3, 4, Jagged1, 2). Notch receptors on the cell surface interact with adjacent Delta and Jagged ligands, triggering sequential proteolytic cleavages. Tumor necrosis factor-alpha-converting enzyme (TACE) or ADAM10 mediates the initial cleavage, followed by the γ-secretase complex executing the second cleavage. This process releases the Notch intracellular domain (NICD) from the cell membrane, allowing its translocation to the nucleus. Inside the nucleus, NICD binds to the CSL transcription factor, displacing co-repressors and recruiting transcriptional activators such as Mastermind-like1 (Maml). This activation leads to the transcription of target genes Hes and Hey, which regulate cellular proliferation and differentiation. Gamma-secretase inhibitors (GSIs) impede the cleavage of Notch receptors by the γ-secretase complex, preventing NICD release and modulating Notch signaling. Therapeutic interventions, including drugs such as RG-4733 and Nirogacestat, act as inhibitors of γ-secretase.
Dysregulation of the Notch signaling pathway significantly contributes to tumorigenesis within the context of pancreatic cancer (68). Mutations that activate Notch receptors (NOTCH1 and NOTCH2) have been detected in a subset of pancreatic cancer cases, leading to ligand-independent activation of Notch signaling (69). Additionally, an increase in the expression of Notch ligands, namely, Jagged1 and Jagged2, further substantiates the dysregulation of this pathway in pancreatic cancer (70). The therapeutic target involves blocking the activity of γ-secretase. Drugs such as RG-4733 and nirogacestat are being tested in clinical trials as inhibitors of γ-secretase (71).
Hedgehog signaling has emerged as a pivotal pathway implicated in advancing pancreatic cancer. While this cascade is vital for embryogenesis and tissue homeostasis, it is associated with pancreatic cancer (72) (Figure 4). The Hedgehog pathway comprises three major components: Hedgehog ligands (Sonic Hedgehog SHH, Indian Hedgehog IHH, and Desert Hedgehog DHH) (73). Patched (PTCH) is a transmembrane receptor, while Smoothened (SMO) is a G protein-coupled receptor-like protein. PTCH inhibits SMO in the absence of Hedgehog ligands (74). However, when Hedgehog ligands bind to PTCH, SMO is released from inhibition, initiating downstream signaling events (75). The aberrant activation of Hedgehog signaling in pancreatic cancer is frequently linked to increased expression of the SHH ligand (76). Furthermore, SHH overexpression is observed in pancreatic cancer precursor lesions (PanIN) and invasive carcinoma (77). Genetic alterations in Hedgehog pathway components, including mutations in SMO and amplifications of GLI1 and GLI2 (downstream transcription factors), contribute to the expression of proproliferative and antiapoptotic genes such as Myc, Bcl-2, and Sox2. Importantly, Hedgehog signaling engages in crosstalk with other pathways, particularly K-Ras and Notch, thereby influencing the behavior of pancreatic cancer cells (78). This activation promotes cancer stem cell characteristics and significantly contributes to tumor initiation, progression, and therapeutic resistance development. Sonidegib and vismodegib function as inhibitors targeting the Smoothened (SMO) protein. These drugs work by interfering with the activity of SMO, a vital component of the Hedgehog signaling pathway (Supplementary Table S1).
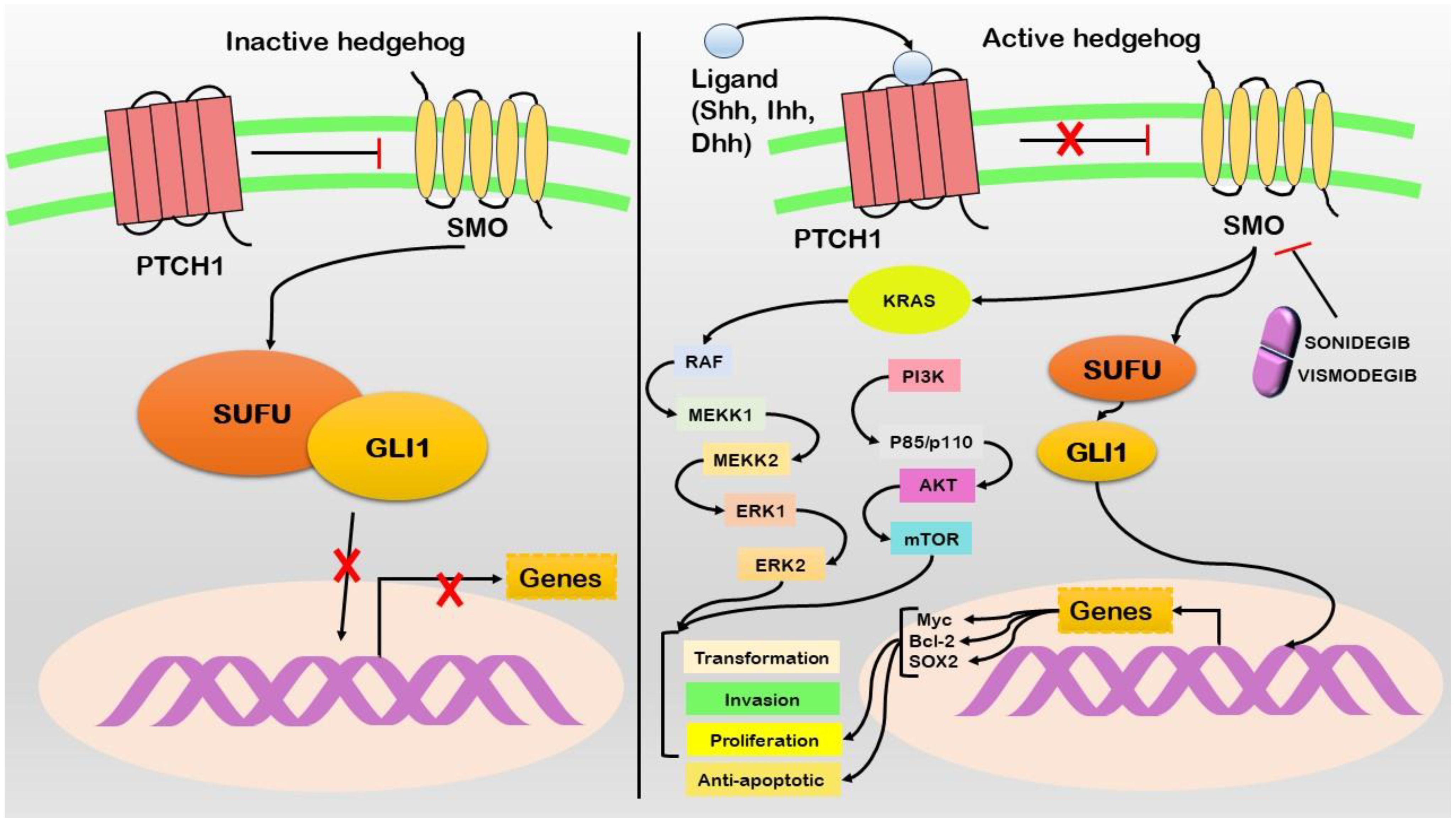
Figure 4 In the absence of Shh ligand (left), the pathway remains inactive with Patched1 (PTCH1) inhibiting Smoothened (SMO), resulting in the sequestration of GLI1 in the cytoplasm via Suppressor of Fused (SUFU). Upon the presence of Shh ligand (right), PTCH1 suppression of SMO is relieved, permitting GLI1 to accumulate in the nucleus. This activation induces the transcription of target genes, promoting various oncogenic properties. Active Hedgehog signaling leads to the activation of KRAS and its downstream signaling cascade. Therapeutic interventions such as Sonidegib and Vismodegib act as SMO inhibitors, thereby disrupting the pathway.
PI3K/AKT/mTOR signaling governs cell survival, proliferation, and metabolism. Its dysregulation is common in pancreatic cancer, contributing significantly to its aggressive phenotype (79). PI3K triggers the activation of this pathway by phosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2) and produces phosphatidylinositol 3,4,5-trisphosphate (PIP3) (80). PIP3 subsequently activates AKT, a serine/threonine kinase that phosphorylates various downstream targets, including the mammalian target of rapamycin (mTOR) (81). In pancreatic cancer, the PI3K/AKT/mTOR pathway frequently undergoes dysregulation due to genetic alterations. These alterations include mutations in PIK3CA, which encodes the catalytic subunit of PI3K; damage to the function of the tumor suppressor phosphatase and tensin homolog (PTEN); and activating mutations in AKT1 (82). These genetic changes result in sustained pathway activation, promoting cell survival, proliferation, and resistance to apoptosis. Furthermore, the PI3K/AKT/mTOR pathway engages in crosstalk with other signaling pathways, including K-Ras and Notch, further contributing to the overall complexity of pancreatic cancer signaling networks (83).
The Wnt/β-catenin signaling pathway is a pivotal regulatory mechanism that governs diverse cellular processes, including cell proliferation, differentiation, and survival. Dysregulation of this pathway has been linked to the initiation and progression of various cancers, including pancreatic cancer (84). Pancreatic cancer exhibits complex molecular alterations, and disturbances in the Wnt/β-catenin signaling pathway significantly contribute to its pathogenesis. In the typical cellular environment of the pancreas, the destruction complex involves APC, Axin, GSK-3β, and CK1, coordinating the destruction of β-catenin (85). However, in pancreatic cancer, various mechanisms contribute to the abnormal initiation of the Wnt pathway. Wnt ligands, particularly Wnt2 and Wnt5a, are frequently overexpressed, initiating signaling through Frizzled receptors and LRP5/6 coreceptors. This binding event disrupts the destruction complex, hindering the phosphorylation and degradation of β-catenin (86). Stable β-catenin then translocates to the nucleus, where it forms a transcriptional complex with TCF/LEF transcription factors (87). This activation prompts the transcription of target genes, including MYC and Cyclin D1, which are pivotal for fostering uncontrolled cell proliferation and survival in pancreatic cancer (88). Genetic mutations further accentuate Wnt pathway dysregulation in pancreatic cancer. Mutations in APC or β-catenin result in constitutive activation of the pathway, emphasizing the genetic keystones of this aberrant signaling cascade (89).
The clinical significance of these molecular insights is highlighted by experimental approaches directing the Wnt/β-catenin pathway in pancreatic cancer (90). Investigations are underway on small molecule inhibitors that disrupt crucial components such as β-catenin or upstream regulators. Nevertheless, translating these promising preclinical discoveries into effective clinical interventions requires thorough examination through clinical trials that are tailored explicitly for patients with pancreatic cancer. Furthermore, research has shown the potential effectiveness of inhibiting the Wnt pathway in preclinical models of pancreatic cancer (91, 92). For instance, inhibiting Wnt signaling has been linked to reduced tumor growth and enhanced survival in murine models (93, 94). These observations offer a compelling rationale for exploring therapies targeting the Wnt pathway in the clinical context.
Pancreatic cancer is characterized by elevated levels of various mitogenic growth factors and their corresponding ligands. This includes heightened expression of epidermal growth factor (EGF) and its associated receptor, EGFR, multiple ligands that engage with EGFR, FGF and its receptor FGFR, insulin-like growth factor (IGF) and its receptor IGFR, platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) (95). These signaling molecules are excessively expressed in pancreatic cancer, contributing to the aggressive nature of the disease.
In neoplastic cells, the activation of EGFR can occur inaccurately through various mechanisms, including ligand-dependent dimerization, point mutations, partial deletions, or overexpression (96). Increased expression of EGFR is linked to structural or numerical alterations of chromosome 7, where the EGFR gene is located (97). The c-ERBB-1 proto-oncogene encodes EGFR, and while in the normal pancreas, c-ERBB-1 is expressed exclusively in the islets of Langerhans, human pancreatic cancer cell lines frequently demonstrate its overexpression, which is observed in up to 85% of ductal adenocarcinomas (98). Pancreatic cancer is characterized by the accumulation of numerous genetic alterations, with early occurrences of KRAS mutations and EGFR gene amplification occurring during disease progression (99). Subsequent alterations involve p16 inactivation, and late changes inactivate the TP53 and SMAD4 genes (100).
Importantly, ligands such as EGF and TGF-α play pivotal roles in EGFR activation. Following ligand binding, EGFR undergoes receptor homo or heterodimerization at the cell surface, followed by internalization. Dimerization leads to phosphorylation of the intracytoplasmic EGFR tyrosine kinase domain, which acts as a binding site for signaling molecules such as RAS (101). Activation of downstream pathways stimulates cellular proliferation, angiogenesis, and metastatic development and inhibits apoptosis (102). PDAC results from multiple mutations, with the initial precursor lesion being intraepithelial pancreatic neoplasia (PanIN). The progression from PanIN to invasive cancer involves sequential steps, starting from PanIN-1 with Kras mutation and telomere shortening to PanIN-2 with p16 inactivation and PanIN-3 with p53 and SMAD4 inactivation, culminating in invasive carcinoma (103). Acinar to ductal metaplasia (ADM) is considered a crucial precursor in PanIN progression (104).
Other noninvasive pancreatic neoplasms include mucinous cystic and intraductal mucinous neoplasms (105). Genome sequencing has identified four genes frequently implicated in PDAC: Kras, CDKNA2A/p16, SMAD4, and TP53 (106). Kras oncogene mutations are predominant in PDAC, and their association with EGFR activation suggests a mechanism in which EGFR stimulation complements oncogenic pathways (107). Kras mutations hinder the ability of the Kras protein to hydrolyze guanosine triphosphate, maintaining the protein in an active signaling state that activates other pathways, such as the Raf and PI3 pathways (108).
PDAC remains one of the most lethal cancer types due to its aggressive nature and resistance to conventional treatments. In the complex domain of pancreatic cancer progression, IGFs and their associated receptors have emerged as central regulators, influencing crucial processes such as angiogenesis, invasion, and cell survival (109). IGFs, particularly IGF-1 and IGF1R, significantly influence cancer biology. The IGF system is pivotal in regulating key processes essential for tumorigenesis and metastasis in various cancers, including pancreatic cancer (110). Increased expression of IGF-1 and IGF1R in PDAC is closely associated with unfavorable clinical outcomes, with elevated levels correlating with poor survival rates and higher tumor grades, establishing them as prognostic indicators for pancreatic cancer patients (111). In vitro investigations employing models of pancreatic cancer have provided valuable insights into the functional role of IGF-1 (112, 113).
Exogenous IGF-1 has been demonstrated to promote the development of pancreatic cancer cells, underscoring its function as a growth factor in disease progression (114). Furthermore, this growth-promoting effect can be counteracted by using antibodies designed to neutralize IGF-1, suggesting a potential avenue for therapeutic intervention. Despite promising preclinical findings, the translation of IGF1R-targeted therapies to clinical success has faced obstacles. Clinical trials, exemplified by the phase III trial investigating ganitumab, an antibody targeting IGF1R in conjunction with gemcitabine for metastatic pancreatic cancer patients, failed to yield a statistically significant improvement in survival (115). Amgen’s discontinuation of the trial underscores the challenges in translating preclinical success into meaningful clinical benefits (116). The setbacks in clinical trials targeting IGF1R in pancreatic cancer have raised critical questions about the complexities of the IGF system in the clinical context. Potential reasons for the lack of success may include adaptive resistance mechanisms, patient population heterogeneity or the influence of the tumor microenvironment. Future research endeavors should focus on identifying the roles of IGF signaling in pancreatic cancer, exploring combination therapies, and identifying potential biomarkers for patient stratification. While IGFs and their receptors drive the aggressive behavior of pancreatic cancer, the translation of knowledge into successful clinical interventions remains a formidable challenge in the field of medical research.
FGFR signaling is pivotal for cellular processes, including proliferation, survival, and angiogenesis. Dysregulation of this pathway has been implicated in various cancers, including pancreatic cancer (117). A cascade of intracellular events occurs upon the binding of fibroblast growth factors (FGFs) to their corresponding FGFRs. This interaction initiates a structural alteration in the receptor, promoting the autophosphorylation of distinct tyrosine residues within the intracellular domain of FGFR. This autophosphorylation activates the receptor, creating docking sites for downstream signaling molecules (118). A crucial downstream target of activated FGFR is FGFR substrate 2 (FRS2). Upon FGF binding, FGFR initiates the phosphorylation of FRS2, a pivotal event in transducing signals to downstream pathways (119). Phosphorylated FRS2 is a scaffold for recruiting and activating components in two principal molecular pathways, the PI3K/Akt pathway and the rat sarcoma (Ras/MAPK) pathway, which are critical cellular signaling pathways (120). As a scaffold, phosphorylated FRS2 facilitates the recruitment and activation of key signaling molecules. In the PI3K/Akt pathway, activated FRS2 promotes the activation of PI3K, generating phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and activating Akt, which is pivotal for cell survival and proliferation (121).
Phosphorylated FRS2 stimulates the Ras/MAPK pathway, which triggers the phosphorylation of mitogen-activated protein kinase kinase (MEK), triggering the activation of MAPK (ERK), which is renowned for its involvement in cellular proliferation and differentiation (122). Upregulation of the FGFR-1 and FGFR-2 receptors and increased expression of their ligands (FGF1-7) have been observed in a subset of pancreatic tumors. This dysregulation contributes to enhanced angiogenesis and mitogenesis, which are critical processes in cancer progression (123). The aberrant activation of FGFR signaling establishes an environment conducive to tumor growth and dissemination. Preclinical models of pancreatic cancer have demonstrated the therapeutic potential of inhibiting FGFR signaling. Approaches such as tyrosine kinase inhibitors, short hairpin RNA (shRNA) targeting FGFRs, and the administration of dovitinib have been explored. Inhibition of FGFR signaling in these models resulted in significant anticancer effects, suggesting that FGFR is a promising therapeutic target for pancreatic cancer (124–126).
VEGF, a potent angiogenic factor, induces endothelial cell proliferation and sustains cell viability through engagement with its receptors, namely, VEGFR-1 and VEGFR-2 (127). In the context of PDAC, dysregulation of VEGF signaling contributes to establishing a proangiogenic microenvironment (128). Although PDAC is not traditionally highly vascularized, increased expression of VEGF mRNA has been consistently detected in tumor samples from PDAC patients. This upregulation correlates with disease progression and increased microvessel density, signifying an essential function for VEGF in fostering an angiogenic phenotype within the TME (129). These findings indicate that increased VEGF levels are associated with more aggressive tumor behavior, higher rates of metastasis, and poorer prognosis. The elevated microvessel density in response to heightened VEGF expression supports the notion that angiogenesis is a dynamic and critical process in PDAC progression (130).
Given the prominent role of VEGF in PDAC, therapeutic interventions targeting the VEGF pathway have garnered attention as potential strategies to prevent tumor growth and metastasis. In murine specimens, TNP-40, an analog of the antiangiogenic agent fumagillin, has demonstrated efficacy in reducing tumor growth and metastasis in PDAC cell lines (131). This preclinical evidence suggests that targeting angiogenesis through agents such as TNP-40 may have therapeutic implications for PDAC management. In preclinical studies involving pancreatic cancer, a viral vector containing PTK 787, a VEGFR tyrosine kinase inhibitor, has shown significant promise in impeding the metastasis and growth of PDAC (132). By specifically targeting the tyrosine kinase activity of VEGFR, PTK 787 interrupts downstream signaling cascades, mitigating the proangiogenic effects induced by VEGF (133). This approach holds the potential for developing targeted therapies that directly interfere with the VEGF–VEGFR axis, thereby impeding angiogenesis and disrupting the tumor’s ability to establish a robust blood supply. VEGF’s influence on endothelial cell proliferation and survival significantly contributes to the angiogenic microenvironment observed in PDAC. As research in this field progresses, the development of targeted therapies aimed at disrupting VEGF-mediated angiogenesis holds promise for improving outcomes in PDAC patients.
The transmembrane receptor, receptor for advanced glycation end products (RAGE or AGER), is a member of the immunoglobulin superfamily and is located in the class III region of the major histocompatibility complex. Activation of this receptor has been linked to the initiation of inflammatory processes, which has implications for a spectrum of persistent ailments, such as hyperglycemia, brain degeneration disorders, and cancer (134). Recent studies have revealed the distinct roles of RAGE in pancreatic tumorigenesis and drug resistance, revealing novel therapeutic possibilities. Studies involving the suppression of RAGE expression, either through knockdown or knockout approaches, have demonstrated a notable delay in the growth of pancreatic tumors driven by oncogenic KRAS (135–137). This finding emphasizes that RAGE is a critical player in pancreatic cancer progression. In addition to its role in tumorigenesis, RAGE has emerged as a factor influencing drug resistance in pancreatic cancer (138). Suppression of RAGE has been associated with a reversal of drug resistance in experimental models (139), suggesting that RAGE, beyond its involvement in tumor initiation and growth, contributes to developing resistance mechanisms that often limit the effectiveness of therapeutic interventions in pancreatic cancer.
RAGE alters the interaction between antiapoptotic pathways, such as the IL6-pSTAT3 pathway, and autophagocytosis in the context of PDAC (140). Research involving the crossbreeding of conditional KRASG12D/+ mice prone to developing pancreatic cancer lesions with RAGE−/− knockout mice revealed a reduction in pancreatic lesions and prolonged survival compared to those of KRASG12D/+ RAGE+/+ mice (141). Another study revealed a progressive increase in RAGE protein levels as pancreatic lesions advanced, suggesting that RAGE is involved in PDAC initiation and disease progression (142). Additionally, heightened expression of RAGE was identified specifically within cancerous lesions, with no such elevation observed in neighboring normal tissue (143).
Two noteworthy RAGE ligands, namely, S100P and high mobility group box 1 (HMGB1), have undergone extensive examination in the context of pancreatic cancer (144). S100P, operating through a RAGE-dependent mechanism, stimulates the proliferation and migration of human pancreatic cancer Panc-1 cells (145). Moreover, S100P has been shown to exhibit protective effects against the cytotoxicity of 5-fluorouracil in Panc-1 cells (146). Additionally, RAGE activation by HMGB1 was linked to enhanced tumor growth, promoting the persistence of cancer cells by upregulating autophagocytosis and inhibiting apoptosis (147).
A substantial proportion of pancreatic cancer-related deaths can be attributed to the pivotal role played by EMT in the rapid progression of metastatic disease (148). Throughout EMT progression, epithelial cells undergo a profound transformation characterized by the loss of epithelial markers such as E-cadherin, occludin, claudin, and laminin-1 while concurrently gaining mesenchymal markers such as N-cadherin, vimentin, and fibronectin (149). This phenotypic shift is a hallmark of EMT and is linked to cancer cell invasion and metastatic potential. Dynamic alterations in cellular identity are essential for the metastatic cascade because they allow cancer cells to detach from the primary tumor, infiltrate surrounding tissues, enter the bloodstream, and colonize distant organs, particularly the liver (150). There are three distinct types of EMT, and their occurrence is context-dependent. Type 3 EMT, which is observable in carcinoma cells, is relevant for invasion and metastasis during tumor development (151). The activation of EMT mechanisms in carcinoma cells underscores its pivotal role in promoting the aggressive and metastatic behavior observed in pancreatic cancer. Hyaluronic acid and collagen are examples of insoluble components (152). Soluble elements in the extracellular matrix, including Wnt, FGF, HGF, Notch, TGF-β family members, TNF-α, and HIF1-α, synergistically contribute to cancer progression by guiding the EMT process. These components create a dynamic microenvironment that helps epithelial cells transdifferentiate into mesenchymal phenotypes (153). Crucial signaling pathways regulating EMT involve activating transcription factors such as Zeb-1 and 2, Snail 1 and 2, and members of the bHLH family (E12, E-47, and Twist). These transcription factors play a central role in composing the molecular changes associated with EMT (154). Furthermore, repression of the E-cadherin encoder (CDH1 gene) has emerged as a shared feature among these transcription factors (155).
TGF-β is a crucial mediator of EMT in a variety of tumors. The conventional TGF-β signaling pathway involves the binding of TGF-β to a type II receptor, which enables the transactivation of type I receptor (TβR I) (156). The serine/threonine kinase TβR I phosphorylates SMAD2, resulting in the association of SMAD2 with SMAD4. After nuclear translocation, this complex regulates target gene transcription (157). The activation of the transcription factors Snail, Zeb-1, Slug, and Twist is pivotal for the TGF-β-mediated induction of EMT (158). In PDAC, TGF-β may engage a noncanonical pathway, including the PI3K, ERK/MAPK, p38, RhoA, JNK, and other signaling pathways (159). EMT responses in the Colo357 pancreatic cancer cell line were not affected by RNA interference-induced SMAD4 knockdown (160). However, in alternative pancreatic cancer cell lines, the induction of TGF-β-mediated EMT was efficiently suppressed by the MEK-1 inhibitor PD98059 (161).
When a Wnt ligand is not present, β-catenin sequestration is regulated through a degradation component comprising Axin, adenomatous polyposis coli, glycogen synthase kinase-3 (GSK-3), and CK-1 (162). This process begins with CK-1 phosphorylating β-catenin at Ser45 (163). GSK-3 activates β-catenin by phosphorylating it at Thr41, Ser33, and Ser37. This phosphorylation event triggers ubiquitination, and subsequently, β-Trcp facilitates the proteasomal degradation of β-catenin (164). The systematic elimination of β-catenin prevents its nuclear buildup, impeding interaction with DNA-bound TCF/LEF complexes and histone deacetylase (HDAC) activity, ultimately suppressing Wnt target genes (165). Wnt ligands bind to the Frizzled and LRP5/6 receptors, causing a complex to develop, phosphorylating LRP5/6, stabilizing Axin, and facilitating GSK-3 complex disassembly. This process inactivates cytosolic β-catenin, allowing it to form a complex with TCF/LEF in the nucleus, thereby regulating genes crucial for cell growth and proliferation (166).
In addition to its role in β-catenin regulation, GSK-3β also promotes the phosphorylation and proteasomal degradation of Snail (167). Conversely, Wnt suppresses GSK-3β activity, causing increased Snail protein levels (168). K-Ras-induced activation of the Wnt/β-catenin pathway upregulates EMT stimulators in cancer cells (169). By decreasing the expression of Slug and Twist, reinstatement of Wnt inhibitory factor 1 causes a reduction in the levels of mesenchymal markers and an increase in epithelial indicators. Inhibition of β-catenin through the use of small hairpin RNA results in increased expression of E-cadherin, coupled with a decrease in the levels of mesenchymal markers such as vimentin, N-cadherin, and MMP-2 (170).
The Notch signaling pathway, which is integral to tissue development and apoptosis, encompasses four Notch receptors and five Notch ligands (Delta-like 1, 3, 4, Jagged-1, and 2) (171). Activation ensues upon the binding of the Notch protein to a neighboring cell’s receptor, initiating proteolytic cleavage facilitated by metalloproteases, TNF-α converting enzymes, and γ-secretase (66). The resulting active Notch intracellular domain fragment (NICD) translocates to the nucleus, where it forms a CSL-NICD complex with the transcription factor CSL (CBF1, a suppressor of Hairless, and Lag-1) (172). Functioning as a coactivator, this complex recruits additional coactivators, including p300, activating Notch target genes pivotal in governing cellular processes such as growth, proliferation, angiogenesis, and programmed cell death (173). Noteworthy target genes implicated in solid and hematological cancers include Cyclin D1, COX-2, Akt, MMP9, ERK, VEGF, c-Myc, mTOR, NF-κB, p53, p27, and p21 (174, 175). The Notch signaling pathway directly induces EMT by activating Slug and Snail-1 (176). The depletion of Notch-2 or midkine suppresses EMT in pancreatic cancer cells through Notch-2-mediated mechanisms (177).
The initiation of EMT in pancreatic cancer involves relationships among distinct molecular entities, each contributing unique functions to the dynamic process (Figure 5). Major surface protease (MSP) collaborates with IGF1 to induce cellular growth and survival, while tumor growth factor β (TGFβ) orchestrates alterations in cell morphology and promotes invasiveness (178). This process is complemented by bone morphogenetic proteins (BMPs), which influence cell differentiation and apoptosis, thus impacting the plasticity of cancer cells undergoing EMT (179). Recepteur d’Origine Nantais (RON) plays a pivotal role in influencing cell motility and invasion by interacting with neuropilin 1 (NRP1), which, in turn, contributes to angiogenesis and neural guidance within the TME (180). Retinoic acid-induced 1 (Ra1) influences cell cycle progression in concert with extracellular signal-regulated kinase (Erk), a key player in signaling cascades that transduce external signals to the nucleus, thereby affecting the cellular changes observed during EMT (181). Histone deacetylases 1 and 2 (HDAC1/2) modulate gene expression through epigenetic regulation, while Msh homeobox 2 (MSX2) influences cell differentiation and migration (182). S100 calcium binding protein A4 (S100A4) impacts cytoskeletal dynamics and motility and is crucial for the migratory aspects of EMT (183). ZO-1 contributes to cell adhesion and polarity through its role in tight junctions (184).
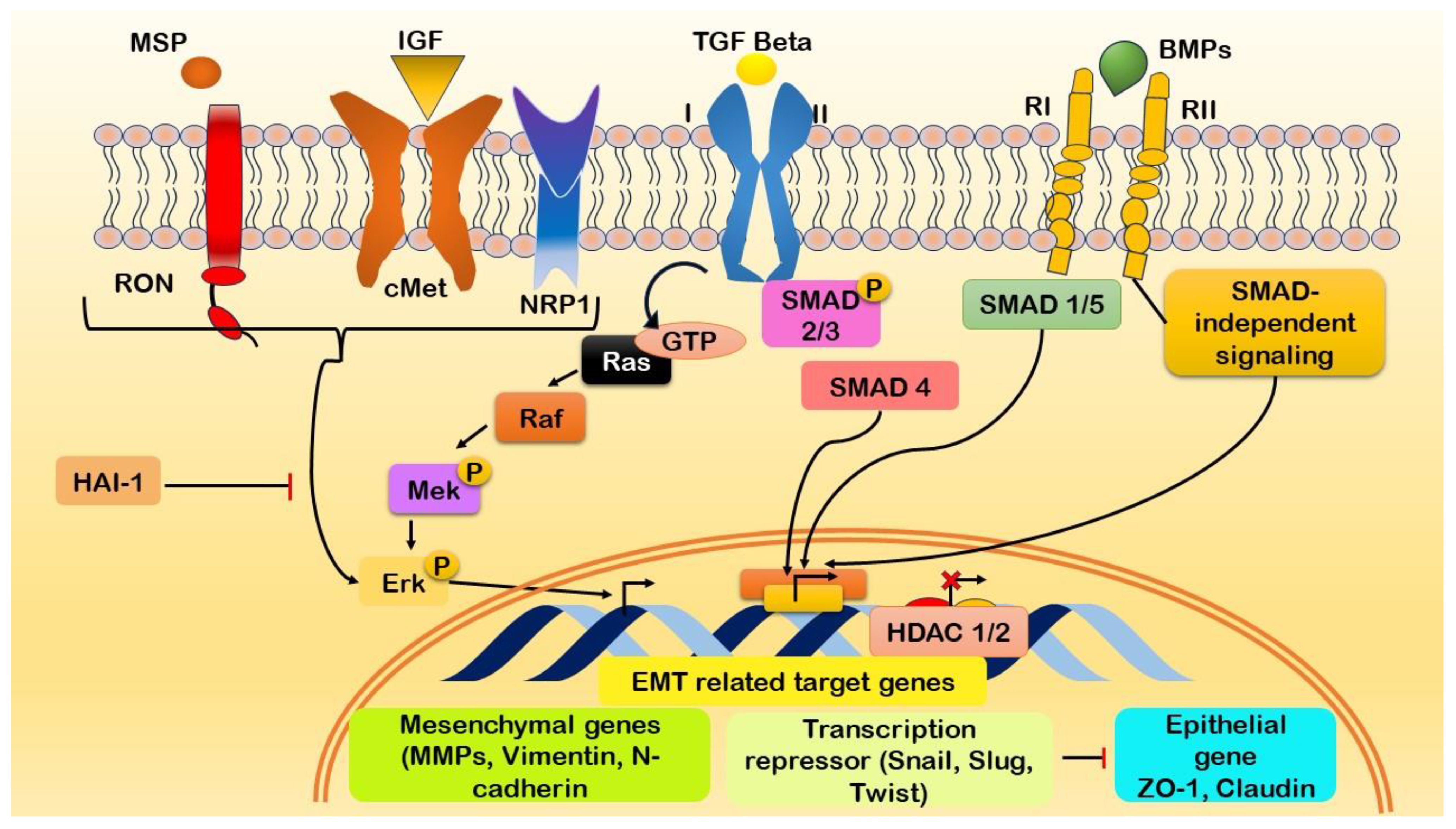
Figure 5 Illustration of the initiation of epithelial-mesenchymal transition (EMT) in pancreatic cancer through the activation of growth factor signaling cascades and the modulation of EMT-associated genes. Interaction between growth factors and their respective receptors initiates the expression of genes associated with EMT. Key molecular players involved in this process include Macrophage-Stimulating Protein (MSP), Insulin-like Growth Factor 1 (IGF1), Transforming Growth Factor-beta (TGFβ), Bone Morphogenetic Proteins (BMPs), Recepteur d’Origine Nantais (RON), Neuropilin-1 (NRP1), Ras-like GTPases (Ra1), Extracellular signal-Regulated Kinases (Erk), Histone Deacetylases 1/2 (HDAC1/2), Msh Homeobox 2 (MSX2), S100 Calcium-Binding Protein A4 (S100A4), and Zonula Occludens-1 (ZO-1). The complex interaction among these molecular components promotes the induction of EMT in pancreatic cancer.
Pancreatic cancer, a formidable challenge in oncology, demands a comprehensive understanding of the underlying molecular pathways. The Hippo signaling network is a conserved system that governs cellular proliferation, organ growth, and regenerative processes (Table 1). At its core are the serine/threonine kinases MST1, MST2, LATS1, and LATS2 (200). MST1 and MST2 phosphorylate and activate the LATS1 and LATS2 kinases in collaboration with SAV1 (201). Subsequently, MOB1 binds to LATS1 and LATS2, leading to the phosphorylation of the Hippo transducers YAP and TAZ (202). This phosphorylation impedes the accumulation of YAP and TAZ in the nucleus and their interaction with TEAD transcription factors (201). When the regulatory module is inactive or when independent stimuli activate YAP/TAZ, these molecules translocate to the nucleus, where they engage with transcription factors (203). This interaction initiates the transcription of target genes, including CTGF, CYR61, ANKRD1, BIRC5, and AXL (204). Mechanical stimuli in the cellular environment (mechano-transduction), soluble substances, and metabolic pathways collectively impact the Hippo signaling pathway. Additionally, the system extensively communicates with other signaling pathways, such as the TGF-beta, Wnt, Sonic Hedgehog, and Notch pathways (205).
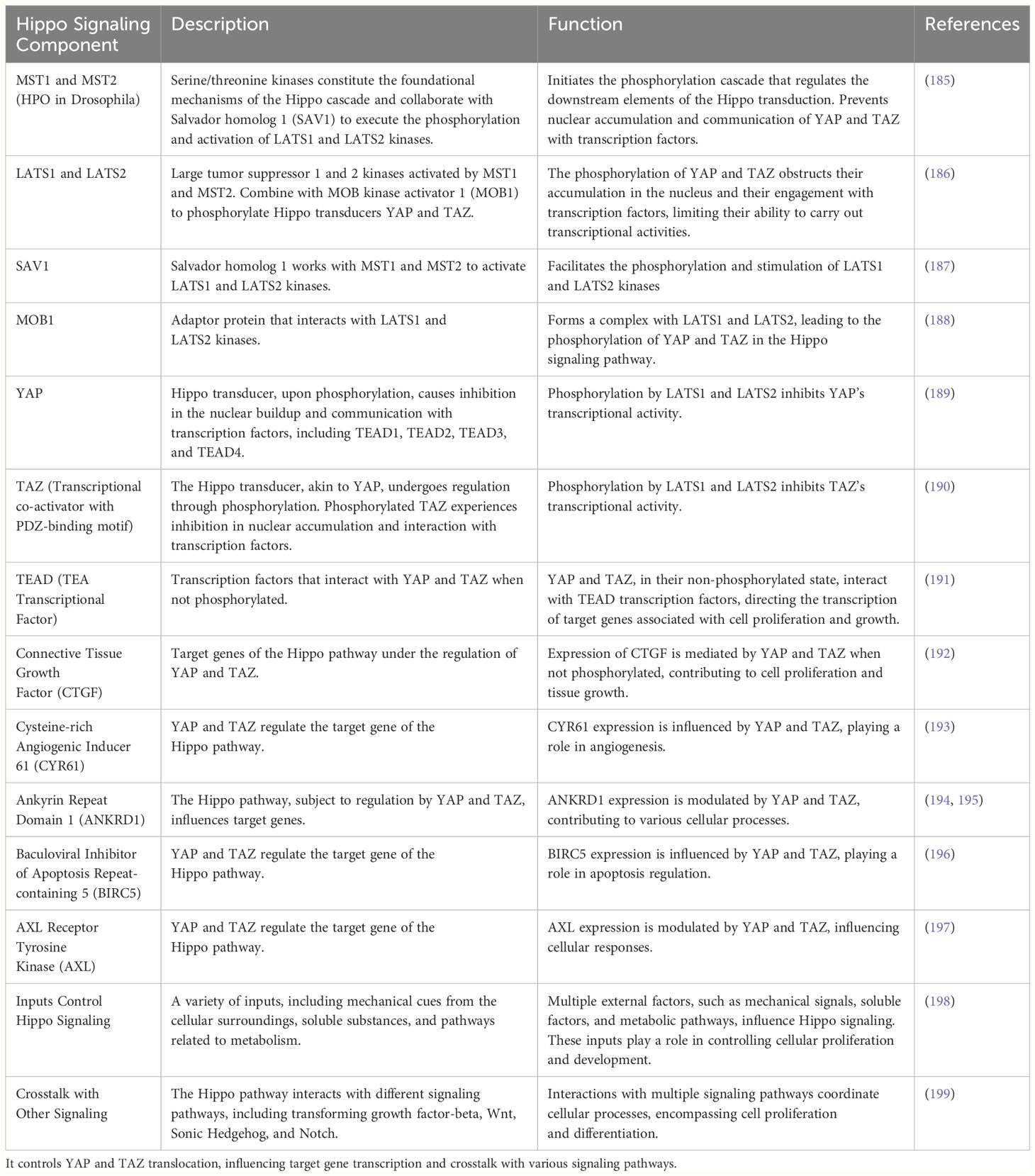
Table 1 The Hippo signaling pathway regulates cell processes involving MST1, MST2, LATS1, and LATS2 kinases.
Snail-1 and Snail-2 are transcription factors that play pivotal roles in regulating the initiation of EMT, a crucial process implicated in the progression and metastasis of pancreatic cancer. These transcription factors are characterized by their conserved C2H2-type zinc finger motifs and the essential Snail1/GFI domain at the amino terminus, which is critical for maintaining the transcriptional suppression of target genes and protein stability (206, 207). In PDAC, Snail and its closely related family member Slug have emerged as key mediators of EMT. Furthermore, Slug is present in 50% of PDAC patients, while Snail expression is detected in a striking 68% of cases (208). Elevated Snail expression levels in pancreatic cancer have been associated with lymph node invasion and distant metastasis, underscoring its role in promoting invasive and metastatic behavior. When pancreatic cancer cell lines are transfected with Snail, they exhibit increased invasive and metastatic potential in orthotopic pancreatic cancer models, manifesting EMT characteristics during the invasive phase of tumor progression (209).
Importantly, the inhibition of Snail amplifies the response to the chemotherapeutic agent gemcitabine and contributes to extended overall survival in a murine model engineered for PDAC (210). This finding highlights the potential therapeutic benefits of targeting Snail in pancreatic cancer treatment. The mechanisms by which Snail exerts its pro-metastatic effects in pancreatic cancer involve suppressing genes crucial for maintaining the epithelial phenotype, such as occludin, E-cadherin, claudin, and cytokeratin-18, while simultaneously promoting the expression of mesenchymal genes like N-cadherin, vimentin, and fibronectin (211). Moreover, Snail governs the expression of genes linked to apoptosis (P53, BID, and DFF40) and cell polarity (Crumbs3, Lgl2, and dlg3), with a particular emphasis on downregulating the key epithelial marker E-cadherin (212).
Numerous studies have examined the Zeb family of transcription factors, demonstrating their important function as strong EMT inducers (213). Interestingly, there is a positive correlation between elevated Zeb-1 expression in the tumor-associated stroma and pancreatic cancer cells and a poor prognosis for individuals with PDAC. Examination of human tissue specimens and pancreatic cancer cell lines revealed a connection between Zeb-1 and the expression of E-cadherin (214). Inhibition of Zeb-1 has been associated with notable decreases in cell migration, tumorigenesis, and dissemination (215). Research indicates that decreased expression of essential components related to epithelial development, cellular adhesion, and cellular polarity is a recognized consequence of heightened Zeb-1 expression (216). Specifically, Zeb-1 selectively engages either HDAC-1/2 or the switch/sucrose nonfermentable chromatin remodeling protein BRG1 at the promoter region of the CDH-1 gene, resulting in a reduction in E-cadherin synthesis (217). Consequently, inhibiting Zeb-1 has emerged as a potentially impactful treatment strategy for individuals with PDAC.
bHLH proteins, including E12, E47, Twist 1, and Twist 2, which are essential EMT players (218), have been investigated. EMT is actively promoted by E47 and E12, which suppress the production of E-cadherin (219). Twists 1 and 2, which have been identified as the primary regulators of EMT during pathogenesis, play important roles (220). Patients with PDAC typically have very weak or no Twist expression in their samples (221). Comparably, whereas Twist expression is enhanced under hypoxic conditions, pancreatic cancer cell lines such as PANC-1, MiaPaCa-2, Capan-1, AsPC-1, and HPAF-2 cells exhibit low Twist expression, suggesting a possible role for Twist in the invasive nature of pancreatic tumors (222). Twist has been linked to decreased E-cadherin expression and increased N-cadherin expression (223). Twist engages with various elements of the Mi2/nucleosome remodeling and deacetylase complex, contributing to the inhibition of E-cadherin transcription (224).
TME is characterized by distinct physical and biochemical properties that promote interactions between stromal and malignant cells to drive metastasis, carcinogenesis, disease progression, and resistance to treatment (225). In addition to the resistance linked to desmoplasia, pancreatic cancer is characterized by a very immunosuppressive environment with several components and processes that obstruct efficient immune responses directed against malignancy (226). Due to the many immunological regulatory cells that enter the pancreatic cancer stroma, the principal processes of the TME are challenging to understand. Important TME constituents include soluble factors, immune cells, acellular stroma, and pancreatic stellate cells (227). Desmoplasia, a condition in which hyperactive cancer-associated fibroblasts deposit abnormal ECM, primarily fibrillar type I collagen, is a characteristic of PDAC (228). Disruption of cell-ECM homeostasis and stromal remodeling are linked to treatment resistance and metastasis during cancer progression (229). A thorough mechanistic understanding of PDAC pathophysiology requires additional sophisticated in vitro and in vivo models owing to the critical interactions between the tumor and the stromal extracellular matrix. The roles of the TME, constituents, and consequences in PDAC are listed in Table 2.
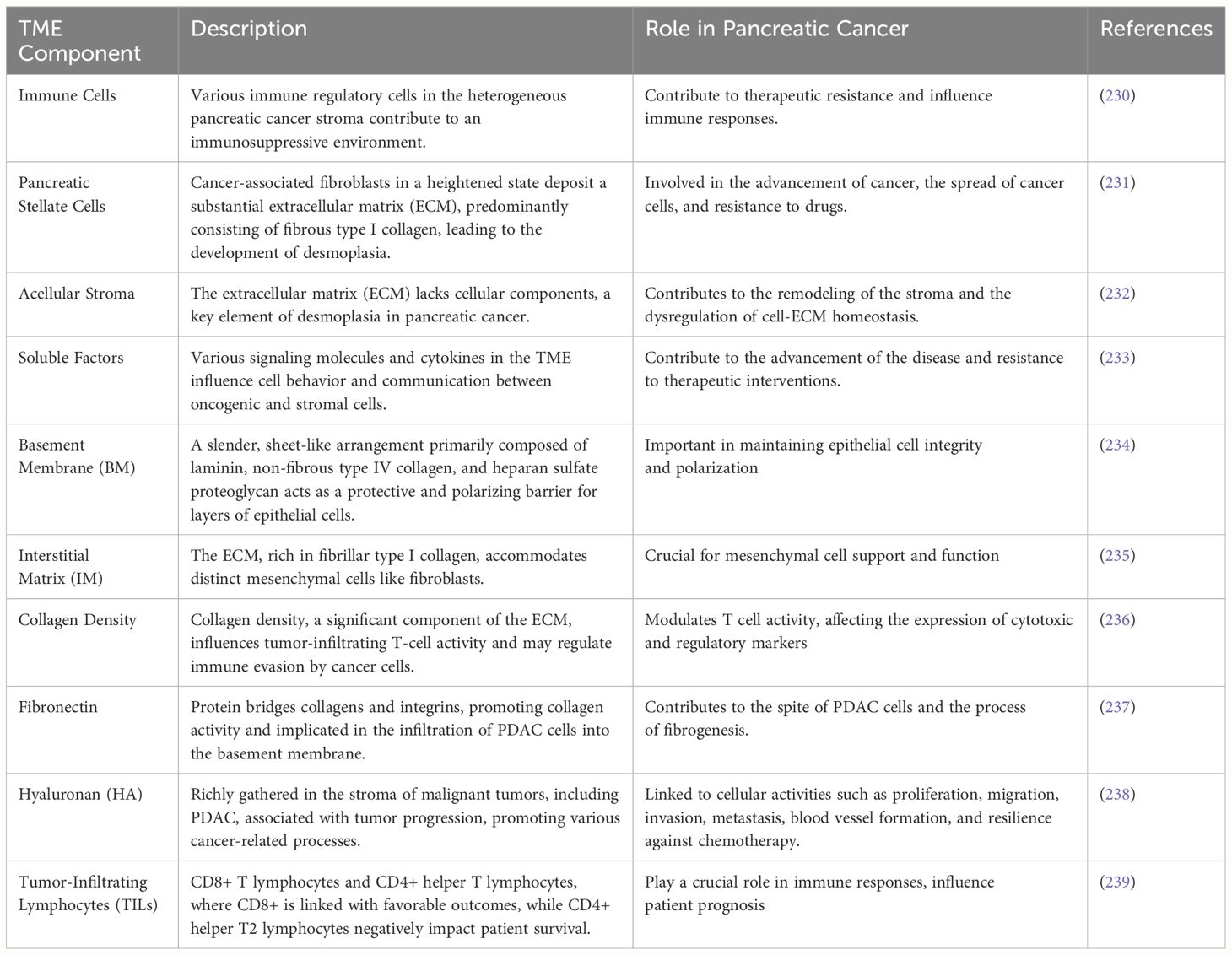
Table 2 Tumor microenvironment characterization, constituents, and consequences in pancreatic ductal adenocarcinoma.
The interstitial matrix (IM) and basement membrane (BM) make up the ECM found in both PDAC and normal tissues (240). BM is a thin, sheet-like structure that provides polarization and protection to epithelial cell layers. It primarily comprises laminin, nonfibrillar type IV collagen, and heparan sulfate proteoglycans (241). In contrast, specific mesenchymal cells, such as fibroblasts, inhabit the IM and are primarily composed of fibrillar type I collagen (242). According to one study, collagen density may play a role in cancer cells’ ability to evade the immune system by acting as a unique anticancer T-cell function controller in three-dimensional T-cell culture (243). The expression of cytotoxic and regulatory markers is influenced by collagen density, which also affects the activity of T lymphocytes that infiltrate tumors (243).
Like collagens, fibronectin has distinct impacts on the biology of prostate cancer and serves as a connecting protein between integrins and collagens, promoting the function of collagens (244). Fibronectin promotes the malignancy and fibrogenesis of PDAC cells, as evidenced by its involvement in pancreatic stellate cell ECM creation and PDAC cell penetration into the basement membrane (245).
In the stroma of malignant tumors, including PDAC, hyaluronan (HA), a significant ECM component, accumulates abundantly. This accumulation is associated with the advancement of tumors, as it stimulates cellular proliferation, movement, infiltration, metastasis, angiogenesis, and resilience to chemotherapy (246). According to research, HA and its receptors are overexpressed in PDAC, and abnormal HA buildup is associated with a poor prognosis (247). Therefore, targeting HA may have therapeutic benefits in the treatment of PDAC. The TME is maintained by ongoing interactions among cells and between cells and the extracellular matrix, and the initiation of interactions between epithelial cells, pancreatic cancer cells, and stromal cells in the TME is critical for drug resistance and the progression of connective tissue in primary and metastatic locations (248).
TME components can promote EMT and angiogenesis, which contribute to the capacity of pancreatic cancer to spread. Furthermore, the TME complicates immunotherapeutic treatments (249). Tumor-infiltrating lymphocytes (TILs), including CD8+ T cells and CD4+ helper T1 lymphocytes, are related to positive outcomes, whereas CD4+ helper T2 lymphocytes are linked to unfavorable patient survival (250). Immune and inflammatory cells play essential roles in the TME of pancreatic cancer, contributing to chemotherapy resilience and serving as early contributors to carcinogenesis and metastasis.
Pancreatic cancer poses a significant challenge in oncology because of its aggressive behavior and restricted treatment modalities. This study examined an integrative strategy that combines conventional medical interventions with complementary and alternative therapies to improve the comprehensive well-being of individuals with pancreatic cancer.
Noncoding RNAs (ncRNAs) are a different family of molecules that play critical regulatory roles in several life processes, including pathological illnesses such as cancer, cardiovascular disease, and neurodegenerative disorders (251). MicroRNAs (miRNAs) and synthetic antagomirs, which have an approximate length of 22 nucleotides, are crucial in the delicate arrangement of cellular processes and significantly influence cellular proliferation, apoptosis, and autophagy (252). Among them is miR-203, which has received attention for its suspected anticancer effects via precise gene expression control (253). Furthermore, the discovery of circulating miRNAs with possible biomarker value offers promise for noninvasive surveillance of the dynamic evolution and severity of pancreatic cancer (254). MiRNAs such as miR17-92 (255) and miR-21 limit cellular growth (256), while miR-126 acts as an antioncogene (257). Additional complexities emerge with miR-15b and miR-155, which are involved in mutation accumulation (258), and with miR-10b and miR-29, which are critical for triggering metastatic pathways (259). The complex interaction includes miRNAs such as let-7d, miR-23b, miR-126, and miR-200c, which promote inflammatory responses (260, 261), and miR-21 and miR17-92, which decrease immune cell clearance (262). Let-7, miR-16, miR-21, and miR-221/222 all play roles in the maintenance of replicative immortality, demonstrating the extensive regulatory networks mediated by these small RNA species (263–266).
MiR-203 has emerged as a crucial regulator in pancreatic cancer, limiting cell invasion and migration through the targeted control of caveolin-1 (267). Its downregulation in pancreatic cancers emphasizes its importance in disease genesis. Other miRNAs, such as miR-21, miR-155, miR-221, miR-222, miR-376a, and miR-301, contribute significantly to tumorigenic qualities by altering the expression of DJ-1 and affecting the PTEN-PI3K/AKT pathway (268). In addition to this complication, miR-203 has dual functions in pancreatic cancer, limiting cell proliferation while simultaneously promoting apoptosis via precise changes in the expression of suppressor of cytokine signaling 3 (SOCS3) (269). However, the specific molecular processes and crucial functions of miR-203 in pancreatic cancer remain unknown.
Pancreatic cancer therapy presents a daunting challenge, as a multimodal strategy that considers the disease stage, the patient’s general health, and the development of research is needed. Surgical intervention is often the first option for resectable tumors, with the possibility of a cure if the cancer is restricted to the pancreas. Adjuvant chemotherapy becomes critical after surgery, with regular use of medicines such as gemcitabine, fluorouracil (5-FU), capecitabine, oxaliplatin, and irinotecan (270). Oxaliplatin, a platinum-based drug, induces cross-linking in DNA, affecting the nucleotide excision repair (NER) cascade and activating the DNA damage response (DDR) pathway (271). Erlotinib, an oral EGFR inhibitor, disrupts essential signaling pathways and is particularly effective in treating tumors with EGFR abnormalities (272).
Gemcitabine targets the deoxycytidine pathway and affects the nucleotide pool, mainly affecting the cell cycle and DNA synthesis. The DNA synthesis and repair route is the primary signaling mechanism affected by gemcitabine (273). Gemcitabine is a nucleoside analog that has structural similarities with DNA. During replication, gemcitabine enters the cell, becomes phosphorylated, and joins the growing DNA chain. This insertion stops the DNA chain from elongating and stops further synthesis from occurring. Gemcitabine thus causes cell cycle arrest in the S phase, the stage at which DNA synthesis occurs (274).
A series of events, such as activating cell cycle checkpoints and DNA damage response pathways, are initiated when DNA replication stalls. Additionally, gemcitabine prevents the manufacture of deoxyribonucleotides, which are necessary building blocks for DNA replication, by inhibiting ribonucleotide reductase (275). Gemcitabine further inhibits DNA synthesis by reducing the intracellular pool of deoxyribonucleotides (276). By targeting the dynamics of microtubules inside cells, nab-paclitaxel affects signaling pathways linked with microtubules (277). The ability of paclitaxel, the active ingredient of nab-paclitaxel, to stabilize microtubules is its primary mode of action. Dynamic structural elements of cytoskeleton microtubules are essential for many cellular functions, including mitosis (278).
In particular, nab-paclitaxel disrupts the normal dynamics and function of microtubules by interfering with their disintegration during mitosis. This perturbation stops the cell cycle in the G2/M phase, triggering apoptosis or programmed cell death (279). Nab-paclitaxel disrupts the mitotic spindle machinery necessary for appropriate chromosomal segregation during cell division by targeting microtubules and altering their regular movements (280). Although nab-paclitaxel primarily affects microtubule stability and the accompanying effects on cell cycle progression, it also indirectly affects several signaling pathways linked to cell survival and division (281). Targeting the thymidylate synthase enzymes 5-fluorouracil (5-FU) and capecitabine—essential drugs for treating different types of cancer—has a similar mechanism of action that involves interfering with DNA synthesis (282). When administered intravenously, 5-FU acts as an antimetabolite, inhibiting DNA replication and repair by impeding the transformation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) (283).
The main protein target of 5-FU is thymidylate synthase. When this enzyme is inhibited, many biological reactions are triggered, including the activation of cell cycle checkpoints, DNA damage response pathways, and death (284). In contrast, capecitabine is an oral prodrug that enters tumor cells and proceeds via enzymatic conversions to produce 5-FU (285). Like 5-FU, which is delivered directly, 5-FU inhibits thymidylate synthase once it is converted to exert its antimetabolite effects (286). The protein target for thymidylate synthase remains constant, causing errors in DNA synthesis and other cellular reactions that result in cell cycle arrest and death (287). Oxaliplatin is a platinum-based chemotherapeutic drug that mainly targets guanine nucleotides in genomic DNA by forming covalent DNA adducts (288). DNA strands become cross-linked due to this contact, making it more difficult to separate during vital biological functions such as transcription and replication. The resulting structural damage causes apoptosis and cell cycle arrest, which enhances the therapeutic effectiveness of oxaliplatin. Although oxaliplatin has a primary effect on DNA, it also indirectly affects cellular proteins involved in DNA repair, namely, those involved in the NER pathway (288).
Proteins in this field include XPC (Xeroderma pigmentosum complementation group C), XPA (Xeroderma pigmentosum complementation group A), and ERCC1 (excision repair cross-complementation group 1) (289). Within the signaling pathway domain, the DDR pathway is activated by oxaliplatin-induced DNA damage. Essential proteins in this pathway, such as the ATM-encoded ataxia-telangiectasia mutated (ATM) protein and the ATR-encoded ataxia-telangiectasia and Rad3-related (ATR) protein, are critical for detecting DNA damage and coordinating biological reactions (290). These defense mechanisms include inducing cell cycle arrest, facilitating DNA repair, and encouraging apoptotic cell death if the damage is not repaired.
As an oral EGFR inhibitor, erlotinib plays a crucial role in cancer therapy by interfering with vital signaling pathways essential for cell survival and proliferation (291). Hepatic metabolism, which is primarily controlled by the cytochrome P450 enzyme system, is involved in its administration, and enzymes such as CYP3A4 and CYP3A5 play important roles (292). This metabolic pathway involves medication interactions and possible differences in drug response caused by hereditary variables. EGFR, encoded by the EGFR gene (ERBB1), is a particular protein target of erlotinib. Erlotinib inhibits EGFR, which interferes with downstream signaling cascades such as the RAS-RAF-MEK-ERK and PI3K-AKT pathways, which are required for cellular processes (293). The effectiveness of this drug is especially noteworthy in tumors with EGFR overexpression or mutations, which contribute to uncontrolled cell proliferation. Understanding erlotinib’s pharmacokinetics, molecular targets, and genetic factors is critical for customizing its usage in treating pancreatic cancer and other malignancies and enhancing therapeutic results.
Pancreatic cancer has always been a complex disease to treat. However, immune checkpoint inhibitors have shown promise in this regard. Among these inhibitors, CTLA-4 and PD-1/PD-L1 inhibitors have attracted much interest (294). T-cell-expressed PD-1 combines with cancer cell-expressed PD-L1 to suppress the immune system (295). Monoclonal antibodies, such as nivolumab and pembrolizumab, obstruct this connection, enabling T cells to attack cancer cells efficiently (296). Similarly, CTLA-4 suppresses T-cell activation by binding with CD28 for binding affinity to antigen-presenting lymphocytes (APCs) (297). An antitumor immune response is promoted, and T-cell activation is enhanced when the CTLA-4 inhibitor ipilimumab interferes with this competition (298).
Combinations of CTLA-4 and PD-1/PD-L1 inhibitors have been studied for potential synergistic effects (299). Although these treatments have potential, they may cause immune-related side effects that require close patient observation. Biomarkers, one of which is PD-L1 expression, help patients choose and predict how well a therapy will work (300). Peptide vaccines, such as GV1001, provide a focused immunotherapeutic strategy for treating pancreatic cancer (301). These vaccines work by identifying tumor-associated antigens (TAAs), including the telomerase-derived peptide GV1001, which targets specific proteins in cancer cells (302). GV1001 has been shown in pancreatic cancer clinical trials to activate cytotoxic T cells, promoting an immune response against cancer cells that display targeted antigens (303). APCs process and deliver GV1001 to T lymphocytes, activating them to recognize and attack cancer cells. This is how the mechanism of antigen presentation works (304). GV1001 also aims to create immunological memory, guaranteeing a focused and long-lasting reaction (305). The possibility of a patient-specific design that enables modification based on unique tumor characteristics is noteworthy in terms of therapeutic concerns.
Using complete cancer cells expressing various antigens, whole-cell vaccines, such as algenpantucel-L, constitute a novel immunotherapy strategy. Algenpantucel-L is composed of irradiated pancreatic cancer cells and stimulates the immune system in a complicated way, affecting T cells, B cells, and APCs. APCs process and present a variety of antigens produced by algenpantucel-L, thereby initiating a thorough immune response. This is the mechanism of stimulation (306). Interestingly, the therapeutic considerations for whole-cell vaccines highlight their objective of concurrently targeting several antigens to elicit a more comprehensive immune response. A complex signaling cascade is used in CAR-T-cell therapy to strengthen the immune response against pancreatic cancer. T cells that have been transformed express the chimeric antigen receptor (CAR) on their surface after being given the CAR. Typically, this synthetic receptor comprises an intracellular signaling domain, a transmembrane domain, and an extracellular domain for antigen recognition (307). Costimulatory domains such as CD28 or 4-1BB (CD137) and components such as CD3ζ are often found in the intracellular signaling domain (308).
The extracellular domain of the CAR binds exclusively to the antigen on the surface of pancreatic cancer cells that express the targeted antigen, such as mesothelin (309). This binding initiates the CAR-T-cell signaling cascade. The transcription of genes linked to T-cell activation and proliferation is ultimately caused by the activation of downstream pathways by intracellular signaling domains, such as the PI3K-Akt and MAPK pathways (310). The activation of γ-chain-associated protein kinase 70 (ZAP-70) and the phosphorylation of CD3ζ are important signaling events that initiate downstream signaling cascades (311). Additional signals from costimulatory domains such as CD28 or 4-1BB improve T-cell activation, proliferation, and survival (312). When these signaling events occur, CAR-T cells produce cytotoxic chemicals, including granzymes and perforin (313). To specifically destroy pancreatic cancer cells, perforin breaks down the membrane of cancer cells, enabling granzymes to enter and cause apoptosis (314). Treatment with cytokines, including drugs such as interleukin-2 (IL-2) and interferon-alpha, is critical for treating pancreatic cancer (315).
The crucial cytokine IL-2 increases T-cell proliferation by activating the JAK-STAT signaling cascade through binding to the IL-2 receptor, which contains the IL-2Rα chain (CD25), IL-2Rβ chain (CD122), and IL-2Rγ chain (CD132) (316). This cascade improves both cell growth and effector functions. Furthermore, NK cells are activated by IL-2, which enhances their antitumor function (317). The complex signaling pathways that mediate the biological effects of IL-2 include those involving the JAK1, JAK3, and STAT proteins (318). Type I interferons, such as interferon-alpha, have anti-proliferative and immunomodulatory effects (319). When interferon-alpha binds to its receptors, such as IFNAR1 and IFNAR2, it triggers the JAK-STAT pathway, which involves STAT, JAK1, and JAK2 (320). This signaling cascade eventually strengthens the immune system’s defense against cancerous cells by controlling gene expression.
Moreover, interferon-alpha acts on several angiogenic factors to suppress angiogenesis (321). Owing to their complex mechanism of action, oncolytic viruses are a potential approach for pancreatic cancer immunotherapy. These viruses are genetically altered to increase and infect cancer cells specifically. They take advantage of the unique biology of cancer cells by focusing on hyperactive signaling pathways and weakened antiviral defenses (322).
Furthermore, viruses such as vaccinia, herpes simplex, and adenoviruses, which are often used in oncolytic virotherapy, may be modified to improve immunogenicity, tumor selectivity, and safety (323). For instance, the adenovirus E1A gene may be altered to enhance tumor selectivity. This gene encodes a protein that interacts with cellular regulators (324). Cancer cells lyse due to the infection process, releasing viral particles that spread the infection to nearby cancer cells. The release of tumor antigens during the lysis process is another benefit of this selective replication. APCs use these tumor antigens, which include proteins such as HER2/neu or carcinoembryonic antigen (CEA), to trigger an immune response (325). These cells stimulate T lymphocytes by processing and presenting antigens, mainly via the JAK-STAT signaling pathway (326).
Herpes simplex viruses may be genetically modified to contain transgenes that improve antitumor immunity, such as GM-CSF, which encodes granulocyte-macrophage colony-stimulating factor (327). Combining treatments with other modalities, such as checkpoint inhibitors such as PD-1 and PD-L1 inhibitors, is important from a clinical standpoint (328). This combination further supports long-term antitumor immunity by boosting the adaptive immune response involving CD8+ cytotoxic T cells (329). The tight ability of oncolytic viruses to limit reproduction in cancer cells is a safety concern and helps to reduce the possibility of nonspecific effects (330).
Different medications and treatments for pancreatic cancer have been tested in clinical trials to evaluate their safety, effectiveness, and possible advantages for patients. The medications listed in Supplementary Table S1 are quickly listed for the provided pancreatic cancer clinical trial information.
This review seeks to provide a comprehensive analysis of PDAC, highlighting the critical molecular pathways involved, such as KRAS, Notch, and Hedgehog, and their implications for disease progression and therapy resistance. Current therapeutic strategies, including surgery, chemotherapy, and radiation, were critically examined, along with emerging treatments like immunotherapy. Despite advancements, significant challenges remain, particularly in overcoming drug resistance and the tumor’s dense stromal environment. The review also explored innovative diagnostic techniques, such as liquid biopsies, which offer a noninvasive approach for early detection, and personalized medicine, which tailors’ treatment to the patient’s genetic profile. The potential of CRISPR/Cas9 for precise genomic editing and computational intelligence for enhancing diagnostic and therapeutic efficacy was highlighted, showing promise for future advancements. The findings highlight the necessity of a multidisciplinary approach to address the complexities of pancreatic adenocarcinoma. By integrating insights from genetic, molecular, and clinical research, the review identifies key challenges and proposes future research directions. These include improving early detection methods, developing more effective therapeutic strategies, and overcoming the tumor’s immunosuppressive microenvironment.
The etiology of pancreatic cancer remains insufficiently understood, necessitating further extensive prospective studies to enhance our comprehension of the associated risk factors. Patients exhibiting a predisposition to familial PDAC could be promising candidates for screening (331). However, consensus is lacking on the optimal age, frequency, and preferred imaging techniques for screening. Conducting thorough retrospective and prospective studies that longitudinally track individuals with familial pancreatic cancer is crucial for untying disease progression and facilitating the implementation of effective screening and treatment strategies. Recognized precursors such as PanIN, IPMN, and MCN offer opportunities for early identification and intervention (332). Implementing appropriate follow-up programs based on extensive retrospective and prospective studies can ensure prompt intervention for susceptible patients and deter superfluous surgical procedures for benign lesions. These studies, conducted over extended periods, will enhance our understanding of disease processes and pinpoint determinants of the risk of these precancerous conditions, opening avenues for targeted screening in specific populations.
The advent of neoadjuvant therapy has improved survival in a few patients, yet challenges persist in identifying those who would benefit most from this approach (333). Ongoing randomized studies are needed to identify the optimal candidates for neoadjuvant therapy. The search for novel biomarkers holds promise for refining decision-making processes in an era of precision medicine, tailoring therapies to specific cases. Surgical excision, which involves vascular resection, is the cornerstone of curative intervention and offers potential benefits in achieving clear margins. However, the survival advantage associated with venous resection warrants further investigation through retrospective studies, shedding light on patient outcomes and contributing valuable insights for future guidelines (334).
Despite progress in neoadjuvant and multimodal therapies, postoperative relapses persist as a formidable challenge, necessitating innovative interventions (335). The complex interaction among neoplastic and stromal components within tumor surroundings adds complexity to the disease. Although surgery remains the primary remedial modality for initial-stage patients, patients with advanced disease require a comprehensive approach involving chemotherapeutic regimens, radiation therapy, and targeted interventions. Promising strategies for immunotherapy are hampered by immune evasion mechanisms (336).
The 5-year overall survival rate of individuals with pancreatic cancer who received FDA-approved chemotherapy and targeted therapies has increased from approximately 2% ten years ago to 11% by 2022 (337). Nevertheless, a deeper understanding of the biological intricacies inherent in PDAC subtypes has facilitated the way for more refined and targeted therapeutic strategies (338). New methodologies in clinical trial design, encompassing drug lead-in, neoadjuvant exploration of investigational agents, and the implementation of platform studies for accelerated evaluation of combinations, are driving progress. Over the subsequent decade, one might expect apparent advancements in clinical outcomes for a more extensive cohort of patients undergoing treatment with tailored combinations of therapeutic agents (339).
The novelty of this review lies in its comprehensive and integrative approach to understanding PDAC, particularly by highlighting emerging areas of research and potential therapeutic strategies that have not been extensively covered in previous literature. The review provides an updated and detailed exploration of critical molecular pathways such as KRAS, Notch, and Hedgehog, emphasizing recent discoveries and their implications for disease progression and therapy resistance. Additionally, the discussion on innovative diagnostic techniques, such as liquid biopsies, represents a significant advancement over conventional biopsy methods. Liquid biopsies offer a noninvasive means for early detection and monitoring of pancreatic cancer, providing real-time insights into tumor dynamics. The review also highlights the promise of personalized medicine, tailored to individual genetic profiles, which can optimize treatment outcomes. Furthermore, it examines the application of CRISPR/Cas9 for precise genomic editing, showcasing a cutting-edge approach to potentially correct oncogenic mutations at their source. Moreover, while previous works have discussed immunotherapy, this review provides analysis of the current challenges, particularly immune evasion mechanisms, and suggests potential strategies to overcome these hurdles. By integrating these novel insights and emerging research areas, the review not only builds upon existing knowledge but also facilitate the way for future research directions that hold promise for significantly improving the diagnosis, treatment, and overall management of PDAC.
PDAC represents a profound clinical challenge in the field of oncology, characterized by an aggressive disease course, high mortality rates, and limited therapeutic options. The prognosis for individuals with PDAC is still poor despite advancements in cancer research and treatment approaches, highlighting the dire need for a thorough comprehension of this form of cancer. A complex relationship between hereditary and environmental variables influences the development of pancreatic cancer. Elucidating the molecular pathways and signaling cascades involved in PDAC is crucial for developing novel therapeutic interventions. It provides a holistic understanding of the critical pathways, such as KRAS, Notch, Hedgehog, and Wnt/β-catenin, that drive tumor growth, metastasis, and therapeutic resistance. Even with a wide range of therapeutic options available, such as radiation therapy, chemotherapy, surgery, and targeted medicines, the overall survival rates for people with pancreatic cancer are still remarkably poor. Pancreatic cancer research is a rapidly evolving field, with numerous promising approaches on the horizon, such as immunotherapy, liquid biopsies, personalized medicine, CRISPR/Cas9 genome editing, and computational intelligence applications. PDAC is a complex disease that requires a multidisciplinary approach involving clinicians, researchers, and experts from various fields. A thorough assessment can help interdisciplinary teams work together more effectively by combining expertise from many fields, creating a common understanding, and pointing out areas where joint efforts can be made to combat this difficult illness.
MM: Conceptualization, Investigation, Methodology, Resources, Validation, Writing – original draft. KA: Conceptualization, Investigation, Project administration, Resources, Writing – review & editing. MA: Formal analysis, Investigation, Methodology, Writing – original draft. SH: Conceptualization, Methodology, Supervision, Validation, Visualization, Writing – original draft. Z: Software, Funding acquisition, Methodology, Project administration, Writing – original draft. GH: Validation, Data curation, Software, Supervision, Writing – review & editing. SI: Investigation, Methodology, Validation, Writing – original draft. AS: Data curation, Investigation, Methodology, Writing – review & editing. IH: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research has been funded by the Central Council for Research in Unani Medicine (CCRUM), Ministry of AYUSH, Government of India (Grant No. 3-69/2020- CCRUM/Tech).
MH thanks Central Council for Research in Unani Medicine (CCRUM), Ministry of AYUSH, Government of India (Grant No. 3-69/2020- CCRUM/Tech).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1427802/full#supplementary-material
1. McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. (2018) 24:4846–61. doi: 10.3748/wjg.v24.i43.4846
2. Desai SA, Patel VP, Bhosle KP, Nagare SD, Thombare KC. The tumor microenvironment: shaping cancer progression and treatment response. J Chemother. (2024) 1–30. doi: 10.1080/1120009X.2023.2300224
3. Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, et al. Targeting the interplay between epithelial-to-mesenchymal-transition and the immune system for effective immunotherapy. Cancers. (2019) 11:714. doi: 10.3390/cancers11050714
4. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in diagnosing, prognosis and treating pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2020) 17:153–68. doi: 10.1038/s41575-019-0245-4
5. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
6. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. (2019) 18:26. doi: 10.1186/s12943-019-0954-x
7. Sano M, Driscoll DR, DeJesus-Monge WE, Quattrochi B, Appleman VA, Ou J, et al. Activation of WNT/β-catenin signaling enhances pancreatic cancer development and the Malignant potential via up-regulation of cyr61. Neoplasia. (2016) 18:785–94. doi: 10.1016/j.neo.2016.11.004
8. Słodkowski M, Wroński M, Karkocha D, Kraj L, Śmigielska K, Jachnis A. Current approaches for the curative-intent surgical treatment of pancreatic ductal adenocarcinoma. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15092584
9. Riedl JM, Posch F, Horvath L, Gantschnigg A, Renneberg F, Schwarzenbacher E, et al. Gemcitabine/nab-Paclitaxel versus FOLFIRINOX for palliative first-line treatment of advanced pancreatic cancer: A propensity score analysis. Eur J Cancer. (2021) 151:3–13. doi: 10.1016/j.ejca.2021.03.040
10. Brown TJ, Reiss KA. PARP inhibitors in pancreatic cancer. Cancer J. (2021) 27:465–75. doi: 10.1097/PPO.0000000000000554
11. Torphy RJ, Zhu Y, Schulick RD. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann Gastroenterol Surg. (2018) 2:274–81. doi: 10.1002/ags3.12176
12. Kamyabi N, Bernard V, Maitra A. Liquid biopsies in pancreatic cancer. Expert Rev Anticancer Ther. (2019) 19:869–78. doi: 10.1080/14737140.2019.1670063
13. Katti A, Diaz BJ, Caragine CM, Sanjana NE, Dow LE. CRISPR in cancer biology and therapy. Nat Rev Cancer. (2022) 22:259–79. doi: 10.1038/s41568-022-00441-w
14. Sagami R, Yamao K, Nakahodo J, Minami R, Tsurusaki M, Murakami K, et al. Pre-operative imaging and pathological diagnosis of localized high-grade pancreatic intra-epithelial neoplasia without invasive carcinoma. Cancers. (2021) 13:945. doi: 10.3390/cancers13050945
15. Pandit N, Yadav TN, Lacoul R, Awale L. Invasive intraductal papillary mucinous neoplasm (IPMN) of the pancreas causing duodenal infiltration and obstruction: a case report. J Gastrointest Cancer. (2020) 51:292–5. doi: 10.1007/s12029-019-00225-w
16. Shen W, Tao G-q, Zhang Y, Cai B, Sun J, Tian Z-q. TGF-β in pancreatic cancer initiation and progression: two sides of the same coin. Cell Bioscience. (2017) 7:39. doi: 10.1186/s13578-017-0168-0
17. Włodarczyk B, Borkowska A, Włodarczyk P, Małecka-Panas E, Gąsiorowska A. Insulin-like growth factor 1 and insulin-like growth factor binding protein 2 serum levels as potential biomarkers in differential diagnosis between chronic pancreatitis and pancreatic adenocarcinoma in reference to pancreatic diabetes. Prz Gastroenterol. (2021) 16:36–42. doi: 10.5114/pg.2020.95091
18. Ndlovu R, Deng LC, Wu J, Li XK, Zhang JS. Fibroblast growth factor 10 in pancreas development and pancreatic cancer. Front Genet. (2018) 9:482. doi: 10.3389/fgene.2018.00482
19. Xu Z, Pang TCY, Liu AC, Pothula SP, Mekapogu AR, Perera CJ, et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: a key element of treatment that limits primary tumour growth and eliminates metastasis. Br J Cancer. (2020) 122:1486–95. doi: 10.1038/s41416-020-0782-1
20. Grapa CM, Mocan T, Gonciar D, Zdrehus C, Mosteanu O, Pop T, et al. Epidermal growth factor receptor and its role in pancreatic cancer treatment mediated by nanoparticles. Int J Nanomedicine. (2019) 14:9693–706. doi: 10.2147/IJN
21. Ortega MA, Pekarek L, Fraile-Martinez O, Garcia-Montero C, Saez MA, Asúnsolo A, et al. Implication of ERBB2 as a predictive tool for survival in patients with pancreatic cancer in histological studies. Curr Oncol. (2022) 29:2442–53. doi: 10.3390/curroncol29040198
22. Thomas G, Chardès T, Gaborit N, Mollevi C, Leconet W, Robert B, et al. HER3 as biomarker and therapeutic target in pancreatic cancer: new insights in pertuzumab therapy in preclinical models. Oncotarget. (2014) 5:7138–48. doi: 10.18632/oncotarget.v5i16
23. Trajkovic-Arsic M, Kalideris E, Siveke JT. The role of insulin and IGF system in pancreatic cancer. J Mol Endocrinol. (2013) 50:R67–74. doi: 10.1530/JME-12-0259
24. Francavilla C, O’Brien CS. Fibroblast growth factor receptor signalling dysregulation and targeting in breast cancer. Open Biol. (2022) 12:210373. doi: 10.1098/rsob.210373
25. Sharma R, Malviya R. Correlation between hypoxia and HGF/c-MET expression in the management of pancreatic cancer. Biochim Biophys Acta Rev Cancer. (2023) 1878:188869. doi: 10.1016/j.bbcan.2023.188869
26. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. (2018) 17:58. doi: 10.1186/s12943-018-0782-4
27. Zhang Z, Zhang H, Liao X, Tsai HI. KRAS mutation: The booster of pancreatic ductal adenocarcinoma transformation and progression. Front Cell Dev Biol. (2023) 11:1147676. doi: 10.3389/fcell.2023.1147676
28. Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. (2014) 142:316–38. doi: 10.1016/j.pharmthera.2013.12.014
29. Pan J, Ho M. Role of glypican-1 in regulating multiple cellular signaling pathways. Am J Physiol Cell Physiol. (2021) 321:C846–c858. doi: 10.1152/ajpcell.00290.2021
30. Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. (2015) 4:215–66. doi: 10.1002/wdev.176
31. Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. (2014) 49:1–15. doi: 10.3109/10409238.2013.838205
32. Gore AJ, Deitz SL, Palam LR, Craven KE, Korc M. Pancreatic cancer-associated retinoblastoma 1 dysfunction enables TGF-β to promote proliferation. J Clin Invest. (2014) 124:338–52. doi: 10.1172/JCI71526
33. Zhang T, Ma C, Zhang Z, Zhang H, Hu H. NF-κB signaling in inflammation and cancer. MedComm (2020). (2021) 2:618–53. doi: 10.1002/mco2.104
34. Zhao Y, Qin C, Zhao B, Wang Y, Li Z, Li T, et al. Pancreatic cancer stemness: dynamic status in Malignant progression. J Exp Clin Cancer Res. (2023) 42:122. doi: 10.1186/s13046-023-02693-2
35. Li J, Peng L, Chen Q, Ye Z, Zhao T, Hou S, et al. Integrin β1 in pancreatic cancer: expressions, functions, and clinical implications. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14143377
36. Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. (2016) 35:575–88. doi: 10.1007/s10555-016-9649-6
37. Chen Z, Guo Y, Zhao D, Zou Q, Yu F, Zhang L, et al. Comprehensive analysis revealed that CDKN2A is a biomarker for immune infiltrates in multiple cancers. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.808208
38. Principe DR, Timbers KE, Atia LG, Koch RM, Rana A. TGFβ Signaling in the pancreatic tumor microenvironment. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13205086
39. Zhang Y, Alexander PB, Wang XF. TGF-β Family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol. (2017) 9. doi: 10.1101/cshperspect.a022145
40. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. (2012) 142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042
41. Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. (2013) 497:638–42. doi: 10.1038/nature12205
42. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduction Targeted Ther. (2021) 6:386. doi: 10.1038/s41392-021-00780-4
43. Pantsar T. The current understanding of KRAS protein structure and dynamics. Comput Struct Biotechnol J. (2020) 18:189–98. doi: 10.1016/j.csbj.2019.12.004
44. Yin G, Kistler S, George SD, Kuhlmann N, Garvey L, Huynh M, et al. A KRAS GTPase K104Q mutant retains downstream signaling by offsetting defects in regulation. J Biol Chem. (2017) 292:4446–56. doi: 10.1074/jbc.M116.762435
45. Li C, Vides A, Kim D, Xue JY, Zhao Y, Lito P. The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science. (2021) 374:197–201. doi: 10.1126/science.abf1730
46. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. (2014) 39:91–100. doi: 10.1016/j.tibs.2013.12.004
47. Haigis KM. KRAS alleles: the devil is in the detail. Trends Cancer. (2017) 3:686–97. doi: 10.1016/j.trecan.2017.08.006
48. Gu Y, Ji Y, Jiang H, Qiu G. Clinical effect of driver mutations of KRAS, CDKN2A/P16, TP53, and SMAD4 in pancreatic cancer: A meta-analysis. Genet Test Mol Biomarkers. (2020) 24:777–88. doi: 10.1089/gtmb.2020.0078
49. Bahar ME, Kim HJ, Kim DR. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther. (2023) 8:455. doi: 10.1038/s41392-023-01705-z
50. Mehra S, Deshpande N, Nagathihalli N. Targeting PI3K pathway in pancreatic ductal adenocarcinoma: rationale and progress. Cancers. (2021) 13(17). doi: 10.3390/cancers13174434
51. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. (2013) 23:406–20. doi: 10.1016/j.ccr.2013.01.023
52. Lin JC, Liu TP, Yang PM. CDKN2A-inactivated pancreatic ductal adenocarcinoma exhibits therapeutic sensitivity to paclitaxel: A bioinformatics study. J Clin Med. (2020) 9. doi: 10.3390/jcm9124019
53. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. (2018) 28:911–25. doi: 10.1016/j.tcb.2018.07.002
54. Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine. (2016) 8:30–9. doi: 10.1016/j.ebiom.2016.04.017
55. Westphalen CB, Olive KP. Genetically engineered mouse models of pancreatic cancer. Cancer J. (2012) 18:502–10. doi: 10.1097/PPO.0b013e31827ab4c4
56. Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. (2018) 14:111–23. doi: 10.7150/ijbs.23230
57. Cheng R, Li F, Zhang M, Xia X, Wu J, Gao X, et al. A novel protein RASON encoded by a lncRNA controls oncogenic RAS signaling in KRAS mutant cancers. Cell Res. (2023) 33:30–45. doi: 10.1038/s41422-022-00726-7
58. Racu ML, Lebrun L, Schiavo AA, Van Campenhout C, De Clercq S, Absil L, et al. The role of SMAD4 inactivation in epithelial-mesenchymal plasticity of pancreatic ductal adenocarcinoma: the missing link? Cancers (Basel). (2022) 14. doi: 10.3390/cancers14040973
59. Voutsadakis IA. Mutations of p53 associated with pancreatic cancer and therapeutic implications. Ann Hepatobiliary Pancreat Surg. (2021) 25:315–27. doi: 10.14701/ahbps.2021.25.3.315
60. Torres MP, Rachagani S, Souchek JJ, Mallya K, Johansson SL, Batra SK. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PloS One. (2013) 8:e80580. doi: 10.1371/journal.pone.0080580
61. Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. (2016) 6:a026104. doi: 10.1101/cshperspect.a026104
62. Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel). (2017) 9. doi: 10.3390/cancers9050042
63. Bannoura SF, Khan HY, Azmi AS. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front Oncol. (2022) 12. doi: 10.3389/fonc.2022.1013902
64. Gao J, Long B, Wang Z. Role of Notch signaling pathway in pancreatic cancer. Am J Cancer Res. (2017) 7:173–86.
65. Kovall RA, Blacklow SC. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr Top Dev Biol. (2010) 92:31–71. doi: 10.1016/S0070-2153(10)92002-4
66. Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. (2000) 5:197–206. doi: 10.1016/S1097-2765(00)80416-5
67. Zema S, Pelullo M, Nardozza F, Felli MP, Screpanti I, Bellavia D. A dynamic role of mastermind-like 1: A journey through the main (Path)ways between development and cancer. Front Cell Dev Biol. (2020) 8. doi: 10.3389/fcell.2020.613557
68. Chung W-C, Xu K. Chapter One - Notch signaling pathway in pancreatic tumorigenesis. In: Emdad L, Atfi A, Gogna R, Trevino JG, Fisher PB, editors. Advances in cancer research. United States: Academic Press (2023). p. 1–36.
69. Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res. (2009) 15:2291–301. doi: 10.1158/1078-0432.CCR-08-2004
70. Song HY, Wang Y, Lan H, Zhang YX. Expression of Notch receptors and their ligands in pancreatic ductal adenocarcinoma. Exp Ther Med. (2018) 16:53–60. doi: 10.3892/etm
71. You WK, Schuetz TJ, Lee SH. Targeting the DLL/notch signaling pathway in cancer: challenges and advances in clinical development. Mol Cancer Ther. (2023) 22:3–11. doi: 10.1158/1535-7163.MCT-22-0243
72. Bai Y, Bai Y, Dong J, Li Q, Jin Y, Chen B, et al. Hedgehog signaling in pancreatic fibrosis and cancer. Med (Baltimore). (2016) 95:e2996. doi: 10.1097/MD.0000000000002996
73. Sasai N, Toriyama M, Kondo T. Hedgehog signal and genetic disorders. Front Genet. (2019) 10. doi: 10.3389/fgene.2019.01103
74. Arensdorf AM, Marada S, Ogden SK. Smoothened regulation: A tale of two signals. Trends Pharmacol Sci. (2016) 37:62–72. doi: 10.1016/j.tips.2015.09.001
75. Qi X, Li X. Mechanistic insights into the generation and transduction of hedgehog signaling. Trends Biochem Sci. (2020) 45:397–410. doi: 10.1016/j.tibs.2020.01.006
76. Gu D, Schlotman KE, Xie J. Deciphering the role of hedgehog signaling in pancreatic cancer. J BioMed Res. (2016) 30:353–60. doi: 10.7555/JBR.30.20150107
77. Nakashima H, Nakamura M, Yamaguchi H, Yamanaka N, Akiyoshi T, Koga K, et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. (2006) 66:7041–9. doi: 10.1158/0008-5472.CAN-05-4588
78. Pietrobono S, Gagliardi S, Stecca B. Non-canonical hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond smoothened. Front Genet. (2019) 10:556. doi: 10.3389/fgene.2019.00556
79. Stanciu S, Ionita-Radu F, Stefani C, Miricescu D, Stanescu S II, Greabu M, et al. Targeting PI3K/AKT/mTOR signaling pathway in pancreatic cancer: from molecular to clinical aspects. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231710132
80. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. (2021) 6:425. doi: 10.1038/s41392-021-00828-5
81. Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am. (2013) 22:641–64. doi: 10.1016/j.soc.2013.06.008
82. Murthy D, Attri KS, Singh PK. Phosphoinositide 3-kinase signaling pathway in pancreatic ductal adenocarcinoma progression, pathogenesis, and therapeutics. Front Physiol. (2018) 9. doi: 10.3389/fphys.2018.00335
83. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. (2023) 22:138. doi: 10.1186/s12943-023-01827-6
84. Ram Makena M, Gatla H, Verlekar D, Sukhavasi S, Pandey MK, Pramanik KC. Wnt/β-catenin signaling: the culprit in pancreatic carcinogenesis and therapeutic resistance. Int J Mol Sci. (2019) 20:4242. doi: 10.3390/ijms20174242
85. Pecoraro C, Faggion B, Balboni B, Carbone D, Peters GJ, Diana P, et al. GSK3β as a novel promising target to overcome chemoresistance in pancreatic cancer. Drug Resistance Updates. (2021) 58:100779. doi: 10.1016/j.drup.2021.100779
86. Ren Q, Chen J, Liu Y. LRP5 and LRP6 in wnt signaling: similarity and divergence. Front Cell Dev Biol. (2021) 9:670960. doi: 10.3389/fcell.2021.670960
87. Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. (2012) 31:2714–36. doi: 10.1038/emboj.2012.150
88. Lecarpentier Y, Schussler O, Hébert JL, Vallée A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front Oncol. (2019) 9:1248. doi: 10.3389/fonc.2019.01248
89. Groenewald W, Lund AH, Gay DM. The role of WNT pathway mutations in cancer development and an overview of therapeutic options. Cells. (2023) 12. doi: 10.3390/cells12070990
90. Dotan E, Cardin DB, Lenz HJ, Messersmith W, O’Neil B, Cohen SJ, et al. Phase ib study of wnt inhibitor ipafricept with gemcitabine and nab-paclitaxel in patients with previously untreated stage IV pancreatic cancer. Clin Cancer Res. (2020) 26:5348–57. doi: 10.1158/1078-0432.CCR-20-0489
91. Wang B, Zou Q, Sun M, Chen J, Wang T, Bai Y, et al. Reversion of trichostatin A resistance via inhibition of the Wnt signaling pathway in human pancreatic cancer cells. Oncol Rep. (2014) 32:2015–22. doi: 10.3892/or.2014.3476
92. Kalantary-Charvadeh A, Hosseini V, Mehdizadeh A, Darabi M. Application of porcupine inhibitors in stem cell fate determination. Chem Biol Drug Des. (2020) 96:1052–68. doi: 10.1111/cbdd.13704
93. Du W, Menjivar RE, Donahue KL, Kadiyala P, Velez-Delgado A, Brown KL, et al. WNT signaling in the tumor microenvironment promotes immunosuppression in murine pancreatic cancer. J Exp Med. (2023) 220. doi: 10.1084/jem.20220503
94. Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA. (2009) 106:1193–8. doi: 10.1073/pnas.0811902106
95. Ozawa F, Friess H, Tempia-Caliera A, Kleeff J, Büchler MW. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen. (2001) 21:27–44. doi: 10.1002/(ISSN)1520-6866
96. Mitchell RA, Luwor RB, Burgess AW. Epidermal growth factor receptor: Structure-function informing the design of anticancer therapeutics. Exp Cell Res. (2018) 371:1–19. doi: 10.1016/j.yexcr.2018.08.009
97. Hodoglugil U, Carrillo MW, Hebert JM, Karachaliou N, Rosell RC, Altman RB, et al. PharmGKB summary: very important pharmacogene information for the epidermal growth factor receptor. Pharmacogenet Genomics. (2013) 23:636–42. doi: 10.1097/FPC.0b013e3283655091
98. Barton CM, Hall PA, Hughes CM, Gullick WJ, Lemoine NR. Transforming growth factor alpha and epidermal growth factor in human pancreatic cancer. J Pathol. (1991) 163:111–6. doi: 10.1002/path.1711630206
99. Lee J, Jang KT, Ki CS, Lim T, Park YS, Lim HY, et al. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer. (2007) 109:1561–9. doi: 10.1002/cncr.22559
100. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
101. Purba ER, Saita EI, Maruyama IN. Activation of the EGF receptor by ligand binding and oncogenic mutations: the “Rotation model. Cells. (2017) 6. doi: 10.20944/preprints201705.0212.v1
102. Kennedy SP, Hastings JF, Han JZR, Croucher DR. The under-appreciated promiscuity of the epidermal growth factor receptor family. Front Cell Dev Biol. (2016) 4. doi: 10.3389/fcell.2016.00088
103. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. (2008) 3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305
104. Marstrand-Daucé L, Lorenzo D, Chassac A, Nicole P, Couvelard A, Haumaitre C. Acinar-to-ductal metaplasia (ADM): on the road to pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24129946
105. Din NU, Zubair M, Abdul-Ghafar J, Ahmad Z. Pancreatic mucinous cystic neoplasms: a clinicopathological study of 11 cases and detailed review of literature. Surg Exp Pathol. (2020) 3:6. doi: 10.1186/s42047-020-0059-2
106. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. (2008) 321:1801–6. doi: 10.1126/science.1164368
107. Schneeweis C, Wirth M, Saur D, Reichert M, Schneider G. Oncogenic KRAS and the EGFR loop in pancreatic carcinogenesis-A connection to licensing nodes. Small GTPases. (2018) 9:457–64. doi: 10.1080/21541248.2016.1262935
108. Ferreira A, Pereira F, Reis C, Oliveira MJ, Sousa MJ, Preto A. Crucial role of oncogenic KRAS mutations in apoptosis and autophagy regulation: therapeutic implications. Cells. (2022) 11. doi: 10.3390/cells11142183
109. Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology. (2002) 63:317–32. doi: 10.1159/000066230
110. Weroha SJ, Haluska P. The insulin-like growth factor system in cancer. Endocrinol Metab Clin North Am. (2012) 41:335–50. doi: 10.1016/j.ecl.2012.04.014
111. Du C, da Silva A, Morales-Oyarvide V, Dias Costa A, Kozak MM, Dunne RF, et al. Insulin-like growth factor-1 receptor expression and disease recurrence and survival in patients with resected pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. (2020) 29:1586–95. doi: 10.1158/1055-9965.EPI-19-1315
112. Makinoshima H, Dezawa M. Pancreatic cancer cells activate CCL5 expression in mesenchymal stromal cells through the insulin-like growth factor-I pathway. FEBS Lett. (2009) 583:3697–703. doi: 10.1016/j.febslet.2009.10.061
113. Hirakawa T, Yashiro M, Doi Y, Kinoshita H, Morisaki T, Fukuoka T, et al. Pancreatic Fibroblasts Stimulate the Motility of Pancreatic Cancer Cells through IGF1/IGF1R Signaling under Hypoxia. PloS One. (2016) 11:e0159912. doi: 10.1371/journal.pone.0159912
114. Mairet-Coello G, Tury A, DiCicco-Bloom E. Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J Neurosci. (2009) 29:775–88. doi: 10.1523/JNEUROSCI.1700-08.2009
115. Fuchs CS, Azevedo S, Okusaka T, Van Laethem JL, Lipton LR, Riess H, et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann Oncol. (2015) 26:921–7. doi: 10.1093/annonc/mdv027
116. Navar AM, Roe MT, White JA, Cannon CP, Lokhnygina Y, Newby LK, et al. Medication discontinuation in the IMPROVE-IT trial. Circ Cardiovasc Qual Outcomes. (2019) 12:e005041. doi: 10.1161/CIRCOUTCOMES.118.005041
117. Kang X, Lin Z, Xu M, Pan J, Wang ZW. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. (2019) 52:e12605. doi: 10.1111/cpr.12605
118. Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. (2000) 6:743–50. doi: 10.1016/S1097-2765(00)00073-3
119. Teven CM, Farina EM, Rivas J, Reid RR. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. (2014) 1:199–213. doi: 10.1016/j.gendis.2014.09.005
120. Pudewell S, Wittich C, Kazemein Jasemi NS, Bazgir F, Ahmadian MR. Accessory proteins of the RAS-MAPK pathway: moving from the side line to the front line. Commun Biol. (2021) 4:696. doi: 10.1038/s42003-021-02149-3
121. Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res. (2006) 12:6652–62. doi: 10.1158/1078-0432.CCR-06-1164
122. Lake D, Corrêa SA, Müller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. (2016) 73:4397–413. doi: 10.1007/s00018-016-2297-8
123. Ferguson HR, Smith MP, Francavilla C. Fibroblast growth factor receptors (FGFRs) and noncanonical partners in cancer signaling. Cells (2021) 10. doi: 10.3390/cells10051201
124. Zhang H, Hylander BL, LeVea C, Repasky EA, Straubinger RM, Adjei AA, et al. Enhanced FGFR signalling predisposes pancreatic cancer to the effect of a potent FGFR inhibitor in preclinical models. Br J Cancer. (2014) 110:320–9. doi: 10.1038/bjc.2013.754
125. Chae YK, Ranganath K, Hammerman PS, Vaklavas C, Mohindra N, Kalyan A, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. (2017) 8:16052–74. doi: 10.18632/oncotarget.v8i9
126. Ruan R, Li L, Li X, Huang C, Zhang Z, Zhong H, et al. Unleashing the potential of combining FGFR inhibitor and immune checkpoint blockade for FGF/FGFR signaling in tumor microenvironment. Mol Cancer. (2023) 22:60. doi: 10.1186/s12943-023-01761-7
127. Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. (2006) 312:549–60. doi: 10.1016/j.yexcr.2005.11.012
128. Annese T, Tamma R, Ruggieri S, Ribatti D. Angiogenesis in pancreatic cancer: pre-clinical and clinical studies. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11030381
129. Tsuzuki Y, Carreira CM, Bockhorn M, Xu L, Jain RK, Fukumura D. Pancreas microenvironment promotes VEGF expression and tumor growth: novel window models for pancreatic tumor angiogenesis and microcirculation. Lab Invest. (2001) 81:1439–51. doi: 10.1038/labinvest.3780357
130. Ali EM, Sheta M, El Mohsen MA. Elevated serum and tissue VEGF associated with poor outcome in breast cancer patients. Alexandria J Med. (2011) 47:217–24. doi: 10.1016/j.ajme.2011.07.003
131. Hotz HG, Reber HA, Hotz B, Sanghavi PC, Yu T, Foitzik T, et al. Angiogenesis inhibitor TNP-470 reduces human pancreatic cancer growth. J Gastrointest Surg. (2001) 5:131–8. doi: 10.1016/S1091-255X(01)80024-X
132. Solorzano CC, Baker CH, Bruns CJ, Killion JJ, Ellis LM, Wood J, et al. Inhibition of growth and metastasis of human pancreatic cancer growing in nude mice by PTK 787/ZK222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases. Cancer Biother Radiopharm. (2001) 16:359–70. doi: 10.1089/108497801753354267
133. Hess-Stumpp H, Haberey M, Thierauch KH. PTK 787/ZK 222584, a tyrosine kinase inhibitor of all known VEGF receptors, represses tumor growth with high efficacy. Chembiochem. (2005) 6:550–7. doi: 10.1002/cbic.200400305
134. Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. (2009) 7:17. doi: 10.1186/1479-5876-7-17
135. Kang R, Hou W, Zhang Q, Chen R, Lee YJ, Bartlett DL, et al. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. (2014) 5:e1480. doi: 10.1038/cddis.2014.445
136. Kang R, Tang D, Livesey KM, Schapiro NE, Lotze MT, Zeh HJ 3rd. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid Redox Signal. (2011) 15:2175–84. doi: 10.1089/ars.2010.3378
137. Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. (2010) 17:666–76. doi: 10.1038/cdd.2009.149
138. Swami P, O’Connell KA, Thiyagarajan S, Crawford A, Patil P, Radhakrishnan P, et al. Inhibition of the receptor for advanced glycation end products enhances the cytotoxic effect of gemcitabine in murine pancreatic tumors. Biomolecules. (2021) 11. doi: 10.3390/biom11040526
139. Li ZY, Chen SY, Weng MH, Yen GC. Ursolic acid restores sensitivity to gemcitabine through the RAGE/NF-κB/MDR1 axis in pancreatic cancer cells and in a mouse xenograft model. J Food Drug Anal. (2021) 29:262–74. doi: 10.38212/2224-6614.3346
140. Kang R, Tang D, Lotze MT, Zeh HJ 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy. (2012) 8:989–91. doi: 10.4161/auto.20258
141. Mahadevan KK, LeBleu VS, Ramirez EV, Chen Y, Li B, Sockwell AM, et al. Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8(+) T cells. Dev Cell. (2023) 58:1562–1577.e8. doi: 10.1016/j.devcel.2023.07.025
142. Swami P, Thiyagarajan S, Vidger A, Indurthi VSK, Vetter SW, Leclerc E. RAGE up-regulation differently affects cell proliferation and migration in pancreatic cancer cells. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21207723
143. Azizian-Farsani F, Abedpoor N, Sheikhha MH, Gure AO, Nasr-Esfahani MH, Ghaedi K. Receptor for advanced glycation end products acts as a fuel to colorectal cancer development. Front Oncol. (2020) 10:552283. doi: 10.3389/fonc.2020.552283
144. Mandarino A, Thiyagarajan S, Martins ACF, Gomes RDS, Vetter SW, Leclerc E. S100s and HMGB1 crosstalk in pancreatic cancer tumors. Biomolecules. (2023) 13. doi: 10.3390/biom13081175
145. Arumugam T, Simeone DM, Golen KV, Logsdon CD. S100P promotes pancreatic cancer growth, survival, and invasion. Clin Cancer Res. (2005) 11:5356–64. doi: 10.1158/1078-0432.CCR-05-0092
146. Dakhel S, Padilla L, Adan J, Masa M, Martinez JM, Roque L, et al. S100P antibody-mediated therapy as a new promising strategy for the treatment of pancreatic cancer. Oncogenesis. (2014) 3:e92. doi: 10.1038/oncsis.2014.7
147. Wu L, Yang L. The function and mechanism of HMGB1 in lung cancer and its potential therapeutic implications. Oncol Lett. (2018) 15:6799–805. doi: 10.3892/ol
148. Wang S, Huang S, Sun YL. Epithelial-mesenchymal transition in pancreatic cancer: A review. BioMed Res Int. (2017) 2017:2646148. doi: 10.1155/2017/2646148
149. Islam MS, Morshed MR, Babu G, Khan MA. The role of inflammations and EMT in carcinogenesis. Adv Cancer Biol - Metastasis. (2022) 5:100055. doi: 10.1016/j.adcanc.2022.100055
150. van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. (2011) 728:23–34. doi: 10.1016/j.mrrev.2011.05.002
151. Ribatti D, Tamma R, Annese T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl Oncol. (2020) 13:100773. doi: 10.1016/j.tranon.2020.100773
152. Sato N, Kohi S, Hirata K, Goggins M. Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. (2016) 107:569–75. doi: 10.1111/cas.12913
153. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. (2006) 7:131–42. doi: 10.1038/nrm1835
154. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. (2008) 10:295–305. doi: 10.1038/ncb1691
155. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. (2000) 2:76–83. doi: 10.1038/35000025
157. Seong HA, Jung H, Ha H. Murine protein serine/threonine kinase 38 stimulates TGF-beta signaling in a kinase-dependent manner via direct phosphorylation of Smad proteins. J Biol Chem. (2010) 285:30959–70. doi: 10.1074/jbc.M110.138370
158. Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. (2005) 132:3151–61. doi: 10.1242/dev.01907
159. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
160. Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. (2005) 25:8108–25. doi: 10.1128/MCB.25.18.8108-8125.2005
161. Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, et al. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. (2001) 61:4222–8.
162. Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. (2013) 5:a007898. doi: 10.1101/cshperspect.a007898
163. Marin O, Bustos VH, Cesaro L, Meggio F, Pagano MA, Antonelli M, et al. A noncanonical sequence phosphorylated by casein kinase 1 in beta-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc Natl Acad Sci USA. (2003) 100:10193–200. doi: 10.1073/pnas.1733909100
164. Hagen T, Vidal-Puig A. Characterisation of the phosphorylation of beta-catenin at the GSK-3 priming site Ser45. Biochem Biophys Res Commun. (2002) 294:324–8. doi: 10.1016/S0006-291X(02)00485-0
165. Sharma A, Mir R, Galande S. Epigenetic regulation of the wnt/β-catenin signaling pathway in cancer. Front Genet. (2021) 12. doi: 10.3389/fgene.2021.681053
166. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduction Targeted Ther. (2022) 7:3. doi: 10.1038/s41392-021-00762-6
167. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. (2014) 5:2881–911. doi: 10.18632/oncotarget.v5i10
168. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. (2004) 6:931–40. doi: 10.1038/ncb1173
169. Tomar VS, Patil V, Somasundaram K. Temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway: implications in glioma therapy. Cell Biol Toxicol. (2020) 36:273–8. doi: 10.1007/s10565-019-09502-7
170. Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, et al. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: signaling, therapeutic implications, and challenges. Cells. (2019) 8. doi: 10.3390/cells8101118
171. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
172. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduction Targeted Ther. (2022) 7:95. doi: 10.1038/s41392-022-00934-y
173. Yao Y, Ni Y, Zhang J, Wang H, Shao S. The role of Notch signaling in gastric carcinoma: molecular pathogenesis and novel therapeutic targets. Oncotarget. (2017) 8:53839–53. doi: 10.18632/oncotarget.v8i32
174. Krebs M, Solimando AG, Kalogirou C, Marquardt A, Frank T, Sokolakis I, et al. miR-221-3p regulates VEGFR2 expression in high-risk prostate cancer and represents an escape mechanism from sunitinib in vitro. J Clin Med. (2020) 9:53839–53. doi: 10.3390/jcm9030670
175. Lamanuzzi A, Saltarella I, Desantis V, Frassanito MA, Leone P, Racanelli V, et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget. (2018) 9:20563–77. doi: 10.18632/oncotarget.v9i29
176. Shao S, Zhao X, Zhang X, Luo M, Zuo X, Huang S, et al. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol Cancer. (2015) 14:28. doi: 10.1186/s12943-015-0295-3
177. Güngör C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, et al. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. (2011) 71:5009–19. doi: 10.1158/0008-5472.CAN-11-0036
178. Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, et al. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. (2018) 6:2. doi: 10.1038/s41413-017-0005-4
179. Ehata S, Miyazono K. Bone morphogenetic protein signaling in cancer; some topics in the recent 10 years. Front Cell Dev Biol. (2022) 10:883523. doi: 10.3389/fcell.2022.883523
180. Kang CM, Babicky ML, Lowy AM. The RON receptor tyrosine kinase in pancreatic cancer pathogenesis and its potential implications for future targeted therapies. Pancreas. (2014) 43:183–9. doi: 10.1097/MPA.0000000000000088
181. Zhang ML, Tao Y, Zhou WQ, Ma PC, Cao YP, He CD, et al. All-trans retinoic acid induces cell-cycle arrest in human cutaneous squamous carcinoma cells by inhibiting the mitogen-activated protein kinase-activated protein 1 pathway. Clin Exp Dermatol. (2014) 39:354–60. doi: 10.1111/ced.12227
182. Kwon DH, Ryu J, Kim YK, Kook H. Roles of histone acetylation modifiers and other epigenetic regulators in vascular calcification. Int J Mol Sci. (2020) 21:354–60. doi: 10.3390/ijms21093246
183. Fei F, Qu J, Zhang M, Li Y, Zhang S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget. (2017) 8:73219–39. doi: 10.18632/oncotarget.v8i42
184. Imafuku K, Iwata H, Natsuga K, Okumura M, Kobayashi Y, Kitahata H, et al. Zonula occludens-1 distribution and barrier functions are affected by epithelial proliferation and turnover rates. Cell Prolif. (2023) 56:e13441. doi: 10.1111/cpr.13441
185. Bae SJ, Luo X. Activation mechanisms of the Hippo kinase signaling cascade. Biosci Rep. (2018) 38. doi: 10.1042/BSR20171469
186. Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. (2015) 6:8357. doi: 10.1038/ncomms9357
187. Jia J, Zhang W, Wang B, Trinko R, Jiang J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. (2003) 17:2514–9. doi: 10.1101/gad.1134003
188. Kim E, Kang JG, Kang MJ, Park JH, Kim YJ, Kweon TH, et al. O-GlcNAcylation on LATS2 disrupts the Hippo pathway by inhibiting its activity. Proc Natl Acad Sci USA (2020) 117:14259–69. doi: 10.1073/pnas.1913469117
189. Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. (2010) 24:862–74. doi: 10.1101/gad.1909210
190. Hong W, Guan KL, The YAP. and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. (2012) 23:785–93. doi: 10.1016/j.semcdb.2012.05.004
191. Zhang H, Liu C-Y, Zha Z-Y, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition*. J Biol Chem. (2009) 284:13355–62. doi: 10.1074/jbc.M900843200
192. Boopathy GTK, Hong W. Role of hippo pathway-YAP/TAZ signaling in angiogenesis. Front Cell Dev Biol. (2019) 7. doi: 10.3389/fcell.2019.00049
193. Shome D, Woedtke Tv, Riedel K, Masur K. The HIPPO transducer YAP and its targets CTGF and cyr61 drive a paracrine signalling in cold atmospheric plasma-mediated wound healing. Oxid Med Cell Longev. (2020) 2020:4910280. doi: 10.1155/2020/4910280
194. Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PloS Genet. (2015) 11:e1005465. doi: 10.1371/journal.pgen.1005465
195. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA. (2016) 113:11525–30. doi: 10.1073/pnas.1613121113
196. Morice S, Danieau G, Rédini F, Brounais-Le-Royer B, Verrecchia F. Hippo/YAP signaling pathway: A promising therapeutic target in bone paediatric cancers? Cancers (Basel). (2020) 12:14259–69. doi: 10.3390/cancers12030645
197. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. (2011) 30:1229–40. doi: 10.1038/onc.2010.504
198. Ehmer U, Sage J. Control of proliferation and cancer growth by the hippo signaling pathway. Mol Cancer Res. (2016) 14:127–40. doi: 10.1158/1541-7786.MCR-15-0305
199. Zheng Y, Pan D. The hippo signaling pathway in development and disease. Dev Cell. (2019) 50:264–82. doi: 10.1016/j.devcel.2019.06.003
200. Fu M, Hu Y, Lan T, Guan KL, Luo T, Luo M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct Target Ther. (2022) 7:376. doi: 10.1038/s41392-022-01191-9
201. Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. (2016) 30:1–17. doi: 10.1101/gad.274027.115
202. Kwon H, Kim J, Jho EH. Role of the Hippo pathway and mechanisms for controlling cellular localization of YAP/TAZ. FEBS J. (2022) 289:5798–818. doi: 10.1111/febs.16091
203. Guo Y, Luo J, Zou H, Liu C, Deng L, Li P. Context-dependent transcriptional regulations of YAP/TAZ in cancer. Cancer Lett. (2022) 527:164–73. doi: 10.1016/j.canlet.2021.12.019
204. Hooglugt A, van der Stoel MM, Boon RA, Huveneers S. Endothelial YAP/TAZ signaling in angiogenesis and tumor vasculature. Front Oncol. (2020) 10:612802. doi: 10.3389/fonc.2020.612802
205. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and Malignancy. Cell Metab. (2019) 29:124–140.e10. doi: 10.1016/j.cmet.2018.09.012
206. Fedotova AA, Bonchuk AN, Mogila VA, Georgiev PG. C2H2 zinc finger proteins: the largest but poorly explored family of higher eukaryotic transcription factors. Acta Naturae. (2017) 9:47–58. doi: 10.32607/20758251-2017-9-2-47-58
207. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. (2010) 29:1803–16. doi: 10.1038/emboj.2010.63
208. Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. (2007) 13:4769–76. doi: 10.1158/1078-0432.CCR-06-2926
209. Yin T, Wang C, Liu T, Zhao G, Zha Y, Yang M. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res. (2007) 141:196–203. doi: 10.1016/j.jss.2006.09.027
210. Koltai T, Reshkin SJ, Carvalho TMA, Di Molfetta D, Greco MR, Alfarouk KO, et al. Resistance to gemcitabine in pancreatic ductal adenocarcinoma: a physiopathologic and pharmacologic review. Cancers (Basel). (2022) 14:1803–16. doi: 10.3390/cancers14102486
211. Wang YL, Zhao XM, Shuai ZF, Li CY, Bai QY, Yu XW, et al. Snail promotes epithelial-mesenchymal transition and invasiveness in human ovarian cancer cells. Int J Clin Exp Med. (2015) 8:7388–93.
212. Wu Y, Zhou BP. Snail: more than EMT. Cell Adh Migr. (2010) 4:199–203. doi: 10.4161/cam.4.2.10943
213. Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. (2009) 66:773–87. doi: 10.1007/s00018-008-8465-8
214. Bronsert P, Kohler I, Timme S, Kiefer S, Werner M, Schilling O, et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery. (2014) 156:97–108. doi: 10.1016/j.surg.2014.02.018
215. Galván JA, Zlobec I, Wartenberg M, Lugli A, Gloor B, Perren A, et al. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br J Cancer. (2015) 112:1944–50. doi: 10.1038/bjc.2015.177
216. Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. (2007) 26:6979–88. doi: 10.1038/sj.onc.1210508
217. Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. (2012) 61:439–48. doi: 10.1136/gutjnl-2011-300060
218. Cubillo E, Diaz-Lopez A, Cuevas EP, Moreno-Bueno G, Peinado H, Montes A, et al. E47 and Id1 interplay in epithelial-mesenchymal transition. PloS One. (2013) 8:e59948. doi: 10.1371/journal.pone.0059948
219. Debnath P, Huirem RS, Dutta P, Palchaudhuri S. Epithelial-mesenchymal transition and its transcription factors. Biosci Rep. (2022) 42:6979–88. doi: 10.1042/BSR20211754
220. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. (2008) 19:4875–87. doi: 10.1091/mbc.e08-05-0506
221. Wang S, Zheng Y, Yang F, Zhu L, Zhu X-Q, Wang Z-F, et al. The molecular biology of pancreatic adenocarcinoma: translational challenges and clinical perspectives. Signal Transduction Targeted Ther. (2021) 6:249. doi: 10.1038/s41392-021-00659-4
222. Monkman JH, Thompson EW, Nagaraj SH. Targeting epithelial mesenchymal plasticity in pancreatic cancer: A compendium of preclinical discovery in a heterogeneous disease. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11111745
223. Sasaki K, Natsugoe S, Ishigami S, Matsumoto M, Okumura H, Setoyama T, et al. Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. (2009) 28:158. doi: 10.1186/1756-9966-28-158
224. Fu J, Qin L, He T, Qin J, Hong J, Wong J, et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. (2011) 21:275–89. doi: 10.1038/cr.2010.118
225. Mustafa M, Habib S, Imtiyaz K, Tufail N, Ahmad R, Hamim B, et al. Characterization of structural, genotoxic, and immunological effects of methyl methanesulfonate (MMS) induced DNA modifications: Implications for inflammation-driven carcinogenesis. Int J Biol Macromol. (2024) 268:131743. doi: 10.1016/j.ijbiomac.2024.131743
226. Sarantis P, Koustas E, Papadimitropoulou A, Papavassiliou AG, Karamouzis MV. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J Gastrointest Oncol. (2020) 12:173–81. doi: 10.4251/wjgo.v12.i2.173
227. Wang S, Li Y, Xing C, Ding C, Zhang H, Chen L, et al. Tumor microenvironment in chemoresistance, metastasis and immunotherapy of pancreatic cancer. Am J Cancer Res. (2020) 10:1937–53.
228. Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. (2009) 7:S44–7. doi: 10.1016/j.cgh.2009.07.039
229. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
230. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol. (2020) 17:527–40. doi: 10.1038/s41571-020-0363-5
231. Belhabib I, Zaghdoudi S, Lac C, Bousquet C, Jean C. Extracellular matrices and cancer-associated fibroblasts: targets for cancer diagnosis and therapy? Cancers (Basel). (2021) 13:44–7. doi: 10.3390/cancers13143466
232. Ferrara B, Pignatelli C, Cossutta M, Citro A, Courty J, Piemonti L. The extracellular matrix in pancreatic cancer: description of a complex network and promising therapeutic options. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13174442
233. Khalaf K, Hana D, Chou JT, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the tumor microenvironment involved in immune resistance and drug resistance. Front Immunol. (2021) 12:656364. doi: 10.3389/fimmu.2021.656364
234. Sekiguchi R, Yamada KM. Basement membranes in development and disease. Curr Top Dev Biol. (2018) 130:143–91. doi: 10.1016/bs.ctdb.2018.02.005
235. D’Arpino MC, Fuchs AG, Sánchez SS, Honoré SM. Extracellular matrix remodeling and TGF-β1/Smad signaling in diabetic colon mucosa. Cell Biol Int. (2018) 42:443–56. doi: 10.1002/cbin.10916
236. De Martino D, Bravo-Cordero JJ. Collagens in cancer: structural regulators and guardians of cancer progression. Cancer Res. (2023) 83:1386–92. doi: 10.1158/0008-5472.CAN-22-2034
237. Perez VM, Kearney JF, Yeh JJ. The PDAC extracellular matrix: A review of the ECM protein composition, tumor cell interaction, and therapeutic strategies. Front Oncol. (2021) 11:751311. doi: 10.3389/fonc.2021.751311
238. Liot S, Balas J, Aubert A, Prigent L, Mercier-Gouy P, Verrier B, et al. Stroma involvement in pancreatic ductal adenocarcinoma: an overview focusing on extracellular matrix proteins. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.612271
239. Rathore AS, Kumar S, Konwar R, Makker A, Negi MP, Goel MM. CD3+, CD4+ & CD8+ tumour infiltrating lymphocytes (TILs) are predictors of favourable survival outcome in infiltrating ductal carcinoma of breast. Indian J Med Res. (2014) 140:361–9.
240. Puls TJ, Tan X, Whittington CF, Voytik-Harbin SL. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PloS One. (2017) 12:e0188870. doi: 10.1371/journal.pone.0188870
241. Chang J, Chaudhuri O. Beyond proteases: Basement membrane mechanics and cancer invasion. J Cell Biol. (2019) 218:2456–69. doi: 10.1083/jcb.201903066
242. Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. (2021) 184:3852–72. doi: 10.1016/j.cell.2021.06.024
243. Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbæk MS, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. (2019) 7:68. doi: 10.1186/s40425-019-0556-6
244. Zhang H, Fredericks T, Xiong G, Qi Y, Rychahou PG, Li JD, et al. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res. (2018) 20:116. doi: 10.1186/s13058-018-1030-y
245. Zeltz C, Orgel J, Gullberg D. Molecular composition and function of integrin-based collagen glues-introducing COLINBRIs. Biochim Biophys Acta. (2014) 1840:2533–48. doi: 10.1016/j.bbagen.2013.12.022
246. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduction Targeted Ther. (2021) 6:153. doi: 10.1038/s41392-021-00544-0
247. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. (2015) 21:3561–8. doi: 10.1158/1078-0432.CCR-14-1051
248. Neophytou CM, Panagi M, Stylianopoulos T, Papageorgis P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13092053
249. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
250. Huertas-Caro CA, Ramírez MA, Rey-Vargas L, Bejarano-Rivera LM, Ballen DF, Nuñez M, et al. Tumor infiltrating lymphocytes (TILs) are a prognosis biomarker in Colombian patients with triple negative breast cancer. Sci Rep. (2023) 13:21324. doi: 10.1038/s41598-023-48300-4
251. Lekka E, Hall J. Noncoding RNAs in disease. FEBS Lett. (2018) 592:2884–900. doi: 10.1002/1873-3468.13182
252. Ranganathan K, Sivasankar V. MicroRNAs - Biology and clinical applications. J Oral Maxillofac Pathol. (2014) 18:229–34. doi: 10.4103/0973-029X.140762
253. Zhao G, Guo Y, Chen Z, Wang Y, Yang C, Dudas A, et al. miR-203 functions as a tumor suppressor by inhibiting epithelial to mesenchymal transition in ovarian cancer. J Cancer Sci Ther. (2015) 7:34–43. doi: 10.4172/1948-5956.1000322
254. Xue J, Jia E, Ren N, Lindsay A, Yu H. Circulating microRNAs as promising diagnostic biomarkers for pancreatic cancer: a systematic review. Onco Targets Ther. (2019) 12:6665–84. doi: 10.2147/OTT
255. Concepcion CP, Bonetti C, Ventura A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. (2012) 18:262–7. doi: 10.1097/PPO.0b013e318258b60a
256. Jung HM, Phillips BL, Patel RS, Cohen DM, Jakymiw A, Kong WW, et al. Keratinization-associated miR-7 and miR-21 regulate tumor suppressor reversion-inducing cysteine-rich protein with kazal motifs (RECK) in oral cancer. J Biol Chem. (2012) 287:29261–72. doi: 10.1074/jbc.M112.366518
257. Tu J, Cheung HH, Lu G, Chan CL, Chen Z, Chan WY. microRNA-126 is a tumor suppressor of granulosa cell tumor mediated by its host gene EGFL7. Front Oncol. (2019) 9:486. doi: 10.3389/fonc.2019.00486
258. Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci USA. (2011) 108:4908–13. doi: 10.1073/pnas.1101795108
259. Sheedy P, Medarova Z. The fundamental role of miR-10b in metastatic cancer. Am J Cancer Res. (2018) 8:1674–88.
260. Yu ML, Wang JF, Wang GK, You XH, Zhao XX, Jing Q, et al. Vascular smooth muscle cell proliferation is influenced by let-7d microRNA and its interaction with KRAS. Circ J. (2011) 75:703–9. doi: 10.1253/circj.CJ-10-0393
262. Xing Y, Wang Z, Lu Z, Xia J, Xie Z, Jiao M, et al. MicroRNAs: immune modulators in cancer immunotherapy. Immunother Adv. (2021) 1:ltab006. doi: 10.1093/immadv/ltab006
263. Ross SA, Davis CD. MicroRNA, nutrition, and cancer prevention. Adv Nutr. (2011) 2:472–85. doi: 10.3945/an.111.001206
264. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
265. Negrini M, Nicoloso MS, Calin GA. MicroRNAs and cancer–new paradigms in molecular oncology. Curr Opin Cell Biol. (2009) 21:470–9. doi: 10.1016/j.ceb.2009.03.002
266. Lee SK, Calin GA. Non-coding RNAs and cancer: new paradigms in oncology. Discovery Med. (2011) 11:245–54.
267. Miao L, Xiong X, Lin Y, Cheng Y, Lu J, Zhang J, et al. miR-203 inhibits tumor cell migration and invasion via caveolin-1 in pancreatic cancer cells. Oncol Lett. (2014) 7:658–62. doi: 10.3892/ol.2014.1807
268. Smolarz B, Durczyński A, Romanowicz H, Hogendorf P. The role of microRNA in pancreatic cancer. Biomedicines. (2021) 9:646–74. doi: 10.3390/biomedicines9101322
269. Lin XM, Chen H, Zhan XL. MiR-203 regulates JAK-STAT pathway in affecting pancreatic cancer cells proliferation and apoptosis by targeting SOCS3. Eur Rev Med Pharmacol Sci. (2019) 23:6906–13. doi: 10.26355/eurrev_201908_18730
270. Fenocchio E, Filippi R, Lombardi P, Quarà V, Milanesio M, Aimar G, et al. Is there a standard adjuvant therapy for resected pancreatic cancer? Cancers (Basel). (2019) 11:245–54. doi: 10.3390/cancers11101547
271. Mustafa M, Ali A, Siddiqui SA, Mir AR, Kausar T, Nayeem SM, et al. Biophysical characterization of structural and conformational changes in methylmethane sulfonate modified DNA leading to the frizzled backbone structure and strand breaks in DNA. J Biomol Struct Dyn. (2022) 40:7598–611. doi: 10.1080/07391102.2021.1899051
272. Alhazzani K, Alsahli M, Alanazi AZ, Algahtani M, Alenezi AA, Alhoshani A, et al. Augmented antitumor effects of erlotinib and cabozantinib on A549 non-small cell lung cancer: In vitro and in vivo studies. Saudi Pharm J. (2023) 31:101756. doi: 10.1016/j.jsps.2023.101756
273. Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel). (2017) 9:6906–13. doi: 10.3390/cancers9110157
274. Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. (2007) 6:1239–48. doi: 10.1158/1535-7163.MCT-06-0633
275. Cerqueira NMFSA, Fernandes PA, Ramos MJ. Understanding ribonucleotide reductase inactivation by gemcitabine. Chem - A Eur J. (2007) 13:8507–15. doi: 10.1002/chem.200700260
276. Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al. Cellular elimination of 2’,2’-difluorodeoxycytidine 5’-triphosphate: a mechanism of self-potentiation. Cancer Res. (1992) 52:533–9.
277. Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. (2014) 13:275–84. doi: 10.1158/1535-7163.MCT-13-0791
278. Foland TB, Dentler WL, Suprenant KA, Gupta ML Jr., Himes RH. Paclitaxel-induced microtubule stabilization causes mitotic block and apoptotic-like cell death in a paclitaxel-sensitive strain of Saccharomyces cerevisiae. Yeast. (2005) 22:971–8. doi: 10.1002/yea.1284
279. Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. (2000) 88:2619–28. doi: 10.1002/(ISSN)1097-0142
280. Ganguly A, Yang H, Cabral F. Paclitaxel-dependent cell lines reveal a novel drug activity. Mol Cancer Ther. (2010) 9:2914–23. doi: 10.1158/1535-7163.MCT-10-0552
281. Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and its evolving role in the management of ovarian cancer. BioMed Res Int. (2015) 2015:413076. doi: 10.1155/2015/413076
282. Azwar S, Seow HF, Abdullah M, Faisal Jabar M, Mohtarrudin N. Recent updates on mechanisms of resistance to 5-fluorouracil and reversal strategies in colon cancer treatment. Biol (Basel). (2021) 10:971–8. doi: 10.3390/biology10090854
283. Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules. (2008) 13:1551–69. doi: 10.3390/molecules13081551
284. Ito SS, Nakagawa Y, Matsubayashi M, Sakaguchi YM, Kobashigawa S, Matsui TK, et al. Inhibition of the ATR kinase enhances 5-FU sensitivity independently of nonhomologous end-joining and homologous recombination repair pathways. J Biol Chem. (2020) 295:12946–61. doi: 10.1074/jbc.RA120.013726
285. Saif MW, Katirtzoglou NA, Syrigos KN. Capecitabine: an overview of the side effects and their management. Anticancer Drugs. (2008) 19:447–64. doi: 10.1097/CAD.0b013e3282f945aa
286. Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PloS One. (2015) 10:e0115686. doi: 10.1371/journal.pone.0115686
287. Siddiqui A, Gollavilli PN, Schwab A, Vazakidou ME, Ersan PG, Ramakrishnan M, et al. Thymidylate synthase maintains the de-differentiated state of triple negative breast cancers. Cell Death Differ. (2019) 26:2223–36. doi: 10.1038/s41418-019-0289-6
288. Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. (2011) 18:18–25. doi: 10.3747/co.v18i1.708
289. Nasrallah NA, Wiese BM, Sears CR. Xeroderma pigmentosum complementation group C (XPC): emerging roles in non-dermatologic Malignancies. Front Oncol. (2022) 12:846965. doi: 10.3389/fonc.2022.846965
290. Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. (2013) 5. doi: 10.1101/cshperspect.a012716
291. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. (2012) 16:15–31. doi: 10.1517/14728222.2011.648617
292. Svedberg A, Vikingsson S, Vikström A, Hornstra N, Kentson M, Branden E, et al. Erlotinib treatment induces cytochrome P450 3A activity in non-small cell lung cancer patients. Br J Clin Pharmacol. (2019) 85:1704–9. doi: 10.1111/bcp.13953
293. Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. (2018) 17:53. doi: 10.1186/s12943-018-0793-1
294. Balsano R, Zanuso V, Pirozzi A, Rimassa L, Bozzarelli S. Pancreatic ductal adenocarcinoma and immune checkpoint inhibitors: the gray curtain of immunotherapy and spikes of lights. Curr Oncol. (2023) 30:3871–85. doi: 10.3390/curroncol30040293
295. Ai L, Chen J, Yan H, He Q, Luo P, Xu Z, et al. Research status and outlook of PD-1/PD-L1 inhibitors for cancer therapy. Drug Des Devel Ther. (2020) 14:3625–49. doi: 10.2147/DDDT.S267433
296. Huang Q, Zheng Y, Gao Z, Yuan L, Sun Y, Chen H. Comparative efficacy and safety of PD-1/PD-L1 inhibitors for patients with solid tumors: A systematic review and bayesian network meta-analysis. J Cancer. (2021) 12:1133–43. doi: 10.7150/jca.49325
297. Chikuma S. CTLA-4, an essential immune-checkpoint for T-cell activation. Curr Top Microbiol Immunol. (2017) 410:99–126. doi: 10.1007/82_2017_61
298. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
299. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
300. Maleki Vareki S, Garrigós C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncology/Hematology. (2017) 116:116–24. doi: 10.1016/j.critrevonc.2017.06.001
301. Kim S, Kim BJ, Kim I, Kim JH, Kim HK, Ryu H, et al. A phase II study of chemotherapy in combination with telomerase peptide vaccine (GV1001) as second-line treatment in patients with metastatic colorectal cancer. J Cancer. (2022) 13:1363–9. doi: 10.7150/jca.70385
302. Mizukoshi E, Kaneko S. Telomerase-targeted cancer immunotherapy. Int J Mol Sci. (2019) 20:99–126. doi: 10.3390/ijms20081823
303. Jo JH, Kim YT, Choi HS, Kim HG, Lee HS, Choi YW, et al. Efficacy of GV1001 with gemcitabine/capecitabine in previously untreated patients with advanced pancreatic ductal adenocarcinoma having high serum eotaxin levels (KG4/2015): an open-label, randomised, Phase 3 trial. Br J Cancer. (2024) 130:43–52. doi: 10.1038/s41416-023-02474-w
304. Park YH, Jung AR, Kim GE, Kim MY, Sung JW, Shin D, et al. GV1001 inhibits cell viability and induces apoptosis in castration-resistant prostate cancer cells through the AKT/NF-κB/VEGF pathway. J Cancer. (2019) 10:6269–77. doi: 10.7150/jca.34859
305. Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. (2012) 1:670–86. doi: 10.4161/onci.20426
306. McCormick KA, Coveler AL, Rossi GR, Vahanian NN, Link C, Chiorean EG. Pancreatic cancer: Update on immunotherapies and algenpantucel-L. Hum Vaccin Immunother. (2016) 12:563–75. doi: 10.1080/21645515.2015.1093264
307. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. (2017) 8:90521–31. doi: 10.18632/oncotarget.v8i52
308. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
309. Zhai X, Mao L, Wu M, Liu J, Yu S. Challenges of anti-mesothelin CAR-T-cell therapy. Cancers (Basel). (2023) 15:6269–77. doi: 10.3390/cancers15051357
310. Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. (2020) 6:eaaz3223. doi: 10.1126/sciadv.aaz3223
311. Ngoenkam J, Schamel WW, Pongcharoen S. Selected signalling proteins recruited to the T-cell receptor-CD3 complex. Immunology. (2018) 153:42–50. doi: 10.1111/imm.12809
312. Philipson BI, O’Connor RS, May MJ, June CH, Albelda SM, Milone MC. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci Signal. (2020) 13:90521–31. doi: 10.1126/scisignal.aay8248
313. Hay ZLZ, Slansky JE. Granzymes: the molecular executors of immune-mediated cytotoxicity. Int J Mol Sci. (2022) 23:413–20. doi: 10.3390/ijms23031833
314. Al Absi A, Wurzer H, Guerin C, Hoffmann C, Moreau F, Mao X, et al. Actin cytoskeleton remodeling drives breast cancer cell escape from natural killer-mediated cytotoxicity. Cancer Res. (2018) 78:5631–43. doi: 10.1158/0008-5472.CAN-18-0441
315. Wagner K, Schulz P, Scholz A, Wiedenmann B, Menrad A. The targeted immunocytokine L19-IL2 efficiently inhibits the growth of orthotopic pancreatic cancer. Clin Cancer Res. (2008) 14:4951–60. doi: 10.1158/1078-0432.CCR-08-0157
316. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
317. Wang KS, Frank DA, Ritz J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood. (2000) 95:3183–90. doi: 10.1182/blood.V95.10.3183
318. Hu X, li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduction Targeted Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
319. Dickow J, Francois S, Kaiserling R-L, Malyshkina A, Drexler I, Westendorf AM, et al. Diverse immunomodulatory effects of individual IFNα Subtypes on virus-specific CD8+ T cell responses. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.02255
320. Zanin N, Lesegno CVd, Lamaze C, Blouin CM. Interferon receptor trafficking and signaling: journey to the cross roads. Front Immunol. (2020) 11:615603. doi: 10.3389/fimmu.2020.615603
321. Rosewicz S, Detjen K, Scholz A, von Marschall Z. Interferon-alpha: regulatory effects on cell cycle and angiogenesis. Neuroendocrinology. (2004) 80(Suppl 1):85–93. doi: 10.1159/000080748
322. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
323. Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. (2008) 8:1581–8. doi: 10.1586/14737140.8.10.1581
324. Wold WS, Toth K. Adenovirus vectors for gene therapy. vaccination Cancer Gene therapy Curr Gene Ther. (2013) 13:421–33. doi: 10.2174/1566523213666131125095046
325. Even-Desrumeaux K, Baty D, Chames P. State of the art in tumor antigen and biomarker discovery. Cancers (Basel). (2011) 3:2554–96. doi: 10.3390/cancers3022554
326. Alrhmoun S, Sennikov S. The role of tumor-associated antigen HER2/neu in tumor development and the different approaches for using it in treatment: many choices and future directions. Cancers (Basel). (2022) 14:85–93. doi: 10.3390/cancers14246173
327. Aldrak N, Alsaab S, Algethami A, Bhere D, Wakimoto H, Shah K, et al. Oncolytic herpes simplex virus-based therapies for cancer. Cells. (2021) 10:642–62. doi: 10.3390/cells10061541
328. Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol. (2022) 29:3044–60. doi: 10.3390/curroncol29050247
329. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. (2020) 10:727–42.
330. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther Oncolytics. (2019) 15:234–47. doi: 10.1016/j.omto.2019.10.007
331. Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. (2013) 62:339–47. doi: 10.1136/gutjnl-2012-303108
332. Lennon AM, Wolfgang CL, Canto MI, Klein AP, Herman JM, Goggins M, et al. The early detection of pancreatic cancer: what will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. (2014) 74:3381–9. doi: 10.1158/0008-5472.CAN-14-0734
333. Sutton JM, Abbott DE. Neoadjuvant therapy for pancreas cancer: past lessons and future therapies. World J Gastroenterol. (2014) 20:15564–79. doi: 10.3748/wjg.v20.i42.15564
334. Schimizzi GV, Jin LX, Davidson J, Krasnick BA, Ethun CG, Pawlik TM, et al. Outcomes after vascular resection during curative-intent resection for hilar cholangiocarcinoma: a multi-institution study from the US extrahepatic biliary Malignancy consortium. HPB (Oxford). (2018) 20:332–9. doi: 10.1016/j.hpb.2017.10.003
335. Wu X, Chau YF, Bai H, Zhuang X, Wang J, Duan J. Progress on neoadjuvant immunotherapy in resectable non-small cell lung cancer and potential biomarkers. Front Oncol. (2022) 12:1099304. doi: 10.3389/fonc.2022.1099304
336. Gajiwala S, Torgeson A, Garrido-Laguna I, Kinsey C, Lloyd S. Combination immunotherapy and radiation therapy strategies for pancreatic cancer-targeting multiple steps in the cancer immunity cycle. J Gastrointest Oncol. (2018) 9:1014–26. doi: 10.21037/jgo
337. Bailey P, Zhou X, An J, Peccerella T, Hu K, Springfeld C, et al. Refining the treatment of pancreatic cancer from big data to improved individual survival. Funct (Oxf). (2023) 4:zqad011. doi: 10.1093/function/zqad011
338. O’Kane GM, Lowery MA. Moving the needle on precision medicine in pancreatic cancer. J Clin Oncol. (2022) 40:2693–705. doi: 10.1200/JCO.21.02514
Keywords: pancreatic adenocarcinoma, therapeutic advancements, tumor microenvironment, neoadjuvant therapies, immunotherapy, clinical manifestations
Citation: Mustafa M, Abbas K, Alam M, Habib S, Zulfareen, Hasan GM, Islam S, Shamsi A and Hassan I (2024) Investigating underlying molecular mechanisms, signaling pathways, emerging therapeutic approaches in pancreatic cancer. Front. Oncol. 14:1427802. doi: 10.3389/fonc.2024.1427802
Received: 04 May 2024; Accepted: 01 July 2024;
Published: 17 July 2024.
Edited by:
Nemat Ali, King Saud University, Saudi ArabiaReviewed by:
Mohd Ishtikhar, Texas A&M University College Station, United StatesCopyright © 2024 Mustafa, Abbas, Alam, Habib, Zulfareen, Hasan, Islam, Shamsi and Hassan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Imtaiyaz Hassan, bWloYXNzYW5Aam1pLmFjLmlu; Anas Shamsi, YW5hcy5zaGFtc2kxOEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.