 Chang Li
Chang Li Liang Han
Liang Han Yuming Song
Yuming Song Rui Liu
Rui Liu- 1Department of VIP Unit, China-Japan Union Hospital of Jilin University, Changchun, Jilin, China
- 2Department of Pathology, China-Japan Union Hospital of Jilin University, Changchun, Jilin, China
Background: Pheochromocytoma is one of the most hereditary human tumors with at least 20 susceptible genes undergoing germline and somatic mutations, and other mutations less than 1% -2%. In recent years, other rare mutations have gradually been discovered to be possibly related to the pathogenesis and metastasis of pheochromocytoma. Most patients with pheochromocytoma experience common symptoms like headaches, palpitations, and sweating, while some may have less common symptoms. The diversity of symptoms, genetic mutations, and limited treatment options make management challenging.
Case presentation: A 53-year-old woman was hospitalized after experiencing episodic epigastric pain for one month. A mass was found in her right adrenal gland and she underwent robot-assisted laparoscopic surgery, revealing a pheochromocytoma. At the 16-month follow-up, multiple metastatic lesions consistent with metastatic pheochromocytoma were found. A germline mutation in the dihydrolipoamide succinyltransferase (DLST) gene (c.330 + 14A>G) was detected, and despite trying chemotherapy and adjuvant therapy, the patient had a limited response with an overall survival of 27 months.
Conclusions: DLST mutation is one of the rare pheochromocytoma-related mutated genes, and genetic sequencing is crucial for effective clinical management.
1 Introduction
Pheochromocytoma (PCC) and paraganglioma (PGL) are collectively known as PPGL (1). Clinical manifestations of hypertension are present in over 90% of patients, while symptoms such as headache, palpitations, and sweating are reported in more than 50% of cases. PPGL has been categorized into metastatic and non-metastatic forms (1). 10% of PCC and 15–35% of PGL are malignant but metastatic diseases are rare (2, 3). PPGL is recognized as one of the most heritable tumors among all human malignancies, with genetic factors accounting for approximately 40% (4). The activation of susceptibility genes in key pathways, including pseudohypoxia, kinase, and Wnt signaling, are identified (5–8). Tumors classified within Cluster 1 typically exhibit a noradrenergic biochemical phenotype and are associated with a heightened risk of sustained hypertension. Gene sequencing, metaphranes, and abdominal imaging are useful for diagnosing PCC (9). Surgery is the preferred treatment, but systemic options are available for inoperable cases with limited effectiveness. Patients with mutation should receive personalized lifelong monitoring (10).
A case study of a female with PCC metastasis occurring 16 months post-surgery was presented, characterized by atypical clinical manifestations. Whole-exome sequencing revealed DLST gene mutations, suggesting a potential association with PCC development. A comprehensive review of literature was conducted to discuss the clinical management.
2 Case presentation
2.1 Medical history and preoperative examination
A 53-year-old female without prior medical history presented with episodic upper abdominal pain in June 2021.The patient exhibited no symptoms of paroxysmal or persistent hypertension, headache, palpitations, sweating, vision loss, body weight loss or other related manifestations. No retinal hemangioma was detected. In July 2021, the patient exhibited normal respiration, heart rate (80 beats/min), and blood pressure (130/80mmHg), with no abnormal findings noted during cardiopulmonary and abdominal examinations. Adrenal enhancement computed tomography (CT) revealed a space occupying lesion in the right adrenal region (about 4.2×5.9cm in size) (Figures 1A, B) and normal lung CT. 18F-fuorodeoxyglucose positron emission tomography (PET)/CT revealed a mass in the right adrenal region with unevenly increased glucose metabolism (maximum cross-sectional area of 5.68×4.81 cm). Auxiliary examinations of the central nervous system, heart, kidney, and pancreas revealed no abnormalities. Metanephrine (MN) and neuron specific enolase (NSE) showed no abnormalities. Blood normetanephrine (NMN) was about three times than the normal upper limit (Supplementary Table 1). CgA concentration in blood and fractionated metanephrines in 24 hour urine weren’t conducted. The patient’s blood routine, liver and kidney function, blood glucose levels, urinary occult blood, and other biochemical indicators showed no significant abnormalities. Normal ACTH rhythm, cortisol rhythm, and renin activity were observed (Supplementary Table 4). Aldosterone levels in the supine position were measured at 219.4pg/ml (reference range 10.0–160.0pg/ml), while orthostatic aldosterone levels were recorded at 415.5pg/ml (reference range 40–310pg/ml).
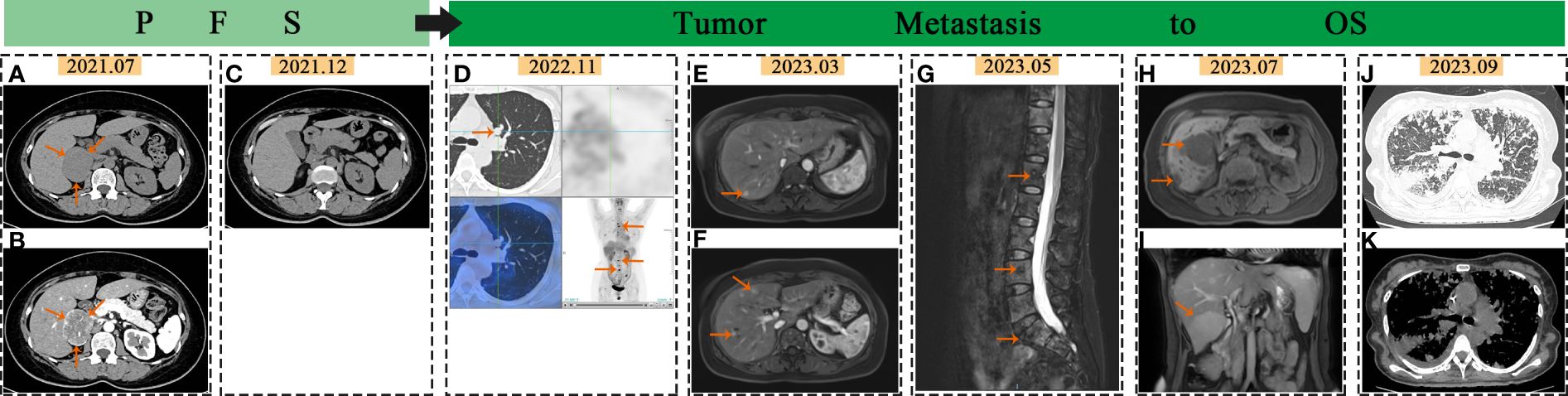
Figure 1 Imaging changes in PCC: In July 2021, the arrows pointed to the adrenal mass on abdominal and enhanced CT scans (A, B). In December 2021, no abnormalities were seen in abdominal CT scans (C). In November 2022, the arrows pointed to lung and bone metastasis on PET-CT (D). In March 2023, the arrows indicated liver metastases on liver MR images (E, F). In May 2023, the arrows pointed to spine metastases on MR images (G). In July 2023, the arrows indicated liver metastases on liver MR images (H, I). In September 2023, CT scan showed extensive lung metastases with inflammation (J, K).
2.2 Surgical treatment and postoperative pathology
To mitigate the risk of cardiovascular complications, the patient was administered oral doxazosin in July 2021, followed by oral bisoprolol prior to surgery. The planned procedures include laparoscopic retroperitoneal lesion resection, right adrenal mass resection, and abdominal adhesiolysis. Intraoperatively, the presence of greater omentum tissue at the inferior margin of the liver and intestinal adhesions was noted. Following dissociation, a mass measuring approximately 5.0 x 5.0 cm, exhibiting moderate activity and in close proximity to the right adrenal gland, was identified. No discernible abnormalities were detected in the liver, stomach, colon, or small intestine. Based on preoperative evaluations and intraoperative observations, the surgical approach was maintained without alterations. The surrounding tissue of the tumor was found to be fully free, allowing for the complete removal of the tumor. Postoperative pathology revealed a retroperitoneal adrenal tumor measuring 5.7 x 4.5 x 4cm and weighing 75g, with incomplete capsular involvement. Histological examination revealed the presence of small nests of chief cells accompanied by small and interspersed blood vessels. Immunohistochemical (IHC) staining indicated positivity for chromogranin A (CgA), synaptophysin (SyN), Ki-67 (5%+), succinate dehydrogenase B (SDHB), NSE, and sustentacular cells (individual cells+) (Figures 2A–D). The patient presented with atypical symptoms and signs of PCC. Based on qualitative diagnostic criteria (elevated blood NMN levels exceeding twice the upper limit of normal) and localization diagnostic methods (adrenal CT, PETCT) conducted prior to surgery, PCC was highly suspected. GAPP score was 5. The final clinical diagnosis was PCC (T3N0M0 stageIII).
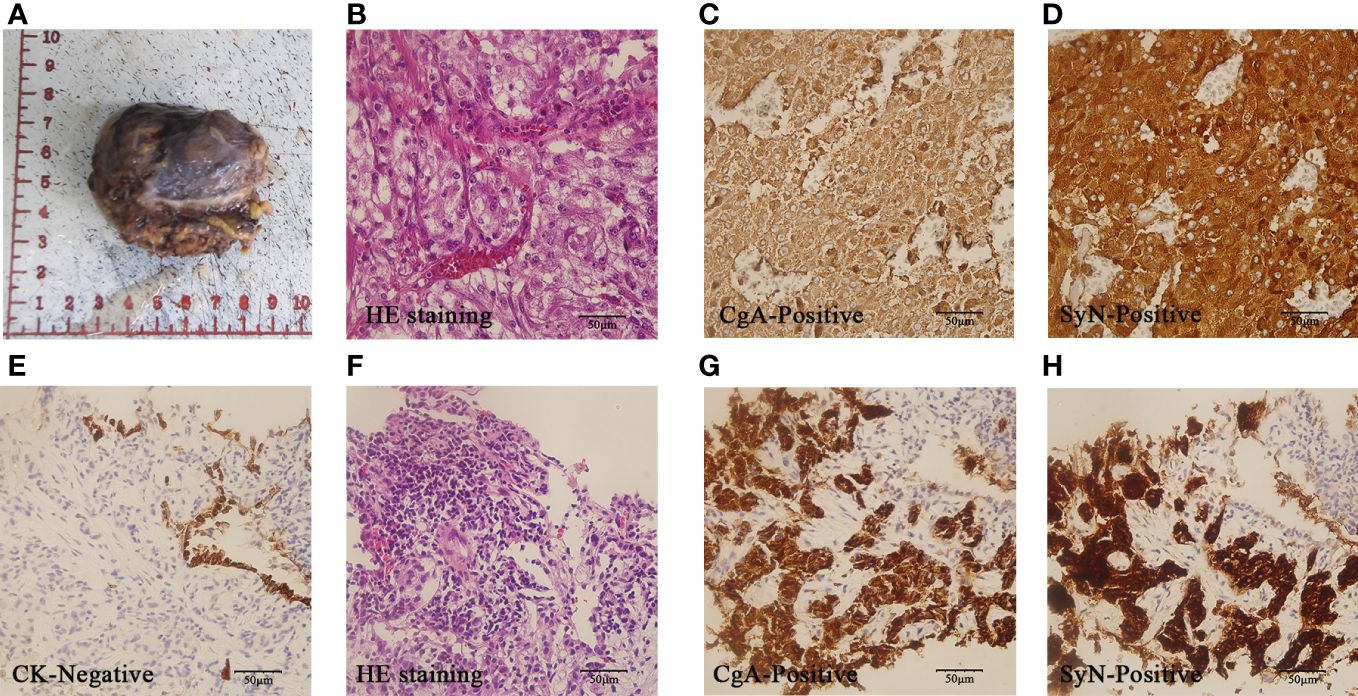
Figure 2 Pathology of PCC after surgery and metastasis:A-D showed pathology images of PCC after surgery. The gross specimen was a right retroperitoneal adrenal mass with incomplete capsule (A). HE staining revealed small nests of chief cells with small and interspersed blood vessels(× 400) (B). Tumor cells demonstrated diffuse CgA positivity (× 400) (C) and SyN positivity(×400) (D). (E–H) displayed pathology images of lung metastases. Lung metastatic cells were negative for CK (× 400) (E). The HE staining showed similar characteristics to the primary tumor (× 400) (F). The lung metastatic cells exhibited diffuse CgA positivity (× 400) (G) and SyN positive (× 400) (H).
2.3 Metastasis and postoperative treatment
The patient remained asymptomatic during routine postoperative follow-up. The adrenal CT was normal 5 months after operation (Figure 1C). Levels of MN, NMN, and NSE were within normal limits. 16 months post-operation,18F-FDG PET/CT imaging revealed increased glucose metabolism in the bilateral lungs and subpleural region. Additionally, heightened glucose metabolism was observed in the sternum, cervical 2, thoracic 4, 5, 7, 8, lumbar 3–5 vertebral bodies, left iliac bone, and right femur (Figure 1D). Lung CT showed multiple metastases. Further imaging with Gallium-68 labeled 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-N-octyl-D-phenylalanine and 18F-Dihydroxyphenylalanine PET/CT demonstrated widespread bone metastases, multiple lung metastases with high expression of growth inhibitory receptors, and liver metastases in both lobes with low expression of growth inhibitory receptors. The lung and bone lesions showed positive results for CgA, SyN, SDHB, vimentin (VIM), CD56, GATA3, and somatostatin receptor 2 (SSTR 2), with Ki-67 levels at 30% in the lung and 5% in the bone, indicating metastatic PCC (Figures 2E–H). Levels of MN, NMN,catecholamine (CA) and NSE were still within normal range.
Whole Exome Sequencing Analysis Reveals: A point mutation in the DLST Gene (c.330 + 14A>G), potentially linked to PGL Type 7, and a mutation in the Cyclin D1 (CCND1) Gene (c.575–13C>T), potentially associated with Von Hippel-Lindau Syndrome(Supplementary Table 2). The patient underwent pre-treatment evaluation and preparation for systemic therapy of metastatic PCC. The evident adverse reactions of nausea and vomiting were observed in patients receiving 68Ga-dotatate PET/CT. Taking into consideration the patient’s physical tolerance, the ultimate treatment regimen consisted of oral temozolomide (300mg administered once daily for 1–5 days, with a treatment course repeated every 28 days) and subcutaneous denosumab (120mg administered once every 4 weeks). Following the completion of one course of oral temozolomide, treatment was discontinued due to notable gastrointestinal symptoms. Subsequent treatment included continuation of the denosumab regimen alongside subcutaneous injections of octreotide acetate microspheres (60mg administered once every 4 weeks), interspersed with traditional Chinese medicine anti-tumor therapy. Subsequent to treatment initiation, the frequency of follow-up assessments was escalated. Blood parameter monitoring was conducted based on clinical status, with imaging evaluations scheduled approximately every two months. The imaging changes of metastatic lesions in March and May 2023 are shown in Figures 1E–G. By July 2023, the patient began experiencing significant dyspnea and intermittent hemoptysis, indicating rapid disease progression (Figures 1H, I). The adverse bleeding reaction associated with tyrosine kinase inhibitors (TKIs) treatment poses a limitation on the utilization of these drugs by the patient. In September 2023, she was hospitalized for worsening breathing difficulties, vomiting, and anemia. Metastatic lesions in the liver and lungs had increased (Figures 1J, K), along with elevated levels of NMN, NE, and NSE(Supplementary Table 1). She was diagnosed with advanced stage IV PCC. Palliative treatment was provided as she was in the terminal stage of the tumor. Supplementary Table 3 showed the detailed timeline, symptoms and the treatment process.
3 Discussion
PPGL have a poor prognosis and limited treatment options. The key to diagnosis lies in appropriate biochemical tests and molecular IHC (11). Approximately 40% of PPGL cases are associated with germline mutations, making genetic testing crucial for early detection of genetic syndromes, follow-up of high-risk patients, and guidance of treatment (12). Half of the mutated genes in PPGL are members of the tricarboxylic acid (TCA) cycle. Recent studies have identified DLST as a component of the rate-limiting enzyme of the TCA cycle, and disruption of DLST has been linked to pseudohypoxia, which contributes to the occurrence and progression of PPGL (13). However, the reported cases are limited in number, and there is lack of comprehensive clinical data. Our patient exhibited DLST point mutations, PCC characterized by atypical clinical symptoms but high malignancy, multiple site metastasis, and a suboptimal response to treatment.
3.1 PCC gene sequencing
There has been a growing recognition of asymptomatic cases of PPGL through familial and germline mutation testing in recent years (9). About 15–17% of patients with PPGL will develop metastasis (14). The natural course of metastatic PPGL is highly heterogeneous, with 5-year survival rates ranging from 40% to 85% (15, 16). Stage IV PPGL has a significantly shorter overall survival (OS) (median OS 8.8 years) compared to stages I-III (17). Current guidelines recommend a comprehensive approach involving simultaneous localization diagnosis, qualitative diagnosis, and genetic counseling to accurately diagnose PCC/PGL (18). PPGL is linked to mutations in 20+ genes, categorized into three groups by TCGA: pseudohypoxia (cluster 1), kinase signaling (cluster 2), and Wnt signaling (cluster 3) (5–8). These mutations cause metabolic and epigenetic imbalances, promoting tumor invasiveness and metastasis (8). Our review focuses on PPGL patients with mutations in these clusters (Table 1).
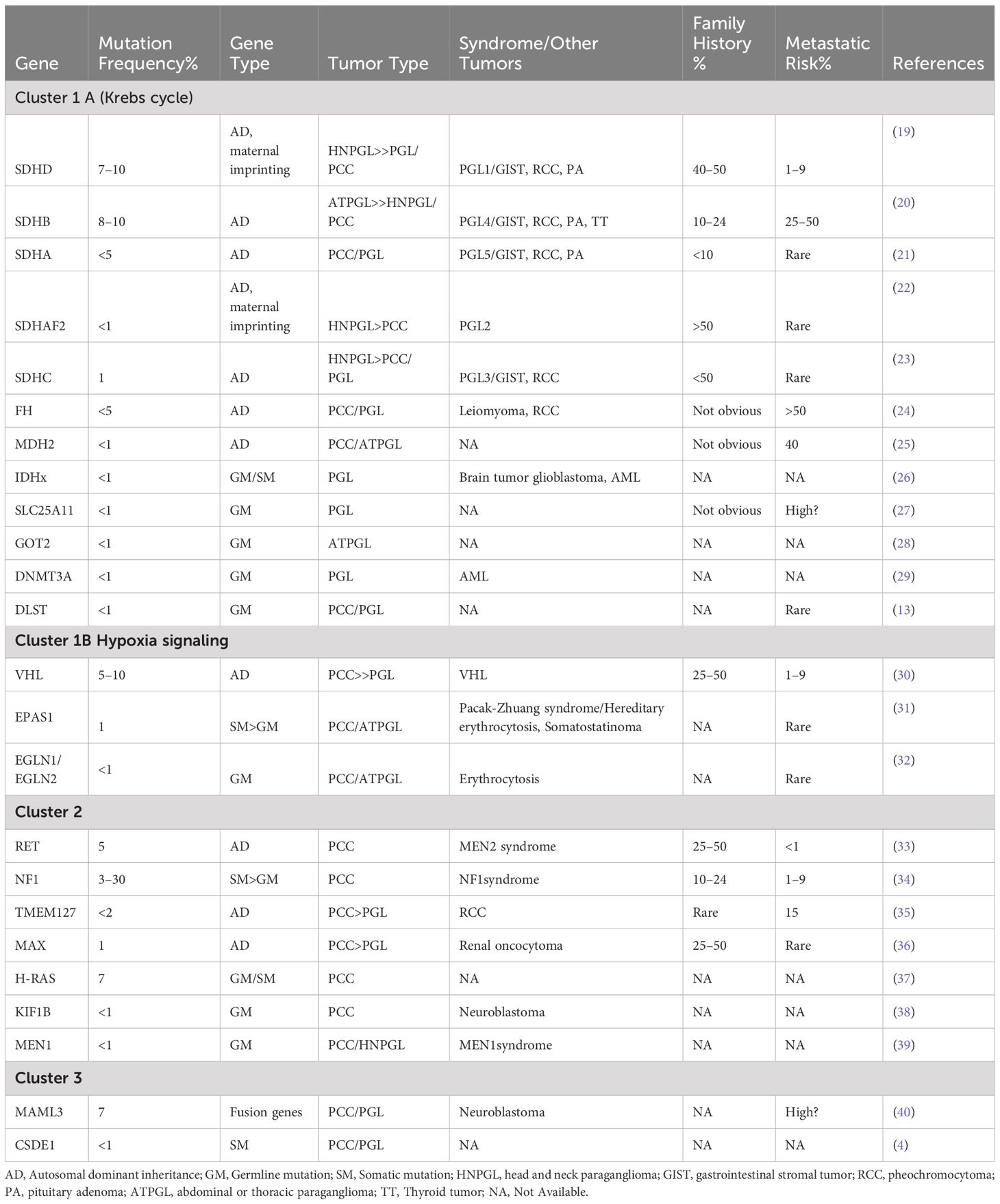
Table 1 Clinical features of PPGL with mutations in susceptibility genes.
In recent years, DLST mutations, accounting for less than 1% of cases, have also been identified as contributing to PPGL. A review of cases reported in the literature on DLST mutations reveals that only three authors have reported a total of 12 patients (Table 2). In 2019, Remacha et al. have described a new PPGL susceptibility gene DLST, which encodes the dihydrolipoamide S-succinyltransferase (13). The dihydrothiamide S-succinyltransferase encoded by the DLST gene is a rate limiting enzyme in the Krebs cycle of cluster 1 subgroup (43). Additionally, Alexandre et al.’s study indicated that DLST likely pathogenic variants may confer susceptibility to PPGL, with a predicted low penetrance (41). The DLST germline variant (p.gly374glu) can cause functional impairment and promote tumorigenesis by increasing α-ketoglutarate levels and activating the pseudohypoxic pathway (13). These patients exhibit sporadic occurrences, with NM abnormalities being more prevalent, and are more likely to develop chest and abdominal PPGL (41). Our case identified a mutation near the splice site of the DLST gene, which may lead to abnormal protein synthesis. The patient did not show typical symptoms of PCC, but NM levels were elevated before surgery and during metastasis, consistent with PCC caused by DLST mutation. There was no evidence of CCND1 gene mutations in the pathogenesis of PCC. The absence of consistent clinical manifestations and test results throughout the disease process of von Hippel Lindau syndrome due to CCND1 gene mutations suggests that the correlation between this specific mutation and the onset of pheochromocytoma was not present in this case (43). The patient’s lack of VHL gene mutations and absence of literature on VHL and DLST dual mutations were noted. Unfortunately, further genetic sequencing and epigenetic analysis of the patient’s family were unsuccessful.
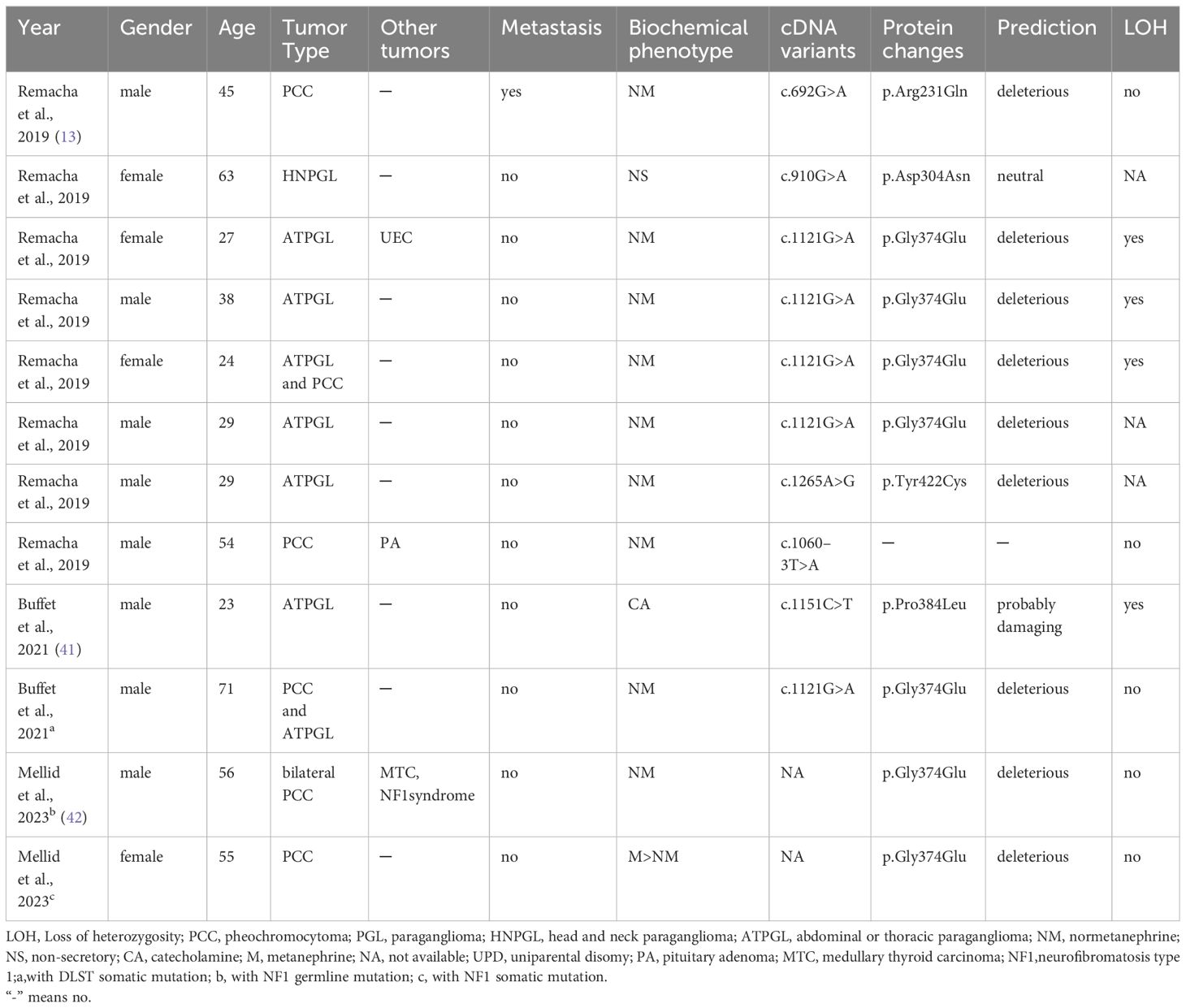
Table 2 Clinical data of DLST mutations in PPGL patients.
3.2 Symptoms, test results, and treatment for PCC
The primary symptom of PPGL is persistent or paroxysmal hypertension with target tissue damage (44). Clinical symptoms can vary from no symptoms to life-threatening events, even with normal blood pressure (45). Our patient initially had mild abdominal pain. Preoperative levels of NMN were found to be significantly elevated, while other blood test results did not show any significant abnormalities. The synthesis, secretion and release of CA are not completely dependent on the adrenal medulla, which may also be the reason why our patient had no typical symptoms despite the increase of NMN (11). Adrenal CT and PETCT imaging supported the diagnosis of PCC without evidence of lesions in other areas. The Endocrine Hypertension Working Group of the European Society for Hypertension recommends minimally invasive adrenalectomy as the preferred surgical approach for PCC, as it can minimize blood loss and shorten postoperative hospitalization (18). While guidelines suggest considering a cesarean section for PCC tumors larger than 5cm,as for our patient the PCC measures approximately 5cm in diameter. The surgical team observed only hepatic omentum tissue and some intestinal adhesions during the procedure, with no apparent abnormalities in the neighboring organs of the tumor. Consequently, they opted for laparoscopic right adrenal mass resection surgery. Surgical intervention has been shown to enhance overall survival (OS) (46). European guidelines suggest postoperative monitoring through blood tests, such as measurement of MN and NMN at 2–6 weeks post-surgery and annually thereafter, as well as imaging studies (CT/MRI) at 3 months, 6 months, and biennially thereafter (18). Another study proposes that patients with SDHA/B PPGL who are at a high risk of metastasis should consider undergoing biochemical tests every 6 to 12 months and imaging every 1 to 2 years (10). Following postoperative normalization of NMN levels, no recurrence or metastasis was detected five months post-operation for our patient. However, distant metastasis (lung, liver, bone) of PCC was discovered 16 months post-surgery. Imaging studies, including lung and liver CT, PETCT, and subsequent pathological examination of lung and bone metastases, provided compelling evidence of multiple site metastasis of PCC. The potential benefit of increasing the frequency of postoperative follow-up, such as every 6 months, for these patients is worth exploring. There is no standardized treatment for metastatic PPGL, but options include surgical resection, targeted radiolabeled carriers, thermal ablation, chemotherapy, and external irradiation (9). Chemotherapy, specifically with drugs like cyclophosphamide, vincristine, and dacarbazine, is preferred for advanced PPGL, especially in rapidly progressing cases. Tumors with mutations in the gene encoding Krebs cycle enzyme may respond better to temozolomide due to reduced expression of methylguanidine DNA methyltransferase (44, 47). Temozolomide chemotherapy was chosen based on the patient’s tolerance, however, due to significant side effects, treatment was discontinued after a single course. Octreotide and denosumab were administered subcutaneously starting at 16 months post-operation. The patient experienced intermittent hemoptysis during the advanced stage of lung metastasis, and TKIs were not utilized. Radionuclide therapy was deemed unsuitable due to the evident adverse effects observed during the relevant radionuclide examination. The patient had a low tolerance to certain treatments and a lower OS rate compared to previous reports on metastatic PPGL.
4 Conclusions
This study presents a case of metastatic PCC with uncommon DLST point mutations, characterized by high malignancy, rapid disease progression, and limited therapeutic efficacy. These cases warrant additional attention in determining the optimal timing for genetic sequencing, enhancing the frequency of monitoring, and developing personalized treatment strategies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of China-Japan Union Hospital of Jilin University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Validation, Writing – original draft. LH: Data curation, Resources, Writing – original draft. YS: Conceptualization, Formal Analysis, Project administration, Visualization, Writing – review & editing. RL: Formal Analysis, Funding acquisition, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by Jilin Province Health Science and Technology Capability Enhancement Project (No. 2022LC119) and Jilin Province health research talents special project (No. 2023SCI32).
Acknowledgments
We thank the patient and their families in this paper for their consent.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1394552/full#supplementary-material
References
1. Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. (2018) 31:1770–86. doi: 10.1038/s41379-018-0110-y
2. Ma XS, Li M, Tong AL, Wang F, Cui YY, Zhang XB, et al. Genetic and clinical profiles of pheochromocytoma and paraganglioma: A single center study. Front Endocrinol. (2020) 11:574662. doi: 10.3389/fendo.2020.574662
3. Martins R, Bugalho MJ. Paragangliomas/pheochromocytomas: clinically oriented genetic testing. Int J Endocrinol. (2014) 2014:794187. doi: 10.1155/2014/794187
4. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson A, Johnson A, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. J Cancer Cell. (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
5. Taieb D, Pacak K. New insights into the nuclear imaging phenotypes of cluster 1 pheochromocytoma and paraganglioma. Trends Endocrinol Metab. (2017) 28:807–17. doi: 10.1016/j.tem.2017.08.001
6. Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr. Rev. (2017) 38:489–515. doi: 10.1210/er.2017–00062
7. Koopman K, Gaal J, de Krijger R. Pheochromocytomas and paragangliomas: new developments with regard to classification, genetics, and cell of origin. J Cancers. (2019) 11:1070. doi: 10.3390/cancers11081070
8. Jochmanova I, Pacak K. Genomic landscape of pheochromocytoma and paraganglioma. J Trends Cancer. (2018) 4:6–9. doi: 10.1016/j.trecan.2017.11.001
9. Neumann H, Young W, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
10. Nolting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr. Rev. (2022) 43:199–239. doi: 10.1210/endrev/bnab019
11. Eisenhofer G, Pamporaki C, Lenders J. Biochemical assessment of pheochromocytoma and paraganglioma. J Endocrine Rev. (2023) 44:862–909. doi: 10.1210/endrev/bnad011
12. Jhawar S, Arakawa Y, Kumar S, Varghese D, Kim Y, Roper N, et al. New insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. J Cancers. (2022) 14:594. doi: 10.3390/cancers14030594
13. Remacha L, Pirman D, Mahoney C, Coloma J, Calsina B, Currás-Freixes M, et al. Recurrent germline DLST mutations in individuals with multiple pheochromocytomas and paragangliomas. J Am J Hum Genet. (2019) 104:651–64. doi: 10.1016/j.ajhg.2019.02.017
14. Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for Malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. (2011) 96:717–25. doi: 10.1210/jc.2010–1946
15. Angelousi A, Peppa M, Chrisoulidou A, Alexandraki K, Berthon A, Faucz FR, et al. Malignant pheochromocytomas/paragangliomas and ectopic hormonal secretion: A case series and review of the literature. Cancers. (2019) 11:724. doi: 10.3390/cancers11050724
16. Bravo EL, Tagle R. Pheochromocytoma: State-of-the-art and future prospects. Endocr. Rev. (2003) 24:539–53. doi: 10.1210/er.2002–0013
17. Jimenez C, Ma J, Gonzalez AR, Varghese J, Zhang M, Perrier N, et al. TNM staging and overall survival in patients with pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab. (2023) 108:1132–42. doi: 10.1210/clinem/dgac677
18. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens. (2020) 38:1443–56. doi: 10.1097/hjh.0000000000002438
19. Taieb D, Wanna GB, Ahmad M, Lussey-Lepoutre C, Perrier N, Noelting S, et al. Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol. (2023) 11:345–61. doi: 10.1016/s2213–8587(23)00038–4
20. Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, Casey RT, et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet. (2018) 55:384–94. doi: 10.1136/jmedgenet-2017–105127
21. Hanson H, Durkie M, Lalloo F, Izatt L, McVeigh TP, Cook JA, et al. K recommendations for SDHA germline genetic testing and surveillance in clinical practice. J Med Genet. (2023) 60:107–11. doi: 10.1136/jmedgenet-2021–108355
22. Lee H, Jeong S, Yu Y, Kang J, Sun H, Rhee J-K, et al. Risk of metastatic pheochromocytoma and paraganglioma in SDHx mutation carriers: a systematic review and updated meta-analysis. J Med Genet. (2020) 57:217–25. doi: 10.1136/jmedgenet-2019–106324
23. Williams ST, Chatzikyriakou P, Carroll PV, McGowan BM, Velusamy A, White G, et al. SDHC phaeochromocytoma and paraganglioma: A UK-wide case series. Clin Endocrinol (Oxf.). (2022) 96:499–512. doi: 10.1111/cen.14594
24. Fuchs TL, Luxford C, Clarkson A, Sheen A, Sioson L, Elston M, et al. A clinicopathologic and molecular analysis of fumarate hydratase-deficient pheochromocytoma and paraganglioma. Am J Surg Pathol. (2023) 47:25–36. doi: 10.1097/pas.0000000000001945
25. Calsina B, Curras-Freixes M, Buffet A, Pons T, Contreras L, Leton R, et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet Med. (2018) 20:1652–62. doi: 10.1038/s41436–018-0068–7
26. Richter S, Gieldon L, Pang Y, Peitzsch M, Thanh H, Leton R, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med. (2019) 21:705–17. doi: 10.1038/s41436–018-0106–5
27. Buffet A, Morin A, Castro-Vega L-J, Habarou F, Lussey-Lepoutre C, Letouze E, et al. Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. (2018) 78:1914–22. doi: 10.1158/0008–5472.Can-17–2463
28. Remacha L, Comino-Mendez I, Richter S, Contreras L, Curras-Freixes M, Pita G, et al. Targeted exome sequencing of krebs cycle genes reveals candidate cancer-predisposing mutations in pheochromocytomas and paragangliomas. Clin Cancer Res. (2017) 23:6315–24. doi: 10.1158/1078–0432.Ccr-16–2250
29. Remacha L, Curras-Freixes M, Torres-Ruiz R, Schiavi F, Torres-Perez R, Calsina B, et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet Med. (2018) 20:1644–51. doi: 10.1038/s41436-018-0003-y
30. Tarade D, Ohh M. The HIF and other quandaries in VHL disease. Oncogene. (2018) 37:139–47. doi: 10.1038/onc.2017.338
31. Rosenblum JS, Wang H, Nazari MA, Zhuang Z, Pacak K. Pacak-Zhuang syndrome: a model providing new insights into tumor syndromes. Endocr. Relat Cancer. (2023) 30:e230050. doi: 10.1530/erc-23–0050
32. Eckardt L, Prange-Barczynska M, Hodson EJ, Fielding JW, Cheng X, Lima JDCC, et al. Developmental role of PHD2 in the pathogenesis of pseudohypoxic pheochromocytoma. Endocr. Relat Cancer. (2021) 28:757–72. doi: 10.1530/erc-21–0211
33. Castinetti F, Waguespack SG, Machens A, Uchino S, Hasse-Lazar K, Sanso G, et al. Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. Lancet Diabetes Endocrinol. (2019) 7:213–20. doi: 10.1016/S2213-8587(18)30336-X
34. Welander J, Soderkvist P, Gimm O. The NF1 gene: a frequent mutational target in sporadic pheochromocytomas and beyond. Endocr. Relat Cancer. (2013) 20:C13–C7. doi: 10.1530/erc-13–0046
35. Armaiz-Pena G, Flores SK, Cheng Z-M, Zhang X, Esquivel E, Poullard N, et al. Genotype-phenotype features of germline variants of the TMEM127 pheochromocytoma susceptibility gene: A 10-year update. J Clin Endocrinol Metab. (2021) 106:E350–E64. doi: 10.1210/clinem/dgaa741
36. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
37. Oudijk L, de Krijger RR, Rapa I, Beuschlein F, de Cubas AA, Tos APD, et al. H-RAS mutations are restricted to sporadic pheochromocytomas lacking specific clinical or pathological features: data from a multi-institutional series. J Clin Endocrinol Metab. (2014) 99:E1376–E80. doi: 10.1210/jc.2013–3879
38. Schlisio S, Kenchappa RS, Vredeveld LCW, George RE, Stewart R, Greulich H, et al. The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. (2008) 22:884–93. doi: 10.1101/gad.1648608
39. Guerin C, Romanet P, Taieb D, Brue T, Lacroix A, Sebag F, et al. Looking beyond the thyroid: advances in the understanding of pheochromocytoma and hyperparathyroidism phenotypes in MEN2 and of non-MEN2 familial forms. Endocr. Relat Cancer.(2018) 25:T15–28doi: 10.1530/erc-17–0266
40. Alzofon N, Koc K, Panwell K, Pozdeyev N, Marshall CB, Albuja-Cruz M, et al. Mastermind like transcriptional coactivator 3 (MAML3) drives neuroendocrine tumor progression. Mol Cancer Res. (2021) 19:1476–85. doi: 10.1158/1541–7786.Mcr-20–0992
41. Buffet A, Zhang J, Rebel H, Corssmit EPM, Jansen JC, Hensen EF, et al. Germline DLST variants promote epigenetic modifications in pheochromocytoma-paraganglioma. J Clin Endocrinol Metab. (2021) 106:459–71. doi: 10.1210/clinem/dgaa819
42. Mellid S, Garcia F, Leandro-Garcia LJ, Diaz-Talavera A, Martinez-Montes AM, Gil E, et al. DLST mutations in pheochromocytoma and paraganglioma cause proteome hyposuccinylation and metabolic remodeling. Cancer Commun. (2023) 43:838–43. doi: 10.1002/cac2.12427
43. Buffet A, Burnichon N, Favier J, Gimenez-Roqueplo A-P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. (2020) 34:101416. doi: 10.1016/j.beem.2020.101416
44. Garcia-Carbonero R, Matute Teresa F, Mercader-Cidoncha E, Mitjavila-Casanovas M, Robledo M, Tena I, et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. J Clin Trans oncology: Off Publ Fed Spanish Oncol Societies Natl Cancer Institute Mexico. (2021) 23:1995–2019. doi: 10.1007/s12094–021-02622–9
45. Zhao L, Zhang T, Meng X, Fan H, Zhang Z, Liu Y, et al. The clinical characteristics of patients with normotension in pheochromocytomas and paragangliomas. J Endocrine. (2023) 80:174–82. doi: 10.1007/s12020–022-03293–4
46. Roman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, et al. Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann Surg. (2018) 268:172–8. doi: 10.1097/sla.0000000000002195
Keywords: pheochromocytoma, neoplasm metastasis, exome sequencing, dihydrolipoamide succinyltransferase, case report
Citation: Li C, Han L, Song Y and Liu R (2024) Case report: A rare DLST mutation in patient with metastatic pheochromocytoma: clinical implications and management challenges. Front. Oncol. 14:1394552. doi: 10.3389/fonc.2024.1394552
Received: 01 March 2024; Accepted: 06 May 2024;
Published: 21 May 2024.
Edited by:
Ichiro Abe, Fukuoka University Chikushi Hospital, JapanReviewed by:
Piotr Glinicki, Centre of Postgraduate Medical Education, PolandYuichi Yoshida, Oita University, Japan
Yoshikiyo Ono, Tohoku University Hospital, Japan
Copyright © 2024 Li, Han, Song and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Liu, bGl1ckBqbHUuZWR1LmNu