
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 11 April 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1383730
 Polina Bellman1Jesus D. Gonzalez-Lugo1
Polina Bellman1Jesus D. Gonzalez-Lugo1 Moazzam Shahzad1,2
Moazzam Shahzad1,2 Muhammad Kashif Amin1
Muhammad Kashif Amin1 Muhammad Fareed Khalid1
Muhammad Fareed Khalid1 Nahid Suleman1
Nahid Suleman1 Nausheen Ahmed1
Nausheen Ahmed1 Anurag K. Singh1Abdulraheem Yacoub1
Anurag K. Singh1Abdulraheem Yacoub1 Da Zhang3
Da Zhang3 Joseph P. McGuirk1
Joseph P. McGuirk1 Muhammad Umair Mushtaq1*
Muhammad Umair Mushtaq1*Vacuoles, E1 syndrome, X-linked, autoinflammatory, somatic (VEXAS) syndrome is a chronic inflammatory disorder that affects various organ systems. It is associated with hematologic malignancies and is generally refractory to therapies. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) may be considered for selected patients. We report a case wherein systemic and hematological manifestations completely resolved in a patient with VEXAS and associated myelodysplastic syndrome (MDS), following the administration of fludarabine and cyclophosphamide as part of the preparation for allo-HSCT. We conducted a systematic literature review and included 86 patients with VEXAS syndrome and associated MDS. Most cases presented with musculoskeletal involvement (71%) and anemia (72%) with lower-risk MDS. Most patients responded to corticosteroids (CS) but had a recurrence of symptoms with CS taper and were refractory to other immunosuppressive agents. Hypomethylating agents and Janus kinase inhibitors achieved a complete response in some cases. Further research is needed to develop more effective treatment strategies.
Vacuoles, E1 syndrome, X-linked, autoinflammatory, somatic (VEXAS) syndrome is a recently reported pathological entity that presents in late adulthood with an inflammatory syndrome, fevers, cytopenias, dysplastic bone marrow, and characteristic cytoplasmic vacuoles in erythroid and myeloid precursors. It is caused by myeloid-restricted somatic missense mutations in ubiquitin-like modifier activating enzyme 1 (UBA1), which is an X-linked gene encoding for the E1 enzyme that initiates ubiquitination of proteins (1). The diagnosis of VEXAS syndrome requires the identification of UBA1 mutations by deoxyribonucleic acid (DNA) sequencing.
It has been estimated that VEXAS syndrome occurs in 1 out of every 4269 men older than 50 years and 1 in 26238 women older than 50 years (2). Most patients with VEXAS syndrome meet clinical criteria for inflammatory syndromes, such as relapsing polychondritis, Sweet syndrome, polyarteritis nodosa, and giant-cell arteritis, among others. Hematological manifestations commonly include Myelodysplastic Syndromes (MDS) or Plasma Cell Dyscrasias, with few other hematological conditions reported in the literature (3–10).
There is no current standard of care treatment for VEXAS syndrome. The inflammatory features can be treated with corticosteroids, immunosuppressants, and in some cases, hematopoietic stem cell transplant (HSCT). Herein, we present a case of successful treatment of VEXAS syndrome and related myelodysplastic syndrome (MDS), achieving a complete response with fludarabine and cyclophosphamide as part of the conditioning regimen in preparation for HSCT. We also conducted a systematic literature review to summarize current evidence regarding clinical presentation, hematological findings, treatment, and outcomes of VEXAS syndrome with associated MDS.
A 66-year-old man with numerous inflammatory manifestations for over 20 years, including recurrent scleritis, relapsing polychondritis, Graves’ disease, bursitis, pyoderma gangrenosum, and leukocytoclastic vasculitis, was evaluated for pancytopenia notable for a white blood cell (WBC) count of 2030/uL with the absolute neutrophil count (ANC) count of 1430/uL, anemia with hemoglobin (Hgb) 9.1 g/dL, mean corpuscular volume of 115 fl and thrombocytopenia with a platelet (PLT) count of 119,000/uL. He had recurrent worsening pancytopenia during inflammatory crises presenting with persistent fatigue, shortness of breath, cough, and fever. On two occasions, his cytopenias worsened to the point of requiring transfusions (initially packed red blood cells (pRBCs), and two months later PLT), though this improved with steroids. For his autoimmune and inflammatory conditions, he had received multiple lines of therapy at an outside institution, including corticosteroids (consistently on prednisone >20 years), methotrexate, dapsone, hydroxychloroquine (several years), rituximab 1 g every two weeks for two courses, adalimumab for a few weeks, tocilizumab (received two monthly infusions but stopped due to worsening symptoms). Several bone marrow biopsies had been performed showing mild dyspoiesis and deletion 20q, increasing in number of cells involved over 10 years from 5% to 24.5%. A bone marrow biopsy was repeated and showed a hypercellular bone marrow with trilineage dyspoiesis, vacuolation of myeloid and erythroid precursor cells, and 1% blasts. Fluorescent in situ hybridization (FISH) and cytogenetics showed deletion 20q with otherwise normal male karyotype. Further testing confirmed a pathogenic Met41Thr (c.122 T>C) mutation in the UBA1 gene consistent with VEXAS Syndrome. Hypomethylating agent was considered but not given due to no increase in blast percentage. He was referred to our clinic for consideration of an allogeneic stem cell transplant. At that time, his Karnofsky Performance Status (KPS) score was 80%, and Eastern Cooperative Oncology Group (ECOG) Performance Status was 1, with no active inflammatory manifestations, apart from intermittent subcutaneous nodules. He remained on 20 mg of prednisone daily and was started on ruxolitinib 5 mg twice daily for systemic symptoms including rash and fever in anticipation of HSCT. His complete blood count was notable for Hgb 7.9 g/dL, PLT 63,000/uL, WBC 6420/uL with ANC 3420/uL. Non-myeloablative conditioning with fludarabine, cyclophosphamide, and total body irradiation (TBI) was planned, followed by HLA-haploidentical peripheral blood stem cell transplantation (PBSCT). After receiving two doses of 30 mg/m2 of fludarabine and cyclophosphamide 14.5 mcg/kg, the patient developed a neutropenic fever. Imaging revealed new ill-defined bilateral pulmonary nodules concerning for an opportunistic fungal infection, and his transplant plan was deferred. He was treated empirically with posaconazole and broad-spectrum antibiotics with the resolution of his fever. Extensive infectious workup was unrevealing. One month after treatment, his cytopenias remarkably improved to Hgb 13.9 g/dL, PLT 265,000/uL, and WBC 5.3 K/uL with ANC 4600 without G-CSF support. His performance status improved to KPS 90% and ECOG 0 with the resolution of all other symptoms while remaining off prednisone. Considering options for further treatment, the patient elected to proceed with haploidentical PBSCT. He underwent a bone marrow biopsy in preparation for HSCT two months after treatment, which demonstrated a complete response (Figure 1). The patient underwent a transplant with conditioning chemotherapy including 3 days of fludarabine 30 mg/m2, 2 days of cyclophosphamide 14.5 mcg/kg, and TBI with 400cGy. He achieved neutrophil engraftment on Day +22. Graft-versus-host disease (GVHD) prophylaxis consisted of cyclophosphamide on Days +3-4, mycophenolate mofetil on Days +5-35, and tacrolimus starting Day +5. Due to suspicion of acute GVHD of the gastrointestinal (GI) tract, tacrolimus was continued beyond Day +60; however, it was stopped at Day +88 with concern for drug-induced thrombotic microangiopathy. Laboratory studies were notable for elevated CH50 and soluble C5b-9 consistent with activation of the terminal complement pathway. Eculizumab was cost-prohibitive, and the patient received narsoplimab via compassionate use. His cytopenias persisted and were not fully explained by major ABO incompatibility (donor AB positive, recipient A positive), vitamin B12 deficiency (206 pg/mL), or medications. The patient received a CD34+ stem cell boost at Day +178. His course was further complicated by chronic GVHD involving the mouth, GI tract, liver, eyes, and nails. This was managed with steroids causing severe steroid myopathy and then with belumosudil with suboptimal response, eventually transitioning to ruxolitinib with no further flares of GVHD symptoms. His 1-year posttransplant bone marrow showed complete response with cytogenetic remission, 0% blasts, and 100% donor cells on FISH for chimerism.
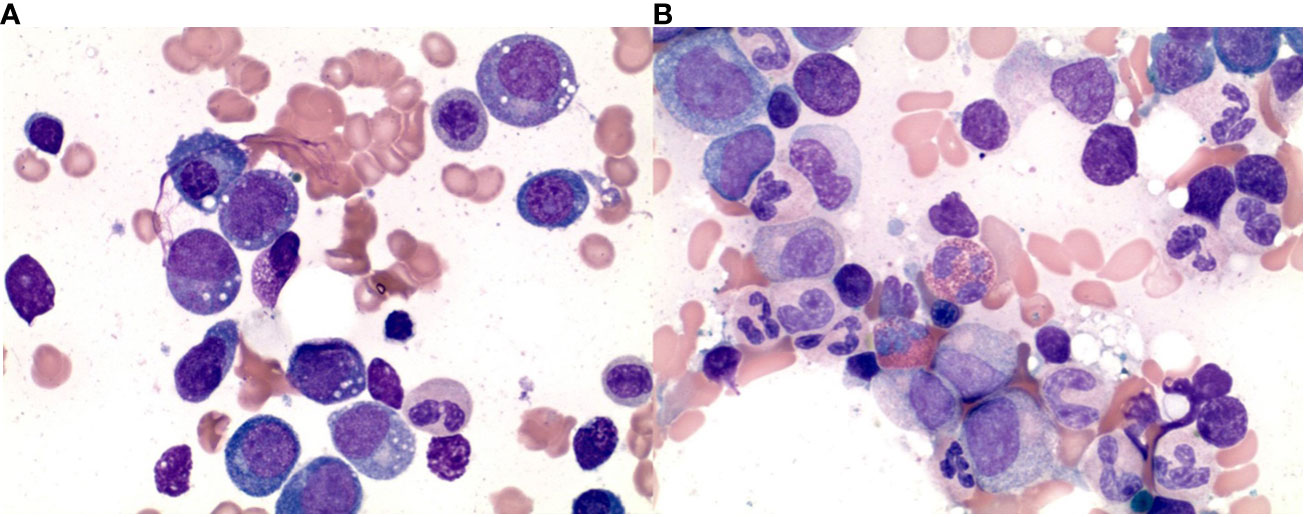
Figure 1 Histopathologic findings in the bone marrow aspirate before and after the treatment. (A) Bone marrow aspirate shows vacuolization in the immature myeloid and erythroid series. (B) Bone marrow aspirate shows absent vacuolization in the immature myeloid and erythroid series after treatment.
We report a case of MDS related to VEXAS syndrome that was successfully treated with fludarabine and cyclophosphamide at the University of Kansas Medical Center. We also performed a systematic review following the Preferred Reporting Items for Systematic Reviews and Meta-Analysis guidelines (PRISMA) guidelines. A literature search was performed on 3 databases (PubMed, Cochrane, and Embase) using the MeSH terms and keywords for “VEXAS syndrome,” “myelodysplastic syndrome,” “MDS”, and “treatment for VEXAS syndrome” from the date of inception to October 2023. We screened 135 articles, and duplicates were removed. Inclusion criteria included original studies (clinical trials, retrospective, and prospective studies), case reports, and case series in all patients with a confirmed diagnosis of VEXAS syndrome with myelodysplasia or other hematological manifestations. Review articles, studies with no treatment given or no information on treatment response, studies with no individual data on patients, and studies in languages other than English, were excluded. A total of 32 studies were included for the review after primary and secondary screening (Figure 2). Data were extracted regarding patient sociodemographic and clinical characteristics, hematological and bone marrow findings, treatment, and treatment response as well as patient outcomes.
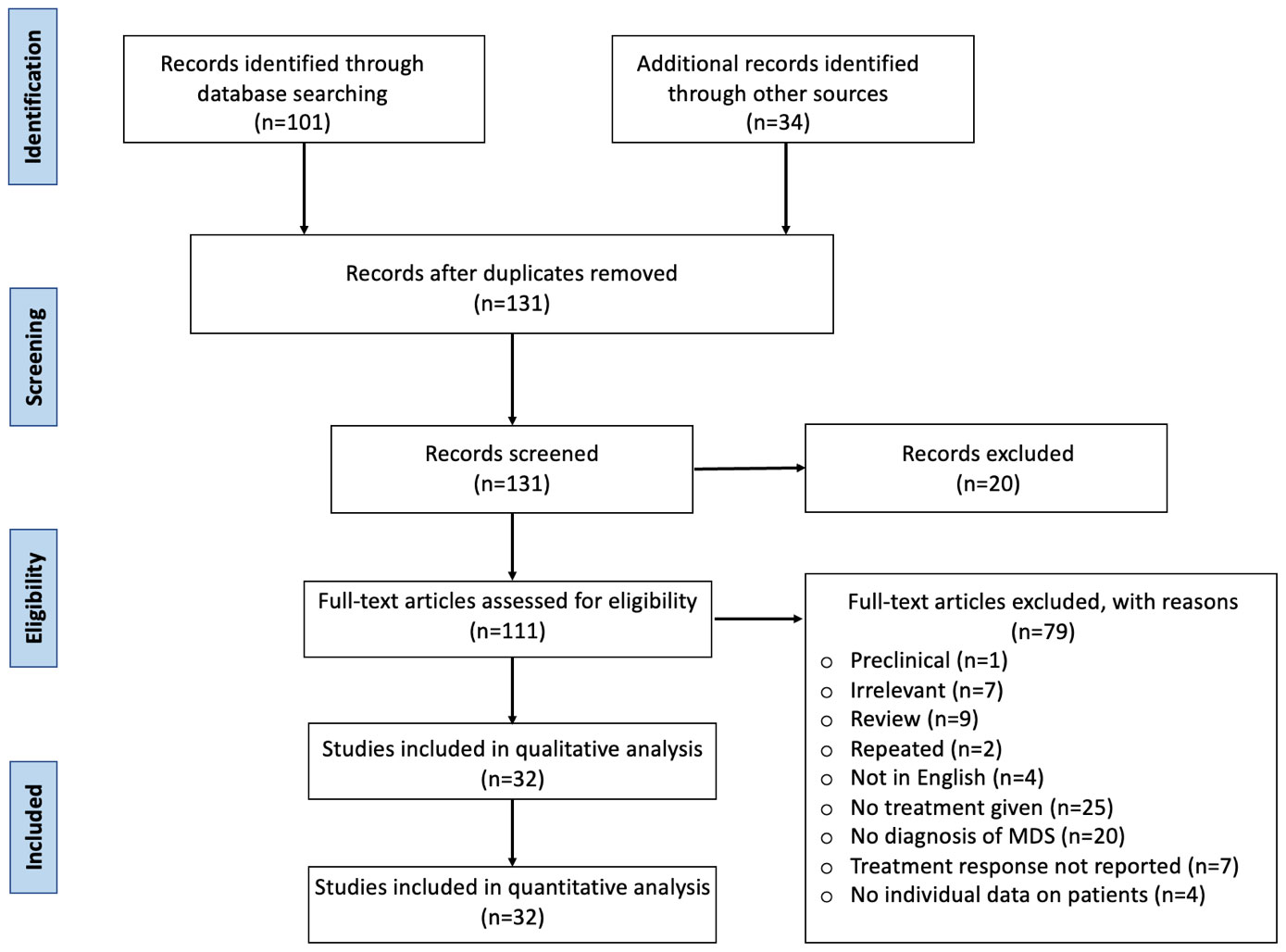
Figure 2 Treatment of VEXAS syndrome: systematic review PRISMA diagram.
VEXAS syndrome is a rare autoinflammatory disease characterized by severe systemic inflammation and various clinical manifestations described by Beck et al. in 2020 (1). It has been observed that patients with VEXAS syndrome have an increased risk of developing MDS (3), which is a clonal disorder of hematopoietic stem cells that leads to ineffective blood cell production. The association between VEXAS syndrome and MDS has been reported in multiple studies, with 25% to 55% of VEXAS patients having underlying MDS (1, 3, 11, 12). Obiorah et al. showed that 10 out of 16 VEXAS patients had hematologic disorders, including MDS, multiple myeloma, monoclonal gammopathy of undetermined significance, or monoclonal B-cell lymphocytosis (6).
VEXAS syndrome is seen almost exclusively in males and is associated with older age (13); however, it has been reported in women as well (14, 15). Interestingly, those women either have monosomy X in the setting of constitutional Turner syndrome or develop an acquired X monosomy in the bone marrow karyotype, thus making them genetically similar to male patients carrying a single X chromosome with a mutation in UBA1 gene resulting in a disease (16). Several mechanisms have been proposed alluding to interaction between UBA1 gene and X chromosome, including X-inactivation escape by UBA1 and skewed X-chromosome inactivation in women (17). The clinical features of VEXAS syndrome are heterogeneous and can include high-grade fever, polychondritis, skin lesions, ocular, pulmonary, and cardiac involvement. In the analyzed cohort of patients with concurrent MDS, the most prevalent features were arthritis, chondritis, or muscle involvement (71%), skin involvement (57%), fever (48%), pulmonary lesions (33%), constitutional symptoms, and thrombosis (both at 27%) (Table 1). 23% of patients met diagnostic criteria for relapsing polychondritis, whereas 14% of patients were diagnosed with Sweet syndrome.
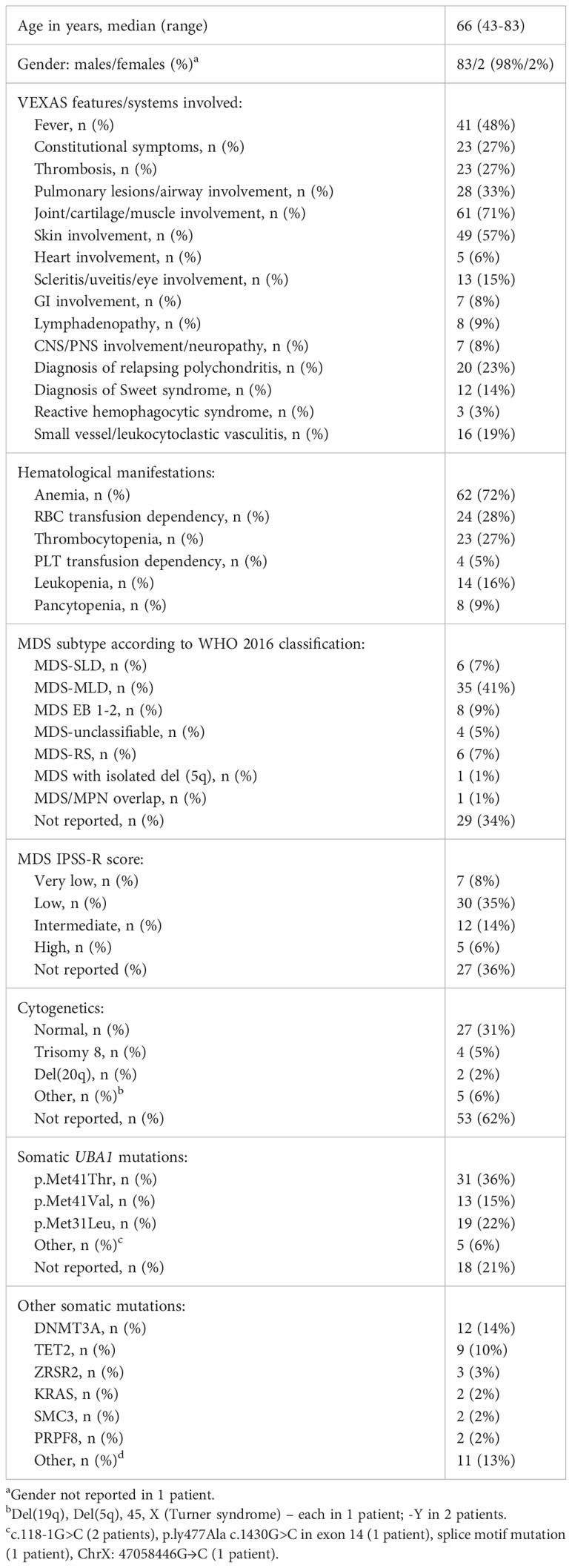
Table 1 Characteristics of patients with VEXAS syndrome with associated MDS (n=86).
There is a high prevalence of anemia in VEXAS patients with associated MDS (72%), with 65% of those patients having macrocytic anemia, and 39% of patients being transfusion dependent. Most patients with VEXAS syndrome develop cytopenias requiring workup with bone marrow biopsy. Examination of bone marrow aspirate in patients with VEXAS syndrome often reveals vacuolation of myeloid and erythroid precursors on bone marrow biopsy (18). Diagnosis of MDS can be challenging in the setting of inflammatory state and therapy. As pointed out by Raajimakers et al., in some patients pancytopenia and myelodysplasia are observed at a time of severe systemic inflammatory exacerbation and were not seen later in the course of the disease when symptoms improved (19).
The clinicopathological and molecular features of MDS associated with VEXAS syndrome in the analyzed cohort are consistent with the previous reports (1, 3, 5, 6). There is a higher frequency of myelodysplastic syndrome with multilineage dysplasia (MDS-MLD), low blast percentages, prevalence of IPSS-R low-risk category, and rare cases of high-risk cytogenetic abnormalities. The limitation of our analysis is a high prevalence of cases without reported MDS subtype, IPSS-R score, and cytogenetics (34%, 36%, and 62%, respectively).
Genetic variants of VEXAS-associated MDS commonly include epigenetic, splicing, and signaling factors such as DNMT3A and TET2, which were observed in 14% and 10%, respectively, which is also consistent with previous reports (19, 20). Loss of function in DNMT3A may contribute to the proinflammatory pathology of VEXAS syndrome as it has been associated with the activation of innate immune inflammatory signaling in myeloid cells (19).
A study by Ferrada et al. including 83 patients with VEXAS syndrome analyzed independent predictors of survival (21). Amino acid substitution of methionine for a valine (p.Met41Val) was associated with decreased survival compared to leucine (p.Met41Leu) and threonine (p.Met41Thr). Transfusion dependence was also associated with higher mortality, whereas ear chondritis was associated with increased survival. In the analyzed cohort, p.Met41Thr was most common (36%), followed by p.Met41Leu (22%) and p.Met41Val (15%). The subtype of UBA1 mutation was not reported in one-fifth of cases.
The treatment of VEXAS syndrome and associated MDS is challenging and not yet well-defined (22). However, there have been some treatment strategies that have shown promise. Diarra et al. reported successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome and severe inflammatory symptoms or MDS (23). Another treatment option is the use of DNA hypomethylating agents such as azacitidine, which has shown efficacy in VEXAS patients with MDS (15, 19, 22, 24–27). In a French registry, clinical responses were observed in 46% of VEXAS patients with MDS after treatment with azacitidine (22). Other potential treatments include anti-IL6 monoclonal antibodies such as tocilizumab (12, 28–31), anti-IL1 receptor antagonists such as anakinra (25, 30, 32), and Janus kinase (JAK) inhibitors including ruxolitinib and others (33–37).
In the analyzed cohort of patients with VEXAS and MDS, almost all patients received corticosteroids (CS) at some point in their disease course (98%), with favorable responses seen in the majority of those patients (94%) (Table 2). Steroid-sparing agents have been used with varying degrees of success: none of the patients achieved a complete response, and most patients required concomitant steroids or combinations with other agents such as rituximab or intravenous immunoglobulin (IVIG). A subset of patients has been exposed to biologic agents targeting IL1-R, IL-6R, IL17, and other, or tumor necrosis factor (TNF) alpha inhibitors, with some patients achieving complete response when those agents were combined with CS. A similar response was seen with JAK inhibitors such as filgotinib, tofacitinib, and upadacitinib. Localized skin reactions were observed with administration of anakinra and tocilizumab (30, 36, 38).

Table 2 Treatment responses in patients with VEXAS syndrome and MDS (n=86).
MDS-directed therapies such as erythropoietin-stimulating agents were attempted in 8% of patients with stable disease as the best response, while hypomethylating agents (HMA) such as azacitidine were used in 41% of patients with high efficacy. In the French VEXAS cohort, azacitidine was effective in 46% of patients (22), which supports the hypothesis that azacitidine may control steroid-dependent inflammatory and autoimmune disorders (50). While allogeneic HSCT is a curative option for VEXAS syndrome that is refractory to immunosuppression and cytokine-inhibiting agents, it is a high-risk treatment modality with associated mortality requiring careful selection of patients. There is currently no evidence guiding the selection of patients with VEXAS syndrome who will benefit from HSCT. Ongoing phase II trial of allogeneic HSCT for subjects with VEXAS syndrome (NCT05027945) may shed light on these guidelines. The development of the Autoinflammatory Disease Alliance (AIDA) registry for patients with VEXAS syndrome will also provide valuable real-world evidence for understanding the natural history of the disease and guiding therapeutic approaches (NCT05200715).
In the presented case, we observed complete resolution of both systemic and hematological manifestations of VEXAS syndrome and associated MDS after administration of 2 doses of conditioning regimen with fludarabine and cyclophosphamide. This was further confirmed by repeat bone marrow biopsy with the disappearance of vacuoles in both myeloid and erythroid precursors. The patient elected to proceed with HSCT as per the previous plan of treatment and is currently beyond 6 months after transplant with complications including pancytopenia requiring stem cell boost and chronic graft-versus-host disease. To our knowledge, this is the first case reporting a complete response using fludarabine and cyclophosphamide in VEXAS syndrome. Further research is needed to better understand the pathogenesis and optimal management of VEXAS syndrome.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
PB: Writing – original draft. JG: Writing – original draft. MS: Writing – original draft. MA: Writing – original draft. MK: Writing – review & editing. NS: Writing – review & editing. NA: Writing – review & editing. AS: Writing – review & editing. AY: Writing – review & editing. DZ: Data curation, Visualization, Writing – review & editing. JPM: Writing – review & editing. MM: Data curation, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
JPM has speaking, consulting and advisory role in Kite, Juno Therapeutics, Allovir, Magenta Therapeutics, EcoR1 Capital, and has research funding from Novartis, Fresenius Biotech, Astellas Pharma, Bellicum Pharmaceuticals, Gamida Cell, Pluristem Therapeutics, Kite and AlloVir.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
2. Beck DB BD, Shah V. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. (2023) 329:318–24. doi: 10.1001/jama.2022.24836
3. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. (2022) 186:564–74. doi: 10.1111/bjd.20805
4. Gurnari C, Pagliuca S, Durkin L, Terkawi L, Awada H, Kongkiatkamon S, et al. Vacuolization of hematopoietic precursors: an enigma with multiple etiologies. Blood. (2021) 137:3685–9. doi: 10.1182/blood.2021010811
5. Grayson PC, Patel BA, Young NS. VEXAS syndrome. Blood. (2021) 137:3591–4. doi: 10.1182/blood.2021011455
6. Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, et al. Benign and Malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. (2021) 5:3203–15. doi: 10.1182/bloodadvances.2021004976
7. Temple M, Kosmider O. VEXAS syndrome: A novelty in MDS landscape. Diagnostics (Basel). (2022) 12(7):1590. doi: 10.3390/diagnostics12071590
8. Matsumoto H, Fujita Y, Fukatsu M, Ikezoe T, Yokose K, Asano T, et al. Case report: coexistence of multiple myeloma and auricular chondritis in VEXAS syndrome. Front Immunol. (2022) 13:897722. doi: 10.3389/fimmu.2022.897722
9. Huang H, Zhang W, Cai W, Liu J, Wang H, Qin T, et al. VEXAS syndrome in myelodysplastic syndrome with autoimmune disorder. Exp Hematol Oncol. (2021) 10:23. doi: 10.1186/s40164-021-00217-2
10. Yildirim F, Erdogan M, Yalcin Mutlu M, Akkuzu G, Ozgur DS, Karaalioglu B, et al. VEXAS syndrome with severe multisystem involvement: Rapid recovery after splenectomy. Int J Rheum Dis. (2023) 26:559–62. doi: 10.1111/1756-185x.14540
11. Poulter JA, Collins JC, Cargo C, De Tute RM, Evans P, Ospina Cardona D, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. (2021) 137:3676–81. doi: 10.1182/blood.2020010286
12. Bourbon E, Heiblig M, Gerfaud Valentin M, Barba T, Durel CA, Lega JC, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. (2021) 137:3682–4. doi: 10.1182/blood.2020010177
13. Khitri M-Y, Guedon AF, Georgin-Lavialle S, Terrier B, Saadoun D, Seguier J, et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: analysis of a French case series of 95 patients. RMD Open. (2022) 8:e002255. doi: 10.1136/rmdopen-2022-002255
14. Arlet J, Terrier B, Kosmider O. Mutant UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2021) 384:2163. doi: 10.1056/NEJMc2102124
15. Stubbins RJ, McGinnis E, Johal B, Chen LY, Wilson L, Cardona DO, et al. VEXAS syndrome in a female patient with constitutional 45,X (Turner syndrome). Haematologica. (2022) 107:1011–3. doi: 10.3324/haematol.2021.280238
16. Beck D, Grayson P, Kastner D. Mutant UBA1 and severe adult-onset autoinflammatory disease. Reply N Engl J Med. (2021) 384:2164–5. doi: 10.1056/NEJMc2102124
17. Luzzatto L, Risitano A, Notaro R. Mutant UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2021) 384:2164. doi: 10.1056/NEJMc2102124
18. Patel N, Dulau-Florea A, Calvo KR. Characteristic bone marrow findings in patients with UBA1 somatic mutations and VEXAS syndrome. Semin Hematol. (2021) 58:204–11. doi: 10.1053/j.seminhematol.2021.10.007
19. Raaijmakers M, Hermans M, Aalbers A, Rijken M, Dalm VASH, van Daele P, et al. Azacytidine treatment for VEXAS syndrome. Hemasphere. (2021) 5:e661. doi: 10.1097/hs9.0000000000000661
20. Manzoni M, Bosi A, Fabris S, Lionetti M, Salerio S, Migliorini AC, et al. Clinical, morphological and clonal progression of VEXAS syndrome in the context of myelodysplasia treated with azacytidine. Clin Hematol Int. (2022) 4:52–5. doi: 10.1007/s44228-022-00002-w
21. Ferrada M, Savic S, Cardona DO. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. (2022) 140:1496–506. doi: 10.1182/blood.2022016985
22. Comont T, Heiblig M, Rivière E, Terriou L, Rossignol J, Bouscary D, et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. (2022) 196:969–74. doi: 10.1111/bjh.17893
23. Diarra A, Duployez N, Fournier E, Preudhomme C, Coiteux V, Magro L, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. (2022) 6:998–1003. doi: 10.1182/bloodadvances.2021004749
24. Kataoka A, Mizumoto C, Kanda J, Iwasaki M, Sakurada M, Oka T, et al. Successful azacitidine therapy for myelodysplastic syndrome associated with VEXAS syndrome. Int J Hematol. (2023) 117(6):919–24. doi: 10.1007/s12185-023-03532-y
25. Delplanque M, Aouba A, Hirsch P, Fenaux P, Graveleau J, Malard F, et al. USAID associated with myeloid neoplasm and VEXAS syndrome: two differential diagnoses of suspected adult onset still's disease in elderly patients. J Clin Med. (2021) 10(23):5586. doi: 10.3390/jcm10235586
26. Cordts I, Hecker JS, Gauck D, Park J, Härtl J, Günthner R, et al. Successful treatment with azacitidine in VEXAS syndrome with prominent myofasciitis. Rheumatol (Oxford). (2022) 61:e117–9. doi: 10.1093/rheumatology/keab866
27. Neupane K, Jayarangaiah A, Zhang Y, Kumar A. VEXAS syndrome with progression of MDS to MDS/MPN overlap syndrome. BMJ Case Rep. (2022) 15(12):e251089. doi: 10.1136/bcr-2022-251089
28. Sakuma M, Tanimura A, Yasui S, Ishiguro K, Kobayashi T, Ohshiro Y, et al. A Case of polychondritis-onset refractory organizing pneumonia with cytopaenia diagnosed as VEXAS syndrome: the disease course of 7 years. Rheumatol (Oxford). (2021) 60:e356–9. doi: 10.1093/rheumatology/keab349
29. Kunishita Y, Kirino Y, Tsuchida N, Maeda A, Sato Y, Takase-Minegishi K, et al. Case report: tocilizumab treatment for VEXAS syndrome with relapsing polychondritis: A single-center, 1-year longitudinal observational study in Japan. Front Immunol. (2022) 13:901063. doi: 10.3389/fimmu.2022.901063
30. van der Made CI, Potjewijd J, Hoogstins A, Willems HPJ, Kwakernaak AJ, de Sevaux RGL, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunol. (2022) 149:432–439.e4. doi: 10.1016/j.jaci.2021.05.014
31. Ross C, Elfassy HL, Makhzoum JP. Somatic mutation in UBA1 and ANCA-associated vasculitis. J Rheumatol. (2021) 48:1626–7. doi: 10.3899/jrheum.210149
32. Lötscher F, Seitz L, Simeunovic H, Sarbu AC, Porret NA, Feldmeyer L, et al. Case report: genetic double strike: VEXAS and TET2-positive myelodysplastic syndrome in a patient with long-standing refractory autoinflammatory disease. Front Immunol. (2021) 12:800149. doi: 10.3389/fimmu.2021.800149
33. Loschi M, Roux C, Sudaka I, Ferrero-Vacher C, Marceau-Renaut A, Duployez N, et al. Allogeneic stem cell transplantation as a curative therapeutic approach for VEXAS syndrome: a case report. Bone Marrow Transplant. (2022) 57:315–8. doi: 10.1038/s41409-021-01544-y
34. Beecher M, Tong J, Halliday L, Hissaria P, Selva D. Recurrent orbital inflammation associated with VEXAS syndrome. Orbit. (2022) 2022:1–4. doi: 10.1080/01676830.2022.2126501
35. Bindoli S, Baggio C, Doria A, Bertoldo E, Sfriso P. JAK inhibitors for the treatment of VEXAS syndrome. Exp Biol Med (Maywood). (2023) 248:394–8. doi: 10.1177/15353702231165030
36. Muratore F, Marvisi C, Castrignanò P, Nicoli D, Farnetti E, Bonanno O, et al. VEXAS syndrome: A case series from a single-center cohort of italian patients with vasculitis. Arthritis Rheumatol. (2022) 74:665–70. doi: 10.1002/art.41992
37. Islam S, Cullen T, Sumpton D, Damodaran A, Heath D, Bosco A, et al. VEXAS syndrome: lessons learnt from an early Australian case series. Intern Med J. (2022) 52:658–62. doi: 10.1111/imj.15742
38. Estes J, Malus M, Wilson L, Grayson PC, Maz M. A case of VEXAS: vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome with co-existing DNA (Cytosine-5)-methyltransferase 3A mutation complicated by localized skin reaction to tocilizumab and azacitidine. Cureus. (2023) 15:e39906. doi: 10.7759/cureus.39906
39. Guerrero-Bermúdez CA, Cardona-Cardona AF, Ariza-Parra EJ, Arostegui JI, Mensa-Vilaro A, Yague J, et al. Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome (VEXAS syndrome) with prominent supraglottic larynx involvement: a case-based review. Clin Rheumatol. (2022) 41:3565–72. doi: 10.1007/s10067-022-06338-1
40. Grey A, Cheong PL, Lee FJ, Abadir E, Favaloro J, Yang S, et al. A case of VEXAS syndrome complicated by hemophagocytic lymphohistiocytosis. J Clin Immunol. (2021) 41:1648–51. doi: 10.1007/s10875-021-01070-y
41. Zhao LP, Schell B, Sebert M, Kim R, Lemaire P, Boy M, et al. Prevalence of UBA1 mutations in MDS/CMML patients with systemic inflammatory and auto-immune disease. Leukemia. (2021) 35:2731–3. doi: 10.1038/s41375-021-01353-8
42. Stiburkova B, Pavelcova K, Belickova M, Magaziner SJ, Collins JC, Werner A, et al. Novel somatic UBA1 variant in a patient with VEXAS syndrome. Arthritis Rheumatol. (2023) 75(7):1285–90. doi: 10.1002/art.42471
43. Yamaguchi H, Kobayashi D, Nakamura G, Aida R, Horii Y, Okamoto T, et al. Acute heart failure due to left common iliac arteriovenous fistula: A case of VEXAS syndrome. Mod Rheumatol Case Rep. (2023) 7:327–33. doi: 10.1093/mrcr/rxac082
44. Koster MJ, Kourelis T, Reichard KK, Kermani TA, Beck DB, Cardona DO, et al. Clinical heterogeneity of the VEXAS syndrome: A case series. Mayo Clin Proc. (2021) 96:2653–9. doi: 10.1016/j.mayocp.2021.06.006
45. Al-Hakim A, Poulter JA, Mahmoud D, Rose AMS, Elcombe S, Lachmann H, et al. Allogeneic haematopoietic stem cell transplantation for VEXAS syndrome: UK experience. Br J Haematol. (2022) 199:777–81. doi: 10.1111/bjh.18488
46. Mangaonkar AA, Langer KJ, Lasho TL, Finke C, Litzow MR, Hogan WJ, et al. Reduced intensity conditioning allogeneic hematopoietic stem cell transplantation in VEXAS syndrome: Data from a prospective series of patients. Am J Hematol. (2023) 98:E28–e31. doi: 10.1002/ajh.26786
47. Mekinian A, Zhao LP, Chevret S, Desseaux K, Pascal L, Comont T, et al. A Phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia. (2022) 36(11):2739–42. doi: 10.1038/s41375-022-01698-8
48. Magnol M, Couvaras L, Degboé Y, Delabesse E, Bulai-Livideanu C, Ruyssen-Witrand A, et al. VEXAS syndrome in a patient with previous spondyloarthritis with a favourable response to intravenous immunoglobulin and anti-IL17 therapy. Rheumatol (Oxford). (2021) 60:e314–5. doi: 10.1093/rheumatology/keab211
49. Kunimoto H, Miura A, Maeda A, Tsuchida N, Uchiyama Y, Kunishita Y, et al. Clinical and genetic features of Japanese cases of MDS associated with VEXAS syndrome. Int J Hematol. (2023) 118:494–502. doi: 10.1007/s12185-023-03598-8
Keywords: myelodysplastic syndrome, VEXAS syndrome, outcomes, allogeneic hematopoietic stem cell transplantation, fludarabine and cyclophosphamide
Citation: Bellman P, Gonzalez-Lugo JD, Shahzad M, Amin MK, Khalid MF, Suleman N, Ahmed N, Singh AK, Yacoub A, Zhang D, McGuirk JP and Mushtaq MU (2024) Successful treatment with fludarabine and cyclophosphamide in a VEXAS syndrome patient with associated myelodysplastic syndrome: a case report and systematic review. Front. Oncol. 14:1383730. doi: 10.3389/fonc.2024.1383730
Received: 07 February 2024; Accepted: 25 March 2024;
Published: 11 April 2024.
Edited by:
Pasquale Niscola, Sant’Eugenio Hospital of Rome, ItalyReviewed by:
Carmelo Gurnari, Cleveland Clinic, United StatesCopyright © 2024 Bellman, Gonzalez-Lugo, Shahzad, Amin, Khalid, Suleman, Ahmed, Singh, Yacoub, Zhang, McGuirk and Mushtaq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Umair Mushtaq, bW11c2h0YXFAa3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.