 Yang Yu
Yang Yu Mengdie Yu2
Mengdie Yu2 Wei Wang
Wei Wang- 1Department of Gastrointestinal Surgery, The First Affiliated Hospital of Guangzhou University of Traditional Chinese Medicine, Guangzhou, Guangdong, China
- 2Guangzhou KingMed Diagnostics Group Co., Ltd., Guangzhou, Guangdong, China
- 3Department of Thyroid and Breast Surgery, Baiyun Hospital, The First Affiliated Hospital of Guangzhou University of Traditional Chinese Medicine, Guangzhou, Guangdong, China
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours, arising mainly from the interstitial cells of Cajal (ICCs) of the gastrointestinal tract. As radiotherapy and chemotherapy are generally ineffective for GISTs, the current primary treatment is surgical resection. However, surgical resection is not choice for most patients. Therefore, new therapeutic strategies are urgently needed. Targeted therapy, represented by tyrosine kinase inhibitors (TKIs), and immunotherapy, represented by immune checkpoint inhibitor therapies and chimeric antigen receptor T-cell immunotherapy (CAR-T), offer new therapeutic options in GISTs and have shown promising treatment responses. In this review, we summarize the molecular classification and immune microenvironment of GISTs and discuss the corresponding targeted therapy and immunotherapy options. This updated knowledge may provide more options for future therapeutic strategies and applications in GISTs.
Introduction
Gastrointestinal stromal tumours (GISTs) arise from gastrointestinal stromal cells, accounting for about 4% to 5% of gastrointestinal malignancies (1). The incidence of GISTs has been increasing worldwide over the years, reflecting in part improvements in diagnostic techniques and a clearer pathological classification (2). Over the past few decades, the treatment of GISTs has mainly relied on conventional therapies such as surgical resection and radiotherapy (3, 4). However, with in-depth research into the pathogenesis of GISTs, there have been significant breakthroughs in the treatment of GISTs, particularly in the areas of targeted therapy and immunotherapy (5, 6).
GISTs typically have mutations in the KIT proto-oncogene, receptor tyrosine kinase (KIT) or platelet-derived growth factor receptor alpha (PDGFRA) genes, leading to uncontrolled cell proliferation through abnormal signalling pathways, which in turn provide therapeutic opportunities to target these molecules (7, 8). Patients with KIT mutations usually respond well to targeted therapies such as imatinib (9). However, some patients may develop resistance after long-term treatment, which is one of the main challenges of targeted therapy.
In contrast to drug-targeted therapy, immunotherapy uses the body’s own immune system to attack cancer cells and includes immune checkpoint inhibitors such as programmed cell death 1 (PDCD1; also known as PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA4) inhibitors (10). These inhibitors prevent cancer cells from evading the immune system, thereby increasing the immune system’s ability to kill cancer cells. Several preclinical studies have shown that immunotherapy can induce a positive therapeutic response in a subset of GIST patients (11, 12).
In this review, we summarize the molecular classification and immune microenvironment of GISTs. We also focus on targeted therapy and immunotherapy for GISTs. A comprehensive and updated knowledge of the characteristics and treatment strategies of GISTs may provide more appropriate options for subsequent therapeutic choices and research.
Molecular classification of GISTs
The molecular classification of GISTs is mainly based on the presence of specific mutations in their tumour cells (Figure 1), mainly including KIT, PDGFRA and succinate dehydrogenase complex iron sulfur (SDH) (Figure 2), which result in the structural activation of specific signalling pathways in a ligand-independent manner, driving the onset and progression of GISTs (Figure 3).
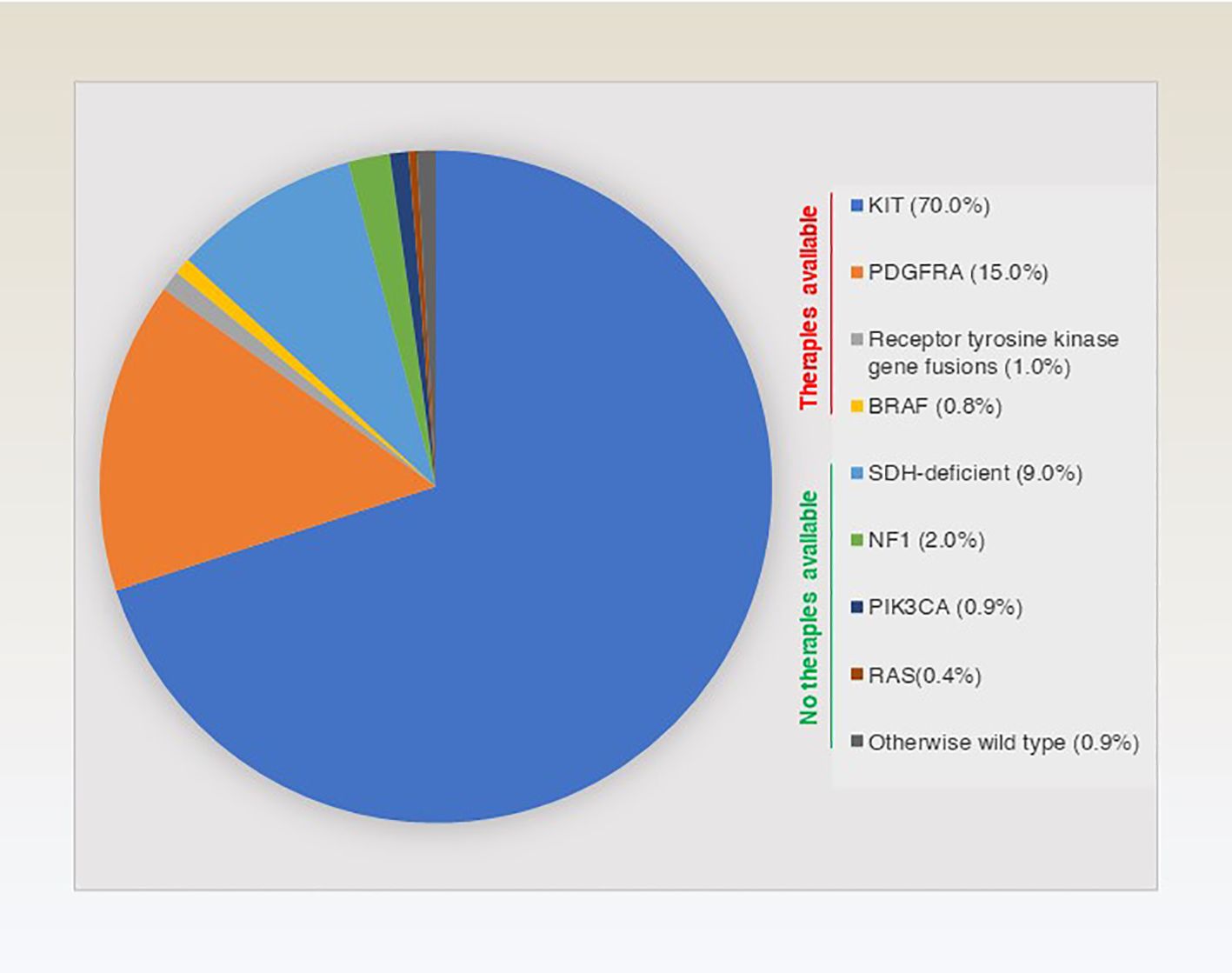
Figure 1 Overview and approximate percentage of molecular typing of genes driving GISTs.
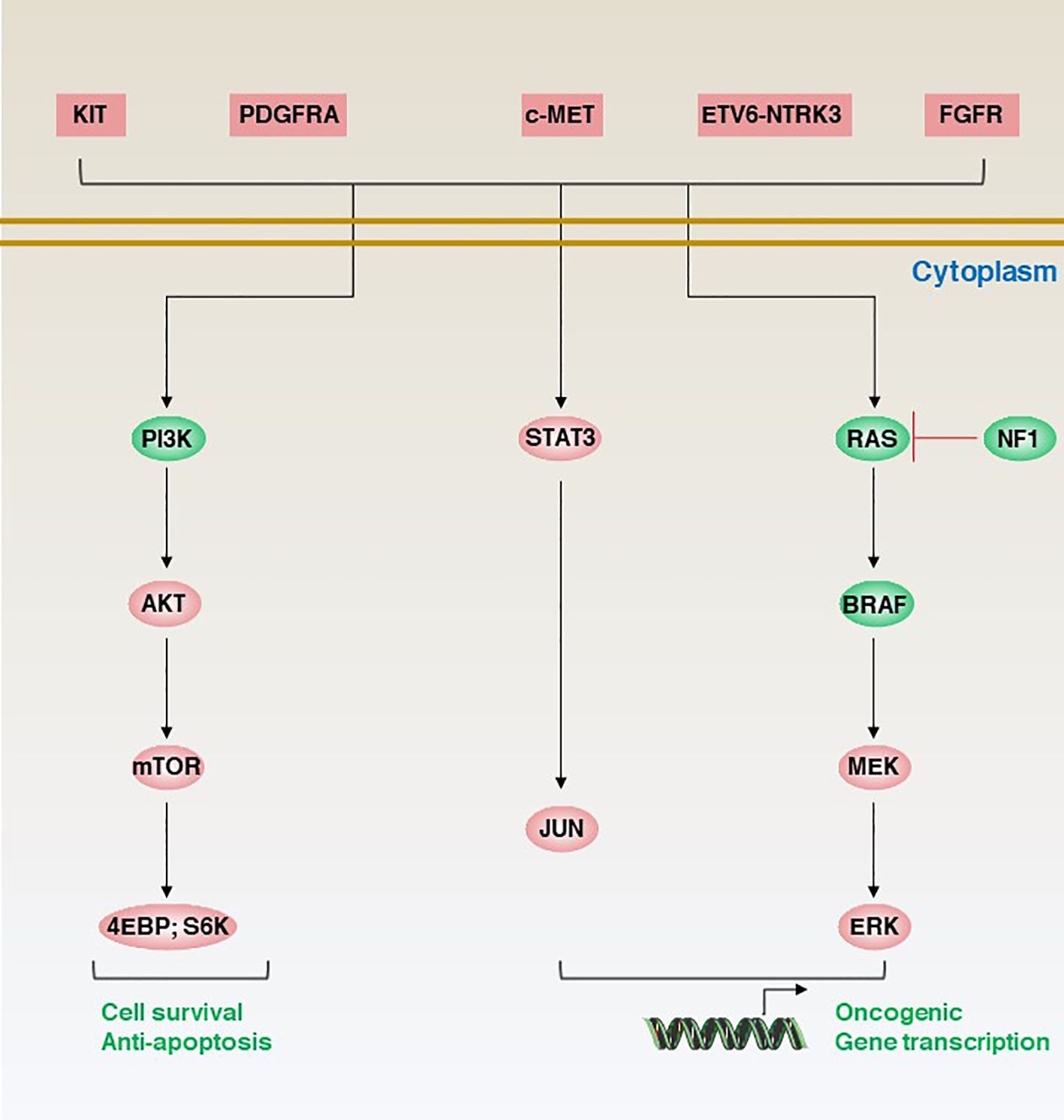
Figure 2 Genetic characteristics and molecular classification of induced GISTs.
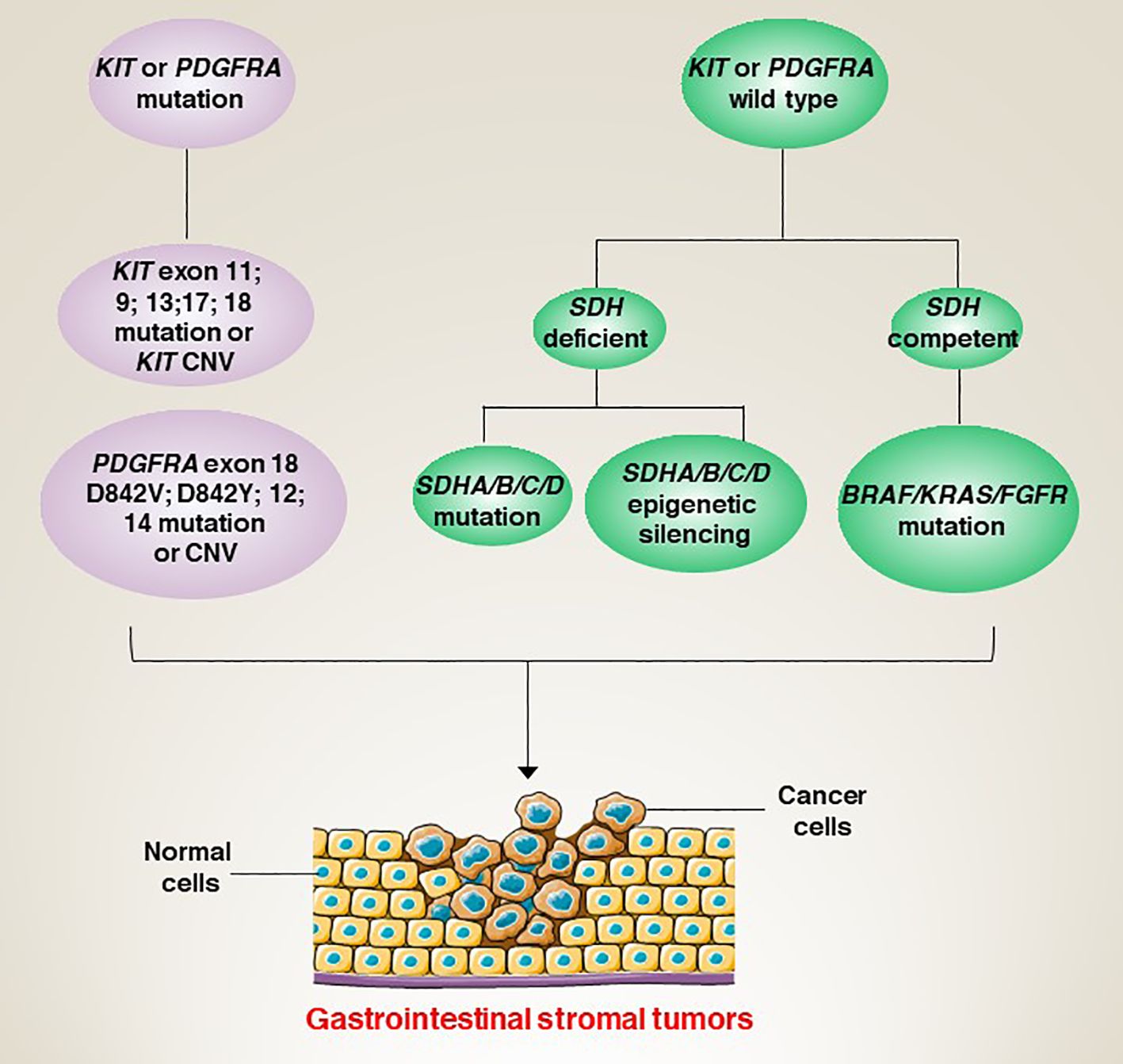
Figure 3 Aberrant genetic alterations in GISTs activate multiple signalling cascades thereby preventing apoptosis and promoting cell survival and proliferation.
KIT
The KIT gene, located on chromosome 4q12, is a proto-oncogene that encodes a type III membrane-penetrating receptor tyrosine kinase protein (13). Under normal physiological conditions, KIT encoded proteins act as cell membrane surface receptors to regulate cell growth and differentiation and are present in a variety of cell types, including embryonic stem cells, haematopoietic stem cells, gastrointestinal stromal cells (interstitial cells of Cajal) and germ cells (14–18). In particular, KIT proteins play a key role in the regulation of gastrointestinal motility and muscle contraction (19). Thus, KIT mutations are a key driver of GISTs.
Depending on the location and type of mutation, KIT gene mutations can be divided into several subtypes. The most common type of mutation occurs in the juxtamembrane domain of the KIT gene, with KIT exon 11 mutations accounting for approximately 50-70% of GIST cases. KIT exon 11 mutations usually involve a deletion, insertion or point mutation in the region of the KIT protein enhancer, resulting in persistent activation of KIT protein kinase activity (20, 21). However, KIT exon 11 mutations are also usually associated with a better prognosis. For example, sequencing of tumour DNA from 104 GIST patients showed a significantly better trend for disease-free survival (DFS) in patients with exon 11 repeat mutations and point mutations compared to other mutations (e.g., deletions, insertions) (22). However, another study showed the opposite conclusion, which may indicate that tumour heterogeneity and the specific type of mutation are critical for prognosis (23). Another common mutant isoform of the KIT gene is KIT exon 9, which accounts for about 10-20% of cases. These mutations result in insertional mutations in the kinase region of the KIT protein, which in turn allows for sustained activation of the kinase activity of the KIT protein (24). In contrast, GISTs with KIT exon 9 mutations tend to have a higher tumour burden and a poorer prognosis (25).
In addition, there are some rare mutant subtypes of the KIT gene, such as KIT exon 13, exon 17 and exon 18 mutations (8, 26, 27). Mutations in each subtype may lead to different clinical features and prognosis of GIST. While KIT gene mutation-dependent aberrant signalling mainly involves mitogen-activated protein kinase (MAPK) and AKT serine/threonine kinase (AKT) (5, 28). In addition to mutations, copy number variation (CNV) in the KIT gene is an important genetic abnormality in the development of GISTs (29).
PDGFRA
PDGFRA TKR is homologous to KIT TKR (30). Similarly, under normal conditions, PDGFRA signalling maintains a normal balance between cell growth and apoptosis through ligand binding and kinase activity (31, 32). However, in GISTs, mutations result in sustained activation of the kinase activity of these receptors. PDGFRA mutations account for approximately 10-15% of GISTs (33). Their mutations mainly affect its kinase structural domain and the most common mutation site is site 842 of exon 18, including point mutations (e.g., D842V, D842Y), deletion and insertion mutations (34). Among these, PDGFRA D842V mutations are the most common, accounting for approximately 70-80% of PDGFRA mutant GISTs (34, 35).
In particular, PDGFRA-mutant GISTs tend to have some unique clinical features and prognosis. First, PDGFRA-mutant GISTs are often found in the stomach, particularly in the gastric antrum and pylorus (36). Second, PDGFRA-mutant GISTs often have lower histological grades and lower proliferation indices, factors associated with a better prognosis (37, 38). In addition, PDGFRA-mutant GISTs tend to have smaller tumour sizes, a lower incidence of metastases and longer survival (39). GISTs with PDGFRA D842V mutations have a better prognosis but are relatively insensitive to conventional KIT inhibitors (e.g., imatinib) and therefore require higher doses of the drug to achieve an effective therapeutic response. For example, a retrospective cohort study of GIST patients with PDGFRA exon 18 mutation showed that 16 GIST patients with D842V mutation received imatinib treatment, and only 2 patients had partial response, indicating that imatinib is not generally applicable to patients with this mutation (40). On the other hand, GISTs with other types of PDGFRA exon 18 mutations (e.g., non-D842V mutations) are usually sensitive to therapeutic agents such as imatinib, have a relatively better prognosis and require lower doses of drugs for maintenance therapy (40). Similar to KIT, CNV (gene dose changes caused by chromosome gain and loss) in the PDGFRA gene is associated with the prognosis of GISTs (41, 42). In addition, the development of GISTs is closely linked to the interaction of other cell types in the tumour microenvironment, such as tumour-associated mesenchymal stromal cells and immune cells (43, 44).
SDH
In addition to KIT/PDGFRA mutations. There is also a small percentage of GISTs (~5-7.5%) whose pathogenesis is closely linked to a deletion of the succinate dehydrogenase (SDH) complex (45). SDH is a mitochondrial enzyme that plays an important role in energy metabolism and oxidative stress in cells (46). Therefore, SDH mutations cause mitochondrial dysfunction, leading to abnormal intracellular energy metabolism and increased oxidative stress, which promotes the growth and division of tumour cells (47, 48). SDH mutations can be classified into four isoforms, namely SDHA, SDHB, SDHC and SDHD (49). Notably, SDH-mutant GISTs tends to occur in younger patients, and the tumours are smaller and less malignant, suggesting that it may have a better prognosis (50). However, SDH-mutant GISTs respond poorly to treatment with conventional targeted agents (such as imatinib) and are relatively sensitive to other treatments, such as chemotherapy (51). In a study based on the molecular and metabolic profiles of a patient-derived SDH-mutant (mSDH) GIST model, temozolomide induced tumour DNA damage and apoptosis in the mSDH GIST model. Importantly, five patients with SDH-mutant GISTs who received temozolomide showed an objective remission rate of 40% and a disease control rate of 100%, suggesting that temozolomide is a promising therapeutic approach (52).
In addition, there is a small percentage of GIST patients who do not have mutations and these patients are referred to as ‘wild-type’ GISTs (53). Therefore, the development of multidisciplinary (such as combined genetic testing and tissue dynamic imaging techniques) and accurate assays to determine the genomic background of different patients is essential for precision therapy.
GIST targeted drug therapy
Targeted therapies have revolutionized the treatment strategies for GISTs over the last few decades. Many drugs have been approved by the U.S Food and Drug Administration (FDA) for the treatment of GISTs, and these drugs mainly target mutant KIT and PDGFRA. In this section, we mainly focus on targeted therapy for KIT or PDGFRA mutations (Figure 4).
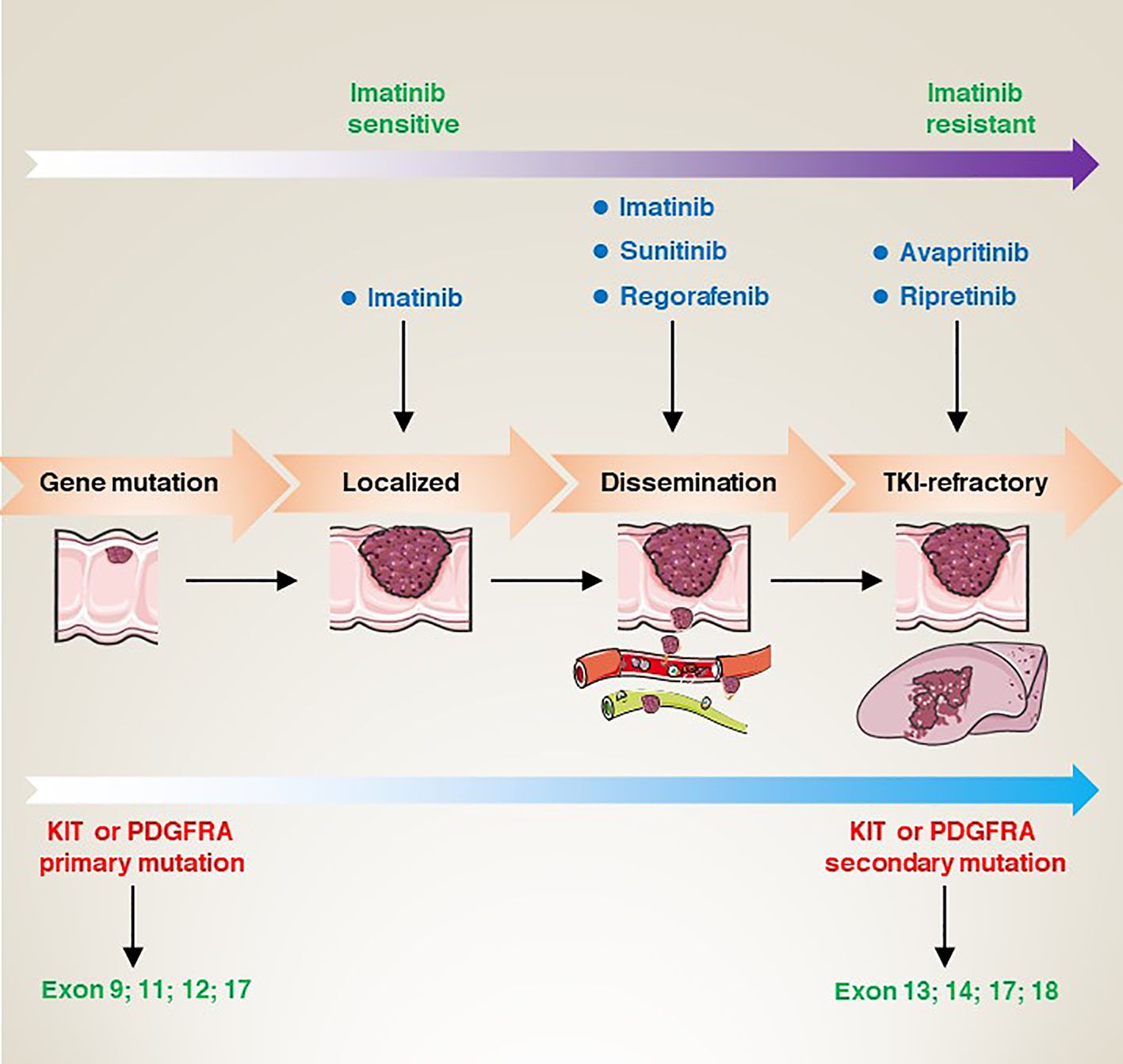
Figure 4 From gene mutation to clinical refractory GISTs, the corresponding site mutation type, the sensitivity changes of imatinib, and the targeted drugs at each stage.
Targeting KIT or PDGFRA
Most GIST patients have acquired mutations in the KIT or PDGFRA genes. Typically, these patients show a favourable response to TKI-targeted therapy. Historically, GISTs were thought to be smooth muscle or neurogenic tumours. However, in 1998, researchers first identified the presence of mutations in the KIT gene in patients with GISTs (7). In subsequent studies, researchers also identified functionally acquired mutations in the PDGFRA gene, which are also strongly associated with the development of GISTs (54). These findings have led to a surge in the development of drugs that target KIT and PDGFRA mutations. Imatinib (IM) became the first FDA-approved first-line treatment for unresectable and metastatic GISTs in 2002 (55). Imatinib selectively inhibits the activity of KIT receptors. Preliminary clinical trial results show that imatinib has a significant therapeutic effect on KIT mutation-positive GISTs patients (55). For KIT/PDGFRA mutant GIST with a high risk of disease recurrence, 3 years of 400 mg/d imatinib adjuvant therapy may prolong relapse-free survival. In addition, for GIST patients at high risk of relapse, the duration of adjuvant therapy with IM (800 mg/d) (3 years) resulted in a longer progression-free survival (the hazard ratio 0.39; 95% CI; 0.22-0.71, P=0.0013) in patients with KIT exon 9 GISTs (56).
However, although imatinib showed good efficacy in most patients, some patients developed resistance or no response. For this reason, after relentless efforts and long-term preclinical studies, sunitinib (SU) was developed as a second-generation targeted agent for the treatment of drug-resistant GISTs patients (57, 58). A randomized controlled trial on the efficacy and safety of sunitinib in patients with advanced gastrointestinal mesenchymal stromal tumours who failed imatinib therapy was evaluated in 2006. The results showed that in 312 patients randomized 2:1 to receive sunitinib (n = 207) or placebo (n = 105), the median time to tumour progression was significantly delayed in patients in the sunitinib group (27.3 weeks) compared to the median time to tumour progression in the control group (6.4 weeks) (57). Subsequently, the FDA re-approved regorafenib as a third-line drug for the clinical treatment of advanced GISTs patients who failed in both IM and SU treatment (59).
In addition, due to the diversity of KIT and PDGFRA mutations in GISTs patients, therapeutic strategies need to be individualized according to the type of mutation (60, 61). For example, PDGFRA D842V mutant GISTs are usually resistant to IM (62). Therefore, ripretinib has been developed as an inhibitor targeting the PDGFRA D842V mutation (63–65). Moreover, several other targeted agents have been used to treat patients with IM-resistant GISTs, such as avapritinib (BLU-285) (66).
As research into GISTs has deepened, IM as the preferred drug has shown good responses in most patients with KIT or PDGFRA mutations, but not complete cures. Patients still have a high chance of developing resistance during follow-up, although SU or regorafenib are approved for the treatment of advanced or resistant GISTs. Thus, drug resistance remains a challenge. To address this problem, exploring different new inhibitors or by combining different drugs and individualized treatment regimens can maximize patient outcomes (67).
The immune microenvironment of GIST
In addition to gene mutations, the tumour immune microenvironment is increasingly considered to play a key role in tumours. In GISTs, tumour-associated macrophages (TAM) and CD3+ tumour-infiltrating lymphocytes (TIL) are the most common immune cells. In addition, natural killer (NK) cells, dendritic cells (DCs), and natural killer T (NKT) cells are also included. In this section, we mainly review how these immune cells are involved in the development of GISTs (Figure 5).
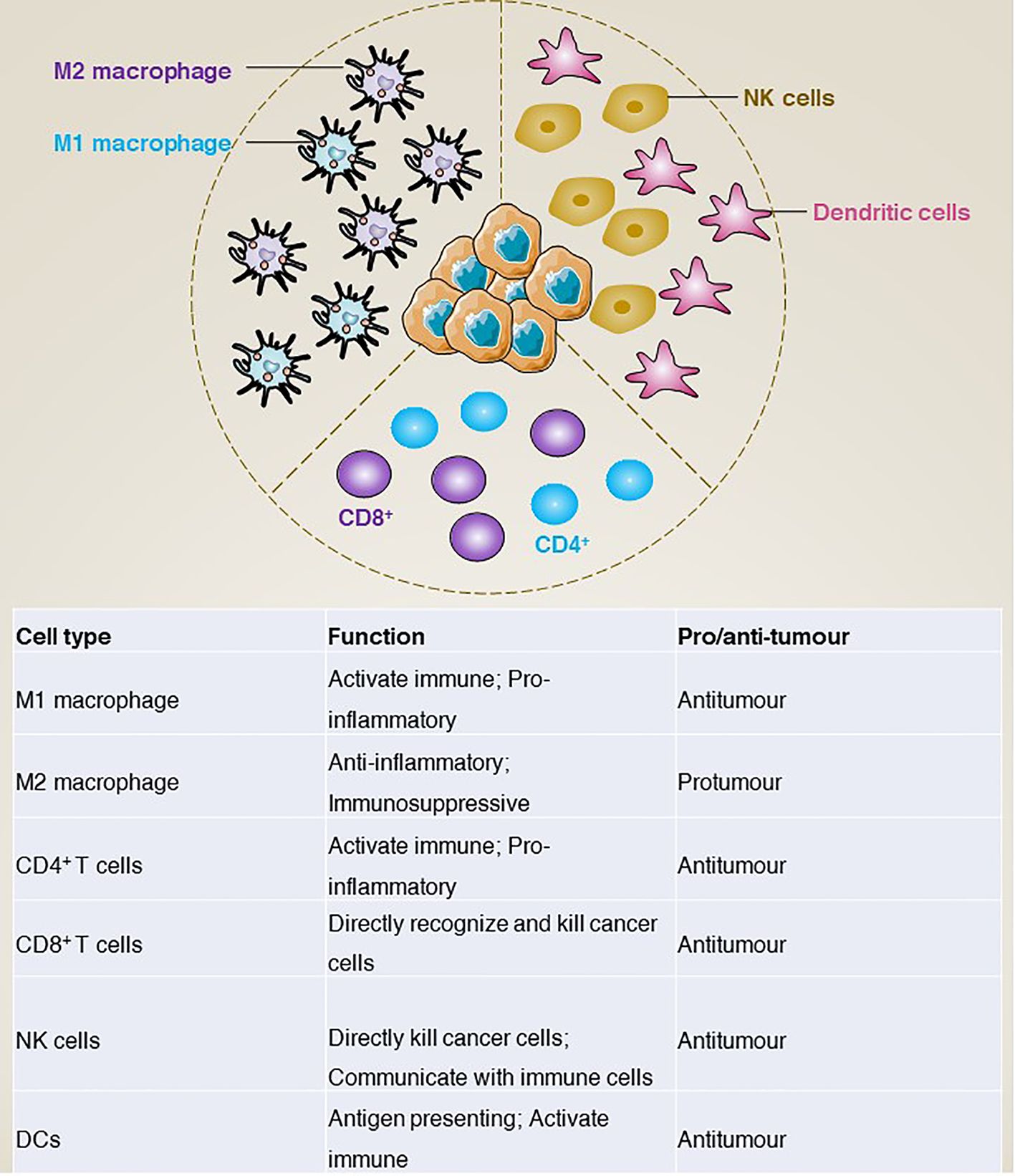
Figure 5 The main immune infiltrating cells and corresponding functions of GISTs microenvironment.
Tumour-associated macrophages
Macrophages are an important class of cells in the immune system, mainly involved in non-specific and innate immune responses (68). In the tumour environment, tumour-associated macrophages can be classified into two main groups according to their surface molecules, origin and functional characteristics: M1 type macrophages and M2 type macrophages (69). Functionally, M1 type macrophages activate the immune response mainly through the production of pro-inflammatory cytokines (e.g., tumour necrosis factor-α [TNFα], interleukin-1β [IL-1β], interleukin-6 [IL-6]) and inhibit tumour growth and spread by phagocytosis and killing of tumour cells (70–73). In addition, M1 macrophages promote the activation of T cells, thereby enhancing specific immune responses (74, 75). In contrast, M2 macrophages produce anti-inflammatory cytokines (e.g., IL-10, transforming growth factor-β [TGFβ]) to modulate the immune response and inhibit T-cell activation and killing (76, 77), thereby promoting tumour evasion of immune surveillance (78). For example, cancer-associated fibroblasts activated by cancer cells release IL-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to induce monocytes to differentiate into M2-like TAMs, activate the immunosuppressive tumour microenvironment and promote GISTs metastasis (79). However, although macrophage polarization in the tumour microenvironment of primary untreated GISTs is controversial, M2 macrophages predominate in metastatic or treated GISTs. For example, imatinib treatment of GISTs induces apoptosis in tumour cells, which further induces TAM M2-like polarization via CCAAT enhancer binding protein beta (CEBPB) (80).
Together, TAMs regulate the development and progression of GISTs through various mechanisms, including promoting tumour growth and dissemination and suppressing the immune response.
Tumour-infiltrating lymphocytes
Tumour-infiltrating T lymphocytes are a class of immune cells that have anti-tumour capacity and are present in tumour tissue (81). According to the phenotypic characteristics, they can be divided into CD4+ and CD8+ T cells (82). CD4+ T cells activate other immune cells and regulate the immune response mainly by releasing cytokines (83). In contrast, CD8+ T cells can directly recognize and kill cancer cells (84). Depending on their function in the tumour microenvironment, tumour-infiltrating T lymphocytes can be further classified into effector T cells (85), regulatory T cells and memory T cells (86), which play the functions of direct tumour killing, suppression of the immune response and recognition of resistance to tumours, respectively.
The presence of abundant tumour-infiltrating immune cells in GISTs has been implicated in the regulation of GISTs through mechanisms such as direct killing of GIST cells, stimulation of immune responses and modulation of immunosuppression. For example, imatinib treatment decreased the frequency of effector CD8+ T cells and increased the frequency of naive CD8+ T cells. Mechanistically, this was mainly due to the production of tumour chemokines and the reduction of intracellular phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic (PI3K) signalling in CD8+ T cells. In contrast, IL15 super-antagonist (IL15SA) in combination with imatinib significantly restored intratumoural effector CD8+ T cell function and intracellular PI3K signalling in CD8+ T cells, thereby enhancing tumour killing (87). Similarly, in spontaneous GISTs, imatinib inhibits the expression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO), which activates CD8+ T cells in the tumour and induces apoptosis of regulatory T cells (T(reg) cells), thereby enhancing the immunotherapeutic effect in a mouse model (88). However, despite the presence of abundant CD8+ infiltration in GISTs, the effect of immune checkpoint inhibition in combination with TKI-targeted therapy in patients with advanced GISTs is rather limited (12).
Together, differences in response to imatinib between patients with early and advanced GIST suggest that there are also different populations of immune cells that play an immunomodulatory role.
Natural killer cells
Tumour-infiltrating natural killer (NK) cells are the first line of defence in the fight against infection and tumour (89). NK cells not only kill tumour cells directly by releasing cytotoxins and cytokines, such as perforin and interferon gamma (90, 91), but also recognize and interact with other immune cells, such as dendritic cells and macrophages, to modulate the GISTs immune response (92, 93). The molecular phenotype of tumour-infiltrating NK cells is closely linked to their function, and their surface molecules include activating and inhibitory receptors such as KIR (killer cell immunoglobulin-like receptor) and killer cell lectin like receptor C1 (KLRC1; also known as NKG2A) -mediated inhibitory signalling (94, 95). These receptors can interact with ligands on the surface of tumour cells to inhibit signalling pathways and prevent NK cell activation. In contrast, killer cell lectin like receptor K1 (KLRK1; also known as NKG2D) (96), CD226 molecule (CD226; also known as DNAM-1) and natural cytotoxicity triggering receptor 1 (NCR1; also known as NKp46) can bind to ligands on the tumour surface and activate NK cells, causing them to release cytotoxins (97, 98). For example, in addition to targeting KIT and PDGFRA, imatinib has also been shown to act on host dendritic cells to enhance anti-tumour effects in vivo by promoting NK cell activation (99).
In addition, NK cell infiltration is strongly associated with the prognosis of GISTs. For example, high expression of the immunosuppressive receptor for NK cells, natural cytotoxicity triggering receptor 3 (NCR3; also known as NKp30), in GISTs is negatively associated with patient survival. Mechanistically, NKp30 expression leads to a reduction in tumour necrosis factor-α (TNF-α) and CD107a release, as well as defects in interferon-γ (IFN-γ) and interleukin-12 (IL-12) secretion in the NK-DC cross-talk, which can be restored by blocking IL-10 (100). Similarly, the ligand for NKp30, natural killer cell cytotoxicity receptor 3 ligand 1 (NCR3LG1; also known as B7-H6), whose soluble form, sB7-H6, was negatively associated with DFS and prognosis in metastatic GISTs (101). While the production of IFN-γ by NK cells in patients treated with imatinib can also be considered an independent predictor of long-term survival in IM-treated advanced GIST (102). In addition, cytokine-secreting CD56 (NCAM1) NK cells were found to be enriched in tumour lesions of imatinib-treated patients and independently predicted progression-free survival (PFS) (103).
In summary, tumour-infiltrating NK cells are involved in the development and prognosis of GISTs through a variety of functions, including direct recognition and killing of tumour cells, production of cytokines and regulation of the immune response. However, the interactions and mechanisms between GISTs and tumour-infiltrating NK cells need to be further investigated in order to develop new immunotherapeutic strategies and improve the outcome of GISTs patients.
Immunotherapy for GIST
In the course of exploring the path of targeting GISTs, from the efficacy of first-line drugs to drug resistance and then continuous iterative updates. However, the efficacy in prolonging the PFS of patients is quite limited, and the targeted treatment of GISTs is in a therapeutic bottleneck. Therefore, immunotherapy, as a new generation of therapies, has begun to have a profound impact on the clinical management of solid tumours. In this section, we focus on immunotherapy based on immune checkpoint inhibition, CAR-T cells and antibody-dependent immunotherapy (Figure 6).
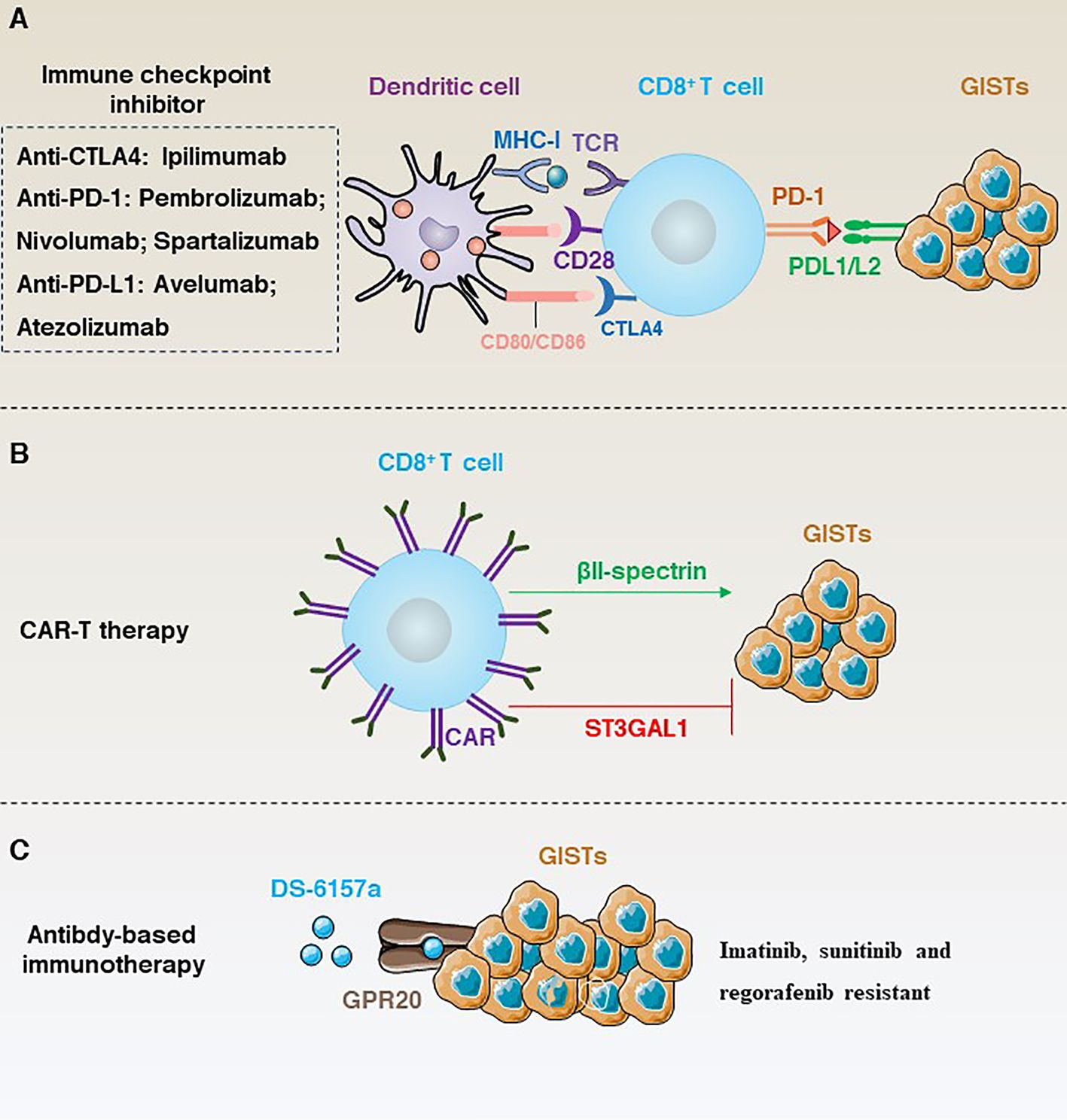
Figure 6 (A) Immune checkpoint inhibition-dependent anti-GISTs immunotherapy and corresponding inhibitors. (B) ST3GAL1 inhibits the localization of CAR-T cells to tumour sites, whereas βII-spectrin enhances the localization of CAR-T cells to tumour sites. (C) DS-6157a targets non-tyrosine kinases GPR20 to inhibit the immunotherapy of drug-resistant GISTs.
Targeting immune checkpoint
Immune checkpoints are a class of inhibitory receptors and associated signals used by the immune system to maintain the body’s immune homeostasis, preventing damage to the body’s own tissues and maintaining tolerance to self-antigens (104). However, tumour cells can also use this mechanism to evade immune surveillance. Therefore, immune checkpoint inhibition can activate the immune system by blocking inhibitory signals at immune checkpoints to enhance its ability to attack tumour cells. Currently, the main immune checkpoints targeted in clinical trials include CTLA-4 (105), PD-1/PD-L1 (106), lymphocyte activating 3 (LAG3) (107), galectin-9 (LGALS9) and hepatitis a virus cellular receptor 2 (HAVCR2; also known as TIM3) (108, 109).
Typically, cancers that respond well to targeted immune checkpoints are characterised by an immune microenvironment infiltrated with high numbers of immune cells and pro-inflammatory cytokines such as CD4+ T cells, CD8+ T cells, interferon (IFN), IL12, IL2, IL23 and TNF-alpha. Cancer cells with this characteristic are called “hot tumours” (110). In contrast, “cold tumours” are characterized by low TIL counts, low PD-L1 expression and infiltration of large numbers of immunosuppressive cells and lack sensitivity to immunotherapy (110). GISTs are often considered to be “cold tumours” (111). Therefore, the use of immune checkpoint therapy in GISTs is relatively limited. However, some studies have shown that immune checkpoint inhibitors may have some benefit in a subset of GISTs patients (Figure 6A). For example, PD-1, LAG3 and TIM3 are significantly upregulated in tumour-infiltrating T cells compared to normal T cells. Importantly, In KitV558Δ/+ mice blockade of PD-1 and PD-L1 alone did not elicit an immune response, whereas the use of imatinib not only targeted KIT, but also inhibited IFNγ-induced upregulation of PD-L1 by inhibiting signal transducer and activator of transcription 1 (STAT1), significantly improving T cell effector function (112).
CD40 is a transmembrane protein that is widely expressed in DCs, B cells and monocytes, and is activated by binding to its ligand CD40LG (also known as CD154), mediating a variety of immune and inflammatory responses that limit therapeutic efficacy in GISTs (113). For example, CD40 is significantly inhibited in tumour-associated macrophages and tumour cells in GIST patients treated with imatinib. However, CD40 inhibition alone had no direct effect on human GISTs, whereas imatinib in combination with a CD40 antagonist significantly inhibited GISTs via the nuclear factor kappa B subunit 1 (NFKB1) pathway in mice carrying a mutation in Kit exon 11 (114).
In addition, the rate-limiting enzyme of human tryptophan metabolism, indoleamine 2,3-dioxygenase (IDO), has been identified as an immune checkpoint. For example, inhibition of IDO expression in tumour cells by imatinib activates CD8+ T cells in the tumour and induces T(reg) cell apoptosis, which, in combination with immunotherapy, significantly enhances the therapeutic effect of imatinib in mice (88). Similarly, a phase 2 clinical trial (NCT02406781) showed that PD-1 inhibition alone was not sufficient to control tumour growth and that a combination of inhibition of the colony stimulating factor 1 receptor and the IDO pathway resulted in more effective tumour control (12).
Together, immunotherapy for GISTs still faces many challenges and requires a better understanding of immune escape mechanisms and biomarkers that predict response to immunotherapy. In addition, individualization of therapy is an important challenge as pathological features and gene expression may differ from patient to patient. Therefore, in-depth analysis of patient data and integration of clinical trial results may improve maximum therapeutic efficacy.
Chimeric antigen receptor T-cell therapy
Chimeric antigen receptor T-cell (CAR-T) is a treatment that modifies a patient’s own T cells so that they can recognize and attack cancer cells (115). The first generation of CAR mainly consisted of structural combinations of CD4 and CD3 (116), but due to the lack of potency a second generation of CAR with co-stimulatory structural domains such as CD28 or TNF receptor superfamily member 9 (TNFRSF9; also known as 4-1BB) is developed (117). CAR-T revolutionized the treatment of haematological cancers in 2010 when Professor Carl June took CAR-T cell therapy into human clinical trials and successfully cured leukaemia patients (116).
Unlike some conventional immunotherapies, such as TIL, CAR-T is not dependent on MHC antigens, and protein-like or lipid antigens specific to the tumour surface can be used as CAR-T targets (118). Given the remarkable effect of CAR-T in the treatment of haematological tumours. Many studies have tried CAR-T in GISTs using KIT/PDGFRA as a target to overcome resistance (Figure 6B). For example, Katz’s team constructed a novel anti-KIT chimeric immune receptors (CIR) with mouse and human T cells and effectively reduced tumour growth rates in a GIST mouse model (119). However, advancing in vitro trials to clinical trials in humans is a long process. To date, CAR-T clinical results in solid tumours have been unsatisfactory. Therefore, research into the negative regulatory mechanisms that modulate CAR-T may provide a solution. For example, a recent study has shown that ST3 β-galactoside α-2,3-glycosyltransferase 1 (ST3GAL1) is a negative regulator of cancer-specific migration of CAR-T cells. ST3GAL1-mediated glycosylation induces spontaneous non-specific tissue chelation of T cells by altering endocytosis of integrin subunit alpha l (ITGAL; also known as LFA-1). In contrast, βII-spectrin, a cytoskeletal molecule expressed on CAR-T cells, enhances the tumour specificity of CAR-T cells to localize to the tumour site and is able to reverse ST3GAL1-mediated non-specific T-cell migration, thereby inhibiting tumour growth in mice (6).
Together, CAR-T may be a potential treatment to overcome GISTs resistance. However, current research is still in preclinical studies, and the exploration of more specific CAR-T targets may lead to further advances in clinical trials.
Antibody-based immunotherapy
TKIs are currently the only approved drugs for the treatment of GISTs, but GISTs are highly susceptible to secondary resistance. In contrast, monoclonal antibody therapy has shown promising therapeutic results in many tumours. Therefore, the development of new monoclonal antibodies targeting KIT, PDGFRA, CD40 and somatostatin receptor type 2 (SSTR2) is worth exploring. For example, a phase II clinical trial analysed the efficacy of nituzumab (N) or nituzumab + ibritumomab (N+I) in 36 patients with refractory GISTs. Results showed that benign responses to therapy and long-term disease control were observed with both N and N+I, but no new safety signals were observed (120). Specifically, the majority of patients treated with N or N+I showed some improvement in median progression-free survival (PFS) and clinical benefit rate (CBR) compared to imatinib treatment.
In addition, targeting non-tyrosine kinases is another important line of research for the treatment of GIST resistance (Figure 6C). For example, G protein-coupled receptor 20 (GPR20) has been identified as an important non-tyrosine kinase that is selectively expressed in GISTs. The anti-GPR20 antibody-drug conjugate DS-6157a showed GPR20 expression-dependent anti-tumour activity in a variety of resistant lines (e.g., imatinib, sunitinib and regorafenib) (121). These results suggest that the exploration of GIST-selective non-tyrosine kinases as therapeutic targets is a potential therapeutic option.
Discussion
GISTs are rare but challenging malignant tumours. Traditional treatments include surgical resection, radiotherapy and chemotherapy (122). However, in recent years, targeted therapy and immunotherapy, as emerging therapeutic strategies, have made some important advances in the treatment of GIST (123–126). First, targeted therapy is an important component of GIST treatment. Some targeted drugs, such as imatinib, sunitinib and regorafenib, block cancer cell proliferation and survival by inhibiting aberrant signal activation mediated by KIT or PDGFRA mutations. These drugs have shown significant efficacy in some GIST patients and are better tolerated than conventional chemotherapy. However, a significant proportion of patients are prone to secondary resistance, which has led to alternative regimens using immunotherapy as a tool. Immune checkpoint inhibitors are one of the most successful immunotherapeutic approaches available and have achieved significant clinical results in a variety of tumours (127).
However, studies in GIST are relatively limited. First, tumour adaptation to KIT/PDGFRA inhibition likely leads to apoptosis evasion and GISTs survival through two intertwined events. Second, GISTs are primarily composed of macrophages and T cells, followed by NK cells and B cells, and it is unclear what role the distribution of other immune cell subtypes plays in the clinical prognosis, evolution and immune evasion of GIST patients. Third, infiltration of M2 macrophages and Treg cells (12, 128), high expression of IDO on GIST cells and immunosuppressive receptors on NK cells, and defective expression of MHC-I on APC. Although CD8+ T cells were enriched in GISTs (111), the proportion of CD8+ T/Treg cells were relatively low. In addition, NK cells were found to be negative for CD69. These factors contribute to the formation of an immunosuppressive microenvironment, which may be responsible for the avoidance of immunotherapy in GISTs. Therefore, the development of a novel therapeutic modality for tumour treatment is needed, and the induction of non-apoptotic-dependent cell death in cancer cells is a promising modality, such as alkaliptosis, cuproptosis and ferroptosis (129–134). Finally, high Ki67 expression is an independent predictor of aggressiveness, malignant potential and poor survival prognosis in GISTs, whereas short-term administration of imatinib activates CD8+ T cells, DC cells and NK cells and inhibits Treg cells, thereby enhancing the host’s anti-tumour immune response. Whereas imatinib was used in the perioperative period and Ki67 index was assessed to improve surgical outcomes (135, 136).
This is despite the fact that at least five currently marketed drugs have resulted in improved prognosis for patients with GISTs. However, the clinical benefits of approved second-, third-, and fourth-line drugs are often unsatisfactory for drug-resistant patients. NB003 is a potent and selective small-molecule tyrosine kinase inhibitor targeting KIT/PDGFRA, which is designed to inhibit a broad spectrum of primary and acquired imatinib-resistant mutations in KIT/PDGFRA (137). Similarly, IDRX-42 has been shown to have significant effects on GISTs with KIT exon 13 mutations (138). However, it should be noted that there are differences in the efficacy of different drugs for different genotypes, and how to effectively differentiate between genotypes can provide the best treatment options for patients with advanced GISTs. And olverembatinib has significant efficacy in the treatment of TKI-resistant SDH-deficient GIST patients, filling the gap in the treatment of SDH-deficient GISTs (139).
In conclusion, the combination strategy of targeted therapy and immunotherapy is worth exploring to further improve the therapeutic efficacy of GISTs (16, 132). The identification of selectively expressed GISTs targets may further improve the response rate and durability of treatment. In addition, research into appropriate biomarkers for early detection is urgently needed to reduce the prevalence of GISTs.
Author contributions
YY: Writing – original draft, Writing – review & editing. MY: Writing – review & editing. LL: Writing – review & editing. ZZ: Writing – review & editing. HZ: Writing – review & editing. YC: Writing – review & editing. ZL: Writing – review & editing. MC: Writing – review & editing. WW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by ‘Double First Class’ and High-level University Discipline Collaborative Innovation Team Project of Guangzhou University of Chinese Medicine (2021xk48). The Supporting Scientific Research Funds of the First Affiliated Hospital of Guangzhou University of Chinese Medicine (09005647001).
Conflict of interest
Author MY was employed by the company Guangzhou KingMed Diagnostics Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
2. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers. (2021) 7:22. doi: 10.1038/s41572-021-00254-5
3. Badic B, Gancel CH, Thereaux J, Joumond A, Bail JP, Meunier B, et al. Surgical and oncological long term outcomes of gastrointestinal stromal tumors (GIST) resection- retrospective cohort study. Int J Surg (London England). (2018) 53:257–61. doi: 10.1016/j.ijsu.2018.03.074
4. Joensuu H, Eriksson M, Collan J, Balk M H, Leyvraz S, Montemurro M. Radiotherapy for GIST progressing during or after tyrosine kinase inhibitor therapy: A prospective study. Radiotherapy oncology: J Eur Soc Ther Radiol Oncol. (2015) 116:233–8. doi: 10.1016/j.radonc.2015.07.025
5. Lu X, Pang Y, Cao H, Liu X, Tu L, Shen Y, et al. Integrated screens identify CDK1 as a therapeutic target in advanced gastrointestinal stromal tumors. Cancer Res. (2021) 81:2481–94. doi: 10.1158/0008-5472.CAN-20-3580
6. Hong Y, Walling BL, Kim HR, Serratelli WS, Lozada JR, Sailer CJ, et al. ST3GAL1 and βII-spectrin pathways control CAR T cell migration to target tumors. Nat Immunol. (2023) 24:1007–19. doi: 10.1038/s41590-023-01498-x
7. Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology. (1998) 115:1090–5. doi: 10.1016/S0016-5085(98)70079-4
8. Sihto H, Sarlomo-Rikala M, Tynninen O, Tanner M, Andersson LC, Franssila K, et al. KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin oncology: Off J Am Soc Clin Oncol. (2005) 23:49–57. doi: 10.1200/JCO.2005.02.093
9. Dagher R, Cohen M, Williams G, Rothmann M, Gobburu J, Robbie , et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable Malignant gastrointestinal stromal tumors. Clin Cancer research: an Off J Am Assoc Cancer Res. (2002) 8:3034–8. doi: 10.1634/theoncologist.2008-0255
10. Dall'Olio FG, Marabelle A, Caramella C, Garcia C, Aldea M, Chaput N, et al. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat Rev Clin Oncol. (2022) 19:75–90. doi: 10.1038/s41571-021-00564-3
11. Pantaleo MA, Tarantino G, Agostinelli C, Urbini M, Nannini M, Saponara M, et al. Immune microenvironment profiling of gastrointestinal stromal tumors (GIST) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology. (2019) 8:e1617588. doi: 10.1080/2162402X.2019.1617588
12. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: A phase 2 clinical trial. JAMA Oncol. (2018) 4:93–7. doi: 10.1001/jamaoncol.2017.1617
13. d'Auriol L, Mattei MG, Andre C, Galibert F. Localization of the human c-kit protooncogene on the q11-q12 region of chromosome 4. Hum Genet. (1988) 78:374–6. doi: 10.1007/BF00291740
14. Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. (1988) 335:88–9. doi: 10.1038/335088a0
15. Hashiyama M, Iwama A, Ohshiro K, Kurozumi K, Yasunaga K, Shimizu Y, et al. Predominant expression of a receptor tyrosine kinase, TIE, in hematopoietic stem cells and B cells. Blood. (1996) 87:93–101. doi: 10.1182/blood.V87.1.93.93
16. Hemming ML, Benson MR, Loycano MA, Anderson JA, Andersen JL, Taddei ML, et al. MOZ and menin-MLL complexes are complementary regulators of chromatin association and transcriptional output in gastrointestinal stromal tumor. Cancer Discovery. (2022) 12:1804–23. doi: 10.1158/2159-8290.CD-21-0646
17. Kinashi T, Escobedo JA, Williams LT, Takatsu K, Springer TA. Receptor tyrosine kinase stimulates cell-matrix adhesion by phosphatidylinositol 3 kinase and phospholipase C-gamma 1 pathways. Blood. (1995) 86:2086–90. doi: 10.1182/blood.V86.6.2086.bloodjournal8662086
18. Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell. (1991) 65:1143–52. doi: 10.1016/0092-8674(91)90010-V
19. Jin B, Ha SE, Wei L, Singh R, Zogg H, Clemmensen B, et al. Colonic motility is improved by the activation of 5-HT(2B) receptors on interstitial cells of cajal in diabetic mice. Gastroenterology. (2021) 161:608–622.e607. doi: 10.1053/j.gastro.2021.04.040
20. Ke H, Kazi JU, Zhao H, Sun J. Germline mutations of KIT in gastrointestinal stromal tumor (GIST) and mastocytosis. Cell bioscience. (2016) 6:55. doi: 10.1186/s13578-016-0120-8
21. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Sci (New York N.Y.). (1998) 279:577–80. doi: 10.1126/science.279.5350.577
22. Braconi C, Bracci R, Bearzi I, Bianchi F, Costagliola A, Catalani R, et al. KIT and PDGFRalpha mutations in 104 patients with gastrointestinal stromal tumors (GISTs): a population-based study. Ann oncology: Off J Eur Soc Med Oncol. (2008) 19:706–10. doi: 10.1093/annonc/mdm503
23. Andersson J, Bümming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology. (2006) 130:1573–81. doi: 10.1053/j.gastro.2006.01.043
24. Klug LR, Khosroyani HM, Kent JD, Heinrich MC. New treatment strategies for advanced-stage gastrointestinal stromal tumours. Nat Rev Clin Oncol. (2022) 19:328–41. doi: 10.1038/s41571-022-00606-4
25. Kim TW, Lee H, Kang YK, Choe MS, Ryu MH, Chang HM, et al. Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer research: an Off J Am Assoc Cancer Res. (2004) 10:3076–81. doi: 10.1158/1078-0432.CCR-03-0581
26. Bachet JB, Landi B, Laurent-Puig P, Italiano A, Le Cesne A, Lévy P, et al. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (GIST) and germline mutation of KIT exon 13. Eur J Cancer (Oxford England: 1990). (2013) 49:2531–41. doi: 10.1016/j.ejca.2013.04.005
27. Ishikawa T, et al. In vivo effect of imatinib on progression of cecal GIST-like tumors in exon 17-type c-kit knock-in mice. Lab investigation; J Tech Methods Pathol. (2009) 89:1161–8. doi: 10.1038/labinvest.2009.78
28. Javidi-Sharifi N, Traer E, Martinez J, Gupta A, Taguchi T, Dunlap J, et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. (2015) 75:880–91. doi: 10.1158/0008-5472.CAN-14-0573
29. Birner P, Beer A, Vinatzer U, Stary S, Höftberger R, Nirtl N, et al. MAPKAP kinase 2 overexpression influences prognosis in gastrointestinal stromal tumors and associates with copy number variations on chromosome 1 and expression of p38 MAP kinase and ETV1. Clin Cancer research: an Off J Am Assoc Cancer Res. (2012) 18:1879–87. doi: 10.1158/1078-0432.CCR-11-2364
30. Gronwald RG, Adler DA, Kelly JD, Disteche CM, Bowen-Pope DF. The human PDGF receptor alpha-subunit gene maps to chromosome 4 in close proximity to c-kit. Hum Genet. (1990) 85:383–5. doi: 10.1007/BF00206767
31. Qian C, Wu Z, Ng RC, Garcia-Barceló MM, Yuan ZW, Wong KKY, et al. Conditional deletion of platelet derived growth factor receptor alpha (Pdgfra) in urorectal mesenchyme causes mesenchyme apoptosis and urorectal developmental anomalies in mice. Cell Death differentiation. (2019) 26:1396–410. doi: 10.1038/s41418-018-0216-2
32. Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci United States America. (2010) 107:6459–64. doi: 10.1073/pnas.0911188107
33. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Sci (New York N.Y.). (2003) 299:708–10. doi: 10.1126/science.1079666
34. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin oncology: Off J Am Soc Clin Oncol. (2005) 23:5357–64. doi: 10.1200/JCO.2005.14.068
35. Jones RL, Serrano C, von Mehren M, George S, Heinrich MC, Kang YK, et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer (Oxford England: 1990). (2021) 145:132–42. doi: 10.1016/j.ejca.2020.12.008
36. Papke DJ Jr., Forgó E, Charville GW, Hornick JL. PDGFRA immunohistochemistry predicts PDGFRA mutations in gastrointestinal stromal tumors. Am J Surg Pathol. (2022) 46:3–10. doi: 10.1097/PAS.0000000000001720
37. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta neuropathologica. (2015) 129:829–48. doi: 10.1007/s00401-015-1432-1
38. Fujimoto K, Arita H, Satomi K, Yamasaki K, Matsushita Y, Nakamura T, et al. TERT promoter mutation status is necessary and sufficient to diagnose IDH-wildtype diffuse astrocytic glioma with molecular features of glioblastoma. Acta neuropathologica. (2021) 142:323–38. doi: 10.1007/s00401-021-02337-9
39. Kang YN, Jung HR, Hwang I. Clinicopathological and immunohistochemical features of gastointestinal stromal tumors. Cancer Res Treat. (2010) 42:135–43. doi: 10.4143/crt.2010.42.3.135
40. Farag S, Somaiah N, Choi H, Heeres B, Wang WL, van Boven H, et al. Clinical characteristics and treatment outcome in a large multicentre observational cohort of PDGFRA exon 18 mutated gastrointestinal stromal tumour patients. Eur J Cancer (Oxford England: 1990). (2017) 76:76–83. doi: 10.1016/j.ejca.2017.02.007
41. Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol (Zurich Switzerland). (2013) 23:565–73. doi: 10.1111/bpa.12043
42. Schoppmann SF, Vinatzer U, Popitsch N, Mittlböck M, Liebmann-Reindl S, Jomrich G, et al. Novel clinically relevant genes in gastrointestinal stromal tumors identified by exome sequencing. Clin Cancer Res. (2013) 19:5329–39. doi: 10.1158/1078-0432.CCR-12-3863
43. Vignaud JM, Marie B, Klein N, Plénat F, Pech M, Borrelly J, et al. The role of platelet-derived growth factor production by tumor-associated macrophages in tumor stroma formation in lung cancer. Cancer Res. (1994) 54:5455–63.
44. Vitiello GA, Bowler TG, Liu M, Medina BD, Zhang JQ, Param NJ, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest. (2019) 129:1863–77. doi: 10.1172/JCI124108
45. Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci United States America. (2011) 108:314–8. doi: 10.1073/pnas.1009199108
46. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e413. doi: 10.1016/j.cell.2016.08.064
47. Goncalves J, Moog S, Morin A, Gentri CG, Müller S, Morrell AP, et al. Loss of SDHB promotes dysregulated iron homeostasis, oxidative stress, and sensitivity to ascorbate. Cancer Res. (2021) 81:3480–94. doi: 10.1158/0008-5472.CAN-20-2936
48. Liu Y, Liu K, Thorne R F, Shi R, Zhang Q, Wu M, et al. Mitochondrial SENP2 regulates the assembly of SDH complex under metabolic stress. Cell Rep. (2023) 42:112041. doi: 10.1016/j.celrep.2023.112041
49. Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Internal Med. (2009) 266:19–42. doi: 10.1111/j.1365-2796.2009.02111.x
50. Somme F, Bender L, Kurtz JE, Gantzer J, Imperiale A. 18F-FDG PET/CT monitoring of tumor response to tyrosine kinase inhibitors and alkylating drugs in an SDH-deficient GIST. Clin Nucl Med. (2021) 46:e515–7. doi: 10.1097/RLU.0000000000003615
51. Chi P, Qin LX, Camacho N, Kelly CM, D'Angelo SP, Dickson MA, et al. Phase ib trial of the combination of imatinib and binimetinib in patients with advanced gastrointestinal stromal tumors. Clin Cancer research: an Off J Am Assoc Cancer Res. (2022) 28:1507–17. doi: 10.1158/1078-0432.CCR-21-3909
52. Yebra M, Bhargava S, Kumar A, Burgoyne AM, Tang CM, Yoon H, et al. Establishment of patient-derived succinate dehydrogenase-deficient gastrointestinal stromal tumor models for predicting therapeutic response. Clin Cancer research: an Off J Am Assoc Cancer Res. (2022) 28:187–200. doi: 10.1158/1078-0432.CCR-21-2092
53. Astolfi A, Indio V, Nannini M, Saponara M, Schipani A, De Leo A, et al. Targeted deep sequencing uncovers cryptic KIT mutations in KIT/PDGFRA/SDH/RAS-P wild-type GIST. Front Oncol. (2020) 10:504. doi: 10.3389/fonc.2020.00504
54. Heinrich MC, Corless CL, Demetri GD, Blanke D, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin oncology: Off J Am Soc Clin Oncol. (2003) 21:4342–9. doi: 10.1200/JCO.2003.04.190
55. Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevi CS, Butterfield JH, et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. (2002) 99:1741–4. doi: 10.1182/blood.V99.5.1741
56. Patel S, Zalcberg JR. Optimizing the dose of imatinib for treatment of gastrointestinal stromal tumours: lessons from the phase 3 trials. Eur J Cancer. (2008) 44:501–9. doi: 10.1016/j.ejca.2007.11.021
57. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verwei JJ, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet (London England). (2006) 368:1329–38. doi: 10.1016/S0140-6736(06)69446-4
58. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. (2018) 24:749–57. doi: 10.1038/s41591-018-0053-3
59. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet (London England). (2013) 381:295–302. doi: 10.1016/S0140-6736(12)61857-1
60. Joensuu H. Adjuvant treatment of GIST: patient selection and treatment strategies. Nat Rev Clin Oncol. (2012) 9:351–8. doi: 10.1038/nrclinonc.2012.74
61. Reyna VF. A scientific theory of gist communication and misinformation resistance, with implications for health, education, and policy. Proc Natl Acad Sci USA. (2021) 118(15):e1912441117. doi: 10.1073/pnas.1912441117
62. Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. (2020) 21:935–46. doi: 10.1016/S1470-2045(20)30269-2
63. Bauer S, Heinrich MC, George S, Zalcberg JR, Serrano C, Gelderblom H, et al. Clinical activity of ripretinib in patients with advanced gastrointestinal stromal tumor harboring heterogeneous KIT/PDGFRA mutations in the phase III INVICTUS study. Clin Cancer research: an Off J Am Assoc Cancer Res. (2021) 27:6333–42. doi: 10.1158/1078-0432.CCR-21-1864
64. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. (2020) 21:923–34. doi: 10.1016/S1470-2045(20)30168-6
65. Smith BD, Kaufman MD, Lu WP, Gupta A, Leary CB, Wise SC, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. (2019) 35:738–751.e739. doi: 10.1016/j.ccell.2019.04.006
66. BLU-285, DCC-2618 show activity against GIST. Cancer Discov. (2017) 7:121–2. doi: 10.1158/2159-8290.CD-NB2016-165
67. Chen F, Kang R, Liu J, Tang D. The V-ATPases in cancer and cell death. Cancer Gene Ther. (2022) 29(11):1529-41. doi: 10.1038/s41417-022-00477-y
68. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
69. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
70. Zhang X, Fan L, Wu J, Xu H, Leung WY, Fu K, et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J Hepatol. (2019) 71:163–74. doi: 10.1016/j.jhep.2019.03.014
71. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
72. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. (2019) 18:42. doi: 10.1186/s12943-019-0988-0
73. Ahirwar DK, Charan M, Mishra S, Verma A K, Shilo K, Ramaswamy B, et al. Slit2 inhibits breast cancer metastasis by activating M1-like phagocytic and antifibrotic macrophages. Cancer Res. (2021) 81:5255–67. doi: 10.1158/0008-5472.CAN-20-3909
74. Klug F, Prakash H, Huber P E, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
75. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. (2017) 32:253–267.e255. doi: 10.1016/j.ccell.2017.07.006
76. Hu J, Deng F, Zhao B, Lin Z, Sun Q, Yang X, et al. Lactobacillus murinus alleviate intestinal ischemia/reperfusion injury through promoting the release of interleukin-10 from M2 macrophages via Toll-like receptor 2 signaling. Microbiome. (2022) 10:38. doi: 10.1186/s40168-022-01227-w
77. Nawaz A, Aminuddin A, Kado T, Takikawa A, Yamamoto S, Tsuneyama K, et al. CD206(+) M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat Commun. (2017) 8:286. doi: 10.1038/s41467-017-00231-1
78. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. (2020) 19:41. doi: 10.1186/s12943-020-01165-x
79. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, et al. Cancer-stimulated CAFs enhance monocyte differentiation and protumoral TAM activation via IL6 and GM-CSF secretion. Clin Cancer research: an Off J Am Assoc Cancer Res. (2018) 24:5407–21. doi: 10.1158/1078-0432.CCR-18-0125
80. Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. (2013) 210:2873–86. doi: 10.1084/jem.20130875
81. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
82. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
83. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
84. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. (2018) 564:268–72. doi: 10.1038/s41586-018-0694-x
85. Levine AG, Mendoza A, Hemmers S, Moltedo B, Nie CRE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. (2017) 546:421–5. doi: 10.1038/nature22360
86. Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. (2002) 2:251–62. doi: 10.1038/nri778
87. Tieniber AD, Hanna AN, Medina BD, Vitiello GA, Etherington MS, Liu M, et al. Tyrosine kinase inhibition alters intratumoral CD8+ T-cell subtype composition and activity. Cancer Immunol Res. (2022) 10:1210–23. doi: 10.1158/2326-6066.CIR-21-1039
88. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. (2011) 17:1094–100. doi: 10.1038/nm.2438
89. Leavy O. Natural killer cells: RAG keeps natural killers fit. Nat Rev Immunol. (2014) 14:716–7. doi: 10.1038/nri3760
90. Prager I, Liesche C, van Ooijen H, Urlau BD, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. (2019) 216:2113–27. doi: 10.1084/jem.20181454
91. Shi Y, Ling B, Zhou Y, Gao T, Feng D, Xiao M, et al. Interferon-gamma expression in natural killer cells and natural killer T cells is suppressed in early pregnancy. Cell Mol Immunol. (2007) 4:389–94.
92. Leno-Durán E, Muñoz-Fernández R, Olivares EG, Tirado-González I. Liaison between natural killer cells and dendritic cells in human gestation. Cell Mol Immunol. (2014) 11:449–55. doi: 10.1038/cmi.2014.36
93. Zhang M, Wen B, Anton OM, Yao Z, Dubois S, Ju W, et al. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc Natl Acad Sci United States America. (2018) 115:E10915–e10924. doi: 10.1073/pnas.1811615115
94. Haroun-Izquierdo A, Vincenti M, Netskar H, van Ooijen H, Zhang B, Bendzick L, et al. Adaptive single-KIR(+)NKG2C(+) NK cells expanded from select superdonors show potent missing-self reactivity and efficiently control HLA-mismatched acute myeloid leukemia. J Immunother Cancer. (2022) 10(11):e005577. doi: 10.1136/jitc-2022-005577
95. Shreeve N, Depierreux D, Hawkes D, Traherne JA, Sovio U, Huhn O, et al. The CD94/NKG2A inhibitory receptor educates uterine NK cells to optimize pregnancy outcomes in humans and mice. Immunity. (2021) 54:1231–1244.e1234. doi: 10.1016/j.immuni.2021.03.021
96. Zhang C, Röder J, Scherer A, Bodden M, Pfeifer Serrahima J, Bhatti A, et al. Bispecific antibody-mediated redirection of NKG2D-CAR natural killer cells facilitates dual targeting and enhances antitumor activity. J immunotherapy Cancer. (2021) 9(10):e002980. doi: 10.1136/jitc-2021-002980
97. Soldierer M, Bister A, Haist C, Thivakaran A, Cengiz SC, Sendker S, et al. Genetic engineering and enrichment of human NK cells for CAR-enhanced immunotherapy of hematological Malignancies. Front Immunol. (2022) 13:847008. doi: 10.3389/fimmu.2022.847008
98. Glasner A, Levi A, Enk J, Isaacson B, Viukov S, Orlanski S, et al. NKp46 receptor-mediated interferon-γ Production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity. (2018) 48:107–119.e104. doi: 10.1016/j.immuni.2017.12.007
99. Borg C, Terme M, Taïe BJ, Ménard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest. (2004) 114:379–88. doi: 10.1172/JCI21102
100. Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. (2011) 17:700–7. doi: 10.1038/nm.2366
101. Rusakiewicz S, Perier A, Semeraro M, Pitt JM, Pogge von Strandmann E, Reiners KS, et al. NKp30 isoforms and NKp30 ligands are predictive biomarkers of response to imatinib mesylate in metastatic GIST patients. Oncoimmunology. (2017) 6:e1137418. doi: 10.1080/2162402X.2015.1137418
102. Ménard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. (2009) 69:3563–9. doi: 10.1158/0008-5472.can-08-3807
103. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. (2013) 73:3499–510. doi: 10.1158/0008-5472.CAN-13-0371
104. Tuong ZK, Loudon K W, Berry B, Richoz N, Jones J, Tan X, et al. Resolving the immune landscape of human prostate at a single-cell level in health and cancer. Cell Rep. (2021) 37:110132. doi: 10.1016/j.celrep.2021.110132
105. Zhang Y, Du X, Liu M, Tang F, Zhang P, Ai C, et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. (2019) 29:609–27. doi: 10.1038/s41422-019-0184-1
106. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
107. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–352.e345. doi: 10.1016/j.immuni.2018.07.007
108. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. (2017) 23:556–67. doi: 10.1038/nm.4314
109. Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. (2021) 595:101–6. doi: 10.1038/s41586-021-03626-9
110. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1
111. Klaver Y, Rijnders M, Oostvogels A, Wijers R, Smid M, Grünhagen D, et al. Differential quantities of immune checkpoint-expressing CD8 T cells in soft tissue sarcoma subtypes. J Immunother Cancer. (2020) 8(2):e000271. doi: 10.1136/jitc-2019-000271
112. Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin Cancer research: an Off J Am Assoc Cancer Res. (2017) 23:454–65. doi: 10.1158/1078-0432.CCR-16-1163
113. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Advanced Drug delivery Rev. (2019) 141:92–103. doi: 10.1016/j.addr.2018.12.005
114. Zhang JQ, Zeng S, Vitiello GA, Seifert AM, Medina BD, Beckman MJ, et al. Macrophages and CD8(+) T cells mediate the antitumor efficacy of combined CD40 ligation and imatinib therapy in gastrointestinal stromal tumors. Cancer Immunol Res. (2018) 6:434–47. doi: 10.1158/2326-6066.CIR-17-0345
115. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
116. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Trans Med. (2012) 4:132ra153. doi: 10.1126/scitranslmed.3003761
117. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. (2021) 9(10):e003354. doi: 10.1136/jitc-2021-003354
118. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
119. Katz SC, Burga RA, Naheed S, Licata LA, Thorn M, Osgood D, et al. Anti-KIT designer T cells for the treatment of gastrointestinal stromal tumor. J Trans Med. (2013) 11:46. doi: 10.1186/1479-5876-11-46
120. Singh AS, Hecht JR, Rosen L, Wainberg ZA, Wang X, Douek M, et al. A randomized phase II study of nivolumab monotherapy or nivolumab combined with ipilimumab in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res: An Off J Am Assoc Cancer Res. (2022) 28:84–94. doi: 10.1158/1078-0432.CCR-21-0878
121. Iida K, Abdelhamid Ahmed AH, Nagatsuma AK, Shibutani T, Yasuda S, Kitamura M, et al. Identification and therapeutic targeting of GPR20, selectively expressed in gastrointestinal stromal tumors, with DS-6157a, a first-in-class antibody-drug conjugate. Cancer Discov. (2021) 11:1508–23. doi: 10.1158/2159-8290.CD-20-1434
122. Kelly CM, Gutierrez Sainz L, Chi P. The management of metastatic GIST: current standard and investigational therapeutics. J Hematol Oncol. (2021) 14:2. doi: 10.1186/s13045-020-01026-6
123. Chen F, Zhu S, Kang R, Tang D, Liu J. ATP6V0D1 promotes alkaliptosis by blocking STAT3-mediated lysosomal pH homeostasis. Cell Rep. (2022) 42:111911. doi: 10.1016/j.celrep.2022.111911
124. Chen F, Cai X, Kang R, Liu J, Tang D. Autophagy-dependent ferroptosis in cancer. Antioxidants Redox Signaling. (2023) 39(1-3):79–101. doi: 10.1089/ars.2022.0202
125. Li B, Chen H, Yang S, Chen F, Xu L, Li Y, et al. Advances in immunology and immunotherapy for mesenchymal gastrointestinal cancers. Mol Cancer. (2023) 22:71. doi: 10.1186/s12943-023-01770-6
126. Blay JY. A decade of tyrosine kinase inhibitor therapy: Historical and current perspectives on targeted therapy for GIST. Cancer Treat Rev. (2011) 37:373–84. doi: 10.1016/j.ctrv.2010.11.003
127. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
128. van Dongen M, Savage ND, Jordanova ES, Briaire-de Bruijn IH, Walburg KV, Ottenhoff TH, et al. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int J Cancer. (2010) 127:899–909. doi: 10.1002/ijc.25113
129. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375:1254–61. doi: 10.1126/science.abf0529
130. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
131. Chen F, Lin J, Kang R, Tang D, Liu J. Alkaliptosis induction counteracts paclitaxel-resistant ovarian cancer cells via ATP6V0D1-mediated ABCB1 inhibition. Mol Carcinog. (2024). doi: 10.1002/mc.23741
132. Chen F, Lu Y, Lin J, Kang R, Liu J. Cholesterol metabolism in cancer and cell death. Antioxidants Redox Signaling. (2023) 39:102–40. doi: 10.1089/ars.2023.0340
133. Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, et al. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med. (2023) 15:eadg3049. doi: 10.1126/scitranslmed.adg3049
134. Chen F, Kang R, Tang D, Liu J. Ferroptosis: principles and significance in health and disease. J Hematol Oncol. (2024) 17:41. doi: 10.1186/s13045-024-01564-3
135. Wu TJ, Lee LY, Yeh CN, Wu PY, Chao TC, Hwang TL, et al. Surgical treatment and prognostic analysis for gastrointestinal stromal tumors (GISTs) of the small intestine: before the era of imatinib mesylate. BMC Gastroenterol. (2006) 6:29. doi: 10.1186/1471-230X-6-29
136. Zovak M, Boban M, Boban L, Cicek S, Madzar Z, Belev B, et al. Significance of surgery for prognosis of GIST in cohort from transitional healthcare settings. Int J Surg. (2014) 12:1167–71. doi: 10.1016/j.ijsu.2014.07.275
137. Cicala CM, Olivares-Rivas I, Aguirre-Carrillo JA, Serrano C. KIT/PDGFRA inhibitors for the treatment of gastrointestinal stromal tumors: getting to the gist of the problem. Expert Opin Investig Drugs. (2024) 33:159–70. doi: 10.1080/13543784.2024.2318317
138. De Sutter L, Wozniak A, Verreet J, Vanleeuw U, De Cock L, Linde N, et al. Antitumor efficacy of the novel KIT inhibitor IDRX-42 (Formerly M4205) in patient- and cell line-derived xenograft models of gastrointestinal stromal tumor (GIST). Clin Cancer Res. (2023) 29:2859–68. doi: 10.1158/1078-0432.CCR-22-3822
139. Jiang Q, Li Z, Qin Y, Li W, Xu N, Liu B, et al. Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial. J Hematol Oncol. (2022) 15:113. doi: 10.1186/s13045-022-01334-z
Keywords: gastrointestinal stromal tumours, immunotherapy, targeted therapy, molecular characteristics, immune microenvironment
Citation: Yu Y, Yu M, Luo L, Zhang Z, Zeng H, Chen Y, Lin Z, Chen M and Wang W (2024) Molecular characteristics and immune microenvironment of gastrointestinal stromal tumours: targets for therapeutic strategies. Front. Oncol. 14:1405727. doi: 10.3389/fonc.2024.1405727
Received: 23 March 2024; Accepted: 24 June 2024;
Published: 12 July 2024.
Edited by:
Lujun Chen, First People’s Hospital of Changzhou, ChinaReviewed by:
Sacheen Kumar, Royal Marsden NHS Foundation Trust, United KingdomZheng Jin Tu, Cleveland Clinic, United States
Copyright © 2024 Yu, Yu, Luo, Zhang, Zeng, Chen, Lin, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang, d2FuZ3dlaTE2NDAwQDE2My5jb20=; Yang Yu, eWFuZy55dTIwMDFAcXEuY29t