 Mariusz A. Wasik
Mariusz A. Wasik Patricia M. Kim
Patricia M. Kim Reza Nejati
Reza Nejati
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 03 April 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1383741
While normal B- and T-lymphocytes require antigenic ligands to become activated via their B- and T-cell receptors (BCR and TCR, respectively), B- and T-cell lymphomas show the broad spectrum of cell activation mechanisms regarding their dependence on BCR or TCR signaling, including loss of such dependence. These mechanisms are generally better understood and characterized for B-cell than for T-cell lymphomas. While some lymphomas, particularly the indolent, low-grade ones remain antigen-driven, other retain dependence on activation of their antigen receptors seemingly in an antigen-independent manner with activating mutations of the receptors playing a role. A large group of lymphomas, however, displays complete antigen receptor independence, which can develop gradually, in a stepwise manner or abruptly, through involvement of powerful oncogenes. Whereas some of the lymphomas undergo activating mutations of genes encoding proteins involved in signaling cascades downstream of the antigen-receptors, others employ activation mechanisms capable of substituting for these BCR- or TCR-dependent signaling pathways, including reliance on signaling pathways physiologically activated by cytokines. Finally, lymphomas can develop cell-lineage infidelity and in the extreme cases drastically rewire their cell activation mechanisms and engage receptors and signaling pathways physiologically active in hematopoietic stem cells or non-lymphoid cells. Such profound reprograming may involve partial cell dedifferentiation or transdifferentiation towards histocytes, dendritic, or mesodermal cells with various degree of cell maturation along these lineages. In this review, we elaborate on these diverse pathogenic mechanisms underlying cell plasticity and signaling reprogramming as well as discuss the related diagnostic and therapeutic implications and challenges.
An unprecedented progress in molecular technologies enabled broad and in-depth studies of normal and malignant cells and tissues. These studies provided us with detailed insights into the mechanisms of carcinogenesis, permitting novel approaches to management of patients with the whole spectrum of malignancies, with lymphomas being a prime example. It also revealed considerable diversity of oncogenic mechanisms as well as a remarkable ability of malignant cells to reprogram intracellular signaling pathways to sustain their survival and proliferation. An in-depth understanding of pathogenic mechanisms activating and sustaining transformed cells at any given stage of lymphoid malignancy, including recognition of their plasticity resulting in reprogramming cell-signaling dependency during lymphoma progression or at its recurrence, should lead to precisely targeted and highly individualized therapies.
As described in-depth elsewhere (1, 2), antigen-driven activation of the BCR and TCR with engagement of downstream signaling pathways is central for proper function, survival, development, and proliferation of normal B- and T-cells. Antigen dependence has been implicated in the pathogenesis of both B-cell (Figure 1) and T-cell lymphomas (Figure 2), particularly the ones from the indolent, low-grade B-cell lymphoma category. The Helicobacter pylori (H. pylori)-driven gastric mucosa-associated lymphoid tissue (MALT) B-cell lymphoma represents the prototypical example of an antigen-dependent lymphoma. Accordingly, therapeutic eradication of H. pylori, results in gastric MALT remission in the majority of patients.
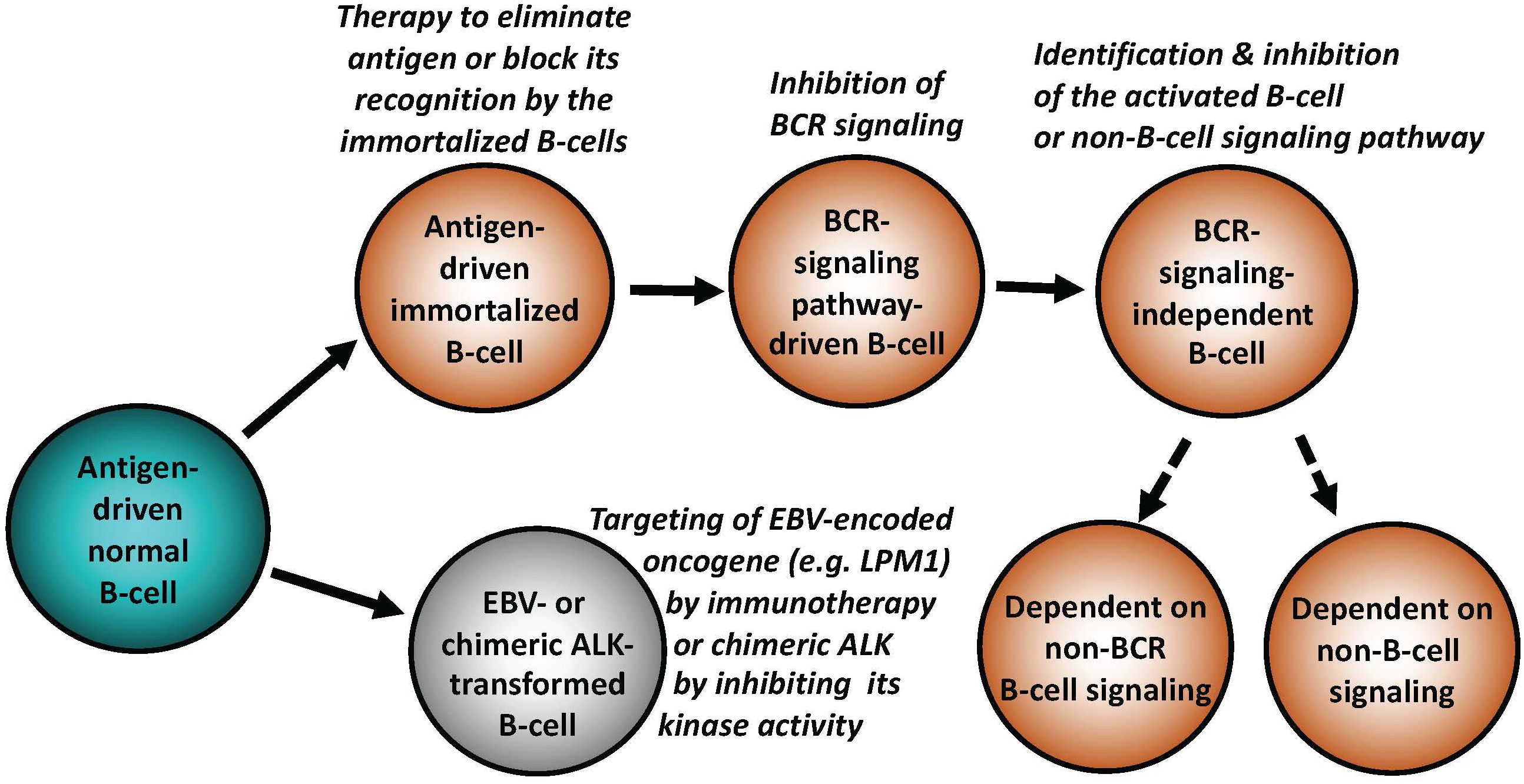
Figure 1 Key mechanisms of malignant cell transformation in B-cell lymphomas. While indolent (“low-grade”) B-cell lymphomas frequently remain antigen dependent, more advanced lymphomas relay on BCR in the antigen-independent manner, signaling pathways down-stream of BCR, non-BCR signaling pathways physiologically activated in normal mature or immature B lymphocytes, or, in extreme cases, develop reliance on signaling pathways normally activated in non-lymphoid types of mesenchymal cells. EBV and chimeric ALK (typically CLTC::ALK for B cells) are able to potently transform normal B lymphocytes. Types of targeted therapies may need to be adjusted to the specific B cell-transforming mechanisms, as listed.
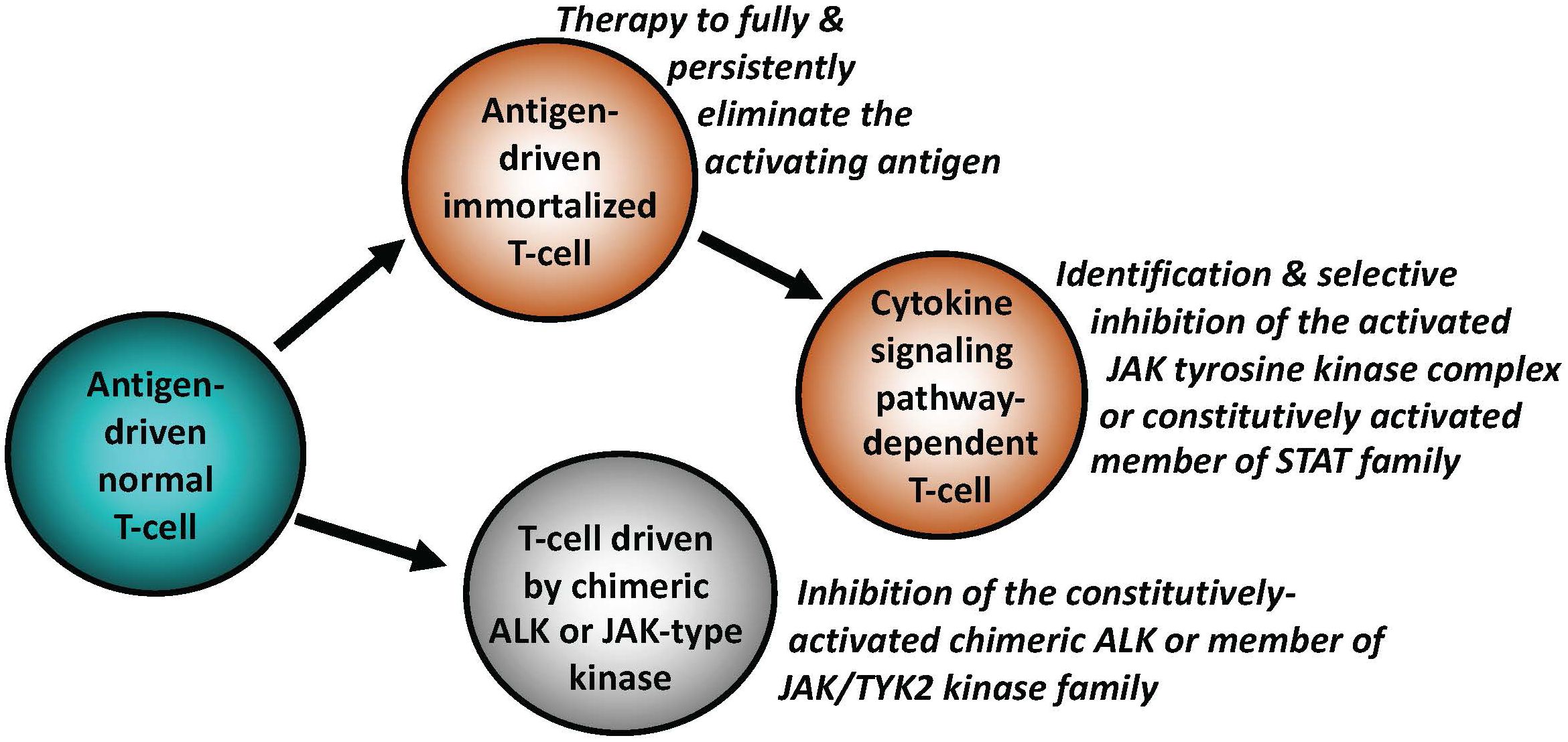
Figure 2 Key mechanisms of malignant cell transformation in T-cell lymphomas. Similar to indolent B-cell lymphomas, a subset of T-cell lymphomas remains antigen-dependent. The antigen-independence can be gained by T cells by activating signaling pathways downstream of cytokine receptors, physiologically required for activation and functional maturation of normal T cells. This occurs foremost through structural genetic alterations of the members of JAK and/or STAT families, resulting in their constitutive activation. Chimeric ALK (typically NPM1::ALK) is able to transform normal T cells by effectively substituting for the persistent JAK signaling. Similar to B-cell lymphomas, the types of targeted therapies may need to be adjusted to the specific T cell-transforming mechanisms, as listed.
The role of gluten in the development of celiac disease and the related enteropathy-associated T-cell lymphoma (EATL) of the small intestine is another example of antigen-dependent lymphoma. However, gluten elimination in EATL appears therapeutically less effective than H. pylori eradication in gastric MALT using antibiotics, possibly because ingestion of gluten in even trace amount may be sufficient to sustain survival of aberrant T cells. Accordingly, ype I of celiac disease considered to be refractory to gluten elimination from the diet (RCD) is characterized by a polyclonal proliferation of seemingly phenotypically and genetically normal T cells, while type II RCD represents a clonal expansion of phenotypically altered T-cell clones, with frequent activating mutations of JAK-STAT pathway (3). These striking differences suggest that the former may, indeed, be a form of aberrant, allergic-type immune response to traces of gluten and the latter may represent progression to a genuine low-grade EATL.
Other lymphomas, documented or postulated to be antigen-driven, include chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) responding to non-muscle myosin heavy chain IIA (4), LDL receptor-related protein-associated protein 1 (LRPAP1)-stimulated mantle cell lymphoma (MCL) (5), hepatitis C virus (HCV)-related splenic marginal zone lymphoma (SMZL), Borrelia burgdorferi-dependent primary cutaneous marginal zone lymphoma (PCMZL), Chlamydia psittaci-activated MALT of ocular adnexa, and Moraxella catarrhalis-promoted nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) (6). The evidence of ongoing immunoglobulin gene somatic hypermutations, preferential selection of specific MHC molecules, and formation of lymphoepithelial lesions in extra nodal, epithelia-associated lymphomas, all further indicate a pathogenic role of antigens in certain types of lymphoma.
In analogy to the curative effects of H. pylori elimination in gastric MALT, identification of the pathogenic antigen in a patient with a type of lymphoma in which antigen-dependence is highly likely, may pave the way for therapy aimed at the elimination, or at least blocking, as it may be the case with autoantigens, of such lymphomagenic antigen (Figures 1, 2).
BCR-mediated cell signaling in the absence of identifiable antigenic ligand gained most attention in the context of diffuse large B-cell lymphoma (DLBCL) (2). Chronic active BCR signaling is highly characteristic of the activated B-cell/non-germinal center (ABC/non-GC) subset of DLBCL. Accordingly, activating mutations of the BCR component CD79B (Y196H) and intracytoplasmic signaling molecule MYD88 (L265P) are a hallmark of the molecularly-defined MCD/C5 subtype within this DLBCL category. It is theorized that these mutations disrupt endocytic internalization of the BCR and permit spontaneous assembly of the signaling myddosome complex resulting in chronic activation of the down-stream signaling pathways, foremost NF-KB pathway. Of note, activating mutations of CARD11, a member of a key signaling complex downstream of the BCR, have also been identified in DLBCL, most likely leading to BCR independence. Accordingly, in mouse models mutated CARD 11 drives B cells into dysregulated plasmablastic differentiation via NF-KB and JNK activation, independently of BCR.
The presence of activating mutations of BCR and its down-stream signaling pathways, particularly the MYD88 mutation, have promising direct therapeutic implications. In early studies, patients with the MCD/(C5) subtype of DLBCL treated with the combination of standard R-CHOP immunochemotherapy and ibrutinib, a first-generation inhibitor of Bruton’s tyrosine kinase (BTK), the key signaling molecule down-stream of BCR, demonstrated long-lasting remissions (7).
It is generally underappreciated that the highly-transformed (high-grade) lymphomas such as DLBCL can undergo cell-signaling reprogramming due to additional mutations of BCR and members of its down-stream signaling pathways, including evidence of development of resistance to the previously applied targeted therapy (8). Consequently, molecular evaluation of DLBCL and other types of lymphoma, both at presentation, and at other clinical stages, particularly after post-therapy recurrence, may provide important insights into the mechanism(s) of lymphoma progression and reveal new therapeutic targets. The increasing accessibility to the extensive DNA and RNA sequencing using formalin-fixed paraffin-embedded tissues, allows for comparative analysis of serial samples from individual patients. This may further characterize the phenomenon of lymphoma reprogramming (9), and enable individualized therapy based on tumor-specific molecular status at a given disease stage (Figure 1).
The growing body of evidence shows that lymphoma cells are capable of becoming fully independent of BCR-mediated intracellular signaling and develop alternative cell-activation triggering mechanisms. Receptor tyrosine kinase-like orphan receptor 1 (ROR1), emerges as a prime example of this kind of lymphoma-cell reprogramming. ROR1, a cell-surface receptor with kinase structure, is normally expressed in the fetal liver and, likely, by cells in the parathyroid, pancreatic islets, adipose tissue, and gastrointestinal tract in adults (10). In hematopoietic cells, its expression is limited to early B-cell precursors (hematogones). Importantly, ROR1 expression has been identified in lymphoid malignancies derived from mature B lymphocytes including CLL/SLL, hairy cell leukemia (HCL), MCL, DLBCL, and follicular lymphoma (FL). ROR1 forms a complex with the CD19, a cell-surface receptor physiologically down-stream of the BCR signaling pathway. ROR1-CD19 complex activates key intracellular signaling pathways (10), including PI3K-AKT, MEK-ERK, and NF-kB (10, 11), effectively substituting for the BCR-induced cell signaling. Ectopic ROR1 expression has relevant translational implications as a target for immunotherapy using either ROR1 antibody-drug conjugate (12), ROR1-CD3 bispecific engager (13), or ROR1-targeting CAR T cells (14). In ongoing clinical trials with the anti-ROR1 CAR T-cells, no significant toxicities to normal tissues have been noted (10).
Furthermore, by bypassing BCR activation, ROR1 leads to BTK-inhibitor resistance in preclinical models of MCL (11). While the existence of this mechanism of resistance to BTK inhibition remains to be confirmed in lymphoma patients, monitoring ROR1 expression may become an important component of managing lymphomas treated with a BTK inhibitor. The need for ROR1 expression monitoring may be particularly applicable to MCL, given that mutations of BTK or its down-stream target PLCγ leading to BTK inhibitor resistance are extremely rare in MCL, in contrast to SLL/CL in which the mutations are highly prevalent (14). Overexpression of transcription factor EGR1 resulting in metabolic reprogramming to oxidative phosphorylation (OXPHOS) has also been recently implicated in BTK-inhibitor resistance in MCL and ABC subtype of DLBCL (15). Of note, malignant cells isolated from such lymphomas become sensitive to OXPHOS inhibitors, suggesting that targeting OXPHOS and EGR1, either separately or in combination, may prove efficacious in patients with lymphomas overexpressing EGR1.
In addition to ROR1, other aberrantly expressed cell-surface receptors may play a pathogenic role in B-cell lymphomas by at least partially substituting for BCR-generated cell signaling. For example, a functional interplay between mutated NOTCH1 and BCR has been identified in CLL/SLL and in ~1/3 of CLL/SLL cases which have undergone large-cell transformation (Richter transformation: RT) (16). This finding suggests that NOTCH1 plays a role in the disease progression and, consequently, may become a therapeutic target, particularly in RT, the clinically aggressive type of lymphoma with poor prognosis. NOTCH1 mutations have been found also in ~1/3 cases of the clinically aggressive blastoid variant of MCL (17), further suggesting a link between NOTCH1 and lymphomas with high-grade features. Furthermore, increased expression of Toll-like receptors (TLR) including TLR9 as well as of WNT ligands, mostly WNT5a, has been identified in a subset of RT cases, in the context of decreased BCR expression (18). This suggests that TLR and WNT signaling compensate for diminished BCR signaling and, consequently, may become therapeutic targets. Finally, AXL, a cell-surface receptor with an intrinsic tyrosine kinase activity, has been implicated in BCR-independent cell signaling (19) and drug resistance (20) in CLL/SLL. Of note, a recent study (21) reports that constitutively activated AXL3 variant is expressed in MCL and its inhibition has proven therapeutically effective in preclinical in vitro and in vivo models.
Although rare, the development of DLBCL with phenotypic and genomic evidence of immaturity following the mature B-cell neoplasms: FL and DLBCL, has been now firmly recognized (22–25). These DLBCL/lymphoblastic B-cell lymphoma/leukemia (LBLL) “chimeras” typically are TdT+, CD10+, CD45low, and cell-surface Ig-negative, attesting to their at least partially immature phenotype. Strikingly all these chimeric TdT+ DLCBCL/LBLL cases reported to date acquired the c-MYC gene translocation, which indicates a critical role of the highly overexpressed c-MYC in this transformation. Translocations of BCL-2 and/or BCL-6 in both pre-and post-dedifferentiation stages are another common feature, which raises the possibility that defects in double-stranded DNA break repair contribute to the emergence of DLCBCL/LBLL. Accordingly, three cases of TdT+ DLBCL/LBLL demonstrated expression of RAG1 (25), the gene encoding for an enzyme inducing DNA breaks to initiate immunoglobulin gene V(D)J recombination in immature B cells.
An in-depth molecular analysis of TdT+ DLBCL/LBLL cases has established a clonal relationship with preceding them FL and DBCL, by both shared rearrangement of immunoglobulin heavy chain (IgH) gene and identical mutations of other genes, with additional mutations seen at the DLBCL/LBLL stage (22–25). Of note, all TdT+ DLBCL/LBLL cases tested so far (22, 23) have displayed somatic hypermutations of the IgH gene, indicating that these lymphomas have indeed dedifferentiated from mature lymphoma cells, rather than have originated from a common, immature malignant progenitor B cell. This unique entity of DLBCL/LBLL poses substantial diagnostic and clinical challenges, as it has a phenotype to large degree mimicking acute B-cell lymphoblastic leukemia (B-ALL) and responds poorly to diverse therapy protocols, including those used for B-ALL.
Transdifferentiation is a striking example of lineage plasticity in malignant cells. Although this phenomenon was originally identified over two decades ago with the description of FL turning into histiocytic/dendritic cell neoplasms (HDCN) (26), it attracted recently much more attention, in the era of targeted kinase-inhibitor and chimeric antigen receptor (CAR) T-cell-based immunotherapy (27). While initially recognized solely by IgH gene rearrangements shared between the preceding FL and ensuing HDCN, recent advances in DNA and RNA sequencing have enabled the identification of clonal relationship between lymphoma and co-occurring or following HDCN, based also on shared profiles of gene translocations and mutations of non-IgH genes (27).
There are currently well-documented cases of HDCN transdifferentiating from not only FL but also CLL/SLL, MCL, MZL, DLBCL (27), and Burkitt lymphoma (BL) (28) with transdifferentiation from FL and CLL/CLL appearing to be the most common. The substantial genomic mutational heterogeneity in these ‘secondary’ HDCN’s likely stems at least in part from the high diversity among the underlying lymphomas. However, in spite of their mutational heterogeneity, comprehensive DNA sequencing studies of these HDCN revealed frequent mutations in members of the MAPK pathway, including KRAS (6/16 cases examined), BRAF (4/16), and MAPK2K1 (3/16). This raises the possibility that MAPK pathway components may become novel therapeutic targets in a large subset of the transdifferentiated HDCN (27).
In addition to morphing into HDCN, transdifferentiation of MCL to poorly differentiated sarcoma with evidence of neuromuscular differentiation has been reported (29, 30), which demonstrates even more striking lineage plasticity potential of malignant lymphocytes. Genome-scale shift in DNA methylation was identified in the sarcoma vs. the preceding MCL cells with >12,000 gene promoters profoundly reprogramming their methylation pattern (29). This sweeping change was associated with silencing of genes involved in B-cell differentiation and partial activation of differentiation programs towards neural and muscle cells (29). While the underlying mechanisms of this profound cell reprograming remain unclear, mutations of retinoblastoma 1 (RB1) and TP53 genes were identified in these transdifferentiated sarcoma cases (29, 30). It is quite intriguing in this context that mutations of these two genes were also detected in poorly differentiated neural tumors transdifferentiated from carcinomas of lung (31) and prostate (32), occurring in response to EGFR inhibition and anti-androgen signaling therapy, respectively. Therefore, it could be surmised that the RB1 and TP53 mutations may play a key role in transdifferentiation of lymphoid and non-lymphoid neoplasms towards neural cell lineage.
Upon transitioning from MCL to sarcoma, the transdifferentiated sarcoma cells gained expression of several non-lymphoid cell receptors and displayed sensitivity to pharmacologic inhibition of one of these receptors, namely FGFR (29). This finding combined with the epigenetics-driven cell reprogramming indicates emergence of newly acquired cell-growth dependency of the transdifferentiated cells. It could be potentially exploited therapeutically in the patient setting by targeting DNA methylation machinery and the new oncogenic drivers such as FGFR. Of note, FGFR1 expression has been recently identified also in advanced MCL, including BTK inhibitor-resistant cases (33). Furthermore, FGFR1 inhibition has shown efficacy in pre-clinical in vitro and in vivo MCL models (33), suggesting that FGFR1 may become a therapeutic target in this type of lymphoma, even without evidence of overt transdifferentiation.
The triggering events leading to transdifferentiation remain mostly unclear. While the lineage change may be spontaneous, therapeutic pressures are more likely to underlie, or accelerate, the process. In analogy to the above-mentioned carcinomas of the lung and prostate transdifferentiating in response to therapies targeting EGFR (31) and androgen (32) signaling, respectively, targeted therapies may be responsible for lymphoma reprogramming from the dependence on BCR signaling. Prolonged exposure to kinase inhibitors such as BTK inhibitors as seen in the MCL case morphed into sarcoma (29), could certainly be a factor promoting BCR/BTK independence including transdifferentiation and a related profound cell-signaling reprograming, as a very drastic mechanism of drug resistance.
CAR T-cell therapy targeting CD19 (CART19) may also play a role in lymphoma reprograming including transdifferentiation by enabling outgrowth of the preexisting non-lymphoid subclone, due to the CART19-mediated eradication of the dominant lymphoma clone expressing CD19 (29). Of interest, comparative analysis of pre- and post-CART19 therapy tissue samples collected from nine patients with high-grade B-cell lymphomas who failed this immunotherapy, revealed not only at least a partial decrease in CD19 expression by eight of the lymphomas but also less frequently seen losses of other B-cell markers CD79A, CD20, and PAX5 (34). These findings suggest a genuine loss of mature B-cell identity in some of the cases, likely promoted by CART19-mediated selection of the preexisting aberrant subclones with at least partial reprogramming away from the B-cell lineage. Accordingly, histiocytic differentiation of a subset of the malignant B cells was noted upon relapse in one case and T-cell differentiation in another one (34), indicating that CART19 therapy can indeed occasionally enable outgrowth of cells displaying transdifferentiation towards a non-B-cell lineage. Finally, the relapsed tumors acquired a number of mutations in genes not typically altered in B-cell lymphomas such as KRAS, MAP2K2, PIK3R1, and PIK3R2, further supporting their loss of B-cell identity and suggesting that targeting PI3K and KRAS in such cases may prove therapeutically effective.
While the establishment of an overt lymphoid malignancy is almost universally a stepwise process, lymphomas expressing anaplastic lymphoma kinase (ALK) and, seemingly, similarly powerful oncogenic kinases (35–37) are a notable exception (Figure 2). ALK, typically expressed as the NPM1-ALK (NPM1::ALK) hybrid protein due to t(2;5) chromosomal translocation resulting in fusion of the involved genes, profoundly reprograms targeted CD4+ T cells by conferring upon these cells features required for a full-fledged malignant cell transformation (38, 39). These NPM1::ALK-imposed oncogenic features are achieved to a large degree through activation of cell signaling pathways triggered in normal T cells by cytokines, foremost activation of transcriptional regulator STAT3 (35). These oncogenic properties include cell immortalization and protection from cell death, enabled at least in part by activation of stem cell-related genes (40), induction of multi-faceted inhibition of anti-tumor immune response (35), epigenetic silencing of TCR- and cytokine-signaling genes acting in the context of NPM1::ALK as tumor suppressors (35), protection from hypoxia (35), and mobilization of the metabolic NAD synthesis salvage pathway to sustain enzymatic activity of the oncogenic ALK (41). Given the above diverse pro-oncogenic effects of NPM1::ALK, it is not surprising that targeting ALK clinically induces complete, long-lasting remissions in response to crizotinib, the first generation ALK inhibitor (42, 43). However, resistance to ALK-inhibition monotherapy has emerged (44), even to the second- and third-generation, highly potent ALK inhibitors, providing rationale for a combination therapy to prevent development of the resistance. ALK inhibition already has, or could be, combined with either standard chemotherapy (45), CD30 antibody-toxin conjugate (46), or, preferably, agents targeting pathways playing a role in ALK-driven lymphomas (35). Similar to NPM1::ALK, another form of chimeric ALK involving clathrin (CLTC::ALK) which is expressed in a distinct subtype of B-cell lymphoma displaying plasmablastic cell morphology and immunophenotype, is also highly responsive to the next generation ALK inhibitors (47), further highlighting the potent oncogenic role of chimeric ALK, regardless of the T- vs. B-cell lineage of the transformed lymphocytes (Figures 1, 2).
ALK-negative anaplastic large cell lymphomas (ALCL) are clinically and molecularly quite heterogeneous. Based on their presentation, they are classified as systemic, primary cutaneous, and breast implant-associated (48). Despite their heterogeneity, common pathogenic mechanisms have emerged. Accordingly, rearrangements of chromosome 6p25.3 involving DUSP22 and IRF4 gene region characterize distinct subset of both cutaneous and systemic ALK-negative ALCL lacking STAT3 activation (48, 49). These chromosome 6p25.3 rearrangements confer an overall better prognosis, according to some studies (48, 49). In other ALK-negative ALCL cases, translocations and related persistent expression of genes encoding tyrosine kinases, to some degree functionally analogous to ALK, has been identified (36, 37, 48). While one of these ALK-like kinases ROS1, is structurally closely related to ALK, the other two:TYK2 and JAK2 are members of the JAK tyrosine kinase family. They all activate similar signaling pathways, foremost STAT3, considered the key transcriptional effector for these chimeric oncogenic kinases (37). Of note, systemic and cutaneous ALCL lacking any of these chromosomal translocations and the related constitutively active fusion kinases, frequently display activating point mutations of JAK1, JAK3, and/or STAT3 (37). Similar point mutations of these genes occur also in breast implant-associated ALCL (48). These findings further point towards the importance of the signaling pathways physiologically activated in normal T cells by certain cytokines for the entire category of ALCL.
Mutation-driven aberrant activation of selected JAKs and STATs has also emerged as a common denominator for T-cell lymphomas beyond ALCL. For example, in addition to the pathognomonic alterations of the TCL1 gene, T-cell prolymphocytic leukemia (T-PLL) frequently displays point mutations of JAK1 (6.3% of cases), JAK3 (36.4%), and STAT5B (18.8%) (50). Furthermore, ~70% of cases show losses of negative regulators of JAKs and STATs including SHP1, SOCS1, and SOCS3 (50), further stressing pathogenic role of constitutively active JAK and STAT signaling in T-PLL. A large subset (40%) of NK-cell large granular lymphocytic leukemia (LGL) harbors mutations of STAT3 gene, the same percentage of T-cell LGL (T-LGL) cases shows mutations of STAT3 and STAT5B genes (51). Mutations of these two oncogenes are also frequently seen in NK and γδ T-cell lymphomas (37). In turn, cytotoxic CD8+ T-cell lymphomas of skin, particularly the primary cutaneous, aggressive and epidermotropic cytotoxic T-cell lymphoma (PCAECyTCL) show alterations of predominantly JAK2 but also JAK3, STAT5B, and SOCS1 genes, among other genomic changes (52, 53). Similarly, a subset of monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) was found to frequently carry mutations in STAT5B (57%), JAK3 (50%), and JAK1 (12.5%). in addition to deletion or expression loss of the SETD2 gene (90%) (54), encoding the histone H3 lysine 36 histone (H3K36) methyltransferase with tumor suppressor gene features. Finally, treatment with ruxolitinib, the kinase inhibitor preferentially impairing activity JAK1 and JAK2, showed therapeutic efficacy against diverse T-cell lymphomas, particularly the ones with evidence of JAK/STAT mutations or even STAT3 activation, foremost in T-LGL (55). These clinical results further indicate oncogenic role of the above-listed JAKs and STATs in the T-cell malignancies and support the notion that the oncogenic forms of JAK family members emerge as attractive therapeutic targets in the whole spectrum of mature T-cell lymphomas and leukemias (Figure 2). Mutated and, hence, constitutively activated members of the STAT family such as STAT3 and STAT5B could also become important therapeutic targets with a number of their direct inhibitors already developed. However, clinical efficacy of these inhibitors still remains to be convincingly proven.
EBV-driven lymphoproliferative disorders, mostly of B-cell origin (56), are another example of a seemingly one-step oncogenesis (Figure 1) in at least some cases with a number of the EBV-encoded genes playing a role in the malignant cell transformation (57). The dominant EBV-encoded oncogenes belong to the late membrane protein (LMP) and Epstein-Barr nuclear antigen (EBNA) families; the former is largely responsible for the considerable immunogenicity of the EBV-dependent lymphoproliferations. This strong immunogenicity limits the occurrence of these disorders to immune-compromised individuals, unless the immunogenicity is greatly diminished. As seen in BL, this can occur via gene translocation-triggered constitutive activation of MYC and mutation of other oncogenes, permitting down-regulation of the presumably highly immunogenic EBV-encoded oncogenic drivers such as LMP1 and LMP2. EBV succeeds in transforming target cells to a large degree by engaging signaling pathways, normally activated by BCR (58) and, hence, by effectively hijacking the physiological mechanisms of B-cell activation. Recent study (59) identified in EBV-positive DLBCL relatively frequent mutations of SOCS1, NOTCH2, and NOTCH1 genes, suggesting that these mutated genes may have a pathogenic role in this type of lymphoid malignancy. While current therapies for in transplant patients affected by the EBV-driven lymphoproliferative disorders typically include tempering immunosuppressive regimens, often combined with CD20-targeting immunotherapy, or multiagent immunochemotherapy (60), the conceptually most advanced clinical trials focus on immunotherapy with EBV-detecting cytotoxic T cells (61) LMP-targeting CAR T cells (62), or anti-EBV vaccines (62).
In summary, in-depth molecular analysis of lymphomas including relatively limited longitudinal studies of tissue samples from consecutive biopsies obtained from the same patients, has revealed remarkable plasticity of malignant lymphoid cells. Lymphomas employ the entire spectrum of signaling mechanisms for survival and growth, spanning from antigen dependence to antigen receptor-dependence in an antigen-independent manner, to reliance on signaling pathways activated independently of BCR (Figure 1) or TCR (Figure 2). While the advanced B-cell lymphomas often depend on mutations of BCR or members of its down-stream signaling pathways, T-cell lymphomas tend to develop activating mutations of selected JAKs and STATs, normally activated by cytokines critical for T-cell development and function. Furthermore, malignant lymphocytes are capable of profound cell-signaling rewiring leading in the most extreme cases to transdifferentiation, i.e. loss of their original lymphoid lineage identity and at least partial acquisition of differentiation towards a different, non-lymphoid mesodermal lineage. The frequency of lymphoma reprogramming is expected to grow in the era of targeted therapies, particularly in monotherapy targeting a member of BCR-dependent cell signaling pathways. An in-depth understanding of these diverse and sometimes complex cell-activating mechanisms including identification of related biomarkers is required for the development of effective and highly individualized therapeutic interventions based not only on the pathogenic features of a given lymphoma at clinical presentation but also at its recurrence.
MW: Conceptualization, Writing – original draft, Writing – review & editing. PK: Writing – original draft, Writing – review & editing. RN: Investigation, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by grants from National Cancer Institute 1R01CA250102 and 1R01CA268601, and funds from the Fox Chase Cancer Center Institute for Cancer Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. (2018) 18:148–67. doi: 10.1038/nrc.2017.121
2. Young RM, Phelan JD, Wilson WH, Staudt LM. Pathogenic B-cell receptor signaling in lymphoid Malignancies: New insights to improve treatment. Immunol Rev. (2019) 291:190–213. doi: 10.1111/imr.12792
3. Soderquist CR, Bhagat G. Cellular and molecular bases of refractory celiac disease. Int Rev Cell Mol Biol. (2021) 358:207–40. doi: 10.1016/bs.ircmb.2020.12.001
4. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. (2013) 34:592–601. doi: 10.1016/j.it.2013.07.002
5. Thurner L, Hartmann S, Fadle N, Kemele M, Bock T, Bewarder M, et al. LRPAP1 is a frequent proliferation-inducing antigen of BCRs of mantle cell lymphomas and can be used for specific therapeutic targeting. Leukemia. (2019) 33:148–58. doi: 10.1038/s41375-018-0182-1
6. Thurner L, Hartmann S, Neumann F, Hoth M, Stilgenbauer S, Küppers R, et al. Role of specific B-cell receptor antigens in lymphomagenesis. Front Oncol. (2020) 10:604685. doi: 10.3389/fonc.2020.604685
7. Wilson WH, Wright GW, Huang DW, Hodkinson B, Balasubramanian S, Fan Y, et al. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell. (2021) 39:1643–53.e3. doi: 10.1016/j.ccell.2021.10.006
8. Kim PM, Nejati R, Lu P, Thakkar D, Mackrides N, Dupoux V, et al. Leukemic presentation and progressive genomic alterations of MCD/C5 diffuse large B-cell lymphoma (DLBCL). Cold Spring Harb Mol Case Stud. (2023) 9. doi: 10.1101/mcs.a006283
9. Wasik MA, Dogan A. Reports from the 2021 SH/EAHP workshop on transformation and transdifferentiation in lymphoid Malignancies. Am J Clin Pathol. (2023) 159:519–21. doi: 10.1093/ajcp/aqac169
10. Kipps TJ. ROR1: an orphan becomes apparent. Blood. (2022) 140:1583–91. doi: 10.1182/blood.2021014760
11. Zhang Q, Wang HY, Liu X, Nunez-Cruz S, Jillab M, Melnikov O, et al. Cutting edge: ROR1/CD19 receptor complex promotes growth of mantle cell lymphoma cells independently of the B cell receptor-BTK signaling pathway. J Immunol. (2019) 203:2043–8. doi: 10.4049/jimmunol.1801327
12. Wang M, Mei M, Barr PM, Barrientos J, de Vos S, Furman R, et al. Zilovertamab vedotin (MK-2140) for the treatment of non-Hodgkin lymphoma: the phase 1 dose escalation and cohort expansion WAVELINE-001 study of an anti-ROR1 antibody-drug conjugate. HemaSphere. (2022) 6):1116–7. doi: 10.1097/01.HS9.0000847788.96420.63
13. Qi J, Li X, Peng H, Cook EM, Dadashian EL, Wiestner A, et al. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc Natl Acad Sci U.S.A. (2018) 115:E5467–E76. doi: 10.1073/pnas.1719905115
14. Bonfiglio S, Sutton LA, Ljungström V, Capasso A, Pandzic T, Weström S, et al. BTK and PLCG2 remain unmutated in one-third of patients with CLL relapsing on ibrutinib. Blood Adv. (2023) 7:2794–806. doi: 10.1182/bloodadvances.2022008821
15. Liu Y, Kimpara S, Hoang NM, Daenthanasanmak A, Li Y, Lu L, et al. EGR1-mediated metabolic reprogramming to oxidative phosphorylation contributes to ibrutinib resistance in B-cell lymphoma. Blood. (2023) 142:1879–94. doi: 10.1182/blood.2023020142
16. Arruga F, Bracciamà V, Vitale N, Vaisitti T, Gizzi K, Yeomans A, et al. Bidirectional linkage between the B-cell receptor and NOTCH1 in chronic lymphocytic leukemia and in Richter's syndrome: therapeutic implications. Leukemia. (2020) 34:462–77. doi: 10.1038/s41375-019-0571-0
17. Khanlari M, Mo H, Kim DH, Sakhdari A, Young KH, Jain P, et al. Blastoid and pleomorphic mantle cell lymphoma demonstrate distinct clinicopathologic and genetic features. Am J Surg Pathol. (2023) 47:849–58. doi: 10.1097/PAS.0000000000002068
18. Nadeu F, Royo R, Massoni-Badosa R, Playa-Albinyana H, Garcia-Torre B, Duran-Ferrer M, et al. Detection of early seeding of Richter transformation in chronic lymphocytic leukemia. Nat Med. (2022) 28:1662–71. doi: 10.1038/s41591-022-01927-8
19. Sinha S, Boysen JC, Chaffee KG, Kabat BF, Slager SL, Parikh SA, et al. Chronic lymphocytic leukemia cells from ibrutinib treated patients are sensitive to Axl receptor tyrosine kinase inhibitor therapy. Oncotarget. (2018) 9:37173–84. doi: 10.18632/oncotarget.v9i98
20. Sinha S, Secreto CR, Boysen JC, Lesnick C, Wang Z, Ding W, et al. Upregulation of AXL and β-catenin in chronic lymphocytic leukemia cells cultured with bone marrow stroma cells is associated with enhanced drug resistance. Blood Cancer J. (2021) 11:37. doi: 10.1038/s41408-021-00426-2
21. Gelebart P, Eriksen Gjerstad M, Benjaminsen S, Han J, Karlsen I, Safont MM, et al. Inhibition of a new AXL isoform, AXL3, induces apoptosis of mantle cell lymphoma cells. Blood. (2023) 142:1478–93. doi: 10.1182/blood.2022015581
22. Belman JP, Meng W, Wang HY, Li J, Strauser HT, Rosenfeld AM, et al. Dramatic increase in gene mutational burden after transformation of follicular lymphoma into TdT. Cold Spring Harb Mol Case Stud. (2020) 6. doi: 10.1101/mcs.a004614
23. Nie K, Redmond D, Eng KW, Zhang T, Cheng S, Mathew S, et al. Mutation landscape, clonal evolution pattern, and potential pathogenic pathways in B-lymphoblastic transformation of follicular lymphoma. Leukemia. (2021) 35:1203–8. doi: 10.1038/s41375-020-01014-2
24. Bhavsar S, Liu YC, Gibson SE, Moore EM, Swerdlow SH. Mutational landscape of TdT+ Large B-cell lymphomas supports their distinction from B-lymphoblastic neoplasms: A multiparameter study of a rare and aggressive entity. Am J Surg Pathol. (2022) 46:71–82. doi: 10.1097/PAS.0000000000001750
25. Duffield AS, Dogan A, Amador C, Cook JR, Czader M, Goodlad JR, et al. Progression of follicular lymphoma and related entities: Report from the 2021 SH/EAHP Workshop. Am J Clin Pathol. (2023) 159:572–97. doi: 10.1093/ajcp/aqad042
26. Feldman AL, Arber DA, Pittaluga S, Martinez A, Burke JS, Raffeld M, et al. Clonally related follicularlymphomas and histiocytic/dendritic cell sarcomas:evidence for transdifferentiation of the follicular lymphoma clone. Blood. (2008) 111:5433–9. doi: 10.1182/blood-2007-11-124792
27. Xiao W, Amador C, Cook JR, Czader M, Dave S, Dogan A, et al. B-cell lineage neoplasms transdifferentiating into histiocytic/dendritic cell neoplasms: diversity, differentiation lineage, genomic alterations, and therapy: Report from the 2021 SH/EAHP Workshop. Am J Clin Pathol. (2023) 159:522–37. doi: 10.1093/ajcp/aqad022
28. Etancelin P, Boussaid I. Two sides of the same coin: transdifferentiation from Burkitt lymphoma to histiocytic sarcoma. Blood. (2023) 142:1576. doi: 10.1182/blood.2023021833
29. Zhang Q, Orlando EJ, Wang HY, Bogusz AM, Liu X, Lacey SF, et al. Transdifferentiation of lymphoma into sarcoma associated with profound reprogramming of the epigenome. Blood. (2020) 136:1980–3. doi: 10.1182/blood.2020005123
30. Kawashima I, Oishi N, Kasai K, Inoue T, Hosokawa E, Nakadate A, et al. Transdifferentiation of mantle cell lymphoma into sarcoma with limited neuromuscular differentiation after conventional chemotherapy. Virchows Arch. (2022) 480:1101–5. doi: 10.1007/s00428-021-03148-9
31. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. (2015) 6:6377. doi: 10.1038/ncomms7377
32. Aggarwal RR, Quigley DA, Huang J, Zhang L, Beer TM, Rettig MB, et al. Whole-genome and transcriptional analysis of treatment-emergent small-cell neuroendocrine prostate cancer demonstrates intraclass heterogeneity. Mol Cancer Res. (2019) 17:1235–40. doi: 10.1158/1541-7786.MCR-18-1101
33. Sircar A, Singh S, Xu-Monette ZY, Coyle KM, Hilton LK, Chavdoula E, et al. Exploiting the fibroblast growth factor receptor-1 vulnerability to therapeutically restrict the MYC-EZH2-CDKN1C axis-driven proliferation in Mantle cell lymphoma. Leukemia. (2023) 37:2094–106. doi: 10.1038/s41375-023-02006-8
34. Laurent C, Syrykh C, Hamon M, Adélaïde J, Guille A, Escudié F, et al. Resistance of B-cell lymphomas to CAR T-cell therapy is associated with genomic tumor changes which can result in transdifferentiation. Am J Surg Pathol. (2022) 46:742–53. doi: 10.1097/PAS.0000000000001834
35. Werner MT, Zhao C, Zhang Q, Wasik MA. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood. (2017) 129:823–31. doi: 10.1182/blood-2016-05-717793
36. Fitzpatrick MJ, Massoth LR, Marcus C, Vergilio JA, Severson E, Duncan D, et al. JAK2 rearrangements are a recurrent alteration in CD30+ Systemic T-cell lymphomas with anaplastic morphology. Am J Surg Pathol. (2021) 45:895–904. doi: 10.1097/PAS.0000000000001708
37. Piccaluga PP, Cascianelli C, Inghirami G. Tyrosine kinases in nodal peripheral T-cell lymphomas. Front Oncol. (2023) 13:1099943. doi: 10.3389/fonc.2023.1099943
38. Zhang Q, Wei F, Wang HY, Liu X, Roy D, Xiong QB, et al. The potent oncogene NPM-ALK mediates Malignant transformation of normal human CD4(+) T lymphocytes. Am J Pathol. (2013) 183:1971–80. doi: 10.1016/j.ajpath.2013.08.030
39. Pawlicki JM, Cookmeyer DL, Maseda D, Everett JK, Wei F, Kong H, et al. NPM-ALK-induced reprogramming of mature TCR-stimulated T cells results in dedifferentiation and Malignant transformation. Cancer Res. (2021) 81:3241–54. doi: 10.1158/0008-5472.CAN-20-2297
40. Zhang Q, Wang HY, Nayak A, Nunez-Cruz S, Slupianek A, Liu X, et al. Induction of transcriptional inhibitor HES1 and the related repression of tumor-suppressor TXNIP are important components of cell-transformation program imposed by oncogenic kinase NPM-ALK. Am J Pathol. (2022) 192:1186–98. doi: 10.1016/j.ajpath.2022.05.005
41. Zhang Q, Basappa J, Wang HY, Nunez-Cruz S, Lobello C, Wang S, et al. Chimeric kinase ALK induces expression of NAMPT and selectively depends on this metabolic enzyme to sustain its own oncogenic function. Leukemia. (2023) 37:2436–47. doi: 10.1038/s41375-023-02038-0
42. Brugières L, Cozic N, Houot R, Rigaud C, Sibon D, Arfi-Rouche J, et al. Efficacy and safety of crizotinib in ALK-positive systemic anaplastic large-cell lymphoma in children, adolescents, and adult patients: results of the French AcSé-crizotinib trial. Eur J Cancer. (2023) 191:112984. doi: 10.1016
43. Rindone G, Aroldi A, Bossi E, Verga L, Zambrotta G, Tarantino S, et al. A monocentric analysis of the long-term safety and efficacy of crizotinib in relapsed/refractory ALK+ lymphomas. Blood Adv. (2023) 7:314–6. doi: 10.1182/bloodadvances.2022007538
44. Wang Y, He J, Xu M, Xue Q, Zhu C, Liu J, et al. Holistic view of ALK TKI resistance in ALK-positive anaplastic large cell lymphoma. Front Oncol. (2022) 12:815654. doi: 10.3389/fonc.2022.815654
45. Lowe EJ, Reilly AF, Lim MS, Gross TG, Saguilig L, Barkauskas DA, et al. Crizotinib in combination with chemotherapy for pediatric patients with ALK+ Anaplastic large-cell lymphoma: the results of children's oncology group trial ANHL12P1. J Clin Oncol. (2023) 41:2043–53. doi: 10.1200/JCO.22.00272
46. Wang R, Li L, Duan A, Li Y, Liu X, Miao Q, et al. Crizotinib enhances anti-CD30-LDM induced antitumor efficacy in NPM-ALK positive anaplastic large cell lymphoma. Cancer Lett. (2019) 448:84–93. doi: 10.1016/j.canlet.2019.02.002
47. Soumerai JD, Rosenthal A, Harkins S, Duffy J, Mecca C, Wang Y, et al. Next-generation ALK inhibitors are highly active in ALK-positive large B-cell lymphoma. Blood. (2022) 140:1822–6. doi: 10.1182/blood.2022015443
48. Vega F, Amador C, Chadburn A, Hsi ED, Slack G, Medeiros LJ, et al. Genetic profiling and biomarkers in peripheral T-cell lymphomas: current role in the diagnostic work-up. Mod Pathol. (2022) 35:306–18. doi: 10.1038/s41379-021-00937-0
49. Sibon D, Bisig B, Bonnet C, Poullot E, Bachy E, Cavalieri D, et al. ALK-negative anaplastic large cell lymphoma with DUSP22 rearrangement has distinctive disease characteristics with better progression-free survival: a LYSA study. Haematologica. (2023) 108:1590–603. doi: 10.3324/haematol.2022.281442
50. Wahnschaffe L, Braun T, Timonen S, Giri AK, Schrader A, Wagle P, et al. JAK/STAT-activating genomic alterations are a hallmark of T-PLL. Cancers (Basel). (2019) 11:1833. doi: 10.3390/cancers11121833
51. Teramo A, Barilà G, Calabretto G, Vicenzetto C, Gasparini VR, Semenzato G, et al. Insights into genetic landscape of large granular lymphocyte leukemia. Front Oncol. (2020) 10:152. doi: 10.3389/fonc.2020.00152
52. Lee K, Evans MG, Yang L, Ng S, Snowden C, Khodadoust M, et al. Primary cytotoxic T-cell lymphomas harbor recurrent targetable alterations in the JAK-STAT pathway. Blood. (2021) 138:2435–40. doi: 10.1182/blood.2021012536
53. Bastidas Torres AN, Cats D, Out-Luiting JJ, Fanoni D, Mei H, Venegoni L, et al. Deregulation of JAK2 signaling underlies primary cutaneous CD8. Haematologica. (2022) 107:702–14. doi: 10.3324/haematol.2020.274506
54. Veloza L, Cavalieri D, Missiaglia E, Ledoux-Pilon A, Bisig B, Pereira B, et al. Monomorphic epitheliotropic intestinal T-cell lymphoma comprises morphologic and genomic heterogeneity impacting outcome. Haematologica. (2023) 108:181–95. doi: 10.3324/haematol.2022.281226
55. Moskowitz AJ, Ghione P, Jacobsen E, Ruan J, Schatz JH, Noor S, et al. A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of JAK/STAT targeting in T-cell lymphomas. Blood. (2021) 138:2828–37. doi: 10.1182/blood.2021013379
56. Quintanilla-Martinez L, Swerdlow SH, Tousseyn T, Barrionuevo C, Nakamura S, Jaffe ES. New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders. Virchows Arch. (2023) 482:227–44. doi: 10.1007/s00428-022-03414-4
57. Dierickx D, Pociupany M, Natkunam Y. Epstein-Barr virus-associated posttransplant lymphoproliferative disorders: new insights in pathogenesis, classification and treatment. Curr Opin Oncol. (2022) 34:413–21. doi: 10.1097/CCO.0000000000000885
58. Fish K, Comoglio F, Shaffer AL 3rd, Ji Y, Pan KT, Scheich S, et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc Natl Acad Sci U.S.A. (2020) 117:26318–27. doi: 10.1073/pnas.2007946117
59. Frontzek F, Staiger AM, Wullenkord R, Grau M, Zapukhlyak M, Kurz KS, et al. Molecular profiling of EBV associated diffuse large B-cell lymphoma. Leukemia. (2023) 37:670–9. doi: 10.1038/s41375-022-01804-w
60. Atallah-Yunes SA, Salman O, Robertson MJ. Post-transplant lymphoproliferative disorder: Update on treatment and novel therapies. Br J Haematol. (2023) 201:383–95. doi: 10.1111
61. Heslop HE, Sharma S, Rooney CM. Adoptive T-cell therapy for epstein-barr virus-related lymphomas. J Clin Oncol. (2021) 39:514–24. doi: 10.1200/JCO.20.01709
Keywords: B-cell lymphoma, T-cell lymphoma, pathogenic signaling plasticity, dedifferentiation, transdifferentiation
Citation: Wasik MA, Kim PM and Nejati R (2024) Diverse and reprogrammable mechanisms of malignant cell transformation in lymphocytes: pathogenetic insights and translational implications. Front. Oncol. 14:1383741. doi: 10.3389/fonc.2024.1383741
Received: 07 February 2024; Accepted: 18 March 2024;
Published: 03 April 2024.
Edited by:
Manlio Ferrarini, University of Genoa, ItalyReviewed by:
Andrea Nicola Mazzarello, University of Genoa, ItalyCopyright © 2024 Wasik, Kim and Nejati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariusz A. Wasik, TWFyaXVzei5XYXNpa0BmY2NjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.