 David Sánchez-Marín
David Sánchez-Marín Macrina Beatriz Silva-Cázares
Macrina Beatriz Silva-Cázares Manuel González-Del Carmen
Manuel González-Del Carmen Alma D. Campos-Parra
Alma D. Campos-Parra
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 08 May 2024
Sec. Molecular and Cellular Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1407511
This article is part of the Research Topic Repurposed Drugs Targeting Cancer Signaling Pathways: Clinical Insights to Improve Oncologic Therapies Volume II View all 10 articles
The diagnosis of thyroid cancer (TC) has increased dramatically in recent years. Papillary TC is the most frequent type and has shown a good prognosis. Conventional treatments for TC are surgery, hormonal therapy, radioactive iodine, chemotherapy, and targeted therapy. However, resistance to treatments is well documented in almost 20% of all cases. Genomic sequencing has provided valuable information to help identify variants that hinder the success of chemotherapy as well as to determine which of those represent potentially druggable targets. There is a plethora of targeted therapies for cancer, most of them directed toward point mutations; however, chromosomal rearrangements that generate fusion genes are becoming relevant in cancer but have been less explored in TC. Therefore, it is relevant to identify new potential inhibitors for genes that are recurrent in the formation of gene fusions. In this review, we focus on describing potentially druggable variants and propose both point variants and fusion genes as targets for drug repositioning in TC.
Thyroid cancer (TC) is the most common malignant tumor of the endocrine system, with 586,202 new cases worldwide in 2020 (1). The overall incidence of TC has increased dramatically in the last 30 years. This increase may be due to overdiagnosis, thanks to improvements made in diagnostic procedures (2). Morphologically and clinically, TC is classified into two main groups: differentiated cancer—comprising papillary and follicular thyroid cancer—and undifferentiated TC, designated anaplastic carcinoma of the thyroid (3). The most prevalent is papillary thyroid cancer (PTC), which accounts for up to 85% and has a good prognosis (5-year survival rate of more than 95%, mainly in patients with stage I or II disease), as does follicular thyroid cancer (FTC), which is less prevalent, accounting for 15% of all cases (4). Patients with poorly differentiated or anaplastic TC, advanced-stage disease, or distant metastases have higher mortality rates (5). Moreover, about 20% of PTC patients manifest disease recurrence because of drug resistance, suggesting a change in treatment approaches. This points out the need to personalize treatments, including drug repositioning (6). Target therapy can be repositioned and offers greater success since it can be customized according to the patient’s genomic alterations. In this review, we highlight therapeutic opportunities for TC, focusing on druggable genes with potential repositioning for personalized therapy.
Radioactive iodine administration and/or surgery remain the first line of treatment for TC; however, for advanced disease, chemotherapy (CT) becomes the systemic option of treatment available (7). Nevertheless, CT constantly faces resistance and severe secondary effects (8). Therefore, it is necessary to overcome resistance by recognizing drug-susceptible mutations, which may lead to the identification of a broad spectrum of target therapies that could be repositioned in TC (Figure 1).
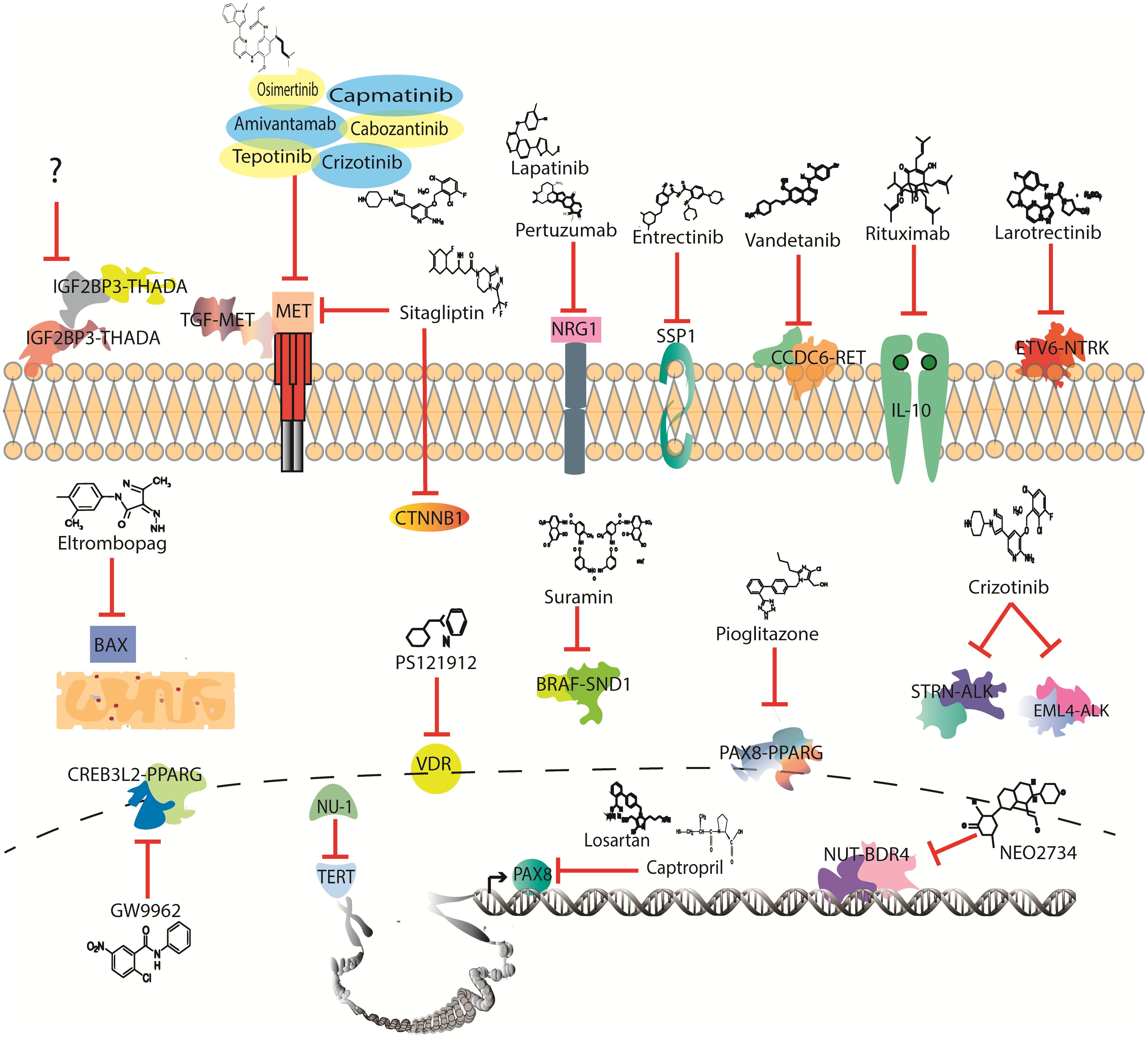
Figure 1 Drugs with potential to be investigated in thyroid cancer clinical trials according to mutational profile.
Next-Generation Sequencing (NGS) has made it possible to sequence the genomes of different types of cancer, which has revealed that around 90% of patients with TC have one or more genetic abnormalities (9). Dysregulation of phosphatidylinositol 3-kinase (PI3KCA) and mitogen-activated protein kinase (MAPK) signaling pathways is mainly affected by point mutations in target genes such as B-Raf proto-oncogene, serine/threonine kinase (BRAF), A-Raf proto-oncogene, serine/threonine kinase (RAS), and ret proto-oncogene (RET) (10). One of the best-documented and highest prevalence point mutations in PTC is BRAF exon 15 p. V600E (45% of all cases), which is associated with poor prognosis and high recurrence (11). The BRAF exon 15 p. V600E variant has constitutively active BRAF serine–threonine leading to the activation of effectors of the MAPK pathway and, consequently, surveillance and proliferation (11). Vemurafenib has shown antitumor activity in patients with BRAF exon 15 p. V600E-positive progressive PTC, representing a potential new therapeutic option (12, 13). Ipilimumab, nivolumab, dabrafenib, and trametinib are also approved target therapy options for BRAF mutations in melanoma (14) that could be repositioned to TC. In addition, drugs blocking phosphatase and tensin homolog (PTEN) and PI3KCA homogenize the font of the letter with that of the rest of the text effects (Table 1).
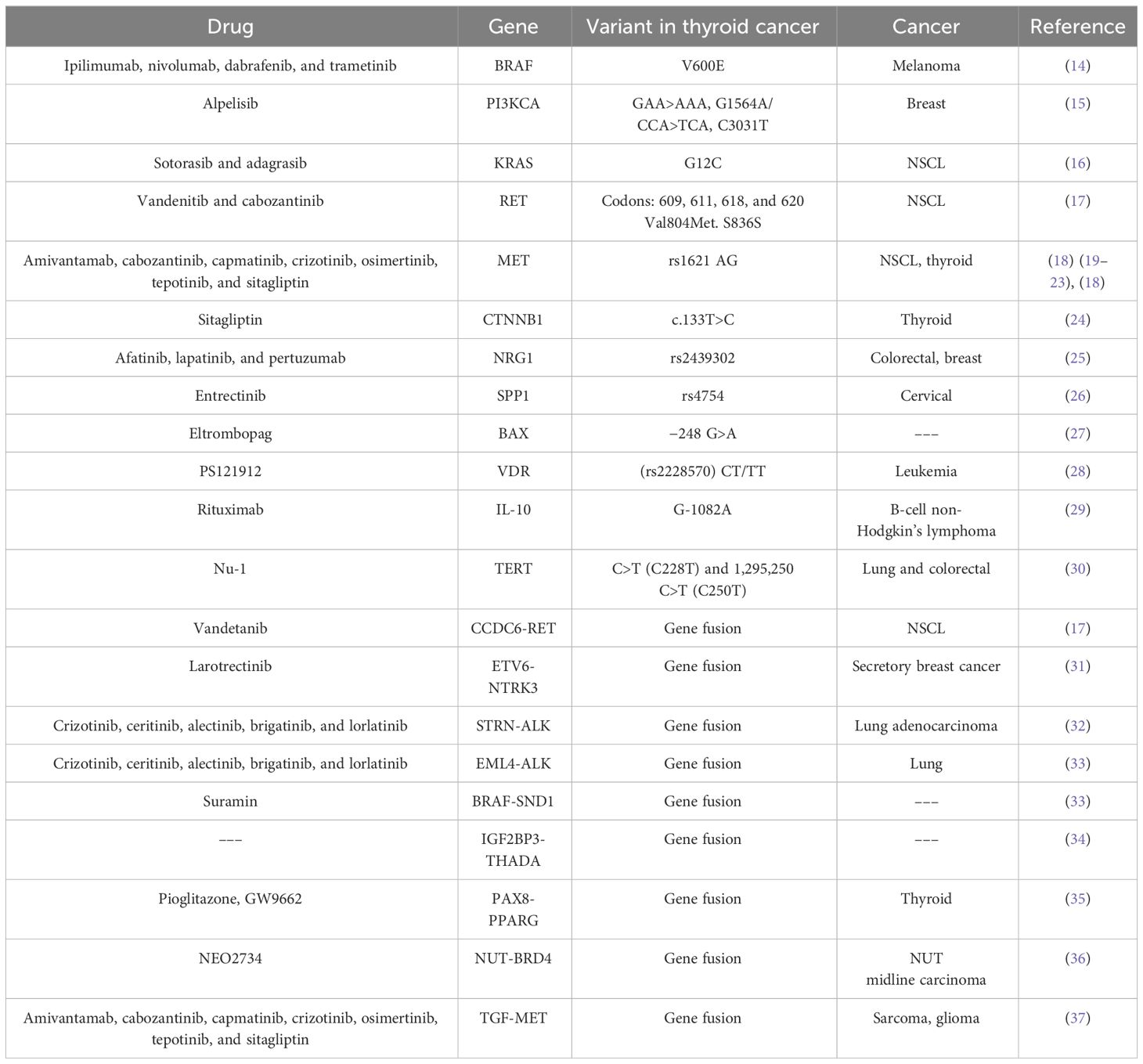
Table 1 Variants in potentially druggable genes in thyroid cancer.
PI3KCA is another gene with several missense mutations in three subtypes of TC: follicular, papillary, and anaplastic. Interestingly, PI3KCA mutations are associated with drug resistance in BRAF exon 15 p. V600E-positive cases. In this scenario, it is worth looking at how alpelisib can counteract the resistance mechanism by diminishing the EPH receptor B2 (EPHB2)-induced signaling (38). Consistent with the latest, PTEN, which has a negative regulatory role in the same pathway, has reported variants in TC (39).
KRAS proto-oncogene, GTPase (KRAS), is a G protein that plays an important role in the PI3KCA/MAPK signaling pathway. Point mutations in KRAS usually occur at codons 12, 13, and 61 and have been found in FTC and PTC at a frequency of 50% and 20%, respectively. These mutations confer a more aggressive phenotype and increase the risk of mortality (40). Sotorasib and adagrasib are KRAS exon 2 p. G12C mutation drugs approved for non-small cell lung cancer (NSCLC) (16, 41). It remains of interest to analyze the effect of these drugs on TC harboring the KRAS exon 2 p. G12C mutation. RET is another gene commonly mutated in PTC and medullary thyroid cancer (MTC), with both large rearrangements and point mutations reported. RET is a receptor tyrosine kinase that regulates growth, survival, migration, and survival, activating multiple intracellular signaling pathways, including PI3KCA/AKT serine/threonine kinase 1 (AKT), MAPK, mitogen-activated protein kinase 8 (JNK), and others. Oncogenic activating point mutations can occur mainly in the extracellular domain, particularly in codon C634 of exon 11, in 609, 611, 618, or 620 of exon 10, and in M918 of exon 16, being RET exon 16 p. M918T mutation the most common and represents more than 75% of all RET somatic mutations found in MTC (42). Selpercatinib and pralsetinib are RET-specific inhibitors approved for the MTC variant and have been well tolerated (43–45). Other multitargeted kinase inhibitors used to inhibit the PI3KCA/AKT/mechanistic target of the rapamycin kinase (MTOR) pathway in MTC are vandenitib and cabozantinib. The first one inhibits RET but also inhibits other kinases such as vascular endothelial growth factor receptor 2 (VEGFR2), vascular endothelial growth factor receptor 3 (VEGFR3), and epidermal growth factor receptor (EGFR), while cabozantinib inhibits RET, vascular endothelial growth factor (VEGF), MET proto-oncogene, receptor tyrosine kinase (MET), and ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) (46). Both inhibitors have shown efficacy and improved overall survival in patients harboring RET exon 16 p. M918T mutation (47, 48).
MET is a receptor tyrosine kinase that has an oncogene role in promoting angiogenesis due to downstream activation of RAS, PI3KCA, and signal transducer and activator of transcription 3 (STAT3) signaling pathways (49). Drugs that inhibit MET are amivantamab, cabozantinib, capmatinib, crizotinib, osimertinib, and tepotinib (50). Particularly, MET has a reported variant in NSCLC that skips the exon 14 and makes the protein constitutively active (51). In TC, it constitutes an inclusion criterion for thyroid gland medullary carcinoma (52, 53). Currently, therapy targeting MET, although only indicated to treat NSCL, represents a potential target in TC. Furthermore, a PTC expression signature has been identified in which three genes are overexpressed, promoting metastasis and being associated with poor prognosis: dipeptidyl peptidase 4 (DPP4), MET, and catenin beta 1 (CTNNB1). The signature is associated with immunosuppression and correlates with tumor infiltration of tumor-associated macrophages, which leads to T-cell exclusion. Interestingly, sitagliptin, an FDA-approved drug to treat diabetes type II, has affinity not only to DPP4 (diabetes target) but also to MET and CTNNB1 (54–56). Moreover, the affinity for MET and CTNNB1 is even higher than FDA-approved inhibitors specific for each of them, like crizotinib and PNU-74654, respectively. Therefore, sitagliptin represents a multidrug therapy window for TC (18).
Paired box 8 (PAX8), a gene implicated in proliferation and migration, is usually overexpressed in TC. Likewise, in high-grade serous ovarian cancer, PAX8 is upregulated (57). Remarkably, losartan and captopril, which are FDA-approved drugs, have been found effective at inhibiting PAX8 expression and function. This evidence suggests potential therapeutic opportunities using losartan and captopril, not only for ovarian cancer but also for TC (57).
Besides the variants reported in the above-mentioned genes, there are also polymorphisms associated with TC (58). For instance, neuregulin 1 (NRG1) acts as an oncogene through its role as a glycoprotein that mediates cell-to-cell signaling (59). In breast cancer, lapatinib may be used to inhibit EGFR and erb-b2 receptor tyrosine kinase 2 (HER2) kinases, two receptors of also relevant function in TC. Nonetheless, resistance is acquired and correlates with an increased expression of NRG1. By trying to overcome it, adding pertuzumab has shown promising results in decreasing NRG1-acquired resistance and tumor progression (25).
Similarly, secreted phosphoprotein 1 (SPP1), an integrin-binding glycophosphoprotein overexpressed in TC that promotes tumorigenesis through the inhibition of differentiation factors of thyroid cells, represents an opportunity for drug repositioning (60, 61). Although there are no current FDA drugs approved for inhibiting SPP1, a recent publication showed a promising inhibitory drug for cervical cancer: entrectinib (26). This represents a highlight, as entrectinib is an FDA-approved drug for NTRK fusions in solid tumors, including TC (62).
As with SPP1, another window of opportunity for targeted treatment is BCL2-associated X, apoptosis regulator (BAX). This gene participates in mitochondrial regulation of cell death; however, in cancer, it contributes to cell death dysregulation (63). Importantly, in TC, BAX has a reported polymorphism positively correlated with PTC, and more importantly, the FDA-approved drug eltrombopag acts as a BAX inhibitor, which drives apoptosis induction (64, 65). SPP1 and BAX are not the only genes in which polymorphism is related to TC. VDR stands for vitamin D receptor and has been associated with cancer development (66). It is not well established if TC contributes to or attenuates tumor growth; however, two polymorphisms, FokI and TaqI, are associated with a more aggressive type, and the heterozygous FokI to metastasis (67). Remarkably, it has been shown that antagonists of vitamin D have therapeutic effects as they inhibit downstream cell cycle proliferation. There is already an insight into potential therapies using VDR as a druggable target. For instance, PS121912 has shown promising therapeutic effects by acting as a selective VDR inhibitor (28).
In the immunology context, several profiles have been described causing differential expression and immune cell proliferation among TC subtypes (68). Interleukin-10 (IL-10) is one of several dysregulated cytokines in TC associated with immunological and apoptosis evasion and aggressiveness (68). This effect is caused by expression induction of BCL2 like 1 (bcl-xL) and BCL2 apoptosis regulator (BcCL2) and resistance to CD95-mediated apoptosis (69, 70). Due to its oncological role, IL-10 figures as a potential therapeutic target. Rituximab has promising inhibitory effects against IL-10 through downregulation of BCL2 and sensitization of B-cell non-Hodgkin’s lymphoma to apoptosis (29). However, resistance constitutes a problem due to broad kinase inhibitor activity and toxicity, which may limit their use and encourage the use of more specific inhibitors (71).
Lastly, telomerase reverse transcriptase (TERT), an enzyme known to be implicated in cancer, has been described as one of the most frequently mutated genes in TC, particularly in its promoter, which causes its overactivation. TERT inhibitors are currently under study, and NU-1 not only sensitizes the cell to chemo- and radiotherapy but also can inhibit proliferation and increase immune activity (30).
From NGS of long DNA fragments, gene fusions have been identified. When two genes conform to a fusion, they either lose or gain function. In cancer, they can contribute to tumor growth due to constitutive activation of an oncogene, such as BCR-ABL (72). Remarkably, some gene fusions are considered drivers, while others contribute to generating more genomic instability and disease development. There are gene fusions that are found across various cancers (73). These features of gene fusions represent an unprecedented opportunity to develop target therapies aimed at providing personalized medicine to patients.
Over 50 gene fusions have been identified in TC, which are mainly conformed by the RET, neurotrophic receptor tyrosine kinase (NTRK), ALK, and BRAF genes (74). These genes are tyrosine kinase overactivated mainly due to kinase retention and overexpression by transcription factors of the parental genes, making them druggable targets (75). Currently, three drugs are being used in clinics to treat TC-targeting gene fusions: pralsetinib, selpercatinib, and larotrectinib (76). The first two are RET inhibitors and were first set as a treatment for both point mutations and gene fusions; however, selpercatinib shows efficacy in specific RET variant genotypes that present pralsetinib resistance. For instance, BaF4/KIF5B-RET shows tumor growth despite treatment with pralsetinib, while selpercatinib can effectively inhibit growth (44, 77). However, as with other variants, these gene fusions are not expressed across all subtypes of cancer, while some therapies face drug resistance and lack of treatment for greater, yet untargeted variants (Figure 1).
For RET, 19 fusions have been described; however, only therapies consisting of RET inhibitors are currently available, leaving the partner genes pharmacologically unexplored (78). This is of great importance as it has been described that the inhibitory sensitivity of several gene fusions varies depending on the partner gene; hence, drug screening should be performed testing not only the most common gene. For instance, the coiled-coil domain containing 6 (CCDC6) is a recurrent gene-forming fusion with RET in lung cancer, where it has shown potential druggability of EGFR inhibitors in combination with RET inhibitors, decreasing resistance to RET inhibitors while also enhancing sensitivity to PARP inhibitors (79). Particularly, the fusion CCDC6-RET is more sensitive to vandetanib due to the off-target inhibitory effect and crosstalk with EGFR pathway activation (80). Furthermore, this fusion and ERC1-RET have not had a response to the RET drug, cabozantinib, supporting the idea of focusing on the second gene as well (81).
Larotrectinib targets the NTRK genes, which are neurotrophic tyrosine kinase receptors. If binding occurs, the protein phosphorylates itself and activates the MAPK pathway. Therefore, as part of a gene fusion, it causes its constitutive activation (82). Several fusions involving NTRK1, NTRK2, and NTRK3 have been described in the lung, colon, brain, head and neck, and TC (83). For this reason, it has been a promising targeted therapy, as the same fusions can occur in several tissues. In TC, larotrectinib is administered to patients diagnosed with the anaplastic subtype, and tumor growth continues despite other treatments (82). An example of this is the ETV6-NTRK3 fusion, which has been described as a driver variant in secretory breast cancer with high efficacy upon larotrectinib treatment (84). However, larotrectinib therapy targets only the NTRK gene, while their partner genes remain untargeted. For instance, sequestosome 1 (SQSTM1) is a gene that conforms to fusions with both NTRKs and plays a role in autophagy, specifically through the AKT/protein kinase AMP-activated catalytic subunit alpha 2 (AMPK)/MTOR signaling reported in PTC (84).
Although only three drugs are being used in TC to target gene fusions, there are several other recurrent genes forming gene fusions that are already targeted in other cancers. On one hand, there is ALK, which is widely known for its oncogenic role, especially as part of gene fusions (85). Currently, ALK fusions do not have a regimen of treatment for TC, but its potential has already been evaluated. For example, STRN-ALK and EML4-ALK are promising targets in TC using the FDA-approved drug crizotinib, among other drugs such as ceritinib, alectinib, brigatinib, and lorlatinib (32, 33).
On the other hand, there are BRAF fusions, and remarkably, despite BRAF having several target drugs, none of them are used to treat TC. Furthermore, among all the gene partners of BRAF, staphylococcal nuclease and Tudor domain containing 1 (SND1), an oncogene in several types of cancer acts in addition to posttranscriptional modifications (86). This is a highlight for novel therapy, as a small molecule called suramin has been identified to inhibit their protein by impairing its interaction with several microRNAs and sensitizing the response to standard chemotherapy (87).
Interestingly, up to five gene fusions are involved in THADA armadillo repeat containing (THADA), which stands for thyroid adenoma-associated gene (Table 1) (74). This gene participates in metabolism and energy storage through the calcium pathway. In cancer, not only fusions but also polymorphisms are associated with the disease development (88). Particularly, it has been described that THADA is necessary to retain CD274 in the Golgi for maturation. On the contrary, if suppressed, the immune response is triggered through the infiltration of CD8 + T cells and increased toxicity (89). In addition to this finding, the IGF2BP3-THADA fusion has been demonstrated to cause overexpression of the partner gene IGF2BP3, leading to sustained growth and invasion through the activation of PI3KCA and MAPK pathways (34, 90). For its part, insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) is associated with a poor prognosis implicated in several mechanisms leading to aberrant metabolism in cancer (91). Currently, there are no inhibitors for THADA; however, the data strongly point out THADA as a potential therapeutic target in TC.
Another gene found in 30%–35% of FTC is PAX8-PPARG, characterized as an oncogene due to its binding to several genomic regions that code for genes related to cell proliferation, apoptosis evasion, and motility (92). Contrary to the case of repurposing losartan to PAX8 alterations, this fusion promotes tumor progression due to the likely loss of functions of peroxisome proliferator-activated receptor gamma (PPARG). When inhibited with pioglitazone, anti-inflammatory effects and growth modulation are observed; however, the function of the gene fusion is not yet fully understood (35). Opposed to this idea, the antitumoral effect of PPARG inhibitor GW9662 has also been described, indicating the existence of independent pathways of PPARG (93). Remarkably, PAX8-PPARG is not the only fusion in TC involving PPARG; there is also CREB3L2-PPARG (94).
NUT-BDR4 is an oncogenic driver fusion that causes a rare type of cancer named NUT midline carcinoma. Bromodomain-containing protein 4 (BDR4) binds to the chromatin, while NUT midline carcinoma family member 1 (NUT) recruits histone acetyltransferase (HAT), promoting the expression of several associated oncogenes (95). This rare fusion has also been described in some TC cases, and it is associated with high expression of CD274 (96). The prognosis is low, with an estimated overall survival of 10 months, while therapy consists of radiotherapy and standard chemotherapy for large tumors. With no targeted therapy available, it is an urgent matter to start studying potential inhibitors for the treatment of these patients (97). Currently, only one inhibitor has been proposed to target the NUT-BDR4 fusion. It consists of a dual inhibitor of bromodomain and extra-terminal motif (BET) proteins and the p300 bromodomain, named NEO2734, with proven inhibition of tumor growth and improvement of overall survival (36).
Lastly, MET not only has point mutations in TC but also a gene fusion. It has been identified that TGF-MET fusion is present in sarcoma, glioma, and TC (37, 98). Interestingly, in sarcoma, tumors that have this variant do not fit into a specific subtype, which is a remarkable finding due to the existence of effective MET inhibitors (50, 98).
It is relevant to recognize that in the era of personalized medicine, drug repositioning has a major impact on oncology. This is possible due to the identification of new therapeutic targets, which can be shared in different diseases and even between cancers. This opens a whole window of opportunity for the use of a plethora of drugs, reducing the time and costs involved in the production of new drugs, which has a positive impact on patients. In this review, we found that several drugs used in different types of cancer can be repositioned to TC, either by the presence of point mutations or by gene fusions. We found an area of opportunity for 13 genes with missense mutations and 10 for gene fusions. Among all these drugs, 22 are FDA-approved drugs, while the remaining five are inhibitors with proven efficacy in in vitro studies, both of which represent a promising area of therapy opportunity. It is the aim of this work to highlight the relevance of the identification of new potential inhibitors for genes that are part of recurrent fusion formation in TC as well as other types of cancer due to the likelihood of their contribution to disease development. Hence, it is of interest to the clinic to elucidate these variants’ potential as biomarkers or prognostic or therapeutic targets.
DS-M: Writing – review & editing, Writing – original draft, Conceptualization. MS-C: Investigation, Formal analysis, Writing – original draft. MG-DC: Writing – original draft, Investigation, Formal analysis. AC-P: Writing – review & editing, Supervision, Resources, Project administration, Methodology, Funding acquisition, Conceptualization, Writing – original draft, Investigation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Instituto de Salud Pública, Universidad Veracruzana, Xalapa, Veracruz, México.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pizzato M, Li M, Vignat J, Laversanne M, Singh D, La Vecchia C, et al. The epidemiological landscape of thyroid cancer worldwide: GLOBOCAN estimates for incidence and mortality rates in 2020. Lancet Diabetes Endocrinol. (2022) 10:264–72. doi: 10.1016/S2213-8587(22)00035-3
2. Xie Z, Zhou J, Zhang X, Li Z. Clinical potential of microbiota in thyroid cancer therapy. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:166971. doi: 10.1016/j.bbadis.2023.166971
3. Cameselle-Teijeiro JM, Sobrinho-Simões M. New WHO classification of thyroid tumors: a pragmatic categorization of thyroid gland neoplasms. Endocrinol Diabetes Nutr (Engl Ed). (2018) 65:133–5. doi: 10.1016/j.endinu.2017.11.012
4. Ganly I, Nixon IJ, Wang LY, Palmer FL, Migliacci JC, Aniss A, et al. Survival from differentiated thyroid cancer: what has age got to do with it? Thyroid. (2015) 25:1106–14. doi: 10.1089/thy.2015.0104
5. Wan Z, Wang B, Yao J, Li Q, Miao X, Jian Y, et al. Predictive factors and clinicopathological characteristics of outcome in poorly differentiated thyroid carcinoma: a single-institution study. Front Oncol. (2023) 13:1102936. doi: 10.3389/fonc.2023.1102936
6. Zhang Y, Xing Z, Liu T, Tang M, Mi L, Zhu J, et al. Targeted therapy and drug resistance in thyroid cancer. Eur J Med Chem. (2022) 238:114500. doi: 10.1016/j.ejmech.2022.114500
7. Nguyen QT, Lee EJ, Huang MG, Park YI, Khullar A, Plodkowski RA. Diagnosis and treatment of patients with thyroid cancer. Am Health Drug Benefits. (2015) 8:30–40.
8. Al-Malky HS, Al Harthi SE, Osman A-MM. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J Oncol Pharm Pract. (2020) 26:434–44. doi: 10.1177/1078155219877931
9. Hsiao SJ, Nikiforov YE. Molecular approaches to thyroid cancer diagnosis. Endocr Relat Cancer. (2014) 21:T301–313. doi: 10.1530/ERC-14-0166
10. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. (2003) 63:1454–7.
11. Abdulhaleem M, Bandargal S, Pusztaszeri MP, Rajab M, Greenspoon H, Krasner JR, et al. The impact of BRAF V600E mutation allele frequency on the histopathological characteristics of thyroid cancer. Cancers (Basel). (2023) 16:113. doi: 10.3390/cancers16010113
12. Brose MS, Cabanillas ME, Cohen EEW, Wirth LJ, Riehl T, Yue H, et al. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1272–82. doi: 10.1016/S1470-2045(16)30166-8
13. Chen D, Su X, Zhu L, Jia H, Han B, Chen H, et al. Papillary thyroid cancer organoids harboring BRAFV600E mutation reveal potentially beneficial effects of BRAF inhibitor-based combination therapies. J Transl Med. (2023) 21:9. doi: 10.1186/s12967-022-03848-z
14. Trojaniello C, Sparano F, Cioli E, Ascierto PA. Sequencing targeted and immune therapy in BRAF-mutant melanoma: lessons learned. Curr Oncol Rep. (2023) 25:623–34. doi: 10.1007/s11912-023-01402-8
15. Chang D-Y, Ma W-L, Lu Y-S. Role of alpelisib in the treatment of PIK3CA-mutated breast cancer: patient selection and clinical perspectives. Ther Clin Risk Manag. (2021) 17:193–207. doi: 10.2147/TCRM.S251668
16. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery. (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167
17. Lee S-H, Lee J-K, Ahn M-J, Kim D-W, Sun J-M, Keam B, et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: a phase II clinical trial. Ann Oncol. (2017) 28:292–7. doi: 10.1093/annonc/mdw559
18. Cheng S-Y, Wu ATH, Batiha GE-S, Ho C-L, Lee J-C, Lukman HY, et al. Identification of DPP4/CTNNB1/MET as a theranostic signature of thyroid cancer and evaluation of the therapeutic potential of sitagliptin. Biol (Basel). (2022) 11:324. doi: 10.3390/biology11020324
19. Grüllich C. Cabozantinib: multi-kinase inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. (2018) 211:67–75. doi: 10.1007/978-3-319-91442-8_5
20. Ramesh S, Cifci A, Javeri S, Minne R, Longhurst CA, Nickel KP, et al. MET inhibitor capmatinib radiosensitizes MET exon 14-mutated and MET-amplified non-small cell lung cancer. bioRxiv. (2023) 2023:10.26.564232. doi: 10.1101/2023.10.26.564232
21. Shalata W, Yakobson A, Weissmann S, Oscar E, Iraqi M, Kian W, et al. Crizotinib in MET exon 14-mutated or MET-amplified in advanced disease non-small cell lung cancer: A retrospective, single institution experience. Oncology. (2022) 100:467–74. doi: 10.1159/000525188
22. Hartmaier RJ, Markovets AA, Ahn MJ, Sequist LV, Han J-Y, Cho BC, et al. Osimertinib + Savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discovery. (2023) 13:98–113. doi: 10.1158/2159-8290.CD-22-0586
23. Lyczak A. [Effect of tetanus toxin on the activity of the 5-hydroxytryptaminergic system in the mouse brain]. Med Dosw Mikrobiol. (1988) 40:155–60.
24. Cadranel J, Liu SV, Duruisseaux M, Branden E, Goto Y, Weinberg BA, et al. Therapeutic potential of afatinib in NRG1 fusion-driven solid tumors: A case series. Oncologist. (2021) 26:7–16. doi: 10.1634/theoncologist.2020-0379
25. Leung W, Roxanis I, Sheldon H, Buffa FM, Li J, Harris AL, et al. Combining lapatinib and pertuzumab to overcome lapatinib resistance due to NRG1-mediated signalling in HER2-amplified breast cancer. Oncotarget. (2015) 6:5678–94. doi: 10.18632/oncotarget.3296
26. Poleboyina PK, Alagumuthu M, Pasha A, Ravinder D, Pasumarthi D, Pawar SC. Entrectinib a plausible inhibitor for osteopontin (SPP1) in cervical cancer-integrated bioinformatic approach. Appl Biochem Biotechnol. (2023) 195:7766–95. doi: 10.1007/s12010-023-04541-7
27. Chen S-M, Feng J-N, Zhao C-K, Yao L-C, Wang L-X, Meng L, et al. A multi-targeting natural product, aiphanol, inhibits tumor growth and metastasis. Am J Cancer Res. (2022) 12:4930–53.
28. Sidhu PS, Teske K, Feleke B, Yuan NY, Guthrie ML, Fernstrum GB, et al. Anticancer activity of VDR-coregulator inhibitor PS121912. Cancer Chemother Pharmacol. (2014) 74:787–98. doi: 10.1007/s00280-014-2549-y
29. Alas S, Emmanouilides C, Bonavida B. Inhibition of interleukin 10 by rituximab results in down-regulation of bcl-2 and sensitization of B-cell non-Hodgkin’s lymphoma to apoptosis. Clin Cancer Res. (2001) 7:709–23.
30. Liu Y, Betori RC, Pagacz J, Frost GB, Efimova EV, Wu D, et al. Targeting telomerase reverse transcriptase with the covalent inhibitor NU-1 confers immunogenic radiation sensitization. Cell Chem Biol. (2022) 29:1517–1531.e7. doi: 10.1016/j.chembiol.2022.09.002
31. Shukla N, Roberts SS, Baki MO, Mushtaq Q, Goss PE, Park BH, et al. Successful targeted therapy of refractory pediatric ETV6-NTRK3 fusion-positive secretory breast carcinoma. JCO Precis Oncol. (2017) 2017:PO.17.00034. doi: 10.1200/PO.17.00034
32. Su C, Jiang Y, Jiang W, Wang H, Liu S, Shao Y, et al. STRN-ALK fusion in lung adenocarcinoma with excellent response upon alectinib treatment: A case report and literature review. Onco Targets Ther. (2020) 13:12515–9. doi: 10.2147/OTT.S282933
33. Mano H. The EML4-ALK oncogene: targeting an essential growth driver in human cancer. Proc Jpn Acad Ser B Phys Biol Sci. (2015) 91:193–201. doi: 10.2183/pjab.91.193
34. Panebianco F, Kelly LM, Liu P, Zhong S, Dacic S, Wang X, et al. THADA fusion is a mechanism of IGF2BP3 activation and IGF1R signaling in thyroid cancer. Proc Natl Acad Sci U.S.A. (2017) 114:2307–12. doi: 10.1073/pnas.1614265114
35. Giordano TJ, Haugen BR, Sherman SI, Shah MH, Caoili EM, Koenig RJ. Pioglitazone therapy of PAX8-PPARγ Fusion protein thyroid carcinoma. J Clin Endocrinol Metab. (2018) 103:1277–81. doi: 10.1210/jc.2017-02533
36. Morrison-Smith CD, Knox TM, Filic I, Soroko KM, Eschle BK, Wilkens MK, et al. Combined targeting of the BRD4-NUT-p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol Cancer Ther. (2020) 19:1406–14. doi: 10.1158/1535-7163.MCT-20-0087
37. Xu T, Wang H, Huang X, Li W, Huang Q, Yan Y, et al. Gene fusion in Malignant glioma: an emerging target for next-generation personalized treatment. Transl Oncol. (2018) 11:609–18. doi: 10.1016/j.tranon.2018.02.020
38. Liu D, Hou P, Liu Z, Wu G, Xing M. Genetic alterations in the phosphoinositide 3-kinase/Akt signaling pathway confer sensitivity of thyroid cancer cells to therapeutic targeting of Akt and mammalian target of rapamycin. Cancer Res. (2009) 69:7311–9. doi: 10.1158/0008-5472.CAN-09-1077
39. Colombo C, Pogliaghi G, Tosi D, Muzza M, Bulfamante G, Persani L, et al. Thyroid cancer harboring PTEN and TP53 mutations: A peculiar molecular and clinical case report. Front Oncol. (2022) 12:949098. doi: 10.3389/fonc.2022.949098
40. Bikas A, Ahmadi S, Pappa T, Marqusee E, Wong K, Nehs MA, et al. Additional oncogenic alterations in RAS-driven differentiated thyroid cancers associate with worse clinicopathologic outcomes. Clin Cancer Res. (2023) 29:2678–85. doi: 10.1158/1078-0432.CCR-23-0278
41. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
42. Ciampi R, Romei C, Pieruzzi L, Tacito A, Molinaro E, Agate L, et al. Classical point mutations of RET, BRAF and RAS oncogenes are not shared in papillary and medullary thyroid cancer occurring simultaneously in the same gland. J Endocrinol Invest. (2017) 40:55–62. doi: 10.1007/s40618-016-0526-5
43. Hadoux J, Elisei R, Brose MS, Hoff AO, Robinson BG, Gao M, et al. Phase 3 trial of selpercatinib in advanced RET-mutant medullary thyroid cancer. N Engl J Med. (2023) 389:1851–61. doi: 10.1056/NEJMoa2309719
44. Wirth LJ, Robinson B, Boni V, Tan DSW, McCoach C, Massarelli E, et al. Patient-reported outcomes with selpercatinib treatment among patients with RET-mutant medullary thyroid cancer in the phase I/II LIBRETTO-001 trial. Oncologist. (2022) 27:13–21. doi: 10.1002/onco.13977
45. Subbiah V, Hu MI, Wirth LJ, Schuler M, Mansfield AS, Curigliano G, et al. Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): a multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol. (2021) 9:491–501. doi: 10.1016/S2213-8587(21)00120-0
46. Gentile C, Martorana A, Lauria A, Bonsignore R. Kinase inhibitors in multitargeted cancer therapy. Curr Med Chem. (2017) 24:1671–86. doi: 10.2174/0929867324666170112112734
47. Fallahi P, Ferrari SM, Elia G, Ragusa F, Paparo SR, Ruffilli I, et al. Evaluating vandetanib in the treatment of medullary thyroid cancer: patient-reported outcomes. Cancer Manag Res. (2019) 11:7893–907. doi: 10.2147/CMAR.S127848
48. Cabanillas ME, de Souza JA, Geyer S, Wirth LJ, Menefee ME, Liu SV, et al. Cabozantinib as salvage therapy for patients with tyrosine kinase inhibitor-refractory differentiated thyroid cancer: results of a multicenter phase II international thyroid oncology group trial. J Clin Oncol. (2017) 35:3315–21. doi: 10.1200/JCO.2017.73.0226
49. You W-K, McDonald DM. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. (2008) 41:833–9. doi: 10.5483/bmbrep.2008.41.12.833
50. Dong Y, Xu J, Sun B, Wang J, Wang Z. MET-targeted therapies and clinical outcomes: A systematic literature review. Mol Diagn Ther. (2022) 26:203–27. doi: 10.1007/s40291-021-00568-w
51. Fujino T, Suda K, Mitsudomi T. Lung cancer with MET exon 14 skipping mutation: genetic feature, current treatments, and future challenges. Lung Cancer (Auckl). (2021) 12:35–50. doi: 10.2147/LCTT.S269307
52. Garcia C, Buffet C, El Khattabi L, Rizk-Rabin M, Perlemoine K, Ragazzon B, et al. MET overexpression and activation favors invasiveness in a model of anaplastic thyroid cancer. Oncotarget. (2019) 10:2320–34. doi: 10.18632/oncotarget.26798
53. Ning L, Yu Y, Liu X, Ai L, Zhang X, Rao W, et al. Association analysis of MET gene polymorphism with papillary thyroid carcinoma in a chinese population. Int J Endocrinol. (2015) 2015:405217. doi: 10.1155/2015/405217
54. Enz N, Vliegen G, De Meester I, Jungraithmayr W. CD26/DPP4 - a potential biomarker and target for cancer therapy. Pharmacol Ther. (2019) 198:135–59. doi: 10.1016/j.pharmthera.2019.02.015
55. Yang X, Liao H-Y, Zhang H-H. Roles of MET in human cancer. Clin Chim Acta. (2022) 525:69–83. doi: 10.1016/j.cca.2021.12.017
56. Shang S, Hua F, Hu Z-W. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. (2017) 8:33972–89. doi: 10.18632/oncotarget.15687
57. Salvi A, Hardy LR, Heath KN, Watry S, Pergande MR, Cologna SM, et al. PAX8 modulates the tumor microenvironment of high grade serous ovarian cancer through changes in the secretome. Neoplasia. (2023) 36:100866. doi: 10.1016/j.neo.2022.100866
58. Guo Y, Zhang W, He R, Zheng C, Liu X, Ge M, et al. Investigating the association between rs2439302 polymorphism and thyroid cancer: A systematic review and meta-analysis. Front Surg. (2022) 9:877206. doi: 10.3389/fsurg.2022.877206
59. Talmage DA. Mechanisms of neuregulin action. Novartis Found Symp. (2008) 289:74–84. doi: 10.1002/9780470751251.ch6
60. Wei T, Bi G, Bian Y, Ruan S, Yuan G, Xie H, et al. The significance of secreted phosphoprotein 1 in multiple human cancers. Front Mol Biosci. (2020) 7:565383. doi: 10.3389/fmolb.2020.565383
61. Ma J, Huang X, Xu J, Li Z, Lai J, Shen Y, et al. SBP1 promotes tumorigenesis of thyroid cancer through TXN/NIS pathway. Mol Med. (2023) 29:121. doi: 10.1186/s10020-023-00700-y
62. Damásio I, Simões-Pereira J, Donato S, Horta M, Cavaco BM, Rito M, et al. Entrectinib in the neoadjuvant setting of anaplastic thyroid cancer: a case report. Eur Thyroid J. (2023) 12:e220179. doi: 10.1530/ETJ-22-0179
63. Huska JD, Lamb HM, Hardwick JM. Overview of BCL-2 family proteins and therapeutic potentials. Methods Mol Biol. (2019) 1877:1–21. doi: 10.1007/978-1-4939-8861-7_1
64. Cardoso-Duarte LCA, Fratelli CF, Pereira ASR, de Souza JNG, de S Freitas R, de Morais RM, et al. BAX gene (-248 G > A) polymorphism in a sample of patients diagnosed with thyroid cancer in the Federal District, Brazil. Int J Biol Markers. (2021) 36:21–6. doi: 10.1177/17246008211057576
65. Spitz AZ, Zacharioudakis E, Reyna DE, Garner TP, Gavathiotis E. Eltrombopag directly inhibits BAX and prevents cell death. Nat Commun. (2021) 12:1134. doi: 10.1038/s41467-021-21224-1
66. Campbell MJ, Trump DL. Vitamin D receptor signaling and cancer. Endocrinol Metab Clin North Am. (2017) 46:1009–38. doi: 10.1016/j.ecl.2017.07.007
67. Cocolos A-M, Muresan A, Caragheorgheopol A, Ghemigian M, Ioachim D, Poiana C. Vitamin D status and VDR polymorphisms as prognostic factors in differentiated thyroid carcinoma. In Vivo. (2022) 36:2434–41. doi: 10.21873/invivo.12977
68. Cunha LL, Domingues GAB, Morari EC, Soares FA, Vassallo J, Ward LS. The immune landscape of the microenvironment of thyroid cancer is closely related to differentiation status. Cancer Cell Int. (2021) 21:387. doi: 10.1186/s12935-021-02084-7
69. Stassi G, Todaro M, Zerilli M, Ricci-Vitiani L, Di Liberto D, Patti M, et al. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res. (2003) 63:6784–90.
70. Todaro M, Zerilli M, Ricci-Vitiani L, Bini M, Perez Alea M, Maria Florena A, et al. Autocrine production of interleukin-4 and interleukin-10 is required for survival and growth of thyroid cancer cells. Cancer Res. (2006) 66:1491–9. doi: 10.1158/0008-5472.CAN-05-2514
71. Ghrenassia E, Mariotte E, Azoulay E. Rituximab-related Severe Toxicity. In: Vincent J-L, editor. Annual Update in Intensive Care and Emergency Medicine 2018. Annual Update in Intensive Care and Emergency Medicine. Springer International Publishing, Cham (2018). p. 579–96. doi: 10.1007/978-3-319-73670-9_43
72. Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. (2015) 15:371–81. doi: 10.1038/nrc3947
73. Yu Y-P, Liu P, Nelson J, Hamilton RL, Bhargava R, Michalopoulos G, et al. Identification of recurrent fusion genes across multiple cancer types. Sci Rep. (2019) 9:1074. doi: 10.1038/s41598-019-38550-6
74. Yakushina VD, Lerner LV, Lavrov AV. Gene fusions in thyroid cancer. Thyroid. (2018) 28:158–67. doi: 10.1089/thy.2017.0318
75. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. (2014) 5:4846. doi: 10.1038/ncomms5846
76. Nacchio M, Pisapia P, Pepe F, Russo G, Vigliar E, Porcelli T, et al. Predictive molecular pathology in metastatic thyroid cancer: the role of RET fusions. Expert Rev Endocrinol Metab. (2022) 17:167–78. doi: 10.1080/17446651.2022.2060819
77. Wu Y, Yan Z, Pan J, Chang X, Huang B, Luo D, et al. Nie X. Partial response to pralsetinib in an advanced pulmonary sarcomatoid carcinoma patient harboring a KIF5B-RET rearrangement: a case report. World J Surg Oncol. (2022) 20:386. doi: 10.1186/s12957-022-02848-z
78. Vodopivec DM, Hu MI. RET kinase inhibitors for RET-altered thyroid cancers. Ther Adv Med Oncol. (2022) 14:17588359221101691. doi: 10.1177/17588359221101691
79. Chang H, Sung JH, Moon SU, Kim HS, Kim JW, Lee JS. EGF induced RET inhibitor resistance in CCDC6-RET lung cancer cells. Yonsei Med J. (2017) 58:9–18. doi: 10.3349/ymj.2017.58.1.9
80. Levinson S, Cagan RL. Drosophila cancer models identify functional differences between ret fusions. Cell Rep. (2016) 16:3052–61. doi: 10.1016/j.celrep.2016.08.019
81. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. (2016) 17:1653–60. doi: 10.1016/S1470-2045(16)30562-9
82. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
83. O’Haire S, Franchini F, Kang Y-J, Steinberg J, Canfell K, Desai J, et al. Systematic review of NTRK 1/2/3 fusion prevalence pan-cancer and across solid tumours. Sci Rep. (2023) 13:4116. doi: 10.1038/s41598-023-31055-3
84. Yu F, Ma R, Liu C, Zhang L, Feng K, Wang M, et al. SQSTM1/p62 promotes cell growth and triggers autophagy in papillary thyroid cancer by regulating the AKT/AMPK/mTOR signaling pathway. Front Oncol. (2021) 11:638701. doi: 10.3389/fonc.2021.638701
85. He Y, Cao L, Wang L, Liu L, Huang Y, Gong X. Metformin inhibits proliferation of human thyroid cancer TPC-1 cells by decreasing LRP2 to suppress the JNK pathway. Onco Targets Ther. (2020) 13:45–50. doi: 10.2147/OTT.S227915
86. Chu Y-H, Wirth LJ, Farahani AA, Nosé V, Faquin WC, Dias-Santagata D, et al. Clinicopathologic features of kinase fusion-related thyroid carcinomas: an integrative analysis with molecular characterization. Mod Pathol. (2020) 33:2458–72. doi: 10.1038/s41379-020-0638-5
87. Lehmusvaara S, Haikarainen T, Saarikettu J, Martinez Nieto G, Silvennoinen O. Inhibition of RNA binding in SND1 increases the levels of miR-1-3p and sensitizes cancer cells to navitoclax. Cancers (Basel). (2022) 14:3100. doi: 10.3390/cancers14133100
88. Moraru A, Cakan-Akdogan G, Strassburger K, Males M, Mueller S, Jabs M, et al. THADA regulates the organismal balance between energy storage and heat production. Dev Cell. (2017) 41:72–81.e6. doi: 10.1016/j.devcel.2017.03.016
89. Li C, Chi H, Deng S, Wang H, Yao H, Wang Y, et al. THADA drives Golgi residency and upregulation of PD-L1 in cancer cells and provides promising target for immunotherapy. J Immunother Cancer. (2021) 9:e002443. doi: 10.1136/jitc-2021-002443
90. Gubbiotti MA, Andrianus S, Baloch Z. THADA-IGF2BP3 fusions detected in fine-needle aspiration specimens of thyroid nodules: An institutional experience. Diagn Cytopathol. (2023) 51:349–55. doi: 10.1002/dc.25113
91. Liu X, Chen J, Chen W, Xu Y, Shen Y, Xu X. Targeting IGF2BP3 in cancer. Int J Mol Sci. (2023) 24:9423. doi: 10.3390/ijms24119423
92. Raman P, Koenig RJ. Pax-8-PPAR-γ fusion protein in thyroid carcinoma. Nat Rev Endocrinol. (2014) 10:616–23. doi: 10.1038/nrendo.2014.115
93. Seargent JM, Yates EA, Gill JH. GW9662, a potent antagonist of PPARgamma, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARgamma agonist rosiglitazone, independently of PPARgamma activation. Br J Pharmacol. (2004) 143:933–7. doi: 10.1038/sj.bjp.0705973
94. Lui W-O, Zeng L, Rehrmann V, Deshpande S, Tretiakova M, Kaplan EL, et al. CREB3L2-PPARgamma fusion mutation identifies a thyroid signaling pathway regulated by intramembrane proteolysis. Cancer Res. (2008) 68:7156–64. doi: 10.1158/0008-5472.CAN-08-1085
95. Lauer UM, Hinterleitner M, Horger M, Ohnesorge PV, Zender L. NUT carcinoma-an underdiagnosed Malignancy. Front Oncol. (2022) 12:914031. doi: 10.3389/fonc.2022.914031
96. Zhou J, Duan M, Jiao Q, Chen C, Xing A, Su P, et al. Primary thyroid NUT carcinoma with high PD-L1 expression and novel massive IGKV gene fusions: A case report with treatment implications and literature review. Front Oncol. (2021) 11:778296. doi: 10.3389/fonc.2021.778296
97. Wang S, Li J, Tong W, Li H, Feng Q, Teng B. Advances in the pathogenesis and treatment of nut carcinoma: a narrative review. Transl Cancer Res. (2020) 9:6505–15. doi: 10.21037/tcr-20-1884
Keywords: thyroid cancer, variants, repurposed drugs, gene fusions, mutations
Citation: Sánchez-Marín D, Silva-Cázares MB, González-Del Carmen M and Campos-Parra AD (2024) Drug repositioning in thyroid cancer: from point mutations to gene fusions. Front. Oncol. 14:1407511. doi: 10.3389/fonc.2024.1407511
Received: 26 March 2024; Accepted: 16 April 2024;
Published: 08 May 2024.
Edited by:
Monica Fedele, National Research Council (CNR), ItalyReviewed by:
Pasquale Pisapia, University of Naples Federico II, ItalyCopyright © 2024 Sánchez-Marín, Silva-Cázares, González-Del Carmen and Campos-Parra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alma D. Campos-Parra, YWxtY2FtcG9zQHV2Lm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.