 Ali Ahmadizad Firouzjaei
Ali Ahmadizad Firouzjaei Samira Mohammadi-Yeganeh
Samira Mohammadi-Yeganeh- Shahid Beheshti University of Medical Sciences, Tehran, Iran
The complex interplay between ferroptosis and efferocytosis in cancer has attracted significant interest recently. Efferocytosis, the process of eliminating apoptotic cells, is essential for preserving tissue homeostasis and reducing inflammation. However, dysregulation of efferocytosis can have profound effects on cancer. Apoptotic cells accumulate because of impaired efferocytosis, which triggers chronic inflammation and the release of pro-inflammatory chemicals. Surprisingly, accumulating evidence suggests that dysregulation of ferroptosis- a form of controlled cell death characterized by lipid peroxidation and the buildup iron-dependent reactive oxygen species (ROS)-can influence efferocytic activities within the tumor microenvironment. Dysfunctional iron metabolism and increased lipid peroxidation, are associated with ferroptosis, resulting in inadequate apoptotic cell clearance. Conversely, apoptotic cells can activate ferroptotic pathways, increasing oxidative stress and inducing cell death in cancer cells. This reciprocal interaction emphasizes the complex relationship between efferocytosis and ferroptosis in cancer biology. Understanding and managing the delicate balance between cell clearance and cell death pathways holds significant therapeutic potential in cancer treatment. Targeting the efferocytosis and ferroptosis pathways may offer new opportunities for improving tumor clearance, reducing inflammation, and sensitizing cancer cells to therapeutic interventions. Further research into the interaction between efferocytosis and ferroptosis in cancer will provide valuable insights for the development of novel therapies aimed at restoring tissue homeostasis and improving patient outcomes.
Higlights
● The dysregulation of efferocytosis in cancer leads to the accumulation of apoptotic cells, resulting in chronic inflammation and the release of pro-inflammatory chemicals.
● Enhanced lipid peroxidation linked to ferroptosis and dysfunctional iron metabolism can impede effective efferocytosis, resulting in inadequate clearance of apoptotic cells.
● Ferroptosis pathways can be triggered by the presence of apoptotic cells, which increase oxidative stress and kill cancer cells.
● The reciprocal interaction between efferocytosis and ferroptosis highlights the complex interplay of these processes in cancer biology.
● Significant therapeutic potential exists for the treatment of cancer if the delicate balance between cell death and clearance pathways is understood and managed.
1 Introduction
In recent years, the intricate relationship between cell clearance and cell death has emerged as a fascinating area of research in cancer biology. Two distinct cellular processes, namely efferocytosis and ferroptosis, have gained considerable attention due to their pivotal roles in maintaining tissue homeostasis and influencing tumor progression. Efferocytosis refers to the clearance of apoptotic cells by phagocytes, while ferroptosis is a regulated form of cell death characterized by lipid peroxidation dependent on iron (1, 2).
Cancer development and progression are often accompanied by an imbalance in these processes, leading to disrupted cell clearance and altered cell death mechanisms (3–5). It is now evident that the interplay between efferocytosis and ferroptosis plays a critical role in shaping the tumor microenvironment and influencing cancer outcomes. Understanding the intricate crosstalk between these two processes is thus essential for uncovering novel therapeutic strategies and improving patient outcomes (6–9).
This article aims to provide a comprehensive overview of the interplay between efferocytosis and ferroptosis in cancer, highlighting the delicate balance between cell clearance and cell death. We will explore the underlying molecular mechanisms governing these processes and discuss their implications in cancer initiation, progression, and therapy resistance. Additionally, we will examine emerging therapeutic approaches targeting efferocytosis and ferroptosis as potential strategies for cancer treatment.
By elucidating the complex relationship between efferocytosis and ferroptosis, this article will contribute to a deeper understanding of the molecular mechanisms underlying cancer biology. Furthermore, it will shed light on novel therapeutic opportunities for modulating cell clearance and cell death pathways, potentially opening new avenues for personalized cancer therapies.
2 Phagocyte recognition, engulfment, and degradation of apoptotic cells
Daily, approximately 0.4% of the estimated 37.2 trillion cells in the adult human body undergo programmed cell death (10). However, even in tissues characterized by high rate of cellular turnover, apoptotic cells (ACs) are remarkably rare, highlighting the extraordinary efficiency and vast capacity of AC clearance. This process, known as “efferocytosis”, is crucial for maintaining tissue homeostasis under normal physiological conditions and for restoring equilibrium after disease (11, 12). During the initial stage of efferocytosis, phagocytes recognize apoptotic cells by detecting “find-me” signals released as cells undergo their final stages of death. These signals consist of a wide range of lipids, proteins, peptides, and complex structures (13, 14). Additionally, apoptotic cells emit “eat-me” signals that appear on their surface, notifying macrophages that they should be consumed and eliminated. This signaling mechanism directs phagocytic cells, such as macrophages, to engulf and remove the dying cell. “Eat-me” signals include a wide range of chemicals such as nucleotides, phosphatidylserine (PS), and calreticulin (CRT) (15–17). Upon recognition of the dying cell, the phagocyte extends pseudopods to surround and engulf it. This process involves intricate cytoskeletal rearrangements driven by actin polymerization, leading to the formation of phagocytic cups that progressively enclose the dead cell. The engulfment is mediated by various phagocyte receptors, including integrins, scavenger receptors, and complement receptors, which interact with ligands and opsonins on the surface of the dying cell (18–20). After capturing the dying cell, the phagocyte orchestrates a well-coordinated process to ensure its degradation. This begins with the fusion of the phagosome, which contains the dying cell, with lysosomes. Lysosomes are rich in enzymes such as lipases, proteases, and nucleases. As a result of this fusion, the contents of the phagosome are subjected to digestion (11, 21, 22). Following the impairment of efferocytosis, a cascade of events can lead to inflammation. Inflammation occurs when neutrophils, having migrated out of blood vessels to the injured or invaded site, undergo programmed cell death. These neutrophils, along with other inflammatory cells, are then engulfed and cleared by macrophages. This crucial process of phagocytosis plays a vital role in restoring tissue homeostasis as the inflammation gradually subsides (Figure 1) (23).
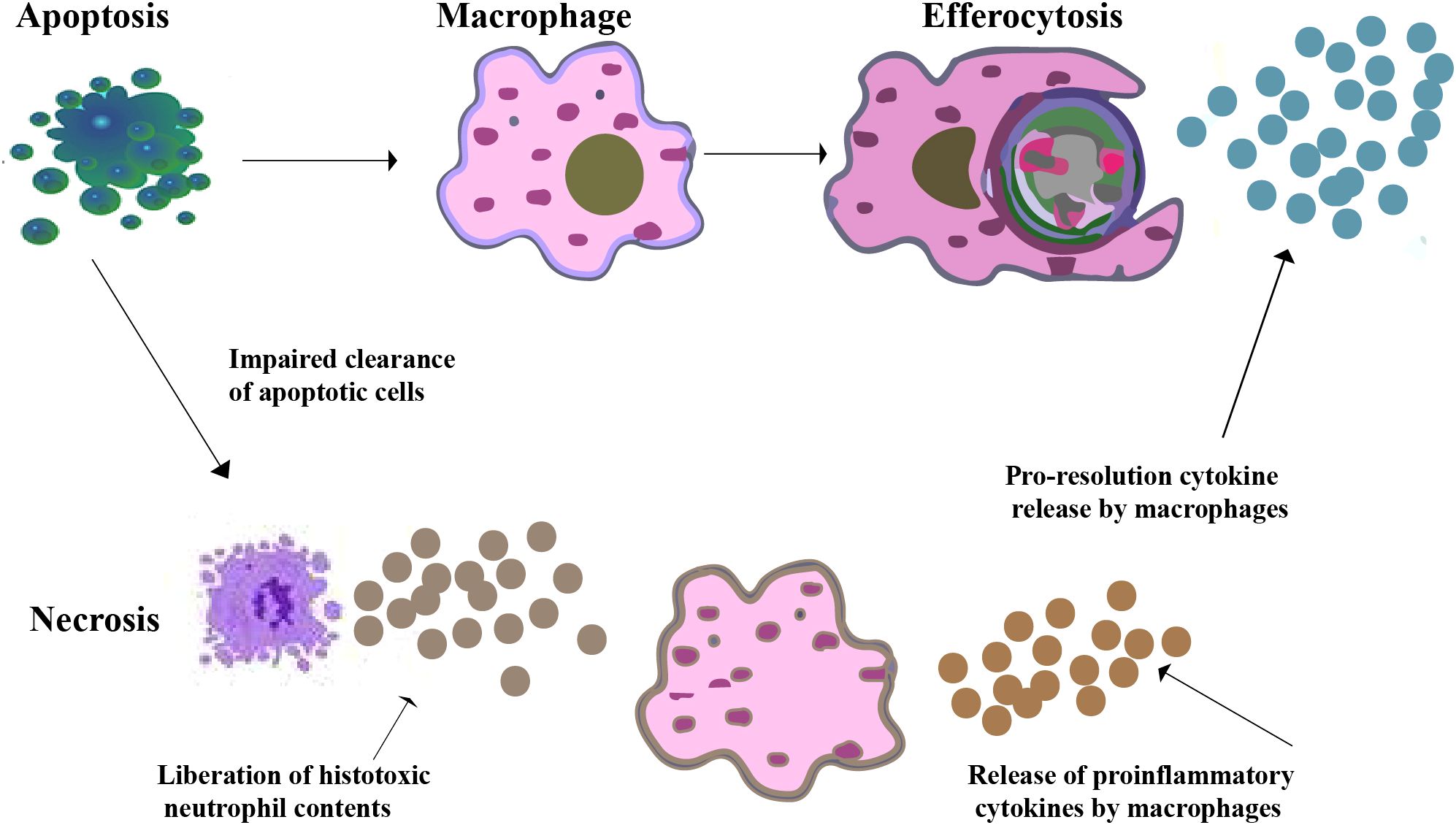
Figure 1. Efferocytosis by macrophages. A macrophage recognizes and phagocytoses an apoptotic neutrophil at an inflammatory site. Pro-resolution cytokines are secreted by macrophages while pro-inflammatory cytokines are downregulated as a result of their contact with apoptotic cells. Apoptotic neutrophils may undergo secondary necrosis if efferocytosis is impaired, which leading to the release of histotoxic substances, cause tissue damage, increased pro-inflammatory signals, and chronic inflammation.
3 Ferroptosis and its unique mechanisms
Ferroptosis is a specific type of regulated cell death caused by the accumulation of lipid peroxides on cell membranes. It differs from apoptosis and other forms of cell death in terms of both morphology and mechanism. Unlike apoptotic cells, ferroptotic cells do not exhibit classic apoptotic features such as condensed chromatin or the production of apoptotic bodies. Instead, ferroptotic cells are characterized by smaller mitochondria with reduced mitochondrial cristae (24, 25). This process requires a delicate balance between ferroptosis-promoting cellular activities and defense systems that prevent ferroptosis. When ferroptosis-promoting actions surpass the antioxidant-buffering capacities of these defense systems, ferroptosis occurs, leading to cell death (26–29). This distinguishes ferroptosis from other forms of controlled cell death, which generally entail the activation of specific executioner proteins. Examples of such pathways include gasdermin D-mediated pyroptosis, MLKL-mediated necroptosis, and caspase-mediated apoptosis. Ferroptosis, however, follows a distinct pathway, highlighting its unique role in regulated cell death (30). Ferroptotic cells display unique profiles of oxidized phospholipids (PL), which are distinct from those of cells undergoing other types of cell death. This unique composition of oxidized phospholipid further sets ferroptosis apart as a distinct mode of cell demise (31, 32). The mechanistic pathways underlying ferroptotic cell death are illustrated in Figure 2, which depicts the key molecular events and signaling cascades involved in the induction and execution of ferroptosis.
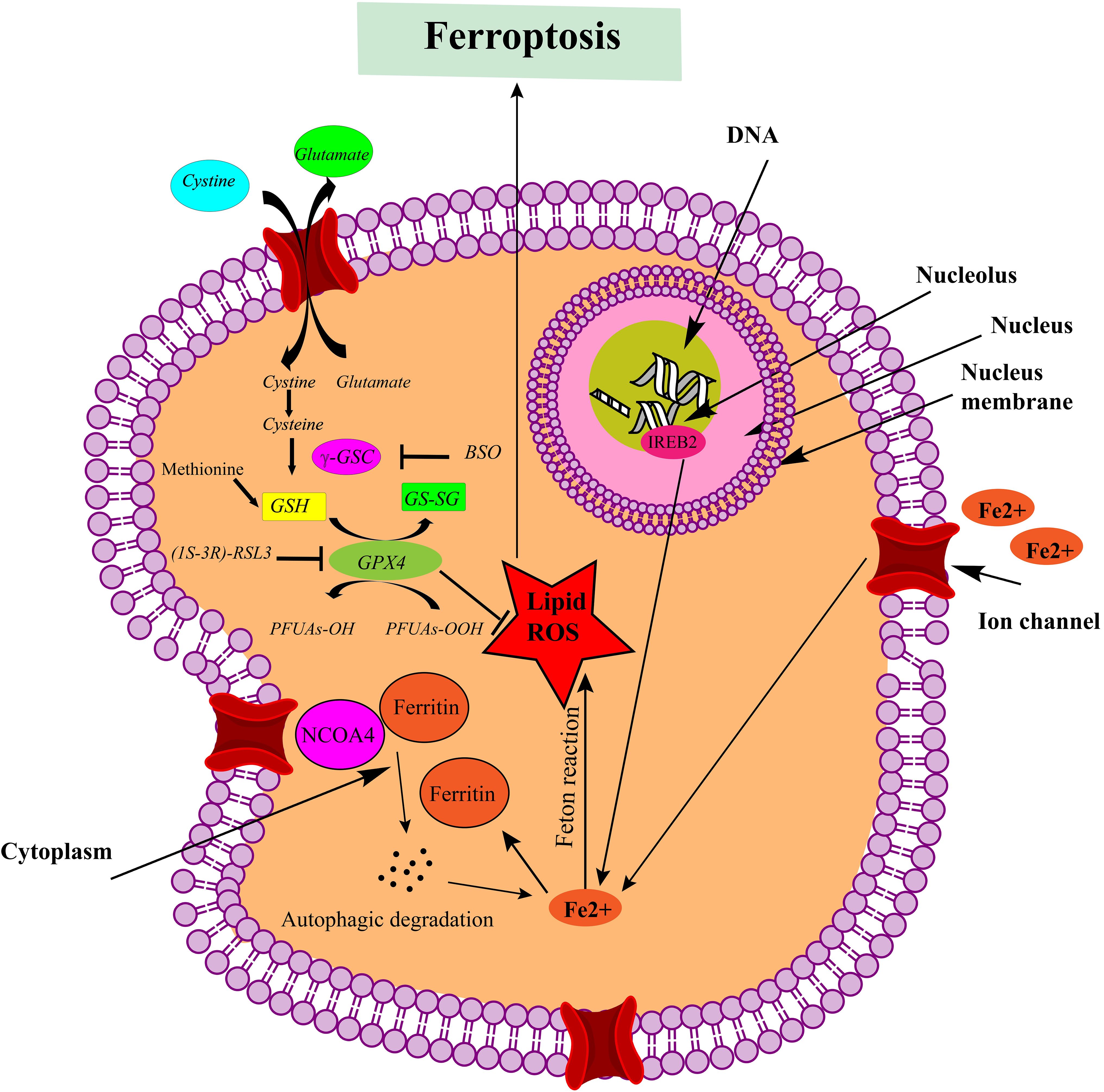
Figure 2. Mechanistic pathways leading to ferroptotic cell death. Ferroptosis is triggered by the inhibition of system Xc- or GPX4 activity, leading to cell death. Lipid ROS are implicated in the ferroptotic process. Notably, peroxidation of polyunsaturated fatty acids (PUFAs) is a significant factor. Excessive iron acts as a fundamental catalyst in the execution of ferroptosis (This figure was created using ChemDraw version 22.2.0.3300, PerkinElmer Informatics.).
4 Efferocytosis and ferroptosis in cancer
Efferocytosis and ferroptosis are two separate cellular mechanisms that have garnered significant interest in cancer biology. Although they represent separate processes, there is growing evidence that they play interrelated role in cancer development and progression. Efferocytosis is an important step for maintaining tissue homeostasis and reducing inflammation. In cancer, the deregulation of efferocytosis can have serious consequences. Impaired efferocytosis results in the buildup of apoptotic cells, resulting in persistent inflammation and the release of pro-inflammatory chemicals (33–35). This inflammatory milieu can promote tumor growth, angiogenesis, and metastasis (36–38). Interestingly, recent research highlights the impact of ferroptosis on efferocytic processes in cancer. Dysfunctional iron metabolism and increased lipid peroxidation associated with ferroptosis can hinder efficient efferocytosis, resulting in poor apoptotic cell clearance (39). Impaired clearance can exacerbate inflammation, further contributing to tumor growth. Conversely, apoptotic cells can activate ferroptotic pathways in cancer cells. The release of components from apoptotic cells, such as oxidized lipids and ROS, can induce oxidative stress and ferroptosis in neighboring cancer cells (40–43). The reciprocal interaction between efferocytosis and ferroptosis underscores their interconnected roles within the tumor microenvironment.
5 Efferocytosis and ferroptosis in tumor biology
Tumor biology encompasses the study of tumors or neoplasms, which are abnormal growths of cells that can occur in various tissues of the body. Understanding tumor biology is crucial for comprehending the mechanisms underlying cancer development, progression, and potential treatment strategies. Efferocytosis and ferroptosis are two important processes in tumor biology that significantly influence tumor development, progression, and response to therapy. Here’s an overview of their roles in tumor biology:
5.1 Efferocytosis in tumor biology
The removal of apoptotic cells by phagocytic cells plays a vital role in tumor biology. This process is essential for preserving tissue homeostasis and regulating the immune response within the tumor microenvironment. Below are some key implications of apoptotic cell clearance in tumor biology:
5.1.1 The impact of efferocytosis on inflammation in the tumor microenvironment
Efficient clearance of apoptotic cells prevents the release of pro-inflammatory molecules and danger signals from dying cells. Failure to clear apoptotic cells can lead to the accumulation of cellular debris, triggering chronic inflammation and activating immune responses. In the context of tumors, persistent inflammation can contribute to tumor progression, angiogenesis, and immune evasion. Proper efferocytosis helps create an immunosuppressive microenvironment and reduces inflammatory responses, thereby influencing tumor growth and metastasis. Additionally, efferocytosis of cancer cells by antigen-presenting cells, such as dendritic cells (DCs), can activate immune responses against tumor antigens. This leads to the priming and activation of tumor-specific T cells, enhancing anti-tumor immunity. Effective efferocytosis promotes the efficient removal of apoptotic cells within the tumor microenvironment, resulting in the polarization of M2-like macrophages, the secretion of wound-healing cytokines (e.g., IL-10, IL-13, TGF-β), and the recruitment of FOXP3+ regulatory T cells. Consequently, a tolerogenic and immunosuppressive tumor microenvironment is established. On the other hand, when efferocytosis is impaired, secondary necrosis occurs, resulting in the release of pro-inflammatory damage-associated molecular patterns (DAMPs). These DAMPs promote the polarization of M1-like macrophages, stimulate the production of pro-inflammatory cytokines (such as TNF, IFN and IL-12), and attract cytotoxic cells, including CD8+ T cells and natural killer cells, which contribute to anti-tumor responses (Figure 3) (22, 44–48).
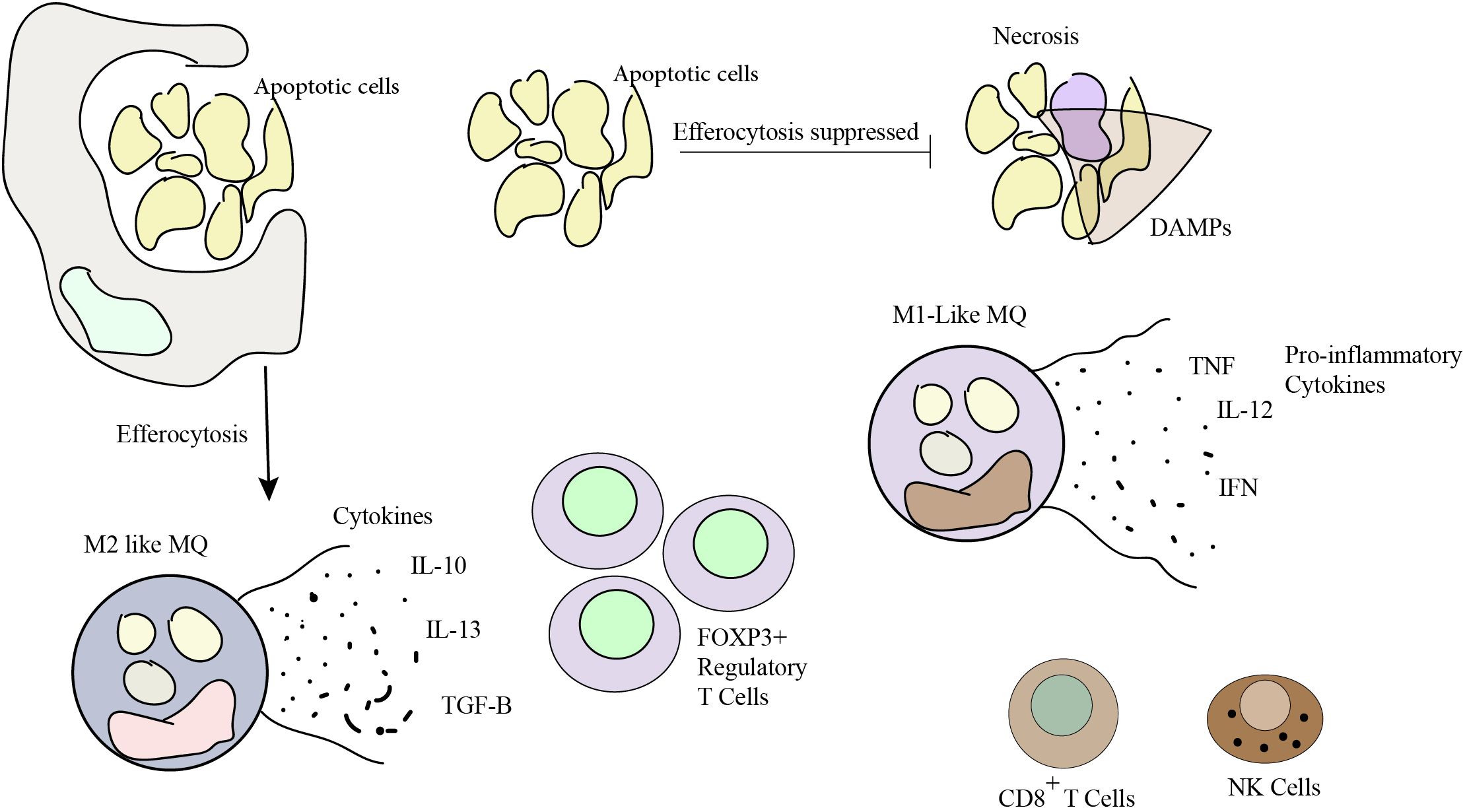
Figure 3. Efferocytosis plays a pivotal role in shaping a pro-tolerogenic tumor microenvironment. A balance in efferocytosis is crucial for sculpting the tumor microenvironment and optimizing immune defenses against cancer. Effective control of efferocytosis holds potential for developing cancer treatments. Efferocytosis that is effective boosts the body’s capacity to fight cancer, while defective efferocytosis causes inflammation and impairs the immune response.
5.1.2 The impact of efferocytosis on tumor antigen release and immune checkpoint expression
Efferocytosis plays a crucial role in recognizing and removing apoptotic tumor cells. Efficient clearance of these dying cells is essential to prevent the release of immunogenic tumor antigens, such as human epidermal growth factor receptor 2 (HER2) and prostate-specific antigen (PSA). These antigens have the potential to trigger an immune response against the tumor (49–52). However, when efferocytosis is impaired, apoptotic tumor cells may not be properly cleared, leading to antigen release and promoting immune activation against the tumor. This defective clearance can have an impact on tumor immune surveillance and influence tumor progression. Moreover, the interaction between phagocytic cells and apoptotic cells also affects immune checkpoint molecules and immune tolerance. When phagocytes engulf apoptotic cells, it can induce the expression of inhibitory immune checkpoint molecules such as PD-L1, LILRB1, TIM3, B7-H3, CD137, and CD47 (53, 54). Increased expression of these immune checkpoints can promote immune tolerance and dampen anti-tumor immune responses (55–57). Thus, the modulation of immune checkpoints through efferocytosis can influence the balance between immune activation and tolerance in the tumor microenvironment.
5.1.3 Metastasis and dormant tumor cells
Efferocytosis of apoptotic tumor cells may impact the metastatic potential of tumors. Efficient clearance of apoptotic cells prevents the accumulation of cellular debris that could stimulate inflammation and promote the recruitment of immune cells involved in metastasis (4, 58, 59). Furthermore, efferocytosis may contribute to the clearance of dormant tumor cells, which can evade immune surveillance and remain latent in distant organs. Impaired efferocytosis can lead to the survival and growth of dormant tumor cells, facilitating metastatic spread. A key factor in the relationship between efferocytosis and metastasis is the origin of macrophages, which influences their localization and function in metastatic environments. Monocyte-derived macrophages (MoMs) and tissue-resident Kupffer cells (KCs) exhibit distinct roles during metastatic progression; while MoMs primarily localize in tumor lesions, KCs are predominantly found at the tumor margins. Studies have revealed that tumor-associated macrophages (TAMs) can exhibit both immunostimulatory and immunosuppressive states within the same tumor (60). Additionally, MoMs accumulate in necrotic regions, where they phagocytose dead resident cells. Inhibiting the phosphatidylserine receptor Mer tyrosine kinase (MerTK) can reverse the immunosuppressive state induced by efferocytosis, as blocking MerTK-mediated phagocytosis of dying cancer cells has been shown to suppress tumor growth in various cancers. In metastatic cancer (61, 62), macrophage accumulation is often observed, accompanied by an immunosuppressive phenotype, that promotes tumor growth and limits the effectiveness of immunotherapy (63, 64). Evidence suggests that both immunostimulatory and immunosuppressive phenotypes of MoMs are present in early and advanced metastatic lesions, with a shift toward immunosuppression occurring at both stages (65). Research by Astuti Y and colleagues indicated that MoMs are constantly attracted to metastatic sites, where they quickly lose their ability to stimulate the immune system. The physiological function of efferocytosis is to shield tissues from immune reactions, which leads to localized immunosuppression. These pathways are used in metastatic cancer to create an immunosuppressive microenvironment, which helps dispersed cancer cells evade immune detection (65).
It has been shown that tissue-resident macrophages are generally more phagocytic than bone marrow-derived macrophages (66). Inhibiting the MerTK can reverse the immunosuppressive state induced by efferocytosis and has been shown to suppress tumor growth in various cancers (46, 67, 68). Notably, MerTK blockade was associated with the accumulation of dead cell debris, activating the STING pathway in macrophages (46).
In summary, Interfering with macrophage efferocytosis or its downstream signaling can inhibit immunosuppressive functions and restore anti-tumor immunity. Targeting macrophage efferocytosis may thus represent a promising therapeutic strategy for patients with metastatic cancer.
5.2 Ferroptosis in tumor biology
Ferroptosis plays an important role in tumor biology, encompassing various aspects that contribute to tumor development, progression, and response to therapy. Below are some key implications of ferroptosis in tumor biology:
5.2.1 Tumor suppression
Ferroptosis acts as a tumor-suppressor mechanism by eliminating cancer cells. Unlike normal cells, many cancer cells exhibit altered iron metabolism and increased sensitivity to ferroptosis. Inducing ferroptosis in cancer cells can suppress tumor growth and inhibit their ability to evade cell death (69). By targeting specific vulnerabilities in cancer cells, such as altered lipid metabolism or weakened antioxidant defenses, ferroptosis-inducing agents show promise as potential therapeutic strategies. One of the key factors in the effectiveness of ferroptosis-based therapeutics is the identification of specific vulnerabilities inherent to cancer cells. For instance, many cancer cells have altered lipid metabolism, leading to an accumulation of polyunsaturated fatty acids (PUFAs) that are prone to oxidative damage. Additionally, these cells often have compromised antioxidant systems, which further predisposes them to ferroptosis. Therefore, ferroptosis-inducing agents that exploit these vulnerabilities could offer novel treatment options for cancer patients. Recent studies have also demonstrated that ferroptosis can be triggered by radiotherapy, one of the most widely used cancer treatments (70). Research suggests that ionizing radiation (IR) induces lipid peroxidation—one of the defining features of ferroptosis—through at least two parallel pathways. First, IR generates reactive oxygen species (ROS), which promote lipid peroxidation. This increase in ROS levels can overwhelm the cellular antioxidant defenses, pushing it toward ferroptosis. Second, IR has been shown to upregulate the expression of acyl-CoA synthetase long-chain family member 4 (ACSL4), a key enzyme involved in the biosynthesis of phospholipids enriched with polyunsaturated fatty acids (PUFA-PLs). These specific lipids are particularly susceptible to peroxidation and thereby facilitating the induction of ferroptosis. By combining radiotherapy with ferroptosis-inducing agents, there is potential to enhance therapeutic outcomes by more effectively targeting and eliminating cancer cells (32, 71, 72). However, IR also triggers the expression of ferroptosis inhibitors such as solute carrier family 7 member 11 (SLC7A11) and glutathione peroxidase 4 (GPX4) as part of an adaptive response. The upregulation of SLC7A11, particularly in the context of IR or KEAP1 deficiency, enhances radioresistance by preventing ferroptosis. By inhibiting SLC7A11 or GPX4 with ferroptosis inducers (FINs), radioresistant cancer cells and xenograft tumors can be made more sensitive to IR (70). The tumor-suppressor role of p53 in ferroptosis has been well studied. Gu and colleagues discovered that p53 can enhance ferroptosis by inhibiting the transcription of system xc–subunit SLC7A11, contributing to p53’s tumor-suppressive properties both in vitro and in vivo. This was observed through a detailed examination of particular lysine acetylation sites on p53 (73, 74). Like p53, BAP1, an epigenetic regulator and tumor suppressor, can also induce ferroptosis by suppressing the expression of SLC7A11 (75). It is unclear, however, how much BAP1’s ferroptosis-promoting activity adds to its tumor-suppressive role, in contrast to p53, whose ferroptosis-promoting activity alone has been proposed to be sufficient to suppress carcinogenesis in vivo (74).
5.2.2 Regulation of tumor microenvironment
Ferroptosis can influence the tumor microenvironment, which encompasses cellular and non-cellular components surrounding the tumor. Tumor-associated fibroblasts and immune cells can undergo ferroptosis, leading to the release of DAMPs and pro-inflammatory signals. This can shape the immune response, promote antitumor immunity, and modulate tumor progression. Additionally, ferroptosis can affect the availability of nutrients, such as iron, and alter the redox balance within the tumor microenvironment (76). The involvement of ferroptosis in antitumor immunity presents a dual role dependent on the specific immune cell type. The promotion of antitumor immunity is facilitated by CD8+ T cells, which secrete interferon-γ (IFNγ), leading to the repression of SLC7A11 expression in cancer cells and subsequently inducing ferroptosis. Consequently, ferroptotic cancer cells emit immunostimulatory signals that enhance DC maturation and boost the phagocytic ability of macrophages, especially M1-like tumor-associated macrophages (TAMs), enabling efficient clearance of ferroptotic cancer cells. This synergy further strengthens CD8+ T cell-mediated tumor suppression. Furthermore, ferroptosis induction, achieved through the inhibition of GPX4 or N-acyl sphingosine amidohydrolase 2 (ASAH2), impairs various immunosuppressive cell types including regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and M2-like TAMs. This impairment results in an augmentation of antitumor immunity. However, it is important to note that ferroptosis also affects certain immune cell subsets such as CD8+ T cells and specific T helper (TH) cell subsets like T follicular helper (TFH) cells, compromising the contribution of ferroptosis to antitumor immunity in those cases (Figure 4) (29).
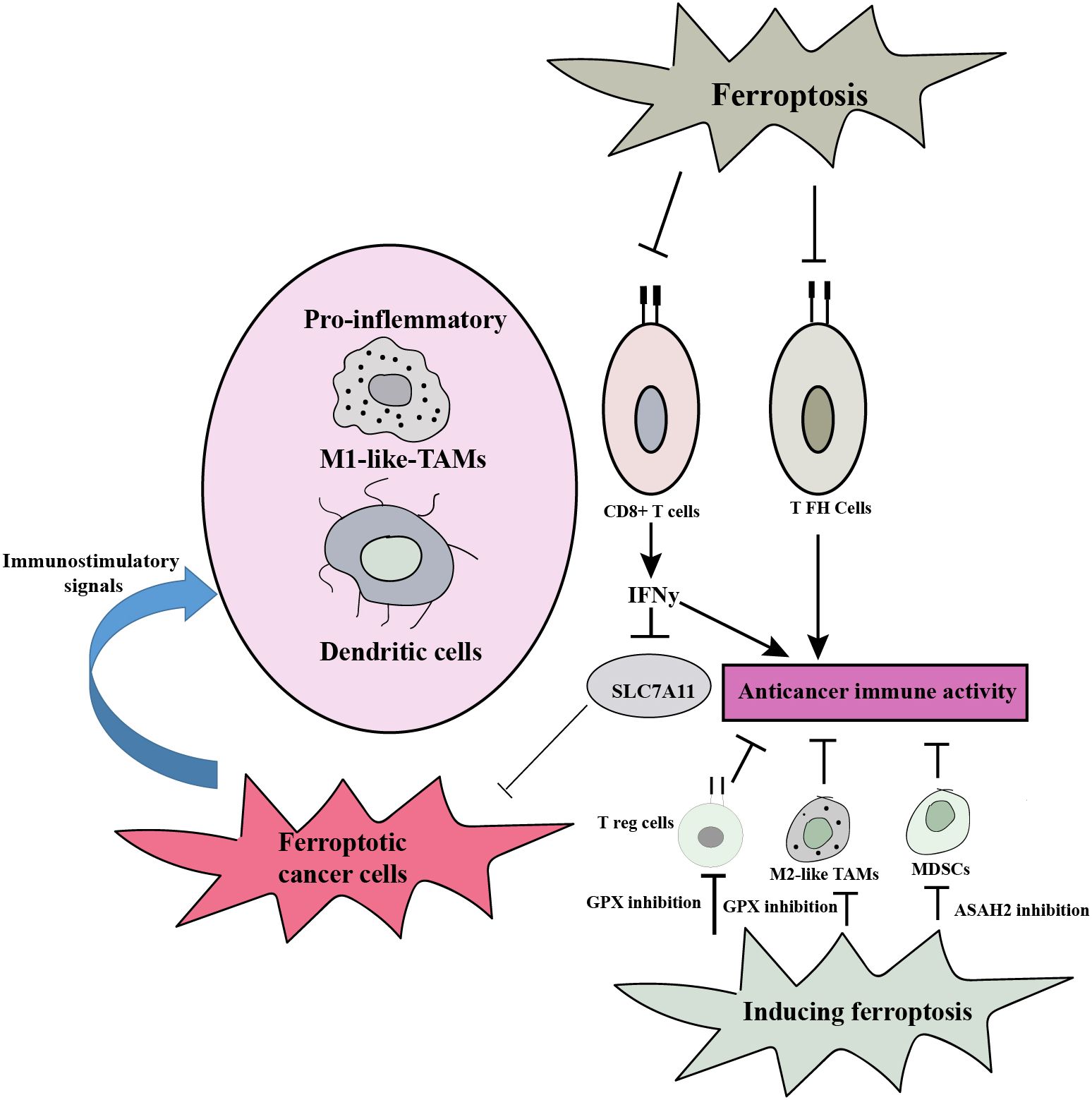
Figure 4. Ferroptosis plays a dual role in antitumor immunity, and the type of immune cell involved determines how it functions. By encouraging ferroptotic cell death in cancer cells and generating immunostimulatory signals that promote DC maturation and M1-like TAM phagocytosis, it can strengthen the immune system’s capacity to fight tumors. By causing cell death in specific immune cell subsets, it can also impair the immunological response. Comprehending these intricate relationships is essential for formulating focused treatment strategies that optimize ferroptosis’ ability to boost antitumor immunity while minimizing any possible negative consequences.
5.2.3 Sensitization of resistant tumor cells
Targeting ferroptosis may be a viable strategy to combat drug resistance and improve the therapeutic efficacy of anticancer treatment, according to an increasing body of clinical evidence. The development of resistance in cancer cells to treatments such as lapatinib, cisplatin, docetaxel, sorafenib, and other drugs can be reversed by ferroptosis inducers (77). Conventional chemotherapy or radiation therapy can cause cancer cells to die when xCT and GPX4 are inhibited (78). Through the consumption of glutathione (GSH) and the inhibition of cystine absorption, the inhibition of xCT increases the susceptibility of cancer cells to anticancer drugs (79, 80). It was discovered that via controlling lipid peroxidation, the therapy-resistant high-mesenchymal cell state aids in the escape from ferroptosis. Ferroptosis results from GPX4 inhibition, which triggers peroxide reactions driven by intracellular iron. Therefore, the high-mesenchymal cell state in cancer cells can be successfully eliminated by inducing ferroptosis (81). Some tumor cells develop resistance to conventional cancer therapies, including chemotherapy and targeted therapies. Ferroptosis represents a promising therapeutic pathway for drug-resistant tumor cells and has attracted attention recently. Interestingly, cancer cells that are resistant to apoptosis or other forms of cell death may still retain sensitivity to ferroptosis. This distinctive feature presents an opportunity to exploit ferroptosis as a therapeutic strategy for overcoming resistance to cancer treatment (82, 83). Combining ferroptosis-inducing agents with existing therapies has the potential to sensitize resistant tumor cells, thereby enhancing the overall efficacy of treatment regimens. By targeting the specific metabolic vulnerabilities of these cells, researchers aim to restore their sensitivity to cell death. For instance, the compound erastin has been identified as a potent inducer of ferroptosis. It functions by activating several critical molecular pathways, including system Xc−, which is involved in cystine uptake and glutamate export, VDAC (voltage-dependent anion channel), and p53, a well-known tumor suppressor protein. The activation of these pathways ultimately leads to effective cancer cell death, highlighting the potential of ferroptosis in future therapeutic approaches (84). Despite the promise of erastin, previous studies have shown that a variety of cancer cells exhibit insensitivity to erastin-induced ferroptosis. This insensitivity poses a challenge to the widespread application of ferroptosis in cancer therapy. To address this limitation, researchers have hypothesized that high-dose vitamin C could serve as a potential inducer of ferroptosis in cancer cells. Further experiments have demonstrated that vitamin C can indeed trigger ferroptosis by depleting GSH, and by promoting the generation of ROS. This finding opens new avenues for using vitamin C, not only as a supplement but also as a therapeutic agent in conjunction with ferroptosis-inducing strategies to enhance the treatment of resistant cancer (85).
5.2.4 Tumor dormancy and metastasis
Metastasis presents a significant challenge in cancer treatment, serving as a critical step in tumor progression. Many cancers, such as triple-negative breast cancer (86, 87), cervical cancer (88), and prostate cancer (89, 90), often metastasize, resulting in treatment-related relapses for numerous patients. The differences between primary tumors and their metastatic counterparts are substantial. According to Li et al., there is a lack of effective preventive and therapeutic strategies, greatly affecting patient survival rates (91). Recent research indicates that ferroptosis may play a role in cancer metastasis. This form of regulated cell death could also influence tumor dormancy, as dormant tumor cells enter a non-proliferative state that allows them to resist standard treatments (92). Inducing ferroptosis in dormant tumor cells may prevent their reactivation and subsequent metastatic outgrowth. Additionally, ferroptosis can influence the metastatic potential of tumor cells by modulating their susceptibility to oxidative stress and redox signaling pathways involved in metastasis (92). Several regulators of ferroptosis impact the metastatic capabilities of cancer cells. For instance, NF2, which encodes Neurofibromin 2 (NF2/Merlin), serves as a tumor suppressor that links external signals to intracellular communication, residing in the plasma membrane, cell cortex, and cytoskeleton (93). Another important regulator is linked to the epithelial-mesenchymal transition (EMT), a process by which epithelial cells lose their adhesive properties and acquire a fibroblast-like phenotype, enhancing their migratory and invasive potential. EMT not only facilitates metastasis but also contributes to treatment resistance. Key transcription factors such as ZEB1, SNAI1, and TWIST1 drive these processes and could serve as targets to combat drug resistance and metastasis (8). xCT is another ferroptosis regulator that significantly influences metastasis. Clinical studies have shown notable differences in recurrence rates between xCT-positive and xCT-negative tumors, alongside a correlation between xCT expression levels and metastatic behavior (94). Overexpression of xCT is often associated with poor prognosis in various cancers, including hepatocarcinoma (95, 96). Hypoxia-inducible factor (HIF) is yet another regulator of ferroptosis. In the hypoxic microenvironment characteristic of many tumors, HIF and related genes are activated, promoting cancer cell invasion and metastasis (97, 98). Additionally, noncoding RNAs, such as miR-9 and miR-137, have been shown to regulate ferroptosis during metastatic processes (99). In summary, ferroptosis influences cancer metastasis through various regulators, and understanding these pathways may provide new avenues for therapeutic intervention.
6 Exploring the relationship between efferocytosis and ferroptosis
The relationship between efferocytosis and ferroptosis is an intriguing area of investigation that is still being explored. While both processes involve distinct cellular mechanisms, some potential connections and interdependencies have been identified:
6.1 Lipid metabolism
Efferocytosis requires dynamic lipid remodeling to facilitate the recognition and engulfment of apoptotic cells (100). This process involves the redistribution of phosphatidylserine (PS) from the inner to the outer leaflet of the apoptotic cell membrane, which serves as an “eat-me” signal for phagocytes (14, 101). Interestingly, alterations in lipid metabolism can also influence the susceptibility of cells to ferroptosis. Certain lipids, such as PUFAs, are prone to peroxidation and can promote ferroptosis (72). Understanding the common lipid pathways and potential regulatory connections between efferocytosis and ferroptosis may provide insights into their relationship.
6.2 Oxidative stress
Ferroptosis is characterized by the accumulation of lipid peroxides, resulting from an imbalance between ROS generation and antioxidant defenses (102). Efferocytosis, particularly when impaired or dysregulated, can lead to the accumulation of apoptotic cells and secondary necrosis (103, 104). This can trigger the release of ROS and pro-inflammatory factors, contributing to oxidative stress and potentially influencing the susceptibility of cells to ferroptosis. Conversely, cellular antioxidant systems, which can be regulated by efferocytosis-related pathways, may impact the sensitivity of cells to both oxidative stress and ferroptosis. Lipid hydroperoxides and the depletion of antioxidants like GSH and GPX4 in the metabolism of amino acids are the key ways in which ROS causes ferroptosis. Since macrophages that have a critical role in efferocytosis are the hub of the metabolism of lipids and iron, their associated proteins are very active and involved in the production and elimination of ROS to keep the equilibrium (39).
Furthermore, ROS produced by macrophages initially influences their recruitment and polarization. Typically, the ROS generated within macrophages drive them to polarize towards the M1 phenotype, leading to an increased release of various inflammatory factors that exacerbate inflammation. This process creates conditions conducive to ferroptosis (105, 106). Findings indicate that ROS can have opposing effects on lipid metabolism during ferroptosis. Research revealed that 15-hydroperoxy-eicosa-tetra-enoyl-phosphatidylethanolamine, produced by 15-lipoxygenase (15-LOX), influences the resistance to ferroptosis in M1 macrophages (107). Further studies showed that ROS, such as superoxide (O2) and nitric oxide (NO•), compete for the same entry points and channels leading to the catalytic site of 15-LOX-2 (108). Additionally, because NO• is small enough to pass through cell membranes, it can inhibit phosphatidic acid (PA)-induced ferroptosis in distant epithelial cells (109).
6.3 Iron metabolism
Iron, as an essential nutrient, plays a vital role in numerous cellular processes, including DNA synthesis, energy production, and enzymatic reactions. However, excessive or inadequate levels of iron can be detrimental to cellular function and contribute to various pathological conditions. Maintaining the delicate equilibrium of iron homeostasis is therefore of utmost importance (110). Iron is essential for numerous cellular processes, including efferocytosis and ferroptosis. Efferocytosis requires iron for the proper functioning of phagocytes involved in engulfing apoptotic cells (111). Iron can affect the fate and function of effector macrophages in efferocytosis, particularly in relation to cell proliferation and differentiation. Macrophages frequently bind iron to ferritin (Ft). The expression of genes related to iron will change based on the macrophage polarization stage (112). Research indicates that iron overload can promote M1 macrophage polarization by elevating levels of M1 markers like IL-6, TNF-α, and IL-1β and lowering levels of M2 producers like TGM2 (113).
Dysregulated iron metabolism is also associated with ferroptosis, as iron can participate in the Fenton reaction, leading to ROS production and lipid peroxidation (114–116). Disruption in iron metabolism can disturb the delicate balance between efferocytosis and ferroptosis, resulting in pathological consequences (69, 111, 117). Understanding how iron levels influence efferocytosis efficiency and susceptibility to ferroptosis can aid in the development of new therapy targets and approaches for a wide range of diseases, including cancer, neurological problems, and cardiovascular maladies. Furthermore, it could lead to novel therapeutic approaches aimed at restoring cellular equilibrium and improving health.
6.4 Inflammatory signaling
Chronic inflammation is associated with various diseases, including cancer (118–120). Efferocytosis can influence the resolution of inflammation by promoting the clearance of apoptotic cells and the release of anti-inflammatory signals (121). Conversely, ferroptosis can lead to the release of pro-inflammatory molecules such as IL-6, iNOS, TNF-α, IL-1β, and COX-2) (122, 123). Interestingly, specific cytokines produced by macrophages can both promote and inhibit ferroptosis. For instance, IL-6, a marker associated with M1 macrophages, enhances lipid peroxidation and disrupts iron homeostasis in bronchial epithelial cells, thereby facilitating ferroptosis (124, 125). It does so by regulating hepcidin, a peptide hormone that affects iron absorption and mobilization in macrophages through the JAK-STAT3 pathway, while also being influenced by the BMP/SMAD signaling pathway (126, 127). TNF-α contributes to ferroptosis by upregulating ACSL3, an enzyme involved in acyl-CoA synthesis, which promotes lipid accumulation and fosters an inflammatory environment conducive to ferroptosis (128). IL-1β can increase the expression of ferroportin (FPN) via the p38-MAPK pathway, potentially leading to excessive iron efflux and accumulation in surrounding nerve cells (129). Furthermore, IL-1β released by macrophages has been shown to upregulate hepcidin transcription by improving the expression of CCAAT enhancer-binding protein (C/EBP) (130, 131) and hepcidin expression through phosphorylated c-Jun N-terminal kinase and its substrates, c-Jun and JunB, ultimately resulting in iron overload and FPN degradation (132). iNOS, although primarily known as a pro-inflammatory factor, has a complex role in ferroptosis. While its activation can produce reactive oxygen and nitrogen species that deplete glutathione and exacerbate lipid peroxidation, studies suggest that inhibiting iNOS may worsen conditions in certain contexts, such as beta-cell death, by promoting lipid peroxidation in the presence of inflammatory cytokines (133–135).
Efficient efferocytosis can limit the release of ROS and lipid peroxides from apoptotic cells, thereby reducing the induction of ferroptosis in neighboring cells. Additionally, apoptotic cells can influence the expression of key molecules involved in ferroptosis, including iron transporters and lipid metabolism enzymes. This modulation potentially sensitizes adjacent cells to ferroptosis (136–140). These findings highlight the intricate interplay and regulatory mechanisms between efferocytosis and ferroptosis, unveiling their impact on cellular responses and fate. Investigating the cross-regulation between inflammatory signaling pathways and the interplay between efferocytosis and ferroptosis could provide important insights into disease pathogenesis.
6.5 Therapeutic implications
The interplay between efferocytosis and ferroptosis has therapeutic implications. Modulating efferocytosis can potentially regulate ferroptosis and vice versa. Enhancing efferocytosis or inhibiting ferroptosis may have therapeutic benefits in certain disease contexts. For example, in cancer, promoting efferocytosis can help clear apoptotic cancer cells and suppress inflammation, while inhibiting ferroptosis can protect normal tissues from oxidative damage (141, 142).
Unraveling the intricate link between efferocytosis and ferroptosis holds promise for understanding the underlying mechanisms of various diseases and exploring novel therapeutic avenues. Future research in this area will likely uncover additional regulatory mechanisms and therapeutic targets, offering opportunities for interventions aimed at restoring tissue homeostasis and mitigating disease progression. The main properties of ferroptosis and efferocytosis are shown in Table 1.
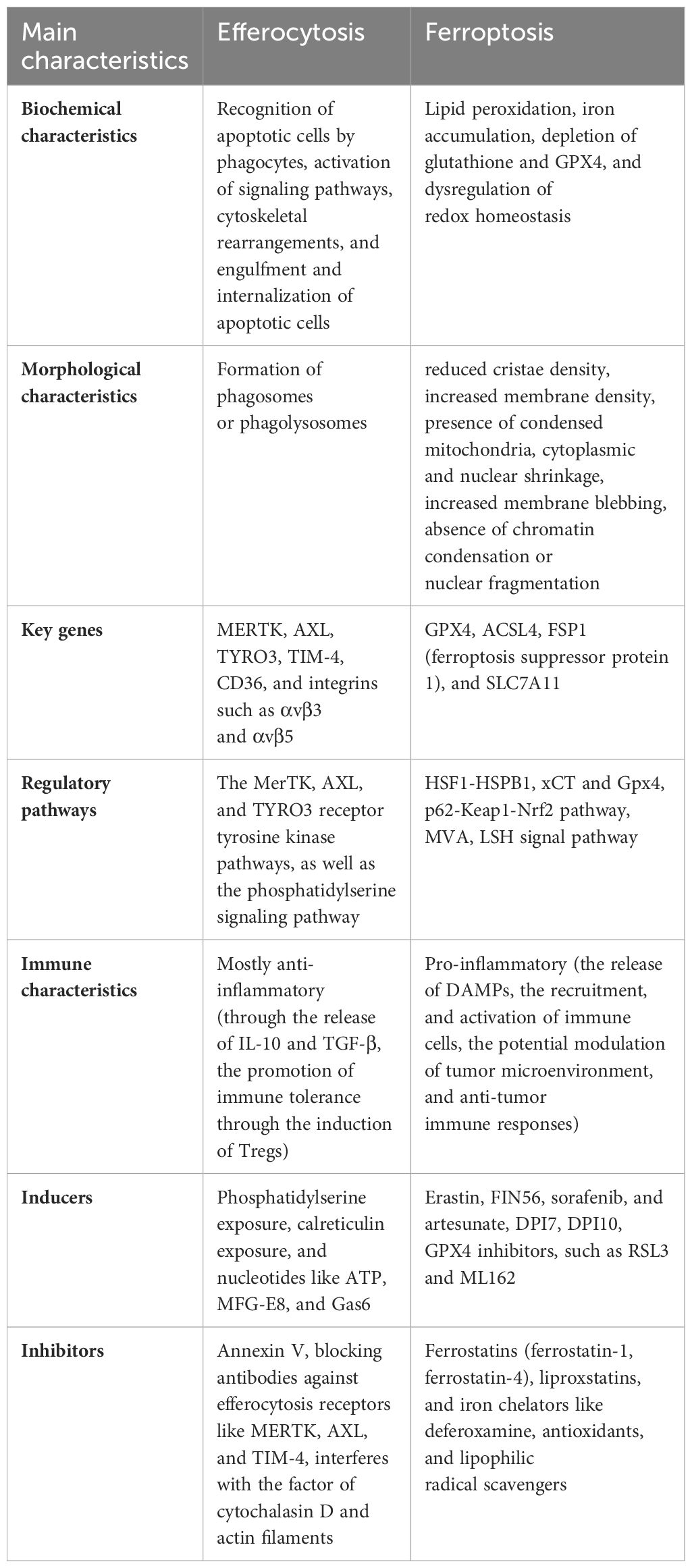
Table 1. The primary characteristics of ferroptosis and efferocytosis.
7 Implications of dysregulated cell clearance and cell death in cancer
Dysregulated cell clearance and cell death processes have profound implications for cancer development and progression (143, 144). The inability to properly clear dying or damaged cells and the dysregulation of cell death pathways can disrupt tissue homeostasis and contribute to various aspects of tumor biology. Dysregulated cell clearance and cell death can lead to the accumulation of apoptotic cells, necrotic debris, and cellular waste in the tumor microenvironment. This sets off an inflammatory reaction that manifests as the release of DAMPs, chemokines, and pro-inflammatory cytokines (145). These molecules act as danger signals, alerting the immune system to tissue damage and cellular stress. Chronic inflammation creates an environment that promotes tumor growth, angiogenesis, and metastasis, providing a favorable niche for cancer cells to thrive (146). Impaired clearance of dying cells hinders the immune system’s capacity to recognize and eliminate cancer cells effectively. This dysfunction not only reduces the immune response to tumors but also fosters an immune-tolerant environment that can further facilitate cancer progression (3, 147). The release of self-antigens from uncleared apoptotic cells can trigger autoimmunity, while the accumulation of apoptotic cells can induce immune tolerance, providing a survival advantage to cancer cells (148–150). Understanding the intricate relationship between impaired cell clearance, immune dysregulation, and cancer progression is crucial for the development of therapeutic strategies aimed at enhancing efferocytosis and restoring immune surveillance to combat cancer effectively. Dysregulated cell clearance and cell death contribute to tumor progression and metastatic spread. Inefficient clearance of apoptotic cells can promote the survival and proliferation of cancer cells, leading to the expansion of the tumor mass. Moreover, the accumulation of necrotic debris and DAMPs can stimulate tumor cell invasion, angiogenesis, and the remodeling of the extracellular matrix, facilitating tumor metastasis (151).
Aberrant cell clearance and modified cell death mechanisms can confer resistance to cancer therapies (152–154). Cancer cells that evade apoptosis or undergo alternative cell death modalities such as autophagy or necrosis may survive treatment interventions. Additionally, impaired clearance of dying cells can lead to the persistence of therapy-induced cellular debris, triggering inflammation and promoting treatment resistance (155).
Dysregulated cell death and impaired clearance can contribute to genomic instability, a hallmark of cancer. Incomplete or defective cell death processes can result in the accumulation of damaged DNA and genomic alterations (156). Genomic instability provides a fertile ground for the acquisition of oncogenic mutations, driving tumor progression and therapeutic resistance.
8 Manipulating efferocytosis and ferroptosis for improved cancer therapy
Manipulating efferocytosis and ferroptosis to improve cancer therapy involves employing various strategies to enhance or inhibit these processes selectively. Here are some approaches being explored to leverage efferocytosis and ferroptosis for improved cancer treatment:
8.1 Enhancing efferocytosis
Promoting efficient clearance of apoptotic cells by enhancing efferocytosis can reduce inflammation and prevent secondary necrosis, which is associated with tumor progression. This can be achieved by targeting signaling pathways involved in efferocytosis, such as the MerTK receptor or the phosphatidylserine (PS) recognition pathway (157–160).
8.1.1 Phagocytic receptor agonists
Phagocytic receptor agonists are compounds that stimulate immune cells, including macrophages and neutrophils, by attaching to particular receptors on their surfaces. These receptors are crucial for identifying and ingesting harmful pathogens, dead cells, and other cellular waste. Among the various types of phagocytic receptors for apoptotic cells are TIM-1, TIM-4, Stabilin-2, and BAI-1, which interact with phosphatidylserine, a molecule present on the surface of dying cells (161–163). Other important receptors include Lactadherin, αVβ3, αVβ5, CD36, and CD14. Notably, Lactadherin and αVβ3 also bind to milk fat globule-epidermal growth factor 8 (MFG-E8) (164), while oxidized lipids act as ligands for CD36 (165). Examples of phagocytic receptor agonists include laminarin, molecules with terminal mannose, and N-Formyl-methioninyl-leucyl-phenylalanine (F-MLF) (166). Terminal mannose is specifically recognized by the mannose receptor, primarily located on macrophages (167). F-MLF activates formylmethionine phagocytic receptors (FPRs), with seven types identified in mice and three in humans (168). Additionally, various synthetic compounds have been developed to activate specific phagocytic receptors. These agonists promote receptor clustering and activate signaling pathways that lead to cytoskeletal rearrangement, enhancing the uptake of apoptotic cells. Moreover, stimulating phagocytic receptors often triggers the release of pro-inflammatory cytokines, further activating the immune response. They can also improve T cell activation by enhancing antigen presentation by phagocytes (169).
Activation of phagocytic receptors, such as MerTK or CD47-SIRPα, can enhance efferocytosis. Agonistic antibodies or small molecules targeting these receptors have been investigated to stimulate phagocytosis of apoptotic cancer cells and promote tumor clearance. In non-small lung cell carcinoma, gastric cancer cells, head and neck cancers, and glioblastoma, there have been observed advantages in targeting the reduction or inhibition of MerTK activity (170, 171).
8.1.2 Lipid mediators
Lipid mediators, such as specialized pro-resolving mediators (SPMs) like resolvins and protectins, can promote efferocytosis and dampen inflammation. Administration of exogenous SPMs or modulation of endogenous SPM production has shown the potential to enhance efferocytosis and limit tumor progression. It was shown that SPMs play a crucial role in actively resolving inflammation. However, when inflammation remains unresolved, chronic inflammation creates a favorable environment that promotes carcinogenesis and cancer progression (172). Conventional cancer therapies can further enhance cancer-related inflammation by inducing extensive tumor cell death, leading to the activation of immune-infiltrating cells such as TAMs (173, 174). Macrophages, with their versatile phenotype, are central players in both inflammation and its active resolution. Exploiting their high plasticity, cancer cells can manipulate macrophages to support tumor progression, immune evasion, and therapy resistance. The impaired resolution of cancer-associated inflammation, mediated by TAMs, can thus reinforce tumor progression. Excitingly, recent evidence suggests that stimulating the pro-resolving functions of macrophages using SPMs can promote inflammation resolution in cancer and enhance the effectiveness of anticancer treatments (175).
8.1.3 Immune checkpoint blockade
Immune checkpoint inhibitors have emerged as promising therapeutic agents in cancer treatment. Specifically, antibodies targeting programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) have demonstrated the potential to not only restore immune responses but also exert a positive influence on efferocytosis. By blocking the PD-1/PD-L1 interaction, these inhibitors effectively inhibit the inhibitory signals that suppress immune cell activity. One of the notable effects of immune checkpoint inhibitors is their ability to enhance the phagocytic activity of immune cells involved in efferocytosis. These inhibitors promote the recognition and clearance of apoptotic cells by immune cells, such as macrophages and dendritic cells, through the upregulation of phagocytic receptors and the improvement of their functional capacity. Consequently, the efficient clearance of apoptotic cells is facilitated, preventing the accumulation of dead cells and the subsequent release of pro-inflammatory molecules. Furthermore, immune checkpoint inhibitors play a crucial role in reinvigorating immune responses against cancer cells. By blocking the PD-1/PD-L1 interaction, they unleash the cytotoxic potential of immune cells, such as T cells, enabling them to effectively target and eliminate tumor cells (176). This enhanced immune response contributes to a more favorable tumor microenvironment, favoring efferocytosis and reducing inflammation. Overall, the administration of immune checkpoint inhibitors, specifically antibodies targeting PD-1 or PD-L1, holds promise in not only restoring immune responses but also promoting efferocytosis and reducing inflammation. The combined effects of immune checkpoint inhibitors on immune cell activity and their ability to restore the balance between immune tolerance and effective tumor recognition make them a valuable approach in cancer therapy (46).
8.1.4 Synergistic approaches
Combining therapies that induce apoptosis, such as chemotherapy or targeted therapies, with agents that enhance efferocytosis can improve treatment response. The clearance of apoptotic cancer cells reduces the release of pro-inflammatory factors and potentially enhances the anti-tumor immune response.
8.2 Inducing ferroptosis
8.2.1 Lipid peroxidation inducers
Small molecules that induce lipid peroxidation and ferroptosis, such as erastin and RSL3, have garnered significant attention as potential anti-cancer agents. These compounds offer a targeted approach to selectively trigger ferroptosis in cancer cells, leading to their demise, while sparing normal cells from excessive damage and toxicity (177, 178). Erastin, for instance, has been extensively studied for its ability to selectively induce ferroptosis in cancer cells. It functions by inhibiting the activity of system xc-, a cystine/glutamate antiporter that plays a crucial role in importing cystine into cells in exchange for glutamate. By blocking system xc-, erastin disrupts the cellular supply of cystine, thereby impairing the synthesis of glutathione, the key antioxidant molecule that protects cells from oxidative stress. Consequently, cancer cells with high metabolic demands and increased dependency on cystine uptake become particularly vulnerable to erastin-induced ferroptosis (179). Similarly, RSL3 acts as a potent inducer of ferroptosis by specifically targeting and inhibiting the activity of GPX4. GPX4 plays a critical role in neutralizing lipid peroxides and suppressing ferroptosis. By inhibiting GPX4, RSL3 disrupts the cellular defense mechanisms against lipid peroxidation, leading to the accumulation of toxic lipid peroxides and subsequent initiation of ferroptosis in cancer cells (180).
8.2.2 Glutathione depletion
Ferroptosis relies on the depletion of intracellular antioxidant systems, primarily glutathione (181, 182). Glutathione acts as a critical line of defense against oxidative stress by neutralizing reactive oxygen species and inhibiting lipid peroxidation, thus preventing ferroptosis. However, inhibiting glutathione synthesis can sensitize cancer cells to ferroptosis-inducing agents and significantly enhance treatment efficacy (183). One approach to deplete intracellular glutathione levels and promote ferroptosis is the use of inhibitors targeting glutathione synthesis. Buthionine sulfoximine (BSO) is a potent inhibitor of γ-glutamylcysteine synthetase, the rate-limiting enzyme in glutathione biosynthesis. By inhibiting this enzyme, BSO impairs the synthesis of glutathione, leading to a reduction in intracellular glutathione levels. Consequently, the decreased availability of glutathione renders cancer cells more susceptible to ferroptosis induction (184, 185). When combined with ferroptosis-inducing agents, such as small molecules that inhibit the activity of GPX4, BSO can significantly potentiate the therapeutic effects. GPX4 is a critical enzyme that suppresses lipid peroxidation and functions as a key regulator of ferroptosis. Inhibition of GPX4 allows the accumulation of lipid peroxides, ultimately leading to the initiation of ferroptosis. By inhibiting glutathione synthesis, BSO disrupts the cellular antioxidant defense system, rendering cancer cells more vulnerable to the accumulation of lipid peroxides and subsequent ferroptotic cell death (186).
8.2.3 Iron-dependent ferroptosis
Modulating iron metabolism and availability is a key strategy to influence ferroptosis. By targeting iron homeostasis, it is possible to regulate intracellular iron levels and subsequently impact the potential for lipid peroxidation and ferroptosis. This approach offers a promising avenue for anti-cancer therapy (187). One approach to modulating iron metabolism is through the use of iron chelators or iron transport inhibitors. Iron chelators such as deferoxamine and deferiprone are compounds that bind to iron ions, forming stable complexes and reducing the availability of free iron within cells. By sequestering iron, chelators limit its participation in Fenton reactions, which generate harmful reactive oxygen species that can trigger lipid peroxidation. Consequently, iron chelators can reduce the potential for lipid peroxidation and, subsequently, ferroptosis induction. In addition to iron chelation, inhibiting iron transporters or channels involved in iron uptake can also limit intracellular iron levels. For instance, inhibiting transferrin receptor 1 (TfR1), which facilitates iron uptake via transferrin-bound iron, can restrict iron availability for cellular processes. This, in turn, can reduce the availability of iron for Fenton reactions and lipid peroxidation, thereby modulating the susceptibility of cancer cells to ferroptosis. In the extracellular environment, Fe3+ binds to transferrin and is internalized into cells via TFR1. Within the endosome, STEAP3 (six-transmembrane epithelial antigen of prostate 3) metalloreductases facilitate the reduction of Fe3+ to Fe2+. Subsequently, divalent metal transporter 1 (DMT1) transports Fe2+ from the endosome to the cytoplasm, where it enters a labile iron pool. To maintain plasma iron levels, the labile iron is exported through the membrane protein ferroportin. Alternatively, excess iron from the labile iron pool can be sequestered in ferritin heteropolymers (such as ferritin heavy chain 1 or ferritin light chain), which represents a redox-inactive form of iron. This storage mechanism serves to safeguard cells and tissues from iron-induced damage. Notably, ferritinophagy, the autophagic degradation of ferritin, contributes to ferroptosis by elevating labile iron levels. The process involves the participation of autophagy-related proteins (Atg5 and Atg7) and nuclear receptor coactivator 4 (NCOA4) (188). Overall, the coordinated regulation of iron metabolism and its impact on ferroptosis are depicted in Figure 5.
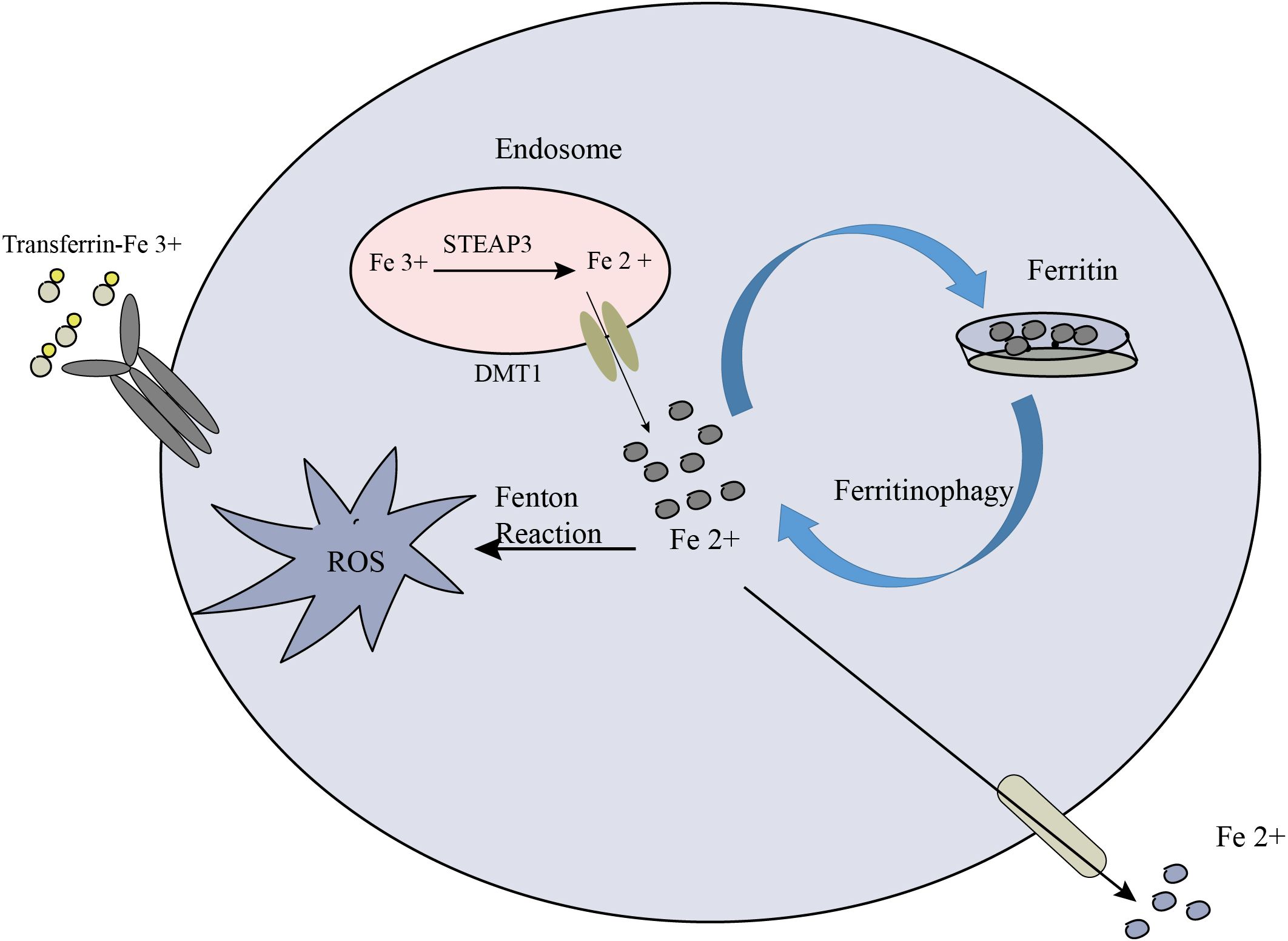
Figure 5. The crucial role of iron metabolism in the process of ferroptosis. Cellular iron homeostasis relies on the coordinated regulation of iron uptake, export, utilization, and storage.
8.2.4 Salinomycin-loaded gold nanoparticles
Gold nanoparticles functionalized with salinomycin have been utilized as a targeted delivery system to induce ferroptosis in cancer cells (189). Salinomycin is a compound known to disrupt cellular processes related to iron metabolism and GSH homeostasis. By encapsulating salinomycin within gold nanoparticles, the drug can be specifically delivered to cancer cells. Once internalized, salinomycin-loaded AuNPs can induce iron accumulation within the cells, leading to increased levels of labile iron. This iron overload disrupts redox balance and exhausts intracellular GSH, a critical component of the cellular antioxidant defense system. As a result, cancer cells become highly susceptible to oxidative damage and ferroptosis (190).
8.2.5 Lipid peroxidation generator with a GSH and iron redox couple
Another approach to induce ferroptosis involves the use of a novel redox couple composed of GSH and iron. GSH, a key intracellular antioxidant, can undergo redox reactions with iron to generate ROS and initiate lipid peroxidation. By manipulating this redox couple, researchers have developed compounds that promote the production of ROS and subsequent lipid peroxidation. This process overwhelms the cellular antioxidant defenses and triggers ferroptosis in cancer cells. This strategy provides a targeted and selective means to induce ferroptosis, exploiting the vulnerabilities of cancer cells with dysregulated iron metabolism and antioxidant systems (191).
8.2.6 Oxygen-boosted phototherapy
Phototherapy techniques, such as photodynamic therapy (PDT), have been combined with strategies to boost oxygen levels within cancer cells. PDT involves the use of light-sensitive compounds, known as photosensitizers, which generate reactive oxygen species upon exposure to specific wavelengths of light. By enhancing the availability of oxygen in the tumor microenvironment, either through oxygen delivery or modulation of hypoxia, the effectiveness of PDT in inducing ferroptosis can be amplified. Increased oxygen levels facilitate the generation of ROS, leading to lipid peroxidation and ferroptotic cell death in cancer cells (192, 193).
8.3 Combination approaches
8.3.1 Efferocytosis-ferroptosis interplay
Synergistic effects may be achieved by combining strategies that enhance efferocytosis with agents that induce ferroptosis. Efficient clearance of apoptotic cancer cells can reduce inflammation and create a microenvironment favorable for ferroptosis induction, leading to enhanced tumor cell death.
8.3.2 Concurrent treatments
Integrating efferocytosis and ferroptosis-targeting approaches with standard treatments, such as chemotherapy, radiation therapy, or immunotherapy, can improve treatment response and overcome resistance mechanisms. Customized combination regimens based on the specific characteristics of the tumor and the patient may optimize therapeutic outcomes.
It is important to note that the manipulation of efferocytosis and ferroptosis in cancer therapy is still an area of active research, and further studies are needed to optimize treatment strategies, understand potential side effects, and identify biomarkers for patient selection. Nonetheless, these approaches offer promising avenues for developing innovative and more effective cancer therapeutics that exploit the intricate interplay between efferocytosis and ferroptosis.
9 Conclusion
The interplay between efferocytosis and ferroptosis has attracted significant interest in cancer research. Efferocytosis is crucial for maintaining tissue balance, while dysregulation can lead to chronic inflammation. Dysregulated ferroptosis, on the other hand, can hinder efficient efferocytosis and impede apoptotic cell clearance. Conversely, apoptotic cells can activate ferroptotic pathways, inducing cell death in cancer cells. Understanding and managing the delicate balance between efferocytosis and ferroptosis could have therapeutic implications in cancer treatment. Targeting these pathways may improve tumor clearance, reduce inflammation, and enhance the effectiveness of existing therapies. Further research in this area is necessary to develop novel therapeutic approaches that restore tissue homeostasis and improve patient outcomes.
Author contributions
AF: Writing – review & editing. SM-Y: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ACs: Apoptotic cells
ACSL4: Acyl-CoA synthetase long-chain family member 4
CRT: Calreticulin
DAMPs: Damage-associated molecular patterns
DMT1: Divalent metal transporter 1
FOXP3: Forkhead box P3
GPX4: Glutathione peroxidase 4
HER2: Human epidermal growth factor receptor 2
HSF1: Heat shock factor-1
HSPB1: Heat shock protein beta-1
IFN: Interferon
IL: Interleukin
Keap1: Keleh-like ECH-associated protein 1
LILRB1: Leukocyte immunoglobulin-like receptor subfamily B member 1
LSH: Lymphoid-specific helicase
MVA: Mevalonate
NCOA4: Nuclear receptor coactivator 4
Nrf2: Nuclear factor erythroid 2-related factor 2
PD-L1: Programmed death-ligand 1
PL: Phospholipids
PS: Phosphatidylserine
PSA: Prostate-specific antigen
PUFAs: polyunsaturated fatty acids
ROS: Reactive oxygen species
RSL3: Ras-selective lethal small molecule 3
SLC7A11: Solute carrier family 7 member 11
STEAP3: Six-transmembrane epithelial antigen of the prostate 3
TFR1: Transferrin receptor 1
TGF-β: Transforming growth factor beta
TIM3: T-cell immunoglobulin and mucin domain- containing protein 3
TNF: Tumor necrosis factor
xCT: System xc-
References
1. Doran AC, Yurdagul A Jr., Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. (2020) 20:254–67. doi: 10.1038/s41577-019-0240-6
2. Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. (2022) 15:174. doi: 10.1186/s13045-022-01392-3
3. Fond AM, Ravichandran KS. Clearance of dying cells by phagocytes: mechanisms and implications for disease pathogenesis. Adv Exp Med Biol. (2016) 930:25–49. doi: 10.1007/978-3-319-39406-0_2
4. Deng J, Yang H, Haak VM, Yang J, Kipper FC, Barksdale C, et al. Eicosanoid regulation of debris-stimulated metastasis. Proc Natl Acad Sci U.S.A. (2021) 12:118. doi: 10.1073/pnas.2107771118
5. Hermetet F, Jacquin E, Launay S, Gaiffe E, Couturier M, Hirchaud F, et al. Efferocytosis of apoptotic human papillomavirus-positive cervical cancer cells by human primary fibroblasts. Biol Cell. (2016) 108:189–204. doi: 10.1111/boc.201500090
6. Lin J, Xu A, Jin J, Zhang M, Lou J, Qian C, et al. MerTK-mediated efferocytosis promotes immune tolerance and tumor progression in osteosarcoma through enhancing M2 polarization and PD-L1 expression. Oncoimmunology. (2022) 11:2024941. doi: 10.1080/2162402X.2021.2024941
7. Lecoultre M, Dutoit V, Walker PR. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J Immunother Cancer. (2020) 8. doi: 10.1136/jitc-2020-001408
8. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
9. Zhao Y, Liu Z, Liu G, Zhang Y, Liu S, Gan D, et al. Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab. (2023) 35:1688–1703.e10. doi: 10.1016/j.cmet.2023.09.004
10. Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An estimation of the number of cells in the human body. Ann Hum Biol. (2013) 40:463–71. doi: 10.3109/03014460.2013.807878
11. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
12. Schilperoort M, Ngai D, Sukka SR, Avrampou K, Shi H, Tabas I. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol Rev. (2023) 319:65–80. doi: 10.1111/imr.v319.1
13. Zhou Y, Yao Y, Deng Y, Shao A. Regulation of efferocytosis as a novel cancer therapy. Cell Commun Signal. (2020) 18:71. doi: 10.1186/s12964-020-00542-9
14. Mahmoudi A, Firouzjaei AA, Darijani F, Navashenaq JG, Taghizadeh E, Darroudi M, et al. Effect of diabetes on efferocytosis process. Mol Biol Rep. (2022) 49:10849–63. doi: 10.1007/s11033-022-07725-2
15. Segawa K, Nagata S. An apoptotic ‘Eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. (2015) 25:639–50. doi: 10.1016/j.tcb.2015.08.003
16. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. (2002) 196:655–65. doi: 10.1084/jem.20020542
17. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. (2005) 123:321–34. doi: 10.1016/j.cell.2005.08.032
18. Richards DM, Endres RG. The mechanism of phagocytosis: two stages of engulfment. Biophys J. (2014) 107:1542–53. doi: 10.1016/j.bpj.2014.07.070
19. Rosales C, Uribe-Querol E. Phagocytosis: A fundamental process in immunity. BioMed Res Int. (2017) 2017:9042851. doi: 10.1155/2017/9042851
20. Wang L, Li H, Tang Y, Yao P. Potential mechanisms and effects of efferocytosis in atherosclerosis. Front Endocrinol (Lausanne). (2020) 11:585285. doi: 10.3389/fendo.2020.585285
21. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. (2005) 122:735–49. doi: 10.1016/j.cell.2005.06.043
22. Werfel TA, Cook RS. Efferocytosis in the tumor microenvironment. Semin Immunopathol. (2018) 40:545–54. doi: 10.1007/s00281-018-0698-5
23. Sendama W. The effect of ageing on the resolution of inflammation. Ageing Res Rev. (2020) 57:101000. doi: 10.1016/j.arr.2019.101000
24. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
25. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
26. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
27. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
28. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
29. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
30. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
31. Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. (2020) 11:922. doi: 10.1038/s41419-020-03118-0
32. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
33. Szondy Z, Garabuczi E, Joós G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol. (2014) 5:354. doi: 10.3389/fimmu.2014.00354
34. Zhang Y, Wang Y, Ding J, Liu P. Efferocytosis in multisystem diseases (Review). Mol Med Rep. (2022) 25. doi: 10.1186/s10020-021-00415-y
35. Mendoza-Reinoso V, Schnepp PM, Baek DY, Rubin JR, Schipani E, Keller ET, et al. Bone marrow macrophages induce inflammation by efferocytosis of apoptotic prostate cancer cells via HIF-1α Stabilization. Cells. (2022) 11. doi: 10.3390/cells11233712
36. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
37. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res. (2020) 39:204. doi: 10.1186/s13046-020-01709-5
38. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
39. Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. (2022) 13:355. doi: 10.1038/s41419-022-04775-z
40. Ye Z, Zhuo Q, Hu Q, Xu X, Liu M, Zhang Z, et al. FBW7-NRA41-SCD1 axis synchronously regulates apoptosis and ferroptosis in pancreatic cancer cells. Redox Biol. (2021) 38:101807. doi: 10.1016/j.redox.2020.101807
41. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. (2022) 13:3676. doi: 10.1038/s41467-022-31218-2
42. Rodriguez R, Schreiber SL, Conrad M. Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol Cell. (2022) 82:728–40. doi: 10.1016/j.molcel.2021.12.001
43. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. (2022) 612:338–46. doi: 10.1038/s41586-022-05443-0
44. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
45. Wei YT, Wang XR, Yan C, Huang F, Zhang Y, Liu X, et al. Thymosin α-1 reverses M2 polarization of tumor-associated macrophages during efferocytosis. Cancer Res. (2022) 82:1991–2002. doi: 10.1158/0008-5472.CAN-21-4260
46. Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y, et al. Blockade of the phagocytic receptor merTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity. (2020) 52:357–373.e9. doi: 10.1016/j.immuni.2020.01.014
47. Di Carlo SE, Raffenne J, Varet H, Ode A, Granados DC, Stein M, et al. Depletion of slow-cycling PDGFRα(+)ADAM12(+) mesenchymal cells promotes antitumor immunity by restricting macrophage efferocytosis. Nat Immunol. (2023) 24:1867–78. doi: 10.1038/s41590-023-01642-7
48. Ghalavand M, Moradi-Chaleshtori M, Dorostkar R, Mohammadi-Yeganeh S, Hashemi SM. Exosomes derived from rapamycin-treated 4T1 breast cancer cells induced polarization of macrophages to M1 phenotype. Biotechnol Appl Biochem. (2023) 70:1754–71. doi: 10.1002/bab.v70.5
49. Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. (2005) 54:187–207. doi: 10.1007/s00262-004-0560-6
50. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. (2021) 21:360–78. doi: 10.1038/s41568-021-00346-0
51. Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. (2022) 15:28. doi: 10.1186/s13045-022-01247-x
52. Ghalavand M, Gouvarchin Ghaleh HE, Khafaei M, Paryan M, Kondori BJ, Nodoushan MM, et al. Effect of calcitriol treated mesenchymal stem cells as an immunomodulation micro-environment on allergic asthma in a mouse model. Endocr Metab Immune Disord Drug Targets. (2023) 23:1096–103. doi: 10.2174/1871530323666230127115847
53. Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Adv Exp Med Biol. (2020) 1248:201–26. doi: 10.1007/978-981-15-3266-5_9
54. Cruz Cruz J, Allison KC, Page LS, Jenkins AJ, Wang X, Earp HS, et al. Inhibiting efferocytosis reverses macrophage-mediated immunosuppression in the leukemia microenvironment. Front Immunol. (2023) 14:1146721. doi: 10.3389/fimmu.2023.1146721
55. Geuijen C, Tacken P, Wang LC, Klooster R, van Loo PF, Zhou J, et al. A human CD137×PD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nat Commun. (2021) 12:4445. doi: 10.1038/s41467-021-24767-5
56. Marciscano AE, Anandasabapathy N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol. (2021) 52:101481. doi: 10.1016/j.smim.2021.101481
57. Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. (2021) 184:4512–4530.e22. doi: 10.1016/j.cell.2021.07.015
58. Weigert A, Mora J, Sekar D, Syed S, Brüne B. Killing is not enough: how apoptosis hijacks tumor-associated macrophages to promote cancer progression. Adv Exp Med Biol. (2016) 930:205–39. doi: 10.1007/978-3-319-39406-0_9
59. Gilligan MM, Gartung A, Sulciner ML, Norris PC, Sukhatme VP, Bielenberg DR, et al. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc Natl Acad Sci U.S.A. (2019) 116:6292–7. doi: 10.1073/pnas.1804000116
60. Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. (2022) 43:546–63. doi: 10.1016/j.it.2022.04.008
61. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. (2016) 18:549–60. doi: 10.1038/ncb3340
62. Tsilimigras DI, Brodt P, Clavien PA, Muschel RJ, D'Angelica MI, Endo I, et al. Liver metastases. Nat Rev Dis Primers. (2021) 7:27. doi: 10.1038/s41572-021-00261-6
63. Doak GR, Schwertfeger KL, Wood DK. Distant relations: macrophage functions in the metastatic niche. Trends Cancer. (2018) 4:445–59. doi: 10.1016/j.trecan.2018.03.011
64. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery. (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
65. Astuti Y, Raymant M, Quaranta V, Clarke K, Abudula M, Smith O, et al. Efferocytosis reprograms the tumor microenvironment to promote pancreatic cancer liver metastasis. Nat Cancer. (2024) 5:774–90. doi: 10.1038/s43018-024-00731-2
66. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. (2017) 47:913–927.e6. doi: 10.1016/j.immuni.2017.10.006
67. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. (2013) 123:3231–42. doi: 10.1172/JCI67655
68. Davra V, Kumar S, Geng K, Calianese D, Mehta D, Gadiyar V, et al. Axl and mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host antitumor immunity. Cancer Res. (2021) 81:698–712. doi: 10.1158/0008-5472.CAN-20-2066
69. Firouzjaei AA, Aghaee-Bakhtiari SH, Tafti A, Sharifi K, Abadi MHJN, Rezaei S, et al. Impact of curcumin on ferroptosis-related genes in colorectal cancer: Insights from in-silico and in-vitro studies. Cell Biochem Funct. (2023) 41:1488–502. doi: 10.1002/cbf.v41.8
70. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
71. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
72. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U.S.A. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
73. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
74. Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. (2016) 17:366–73. doi: 10.1016/j.celrep.2016.09.022
75. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. (2018) 20:1181–92. doi: 10.1038/s41556-018-0178-0
76. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
77. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) 547:453–7. doi: 10.1038/nature23007
78. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
79. Yoshikawa M, Tsuchihashi K, Ishimoto T, Yae T, Motohara T, Sugihara E, et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. (2013) 73:1855–66. doi: 10.1158/0008-5472.CAN-12-3609-T
80. Liu J, Li D, Dang L, Liang C, Guo B, Lu C, et al. Osteoclastic miR-214 targets TRAF3 to contribute to osteolytic bone metastasis of breast cancer. Sci Rep. (2017) 7:40487. doi: 10.1038/srep40487
81. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
82. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. (2022) 82:2215–27. doi: 10.1016/j.molcel.2022.03.022
83. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
84. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
85. Liu Y, Huang P, Li Z, Xu C, Wang H, Jia B, et al. Vitamin C sensitizes pancreatic cancer cells to erastin-induced ferroptosis by activating the AMPK/nrf2/HMOX1 pathway. Oxid Med Cell Longev. (2022) 2022:5361241. doi: 10.1155/2022/5361241
86. Gaffan J, Dacre J, Jones A. Educating undergraduate medical students about oncology: a literature review. J Clin Oncol. (2006) 24:1932–9. doi: 10.1200/JCO.2005.02.6617
87. Pei S, Yang X, Wang H, Zhang H, Zhou B, Zhang D, et al. Plantamajoside, a potential anti-tumor herbal medicine inhibits breast cancer growth and pulmonary metastasis by decreasing the activity of matrix metalloproteinase-9 and -2. BMC Cancer. (2015) 15:965. doi: 10.1186/s12885-015-1960-z
88. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. (2013) 49:1374–403. doi: 10.1016/j.ejca.2012.12.027
89. Norum J, Nieder C. Treatments for metastatic prostate cancer (mPC): A review of costing evidence. Pharmacoeconomics. (2017) 35:1223–36. doi: 10.1007/s40273-017-0555-8
90. Peng S, Yi Z, Liu M. Ailanthone: a new potential drug for castration-resistant prostate cancer. Chin J Cancer. (2017) 36:25. doi: 10.1186/s40880-017-0194-7
91. Li J, Meng H, Bai Y, Wang K. Regulation of lncRNA and its role in cancer metastasis. Oncol Res. (2016) 23:205–17. doi: 10.3727/096504016X14549667334007
92. Gomatou G, Syrigos N, Vathiotis IA, Kotteas EA. Tumor dormancy: implications for invasion and metastasis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22094862
93. Meng F, Yu Z, Zhang D, Chen S, Guan H, Zhou R, et al. Induced phase separation of mutant NF2 imprisons the cGAS-STING machinery to abrogate antitumor immunity. Mol Cell. (2021) 81:4147–4164.e7. doi: 10.1016/j.molcel.2021.07.040
94. Sugano K, Maeda K, Ohtani H, Nagahara H, Shibutani M, Hirakawa K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. (2015) 35:677–82.
95. Chen RS, Song YM, Zhou ZY, Tong T, Li Y, Fu M, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. (2009) 28:599–609. doi: 10.1038/onc.2008.414
96. Lee JR, Roh JL, Lee SM, Park Y, Cho KJ, Choi SH, et al. Overexpression of cysteine-glutamate transporter and CD44 for prediction of recurrence and survival in patients with oral cavity squamous cell carcinoma. Head Neck. (2018) 40:2340–6. doi: 10.1002/hed.v40.11
97. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
98. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. (2016) 352:175–80. doi: 10.1126/science.aaf4405
99. Liu Y, Chen Q, Zhu Y, Wang T, Ye L, Han L, et al. Non-coding RNAs in necroptosis, pyroptosis and ferroptosis in cancer metastasis. Cell Death Discovery. (2021) 7:210. doi: 10.1038/s41420-021-00596-9
100. Teplova I, Lozy F, Price S, Singh S, Barnard N, Cardiff RD, et al. ATG proteins mediate efferocytosis and suppress inflammation in mammary involution. Autophagy. (2013) 9:459–75. doi: 10.4161/auto.23164
101. Lemke G. How macrophages deal with death. Nat Rev Immunol. (2019) 19:539–49. doi: 10.1038/s41577-019-0167-y
102. Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. (2018) 32:602–19. doi: 10.1101/gad.314674.118
103. Tajbakhsh A, Gheibihayat SM, Askari H, Savardashtaki A, Pirro M, Johnston TP, et al. Statin-regulated phagocytosis and efferocytosis in physiological and pathological conditions. Pharmacol Ther. (2022) 238:108282. doi: 10.1016/j.pharmthera.2022.108282
104. Tajbakhsh A, Kovanen PT, Rezaee M, Banach M, Moallem SA, Sahebkar A, et al. Regulation of efferocytosis by caspase-dependent apoptotic cell death in atherosclerosis. Int J Biochem Cell Biol. (2020) 120:105684. doi: 10.1016/j.biocel.2020.105684
105. Yuan Y, Chen Y, Peng T, Li L, Zhu W, Liu F, et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin Sci (Lond). (2019) 133:1759–77. doi: 10.1042/CS20190672
106. Zhang B, Yang Y, Yi J, Zhao Z, Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res. (2021) 56:991–1005. doi: 10.1111/jre.12912
107. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
108. Mikulska-Ruminska K, Anthonymuthu TS, Levkina A, Shrivastava IH, Kapralov AA, Bayır H, et al. NO(•) represses the oxygenation of arachidonoyl PE by 15LOX/PEBP1: mechanism and role in ferroptosis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22105253
109. Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO(•) sabotage of theft-ferroptosis. Redox Biol. (2021) 45:102045. doi: 10.1016/j.redox.2021.102045
110. ScIndia Ph DY, Leeds Md J, Swaminathan Md S. Iron homeostasis in healthy kidney and its role in acute kidney injury. Semin Nephrol. (2019) 39:76–84. doi: 10.1016/j.semnephrol.2018.10.006
111. Gao J, Huang C, Kong L, Zhou W, Sun M, Wei T, et al. SIRT3 regulates clearance of apoptotic cardiomyocytes by deacetylating frataxin. Circ Res. (2023) 133:631–47. doi: 10.1161/CIRCRESAHA.123.323160
112. Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F. Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta. (2016) 1862:1137–46. doi: 10.1016/j.bbadis.2016.03.004
113. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol. (2019) 105:1015–26. doi: 10.1002/JLB.3A0318-108R
114. Kajarabille N, Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20194968
115. Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. (2022) 289:7038–50. doi: 10.1111/febs.v289.22
116. Han S, Lin F, Qi Y, Liu C, Zhou L, Xia Y, et al. HO-1 contributes to luteolin-triggered ferroptosis in clear cell renal cell carcinoma via increasing the labile iron pool and promoting lipid peroxidation. Oxid Med Cell Longev. (2022) 2022:3846217. doi: 10.1155/2022/3846217
117. Delbosc S, Bayles RG, Laschet J, Ollivier V, Ho-Tin-Noé B, Touat Z, et al. Erythrocyte efferocytosis by the arterial wall promotes oxidation in early-stage atheroma in humans. Front Cardiovasc Med. (2017) 4:43. doi: 10.3389/fcvm.2017.00043
118. Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge sword for cancer and other age-related diseases. Front Immunol. (2018) 9:2160. doi: 10.3389/fimmu.2018.02160
119. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
120. Korbecki J, Simińska D, Gąssowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, et al. Chronic and cycling hypoxia: drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: A review of the molecular mechanisms. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms221910701
121. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004
122. Coradduzza D, Congiargiu A, Chen Z, Zinellu A, Carru C, Medici S. Ferroptosis and senescence: A systematic review. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24043658
123. Mazhar M, Din AU, Ali H, Yang G, Ren W, Wang L, et al. Implication of ferroptosis in aging. Cell Death Discovery. (2021) 7:149. doi: 10.1038/s41420-021-00553-6
124. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered. (2021) 12:5279–88. doi: 10.1080/21655979.2021.1964158
125. Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol. (2016) 23:189–97. doi: 10.1097/MOH.0000000000000236
126. Ren F, Yang Y, Wu K, Zhao T, Shi Y, Song M, et al. The effects of dandelion polysaccharides on iron metabolism by regulating hepcidin via JAK/STAT signaling pathway. Oxid Med Cell Longev. (2021) 2021:7184760. doi: 10.1155/2021/7184760
127. Li B, Gong J, Sheng S, Lu M, Guo S, Zhao X, et al. Increased hepcidin in hemorrhagic plaques correlates with iron-stimulated IL-6/STAT3 pathway activation in macrophages. Biochem Biophys Res Commun. (2019) 515:394–400. doi: 10.1016/j.bbrc.2019.05.123
128. Jung HS, Shimizu-Albergine M, Shen X, Kramer F, Shao D, Vivekanandan-Giri A, et al. TNF-α induces acyl-CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res. (2020) 61:33–44. doi: 10.1194/jlr.RA119000256
129. Persichini T, Maio N, di Patti MC, Rizzo G, Toscano S, Colasanti M, et al. Interleukin-1β induces ceruloplasmin and ferroportin-1 gene expression via MAP kinases and C/EBPβ, AP-1, and NF-κB activation. Neurosci Lett. (2010) 484:133–8. doi: 10.1016/j.neulet.2010.08.034
130. Shanmugam NK, Chen K, Cherayil BJ. Commensal bacteria-induced interleukin 1β (IL-1β) secreted by macrophages up-regulates hepcidin expression in hepatocytes by activating the bone morphogenetic protein signaling pathway. J Biol Chem. (2015) 290:30637–47. doi: 10.1074/jbc.M115.689190
131. Kanamori Y, Murakami M, Sugiyama M, Hashimoto O, Matsui T, Funaba M. Interleukin-1β (IL-1β) transcriptionally activates hepcidin by inducing CCAAT enhancer-binding protein δ (C/EBPδ) expression in hepatocytes. J Biol Chem. (2017) 292:10275–87. doi: 10.1074/jbc.M116.770974
132. Kanamori Y, Murakami M, Matsui T, Funaba M. JNK facilitates IL-1β-induced hepcidin transcription via JunB activation. Cytokine. (2018) 111:295–302. doi: 10.1016/j.cyto.2018.09.014
133. Wen Y, Chen H, Zhang L, Wu M, Zhang F, Yang D, et al. Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple-negative breast cancer cells. Free Radic Biol Med. (2021) 173:41–51. doi: 10.1016/j.freeradbiomed.2021.07.019
134. Jiang L, Zheng H, Lyu Q, Hayashi S, Sato K, Sekido Y, et al. Lysosomal nitric oxide determines transition from autophagy to ferroptosis after exposure to plasma-activated Ringer’s lactate. Redox Biol. (2021) 43:101989. doi: 10.1016/j.redox.2021.101989
135. Krümmel B, Plötz T, Jörns A, Lenzen S, Mehmeti I. The central role of glutathione peroxidase 4 in the regulation of ferroptosis and its implications for pro-inflammatory cytokine-mediated beta-cell death. Biochim Biophys Acta Mol Basis Dis. (2021) 1867:166114. doi: 10.1016/j.bbadis.2021.166114
136. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. (2019) 20:40–9. doi: 10.1038/s41590-018-0249-1
137. Glinton KE, Ma W, Lantz C, Grigoryeva LS, DeBerge M, Liu X, et al. Macrophage-produced VEGFC is induced by efferocytosis to ameliorate cardiac injury and inflammation. J Clin Invest. (2022) 132. doi: 10.1172/JCI140685
138. Deng L, He S, Guo N, Tian W, Zhang W, Luo L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflammation Res. (2023) 72:281–99. doi: 10.1007/s00011-022-01672-1
139. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. (2021) 218. doi: 10.1084/jem.20210518
140. Pan Y, Li J, Wang J, Jiang Q, Yang J, Dou H, et al. Ferroptotic MSCs protect mice against sepsis via promoting macrophage efferocytosis. Cell Death Dis. (2022) 13:825. doi: 10.1038/s41419-022-05264-z
141. Werfel TA. Assessment of the immune response to tumor cell apoptosis and efferocytosis. Methods Mol Biol. (2022) 2543:45–55. doi: 10.1007/978-1-0716-2553-8_5
142. Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. (2022) 808:145968. doi: 10.1016/j.gene.2021.145968
143. Schneider K, Arandjelovic S. Apoptotic cell clearance components in inflammatory arthritis. Immunol Rev. (2023) 319:142–50. doi: 10.1111/imr.v319.1
144. Li S, Guo W, Wu H. The role of post-translational modifications in the regulation of MCL1. Cell Signal. (2021) 81:109933. doi: 10.1016/j.cellsig.2021.109933
145. Fiorilla I, Martinotti S, Todesco AM, Bonsignore G, Cavaletto M, Patrone M, et al. Chronic inflammation, oxidative stress and metabolic plasticity: three players driving the pro-tumorigenic microenvironment in Malignant mesothelioma. Cells. (2023) 12:2048. doi: 10.3390/cells12162048
146. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. (2014) 2014:149185. doi: 10.1155/2014/149185
147. Palacio L, Goyer ML, Maggiorani D, Espinosa A, Villeneuve N, Bourbonnais S, et al. Restored immune cell functions upon clearance of senescence in the irradiated splenic environment. Aging Cell. (2019) 18:e12971. doi: 10.1111/acel.2019.18.issue-4
148. Akagbosu B, Tayyebi Z, Shibu G, Paucar Iza YA, Deep D, Parisotto YF, et al. Novel antigen-presenting cell imparts T(reg)-dependent tolerance to gut microbiota. Nature. (2022) 610:752–60. doi: 10.1038/s41586-022-05309-5
149. Benlaribi R, Gou Q, Takaba H. Thymic self-antigen expression for immune tolerance and surveillance. Inflammation Regener. (2022) 42:28. doi: 10.1186/s41232-022-00211-z
150. Sakowska J, Arcimowicz Ł, Jankowiak M, Papak I, Markiewicz A, Dziubek K, et al. Autoimmunity and cancer-two sides of the same coin. Front Immunol. (2022) 13:793234. doi: 10.3389/fimmu.2022.793234
151. Otazu GK, Dayyani M, Badie B. Role of RAGE and its ligands on inflammatory responses to brain tumors. Front Cell Neurosci. (2021) 15:770472. doi: 10.3389/fncel.2021.770472
152. Cheng L, Weng B, Jia C, Zhang L, Hu B, Deng L, et al. The expression and significance of efferocytosis and immune checkpoint related molecules in pancancer samples and the correlation of their expression with anticancer drug sensitivity. Front Pharmacol. (2022) 13:977025. doi: 10.3389/fphar.2022.977025
153. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. (2022) 21:47. doi: 10.1186/s12943-022-01530-y
154. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
155. Felton JM, Lucas CD, Rossi AG, Dransfield I. Eosinophils in the lung - modulating apoptosis and efferocytosis in airway inflammation. Front Immunol. (2014) 5:302. doi: 10.3389/fimmu.2014.00302
156. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
157. Sakhno LV, Shevela EY, Tikhonova MA, Maksimova AA, Tyrinova TV, Ostanin AA, et al. Efferocytosis modulates arginase-1 and tyrosine kinase mer expression in GM-CSF-differentiated human macrophages. Bull Exp Biol Med. (2021) 170:778–81. doi: 10.1007/s10517-021-05153-z
158. Schilperoort M, Ngai D, Katerelos M, Power DA, Tabas I. PFKFB2-mediated glycolysis promotes lactate-driven continual efferocytosis by macrophages. Nat Metab. (2023) 5:431–44. doi: 10.1038/s42255-023-00736-8
159. Kimani SG, Geng K, Kasikara C, Kumar S, Sriram G, Wu Y, et al. Contribution of defective PS recognition and efferocytosis to chronic inflammation and autoimmunity. Front Immunol. (2014) 5:566. doi: 10.3389/fimmu.2014.00566
160. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. (2016) 23:962–78. doi: 10.1038/cdd.2016.11
161. Kobayashi N, Karisola P, Peña-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. (2007) 27:927–40. doi: 10.1016/j.immuni.2007.11.011
162. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. (2008) 15:192–201. doi: 10.1038/sj.cdd.4402242
163. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. (2007) 450:430–4. doi: 10.1038/nature06329
164. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. (2002) 417:182–7. doi: 10.1038/417182a
165. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. (2006) 203:2613–25. doi: 10.1084/jem.20060370
166. Vetvicka V. Glucan-immunostimulant, adjuvant, potential drug. World J Clin Oncol. (2011) 2:115–9. doi: 10.5306/wjco.v2.i2.115
167. Napper CE, Dyson MH, Taylor ME. An extended conformation of the macrophage mannose receptor. J Biol Chem. (2001) 276:14759–66. doi: 10.1074/jbc.M100425200
168. Dufton N, Perretti M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol Ther. (2010) 127:175–88. doi: 10.1016/j.pharmthera.2010.04.010
169. Janotová T, Jalovecká M, Auerová M, Švecová I, Bruzlová P, Maierová V, et al. The use of anchored agonists of phagocytic receptors for cancer immunotherapy: B16-F10 murine melanoma model. PloS One. (2014) 9:e85222. doi: 10.1371/journal.pone.0085222
170. Cummings CT, Deryckere D, Earp HS, Graham DK. Molecular pathways: MERTK signaling in cancer. Clin Cancer Res. (2013) 19:5275–80. doi: 10.1158/1078-0432.CCR-12-1451
171. Park M, Kang KW. Phosphatidylserine receptor-targeting therapies for the treatment of cancer. Arch Pharm Res. (2019) 42:617–28. doi: 10.1007/s12272-019-01167-4
172. Irún P, Carrera-Lasfuentes P, Sánchez-Luengo M, Belio Ú, Domper-Arnal MJ, Higuera GA, et al. Pharmacokinetics and changes in lipid mediator profiling after consumption of specialized pro-resolving lipid-mediator-enriched marine oil in healthy subjects. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms242216143
173. Chen YJ, Li GN, Li XJ, Wei LX, Fu MJ, Cheng ZL, et al. Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapy. Sci Adv. (2023) 9:eadg0654. doi: 10.1126/sciadv.adg0654
174. Wang H, Tian T, Zhang J. Tumor-associated macrophages (TAMs) in colorectal cancer (CRC): from mechanism to therapy and prognosis. Int J Mol Sci. (2021) 22:8470. doi: 10.3390/ijms22168470
175. Lavy M, Gauttier V, Poirier N, Barillé-Nion S, Blanquart C. Specialized pro-resolving mediators mitigate cancer-related inflammation: role of tumor-associated macrophages and therapeutic opportunities. Front Immunol. (2021) 12:702785. doi: 10.3389/fimmu.2021.702785
176. Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell. (2022) 185:4049–4066.e25. doi: 10.1016/j.cell.2022.09.020
177. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. (2019) 26:623–633.e9. doi: 10.1016/j.chembiol.2019.01.008
178. Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. (2023) 62:102703. doi: 10.1016/j.redox.2023.102703
179. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The role of erastin in ferroptosis and its prospects in cancer therapy. Onco Targets Ther. (2020) 13:5429–41. doi: 10.2147/OTT.S254995
180. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. (2018) 9:1371. doi: 10.3389/fphar.2018.01371
181. Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan X, et al. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials. (2021) 277:121110. doi: 10.1016/j.biomaterials.2021.121110
182. Sekhar KR, Hanna DN, Cyr S, Baechle JJ, Kuravi S, Balusu R, et al. Glutathione peroxidase 4 inhibition induces ferroptosis and mTOR pathway suppression in thyroid cancer. Sci Rep. (2022) 12:19396. doi: 10.1038/s41598-022-23906-2
183. Luo Y, Yan P, Li X, Hou J, Wang Y, Zhou S, et al. pH-sensitive polymeric vesicles for GOx/BSO delivery and synergetic starvation-ferroptosis therapy of tumor. Biomacromolecules. (2021) 22:4383–94. doi: 10.1021/acs.biomac.1c00960
184. Wei Y, Wang Z, Yang J, Xu R, Deng H, Ma S, et al. Reactive oxygen species/photothermal therapy dual-triggered biomimetic gold nanocages nanoplatform for combination cancer therapy via ferroptosis and tumor-associated macrophage repolarization mechanism. J Colloid Interface Sci. (2022) 606:1950–65. doi: 10.1016/j.jcis.2021.09.160
185. Wang C, Wang J, Pan X, Yu S, Chen M, Gao Y, et al. Reversing ferroptosis resistance by MOFs through regulation intracellular redox homeostasis. Asian J Pharm Sci. (2023) 18:100770. doi: 10.1016/j.ajps.2022.11.004
186. Rao Z, Xia Y, Jia Q, Zhu Y, Wang L, Liu G, et al. Iron-based metal-organic framework co-loaded with buthionine sulfoximine and oxaliplatin for enhanced cancer chemo-ferrotherapy via sustainable glutathione elimination. J Nanobiotechnol. (2023) 21:265. doi: 10.1186/s12951-023-01998-w
187. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the “Iron maiden” cell death. Cells. (2020) 9. doi: 10.3390/cells9061505
188. Hao S, Liang B, Huang Q, Dong S, Wu Z, He W, et al. Metabolic networks in ferroptosis. Oncol Lett. (2018) 15:5405–11. doi: 10.3892/ol.2018.8066
189. Zhao Y, Zhao W, Lim YC, Liu T. Salinomycin-loaded gold nanoparticles for treating cancer stem cells by ferroptosis-induced cell death. Mol Pharm. (2019) 16:2532–9. doi: 10.1021/acs.molpharmaceut.9b00132
190. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano. (2020) 14:3414–25. doi: 10.1021/acsnano.9b09426
191. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
192. He Z, Zhou H, Zhang Y, Du X, Liu S, Ji J, et al. Oxygen-boosted biomimetic nanoplatform for synergetic phototherapy/ferroptosis activation and reversal of immune-suppressed tumor microenvironment. Biomaterials. (2022) 290:121832. doi: 10.1016/j.biomaterials.2022.121832
Keywords: cancer, efferocytosis, ferroptosis, inflammation, tissue homeostasis
Citation: Firouzjaei AA and Mohammadi-Yeganeh S (2024) The intricate interplay between ferroptosis and efferocytosis in cancer: unraveling novel insights and therapeutic opportunities. Front. Oncol. 14:1424218. doi: 10.3389/fonc.2024.1424218
Received: 01 May 2024; Accepted: 04 October 2024;
Published: 31 October 2024.
Edited by:
Luisa Lanfrancone, European Institute of Oncology (IEO), ItalyReviewed by:
Praveen Koganti, Sanford Burnham Prebys Medical Discovery Institute, United StatesChuanlong Zhang, Capital Medical University, China
Copyright © 2024 Firouzjaei and Mohammadi-Yeganeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samira Mohammadi-Yeganeh, c215ZWdhbmVoQGdtYWlsLmNvbQ==
†Present address: Ali Ahmadizad, Bioinformatics Research Center, Mashhad University of Medical Sciences, Mashhad, Iran