 Swarn Arya
Swarn Arya Zainab Shahid
Zainab Shahid- 1Infectious Disease Service, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 2Department of Medicine, Weill Cornell School of Medicine, New York, NY, United States
Chimeric antigen receptor-modified T cell (CAR T-cell) therapy has revolutionized the management of hematological malignancies. In addition to impressive malignancy-related outcomes, CAR T-cell therapy has significant toxicity-related adverse events, including cytokine release syndrome (CRS), immune effector cell associated neurotoxicity syndrome (ICANS), immune effector cell-associated hematotoxicity (ICAHT), and opportunistic infections. Different CAR T-cell targets have different epidemiology and risk factors for infection, and these targets result in different long-term immunodeficiency states due to their distinct on-target and off- tumor effects. These effects are exacerbated by the use of multimodal immunosuppression in the management of CRS and ICANS. The most effective course of action for managing infectious complications involves determining screening, prophylactic, and monitoring strategies and understanding the role of immunoglobulin replacement and re-vaccination strategies. This involves considering the nature of prior immunomodulating therapies, underlying malignancy, the CAR T-cell target, and the development and management of related adverse events. In conclusion, we now have an increasing understanding of infection management for CAR T-cell recipients. As additional effector cells and CAR T-cell targets become available, infection management strategies will continue to evolve.
1 Introduction
The chimeric antigen receptor-modified T cell (CAR T-cell) therapy field has rapidly expanded since the US Food and Drug Administration (FDA) first approved CD19-targeted CAR T-cells for patients with relapsed refractory B lymphoid malignancies in 2017 (1, 2). There are 6 FDA-approved CAR-T products (four CD19 CAR T-cell products and two B cell maturation [BCMA] CAR T-cells) for hematological malignancies; several are under investigation for other malignancies (1).
Clinical trials have demonstrated encouraging outcomes regarding hematological malignancy-related outcomes, albeit accompanied by infectious complications and toxicity-related adverse events (3). The burden of infection among CAR T-cell therapy recipients remains a critical consideration in managing these patients. Most patients are at high infection risk due to their underlying malignancies, prior lines of cancer-directed treatment, and pre-CAR T-cell lymphodepletion (2, 4, 5). Furthermore, the intensive lymphodepleting chemotherapy given before CAR-T infusion exacerbates immune system suppression, heightening susceptibility to opportunistic infections. Post infusion, immune dysregulation can lead to cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and hemophagocytic lymphohistiocytosis (HLH) -like syndrome; their treatments further predispose to infection (6–8). The post- CAR T-cell period is marked by varying degrees of lymphopenia, B-cell depletion, and hypogammaglobulinemia, which may influence long-term susceptibility to infection (5).
Prophylactic and monitoring measures based on an understanding of infection epidemiology are part of the efforts to lessen the burden of infections among CAR-T recipients. Due to the absence of comprehensive data, prophylactic and management strategies are based on consensus guidelines largely extrapolated from the post-hematopoietic stem cell transplant (HSCT) population (4, 9), and approaches vary widely by institution. This review aims to comprehensively analyze the epidemiology and risk factors associated with infections following CAR T-cell therapy in patients with hematological malignancy. It explores strategies for managing infectious risks associated with treatment-related toxicities and offers suggestions for screening, prophylaxis, immunoglobulin replacement therapy (IgRT), and vaccinations. Further research is needed in all these domains to expand our knowledge and optimize clinical practice.
2 Epidemiology of infections
The incidence of infection in patients receiving CAR T-cell therapy varies widely across prospective and retrospective trials, likely reflecting differences amongst patient populations, CAR T-cell related factors, definitions of infection, and follow-up duration. Epidemiology varies with CAR T-cell target and with time since cell infusion (day 0) due to the chronological evolution of infectious risk factors (4). For this review, epidemiology will be discussed in the context of two broad time points: those occurring in the early period, between days 0 and 30, and those occurring in the late period, beyond day 30. Overall, across the largest observational studies of infectious risk, infections were reported in 19–69% of patients after CD19-directed CAR T-cell therapy (2, 5), and in 42%-69% of patients after BCMA-directed CAR T-cell therapy; life-threatening infections are infrequently reported (2). A recent meta-analysis reported the pooled incidence of infection-related mortality as 1% (95% CI 0.01–0.02) and comparable amongst hematological malignancies (MM, ALL, NHL) (10).
2.1 Early infections: day 0 to day 30
Cohort studies evaluating infectious complications after CD19-directed CAR T-cell therapy report a higher incidence of infections within the first month and a subsequent decrease in the following months (11–20). The largest study of infectious complications after CD19-directed CAR T-cell therapy to date followed 133 patients with various malignancies (ALL, CLL, NHL) for up to 3 months after infusion; infection density was reported at 1.19 infections for every 100 days at risk within the first 28 days after cell infusion, and 0.67 between days 29 and 90 (21). The frequency of serious infection (grade 3 or higher) varies between 5–32% across the largest trials (2). Most documented early infections are bacterial, including both bacteremias and site infections; clostridium difficile colitis has specifically been identified in multiple trials (12, 13). The second most common infection is viral infections, of which respiratory viral infections account for the majority12-14. Hill et al. reported that infections, if present before lymphodepletion, can progress after CAR T-cell infusion (bacterial sinusitis, invasive fungal sinusitis, perirectal abscess) (11).
Patterns of early infection after BCMA-targeted CAR T-cell therapy differ somewhat from those encountered after CD19-directed therapy, likely reflecting differences in infectious risk attributable to patients’ baseline malignancies. Infectious risk is reported to be highest in the first 30–100 days and decreases thereafter (22, 23). Most infections are mild to moderate in severity and involve the respiratory tract. When a microbiologic diagnosis is made, bacterial and viral pathogens are identified, but viral etiologies are slightly more common (22, 23). Severe infections are infrequently reported; by one report, most serious infections and bacterial bloodstream infections occurred in the first 28 days after BCMA CAR-T infusion (23).
2.2 Late infections (beyond day 30)
CD19-directed CAR T-cell therapy results in the depletion of endogenous B-cells and hypogammaglobulinemia, which lasts for an unclear duration and may impact long-term risk for infectious complications (19, 24). Overall infection density decreased over time by one report; the incidence decreased from 11.7 infections per 1000 person-days in the first 30 days to 2.3 between days 31 and 90, and incidence continued to decrease over time (16). Cordeiro et al. reported infection density beyond day 90 was 0.55 infections/100 days at risk, or 2.08 per patient-year (25). The most common infections reported are of the respiratory tract; in some reports, viral etiologies predominate (13, 16, 21); in other reports, bacterial etiologies remain important causes of infection (12, 25). Bacteremias (21)and other bacterial site infections, particularly those of the urinary tract (12), also continue to be reported. In another cohort of 60 diffuse large b-cell lymphoma (DLBCL) patients, 37% of patients who developed bacterial infections after day 30 had documented bacterial infections between days 0 and 30 after CD19-directed therapy (12). Most infections are mild-moderate in severity and managed in the outpatient setting (12, 13, 16, 25).
The cumulative incidence of infection also declines over time among patients with multiple myeloma treated with BMCA-directed CAR-T therapy (23, 26). Bacterial infections continue to be reported between days 31 and 100, but viral infections predominate after that, mostly mild to moderate in severity (23, 26, 27). Amongst bacterial infections, site infections, specifically pneumonia and sinusitis, were most frequently reported in the late period by two reports (27, 28). The epidemiology of less common infections and risk factors are discussed later in the article.
Key points:
-The risk of infection is higher in the early risk period compared to the late risk period.
-Bacterial infections are relatively common during the neutropenic phase, then viral infections are more common. BCMA CAR-T recipients have a comparable incidence of viral infections to bacterial infections in the early risk period.
-Data on long-term infection incidence and risk is still evolving.
3 Factors associated with infectious risk
Factors influencing infection risk in CAR T-cell therapy recipients can relate to the host at baseline or the intervention; only a few known factors are modifiable. We will examine infectious risk in the context of two broad categories: pre-CAR T-cell infusion and CAR-T/post-CAR T-cell infusion. The post-CAR-T period can have early and late risk factors. Data comes from small, single-center experiences with heterogeneous cohorts and varying patterns of prophylaxis and management, which limits cross-trial comparisons. However, several distinct patterns do emerge.
3.1 Pre-CAR-T risk factors
CAR T-cell therapy recipients often are heavily treated and can have varying degrees of pre-existing cytopenia, decreased bone marrow reserve, and hypogammaglobulinemia. The burden of pretreatment (>3 prior lines) has been identified as a risk factor for infection regardless of baseline malignancy and CAR T-cell target (15, 17, 21, 26). History of allogeneic HCT was identified as a risk factor for early bacterial and viral infections in a cohort of 84 pediatric and young adult patients with R/R ALL (14). Impaired baseline performance status is associated with increased infectious risk after CAR T-cell therapy (12, 26). Finally, a history of infection 30–100 days before lymphodepletion has been associated with increased infectious risk post in the early period after cell therapy in multiple studies (12, 15, 26); Hill et al. reported that severe early infections present prior to lymphodepletion progressed after CAR T-cell infusion (11). Bridging chemotherapy, which is at times given for disease control during the CAR-T manufacturing period, was identified as a risk factor for severe infection before day 30 amongst 85 adults receiving CD19-targeted CAR T-cell therapy for DLBCL (16). This relationship was also noted by Kambhampati et al. in a cohort of 56 adults with MM undergoing BCMA-directed CAR T-cell therapy (26).
Underlying malignancy type has shown to play a role: a recent systematic review of 41 studies with 3199 patients receiving CAR-T therapy for hematological malignancy identified multiple myeloma patients as those at the highest risk for bacterial and viral infections as compared to those with ALL or NHL (29). Mikkilineni et al., in a retrospective analysis of 162 children and adults with a variety of malignancies treated with CAR T-cells directed against a variety of targets (CD19, CD22, GD2, BCMA), also identified those with multiple myeloma as the group with the highest risk for infection (15). Among CD19-directed CAR T-cell therapy, Hill et al. identified diagnosis of ALL as a risk factor for infectious complication within 90 days among 133 patients (ALL, NHL, CLL) (11).
Baseline hypogammaglobulinemia (IgG<400mg/dL) has been reported amongst CD19- and BCMA-directed CAR T-cell therapy recipients (11, 23–26). However, the impact of this characteristic on overall infectious risk is unclear. While some reports detect an association between baseline hypogammaglobulinemia and infectious risk after CD19-directed CAR T-cell therapy (12, 24), one large study did not (11). Moreover, baseline hypogammaglobulinemia has been reported in up to 88% of patients before receiving BCMA-directed therapy (23), and this has not been found to correlate with post-CAR-T infectious risk consistently (26).
Baseline neutropenia (absolute neutrophil count, ANC<500cell/mm3) has been shown to increase infectious risk in the early period after CAR T-cell therapy (11, 15, 18, 30). Recently, the CAR-HEMATOTOX (HT) score model evaluated baseline marrow reserve (platelets, hemoglobin, ANC) and baseline inflammatory markers (ferritin level and CRP) as predictors of post-CAR-T prolonged neutropenia and clinical outcomes, including infection, where high HT score was found to be associated with high risk of severe infection (30–32) Table 1. This tool might become helpful in identifying patients at high risk for post-CAR-T complications, including infections.
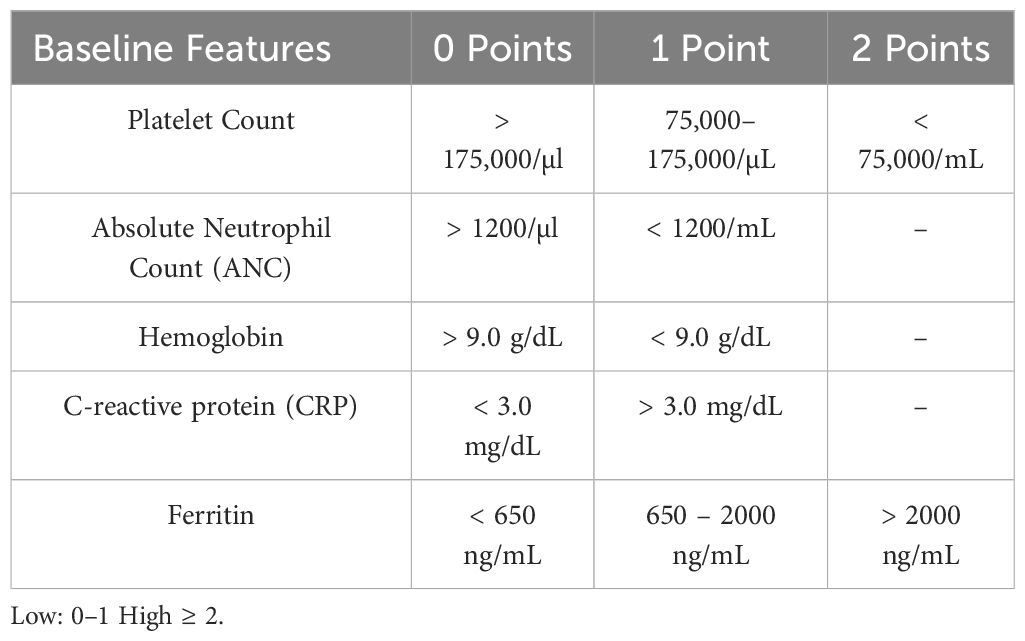
Table 1 CAR-HEMATOTOX (HT) Score Model for Pre CAR-T Risk Assessment (31).
Key points:
-Prior lines of therapy, history of allogeneic HCT, underlying malignancy, performance status, baseline hypogammaglobulinemia, pancytopenia, and inflammatory markers have been associated with increased baseline before CAR-T.
-Baseline risk factor assessment is important to identify high-risk patients before CAR-T infusion.
3.2 CAR-T/Post CAR-T related risk factors
Lymphodepletion (LD) before CAR T-cell infusion attenuates the immune response to CAR T-cells, allowing for robust engraftment and anti-tumor effect (33, 34). Cyclophosphamide with fludarabine has been associated with lower infectious risk than other lymphodepletion regimens (11, 24). Notably, early infections most often occur at times of neutropenia (13), which may be pre-existing or brought on by bridging chemotherapy and/or LD prior to CAR T-cell infusion.
A high CAR T-cell dose (2 x 10^7 cells/kg) is associated with an increased risk for infection (11). This may be due to the reported relationship between higher CAR T-cell dose and CRS development and severity. This study identified an increased hazard for infection with each increase in CRS severity category (11). Multiple other studies have demonstrated this relationship between CRS severity and infectious risk (13, 24, 35). Park et al. identified CRS of grade 3 or higher as an independent risk factor for infection (adjusted hazard ratio 2.67, p = .05), particularly with bloodstream infection (13). Still, this relationship has not been replicated in other studies (12).
Post CAR T-cell therapy, the use of steroids and tocilizumab in the management of CRS is not consistently shown to be associated with increased infection density. Hill et al. did not identify treatment with corticosteroids as a risk factor for infectious complications, and there was insufficient evidence to ascertain any correlation between tocilizumab duration or dose and risk for infectious complications (11). However, other studies have found that CRS, tocilizumab use, and corticosteroid use were associated with increased infectious risk after BCMA-directed CAR T-cell therapy (23) and in the first 30 days after CD19-directed CAR T-cell therapy (16). It is observed that most infections occur after the onset of CRS and do not appear to precipitate or exacerbate it (11). ICANS grade >2 has been identified as a risk factor for infection after CD19-directed therapy in multiple studies (11, 12, 16). The mechanisms by which CRS and ICANS may predispose to infection are unclear; pre-clinical studies have shown that chronic CAR signaling may induce early exhaustion of T cells, but there is currently no evidence to suggest that this results in clinically significant immune dysfunction (36). Also, severe CRS is associated with hematological toxicity, which can indirectly increase infection risk (37). While high-dose systemic corticosteroids are well known to predispose to infection (12, 15, 16, 19, 30), the effect of limited doses of tocilizumab and/or anakinra on overall infectious risk is less clear (38). Long-term treatment with tocilizumab has been shown to increase susceptibility to opportunistic infections in rheumatoid arthritis patients treated with these agents (39).
Hematological toxicity, also known as Immune Effector Cell-Associated Hematotoxicity (ICAHT), is recognized as an important toxicity attributable to CAR T-cell therapy regardless of target (40). Cytopenia persists beyond the immediate post-CAR-T phase, and count recovery often follows a nonlinear trajectory–intermittent recovery is often followed by subsequent dips (40). Patients can develop severe bone marrow aplasia, often refractory to growth factor support. ICAHT is divided into early (day 0–30) and late (day +30) based on the depth and duration of neutropenia (40). ICAHT has come to be recognized as a novel toxicity category of CAR T-cell therapy (40). A real-world experience applying the grading system to a cohort of 549 patients treated with BCMA- or CD19-directed CAR T-cells for refractory B-cell malignancies (MM, DLBCL, MCL) found that severe ICAHT was associated with a higher rate of severe infections and inferior survival outcomes (41). Among ICAHT, late neutropenia has been reported in multiple trials involving BCMA-directed and CD19-directed CAR T-cell therapy and can occur in a biphasic pattern with an intermediate recovery period (25, 26, 42, 43). In multiple studies, prolonged neutropenia due either to CAR-T-related factors or persistent disease has been shown to increase the risk for late infections (11, 26). CD4 lymphopenia (CD4 < 200 cell/mm3) is also frequently reported, lasting beyond day 30 (42) and up to 1 year (16) after CD19-directed CAR T-cell therapy and up to 9–12 months after BCMA-directed therapy (26). However, the impact of lymphopenia on overall infectious risk still needs further exploration. One study in a cohort that received CD19-directed therapy for DLBCL detected no relationship between CD4 and CD8 count at 30 days and infectious risk over 1 year (27). However, another study did detect a trend toward increased infectious risk with post-CAR-T lymphopenia after BCMA-directed therapy (22).
Hypogammaglobulinemia (IgG, <400mg/dL) affects 16–40% of patients before CAR T-cell therapy, and levels may decrease further after cell infusion and remain low for months or even years (11, 16, 20, 26). Amongst pediatric and young adult populations, CD19-directed therapy is associated with prolonged B-cell aplasia in up to two-thirds of patients and can persist for up to 5 years (44, 45). In a real-world experience, as many as half of patients received immunoglobulin replacement therapy (IGRT) for IgG<400 mg/dL in the post-CAR T-cell period (46). The reported severity and duration of hypogammaglobulinemia differs amongst adult populations receiving CD19-directed therapy; in one study, about half of patients had IgG >400mg/dL at the 1-year time point (26) after BCMA-directed CAR T-cell therapy, IgG<300 mg/dL in 70% of patients between 30–90 days and in 41% of patients after 1 year. Hill et al. reported that among 39 patients, 22 (56%) had a total IgG concentration <400 mg/dL at any time post CD19 CAR-T, and the cumulative incidence of an IgG concentration <400 mg/dL by 3 and 12 months post–CD19-CARTx was 36% and 60%, respectively (47). The significance of prolonged hypogammaglobulinemia on infectious risk remains unclear and may vary with CAR-T target. BCMA and CD19 are expressed on normal B cells at different stages of differentiation; CD19 is expressed on B cells at earlier stages and is lacking from long-lived plasma cells, which maintain stable concentrations of antigen-specific antibodies (48). Targeting CD19, therefore, leads to B-cell aplasia and hypogammaglobulinemia, but pathogen-specific IgG levels may be maintained. This was illustrated by the persistence of seroprotective levels of measles antibody independent of total immunoglobulin level after CD19-directed CAR T-cell therapy in one study (47). In agreement with this finding, evidence suggests that infectious risk may not correlate with lower IgG levels in this population (20, 47). In contrast, BCMA is expressed on plasma cells, so targeting this would be expected to lead to more severe hypogammaglobulinemia with a decline in pathogen-specific antibodies. Loss of immunoglobulin diversity and pathogen-specific immunity after BCMA-directed CAR T-cell therapy has been demonstrated in two cohort studies to date (22, 49). Kambhampati et al. also found a trend toward more infections during times of profound hypogammaglobulinemia after BCMA-directed therapy (22).
None of the risk variables listed above has an established attributable risk. The interaction and cumulative risk of the aforementioned variables may impact the patient’s overall infection risk. Thus, it is important to evaluate each situation carefully to optimize screening and preventive measures.
Key points:
- Risk factors associated with and after CAR-T infections include the type of lymphodepletion regimen, the dose of CAR T-cells, CRS post-infusion, use of systemic steroids, ICAHT, and hypogammaglobulinemia.
- Management of ICAHT and hypogammaglobulinemia management may modify infection risk after CAR-T infusion; prospective trials are needed to support this approach.
3.3 Multidrug-resistant organisms and antibiotic utilization
Antibiotic utilization post-CAR-T infection remains high (50), given that the most common infections in the early post-CAR-T period are bacterial, and most IEC-associated toxicities can present with fevers. The epidemiology of MDRO infections among CAR-T recipients is unknown; Yang J et al. showed poor 1-year clinical outcomes associated with Carbapenem-resistant organism infections among these patients (51). The utility of MDRO screening to assess MDRO colonization [as studied among HCT recipients (52)] and its impact on clinical outcomes still needs further investigation for CAR-T recipients. Similarly, antibiotic-associated microbiome dysbiosis has been associated with poor response to CAR-T therapy and increased toxicity (53, 54). These findings highlight the unmet need to study MDROs and antimicrobial stewardship in these complex settings.
3.4 Fungal infections and their risk factors
Despite multifactorial immune suppression, both mold and non-mold infections are infrequently reported as complications of CAR T-cell therapy (11, 55). Based upon cohort studies, epidemiology varies with CAR-T target, possibly due to unique pre- and post-CAR T-cell period features.
A review of published studies in 2021 by Garner et al. among CD-19-directed CAR T-cell recipients reported 1–10% incidence of yeast and 0–7% incidence of mold infections; most fungal occurred within the first 30 days and often represented breakthrough yeast infections in patients receiving fluconazole or echinocandin prophylaxis (56). Earlier, Hill et al. reported a 3% incidence of fungal infections between days 0 and 28 amongst 133 patients who all received fluconazole prophylaxis during the period of neutropenia after CD19-directed CAR T-cell therapy; all patients with fungal infections were reported to have been treated for CRS or ICANS with tocilizumab and/or corticosteroids (11). Incidence of fungal infections declined between days 28 and 90; late fungal infections were noted to have occurred in patients who had undergone prior allogeneic HSCT. Invasive mold infections were documented in both early and late periods but remain rare (11). This pattern of early-period fungemia while on echinocandin prophylaxis has been described by Park et al., and others have also found low-frequency invasive mold infections in both early and late periods (12, 13). More data is needed, but according to these results, it seems that a higher net burden of immunosuppression—development of CRS/ICANS (11, 16), HLH (35), treatment with tocilizumab or corticosteroids (11), other immunomodulating agents, and higher burden of prior treatment (>5 prior lines of therapy) (16), and history of HSCT (11)correlates with risk for fungal infection after CAR T-cell therapy.
While the low incidence of fungal infections reported in studies suggests the efficacy of antifungal prophylaxis (11–13), one large study reported a similarly low 2.9% incidence of invasive fungal infections at 1-year follow-up amongst 280 CD19 CAR T-cell patients with NHL who did not receive any antifungal prophylaxis (35). That cohort was also reported to have a high (41%) prevalence of severe delayed neutropenia, which may have been expected to increase susceptibility to fungal infection. Five of eight fungal infections reported occurred before day 100, including non-mold and mold infections. Of the three invasive mold infections reported, all were diagnosed by day 100 (35). Two of these infections occurred in patients with CLL and had received ibrutinib (35), which is an independent risk factor for mold infection (57, 58). Garner et al., in their review of invasive fungal disease, also noted that 73% of invasive mold infections occurred in patients with B-ALL or CLL (56). Studies to date have not been sufficiently powered to detect differences in risk for mold infection by different malignancy types. However, the underlying immune deficits associated with CLL and B-ALL and treatment with Bruton’s tyrosine kinase (BTK) inhibitors likely impact infectious risk in these patients (59, 60).
The epidemiology of fungal infections after BCMA-directed CAR T-cell therapy is less well-characterized. The overall incidence of fungal infection at 6 months of follow-up amongst patients who received some form of antifungal prophylaxis during their period of neutropenia has been reported at 4% (49) and 6% (22). Cumulative incidence declines with distance from the date of infusion (49), although one report recorded fungal infections, including invasive mold infections, occurred beyond 30 days after CAR T-cell infusion (11). Other reports also identified invasive mold infections in the early period (27, 49). High-grade CRS2516 and severe prolonged neutropenia (49) are possible risk factors for mold infections in the early and late periods, respectively.
3.5 Pneumocystis jirovecii pneumonia and its risk factors
PJP has been reported in patients beyond 3 months after CD19-directed CAR T-cell infusion and typically occurs after PJP prophylaxis has been discontinued (12, 35) or when prescribed prophylaxis has not been appropriately taken (35). According to one report, most cases of PJP occurred in patients with CD4 <200 cells/mm (35). A recent report of a real-world research network database showed that among 1107 Cd-19 CAR-T patients and 280 BCMA CAR-T patients, the incidence of PJP pneumonia was 1.7% and 1.4%, respectively. Patients who developed PJP had a higher likelihood of prior dexamethasone usage (65% versus 43%, p=0.02) and a reduced duration of trimethoprim/sulfamethoxazole (TMP/SMX) prophylaxis (median 9 weeks [range 1 to 45] versus 19 weeks [range 1 to 106], p=0.002. There was no difference in overall survival among patients with and without PJP (median 518 days vs not-reached, HR 1.61, 95%CI 0.91 to 2.88) (61). PJP incidence remains low likely related to widespread use of prophylaxis for a reported 3–6 months starting after neutrophil recovery and/or low rates of sustained CD4 lymphopenia among BCMA CAR-T, though further data is needed (22, 49).
3.6 Cytomegalovirus infection and its risk factors
The epidemiology of CMV infection after CAR T-cell therapy is poorly understood due to inconsistencies in routine surveillance practices. One prospective trial of 72 adult CMV seropositive patients receiving CD19-, CD20-, or BCMA-targeted CAR T-cell therapy identified a 27% (95% CI 16.8–38.2) cumulative incidence of CMV viremia (62). No end-organ disease was observed in that cohort, although 5 patients received preemptive therapy. BCMA-directed CAR T-cell therapy and corticosteroid use for >3 days were significantly associated with CMV reactivation (62). Another study reported a 10% incidence of CMV viremia among 61 BCMA CAR-T recipients in the first 6 months after cell infusion; end-organ involvement was uncommon but was reported in 3 cases, including gastrointestinal and possible lung involvement (63).
In one recent study, CMV accounted for 11% of all documented viral infections in the first year after CD19-directed therapy for DLBCL and was one of the most common viral infections in the study period (64). ICANS grade 3 or 4, CRS grade 3 or 4, anakinra use for treating CRS/ICANS, and higher cumulative doses of steroids within the first 30 days after cell infusion were all associated with CMV reactivation in this report.
In one large study by Marquez-Algaba et al. among 95 CMV-seropositive patients receiving CD19 CAR T-cell therapy for aggressive B cell lymphoma, 42(44%) patients had at least one positive serum CMV viral PCR; only 7 patients received preemptive antiviral treatment, and no CMV end-organ disease was reported (65). Dexamethasone treatment was the sole independent risk factor associated with CMV viremia > 1000 IU/mL in the study (65). In another cohort, amongst 133 patients with various B cell malignancies receiving CD19-directed therapy, there was one case of CMV pneumonia that occurred between days 29 and 90 in a patient with B-ALL (11). A recent meta-analysis identified an increased incidence of CMV reactivation in NHL patients; there were 39 cases in 949 patients, most of which occurred in the late period (10). Fareed et al. reported among 230 CD-19 CAR-T recipients, 10% developed clinically significant CMV infection. CMV infection was observed more among the female gender, with low ANC and monocyte count at day 30, grade 2 or higher CRS or ICANS requiring higher doses of steroids with higher mortality (66). These results demonstrate that CMV can cause disease in specific high-risk groups after CAR T-cell therapy. However, the significance of CMV viremia in the absence of end-organ disease still needs to be further explored.
3.7 Herpes simplex virus and herpes zoster virus infection and its risk factors
Reports of (HSV) and (VZV) reactivations after CAR T-cell therapy are infrequent, perhaps owing to the widespread use of antiviral prophylaxis. Reactivations have been reported after both CD19 and BCMA-directed therapies after discontinuing prophylaxis or in the setting of nonadherence with the prophylactic regimen (11, 13, 24, 28). However, delayed reactivations have also been reported despite the appropriate use of prophylaxis. There were two cases (3% incidence) of herpes zoster reactivation while on acyclovir prophylaxis in one study; both cases occurred after day 30 post-CAR T-cell infusion in patients who had received CD19 CAR T cells for DLBCL (11). A recent meta-analysis detected an increased signal for HSV/VZV reactivations in the late period in those with NHL (10). End-organ disease is not often reported to our knowledge; there was one reported case of fatal HSV pneumonia after BCMA-directed therapy in the setting of severe CRS and acyclovir resistance (67).
3.8 Human herpes virus 6 infection and its risk factors
Multiple cases of HHV-6 encephalitis in CAR-T recipients have been reported in the literature (19, 68–72). In one recent study, HHV-6 accounted for 8% of all documented viral infections in the first year after CD19-directed therapy for DLBCL (64). Several reported cases occurred in the context of mental status changes that initially responded to steroids but then relapsed and lasted beyond the typical time course of ICANS, prompting further infectious workup (68, 69). There has also been one case report of fatal HHV-6 myelitis following CD19-targeted CAR T-cell therapy in a patient who developed CRS and ICANS that initially responded to corticosteroids with subsequent development of ascending flaccid paralysis (73). Younger age, more prior lines of therapy, including allogeneic HSCT, and receipt of systemic corticosteroids may also increase susceptibility and lower the threshold for investigation (69, 71). Recently, Lareau et al. reported that HHV-6 can be reactivated among cultured T cells; implications and significance of this finding still need to be determined (74).
3.9 Adenovirus infections and its risk factors
There are no studies on the incidence of adenovirus infection in patients treated with CAR T-cells. Logue et al. reported one case of adenovirus viremia in a patient with DLBCL within 30 days of receiving CD19-directed CAR T-cell therapy. Still, the clinical significance of this finding was unclear (16). There are case reports of hemorrhagic cystitis cases associated with adenovirus infection (75). Further study is needed to elucidate the epidemiology and manifestations of adenovirus infection in the post-CAR T-cell period.
3.10 Polyomavirus (BKv) infection and its risk factors
There are anecdotal reports of BK virus infections and hemorrhagic cystitis after CD19-directed CAR T-cell therapy (11, 13). There are no reports of such infections associated with BCMA-directed CAR T-cell therapy. Case reports of late development of progressive multifocal leukoencephalopathy associated with JV virus have also been described (76, 77). Given the lack of conclusive data on the risk and impact of reactivation, the index of suspicion should be high in the context of hemorrhagic cystitis or atypical neurological symptoms with multifocal demyelination.
4 Prevention of infections
Preventive strategies can also be divided into two-time points, pre-CAR-T, and post-CAR-T, keeping the risk modifiable risk factors in mind. Screening and prophylaxis strategies are determined before CAR T-cell therapy. In contrast, post- CAR-T strategies are focused on reducing infection risk by infection surveillance when needed, ongoing chemoprophylaxis, immunoglobulin replacement therapy (IGRT), and vaccination administration.
4.1 Baseline screening pre CAR T-cell therapy
Infection screening before CAR T-cell therapy is an important risk assessment component. HIV, HBV, and HCV serologic testing with reflex nucleic acid testing is recommended for all patients (4). False positive HIV nucleic acid amplification tests (NAAT) may be seen in CAR-T recipients when the CAR-T product was generated using lentiviral vectors, owing to the use of conserved regions of HIV-1 (78). Products prepared with murine gamma-retroviral vectors are not thought to carry the same risk. In the case of a true false positive test, confirmatory fourth-generation HIV-1/2 antibody and p24 antigen testing returns negative, and HIV-1 RNA may be detectable at a low level, possibly due to circulating cell-free DNA (78). No further intervention or monitoring is required in these cases, and CAR T-cell therapy may proceed as planned (78). Patients with HIV infection were excluded from clinical trials, so the safety of CAR T-cell therapy in the HIV-positive population is poorly understood (79). There are, however, published reports of successful treatment of HIV-infected patients with DLBCL with CD19-targeted CAR T-cell therapy (80, 81).
Patients with HCV viremia (chronic HCV infection) should be considered for antiviral treatment if compatible with liver function and overall clinical situation (82). If liver test abnormalities develop during treatment, then the potential role of HCV and other hepatotropic viruses should be assessed. Notably, hepatitis E has been reported to cause chronic disease in immunocompromised patients (83). Prophylaxis and surveillance strategies in the case of positive hepatitis B serologies are discussed in detail in the next section. Serologic screening for HSV1/2 and VZV is recommended in those not already receiving antiviral prophylaxis; this is also important for future consideration of VZV vaccination (4). Screening for Mycobacterium tuberculosis (MTB) should be considered in patients with risk factors for exposure (84). Tocilizumab use is independently associated with an increased risk for MTB infections in rheumatoid arthritis patients [91]; however, the impact of the short-term dosing utilized in CRS management has not yet been reported. Baseline toxoplasma serologies may be considered on a case-by-case basis (4, 9). Finally, screening for antibodies to Strongyloides stercoralis or empiric treatment with ivermectin should be considered in patients with a history of time spent in tropical or subtropical regions, given the risk for reactivation due to high-dose corticosteroids and/or tocilizumab (4, 9).
A complete history and physical exam should be performed to evaluate for active infections before lymphodepletion. If the evaluation concerns an infection, a more directed workup should be performed as indicated by the clinical situation.
4.2 Prophylaxis and monitoring post CAR T-Cell infusion:
Based on the current literature, the recommendations below represent our opinion; further prospective studies are needed to optimize these practices.
The role of antibacterial prophylaxis in this population is unclear; fluoroquinolones are often employed, but practice patterns vary widely across institutions. A large retrospective analysis of patients with DLBCL receiving CD19 CAR T-cells identified a significant reduction of severe bacterial infections with fluoroquinolone prophylaxis in patients who were categorized as CAR-HEMATOTOXhigh but not in those who were CAR-HEMATOTOXlow at baseline (41). A risk-adapted approach may be the optimal strategy, but more studies are needed, and institutional guidelines vary. Multiple studies have reported early bacterial infections with gram-negative organisms with acquired or intrinsic fluoroquinolone resistance, regardless of fluoroquinolone prophylaxis patterns (11, 13). It is unclear how much antibacterial prophylaxis strategies should be adjusted to account for this. We suggest using fluoroquinolone prophylaxis when ANC<500 cells/mm3. Optimal infection prophylaxis strategies for those with severe ICAHT require further study. There may be a role in closely monitoring patients with high CAR-HEMATOTOX scores before lymphodepletion or among those who develop severe ICAHT post-CAR T-cell therapy (41).
The role of routine antifungal prophylaxis is also debated, given the low reported incidences of non-mold infections with or without prophylaxis (5, 11, 13, 16, 35, 49, 85). Until further data are available, we recommend fluconazole prophylaxis during periods of neutropenia in all CAR T-cell recipients. Anti-mold prophylaxis should be considered in select high-risk patients for invasive mold infection. We would recommend the use of a mold-active azole in those with a history of invasive fungal infection (IFI), ANC < 500 cell/mm3 for >21 days present prior to CAR T-cell therapy or developing after infusion, and treatment with high-dose steroids and for longer duration. Duration of anti-mold prophylaxis should be determined on a case-by-case basis; in the presence of multiple risk factors, extending prophylaxis beyond ANC recovery (ANC > 500 cell/mm3 for 3 consecutive days) may be appropriate.
PJP prophylaxis has also been widely adopted, though opinions on the optimal duration of prophylaxis remain mixed. Given reports of PJP infections diagnosed beyond 3–6 months after cell infusion in the context of prolonged CD4 lymphopenia and prophylaxis discontinuation or incomplete adherence (5, 12, 35), we would suggest starting prophylaxis upon initiation of lymphodepleting chemotherapy and continuing for at least 6 months or until CD4 count >200 cell/mm3. HSV/VZV prophylaxis with acyclovir or valacyclovir has been widely adopted and recommended.
Entecavir is recommended when HBsAg is positive and/or when HBV DNA is detectable before receiving CAR T therapy and should be continued for at least 6–12 months from infusion due to the risk of reactivation following B cell depletion (4, 9). If anti-HBc is positive but surface antigen and DNA are negative, antiviral prophylaxis OR lab monitoring with liver function tests and HBV DNA every 1–3 months can be performed. Those with anti-HBc+ and anti-HBs+ likely have a lower risk of reactivation. A more conservative approach may be appropriate in multiple myeloma patients given their more severe humoral immunodeficiency, including pathogen-specific immunoglobulin deficiency, which is depleted after BCMA CAR T-cell therapy (49). Table 2 summarizes recommendations for antimicrobial prophylaxis.
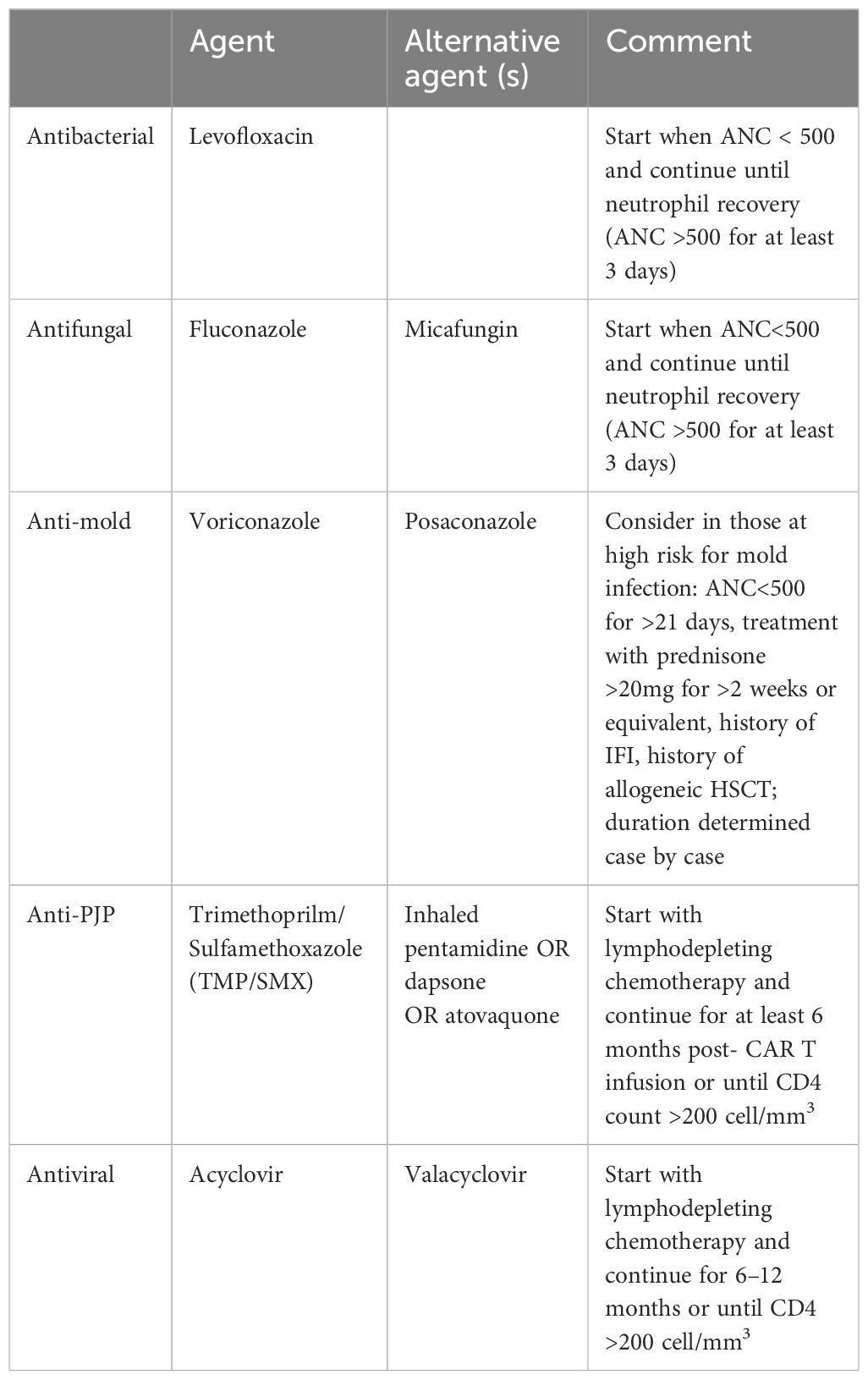
4.3 CMV monitoring, prophylaxis, and pre-emptive therapy post CAR-T infusion
The role of CMV monitoring remains a topic of debate. Still, there are reports of both CMV viremia and end-organ disease occurring after CD19- and BCMA-directed CAR T-cell therapy (11, 28, 65, 91). We suggest checking baseline CMV serostatus and serum PCR in all patients proceeding with CAR T-cell therapy. Those with a negative CMV assessment before CAR T-cell infusion and who do not require systemic steroids for related complications would likely not benefit from regular monitoring.
Those with a positive CMV assessment and/or those who require >3 days of high-dose systemic steroids and have received BCMA-directed therapy may benefit from weekly CMV monitoring during the first 6 weeks after CAR T-cell therapy. This recommendation is based upon limited data on the kinetics of CMV reactivation after CAR T-cell therapy (62), the duration of monitoring should be adjusted on a case-by-case basis. There is insufficient data to suggest prophylactic strategies in this setting. The optimal role and threshold for pre-emptive therapy also remain unclear and must be balanced against the risks of therapy-related toxicity, including further bone marrow suppression.
Allogeneic CAR T-cell therapy (allo CAR-T) is an active research area and may carry unique risk factors for CMV and other viral reactivations. Chemotherapy ahead of allo CAR-T, or lymphodepletion in preparation, may require novel strategies such as using alemtuzumab to mitigate the risk of GVHD and promote engraftment and expansion of the infused cells (92, 93). The resultant deeper and more prolonged lymphodepletion would pose an important risk factor for viral reactivation (93).
5 CRS, ICANS, and fever after CAR-T infusion
CAR T-cell infusion can result in CRS, manifesting as fever, capillary leak, and end-organ dysfunction (94–96). The typical time frame for CRS presentation is 2–7 days after CAR T-cell infusion, but it may occur within hours or up to 10–15 days after infusion (97). ASTCT consensus grading ranges from grade 1 to 5, reflecting a spectrum of presentations from fever and constitutional symptoms to critical illness requiring invasive monitoring, pressors, and ventilatory support (97). ICANS is regarded as a separate clinical entity characterized by encephalopathy that can be progressive, language disturbances, motor weakness, seizures, and cerebral edema (3, 95, 96). ICANS typically present later than CRS, with a typical time to onset of 4 to 19 days after receiving CAR T-cell infusion (98). While the two entities do not always occur in the same host, severe ICANS is unlikely to be seen without severe CRS (95, 96).
Both CRS and ICANS can mimic infections. Severe CRS may present as septic shock requiring invasive monitoring and is managed with high doses of steroids; critical illness and steroids can cause neurologic symptoms (3, 95–97, 99), which may be difficult to distinguish from ICANS. Moreover, managing CRS and ICANS involves multimodal systemic immunosuppression, which can mask the clinical presentation of infection. Corticosteroids and anti-IL-6 therapy with tocilizumab remain cornerstones of CRS management, while corticosteroids are the treatment of choice for ICANS with the addition of anti-IL-1 therapy with anakinra in refractory cases (3, 94–96). More recently, there has been a paradigm shift towards pre-emptive use of these modalities to prevent progression to higher-grade manifestations (98, 100).
Given the ambiguity of the clinical presentation, it is crucial to initiate broad-spectrum antibiotics promptly and to investigate reversible infectious causes as part of the initial assessment. In practice, broad-spectrum antibiotics are generally administered in the setting of CRS after CAR-T infusion in the setting of febrile neutropenia, hemodynamic instability, and hypoxia (4, 8). Empiric coverage should consider the patient’s history of prophylaxis, and local antibiogram and infection work-up should be initiated. Infectious disease consultation should be considered early, especially when the clinical picture is complex. There is increasing awareness of the adverse effects of broad-spectrum antimicrobials, including microbiome dysbiosis, the emergence of multidrug resistance organisms, and Clostridium difficile infection (101, 102). To mitigate these risks, we advocate for diligent de-escalation strategies. Consideration should be given to stopping broad-spectrum antibiotics in patients with neutropenia who have been afebrile for 72 hours and remain without clinical or microbiologic source of infection (103–105).
6 Emergent hemophagocytic lymphohistiocytosis-like toxicities and infections
Immune effector cell-associated HLH-like syndrome (IEC-HS) has been described post-CD-19 directed CAR-T infusion, with severe and fulminant cases occurring in <1% of patients (6). Earlier reports characterized IEC-HS progressing from cases of severe CRS, but recently, there has been increasing recognition of delayed HLH-like toxicities (7). IEC-HS is also becoming more apparent after BCMA CAR T-cell therapy (105). The definition of IEC-HS is outside the scope of this paper; it is important to recognize that the clinical presentation can mimic infection and that infection can co-occur with this entity. We recommend initiating appropriate antimicrobials while infectious work-up is underway. Treatment of IEC-HS involves extensive immunosuppression, thus increasing overall infection risk. Table 3 highlights the immunosuppressive agents and the infections associated with them. As treatment strategies for CRS, ICANS, and IEC-HS evolve, it is imperative to keep the infectious risks attributed to these agents in mind and consider adjusting the prophylaxis strategy to account for their use.
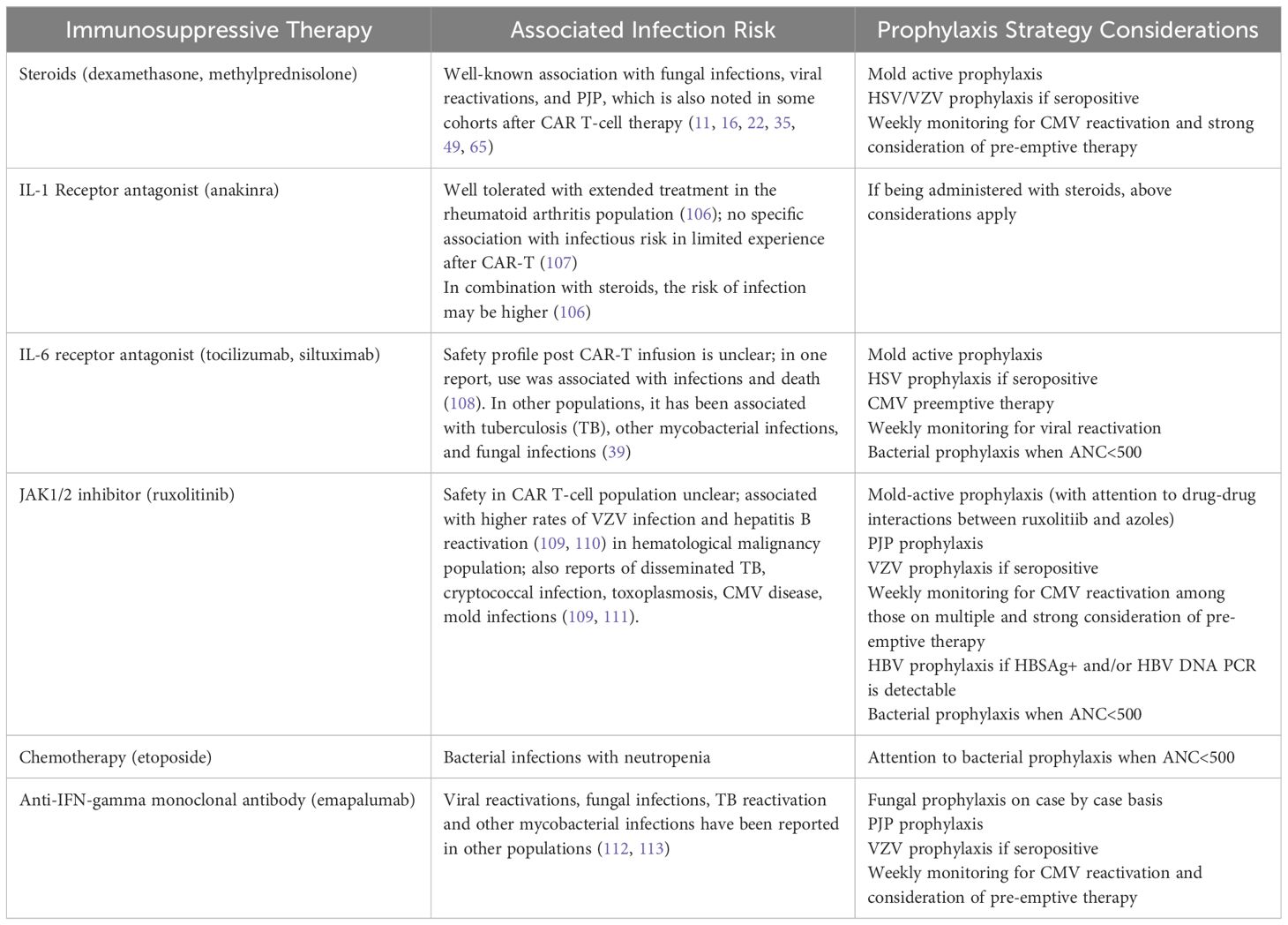
Table 3 Immunosuppressive Agents Used for Treatment Of CRS/ICANS/HLH and their Associated Infection.
7 Post-infusion hypogammaglobulinemia and IgRT
Although prophylactic IgG has received regulatory approval in specific immunocompromised groups, less extensive data indicates similar effectiveness in CAR T-cell recipients5510. IgRT has primarily been established as beneficial in preventing serious bacterial infections; there is less evidence to support its use in the prevention of viral infections, which are more frequently reported as late complications of CAR T-cell therapy (48). In fact, studies to date have failed to reliably show an association between hypogammaglobulinemia and infectious risk after CAR T-cell therapy regardless of targe (47, 48), and there is similarly mixed data on the efficacy of IgRT in these populations (22).
However, based on the pathophysiology of humoral immunodeficiency after CAR T-cell infusion, there is consensus that IgG levels should be monitored both pre-CAR T-cell infusion and monthly for at least 3 months after infusion (4, 5). A threshold IgG level of 400 mg/dL is frequently used to initiate IGRT in adults (4, 5). Adverse events are infrequently reported after IVIG infusion but do include mild and occasionally severe infusion reactions, and delayed toxicities including thrombosis that may manifest as ischemic stroke or myocardial infarction, renal failure, and transient hemolytic anemia (48, 114). IVIG is also associated with significant costs and accessibility issues, so use should be judicious and guided by a multidisciplinary team effort (48).
Given mixed data on the impact of hypogammaglobulinemia on infectious risk after CD19-directed CAR T-cell therapy, as well as the preservation of pathogen-specific antibodies detected after CD19-directed therapy, we would suggest a tailored approach to IgRT in this subset of CAR-T recipients. It may be appropriate to reserve IgRT for patients with recurrent and/or severe infections and IgG < 400 mg/dL before cell infusion and in the first 3–6 months after CD19-targeted therapy. In those with IgG levels between 400 mg/dL and 600 mg/dL, IGRT may also be considered for severe or recurrent infections. If there are recurrent infections in normal serum immunoglobulin levels, testing for functional humoral immune dysfunction, e.g., vaccine response, may be helpful (4, 114). A more conservative approach may be appropriate in BCMA CAR T-cell recipients given their more profound depletion of pathogen-specific immunity (22, 49); IgRT may be considered in those with IgG< 400 mg/dL, even without evidence of recurrent infection with caution (48). In these patients, distinguishing between normal IgG and paraprotein with serum protein electrophoresis is important. Prospective trials are needed to optimize these strategies.
8 Vaccinations for CAR T-cell recipients
B-cell aplasia and hypogammaglobulinemia can increase susceptibility to infection by vaccine-preventable encapsulated bacteria such as Haemophilus influenzae type B, Neisseria meningitidis, and Streptococcus pneumoniae. Appropriate vaccination has the potential to prevent infections, decrease their severity, mitigate the need for IgRT, and improve survival and quality of life (4).
Walti et al. identified decreased seroprotection levels against S. pneumoniae and H. influenzae type B after CD19-directed therapy (115). In this same study, levels of IgG against measles, tetanus toxin, Epstein-Barr virus, varicella-zoster virus, and herpes simplex virus remained detectable despite decreased total serum IgG and B-cell aplasia (115). Among those who attained CR, the proportion of participants with seroprotective IgG titers to vaccine-preventable infections was comparable to population-based seroprevalence data without re-vaccination (115). In contrast, a small cross-sectional study suggested that BCMA CAR T-cell recipients are less likely to have seroprotective IgG titers to vaccine-preventable infections (49). It remains unclear whether this reflects CAR T-cell therapy’s effects or indicates baseline humoral immunodeficiency; in that same study, measles-specific IgG was present in only 16% of patients before cell infusion (49).
The optimal timing of vaccination after CAR T-cell therapy is unclear, as the timing of cellular and humoral immunity reconstitution after CAR T-cell therapy can vary widely (2, 9). However, a recent study reported that neither B-cell aplasia nor hypogammaglobulinemia reduced influenza vaccine immunogenicity after CAR T-cell therapy (116), suggesting that strictly following these markers to assess for immune reconstitution may create unnecessary delays in vaccination. Another study noted recovery of seroprotective measles IgG level by 114 days after CD19 CAR T-cell therapy without vaccination and coinciding with CD19+ B-cell recovery (47), suggesting that immune recovery may obviate the need for aggressive re-vaccination strategies in select groups. Optimal strategies may need to be considered based on the CAR-T target.
Vaccination against influenza during flu season should be considered at least two weeks before lymphodepletion; additional vaccination before cell infusion is likely of low utility given the impending severe immunosuppression. Clinical practice for revaccination currently follows protocols used in HCT recipients, though the need for revaccination for all previously completed vaccine series remains unclear (4, 48, 117, 118). In general, for patients who are in remission and not planned to receive further T-cell and/or B-cell depleting therapies, killed/inactivated vaccinations should be considered starting at least 6 months after CAR T-cell infusion, and live and adjuvant vaccines should be considered at least 1 year after cell infusion. IgRT may interfere with the efficacy of live vaccines, so these should generally be delayed for at least 9 months after the most recent IgRT (4, 48). This recommendation is based on the guidelines for vaccination of immunocompromised hosts (118, 119) and the kinetics of immune reconstitution after CAR T-cell therapy (11, 120, 121). SARs-CoV-2 vaccination can be started after 90 days given (122).
Based on the epidemiology of infections after CAR T-cell therapy and known effects of B-cell aplasia and hypogammaglobulinemia, key vaccines to consider include annual influenza, Streptococcus pneumoniae, Haemophilus influenzae type b, Corynebacterium diphtheriae and Clostridium tetani toxins, Bordetella pertussis, and hepatitis A and B viruses. For patients 50 years old who are seropositive for VZV or have a history of shingles, recombinant zoster vaccine (Shingrix) should also be considered. Conjugated vaccines should be used, when possible, given higher response rates in immunocompromised patients (123). Measuring vaccine responses may be helpful to assess the utility of additional vaccination on a case-by-case basis (4).
For respiratory viruses, COVID-19 and RSV vaccines (age-dependent indication) should also be considered in addition to the influenza vaccine (124, 125). Data regarding the durability of seroprotection provided by pre-infusion COVID vaccination and the immunogenicity of mRNA-based COVID vaccines after CAR T-cell therapy are mixed (126). We suggest following the CDC’s guidance on COVID-19 vaccines for moderately and severely immunocompromised people (127). The efficacy of RSV vaccines in CAR-T recipients is unknown. Table 4 highlights the vaccine recommendations for CAR-T recipients.
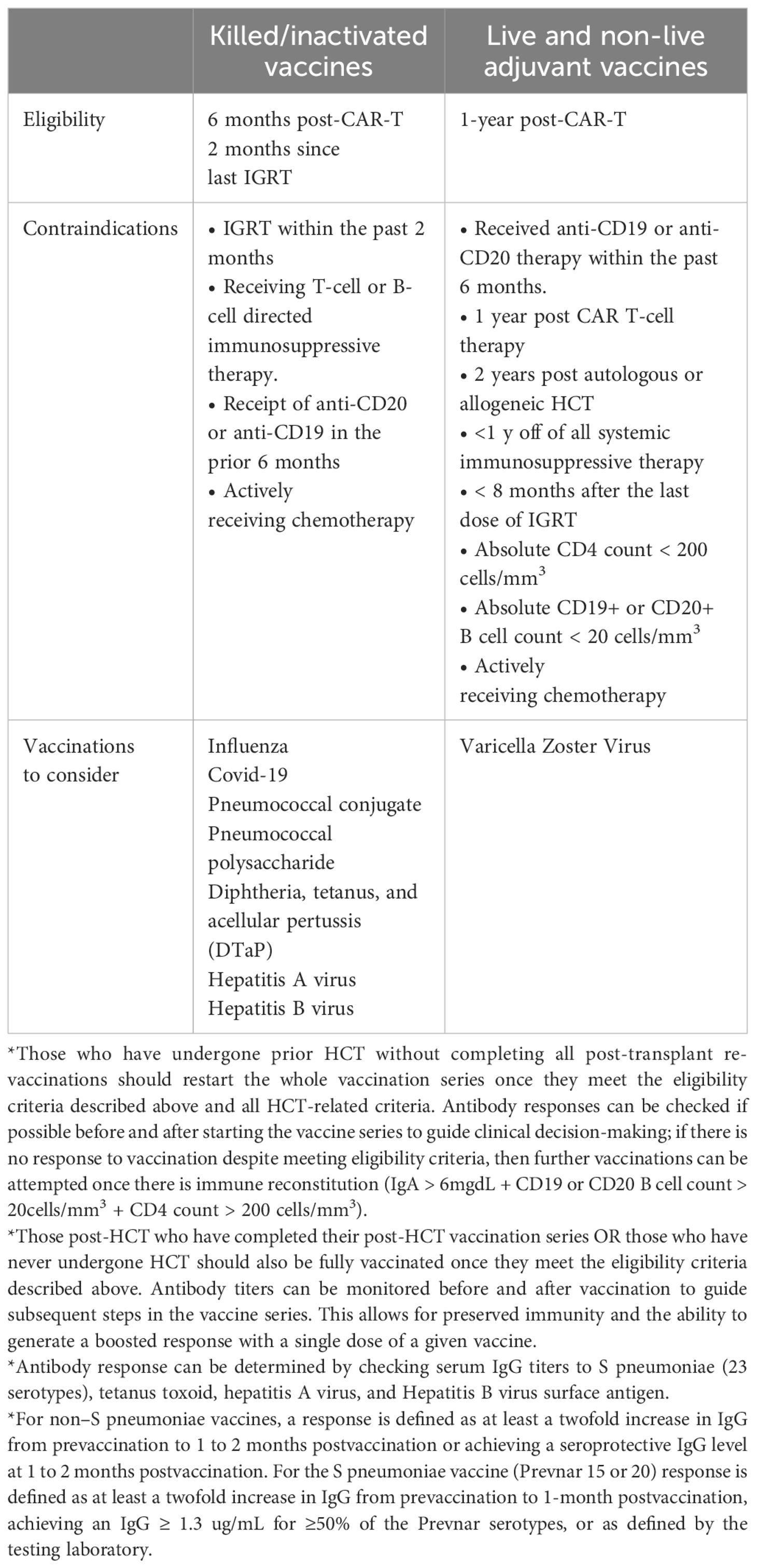
Table 4 Vaccines For CAR T-cell Therapy Recipients.
9 Future directions and knowledge gaps
The field of CAR T-cell therapy is continuously evolving, with the development of newer targets, combination therapies, newer CAR-T designs, and off-the-shelf products [121], and important gaps remain in our understanding of short and long-term infectious complications. As new CAR T-cell targets are introduced to the market for hematological malignancies as well as for solid tumors, and potentially for use in autoimmune diseases and suppression of rejection in organ transplant (82), transparency and completeness of reporting about infectious complications of these therapies will remain critical as we seek to devise strategies to mitigate risks (128). An example is the development of allogeneic (off-the-shelf) CAR T-cell therapy; these products come with the risk of rejection and development of graft versus host disease (GVHD) and may require unique pre-infusion LD and post-infusion immunosuppression strategies (129). Allogeneic T cell-based products have entered Phase I and II clinical trials, and different effector cell types, such as NK cells and macrophages, are also under investigation (129). Dual-targeted CAR T-cells, for example, targeting CD19 and CD22 for treating R/R aggressive B-cell lymphomas, are also in development (130). CAR-T therapy for Acute Myeloid Leukemia can lead to myeloablation (131). Understanding infection incidence and risk associated with these novel treatments is vital to developing mitigating strategies.
The infection burden of DNA viruses post CAR T-cell therapy is still evolving (63, 74, 132, 133), and might vary among CAR T-cell targets. Prospective studies are important to understand CAR T-cell recipients’ specific pathogen and infection burden. Similarly, long-term infection data are lacking. Extended research is necessary to evaluate the frequency of late-onset infections and the influence of persistent immune dysfunction on the risk of infections among individuals treated with CAR T-cell therapy.
Microbiome dysbiosis among CAR T-cell recipients is associated with poor clinical outcomes, including CAR-T toxicities and response to therapy (53). Similarly, a non-antibiotic-disrupted gut microbiome is associated with improved clinical response to CD-19 CAR T-cell therapy (134). This highlights the importance of developing strategies for early identification of infection mimickers post CAR T-cell and prompt de-escalation of broad-spectrum antimicrobials when appropriate.
In conclusion, infections remain an important concern among CAR T-cell recipients, and our understanding of infection dynamics among different settings is still evolving. Addressing these knowledge gaps is imperative to improving patient outcomes. Additionally, ongoing research efforts should prioritize the establishment of standardized guidelines for monitoring infections, implementing prophylactic measures, and administering treatments that cater to the unique requirements of CAR T-cell recipients. This approach will ultimately enhance our patients’ overall quality of care and prognosis.
Author contributions
ZS: Conceptualization, Methodology, Supervision, Writing – original draft, Writing – review & editing. SA: Data curation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors express their deep appreciation to the Leon Levine Foundation and the Kerry and Simone Vickar Family Foundation for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel). (2023) 15(3):663. doi: 10.3390/cancers15030663
2. Kampouri E, Little JS, Rejeski K, Manuel O, Hammond SP, Hill JA. Infections after chimeric antigen receptor (CAR)-T-cell therapy for hematologic Malignancies. Transpl Infect Dis. (2023) 25:e14157. doi: 10.1111/tid.14157
3. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
4. Hill JA, Seo SK. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell Malignancies. Blood. (2020) 136(8):925–35. doi: 10.1182/blood.2019004000
5. Wudhikarn K, Perales MA. Infectious complications, immune reconstitution, and infection prophylaxis after CD19 chimeric antigen receptor T-cell therapy. Bone Marrow Transplantation. (2022) 57:1477–88. doi: 10.1038/s41409-022-01756-w
6. Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther. (2023) 29:438.e1–438.e16. doi: 10.1016/j.jtct.2023.03.006
7. Hashmi H, Bachmeier C, Chavez JC, Song J, Hussaini M, Krivenko G, et al. Haemophagocytic lymphohistiocytosis has variable time to onset following CD19 chimeric antigen receptor T cell therapy. Br J Haematol. (2019) 187(2):e35–8. doi: 10.1111/bjh.16155
8. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
9. Los-Arcos I, Iacoboni G, Aguilar-Guisado M, Alsina-Manrique L, Díaz de Heredia C, Fortuny-Guasch C, et al. Recommendations for screening, monitoring, prevention, and prophylaxis of infections in adult and pediatric patients receiving CAR T-cell therapy: a position paper. Infection. (2021) 49(2):215–31. doi: 10.1007/s15010–020-01521–5
10. Reynolds GK, Sim B, Spelman T, Thomas A, Longhitano A, Anderson MA, et al. Infections in haematology patients treated with CAR-T therapies: A systematic review and meta-analysis. Crit Rev Oncol Hematol. (2023) 192:104134. doi: 10.1016/j.critrevonc.2023.104134
11. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. (2018) 131(1):121–30. doi: 10.1182/blood-2017–07-793760
12. Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection during the first year in patients treated with CD19 CAR T-cells for diffuse large B cell lymphoma. Blood Cancer J. (2020) 10(8):79. doi: 10.1038/s41408–020-00346–7
13. Park JH, Romero FA, Taur Y, Sadelain M, Brentjens RJ, Hohl TM, et al. Cytokine release syndrome grade as a predictive marker for infections in patients with relapsed or refractory b-cell acute lymphoblastic leukemia treated with chimeric antigen receptor T cells. Clin Infect Diseases. (2018) 67:533–40. doi: 10.1093/cid/ciy152
14. Vora SB, Waghmare A, Englund JA, Hill JA, Gardner R. Infectious complications following CD19 CAR T cell immunotherapy for children and young adults with refractory ALL. Biol Blood Marrow Transplantation. (2019) 25:S355–6. doi: 10.1016/j.bbmt.2018.12.575
15. Mikkilineni L, Yates B, Steinberg SM, Shahani SA, Molina JC, Palmore T, et al. Infectious complications of CAR T-cell therapy across novel antigen targets in the first 30 days. Blood Advances. (2021) 5(23):5312–22. doi: 10.1182/bloodadvances.2021004896
16. Logue JM, Zucchetti E, Bachmeier CA, Krivenko GS, Larson V, Ninh D, et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica. (2021) 106(4):978–86. doi: 10.3324/haematol.2019.238634
17. Zhu F, Hu Y, Guoqing W, Huang H, Wu W. Incidence and risk factors associated with infection after chimeric antigen receptor T cell therapy for relapsed/refractory B-cell Malignancies. Blood. (2019) 134. doi: 10.1182/blood-2019–129649
18. Zhu F, Wei G, Liu Y, Zhou H, Wu W, Yang L, et al. Incidence and risk factors associated with infection after chimeric antigen receptor T cell therapy for relapsed/refractory B-cell Malignancies. Cell Transplant. (2021) 30:9636897211025503. doi: 10.1177/09636897211025503
19. Baird JH, Epstein DJ, Tamaresis JS, Ehlinger Z, Spiegel JY, Craig J, et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood Advances. (2021) 5(1):143–55. doi: 10.1182/bloodadvances.2020002732
20. Luo H, Wang N, Huang L, Zhou X, Jin J, Li C, et al. Inflammatory signatures for quick diagnosis of life-threatening infection during the CAR T-cell therapy. J ImmunoTherapy Cancer. (2019) 7(1):271. doi: 10.1186/s40425-019-0767-x
21. Hill J, Li D, Hay K, Green ML, Riddell S, Maloney D, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T cell immunotherapy. Open Forum Infect Dis. (2017) 4:S698–9. doi: 10.1093/ofid/ofx163.1875
22. Kambhampati S, Sheng Y, Huang CY, Bylsma S, Lo M, Kennedy V, et al. Infectious complications in patients with relapsed refractory multiple myeloma after BCMA CAR T-cell therapy. Blood Adv. (2022) 6(7):2045–54. doi: 10.1182/bloodadvances.2020004079
23. Josyula S, Pont MJ, Dasgupta S, Song X, Thomas S, Pepper G, et al. Pathogen-Specific Humoral Immunity and Infections in B Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy Recipients with Multiple Myeloma23. Transplant Cell Ther. (2021) 28(6):304.e1–304.e9. doi: 10.1016/j.jtct.2022.03.005
24. Vora SB, Waghmare A, Englund JA, Qu P, Gardner RA, Hill JA. Infectious complications following CD19 chimeric antigen receptor t-cell therapy for children, adolescents, and young adults. Open Forum Infect Diseases. (2020) 7:ofaa121. doi: 10.1093/OFID/OFAA121
25. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol Blood Marrow Transplant. (2020) 26(1):26–33. doi: 10.1016/j.bbmt.2019.08.003
26. Kambhampati S, Fakhri B, Sheng Y, Huang C-Y, Byslma S, Natsuhara K, et al. Infectious complications of BCMA-targeted and CD19-targeted chimeric antigen receptor T-cell immunotherapy. Blood. (2020) 136:4–5. doi: 10.1182/blood-2020–138940
27. Logue JM, Peres LC, Hashmi H, Colin-Leitzinger CM, Shrewsbury AM, Hosoya H, et al. Early cytopenias and infections after standard of care idecabtagene vicleucel in relapsed or refractory multiple myeloma. Blood Advances. (2022) 6(24):6109–19. doi: 10.1182/bloodadvances.2022008320
28. Wang Y, Li C, Xia J, Li P, Cao J, Pan B, et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. (2021) 5(23):5290–9. doi: 10.1182/bloodadvances.2021004603
29. Reynolds G, Hall VG, Teh BW. Vaccine schedule recommendations and updates for patients with hematologic Malignancy post-hematopoietic cell transplant or CAR T-cell therapy. Transplant Infect Dis. (2023) 25:e14109. doi: 10.1111/tid.14109
30. Rejeski K, Perez A, Iacoboni G, Penack O, Bücklein V, Jentzsch L, et al. The CAR-HEMATOTOX risk-stratifies patients for severe infections and disease progression after CD19 CAR-T in R/R LBCL. J Immunother Cancer. (2022) 10(5):e004475. doi: 10.1136/jitc-2021–004475
31. Rejeski K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, et al. CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood. (2021) 138(24):2499–513. doi: 10.1182/blood.2020010543
32. Rejeski K, Wang Y, Albanyan O, Munoz J, Sesques P, Iacoboni G, et al. The CAR-HEMATOTOX score identifies patients at high risk for hematological toxicity, infectious complications, and poor treatment outcomes following brexucabtagene autoleucel for relapsed or refractory MCL. Am J Hematol. (2023) 98(11):1699–710. doi: 10.1002/ajh.27056
33. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621
34. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126(6):2123–38. doi: 10.1172/jci85309
35. Little JS, Aleissa MM, Beluch K, Gonzalez-Bocco IH, Marty FM, Manne-Goehler J, et al. Low incidence of invasive fungal disease following CD19 chimeric antigen receptor T-cell therapy for non-Hodgkin lymphoma. Blood Advances. (2022) 6(16):4821–30. doi: 10.1182/bloodadvances.2022007474
36. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21(6):581–90. doi: 10.1038/nm.3838
37. Juluri KR, Wu QV, Voutsinas J, Hou J, Hirayama AV, Mullane E, et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. (2022) 6(7):2055–68. doi: 10.1182/bloodadvances.2020004142
38. Frigault MJ, Nikiforow S, Mansour MK, Hu ZH, Horowitz MM, Riches ML, et al. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: implications for COVID-19? Blood. (2020) 136(1):137–9. doi: 10.1182/blood.2020006216
39. Schiff MH, Kremer JM, Jahreis A, Vernon E, Isaacs JD, van Vollenhoven RF. Integrated safety in tocilizumab clinical trials. Arthritis Res Ther. (2011) 13(5):R141. doi: 10.1186/ar3455
40. Rejeski K, Subklewe M, Aljurf M, Bachy E, Balduzzi A, Barba P, et al. Immune effector cell-associated hematotoxicity: EHA/EBMT consensus grading and best practice recommendations. Blood. (2023) 142(10):865–77. doi: 10.1182/blood.2023020578
41. Rejeski K, Wang Y, Hansen DK, Iacoboni G, Bachy E, Bansal R, et al. Applying the EHA/EBMT grading for ICAHT after CAR-T: comparative incidence and association with infections and mortality. Blood Adv. (2024) 8(8):1857–68. doi: 10.1182/bloodadvances.2023011767
42. Little JS, Hammond SP, Jacobson C, Aleissa MM. Bacterial infections in the setting of late neutropenia following CD19 chimeric antigen receptor T-cell (CAR-T) therapy for non-hodgkin lymphoma (NHL). Open Forum Infect Dis. (2022) 9. doi: 10.1093/ofid/ofac492.1719
43. Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. (2019) 54(10):1643–50. doi: 10.1038/s41409–019-0487–3
44. Levine JE, Grupp SA, Pulsipher MA, Dietz AC, Rives S, Myers GD, et al. Pooled safety analysis of tisagenlecleucel in children and young adults with B cell acute lymphoblastic leukemia. J Immunother Cancer. (2021) 9(8):e002287. doi: 10.1136/jitc-2020–002287
45. Arnold DE, Maude SL, Callahan CA, DiNofia AM, Grupp SA, Heimall JR. Subcutaneous immunoglobulin replacement following CD19-specific chimeric antigen receptor T-cell therapy for B-cell acute lymphoblastic leukemia in pediatric patients. Pediatr Blood Cancer. (2020) 67:e28092. doi: 10.1002/pbc.28092
46. Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. (2020) 4(21):5414–24. doi: 10.1182/bloodadvances.2020003092
47. Hill JA, Krantz EM, Hay KA, Dasgupta S, Stevens-Ayers T, Bender Ignacio RA, et al. Durable preservation of antiviral antibodies after CD19-directed chimeric antigen receptor T-cell immunotherapy. Blood Advances. (2019) 3(22):3590–601. doi: 10.1182/bloodadvances.2019000717
48. Kampouri E, Walti CS, Gauthier J, Hill JA. Managing hypogammaglobulinemia in patients treated with CAR-T-cell therapy: key points for clinicians. Expert Rev Hematol. (2022) 15:305–20. doi: 10.1080/17474086.2022.2063833
49. Josyula S, Pont MJ, Dasgupta S, Song X, Thomas S, Pepper G, et al. Pathogen-specific humoral immunity and infections in B cell maturation antigen-directed chimeric antigen receptor T cell therapy recipients with multiple myeloma. Transplant Cell Ther. (2022) 28(6):304.e1–9. doi: 10.1016/j.jtct.2022.03.005
50. Nagel J, Boeser KD, Hale C, Harris A, Jain R, Postelnick M, et al. 1015. A multicenter evaluation of antimicrobial utilization in hospitalized bone marrow transplant (BMT) and T-cell therapy (CAR-T) patients, and the impact on rates of C. difficile infection (CDI). Open Forum Infect Diseases. (2023) 10. doi: 10.1093/ofid/ofad500.046
51. Yang J, Hu H, Zhu X, Zou S, Song J, Liu D, et al. CRO infection and the Use of MRSA-active Medication for Prophylaxis affect the Prognosis of Patients with Hematological Malignancies after CAR-T Infusion. Int J Antimicrob Agents. (2023) 62:106874. doi: 10.1016/j.ijantimicag.2023.106874
52. Scheich S, Lindner S, Koenig R, Reinheimer C, Wichelhaus TA, Hogardt M, et al. Clinical impact of colonization with multidrug-resistant organisms on outcome after allogeneic stem cell transplantation in patients with acute myeloid leukemia. Cancer. (2018) 124(2):286–96. doi: 10.1002/cncr.31045
53. Smith M, Dai A, Ghilardi G, Amelsberg KV, Devlin SM, Pajarillo R, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med. (2022) 28(4):713–23. doi: 10.1038/s41591–022-01702–9
54. Abid MB, Shah NN, Maatman TC, Hari PN. Gut microbiome and CAR-T therapy. Exp Hematol Oncol. (2019) 8:31. doi: 10.1186/s40164–019-0155–8
55. Haidar G, Dorritie K, Farah R, Bogdanovich T, Nguyen MH, Samanta P. Invasive mold infections after chimeric antigen receptor-modified T-cell therapy: A case series, review of the literature, and implications for prophylaxis. Clin Infect Dis. (2020) 71:672–6. doi: 10.1093/cid/ciz1127
56. Garner W, Samanta P, Haidar G. Invasive fungal infections after anti-cd19 chimeric antigen receptor-modified t-cell therapy: State of the evidence and future directions. J Fungi. (2021) 7:1–15. doi: 10.3390/jof7020156
57. Chamilos G, Lionakis MS, Kontoyiannis DP. Call for action: invasive fungal infections associated with ibrutinib and other small molecule kinase inhibitors targeting immune signaling pathways. Clin Infect Dis. (2018) 66:140–8. doi: 10.1093/cid/cix687
58. Little JS, Weiss ZF, Hammond SP. Invasive fungal infections and targeted therapies in hematological Malignancies. J Fungi (Basel). (2021) 7. doi: 10.3390/jof7121058
59. Ghez D, Calleja A, Protin C, Baron M, Ledoux MP, Damaj G, et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood. (2018) 131(17):1955–9. doi: 10.1182/blood-2017–11-818286
60. Tillman BF, Pauff JM, Satyanarayana G, Talbott M, Warner JL. Systematic review of infectious events with the Bruton tyrosine kinase inhibitor ibrutinib in the treatment of hematologic Malignancies. Eur J Haematol. (2018) 100:325–34. doi: 10.1111/ejh.13020
61. Wang J, Daunov M, Cooper BW. Incidence and outcomes of pneumocystis pneumonia following chimeric antigen receptor T-cell therapy: A real-world analysis. Blood. (2023) 142:6897–7. doi: 10.1182/blood-2023–190785
62. Kampouri E, Ibrahimi SS, Xie H, Wong ER, Hecht JB, Sekhon MK, et al. CMV reactivation and CMV-specific cell-mediated immunity after chimeric antigen receptor T-cell therapy. Clin Infect Dis. (2023) 78(4):1022–32. doi: 10.1093/cid/ciad708
63. Chen G, Herr M, Nowak J, Ho C, Almyroudis N, Attwood K, et al. Cytomegalovirus reactivation after CD19 CAR T-cell therapy is clinically significant. Haematologica. (2023) 108(2):615–20. doi: 10.3324/haematol.2022.281719
64. Khawaja F, Sassine J, Handley G, Iyer SP, Ramdial J, Ahmed S, et al. Herpesviruses infections in CAR T cell recipients. Transplantation and Cellular Therapy. (2022) 28:S381–S382. doi: 10.1016/S2666-6367(22)00649-2
65. Márquez-Algaba E, Iacoboni G, Pernas B, Esperalba J, Los Arcos I, Navarro V, et al. Impact of cytomegalovirus replication in patients with aggressive B cell lymphoma treated with chimeric antigen receptor T cell therapy. Transplant Cell Ther. (2022) 28(12):851. doi: 10.1016/j.jtct.2022.09.007
66. Khawaja F, Prakash R, Sassine J, Handley G, VanWieren T, Angelidakis G, et al. Cytomegalovirus (CMV) Reactivation within in the First Year after Chimeric Antigen Receptor (CAR) T Cell Therapy: Experience from the First Two Years at a Major Cancer Center. Blood. (2022) 140:7533–5. doi: 10.1182/blood-2022–167908
67. Heldman MR, Ma J, Gauthier J, O'Hara RA, Cowan AJ, Yoke LM, et al. CMV and HSV pneumonia after immunosuppressive agents for treatment of cytokine release syndrome due to chimeric antigen receptor-modified T (CAR-T)-cell immunotherapy. J Immunother. (2021) 44(9):351–4. doi: 10.1097/CJI.0000000000000388
68. Spanjaart AM, van der Valk FM, van Rooijen G, Brouwer MC, Kersten MJ. Confused about confusion. N Engl J Med. (2022) 386:80–7. doi: 10.1056/NEJMcps2114818
69. Rebechi MT, Bork JT, Riedel DJ. HHV-6 encephalitis after chimeric antigen receptor T-cell therapy (CAR-T): 2 case reports and a brief review of the literature. Open Forum Infect Dis. (2021) 8:ofab470. doi: 10.1093/ofid/ofab470
70. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. (2019) 20(1):31–42. doi: 10.1016/s1470–2045(18)30864–7
71. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
72. Shah M, Kuhnl A, Shields G, Sudhanva M, Metaxa V, Wong S, et al. Human herpesvirus 6 encephalitis following axicabtagene ciloleucel treatment for refractory diffuse large B cell lymphoma. Hemasphere. (2021) 5(3):e535. doi: 10.1097/hs9.0000000000000535
73. Handley G, Khawaja F, Kondapi DS, Lee HJ, Kaufman GP, Neelapu SS, et al. Human herpesvirus 6 myelitis after chimeric antigen receptor T-cell therapy. Int J Infect Dis. (2021) 112:327–9. doi: 10.1016/j.ijid.2021.09.061
74. Lareau CA, Yin Y, Maurer K, Sandor KD, Daniel B, Yagnik G, et al. Latent human herpesvirus 6 is reactivated in CAR T cells. Nature. (2023) 623(7987):608–15. doi: 10.1038/s41586–023-06704–2
75. Khan AM, Ajmal Z, Tuz Zahra F, Ramani A, Zackon I. Hemorrhagic cystitis secondary to adenovirus and BK virus infection in a diffuse large B-cell lymphoma patient with recent CAR T-cell therapy. Case Rep Hematol. (2020) 2020:6621967. doi: 10.1155/2020/6621967
76. Ahrendsen JT, Sehgal K, Sarangi S, Uhlmann EJ, Varma H, Arnason J, et al. Progressive multifocal leukoencephalopathy after chimeric antigen receptor T-cell therapy for recurrent non-hodgkin lymphoma. J Hematol. (2021) 10:212–6. doi: 10.14740/jh903
77. Mackenzie S, Shafat M, Roddy H, Hyare H, Neill L, Marzolini MAV, et al. Pembrolizumab for the treatment of progressive multifocal leukoencephalopathy following anti-CD19 CAR-T therapy: a case report. EJHaem. (2021) 2(4):848–53. doi: 10.1002/jha2.274
78. Villalba JA, Maus MV, Frigault MJ, Zaffini R, Gandhi RT, Rosenberg ES, et al. False-positive human immunodeficiency virus test results in patients receiving lentivirus-based chimeric antigen receptor T-cell therapy: case report, review of the literature, and proposed recommendations. J Infect Dis. (2022) 225:1933–6. doi: 10.1093/infdis/jiab605
79. Hayden PJ, Sirait T, Koster L, Snowden JA, Yakoub-Agha I. An international survey on the management of patients receiving CAR T-cell therapy for haematological Malignancies on behalf of the Chronic Malignancies Working Party of EBMT. Curr Res Trans Med. (2019) 67:79–88. doi: 10.1016/j.retram.2019.05.002
80. Abramson JS, Irwin KE, Frigault MJ, Dietrich J, McGree B, Jordan JT, et al. Successful anti-CD19 CAR T-cell therapy in HIV-infected patients with refractory high-grade B-cell lymphoma. Cancer. (2019) 125(21):3692–8. doi: 10.1002/cncr.32411
81. Abbasi A, Peeke S, Shah N, Mustafa J, Khatun F, Lombardo A, et al. Axicabtagene ciloleucel CD19 CAR-T cell therapy results in high rates of systemic and neurologic remissions in ten patients with refractory large B cell lymphoma including two with HIV and viral hepatitis. J Hematol Oncol. (2020) 13(1):1. doi: 10.1186/s13045-019-0838-y
82. EASL. Recommendations on treatment of hepatitis C 2018. J Hepatol. (2018) 69:461–511. doi: 10.1016/j.jhep.2018.03.026
83. Mallet V, van Bömmel F, Doerig C, Pischke S, Hermine O, Locasciulli A, et al. Management of viral hepatitis in patients with haematological Malignancy and in patients undergoing haemopoietic stem cell transplantation: recommendations of the 5th European Conference on Infections in Leukaemia (ECIL-5). Lancet Infect Dis. (2016) 16(5):606–17. doi: 10.1016/s1473–3099(16)00118–3
84. Available online at: https://wwwnc.cdc.gov/travel/yellowbook/2024/infections-diseases/tuberculosis#epi.
85. Balmaceda N, Aziz M, Chandrasekar VT, McClune B, Kambhampati S, Shune L, et al. Infection risks in multiple myeloma: a systematic review and meta-analysis of randomized trials from 2015 to 2019. BMC Cancer. (2021) 21(1):730. doi: 10.1186/s12885-021-08451-x
86. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. (2022) 33(3):259–75. doi: 10.1016/j.annonc.2021.12.003
87. Wudhikarn K, Perales MA. Infectious complications, immune reconstitution, and prophylaxis after CD19 chimeric antigen receptor T-cell therapy. Bone Marrow Transplant. (2022) 57:1477–88. doi: 10.1038/s41409–022-01756-w
88. Los-Arcos I, Iacoboni G, Aguilar-Guisado M. Recommendations for screening, monitoring, prevention and prophylaxis of infections in adult and pediatric patients receiving CAR T-cell therapy: a position paper. Infection. (2021) 49:215–31. doi: 10.1007/s15010–020-01521–5
89. Stewart AG, Henden AS. Infectious complications of CAR T-cell therapy: a clinical update. Ther Adv Infect Dis. (2021) 8(1):1–22. doi: 10.1177/20499361211036773
90. Kampouri E, Little JS, Rejeski K, Manuel O, Hammond SP, Hill JA. Infections after chimeric antigen receptor (CAR)-T-cell therapy for hematologic malignancies. Transpl Infect Dis. (2023) 25(Suppl 1):S1–S17. doi: 10.1111/tid.141576
91. Zu C, Xu Y, Wang Y, Zhang M, Zhao H, Fang X, et al. Cytomegalovirus retinitis and retinal detachment following chimeric antigen receptor T cell therapy for relapsed/refractory multiple myeloma. Curr Oncol. (2022) 29(2):490–6. doi: 10.3390/curroncol29020044
92. Qasim W. Allogeneic CAR T cell therapies for leukemia. Am J Hematol. (2019) 94:S50–s54. doi: 10.1002/ajh.25399
93. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573–019-0051–2
94. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016–04-703751
95. Penack O, Koenecke C. Complications after CD19+ CAR T-cell therapy. Cancers. (2020) 12:3445. doi: 10.3390/cancers12113445
96. Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. (2021) 56:552–66. doi: 10.1038/s41409–020-01134–4
97. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25(4):625–38. doi: 10.1016/j.bbmt.2018.12.758
98. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. (2019) 134(24):2149–58. doi: 10.1182/blood.2019001463
99. Thompson JA, Schneider BJ, Brahmer J, Achufusi A, Armand P, Berkenstock MK, et al. Management of immunotherapy-related toxicities, version 1.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2022) 20(4):387–405. doi: 10.6004/jnccn.2022.0020
100. Banerjee R, Marsal J, Huang CY, Lo M, Kambhampati S, Kennedy VE, et al. Early time-to-tocilizumab after B cell maturation antigen-directed chimeric antigen receptor T cell therapy in myeloma. Transplant Cell Ther. (2021) 27(6):477. doi: 10.1016/j.jtct.2021.03.004
101. Robilotti E, Holubar M, Seo SK, Deresinski S. Feasibility and applicability of antimicrobial stewardship in immunocompromised patients. Curr Opin Infect Dis. (2017) 30:346–53. doi: 10.1097/qco.0000000000000380
102. Puerta-Alcalde P, Cardozo C, Marco F, Suárez-Lledó M, Moreno E, Morata L, et al. Changing epidemiology of bloodstream infection in a 25-years hematopoietic stem cell transplant program: current challenges and pitfalls on empiric antibiotic treatment impacting outcomes. Bone Marrow Transplant. (2020) 55:603–12. doi: 10.1038/s41409–019-0701–3
103. Shahid Z, Epstein DJ. Noninfectious causes of fever in hematologic Malignancies. Are antibiotics still indicated? Curr Opin Infect Dis. (2023) 36:209–17. doi: 10.1097/qco.0000000000000940
104. Rearigh L, Stohs E, Freifeld A, Zimmer A. De-escalation of empiric broad spectrum antibiotics in hematopoietic stem cell transplant recipients with febrile neutropenia. Ann Hematol. (2020) 99:1917–24. doi: 10.1007/s00277–020-04132–0
105. Gustinetti G, Raiola AM, Varaldo R, Galaverna F, Gualandi F, Del Bono V, et al. De-escalation and discontinuation of empirical antibiotic treatment in a cohort of allogeneic hematopoietic stem cell transplantation recipients during the pre-engraftment period. Biol Blood Marrow Transplant. (2018) 24(8):1721–6. doi: 10.1016/j.bbmt.2018.03.018
106. Fleischmann RM, Tesser J, Schiff MH, Schechtman J, Burmester GR, Bennett R, et al. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis. (2006) 65(8):1006–12. doi: 10.1136/ard.2005.048371
107. Wehrli M, Gallagher K, Chen YB, Leick MB, McAfee SL, El-Jawahri AR, et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J Immunother Cancer. (2022) 10(1):e003847. doi: 10.1136/jitc-2021–003847
108. Kim JY, Kim M, Park JK, Lee EB, Park JW, Hong J. Limited efficacy of tocilizumab in adult patients with secondary hemophagocytic lymphohistiocytosis: a retrospective cohort study. Orphanet J Rare Dis. (2022) 17:363. doi: 10.1186/s13023–022-02516–1
109. Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: A systematic review and meta-analysis. Am J Hematol. (2018) 93:339–47. doi: 10.1002/ajh.24976
110. Gill H, Leung GMK, Seto WK, Kwong YL. Risk of viral reactivation in patients with occult hepatitis B virus infection during ruxolitinib treatment. Ann Hematol. (2019) 98:215–8. doi: 10.1007/s00277–018-3405–7
111. Sylvine P, Thomas S, Pirayeh E. Infections associated with ruxolitinib: study in the French Pharmacovigilance database. Ann Hematol. (2018) 97:913–4. doi: 10.1007/s00277–018-3242–8
112. Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. (2004) 364(9451):2113–21. doi: 10.1016/s0140–6736(04)17552–1
113. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382(19):1811–22. doi: 10.1056/NEJMoa1911326
114. Hill JA, Giralt S, Torgerson TR, Lazarus HM. CAR-T – and a side order of IgG, to go? – Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. (2019) 38. doi: 10.1016/j.blre.2019.100596
115. Walti CS, Krantz EM, Maalouf J, Boonyaratanakornkit J, Keane-Candib J, Joncas-Schronce L, et al. Antibodies against vaccine-preventable infections after CAR-T cell therapy for B cell Malignancies. JCI Insight. (2021) 6(11):e146743. doi: 10.1172/jci.insight.146743
116. Walti CS, Loes AN, Shuey K, Krantz EM, Boonyaratanakornkit J, Keane-Candib J, et al. Humoral immunogenicity of the seasonal influenza vaccine before and after CAR-T-cell therapy: A prospective observational study. J ImmunoTherapy Cancer. (2021) 9(10):e003428. doi: 10.1136/jitc-2021–003428
117. Hill JA, Giralt S, Torgerson TR, Lazarus HM. CAR-T - and a side order of IgG, to go? - Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. (2019) 38:100596. doi: 10.1016/j.blre.2019.100596
118. Mikulska M, Cesaro S, de Lavallade H, Di Blasi R, Einarsdottir S, Gallo G, et al. Vaccination of patients with haematological Malignancies who did not have transplantations: guidelines from the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis. (2019) 19(6):e188–99. doi: 10.1016/s1473–3099(18)30601–7
119. Cordonnier C, Einarsdottir S, Cesaro S, Di Blasi R, Mikulska M, Rieger C, et al. Vaccination of haemopoietic stem cell transplant recipients: guidelines of the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis. (2019) 19(6):e200–12. doi: 10.1016/s1473–3099(18)30600–5
120. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. (2022) 386(7):640–54. doi: 10.1056/NEJMoa2116133
121. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377(26):2545–54. doi: 10.1056/NEJMoa1708566
122. Hill JA, Martens MJ, Young JH, Bhavsar K, Kou J, Chen M, et al. SARS-CoV-2 vaccination in the first year after hematopoietic cell transplant or chimeric antigen receptor T cell therapy: A prospective, multicenter, observational study (BMT CTN 2101). Clin Infect Dis. (2024) 27:ciae291. doi: 10.1093/cid/ciae291.
123. Antin JH, Guinan EC, Avigan D, Soiffer RJ, Joyce RM, Martin VJ, et al. Protective antibody responses to pneumococcal conjugate vaccine after autologous hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2005) 11:213–22. doi: 10.1016/j.bbmt.2004.12.330
124. Melgar M, Britton A, Roper LE, Talbot HK, Long SS, Kotton CN, et al. Use of respiratory syncytial virus vaccines in older adults: recommendations of the advisory committee on immunization practices — United states, 2023. MMWR Morb Mortal Wkly Rep. (2023) 72(29):793–801. doi: 10.15585/mmwr.mm7229a4
125. Khawaja F, Papanicolaou G, Dadwal S, Pergam SA, Wingard JR, Boghdadly ZE, et al. Frequently asked questions on coronavirus disease 2019 vaccination for hematopoietic cell transplantation and chimeric antigen receptor T-cell recipients from the american society for transplantation and cellular therapy and the american society of hematology. Transplant Cell Ther. (2023) 29(1):10–8. doi: 10.1016/j.jtct.2022.10.010
126. Uyemura BS, Abid MA, Suelzer E, Abid MB. Efficacy of SARS-CoV-2 primary and booster vaccine doses in CAR-T recipients – targeting the target antigen. Bone Marrow Transplant. (2022) 57:1727–31. doi: 10.1038/s41409–022-01795–3
127. Available online at: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/recommendations/immuno.html#:~:text=Learn%20more.&text=CDC%20recommends%20the%202023%E2%80%932024,2024%20updated%20COVID%2D19%20vaccine.
128. Teh BW, Mikulska M, Averbuch D, de la Camara R, Hirsch HH, Akova M, et al. Consensus position statement on advancing the standardised reporting of infection events in immunocompromised patients. Lancet Infect Dis. (2024) 24(1):e59–68. doi: 10.1016/s1473–3099(23)00377–8
129. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
130. Wei G, Zhang Y, Zhao H, Wang Y, Liu Y, Liang B, et al. CD19/CD22 dual-targeted CAR T-cell therapy for relapsed/refractory aggressive B-cell lymphoma: A safety and efficacy study. Cancer Immunol Res. (2021) 9(9):1061–70. doi: 10.1158/2326–6066.Cir-20–0675
131. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. (2020) 10:697. doi: 10.3389/fonc.2020.00697
132. Kunte AS, Gagelmann N, Badbaran A, Berger C, Massoud R, Klyuchnikov E, et al. Viral reactivation and immune reconstitution after CAR-T cell treatment in patients with hematologic Malignancies. Blood. (2022) 140:7531–2. doi: 10.1182/blood-2022–168667
133. Wang D, Mao X, Que Y, Xu M, Cheng Y, Huang L, et al. Viral infection/reactivation during long-term follow-up in multiple myeloma patients with anti-BCMA CAR therapy. Blood Cancer J. (2021) 11(10):168. doi: 10.1038/s41408–021-00563–8
Keywords: infectious complications, chimeric antigen receptor T-cell therapy, infection management, immunoglobulin replacement therapy, vaccinations
Citation: Arya S and Shahid Z (2024) Overview of infectious complications among CAR T- cell therapy recipients. Front. Oncol. 14:1398078. doi: 10.3389/fonc.2024.1398078
Received: 08 March 2024; Accepted: 04 June 2024;
Published: 03 July 2024.
Edited by:
Jose-Maria Ribera, Germans Trias i Pujol Health Science Research Institute (IGTP), SpainReviewed by:
Yifan Pang, Levine Cancer Institute, United StatesCopyright © 2024 Arya and Shahid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zainab Shahid, c2hhaGlkekBtc2tjYy5vcmc=