 D. Isabel Petrescu
D. Isabel Petrescu Jason T. Yustein1*
Jason T. Yustein1* Atreyi Dasgupta
Atreyi Dasgupta- 1Aflac Cancer and Blood Disorders Center, Emory University, Atlanta, GA, United States
- 2The Faris D. Virani Ewing Sarcoma Center, Baylor College of Medicine, Texas Children’s Cancer and Hematology Centers, Houston, TX, United States
Sarcomas comprise between 10–15% of all pediatric malignancies. Osteosarcoma and Ewing sarcoma are the two most common pediatric bone tumors diagnosed in children and young adults. These tumors are commonly treated with surgery and/or radiation therapy and combination chemotherapy. However, there is a strong need for the development and utilization of targeted therapeutic methods to improve patient outcomes. Towards accomplishing this goal, pre-clinical models for these unique malignancies are of particular importance to design and test experimental therapeutic strategies prior to being introduced to patients due to their origination site and propensity to metastasize. Pre-clinical models offer several advantages for the study of pediatric sarcomas with unique benefits and shortcomings dependent on the type of model. This review addresses the types of pre-clinical models available for the study of pediatric solid tumors, with special attention to the bone sarcomas osteosarcoma and Ewing sarcoma.
1 Introduction
Cancer is the second most common cause of death in children aged 0–14 in the United States. In 2023, nearly 10,000 children from birth to 14 years of age and more than 5,000 adolescents from 15 to 19 years of age were expected to be diagnosed (1). Pediatric solid tumors are a subcategory that constitutes approximately 40% of pediatric cancers (2). Common types include brain tumors, neuroblastoma, Wilms tumors, germ cell tumors, rhabdomyosarcoma, Ewing sarcoma (EwS), and osteosarcoma (OS) (Table 1) (3). Although these are the most common types of pediatric solid tumors, these are still classified as pediatric rare cancers, as are all other types of pediatric cancer. Within the past decade, there has been a strong scientific focus on genes and genomics, including a renewed interest to determine how each individual’s body and their tissues and organs contribute a crucial role in the development of cancer. Efforts are now focusing on how to manipulate and exploit this special microenvironment to improve chemotherapy along with how to adapt tumor cells to be more susceptible to targeted inhibitors. There is a tremendous amount of evidence that it is not only the tumor cells, but also the host cells that can be reprogrammed to significantly contribute to tumor development and growth. Thus, it is imperative to study the tumor in its environment – both at the micro and macro scale. To do so, it is critical to employ model systems that can best mimic the natural, physiological state. These models not only help our understanding of tumor biology, but they also provide a platform for experimental therapeutic strategies. In this review, we address the existing and innovative preclinical models and their benefits and shortcomings for the study of these types of cancers with a focus on bone sarcomas.
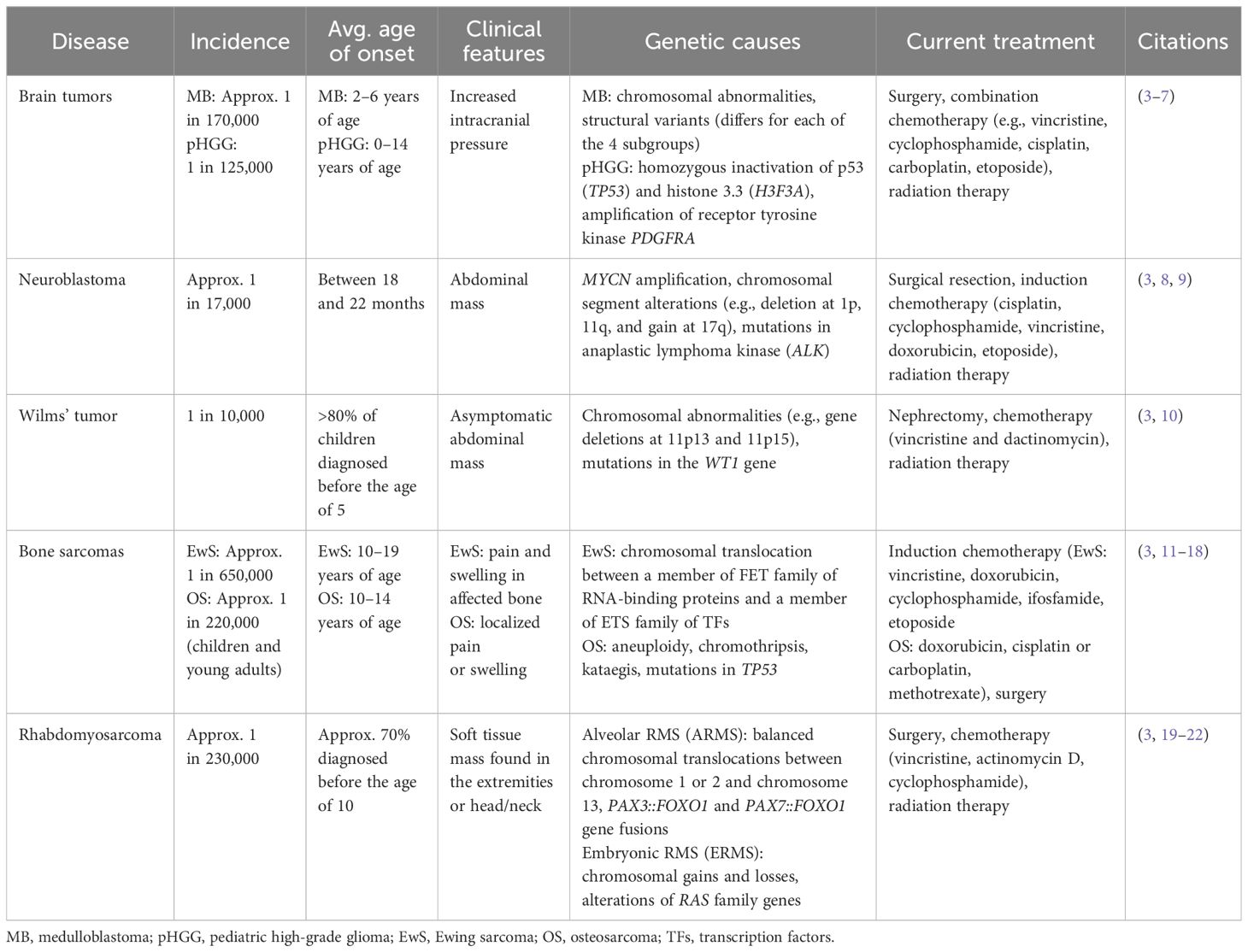
Table 1 Characteristics of common pediatric solid tumors.
1.1 Pediatric solid tumors and need for preclinical models
A unique challenge with pediatric solid tumors is that symptoms can mimic common childhood illnesses, requiring careful attention from primary care providers to diagnose a solid tumor malignancy in children and adolescents (2). Unlike other solid tumors, the most common initially presenting symptom is pain or swelling for bone sarcomas (23). Furthermore, the current treatment strategy for most pediatric solid tumors remains limited to surgical resection, radiation therapy, and chemotherapy. Even with recent advancements in therapeutic options, there has been limited improvement in survival for most pediatric cancers, including solid malignancies such as metastatic sarcomas. This highlights the necessity to advance pre-clinical models of pediatric solid tumors for the purpose to identify new therapeutic targets and improve patient outcomes and survival.
An advantage of preclinical models is their utility as a method for testing new therapeutic targets. This is especially imperative for less common pediatric cancers for which specific therapeutic agents have yet to be identified (24). Due to the rarity of pediatric cancers, relatively low numbers of children are available as subjects for clinical trials assessing experimental therapies. For those that are available, their disease state is likely to be advanced or resistant to treatment (25). As an example, with the genomic complexity of OS, identifying therapeutic approaches that can be broadly applicable to most patients becomes challenging (11). Therefore, using complementary preclinical models is especially important to increase relevance of identified targets and therapeutic potential and to overcome the intrinsic disadvantages or challenges of each type of model when used in isolation.
1.2 Established models
Types of preclinical models can be broadly divided into the categories of in vivo and in vitro. In vivo models include animal models (spontaneous and xenografts), genetically engineered murine models (GEMMs), and patient-derived xenografts (PDXs). By contrast, in vitro models include cell culture, co-culture, organoids, and 3D model systems. In vitro models are similar in their function and use across most solid tumor types, whereas the various in vivo animal models can differ by type of sarcoma. For the purpose of this review, we will limit our focus to address in vivo models for the bone sarcomas osteosarcoma and Ewing sarcoma. A summary of the common in vitro (cell culture) and in vivo models of osteosarcoma and Ewing sarcoma is provided in Figure 1 (OS) and Figure 2 (EwS).
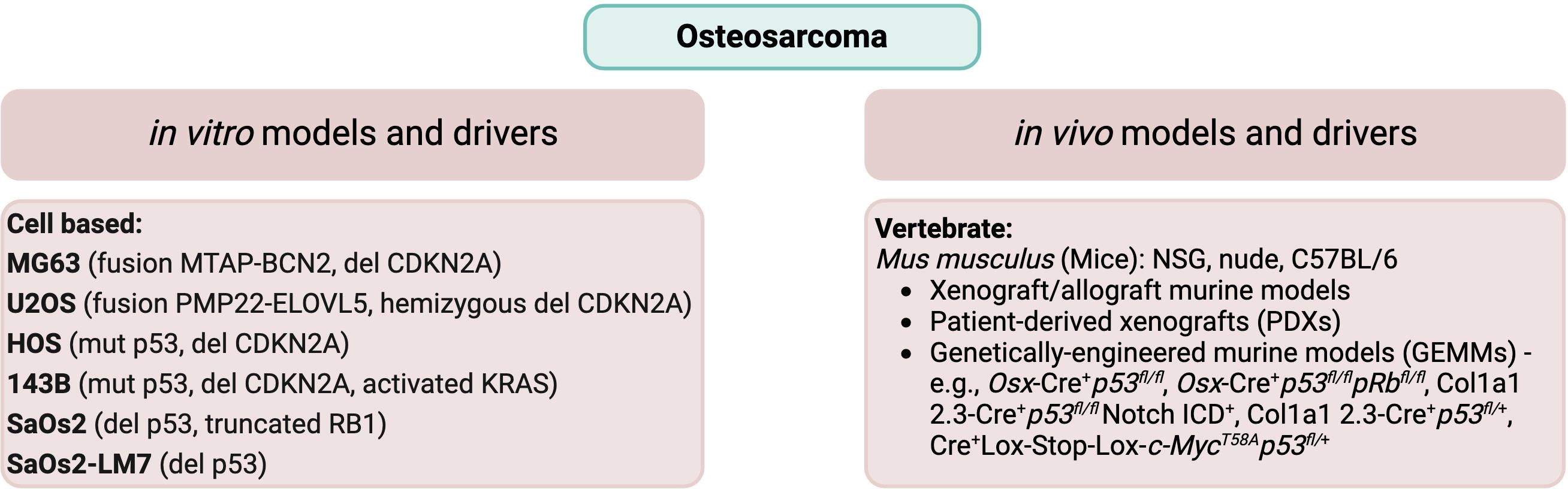
Figure 1 Common in vitro cell lines and in vivo animal models of osteosarcoma. Commonly used cell lines, including their genomic characteristics (26, 27), and in vivo animal models (28–30) are listed for osteosarcoma.
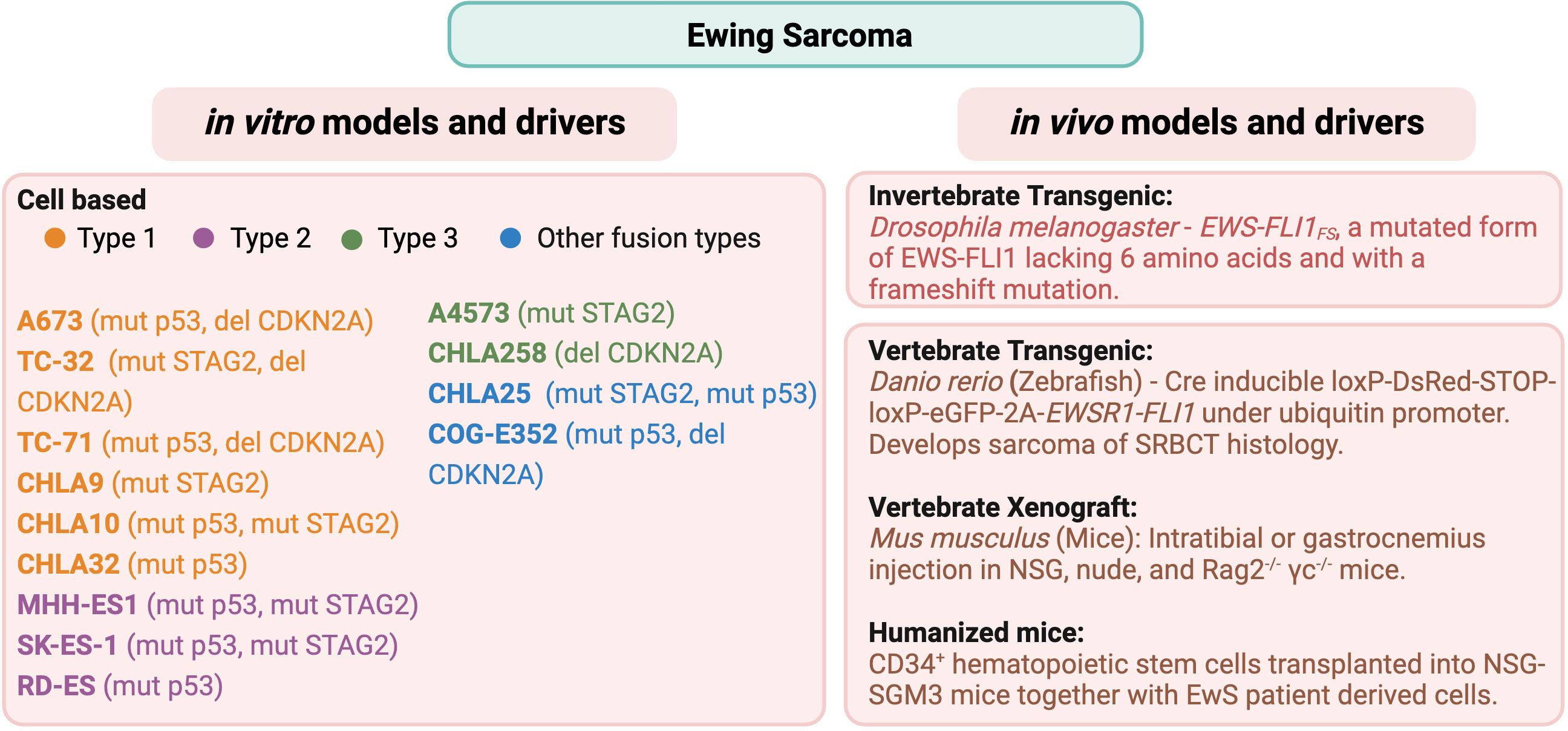
Figure 2 Common in vitro cell lines and in vivo animal models of Ewing sarcoma. Commonly used cell lines, including their fusion type (31) and genomic characteristics (32–36), and in vivo animal models (37–42) are listed for Ewing sarcoma.
2 Osteosarcoma
OS is the most common primary malignant bone tumor, with bimodal peak incidence in adolescents and adults 60 years of age and older (43). The rate of childhood and adolescent OS is approximately 3 to 4.5 cases/million each year, with tumors most commonly developing in the lower long bones such as the femur, tibia, and fibula, as well as the humerus (44). OS tumors appear as a lytic intramedullary lesion that often invade and destroy the bony cortex and extend into nearby soft tissues. The major presenting sign is pain in the affected area (45). Approximately 20% of cases will present with metastatic disease, most commonly to the lungs. Metastases can also be found in additional bone, soft tissue, or lymph node lesions (12). The major treatment methods are surgery and chemotherapy. However, for OS patients with metastatic disease, the 5-year survival rate significantly decreases from approximately 70% to less than 40%, underscoring the need for novel therapeutic strategies (28). Patient-derived primary tumor cultures and genetically engineered murine models are frequently used pre-clinical models, which are complemented with available resources from the study of canine osteosarcoma, arising spontaneously in large breed dogs (11), to help gain insights into the molecular pathogenesis of the disease as well as assessing novel therapeutic regimens.
Studying canine osteosarcoma has proven to be valuable towards further understanding the molecular pathogenesis of OS and identification of potential therapeutic options for human osteosarcoma patients. There is evidence to suggest that transcriptomic and clinical patterns are comparable between canine and human OS. Of particular interest, the proportion of macrophage subtypes M0 and M2 present in the TME is similar between canine and human OS tumors (46). Furthermore, clinical trials of novel therapeutics are possible to be tested in canines before being approved for use in human patients (47). As an example, the drug auranofin (AF), an inhibitor of thioredoxin reductases (TrxRs), had been used in a clinical trial to evaluate its safety and efficacy in addition to the regular standard of care for the treatment of canine OS. The research findings demonstrated improved survival in male dogs when AF was combined with the standard of care treatment (48).
2.1 Molecular features
OS demonstrates genomic complexity without having characteristic features shared by every tumor. Nevertheless, there can be common features. Aneuploidy is a feature of many OS tumors. Furthermore, many tumors have been found to demonstrate chromosomal structural rearrangements, copy number gain, and copy number loss (45). Gene mutations in OS include TP53, which is the most frequently mutated, RB1, ATRX, and DLG2 (11). Patients with Li-Fraumeni syndrome have a germline mutation in p53, resulting in an increased risk of developing OS (45). Similarly, patients with the inherited familial syndromes hereditary retinoblastoma, Rothmund-Thomson, Bloom or Warner syndrome have driver mutations for OS in RB1, RECQL4, BLM, and WRN, respectively (28). Chromosomal copy number gain is observed for sections of chromosomes 1p, 1q, 6p, 8q, and 17p. Conversely, copy number loss may include sections of chromosomes 3q, 6q, 9, 10, 13, 17p, and 18q. The oncogene MYC is found on chromosome 8q and has been found to be frequently amplified in OS (26). Several studies have also found associations with disease incidence at various loci, including single nucleotide polymorphisms (SNPs) within genes important for growth and development such as IGF2R, encoding the IGF2 receptor, FGFR3, encoding fibroblast growth receptor 3, and the genes MDM2, encoding the MDM2 p53 binding protein homolog with a role in the regulation of p53 function, and TGFBR1, encoding transforming growth factor-β receptor type 1 with a role in the regulation of cell proliferation (49). The dominant genomic drivers for several of the most commonly used OS cell lines are listed in Figure 1.
3 Ewing sarcoma
Ewing sarcoma (EwS) is a rare but aggressive bone cancer among children, adolescents, and young adults with an annual incidence rate in the United States of approximately 1 case per 1.5 million (50). Histologically, EwS appears as small round blue cells with a prominent nucleus, with tumors arising primarily in the pelvis and the proximal long bones: femur, tibia, and ribs (50). Other extraosseous sites include the thoracic wall, pleural spaces, and cervical and gluteal muscles (13). EwS cells commonly express CD99 on the plasma membrane and contain glycogen-rich cytoplasmic content (51). In radiological findings, EwS lesions appear as lytic, described as “moth eaten”, with frequent appearance of bone sclerosis, and newly formed, elevated periosteum appearing as lamellar (onion skin) or as appearance of displaced periosteum (Codman’s triangle) (50, 52).
With the advent of chemotherapy, the overall survival rate for children with localized EwS disease has increased to 70–75%. However, at least 25% of these patients will relapse, even after completion of the initial treatment plan. Another 25% of patients presenting with metastatic disease at the time of diagnosis have an abysmal 20–25% survival rate (53). This rate has not improved even after decades of research. Consequently, since the introduction of etoposide, no new therapeutic has been introduced to the standard treatment regimen (54). Thus, there is a serious need for novel approaches and therapeutic targets to be discovered.
3.1 Molecular features
Ewing sarcoma is characterized by the translocation of t(11;22)(q24;q12), resulting in the fusion of FET family of RNA binding proteins (EWSR1, FUS, and TAF15) to ETS (E-26 specific) family of proteins (FLI1, FEV, ERG, FEV, ETV1, ETV4, and ETV5) that act as transcription factors (55–57). The most common fusion protein, EWS::FLI1, arises from the fusion of Ewing sarcoma breakpoint region 1 protein (EWSR1) to Friend leukemia integration 1 transcription factor (FLI1) (31). EWS::FLI1 is the oncogenic driver for EwS in 85% of cases (13, 58). The EWS::FLI1 fusion protein is localized to the nucleus and binds DNA with the same sequence specificity as the parental FLI1 protein. Moreover, EWS-FLI1 acts as an efficient sequence-specific transcriptional activator, dependent on a transcriptional activation domain within the EWS sequence (59). Based upon the specific rearrangement of the EWS::FLI1 fusion protein, the type of EwS can be further classified. In more than 50% of cases, fusion of EWS exon 7 to FLI1 exon 6 (type 1) or to FLI1 exon 5 (type 2) is observed. The third most common fusion is between EWS exon 10 and FLI1 exon 5 (type 3). Lastly, fusion can be between EWS exon 10 and FLI1 exon 6 (type 4) (31). Based upon the genomic breakpoint, 12 types of EWS::FLI1 chimeric transcripts have been observed in patient samples (60).
EWS::FLI1 has been shown to bind to two different types of sites. The first site is at enhancers containing the ETS-2 binding motif, with a single GGAA as the core sequence. This is the binding site for the wild-type FLI1 protein. Compared to the FLI1 protein, the chimeric protein acts as a much more potent transcriptional activator, transforming NIH3T3 cells (61). The second site is specific to EWS-FLI1, consisting of microsatellite sequences of tandem GGAA repeats (GGAAμSats) (62), seen in many downstream target genes required for EwS tumorigenesis such as NROB1 (63). Transcriptional activity of EWS::FLI1 often leads to the formation of R loops made of 3 strands of DNA-RNA hybrid with single-stranded non-template DNA. Accumulation of R-loops can cause replication fork collapse, DNA damage, and increased activity in the Ataxia-telangiectasia mutated and Rad3-related (ATR) pathway (64). Indeed, due to these features, EwS demonstrates increased sensitivity to DNA damage repair inhibitors such as poly(ADP-ribose) polymerase (PARP) inhibitors. Thus, PARP and other DNA damage repair inhibitors are being investigated for targeted therapy in EwS (64, 65). Nonetheless, the genomic landscape of EwS is relatively stable with concomitant genetic alterations found only in a few genes such as mutations in TP53, STAG2, and deletion in CDKN2A as analyzed by several groups (32–36). We have mentioned the dominant genomic drivers for some of the most commonly referenced cell lines in Figure 2.
Microarray and chromatin immunoprecipitation analysis revealed EWS::FLI1 to be a direct or indirect transcriptional activator as well as a repressor for downstream targets (66, 67). Some of the direct activation targets are the neuronally expressed transcriptional repressor NKX2–2 (68); nuclear receptor NROB1, that plays a critical role in osteoblast differentiation and skeletal development (69); GLI1 (70), a member of the Sonic Hedgehog pathway; Id2, cMYC, CCND1 (71); EZH2 (72); and repression of the tumor suppressor FOXO1 (73). Other than direct transcriptional regulation, EWS::FLI1 contributes to sarcomagenesis by regulating alternative splicing (74, 75) and chromatin remodeling (76, 77).
Even though EWS::FLI1 hypothetically presents itself as a clear therapeutic target, a reliable, efficient inhibitor is still lacking. This remains a problem despite the detailed characterization of EWS::FLI1 as an aberrant transcription factor and a potent transforming gene (78–80). A major hindrance to developing a therapeutic inhibitor is the lack of EWS::FLI1-driven EwS murine models.
Ewing sarcoma appears to be a cancer specific to humans only. Many of the murine orthologs of EWS::FLI1 target genes lack GGAA binding sites (62). This, added with two other primary reasons perhaps explain why there remains a considerable challenge towards developing a reliable mouse model for Ewing sarcoma: 1) the lack of precision around its cell of origin; 2) although EWS::FLI1 appears to be the oncogenic driver, its overexpression results in cell death.
To design a model, one needs to have a definitive and rigorous understanding of the cell of origin, but for Ewing sarcoma it has been difficult to identify lineage-specific differentiation.
3.2 Cell of origin
Contrary to osteosarcoma, Ewing sarcoma lacks lineage-specific differentiation. The tumor is histologically classified as being formed from sheets of undifferentiated small round basophilic cells. Thus, the cell of origin of Ewing sarcoma is mired in controversy. It was first described by Dr. James Ewing in 1921 (81). Based upon cellular morphology, frequent presence of cells adjacent to vascular channels, and the presence of very little stroma, he described the undifferentiated tumor in the radius of a 14-year-old girl as “diffuse endothelioma of bone”, thus attributing an endothelial origin. Around the same time, a different school of thought identified similarities with neuroblastoma (82). Around 50 years later, based upon features resembling developing myelocytes, a myelogenous origin was suggested by Kadin and Bensch (82). One of the earlier theories suggested the origin was from primitive neuroectodermal cells, due to its histological resemblance to the tumor cells and expression of neuron-specific enolase (NSE) and S-100 markers in some EwS tumors (83–87). Gene expression patterns from ectopic expression of EWS::FLI1 in undifferentiated neural crest stem cells indicate similar patterns to that of EWS-FLI1 target genes (88). Hence the term primitive neuroectodermal tumors (PNETs) was used interchangeably with EwS. In 2013, WHO classified PNETs and EwS in one pathological entity under the umbrella term Ewing sarcoma family of tumors (ESFTs). WHO classification of tumors of soft tissue and bone in 2020 added a new category of “undifferentiated small round cell sarcomas of bone and soft tissue tumors” that now includes Ewing sarcoma and three new entities: EWSR1::non-ETS fusions, CIC-rearranged sarcoma, and sarcomas with BCOR genetic alterations along with prototypical Ewing sarcoma arising in bone or soft tissue (89). An alternative, more modern proposal for the cell of origin of EwS is the mesenchymal stem cell (90, 91). This is supported by the fact that knock-down of EWS::FLI1 in several different cell lines leads to a more distinct mesenchymal stem cell gene expression pattern (91). Additionally, EWS::FLI1 by itself can transform primary bone marrow derived MSCs and develop tumor with EwS features including small round cells, dependence on IGF-1, and expression of EWS::FLI1 target genes (92).
In attempts to generate a mouse model, three types of primary cell lines have been identified as permissive for EWS::FLI1 expression. These are mesenchymal stem cells (92, 93), neural crest stem cells (88), and embryonic osteochondrogenic progenitor cells (94, 95).
4 In vitro models
Types of in vitro models are comparable across various types of solid tumors, including the bone sarcomas osteosarcoma and Ewing sarcoma, highlighted in this review. In vitro models include two-dimensional (2D) cell culture, co-culture between tumor cells and cell types within the tumor microenvironment (TME), three-dimensional (3D) organoids/spheroids, and bioengineered 3D culture models.
4.1 Cell culture
Cell culture is widely used as a pre-clinical model across many cancer types, including for the study of pediatric solid tumors. A major advantage of cell culture is the relative ease of use, allowing novel therapeutic agents to be tested quickly and meticulously (96). Other advantages of cell culture are the use of standardized methods, the potential for rapid cell growth, and growth in translucent media, allowing cells to be monitored by light microscopy in real-time. In addition, cell culture is cost-effective does not require special equipment (97). The first cell lines for solid tumors were established in 1973 from tumor explants, with cell lines successfully developed for kidney, lung, and epidermoid carcinoma; rhabdomyosarcoma; glioblastoma and astrocytoma in the brain; and melanoma (98). In the decades since, the American Type Culture Collection (ATCC) (https://www.atcc.org/) has collected dozens of cell lines that are beneficial for the study of pediatric solid tumors. With the purpose to identify novel therapeutic targets, Shen et al. treated a panel of 19 well-characterized cell lines with more than 3800 unique chemical compounds (99). These cell lines were derived from solid tumors such as Ewing sarcoma, rhabdomyosarcoma, osteosarcoma, neuroblastoma, and central nervous system tumors (e.g., medulloblastoma, glioblastoma, atypical teratoid rhabdoid tumor). After completing dose-response curves for all compounds, a total of 62 compounds were found to be effective against 17 of the 19 cell lines. These classes of compounds included antineoplastic agents and immunosuppressants, antiparasitics, and anti-inflammatories, which had not been previously known to be effective against pediatric cancer cell lines. The antiparasitics mebendazole and difluoromethylornithine (DFMO) are being tested in clinical trials for pediatric brain tumors (NCT02644291) and neuroblastomas (NCT02395666) (99). Characterization of osteosarcoma cell lines could confirm differentiation capacity as well as tumor formation capacity in vivo, indicative of their utility as a strong representative model of the disease. However, a drawback of in vitro cell culture arises from the absence of stroma or the extra-cellular matrix. As a result, the microenvironment and tissue architecture of the tumor fails to be adequately represented, requiring other methods to supplement the cell culture model to study these features (100). A common in vitro method to study the interaction between tumor cells and immune cells within the tumor microenvironment (TME) is co-culture techniques (101). In this model, tumor cells are plated and grown in culture along with immune cell types.
4.2 Tumor microenvironment/co-culture
The multicellular co-culture model is considered advantageous for providing an approximate imitation of the communication network between the tumor and its microenvironment. A study into healthy human fibroblasts (HFs) co-cultured with the human osteosarcoma cell line MG-63 demonstrated morphological changes in the cell types compared to when HFs and MG-63 cells were grown independently. Furthermore, expression of biomarkers (i.e., YKL-40, VEGF, MMP1) was found to differ in the co-culture, with elevated expression of YKL-40 in HFs and VEGF in MG-63 (102). Possible methods for co-culture plating include direct or indirect mechanisms. With direct co-culture, cell types are layered on top of each other to allow for physical contact whereas with indirect co-culture, the different cell types are separated by porous membranes, such that non-contact-mediated signaling through secreted soluble factors can be investigated (101). For greater specificity and control of spatial arrangement, microfluidic techniques can be used to co-culture cancer cells with fibroblasts. As an example, tumor spheroids grown in co-culture with cancer fibroblasts in a microfluidic chip-based culture model were more resistant to the chemotherapeutic drug paclitaxel (103). Additionally, the tumor microenvironment can be investigated at sites of metastasis as well as the primary tumor. Mendoza et al. developed an ex vivo method called the pulmonary metastasis assay (PuMA) to study pulmonary metastasis, which is a common outcome of Ewing sarcoma and osteosarcoma disease progression. For this assay, GFP-expressing cancer cells proliferate in lung tissue with various other cell types present in the tissue culture such as migratory cells, type I and II pneumocytes, alveolar macrophages, vascular endothelial cells, fibroblasts, and red blood cells. This assay can be used to compare the metastatic ability of different human and murine cancer cell lines and to identify lung microenvironments conducive to metastasis (104). Taylor et al. investigated the role of EwS tumor cells in osteoclast formation and the osteolysis observed in EwS using a co-culture model system. EwS cell lines co-cultured with CD14+ monocytes primed with macrophage-colony stimulating factor (M-CSF) resulted in the formation of multinucleated cells positive for the osteoclast functional marker TRAP (tartrate-resistant acid phosphatase) (105, 106). Moreover, the co-culture of M-CSF-primed monocytes with EwS cell lines was able to cause lacunar resorption, a hallmark characteristic of osteoclasts. These results indicate that osteoclast-like cells are able to be formed from tumor-associated macrophages present within the EwS tissue cultures (106).
4.3 Organoids/spheroids
A recently developed in vitro model is three-dimensional (3D) culture systems to overcome challenges with 2D cell culture. The major advantage of 3D organoid/spheroid cultures is the potential to capture the complexity of the tissue environment in which a tumor resides (107). 3D spheroids can be formed by seeding a known density of cells into a 3D matrix scaffold, with the scaffold consisting of pores that are sufficiently small to restrict the size of the spheroids. The porous scaffold is supplemented with complete media then the plated cells are given time to grow into spheroids (108). Lawlor et al. developed spheroids of EwS cell lines TC32, A4574 and 5838 to investigate the roles of the RAS-RAF1-MEK-ERK 1/2 mitogen-activated protein kinase (MAPK) and phosphatidyl inositide-3-kinase (PI3K)-AKT pathways in the regulation of cell proliferation specifically regarding the regulation of cyclin D1 expression. Their results indicated that cyclin D1 protein expression required serum stimulation and cell-cell adhesion within the spheroid cultures grown in suspension. Moreover, the ERK1 and ERK2 kinases were found to be constitutively active in TC32 cells placed in suspension, suggestive of increased phosphorylation compared to monolayer cultures. Similar results were seen in 5838 and A4573 spheroids (109). He et al. described two distinct methods to establish OS organoids from primary tumors or lung metastases isolated from patients (110). The first method (“Cut/EnBloc”) used an inner transwell insert with a bottom layer of collagen gel matrix and minced tissue samples resuspended in collagen layered on top, with the transwell insert placed inside of a tissue culture dish. The second method (“Single-cell”) established each organoid from a single-cell suspension arising from the tissue samples. For this method, minced tissue samples were digested followed by treatment wth DNaseI and red blood cell lysis buffer. Lastly, cells were filtered through a cell strainer to recover single-cell suspensions, which were then embedded in Matrigel and allowed to solidify. The single-cell method was successful to establish organoids either from lung metastatic tissues or OS primary tumors. The authors concluded that the Cut/EnBloc method was more effective for establishing organoids from OS lung metastases whereas the single cell method was more successful for culturing organoids from primary OS tumor samples (110). A common function of 3D cell culture is to study the interaction between cells and the extracellular matrix (ECM). This can be particularly useful for testing drug efficiency by identifying whether the ECM prevents drug diffusion to cells (111). Benefits of spheroids are that their generation and use is simple and inexpensive. However, using spheroids to model tumor cells in 3D presents challenges for high-throughput screening, such as drug testing, and requires precise control of droplet size and size uniformity when developing the spheroids (112).
4.4 Engineered 3D culture
An emerging area of study in cancer research is the development of engineered ex vivo 3D cell culture models. This type of model is advantageous for its ability to recapitulate the shape and environment of the in vivo tumor. Each component of the TME can be experimentally modified to determine its effect on the tumor cells. Within 3D culture models, cancer cells, endothelial cells, and other stromal cells are co-cultured in a spatially relevant system representative of the native tumor environment. In this manner, hypoxia and the release of angiogenic factors in response to hypoxia can be examined and modified to mimic levels found in human tumors (97). To provide an example, cancer stem cells (CSCs) are a cell type present in cancer tissues that have the ability for self-renewal or differentiation into tumor cells. The 3D culture model is beneficial for understanding the interplay between CSCs and their niche in the tumor microenvironment (113). An innovative research study from Molina et al. involved engineering a 3D bone tumor niche using mesenchymal stem cells (MSCs) cultured on electrospun poly(ε-caprolactone) (PCL) scaffolds to imitate bone-like architecture, ECM, and mineralization with the goal to understand how components of the 3D environment would affect EwS cell growth, morphology, and signaling activation (114). Similarly, Bassi et al. designed two hydroxyapatite-based bone-mimicking scaffolds to focus on the CSC niche within the OS TME. The system consisted of 3D scaffolds together with enriched CSCs obtained by sarcosphere-forming culture originating from the OS cell lines MG-63 and SaOS2. The sarcospheres grown in the 3D scaffolds maintained their circular morphology, representative of the scaffold-free sarcospheres of the parental cells, while embedding within the scaffold matrix. Furthermore, quantitative PCR to analyze the expression profile of genes involved in stemness and niche communication between OS cells and CSCs revealed upregulation of the stemness genes NANOG and OCT-4 and the signaling gene IL-6 in the SaOS2 3D scaffold-based sarcosphere models in comparison to the scaffold-free sarcospheres. Likewise, the expression of NANOG and the signaling genes NOTCH-1 and HIF-1α was upregulated in the MG-63 3D scaffold-based sarcosphere models (115).
An exciting new area of research is the organ-on-chip model, also known as microfabrication. In this system, miniaturized organs or tissues are designed in a chip-like array, similar to a computer processor, that can be used for drug modeling. The advantages of the organ-on-chip model are its 3D design, modularity, versatility to represent different organ systems, and the possibility for rapid manufacturing of organ environments (116). Lu et al. designed an osteosarcoma-on-a-chip model by using a decellularized osteosarcoma extracellular matrix (dOsEM) that was loaded with extracellular vesicles from human bone marrow-derived stem cells (hBMSC-EVs) along with OS cells. These cell types were used as a bioink to create a micro-osteosarcoma (micro-OS) via 3D printing. The micro-OS was combined with a microfluidic system to establish an OS-on-a-chip (OOC) (117). After each of the multiple components was prepared and characterized, characterization of the OOC indicated that the OOC could mimic the OS bone marrow niche and exhibit high OS aggressiveness through activation of CXCL12/CXCR4 signaling to maintain PI3K/AKT-mediated proliferation and metastasis. Additionally, doxorubicin treatment of the OOC successfully simulated concentration changes of doxorubicin necessary to limit the viability of patient-derived OS cells in vivo (117).
5 Animal models
In OS, pre-clinical animal models are predominantly murine models. One specific advantage of murine models is that this type of model presents an invaluable method to study the metastatic cascade and to understand the sequential order of events for the development and metastatic spread of OS (14). Furthermore, various types of models can be combined in a study to address a hypothesis from multiple directions. As an example, Zhang et al. used a combination of in-vitro human OS cell lines along with three-dimensional (3D) culture models and tumor-bearing mice to identify the cyclin-dependent kinase 7 (CDK7) as a key player in OS growth and metastasis, resulting in the conclusion that targeting CDK7 or its target glucose-related protein 78-kDa (GRP78) presents a potential treatment strategy for OS (118).
In Ewing sarcoma, commonly used animal models are immunodeficient mice (Mus musculus). To a lesser extent, fruit flies (Drosophila melanogaster) and zebrafish (Danio rerio) may also be used as animal models to study Ewing sarcoma. Each of these models offers unique advantages and disadvantages. However, challenges remain with establishing animal models to accurately represent EwS tumors.
5.1 Xenograft/allograft murine models
In xenograft murine models, human cell lines are injected into immunodeficient mice, such as non-obese diabetic/severe combined immunodeficient (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) (https://www.jax.org/strain/005557), commonly referred to as NSG or NOD scid gamma, NOD Rag gamma (NRG) (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ) (https://www.jax.org/strain/007799), and athymic immunosuppressed nude (NU/J) (https://www.jax.org/strain/002019) mice (119). Conversely, murine cell lines are injected into immunocompetent mice to establish allograft murine models (120). For this type of model, careful consideration for the site of injection is needed based on the experimental question. Each site of injection poses benefits and shortfalls for modeling OS tumor growth and progression (14). For example, injection subcutaneously (heterotopic) versus at the corresponding initial anatomical position for the tumor (orthotopic) affects the strength of the model to represent the complex process of carcinogenesis (121). Di Fiore et al. designed an OS model of CSCs by treating human OS MG-63 cells with 3-aminobenzamide (3-AB), a potent inhibitor of poly(ADP-ribose)-polymerase (PARP), which had resulted in massive cell death and the enrichment of a new CSC population, termed 3AB-OS. The 3AB-OS CSCs and MG-63 cells were injected into athymic nude mice to establish xenografts and determine the tumor-initiating capacity of each cell line. The results demonstrated that the 3AB-OS cells had the ability to form tumors in vivo whereas the MG-63 cells did not. Furthermore, the presence of Matrigel and the length of time for engraftment was demonstrated to affect tumor volume (122). In addition, for OS, the use of syngeneic murine cell lines in immunocompetent mice include MOS-J, K7M2, F420, K12, and POS-1 (37, 38, 123) provide an in vivo model with an intact, representative immune microenvironment (39),, which is not feasible when human OS cell lines are injected into immunodeficient mice.
Most EwS pre-clinical research depends upon developing xenograft tumors in immunocompromised mice. Although many researchers use subcutaneous, a more physiologically relevant orthotopic injection is intratibial or intrafemoral. For EwS cell lines that were originally extraosseous, gastrocnemius or intramuscular injections can be done. Several established EwS cell lines such as TC71, TC32, A673, RDES, SK-N-MC (124) are able to grow, develop, and metastasize in immunocompromised murine models. In these models, the primary site of metastasis is the lungs or secondary bone (125, 126).
Xenograft models allow for the characterization and targeting of tumor inherent properties. Thus, this type of model is suitable for pre-clinical drug testing, including bioavailability, for gene specific knock-in and knock-down effects, and for testing the efficacy of genetically-engineered chimeric antigen receptor T (CAR-T) cells. Visualizing the cell lines with luciferase or fluorescent proteins allows for live cell tracing and monitoring of metastatic spread. Using this method, we and several other research groups have shown distal metastasis typical for EwS in secondary bone, lungs, and liver. In addition, xenograft murine models were used to harvest organs and characterize ex vivo lung-specific metastasis in EwS (127, 128). Further, xenografts have been reported to be used to test and characterize cell lines that may be developed to be chemoresistant (129–131), radiation-adapted (132), or designed as organ-specific metastatic cell lines (128).
5.2 Patient-derived xenografts
Patient-derived xenografts (PDXs) are advantageous for their ability to recreate the cellular, molecular, and histologic characteristics of the primary tumor. Furthermore, cell lines can be expanded from PDXs, with several possible uses, such as high throughput combinatorial drug screening (133). PDXs are established by transplanting biopsies from human tumors in immunodeficient mice (134) (Figure 3). However, success rates for the establishment of PDXs varies widely, depending on tumor type, tumor stage, mouse strain, and site of implantation.
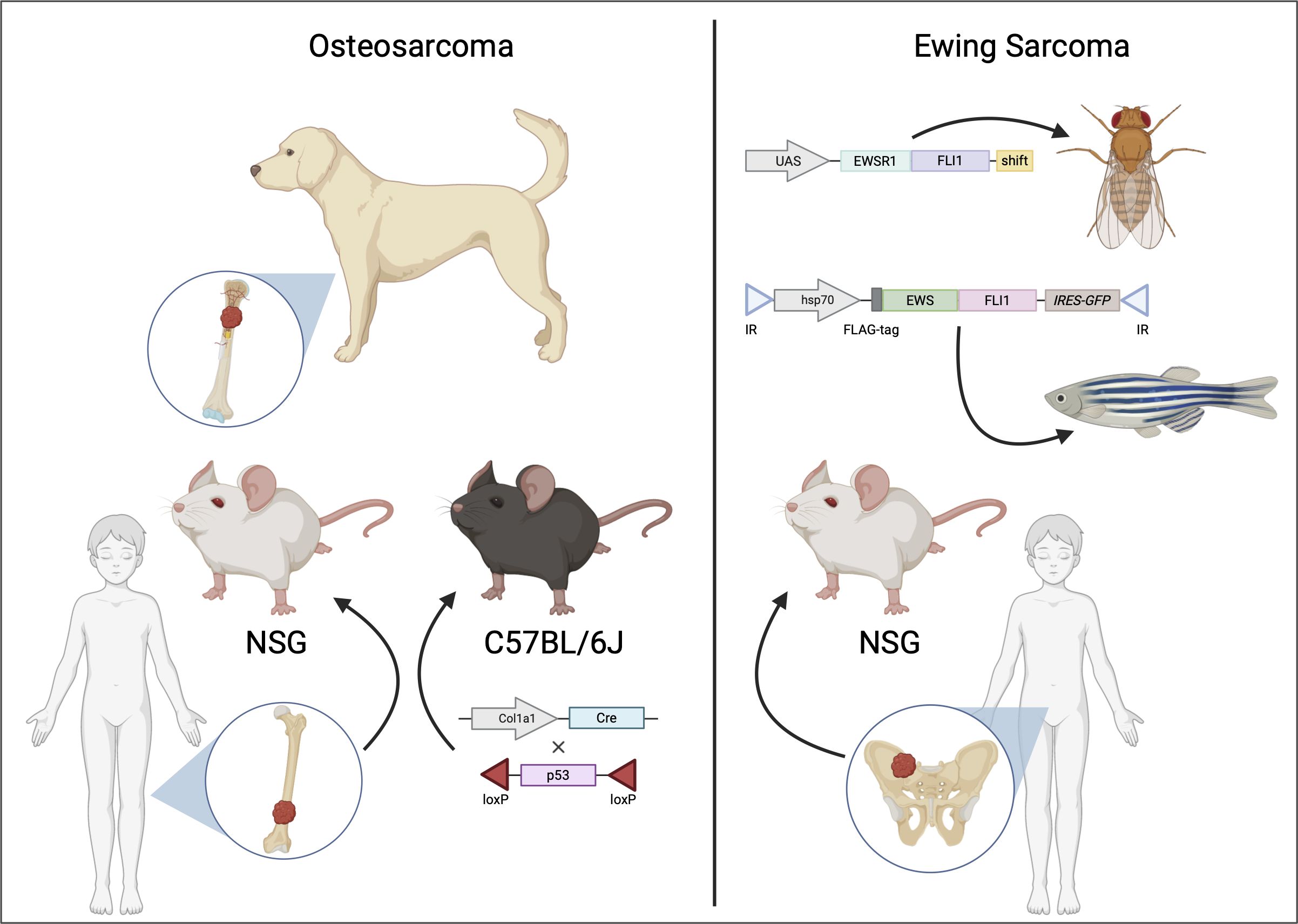
Figure 3 Animal models of osteosarcoma and Ewing sarcoma. Osteosarcoma (shown on the left) is often modeled using genetically modified murine models (GEMMs) [adapted from (135)] and patient-derived xenografts (PDXs) implanted in immunodeficient mice. Additionally, canine osteosarcoma closely resembles the human disease, providing insights into potential therapeutic options for human osteosarcoma. For Ewing sarcoma (shown on the right), in vivo pre-clinical models include PDXs or transgenic models such as Drosophila melanogaster [adapted from (42)] or zebrafish [adapted from (136)].
Global efforts to develop PDXs for research and pre-clinical models include the EuroPDX consortium (www.europdx.eu/), the Public Repository of Xenografts (www.proxe.org), and the Patient-Derived Models Repository (www.pdmr.cancer.gov/) from the National Cancer Institute (NCI) (137). Specific to pediatric solid tumors, St. Jude Children’s Research Hospital has established the Childhood Solid Tumor Network (CSTN) repository of PDXs (www.cstn.stjude.cloud/). Another available repository is the Baylor College of Medicine PDX Portal (www.pdxportal.research.bcm.edu/pdxportal/).
PDXs were used as in vivo models to measure the drug response to vincristine or cyclophosphamide in pediatric solid tumors, including Wilms tumors, sarcomas, brain tumors, and rhabdoid tumors. The results of this study indicated broad-spectrum activity of vincristine and cyclophosphamide across cancer types, providing encouraging evidence to use in vivo PDX models for the identification and validation of novel therapeutics (25). With regards to the study of OS, many groups have developed orthotopic OS models, termed patient-derived orthotopic xenograft (PDOX) models, so that the transplanted tissue can grow at a location that most closely resembles the site of spontaneous disease in patients (26, 138). One of the disadvantages of the PDX model is the inherent inability to study immune system interactions and the tumor microenvironment due to the requirement of immunodeficient mice in order to establish the model (139). Although PDXs in immunocompromised mice lack the developmental pathway and the systemic environment of a transgenic model, compared to cell-derived xenografts (CDXs), they are better able to retain crucial tumor microenvironment, which has a significant role in tumor growth and development, partially due to tumor cell infiltration and the stromal component of the tumor tissue. Indeed, when compared to clinical patient samples, gene expression profiling from different PDX models shows a closer similarity than CDXs (140). This result remained consistent over several passages in mice (141, 142). PDXs can circumvent issues seen with low clinical correlation when chemotherapy agents are tested on cell-derived models (143). In addition, orthotopic transplantation of PDXs followed by hindlimb amputation provides a method to mimic surgical resection in OS patients and to allow time for the development of spontaneous metastases, such that therapies can be identified to target distant metastases (144).
Using mostly NSG or nude mice (5–10 weeks old), several groups (137, 140–142, 145, 146) have generated Ewing sarcoma PDXs from both local as well as metastatic samples using incisional or percutaneous biopsies in patients with varying success rate of engraftment of 25–45% (137, 141, 145). The rate of engraftment was positively correlated with relapse in newly diagnosed EwS patients (145).
Drawbacks to this type of model include the fact that over time, tumor infiltrates from patient samples can be replaced by host immune and stromal cells resulting in differential gene expression patterns between the primary patient samples and serial implants in mice. With successive implantation in mice, there is a high chance of clonal selection to occur, with expansion of the most aggressive subpopulations to dominate the tumor cell population. Additionally, there can be cross-species signaling mismatch between signaling molecules of the host and the human receptors present in the tumor (140, 145).
5.3 Transgenic models
For transgenic models of OS, the most commonly used model is genetically engineered murine models (GEMMs) (Figure 3). Due to the fact that GEMMs are not easily feasible for EwS, available transgenic models are the fruit fly (Drosophila melanogaster) and the zebrafish (Danio rerio) (Figures 2, 3).
5.3.1 Genetically engineered murine models
Genetically engineered murine models (GEMMs) (Figure 3) are a common method to study tumor biology for several types of cancer, including OS. Advantages of murine models include the small size of mice, that mice reproduce quickly and produce large litters, and with technical advancements, including CRISPR/Cas9, mice can be genetically manipulated with relative ease. Types of mouse models can be grouped into various categories, depending on the intended purpose of the model. These categories include transgenic and gene-targeting approaches for either loss-of-function or gain of function studies (147). Although there are numerous advantages, a disadvantage of GEMMs is that they present a major commitment in the cost, effort, and time required to develop and maintain these models (139).
Specifically in the context of OS, one of the benefits of GEMMs is their potential use for gaining further insights into genetic and molecular mechanisms of OS development, such as by identifying driver mutations and/or mutations that co-exist in both normal and tumor cells (28). As an example, a murine model using conditional (floxed) alleles of both p53 and pRb or p53 alone to allow for tissue-restricted gene inactivation of osteoblasts following Cre expression resulted in the development of murine OS, indicating the requirement for disruption of p53 (29). Another study using GEMMs had identified a role for Wnt signaling in the development of OS through the activity of c-Fos and Loxl2. c-Fos, a member of the Activator Protein-1 (AP-1) transcription factor complex binds to the promoter of each of the Wnt ligands Wnt7b and Wnt9a, which is followed by expression of the collagen-modifying enzyme Loxl2 dependent on the transcription factors Zeb1 and Zeb2. c-Fos GEMMs treated with the pan-Lox inhibitor BAPN or specific Loxl2 blocking antibodies exhibit delayed OS development (148). In like manner, osteosarcoma-specific GEMMs were established utilizing an osteoblast-specific Cre allele crossed with floxed p53 or Lox-Stop-Lox (LSL) R172H-mutated p53 alleles. These GEMMs represent a closely representative model of human OS tumor development and progression. Microarray analysis of both localized and metastatic tumors developed from these models revealed downregulation of the naked cuticle homolog 2 (NKD2) gene, a negative regulator of Wnt signaling, that was found to be important for the suppression of OS tumor growth and metastasis (135). In addition, approximately 20–30% of OS tumors exhibit amplification of chromosome 8q24, the chromosomal location of the oncogene c-Myc, which correlates with a poor prognosis (30, 149, 150). Recently, an osteoblast-specific Myc knock-in OS model was generated and molecularly characterized by Nirala et al. (30). This model leads to acceleration of tumor development, with a high incidence of metastasis and homologous gene signatures seen in human tumors (30).
Ewing sarcoma is a tumor occurring only in humans, and there are no records of EwS in mice or any other species (13, 95, 151). Thus far there have been no reliable, viable murine models that recapitulate Ewing sarcoma development and progression. Several attempts at generating murine models in recent decades have either caused embryonic lethality, developmental defects, or failed to create any reliable genetically engineered mouse model for EwS that could consistently reciprocate the histological, epigenetic, or proteomic features attributed to EwS (152). The lack of a clear lineage-specific promoter confounds the issue further, even though the uniquely expressed translocated protein EWS::FLI1 is the oncogenic driver. A second set of problems arises from the fact that the cis-regulatory enhancers with GGAA microsatellites, the binding site for EWS::FLI1, are poorly conserved (13). Transgenic expression of EWS::FLI1 under a Prx1 promoter – a promoter that is activated in undifferentiated mesenchyme transitioning to osteoblasts during early differentiation in the developing limbs (40, 153), resulted in abnormal development with no tumor formation (41). Using Osterix (osteoclast-specific) or Mx1-Cre conditional models, development of erythroid or myeloid leukemias were observed, but development of sarcomas failed to occur (95, 154). Attempts with other promoters targeting EWS::FLI1 or Cre expression include Runx2, Col1a2.3, Col1a3.6, CAG, Nse, NEFL, Dermo1, P0, and Sox9 (95). Together, it would appear the co-occurrence of several factors that are yet to be discovered, including the cell of origin, the development phase, promoter leakiness, other co-factors, binding sites, other oncogenic molecules are critical for Ewing sarcoma to develop.
5.3.2 Drosophila model
Although development of a realistic model is not physiologically or anatomically permissive in Drosophila, there is a strong conservation of molecular pathways with humans. Thus, there is potential in using a Drosophila model to reproduce EWS::FLI1 regulated oncogenic pathways (Figure 3). A recent report used a spontaneous mutant variant – EWS::FLI1FS to create transgenic lines in Drosophila (42). EWS::FLI1FS is a truncated protein with six amino acid loss with a C-terminal frameshift retaining the entire EWSR1 and most of the FLI1 including the DNA binding domain. Using this construct, the group could circumvent EWS::FLI1-mediated cellular toxicity and induction of apoptosis. Proteogenomic analyses revealed EWS::FLI1FS interacts with known homologues of EWS::FLI1 interacting proteins, including transcription, chromatin remodeling, and spliceosome factors. This model could also demonstrate activation of transcription from the two types of EWS::FLI1 binding sites.
5.3.3 Zebrafish
Zebrafish (Danio rerio) has approximately 70% overlap with the human genome (155). They are almost transparent; hence, internal development can be easily observed. Hundreds of embryos that develop outside the mother’s body can be studied for a limited cost. Thus, they provide an attractive model to trace early-stage tumor development. In one study from Leacock et al., in p53-deficient embryos, transposon mediated mosaic expression of EWS::FLI1 (Figure 3) developed into peripheral nerve sheath tumors and in rare numbers, into leukemia-like or small round blue cell tumors (SRBCT), partly resembling EwS in histological features and gene expression patterns. The limitations of this model were low penetrance and the co-occurrence of other tumors (136). In a more recent study, a zebrafish model based upon Cre-inducible expression in a wild-type background reported high penetrance with rapid onset of SRBCTs. The EWS::FLI1-expressing tumors were positive for EwS markers such as CD99 and transcriptional targets such as activation of MAPK/ERK pathway (156).
5.4 Humanized murine models
In the absence of GEMMs for EwS, attempts have been made by several research groups to use humanized murine models, particularly in the context of studying tumor immunotherapy and the TME of EwS (157). In these studies, fresh human cord blood CD34+ hematopoietic stem cells were intratibially injected into 3–4 weeks old NSG-SGM3 mice along with EwS patient-derived cells. Reconstitution of T cells, B cells, natural killer (NK) cells and monocytes could be observed (157). The main caveats of humanized mouse models are the cost-prohibitory aspect along with the technical and regulatory challenges they can pose. One important disadvantage of this model includes the species difference between each cytokine and its corresponding receptor, resulting in a significantly reduced level of myeloid and NK cells due to the lack of crosstalk (158). Humanized mice have a reduced life span and can develop anemia and other pathological conditions due to an imperfect immune system (157, 159). Further advancements in this field and subsequent utilization of these models are an active area of research.
6 Applicability
The benefits of preclinical models can be directly measurable by considering that therapeutics tested using preclinical models have been approved for clinical trials and translated into the clinical setting for the treatment of patients with pediatric solid tumors. One such small molecule is cabozantinib (CBZ) in a phase II clinical trial (NCT02243605) completed for patients with relapsed EwS or OS. The cabozantinib in patients with advanced Ewing sarcoma and osteosarcoma (CABONE) phase II clinical trial, conducted as a collaboration between the National Cancer Institute (NCI) and the French Sarcoma Group, recruited patients from ten centers in the French Sarcoma Group, with 45 patients for each sarcoma. Most patients with EwS and 50% of OS patients treated with cabozantinib exhibited tumor shrinkage (160). CBZ is a tyrosine kinase inhibitor that targets multiple tyrosine kinase receptors, including vascular endothelial growth factor 2 (VEGF2), MET, and rearranged during transfection (RET) proto-oncogene (161, 162). Given that MET has been found to be overexpressed in OS, Fioramonti et al. evaluated the effect of CBZ on OS using in vitro OS cell line models or co-culture models of OS cells with bone cells to reproduce the tumor microenvironment interactions. Their results indicated that CBZ treatment could inhibit OS cell proliferation in a co-culture model system through a mechanism dependent on the expression of receptor activator of nuclear factor κB (RANK) by the OS cells. CBZ treatment had also inhibited OS proliferation and cell migration, an indicator of invasion and metastasis in tumor cells, when OS cells were cultured independently (161).
In Ewing sarcoma, attempts to directly target the tumor-specific EWS::FLI1 protein using the inhibitor TK216 in patients with recurrent/refractory EwS (NCT05046314) is now in a phase II clinical trial. TK216 is an analog of YK-4–279, an inhibitor that blocks RNA helicase A binding to EWS::FLI1, which had been identified from a drug screen. TK216 was tested in pre-clinical models to induce apoptosis in EwS cells and to inhibit tumor growth in murine xenograft models (163). From pre-clinical studies, insulin-like growth factor-1 receptor (IGF1R) was found to be an important therapeutic target in EwS (164). Although several phase I trials showed moderate effect in a small number of patients, successfully transitioning to phase II trials include the combination of IGF1R inhibitor (cixutumumab) and an mTOR inhibitor (temsirolimus) for patients with refractory EwS (NCT01614795). This trial showed a promising response rate of 29% (165). Cell line models have demonstrated a significant association between presence of EWS-FLI1 and sensitivity to PARP inhibitors such as olaparib (65). However, xenograft models identified the limitation of olaparib as a single agent while highlighting its efficacy in combination with other agents (166, 167). Similarly, clinical trials have not shown significant efficacy for the PARP inhibitors olaparib or talazoparib as single agents (NCT01583543, NCT02116777) and demonstrated moderate success in combination with irinotecan alone or with temozolomide (NCT04901702).
Many other chemotherapeutics currently in clinical trials have been previously tested and validated using pre-clinical models. Nonetheless, there are many chemotherapeutics that fail to transition from pre-clinical models into clinical trials beyond phase I. As such, there is a special importance for pre-clinical models to closely resemble the spontaneous disease in order for chemotherapeutics and targeted agents to be strong candidates for treatment options before being tested in clinical trials and introduced to patients. With this in mind, it is especially important to account for pharmacokinetically clinically equivalent drug doses with treatment of human tumor xenografts such that similar results can be observed when testing the drugs in a clinical setting (168). Studies making use of multiple types of pre-clinical models are promising so that shortfalls with one type of model can be overcome with the use of complementary models.
7 Discussion
Over the past couple of decades there has been an explosion in advancements in genomic analysis of patient tumors and identification of genetic mutations or alterations that can be candidate therapeutic targets. While these tremendous advancements in profiling patient tumors have allowed for the development of personalized medicine and a greater understanding into cancer biology, the validation of these genetic aberrations as therapeutic targets relies heavily on the development and applications of preclinical models that represent the molecular, cellular, and biophysical landscapes of the specific tumor types.
As we have outlined and described above for pediatric bone sarcomas, there has been the development and characterization of numerous in vitro and in vivo preclinical models ranging from direct patient tumor xenografts and cell lines to 3D organoid and microfluidic platforms to genetically engineered murine, zebrafish, and even Drosophila models. The establishment of these models has not only allowed further molecular profiling of patient tumors, but also the development of complementary, cross species comparative oncology studies to further identify and define key genomic features of individual cancer types that are candidate therapeutic targets and models that can be used for preclinical testing.
However, the field is still actively working towards improving these approaches in model development to enhance their representation of both tumor intrinsic and extrinsic (e.g., tumor immune microenvironment, cancer-associated fibroblasts, etc.) patient tumor biology, at different stages of their development, which will allow for more efficient and accurate identification of effective therapies. As it has been an exciting era in molecular biology and tumor genomics, it has been an equally stimulating time in biomedical engineering and preclinical animal models, with innovative technical approaches that have improved our ability to accurately model both tumor intrinsic and extrinsic tumor development and evolution. Applying the armamentarium of spatial and single cell multi-omics analysis to these models has already increased our insights into patient tumor biology. Further advancements in technology and integration of preclinical models will undoubtedly improve our ability to provide real time patient tumor profiling and testing of individualized therapies that will lead to improved outcomes for those conditions in dire need of improved therapeutic interventions.
Author contributions
DIP: Conceptualization, Visualization, Writing – original draft, Writing – review & editing, Investigation. JY: Conceptualization, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing. AD: Conceptualization, Funding acquisition, Visualization, Writing – original draft, Writing – review & editing, Investigation, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. DIP was supported by National Institutes of Health grant GM149422. JY and AD were supported by National Institutes of Health/National Cancer Institute grants NIH 1R21CA260331 and NIH 1R21CA267914.
Acknowledgments
Figures were created using BioRender.com under the Emory University School of Medicine (SOM) institutional license.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
2. Allen-Rhoades W, Whittle SB, Rainusso N. Pediatric solid tumors of infancy: an overview. Pediatr Rev. (2018) 39:57–67. doi: 10.1542/pir.2017-0057
3. Kline NE, Sevier N. Solid tumors in children. J Pediatr Nurs. (2003) 18:96–102. doi: 10.1053/jpdn.2003.12
4. Glod J, Rahme GJ, Kaur H, Raabe EH, Hwang EI, Israel MA. Pediatric brain tumors: current knowledge and therapeutic opportunities. J Pediatr Hematol Oncol. (2016) 38:249–60. doi: 10.1097/MPH.0000000000000551
5. Abedalthagafi M, Mobark N, Al-Rashed M, AlHarbi M. Epigenomics and immunotherapeutic advances in pediatric brain tumors. NPJ Precis Oncol. (2021) 5:34. doi: 10.1038/s41698-021-00173-4
6. Patterson JD, Henson JC, Breese RO, Bielamowicz KJ, Rodriguez A. CAR T cell therapy for pediatric brain tumors. Front Oncol. (2020) 10:1582. doi: 10.3389/fonc.2020.01582
7. Liu KW, Pajtler KW, Worst BC, Pfister SM, Wechsler-Reya RJ. Molecular mechanisms and therapeutic targets in pediatric brain tumors. Sci Signal. (2017) 10:1–19. doi: 10.1126/scisignal.aaf7593
8. Newman EA, Abdessalam S, Aldrink JH, Austin M, Heaton TE, Bruny J, et al. Update on neuroblastoma. J Pediatr Surg. (2019) 54:383–9. doi: 10.1016/j.jpedsurg.2018.09.004
9. Li J, Thompson TD, Miller JW, Pollack LA, Stewart SL. Cancer incidence among children and adolescents in the United States, 2001-2003. Pediatrics. (2008) 121:e1470–7. doi: 10.1542/peds.2007-2964
10. Szychot E, Apps J, Pritchard-Jones K. Wilms' tumor: biology, diagnosis and treatment. Transl Pediatr. (2014) 3:12–24. doi: 10.3978/j.issn.2224-4336.2014.01.09
11. Castillo-Tandazo W, Mutsaers AJ, Walkley CR. Osteosarcoma in the post genome era: preclinical models and approaches to identify tractable therapeutic targets. Curr Osteoporos Rep. (2019) 17:343–52. doi: 10.1007/s11914-019-00534-w
12. Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. (2018) 18:39–50. doi: 10.1080/14737140.2018.1413939
13. Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Alava E, Kovar H, et al. Ewing sarcoma. Nat Rev Dis Primers. (2018) 4:5. doi: 10.1038/s41572-018-0003-x
14. Beck J, Ren L, Huang S, Berger E, Bardales K, Mannheimer J, et al. Canine and murine models of osteosarcoma. Vet Pathol. (2022) 59:399–414. doi: 10.1177/03009858221083038
15. Dupuy M, Lamoureux F, Mullard M, Postec A, Regnier L, Baud'huin M, et al. Ewing sarcoma from molecular biology to the clinic. Front Cell Dev Biol. (2023) 11:1248753. doi: 10.3389/fcell.2023.1248753
16. Spector LG, Hubbard AK, Diessner BJ, Machiela MJ, Webber BR, Schiffman JD. Comparative international incidence of Ewing sarcoma 1988 to 2012. Int J Cancer. (2021) 149:1054–66. doi: 10.1002/ijc.33674
17. Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. (2015) 33:3036–46. doi: 10.1200/JCO.2014.59.5256
18. Nie Z, Peng H. Osteosarcoma in patients below 25 years of age: An observational study of incidence, metastasis, treatment and outcomes. Oncol Lett. (2018) 16:6502–14. doi: 10.3892/ol
19. Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. (2019) 5:1. doi: 10.1038/s41572-018-0051-2
20. Miwa S, Yamamoto N, Hayashi K, Takeuchi A, Igarashi K, Tsuchiya H. Recent advances and challenges in the treatment of rhabdomyosarcoma. Cancers. (2020) 12:1–18. doi: 10.3390/cancers12071758
21. Amer KM, Thomson JE, Congiusta D, Dobitsch A, Chaudhry A, Li M, et al. Epidemiology, incidence, and survival of rhabdomyosarcoma subtypes: SEER and ICES database analysis. J Orthop Res. (2019) 37:2226–30. doi: 10.1002/jor.24387
22. Agaram NP. Evolving classification of rhabdomyosarcoma. Histopathology. (2022) 80:98–108. doi: 10.1111/his.14449
23. Reed DR, Hayashi M, Wagner L, Binitie O, Steppan DA, Brohl AS, et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. (2017) 123:2206–18. doi: 10.1002/cncr.30589
24. Laetsch TW, DuBois SG, Bender JG, Macy ME, Moreno L. Opportunities and challenges in drug development for pediatric cancers. Cancer Discovery. (2021) 11:545–59. doi: 10.1158/2159-8290.CD-20-0779
25. Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. (2007) 49:928–40. doi: 10.1002/pbc.21078
26. Schott C, Shah AT, Sweet-Cordero EA. Genomic complexity of osteosarcoma and its implication for preclinical and clinical targeted therapies. Adv Exp Med Biol. (2020) 1258:1–19. doi: 10.1007/978-3-030-43085-6_1
27. Ottaviano L, Schaefer KL, Gajewski M, Huckenbeck W, Baldus S, Rogel U, et al. Molecular characterization of commonly used cell lines for bone tumor research: a trans-European EuroBoNet effort. Genes Chromosomes Cancer. (2010) 49:40–51. doi: 10.1002/gcc.20717
28. Rickel K, Fang F, Tao J. Molecular genetics of osteosarcoma. Bone. (2017) 102:69–79. doi: 10.1016/j.bone.2016.10.017
29. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. (2008) 22:1662–76. doi: 10.1101/gad.1656808
30. Nirala BK, Patel TD, Kurenbekova L, Shuck R, Dasgupta A, Rainusso N, et al. MYC regulates CSF1 expression via microRNA 17/20a to modulate tumor-associated macrophages in osteosarcoma. JCI Insight. (2023) 8:1–18. doi: 10.1172/jci.insight.164947
31. Zoubek A, Dockhorn-Dworniczak B, Delattre O, Christiansen H, Niggli F, Gatterer-Menz I, et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol. (1996) 14:1245–51. doi: 10.1200/JCO.1996.14.4.1245
32. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discovery. (2014) 4:1342–53. doi: 10.1158/2159-8290.CD-14-0622
33. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PloS Genet. (2014) 10:e1004475. doi: 10.1371/journal.pgen.1004475
34. Brohl AS, Sindiri S, Wei JS, Milewski D, Chou HC, Song YK, et al. Immuno-transcriptomic profiling of extracranial pediatric solid Malignancies. Cell Rep. (2021) 37:110047. doi: 10.1016/j.celrep.2021.110047
35. Orth MF, Surdez D, Faehling T, Ehlers AC, Marchetto A, Grossetete S, et al. Systematic multi-omics cell line profiling uncovers principles of Ewing sarcoma fusion oncogene-mediated gene regulation. Cell Rep. (2022) 41:111761. doi: 10.1016/j.celrep.2022.111761
36. Surdez D, Zaidi S, Grossetete S, Laud-Duval K, Ferre AS, Mous L, et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell. (2021) 39:810–26.e9. doi: 10.1016/j.ccell.2021.04.001
37. Bertin H, Guilho R, Brion R, Amiaud J, Battaglia S, Moreau A, et al. Jaw osteosarcoma models in mice: first description. J Transl Med. (2019) 17:56. doi: 10.1186/s12967-019-1807-5
38. Ory B, Heymann MF, Kamijo A, Gouin F, Heymann D, Redini F. Zoledronic acid suppresses lung metastases and prolongs overall survival of osteosarcoma-bearing mice. Cancer. (2005) 104:2522–9. doi: 10.1002/cncr.21530
39. Fan TM. Animal models of osteosarcoma. Expert Rev Anticancer Ther. (2010) 10:1327–38. doi: 10.1586/era.10.107
40. Ouyang Z, Chen Z, Ishikawa M, Yue X, Kawanami A, Leahy P, et al. Prx1 and 3.2kb Col1a1 promoters target distinct bone cell populations in transgenic mice. Bone. (2014) 58:136–45. doi: 10.1016/j.bone.2013.10.016
41. Lin PP, Pandey MK, Jin F, Xiong S, Deavers M, Parant JM, et al. EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer Res. (2008) 68:8968–75. doi: 10.1158/0008-5472.CAN-08-0573
42. Molnar C, Reina J, Herrero A, Heinen JP, Mendiz V, Bonnal S, et al. Human EWS-FLI protein recapitulates in Drosophila the neomorphic functions that induce Ewing sarcoma tumorigenesis. PNAS Nexus. (2022) 1:pgac222. doi: 10.1093/pnasnexus/pgac222
43. Beird HC, Bielack SS, Flanagan AM, Gill J, Heymann D, Janeway KA, et al. Osteosarcoma. Nat Rev Dis Primers. (2022) 8:77. doi: 10.1038/s41572-022-00409-y
44. Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. (2011) 2011:548151. doi: 10.1155/2011/548151
45. Ragland BD, Bell WC, Lopez RR, Siegal GP. Cytogenetics and molecular biology of osteosarcoma. Lab Invest. (2002) 82:365–73. doi: 10.1038/labinvest.3780431
46. Mannheimer JD, Tawa G, Gerhold D, Braisted J, Sayers CM, McEachron TA, et al. Transcriptional profiling of canine osteosarcoma identifies prognostic gene expression signatures with translational value for humans. Commun Biol. (2023) 6:856. doi: 10.1038/s42003-023-05208-z
47. Rodriguez CO Jr. Using canine osteosarcoma as a model to assess efficacy of novel therapies: can old dogs teach us new tricks? Adv Exp Med Biol. (2014) 804:237–56. doi: 10.1007/978-3-319-04843-7_13
48. Endo-Munoz L, Bennett TC, Topkas E, Wu SY, Thamm DH, Brockley L, et al. Auranofin improves overall survival when combined with standard of care in a pilot study involving dogs with osteosarcoma. Vet Comp Oncol. (2020) 18:206–13. doi: 10.1111/vco.12533
49. Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy. Nat Rev Endocrinol. (2017) 13:480–91. doi: 10.1038/nrendo.2017.16
50. Riggi N, Suva ML, Stamenkovic I. Ewing's sarcoma. N Engl J Med. (2021) 384:154–64. doi: 10.1056/NEJMra2028910
51. Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol. (2012) 7:145–59. doi: 10.1146/annurev-pathol-011110-130237
52. Granowetter L, West DC. The Ewing's sarcoma family of tumors: Ewing's sarcoma and peripheral primitive neuroectodermal tumor of bone and soft tissue. Cancer Treat Res. (1997) 92:253–308. doi: 10.1007/978-1-4615-5767-8_9
53. Esiashvili N, Goodman M, Marcus RB Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. (2008) 30:425–30. doi: 10.1097/MPH.0b013e31816e22f3
54. Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. (2003) 348:694–701. doi: 10.1056/NEJMoa020890
55. Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A. Translocation involving chromosome 22 in Ewing's sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. (1984) 12:21–5. doi: 10.1016/0165-4608(84)90003-7
56. Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir GM. Chromosome study of Ewing's sarcoma (ES) cell lines. Consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet Cytogenet. (1984) 12:1–19. doi: 10.1016/0165-4608(84)90002-5
57. Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir G. [Chromosomal translocation (11; 22) in cell lines of Ewing's sarcoma]. C R Seances Acad Sci III. (1983) 296:1101–3.
58. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. (1992) 359:162–5. doi: 10.1038/359162a0
59. Bailly RA, Bosselut R, Zucman J, Cormier F, Delattre O, Roussel M, et al. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol Cell Biol. (1994) 14:3230–41. doi: 10.1128/MCB.14.5.3230
60. Lin PP, Brody RI, Hamelin AC, Bradner JE, Healey JH, Ladanyi M. Differential transactivation by alternative EWS-FLI1 fusion proteins correlates with clinical heterogeneity in Ewing's sarcoma. Cancer Res. (1999) 59:1428–32. doi: 10.1371/journal.pone.0004932
61. Guillon N, Tirode F, Boeva V, Zynovyev A, Barillot E, Delattre O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PloS One. (2009) 4:e4932. doi: 10.1371/journal.pone.0004932
62. Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proc Natl Acad Sci U S A. (2008) 105:10149–54. doi: 10.1073/pnas.0801073105
63. Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing's sarcoma. Mol Cancer Res. (2006) 4:851–9. doi: 10.1158/1541-7786.MCR-06-0090
64. Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. (2018) 555:387–91. doi: 10.1038/nature25748
65. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. (2012) 483:570–5. doi: 10.1038/nature11005
66. Hancock JD, Lessnick SL. A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle. (2008) 7:250–6. doi: 10.4161/cc.7.2.5229
67. Kauer M, Ban J, Kofler R, Walker B, Davis S, Meltzer P, et al. A molecular function map of Ewing's sarcoma. PloS One. (2009) 4:e5415. doi: 10.1371/journal.pone.0005415
68. Fadul J, Bell R, Hoffman LM, Beckerle MC, Engel ME, Lessnick SL. EWS/FLI utilizes NKX2-2 to repress mesenchymal features of Ewing sarcoma. Genes Cancer. (2015) 6:129–43. doi: 10.18632/genesandcancer.v6i3-4
69. Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing's sarcoma. Cancer Res. (2009) 69:9047–55. doi: 10.1158/0008-5472.CAN-09-1540
70. Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem. (2009) 284:9074–82. doi: 10.1074/jbc.M806233200
71. Fukuma M, Okita H, Hata J, Umezawa A. Upregulation of Id2, an oncogenic helix-loop-helix protein, is mediated by the chimeric EWS/ets protein in Ewing sarcoma. Oncogene. (2003) 22:1–9. doi: 10.1038/sj.onc.1206055
72. Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. (2009) 106:5324–9. doi: 10.1073/pnas.0810759106
73. Yang L, Hu HM, Zielinska-Kwiatkowska A, Chansky HA. FOXO1 is a direct target of EWS-Fli1 oncogenic fusion protein in Ewing's sarcoma cells. Biochem Biophys Res Commun. (2010) 402:129–34. doi: 10.1016/j.bbrc.2010.09.129
74. Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc Natl Acad Sci U S A. (2015) 112:E1307–16. doi: 10.1073/pnas.1500536112
75. Knoop LL, Baker SJ. EWS/FLI alters 5'-splice site selection. J Biol Chem. (2001) 276:22317–22. doi: 10.1074/jbc.M008950200
76. Showpnil IA, Selich-Anderson J, Taslim C, Boone MA, Crow JC, Theisen ER, et al. EWS/FLI mediated reprogramming of 3D chromatin promotes an altered transcriptional state in Ewing sarcoma. Nucleic Acids Res. (2022) 50:9814–37. doi: 10.1093/nar/gkac747
77. Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. (2014) 26:668–81. doi: 10.1016/j.ccell.2014.10.004
78. May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A. (1993) 90:5752–6. doi: 10.1073/pnas.90.12.5752
79. May WA, Denny CT. Biology of EWS/FLI and related fusion genes in Ewing's sarcoma and primitive neuroectodermal tumor. Curr Top Microbiol Immunol. (1997) 220:143–50.
80. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, et al. The Ewing's sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. (1993) 13:7393–8. doi: 10.1128/mcb.13.12.7393
82. Kadin ME, Bensch KG. On the origin of Ewing's tumor. Cancer. (1971) 27:257–73. doi: 10.1002/(ISSN)1097-0142
83. Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing's sarcoma of bone. Am J Pathol. (1987) 127:507–18.
84. Suh CH, Ordonez NG, Hicks J, Mackay B. Ultrastructure of the Ewing's sarcoma family of tumors. Ultrastruct Pathol. (2002) 26:67–76. doi: 10.1080/01913120252959236
85. Teitell MA, Thompson AD, Sorensen PH, Shimada H, Triche TJ, Denny CT. EWS/ETS fusion genes induce epithelial and neuroectodermal differentiation in NIH 3T3 fibroblasts. Lab Invest. (1999) 79:1535–43.
86. Zhou R, Xu A, Tu J, Liu M, Gingold JA, Zhao R, et al. Modeling osteosarcoma using Li-Fraumeni syndrome patient-derived induced pluripotent stem cells. J Vis Exp. (2018) 136:1–11. doi: 10.3791/57664
87. Franchi A, Pasquinelli G, Cenacchi G, Della Rocca C, Gambini C, Bisceglia M, et al. Immunohistochemical and ultrastructural investigation of neural differentiation in Ewing sarcoma/PNET of bone and soft tissues. Ultrastruct Pathol. (2001) 25:219–25. doi: 10.1080/01913120120194
88. von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, et al. Modeling initiation of Ewing sarcoma in human neural crest cells. PloS One. (2011) 6:e19305. doi: 10.1371/journal.pone.0019305
89. Hwang S, Hameed M, Kransdorf M. The 2020 World Health Organization classification of bone tumors: what radiologists should know. Skeletal Radiol. (2023) 52:329–48. doi: 10.1007/s00256-022-04093-7
90. Lin PP, Wang Y, Lozano G. Mesenchymal stem cells and the origin of Ewing's sarcoma. Sarcoma. (2011) 2011:1–8. doi: 10.1155/2011/276463
91. Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. (2007) 11:421–9. doi: 10.1016/j.ccr.2007.02.027
92. Riggi N, Cironi L, Provero P, Suva ML, Kaloulis K, Garcia-Echeverria C, et al. Development of Ewing's sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. (2005) 65:11459–68. doi: 10.1158/0008-5472.CAN-05-1696
93. Riggi N, Suva ML, Suva D, Cironi L, Provero P, Tercier S, et al. EWS-FLI-1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. (2008) 68:2176–85. doi: 10.1158/0008-5472.CAN-07-1761
94. Tanaka M, Yamazaki Y, Kanno Y, Igarashi K, Aisaki K, Kanno J, et al. Ewing's sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J Clin Invest. (2014) 124:3061–74. doi: 10.1172/JCI72399
95. Minas TZ, Surdez D, Javaheri T, Tanaka M, Howarth M, Kang HJ, et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget. (2017) 8:34141–63. doi: 10.18632/oncotarget.v8i21
96. Allen DD, Caviedes R, Cardenas AM, Shimahara T, Segura-Aguilar J, Caviedes PA. Cell lines as in vitro models for drug screening and toxicity studies. Drug Dev Ind Pharm. (2005) 31:757–68. doi: 10.1080/03639040500216246
97. Lamhamedi-Cherradi SE, Santoro M, Ramammoorthy V, Menegaz BA, Bartholomeusz G, Iles LR, et al. 3D tissue-engineered model of Ewing's sarcoma. Adv Drug Delivery Rev. (2014) 79-80:155–71. doi: 10.1016/j.addr.2014.07.012
98. Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. (1973) 51:1417–23. doi: 10.1093/jnci/51.5.1417
99. Shen M, Asawa R, Zhang YQ, Cunningham E, Sun H, Tropsha A, et al. Quantitative high-throughput phenotypic screening of pediatric cancer cell lines identifies multiple opportunities for drug repurposing. Oncotarget. (2018) 9:4758–72. doi: 10.18632/oncotarget.v9i4
100. Mohseny AB, MaChado I, Cai Y, Schaefer KL, Serra M, Hogendoorn PC, et al. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab Invest. (2011) 91:1195–205. doi: 10.1038/labinvest.2011.72
101. Miki Y, Ono K, Hata S, Suzuki T, Kumamoto H, Sasano H. The advantages of co-culture over mono cell culture in simulating in vivo environment. J Steroid Biochem Mol Biol. (2012) 131:68–75. doi: 10.1016/j.jsbmb.2011.12.004
102. Salvatore V, Teti G, Bolzani S, Focaroli S, Durante S, Mazzotti MC, et al. Simulating tumor microenvironment: changes in protein expression in an in vitro co-culture system. Cancer Cell Int. (2014) 14:40. doi: 10.1186/1475-2867-14-40
103. Jeong SY, Lee JH, Shin Y, Chung S, Kuh HJ. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PloS One. (2016) 11:e0159013. doi: 10.1371/journal.pone.0159013
104. Mendoza A, Hong SH, Osborne T, Khan MA, Campbell K, Briggs J, et al. Modeling metastasis biology and therapy in real time in the mouse lung. J Clin Invest. (2010) 120:2979–88. doi: 10.1172/JCI40252
105. Minkin C. Bone acid phosphatase: tartrate-resistant acid phosphatase as a marker of osteoclast function. Calcif Tissue Int. (1982) 34:285–90. doi: 10.1007/BF02411252
106. Taylor R, Knowles HJ, Athanasou NA. Ewing sarcoma cells express RANKL and support osteoclastogenesis. J Pathol. (2011) 225:195–202. doi: 10.1002/path.2869
107. Chaicharoenaudomrung N, Kunhorm P, Noisa P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J Stem Cells. (2019) 11:1065–83. doi: 10.4252/wjsc.v11.i12.1065
108. Godugu C, Patel AR, Desai U, Andey T, Sams A, Singh M. AlgiMatrix based 3D cell culture system as an in-vitro tumor model for anticancer studies. PloS One. (2013) 8:e53708. doi: 10.1371/journal.pone.0053708
109. Lawlor ER, Scheel C, Irving J, Sorensen PH. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene. (2002) 21:307–18. doi: 10.1038/sj.onc.1205053
110. He A, Huang Y, Cheng W, Zhang D, He W, Bai Y, et al. Organoid culture system for patient-derived lung metastatic osteosarcoma. Med Oncol. (2020) 37:105. doi: 10.1007/s12032-020-01429-y
111. Valente KP, Khetani S, Kolahchi AR, Sanati-Nezhad A, Suleman A, Akbari M. Microfluidic technologies for anticancer drug studies. Drug Discovery Today. (2017) 22:1654–70. doi: 10.1016/j.drudis.2017.06.010
112. Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater Today. (2015) 18:539–53. doi: 10.1016/j.mattod.2015.05.002
113. He J, Xiong L, Li Q, Lin L, Miao X, Yan S, et al. 3D modeling of cancer stem cell niche. Oncotarget. (2018) 9:1326–45. doi: 10.18632/oncotarget.v9i1
114. Molina ER, Chim LK, Salazar MC, Koons GL, Menegaz BA, Ruiz-Velasco A, et al. 3D tissue-engineered tumor model for Ewing's sarcoma that incorporates bone-like ECM and mineralization. ACS Biomater Sci Eng. (2020) 6:539–52. doi: 10.1021/acsbiomaterials.9b01068
115. Bassi G, Panseri S, Dozio SM, Sandri M, Campodoni E, Dapporto M, et al. Scaffold-based 3D cellular models mimicking the heterogeneity of osteosarcoma stem cell niche. Sci Rep. (2020) 10:22294. doi: 10.1038/s41598-020-79448-y
116. Mittal R, Woo FW, Castro CS, Cohen MA, Karanxha J, Mittal J, et al. Organ-on-chip models: Implications in drug discovery and clinical applications. J Cell Physiol. (2019) 234:8352–80. doi: 10.1002/jcp.27729
117. Lu Z, Miao X, Zhang C, Sun B, Skardal A, Atala A, et al. An osteosarcoma-on-a-chip model for studying osteosarcoma matrix-cell interactions and drug responses. Bioact Mater. (2024) 34:1–16. doi: 10.1016/j.bioactmat.2023.12.005
118. Zhang T, Li J, Yang M, Ma X, Wang Z, Ma X, et al. CDK7/GRP78 signaling axis contributes to tumor growth and metastasis in osteosarcoma. Oncogene. (2022) 41:4524–36. doi: 10.1038/s41388-022-02446-z
119. Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harb Protoc. (2014) 2014:694–708. doi: 10.1101/pdb.top073585
120. Masuda J, Shigehiro T, Matsumoto T, Satoh A, Mizutani A, Umemura C, et al. Cytokine expression and macrophage localization in xenograft and allograft tumor models stimulated with lipopolysaccharide. Int J Mol Sci. (2018) 19:1–12. doi: 10.3390/ijms19041261
121. Ireson CR, Alavijeh MS, Palmer AM, Fowler ER, Jones HJ. The role of mouse tumour models in the discovery and development of anticancer drugs. Br J Cancer. (2019) 121:101–8. doi: 10.1038/s41416-019-0495-5
122. Di Fiore R, Guercio A, Puleio R, Di Marco P, Drago-Ferrante R, D'Anneo A, et al. Modeling human osteosarcoma in mice through 3AB-OS cancer stem cell xenografts. J Cell Biochem. (2012) 113:3380–92. doi: 10.1002/jcb.24214
123. Ek ET, Dass CR, Choong PF. Commonly used mouse models of osteosarcoma. Crit Rev Oncol Hematol. (2006) 60:1–8. doi: 10.1016/j.critrevonc.2006.03.006
124. Cidre-Aranaz F, Ohmura S. Tumor growth analysis of Ewing sarcoma cell lines using subcutaneous xenografts in mice. Methods Mol Biol. (2021) 2226:191–9. doi: 10.1007/978-1-0716-1020-6
125. Cerra C, Harris MA, Hawkins CJ. Establishment and characterisation of metastatic extraskeletal Ewing sarcoma mouse models. In Vivo. (2021) 35:3097–106. doi: 10.21873/invivo.12604
126. Li M, Chen C. Regulation of metastasis in Ewing sarcoma. Cancers. (2022) 14:1–15. doi: 10.3390/cancers14194902
127. Scopim-Ribeiro R, Lizardo MM, Zhang HF, Dhez AC, Hughes CS, Sorensen PH. NSG Mice Facilitate ex vivo Characterization of Ewing Sarcoma Lung Metastasis Using the PuMA Model. Front Oncol. (2021) 11:645757. doi: 10.3389/fonc.2021.645757
128. Dasgupta A, Kurenbekova L, Patel TD, Rajapakshe K, Ghosal G, Nirala B, et al. Modeling Ewing sarcoma lung metastasis. Curr Protoc. (2023) 3:e670. doi: 10.1002/cpz1.670
129. Horbach L, Sinigaglia M, Da Silva CA, Olguins DB, Gregianin LJ, Brunetto AL, et al. Gene expression changes associated with chemotherapy resistance in Ewing sarcoma cells. Mol Clin Oncol. (2018) 8:719–24. doi: 10.3892/mco
130. Conn E, Hour S, Allegakoen D, Graham G, Petro J, Kouassi-Brou M, et al. Development of an Ewing sarcoma cell line with resistance to EWS−FLI1 inhibitor YK−4−279. Mol Med Rep. (2020) 21:1667–75. doi: 10.3892/mmr
131. May WA, Grigoryan RS, Keshelava N, Cabral DJ, Christensen LL, Jenabi J, et al. Characterization and drug resistance patterns of Ewing's sarcoma family tumor cell lines. PloS One. (2013) 8:e80060. doi: 10.1371/journal.pone.0080060
132. Shapiro MC, Tang T, Dasgupta A, Kurenbekova L, Shuck R, Gaber MW, et al. In vitro and in vivo characterization of a preclinical irradiation-adapted model for Ewing sarcoma. Int J Radiat Oncol Biol Phys. (2018) 101:118–27. doi: 10.1016/j.ijrobp.2018.01.095
133. Loh AHP, Stewart E, Bradley CL, Chen X, Daryani V, Stewart CF, et al. Combinatorial screening using orthotopic patient derived xenograft-expanded early phase cultures of osteosarcoma identify novel therapeutic drug combinations. Cancer Lett. (2019) 442:262–70. doi: 10.1016/j.canlet.2018.10.033
134. Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med. (2017) 9:137–53. doi: 10.15252/emmm.201606857
135. Zhao S, Kurenbekova L, Gao Y, Roos A, Creighton CJ, Rao P, et al. NKD2, a negative regulator of Wnt signaling, suppresses tumor growth and metastasis in osteosarcoma. Oncogene. (2015) 34:5069–79. doi: 10.1038/onc.2014.429
136. Leacock SW, Basse AN, Chandler GL, Kirk AM, Rakheja D, Amatruda JF. A zebrafish transgenic model of Ewing's sarcoma reveals conserved mediators of EWS-FLI1 tumorigenesis. Dis Model Mech. (2012) 5:95–106. doi: 10.1242/dmm.007401
137. Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. (2017) 549:96–100. doi: 10.1038/nature23647
138. Higuchi T, Igarashi K, Yamamoto N, Hayashi K, Kimura H, Miwa S, et al. Osteosarcoma patient-derived orthotopic xenograft (PDOX) models used to identify novel and effective therapeutics: A review. Anticancer Res. (2021) 41:5865–71. doi: 10.21873/anticanres.15406
139. Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. (2014) 87:150–61. doi: 10.1016/j.bcp.2013.06.020
140. Surdez D, Landuzzi L, Scotlandi K, Manara MC. Ewing sarcoma PDX models. Methods Mol Biol. (2021) 2226:223–42. doi: 10.1007/978-1-0716-1020-6_18
141. Nanni P, Landuzzi L, Manara MC, Righi A, Nicoletti G, Cristalli C, et al. Bone sarcoma patient-derived xenografts are faithful and stable preclinical models for molecular and therapeutic investigations. Sci Rep. (2019) 9:12174. doi: 10.1038/s41598-019-48634-y
142. Rainusso N, Cleveland H, Hernandez JA, Quintanilla NM, Hicks J, Vasudevan S, et al. Generation of patient-derived tumor xenografts from percutaneous tumor biopsies in children with bone sarcomas. Pediatr Blood Cancer. (2019) 66:e27579. doi: 10.1002/pbc.27579
143. Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. (2001) 84:1424–31. doi: 10.1054/bjoc.2001.1796
144. Goldstein SD, Hayashi M, Albert CM, Jackson KW, Loeb DM. An orthotopic xenograft model with survival hindlimb amputation allows investigation of the effect of tumor microenvironment on sarcoma metastasis. Clin Exp Metastasis. (2015) 32:703–15. doi: 10.1007/s10585-015-9738-x
145. Castillo-Ecija H, Pascual-Pasto G, Perez-Jaume S, Resa-Pares C, Vila-Ubach M, Monterrubio C, et al. Prognostic value of patient-derived xenograft engraftment in pediatric sarcomas. J Pathol Clin Res. (2021) 7:338–49. doi: 10.1002/cjp2.210
146. Domenici G, Eduardo R, Castillo-Ecija H, Orive G, Montero Carcaboso A, Brito C. PDX-derived Ewing's sarcoma cells retain high viability and disease phenotype in alginate encapsulated spheroid cultures. Cancers. (2021) 13:1–18. doi: 10.3390/cancers13040879
147. Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol. (2011) 6:95–119. doi: 10.1146/annurev.pathol.3.121806.154244
148. Matsuoka K, Bakiri L, Wolff LI, Linder M, Mikels-Vigdal A, Patino-Garcia A, et al. Wnt signaling and Loxl2 promote aggressive osteosarcoma. Cell Res. (2020) 30:885–901. doi: 10.1038/s41422-020-0370-1
149. De Noon S, Ijaz J, Coorens TH, Amary F, Ye H, Strobl A, et al. MYC amplifications are common events in childhood osteosarcoma. J Pathol Clin Res. (2021) 7:425–31. doi: 10.1002/cjp2.219
150. Marinoff AE, Spurr LF, Fong C, Li YY, Forrest SJ, Ward A, et al. Clinical targeted next-generation panel sequencing reveals MYC amplification is a poor prognostic factor in osteosarcoma. JCO Precis Oncol. (2023) 7:e2200334. doi: 10.1200/PO.22.00334
151. Sharkey FE, Fogh J. Incidence and pathological features of spontaneous tumors in athymic nude mice. Cancer Res. (1979) 39:833–9.
152. Ramachandran B, Rajkumar T, Gopisetty G. Challenges in modeling EWS-FLI1-driven transgenic mouse model for Ewing sarcoma. Am J Transl Res. (2021) 13:12181–94.
153. Kuratani S, Martin JF, Wawersik S, Lilly B, Eichele G, Olson EN. The expression pattern of the chick homeobox gene gMHox suggests a role in patterning of the limbs and face and in compartmentalization of somites. Dev Biol. (1994) 161:357–69. doi: 10.1006/dbio.1994.1037
154. Torchia EC, Boyd K, Rehg JE, Qu C, Baker SJ. EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol Cell Biol. (2007) 27:7918–34. doi: 10.1128/MCB.00099-07
155. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. (2013) 496:498–503. doi: 10.1038/nature12111
156. Vasileva E, Warren M, Triche TJ, Amatruda JF. Dysregulated heparan sulfate proteoglycan metabolism promotes Ewing sarcoma tumor growth. Elife. (2022) 11. doi: 10.7554/eLife.69734
157. Luo W, Hoang H, Liao Y, Pan J, Ayello J, Cairo MS. A humanized orthotopic mouse model for preclinical evaluation of immunotherapy in Ewing sarcoma. Front Immunol. (2023) 14:1277987. doi: 10.3389/fimmu.2023.1277987
158. Tian H, Lyu Y, Yang YG, Hu Z. Humanized rodent models for cancer research. Front Oncol. (2020) 10:1696. doi: 10.3389/fonc.2020.01696
159. Brehm MA, Shultz LD, Luban J, Greiner DL. Overcoming current limitations in humanized mouse research. J Infect Dis. (2013) 208 Suppl 2:S125–30. doi: 10.1093/infdis/jit319
160. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2020) 21:446–55. doi: 10.1016/S1470-2045(19)30825-3
161. Fioramonti M, Fausti V, Pantano F, Iuliani M, Ribelli G, Lotti F, et al. Cabozantinib affects osteosarcoma growth through A direct effect on tumor cells and modifications in bone microenvironment. Sci Rep. (2018) 8:4177. doi: 10.1038/s41598-018-22469-5
162. Hattinger CM, Patrizio MP, Magagnoli F, Luppi S, Serra M. An update on emerging drugs in osteosarcoma: towards tailored therapies? Expert Opin Emerg Drugs. (2019) 24:153–71. doi: 10.1080/14728214.2019.1654455
163. Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med. (2009) 15:750–6. doi: 10.1038/nm.1983
164. Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J Biol Chem. (1997) 272:30822–7. doi: 10.1074/jbc.272.49.30822
165. Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res. (2012) 18:2625–31. doi: 10.1158/1078-0432.CCR-12-0061
166. Smith MA, Reynolds CP, Kang MH, Kolb EA, Gorlick R, Carol H, et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res. (2015) 21:819–32. doi: 10.1158/1078-0432.CCR-14-2572
167. Norris RE, Adamson PC, Nguyen VT, Fox E. Preclinical evaluation of the PARP inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatr Blood Cancer. (2014) 61:145–50. doi: 10.1002/pbc.24697
Keywords: preclinical models, cancer therapy, pediatric cancer, rare cancer, solid tumors, bone sarcomas, Ewing sarcoma, osteosarcoma
Citation: Petrescu DI, Yustein JT and Dasgupta A (2024) Preclinical models for the study of pediatric solid tumors: focus on bone sarcomas. Front. Oncol. 14:1388484. doi: 10.3389/fonc.2024.1388484
Received: 19 February 2024; Accepted: 01 July 2024;
Published: 18 July 2024.
Edited by:
Joanna Kitlinska, Georgetown University, United StatesReviewed by:
Massimo Broggini, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyFrédéric Lézot, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2024 Petrescu, Yustein and Dasgupta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason T. Yustein, amFzb24ueXVzdGVpbkBlbW9yeS5lZHU=; Atreyi Dasgupta, YXRyZXlpLmRhc2d1cHRhQGJjbS5lZHU=