 Jun Qian
Jun Qian Min Peng
Min Peng Yanan Li
Yanan Li Wei Liu
Wei Liu Xinwei Zou
Xinwei Zou Huafei Chen
Huafei Chen Sujuan Zhou
Sujuan Zhou Sheng Xiao
Sheng Xiao Jinhua Zhou
Jinhua Zhou- 1Department of Gynecology and Obstetrics, The First Affiliated Hospital of Soochow University, Suzhou, China
- 2Molecular Genetics Laboratory, Suzhou Sano Precision Medicine Ltd., Suzhou, China
- 3Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
Background: Genome instability plays a crucial role in promoting tumor development. Germline mutations in genes responsible for DNA repair are often associated with familial cancer syndromes. A noticeable exception is the CHEK1 gene. Despite its well-established role in homologous recombination, germline mutations in CHEK1 are rarely reported.
Case presentation: In this report, we present a patient diagnosed with ovarian clear cell carcinoma who has a family history of cancer. Her relatives include a grandfather with esophageal cancer, a father with gastric cancer, and an uncle with a brain tumor. The patient carried a typical genomic profile of clear cell carcinoma including mutations in KRAS, PPP2R1A, and PIK3R1. Importantly, her paired peripheral blood cells harbored a germline CHEK1 mutation, CHEK1 exon 6 c.613 + 2T>C, which was also found in her father. Unfortunately, the CHEK1 status of her grandfather and uncle remains unknown due to the unavailability of their specimens. Further evaluation via RT-PCR confirmed a splicing error in the CHEK1 gene, resulting in truncation at the kinase domain region, indicative of a loss-of-function mutation.
Conclusion: This case highlights a rare germline CHEK1 mutation within a family with a history of cancer. The confirmed splicing error at the mRNA level underscores the functional consequences of this mutation. Documenting such cases is vital for future evaluation of inheritance patterns, clinical penetrance of the mutation, and its association with specific cancer types.
1 Introduction
DNA repair involves complex mechanisms and intricate pathways. Both checkpoint kinase 1 and 2 (CHEK1, CHEK2) play crucial roles in DNA repair and safeguarding genomic stability during the cell cycle (1). They serve as vital checkpoints that, when activated, temporarily halt the cell cycle, allowing time for DNA repair or triggering apoptosis when DNA damage becomes irreparable. Specifically, CHEK1 is activated by the DNA damage sensor known as the ataxia telangiectasia and Rad3-related protein (ATR) in response to single-strand DNA breaks during the S phase of the cell cycle. Subsequently, CHEK1 phosphorylates CDC25A and CDC25C, leading to the inhibition of cyclin-dependent kinases (CDKs) and cell cycle arrest (2, 3). CHEK2 is activated when double-strand DNA damage is detected by Ataxia-telangiectasia mutated (ATM) during the G1 phase of the cell cycle (4). The activated CHEK2 kinase phosphorylates and stabilizes p53 (ATM-CHEK2-P53 axis), leading to p21 expression (5–7). p21 is a potent CDK inhibitor that blocks the cell cycle progress (8).
Germline mutations in genes involved in DNA repair pathways are associated with genome instability and an increased risk of cancer development. For example, individuals with Li-Fraumeni syndrome are predisposed to various cancers, including leukemia, sarcomas, brain tumors, adrenocortical carcinoma, and other solid tumors, often manifesting at a young age (9). While TP53 germline mutations are frequently associated with Li-Fraumeni syndrome, some individuals with TP53-negative Li-Fraumeni syndrome harbor germline mutations in CHEK2 (10, 11). Germline mutation of CHEK1, however, is rarely documented in inherited cancers. This report presents the discovery of a novel CHEK1 splicing mutation within a family with multiple cancer patients. This mutation results in a splicing error and a frameshift coding sequence alteration, confirming its inactivation nature.
2 Case presentation
During her annual physical examination, a 57-year-old female presented with pelvic effusion and a cystic mass measuring 119 × 126 × 100 mm, which exhibited nodular protrusions into the cavity as revealed by gynecological B-ultrasound. A benign ovarian tumor was suspected at the time. However, two months later, an MRI evaluation indicated a progressive enlargement of the mass (Figure 1A). Further serum examination showed elevated levels of tumor biomarkers: CA125 at 49.80 U/mL (normal range 0.1 ~35 U/ml) and HE4 at 3250 pmol/L (normal range <70 pmol/L for premenopause women and <140 pmol/L for post-menopause women). The patient underwent a transabdominal total hysterectomy, bilateral salpingo-oophorectomy, omentectomy, and pelvic and paraaortic lymphadenectomy. Hematoxylin and eosin (H&E) staining of formalin-fixed paraffin-embedded (FFPE) sections from the right ovarian appendage revealed a clear cell carcinoma with papillary and focal solid growth patterns (Figure 1B). Tumor cells contained clear cytoplasm, uniform nuclear atypia, prominent nucleoli, and occasional mitosis. Immunohistochemistry of the tumor cells was positive for ER (90% +), HNF1-β, PAX-8, p53(diffuse +), p16 (partial +), Ki-67 (30% +), and negative for PR, Vimentin, WT1, and Napsin A. Based on these findings, a diagnosis of ovarian clear cell carcinoma (OCCC) was established.
Targeted DNA NGS analysis of 638 cancer-related genes, combined with whole-genome single-nucleotide polymorphism (SNP) examination, was performed on both tumor and paired blood specimens. This analysis revealed somatic mutations, including KRAS exon 2 c.35G>T p.G12V (26.7%), PPP2R1A exon 5 c.547C>T p.R183W (18.1%), and PIK3R1 exon 13 c.1721_1727del p.K575Efs*5 (21%) (Figure 1C). These somatic mutations align with the characteristic profile of ovarian clear cell carcinoma. Additionally, a CHEK1 exon 6 c.613 + 2T>C mutation was identified in both the tumor and normal blood cells, confirming its germline origin. The timeline summarized the major clinical events of the patient (Figure 1D).
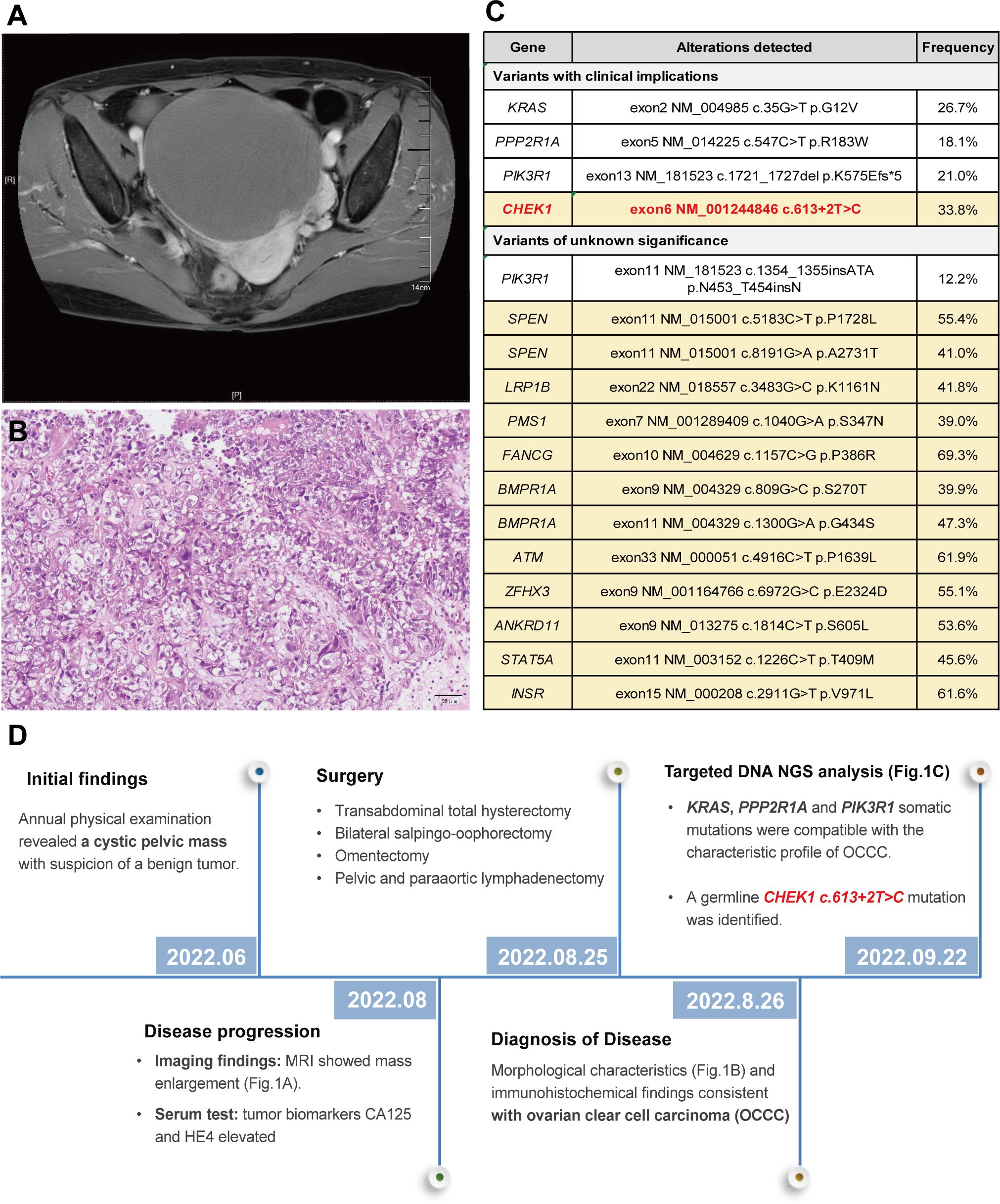
Figure 1 MRI revealed a pelvic cystic mass (A); histological examination of the tumor demonstrated a clear cell morphology with papillary and focal solid growth patterns, consistent with the diagnosis of ovarian clear cell carcinoma (OCCC) (B); Genomic profiling of the tumor exhibited a somatic mutation pattern consistent with the OCCC. Germline mutations, including CHEK1, were detected in the peripheral blood specimen (highlighted in yellow) (C); and a timeline summarizing the key clinical events of the proband (D).
The patient reported a family history of cancer, spanning esophageal cancer in her grandfather, gastric cancer in her father, a brain tumor in her uncle, and renal cell carcinoma in her mother. Her grandfather (I-1) and uncle (II-3) were diagnosed with esophageal cancer and a brain tumor, respectively, based on imaging without biopsy, and both succumbed to the disease without undergoing surgery. Her father (II-1) had stage 2A gastric adenocarcinoma in 1995, which was successfully treated with surgery. The patient’s mother (II-2) underwent left-sided renal cancer surgery in 2007, with a postoperative histopathologic diagnosis of clear cell carcinoma. As of genetic counseling in February 2023, her other siblings had no reported history of cancer. To assess the presence of the CHEK1 mutation in family members, a PCR reaction was performed to amplify the DNA fragment containing the mutation site using gene-specific primers (CHEK1_DNA_F: GCTCCAGAACTTCTGAAGAGAAG; CHEK1_DNA_R: GCTGCAGTGAGCTATAACAGC). The PCR product was subjected to direct Sanger sequencing. These studies detected the same CHEK1 mutation in her father, who was diagnosed with gastric cancer at the age of 64, and two siblings, both of whom remained tumor-free at the ages of 60 and 68, respectively. Unfortunately, no specimens were available to evaluate the mutation status of her deceased grandfather, who was diagnosed with esophageal cancer at the age of 80, and her uncle, who was diagnosed with a brain tumor at the age of 72 (Figure 2).
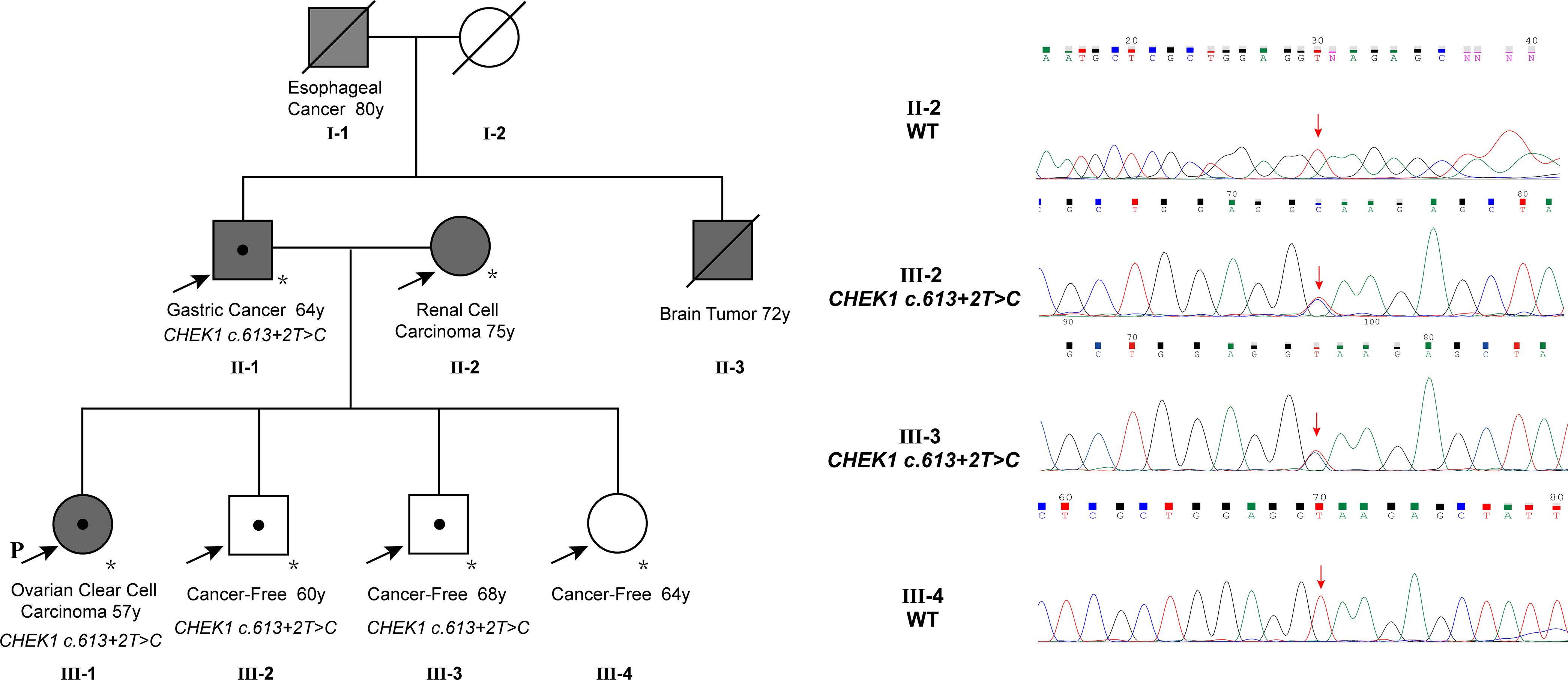
Figure 2 The family pedigree showed multiple members with cancer, including the proband with ovarian cancer at 57, the father with gastric cancer at 64, an uncle with a brain tumor at 72, the grandfather with esophageal cancer at 80, and the mother with renal cell carcinoma at 75. The CHEK1 c.613 + 2T>C mutation was confirmed in the proband, the father, and two siblings. Note that the mutation in the father was validated using NGS instead of Sanger sequencing due to poor sequencing results from the PCR product, likely attributed to low DNA quality from the very old tumor block.
The CHEK1 exon 6 c.613 + 2T>C mutation was not documented in any cancer databases or in gnomAD. To determine whether this mutation impacts RNA splicing, an RT-PCR analysis was performed using peripheral blood from the proband. This assay amplified a fragment encompassing the splicing sites of exons 6 and 7 (CHEK1_RNA_F: 5’-GCTCCAGAACTTCTGAAGAGAAG; CHEK1_RNA_R: 5’-CTTGCTGATGGATTCTCAACT). Sanger sequencing of this PCR product confirmed that the CHEK1 c.613 + 2T>G mutation disrupted the donor splice site of exon 6, leading to the utilization of an alternative splice site located 20 bp downstream of exon 6 (Figure 3A). A similar aberrant CHEK1 transcript was confirmed by a RNAseq analysis (Figure 3B). This resulted in a reading frame shift after amino acid residue 204 of CHEK1 and the presence of a premature translational stop codon at 41 amino acid residues downstream of the mutated nucleotide (truncated CHEK1 p. E205Gfs*41) (Figure 3C).
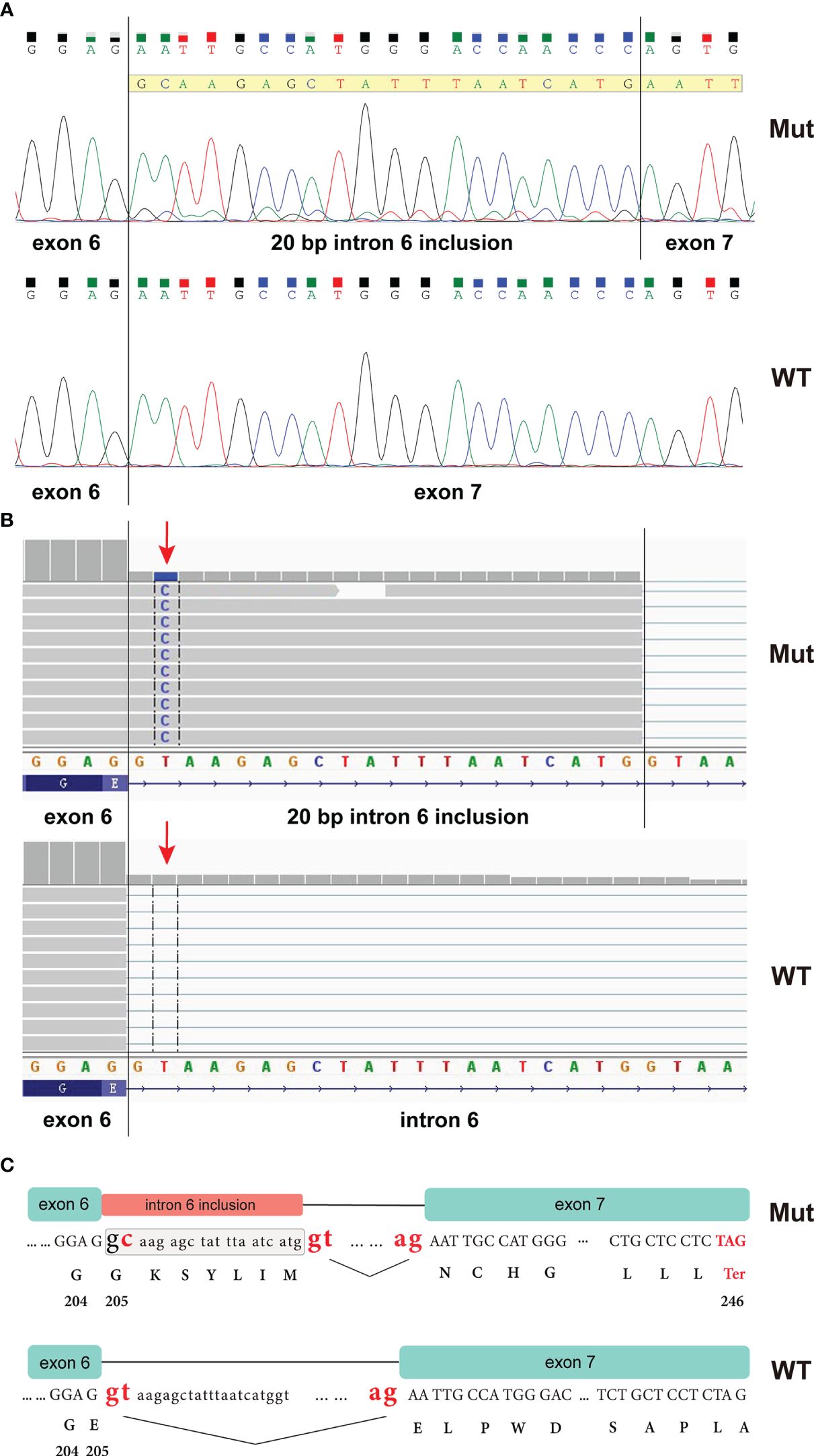
Figure 3 RT-PCR analysis of peripheral blood from the proband revealed that the CHEK1 c.613 + 2T>C mutation caused a splicing error, resulting in the inclusion of a 20 bp intronic sequence in the CHEK1 transcript (A); an RNA NGS assay demonstrated a similar aberrant CHEK1 transcript due to the mutation at the splicing site (arrow) (B); and an ideogram illustrated the wild-type splicing donor and acceptor sites of CHEK1 intron 6, along with the alternative splicing donor and acceptor sites from the mutated allele. This alternative splicing event leads to the formation of a premature stop codon (C).
3 Discussion
CHEK1 is a serine/threonine kinase composed of an N-terminal kinase domain (amino acid residues 8-265) and a C-terminal regulatory domain (residues 317-476). The C-terminal regulatory domain of CHEK1 is self-inhibitory, interacting with the N-terminal to block the kinase activity of CHEK1 (12, 13). Upon encountering DNA damage or replication stress signals, the ATR kinase becomes activated, phosphorylating multiple serine/glutamine residues within the C-terminal of CHEK1, especially Ser317 and Ser345 residues. This phosphorylation event leads to the dissociation of the N- and C-terminal structural domains, activating CHEK1 (13–16). It has been demonstrated that the catalytic activity of the N-terminal kinase domain alone is significantly higher than that of the full-length CHEK1 protein in vitro (17). However, the integrity of the C-terminal regulatory domain is crucial for the proper in vivo functioning of the CHEK1 protein. In a study conducted by Ning et al. (12), the effects of various CHEK1 truncating variants on cell cycle regulation were investigated. The CΔ368 variant (residues 368-476 truncation) exhibited higher catalytic activity compared to full-length CHEK1, while CΔ288 (residues 288-476 truncation) had nearly lost its entire catalytic activity. Therefore, the C-terminal regulatory domain of CHEK1 contains not only inhibitory elements but also essential positive regulatory elements for catalytic activity (13–16). The CHEK1 c.613 + 2T>G splice variant reported in this study was predicted to generate a truncating protein p.E205Gfs*41, resulting in a partial loss of the N-terminal kinase domain (residues 205-265) and the complete deletion of the C-terminal, thus predicting a loss-of-function mutation.
The role of CHEK1 in DNA repair through homologous recombination is well-established. For instance, knocking down CHEK1 has been shown to result in deficient homologous recombination repair, confirming its function as a BRCA-like tumor suppressor (18). In addition, CHEK1 heterozygous deletion has been observed to accelerate tumorigenesis in WNT1 transgenic oncogenic mice compared to wild-type mice (19), as well as in chemically induced skin papilloma formation (20). During the early stages of tumorigenesis, partial deletion of CHEK1 has been linked to genomic instability, which in turn accelerates tumor development (19, 21, 22). However, the status of CHEK1 as a bona fide tumor suppressor remains controversial. CHEK1 is generally overexpressed in various cancers, including ovarian, breast (23), cervical cancer, and brain cancers (24), where tumor cells gain a survival advantage by enhancing checkpoints to facilitate DNA damage repair (25, 26). Elevated CHEK1 levels (both protein and mRNA) have been associated with chemoresistance (27–29), and many clinical trials are evaluating CHEK1 inhibitors in combination with chemotherapy (3, 26). On the other hand, loss-of-function variants have only been identified in a few cancers, such as gastric (30), colorectal (31, 32), and endometrial cancers (31, 33) with MSI-H features. Therefore, while germline CHEK1 mutations with loss-of-function potential may contribute to genome instability, cell cycle deregulation, and tumor development, somatic tumor cells with functional CHEK1 or gain-of-function CHEK1 alterations may assist in tumor cell survival by promoting lethal DNA damage repair.
Epithelial ovarian cancer is categorized into “Type I” and “Type II” subtypes (34). Our proband was diagnosed with OCCC, which is a Type I ovarian cancer and typically follows a relatively indolent course, often progressing through multiple steps from atypical hyperplasia, precancerous lesions, and borderline tumors (34, 35). Hereditary ovarian cancer associated with BRCA1/2 mutations usually manifests as Type II tumors, characterized by rapid disease progression and high aggressiveness (35–38). However, OCCC is rarely linked to germline BRCA1/2 mutations (39, 40). OCCC is hypothesized to arise from benign ectopic endometrial tissue on the ovary (41, 42), with approximately 50% to 74% of OCCC cases associated with endometriosis. It is suggested that the inflammatory and oxidative stress responses induced by endometriosis contribute to DNA damage and the development of malignancy (42). The most frequent genomic alterations in OCCC involve somatic mutations in ARID1A and PIK3CA, with loss of ARID1A function considered one of the earliest events, occurring in atypical endometriosis (43–45), and contributing to genomic instability. However, ARID1A loss alone is insufficient for tumor formation, typically occurring concomitantly with activation of the PI3K/AKT/mTOR pathway. ARID1A and PIK3CA alterations coexist in 20% to 56% of OCCC cases (46). Mouse models harboring both ARID1A and PIK3CA mutations develop tumors that phenotypically and molecularly resemble human OCCC (47), confirming the pivotal role of these pathways in OCCC pathogenesis. Our proband had no history of endometriosis or ARID1A mutation. We hypothesize that germline haploinsufficiency of CHEK1, as an alternative early genomic instability event, contributed to OCCC oncogenesis by cooperating with PI3K pathway activation. Notably, in the DNA damage response signaling, ARID1A interacts with ATR and is recruited to double-strand breaks to sustain DNA damage signaling (48). Defective ARID1A may impair DNA damage-induced ATR activation and its downstream signaling, particularly involving CHEK1 (48). Thus, functional defects of the CHEK1 may partially overlap with ARID1A in initiating events within this pathway.
While the CHEK1 gene is included in some commercial NGS panels designed for assessing hereditary cancer risk (49), the available data regarding germline CHEK1 mutations and cancer risk are limited. In a study involving 48 women with inherited ovarian cancer lacking BRCA1 or BRCA2 mutations, a CHEK1 exon 7 c.1564-1565insA frameshift mutation was identified (50). Additionally, in a group of 246 prostate cancer patients with a cancer history recommended for germline mutation testing by NCCN guidelines, a germline CHEK1 “stop-gain” mutation was detected (51). However, neither study provided comprehensive mutation tracking within family members. The rarity of germline CHEK1 mutations may underscore its significance in embryonic and organizational development. In mouse models, homozygous deletion of CHEK1 resulted in embryonic death (19, 52), whereas mice with heterozygous loss of CHEK1 were viable but exhibited haploinsufficiency, showing increased accumulation of DNA damage, cell cycle dysregulation, increased spontaneous cell death, and defects in tissue development (19, 53, 54). Moreover, CHEK1 plays a crucial role in maintaining functional hematopoiesis, as CHEK1 haploinsufficiency leads to anemia and abnormal erythropoiesis in mice (55). Chemical inhibition of CHEK1 induced hematopoietic stem cell and progenitor cell death in both mice and humans (56). Recent studies have reported several germline heterozygous mutations occurring in the C-terminal regulatory domain of CHEK1, including three missense variants (R379Q, R420K, and R442Q) and a truncating variant (F441fs*). These mutations were demonstrated to be gain-of-function, with increased kinase activity of CHEK1 causing arrested fertilized ovum division and resulting in infertility in human females (57–59).
In summary, we report a rare germline inactivation mutation of CHEK1 in a family with a history of cancer, and we confirmed that this mutation led to splicing errors at the mRNA level. Although data on the prevalence of CHEK1 germline mutations in inherited cancer is limited, along with their clinical penetrance and association with specific cancer types, documenting these mutations holds significant value for future assessments and conclusions.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the individual(s) and minor(s)’ legal guardian/next of kin to participate in this study. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JQ: Data curation, Writing – original draft, Writing – review & editing, Investigation. MP: Writing – review & editing, Writing – original draft, Investigation, Validation. YL: Investigation, Writing – original draft. WL: Software, Validation, Writing – review & editing, Data curation. XZ: Writing – review & editing, Data curation, Validation. HC: Methodology, Resources, Writing – review & editing. SZ: Methodology, Resources, Writing – review & editing. SX: Conceptualization, Supervision, Writing – review & editing, Project administration. JZ: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
Authors YL, HC, and SZ were employed by the company Suzhou Sano Precision Medicine Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. (2003) 3:421–9. doi: 10.1016/S1535-6108(03)00110-7
2. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 axis in cancer therapy. Cancers. (2017) 9:41. doi: 10.3390/cancers9050041
3. Gorecki L, Andrs M, Korabecny J. Clinical candidates targeting the ATR-CHK1-WEE1 axis in cancer. Cancers (Basel). (2021) 13(4):795. doi: 10.3390/cancers13040795
4. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U.S.A. (2000) 97:10389–94. doi: 10.1073/pnas.190030497
5. Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. (1998) 282:1893–7. doi: 10.1126/science.282.5395.1893
6. Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. (2001) 410:842–7. doi: 10.1038/35071124
7. Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy – two sides of the same coin? Nat Rev Cancer. (2007) 7:925–36. doi: 10.1038/nrc2251
8. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. (1998) 282:1497–501. doi: 10.1126/science.282.5393.1497
9. Rocca V, Blandino G, D'Antona L, Iuliano R, Di Agostino S. Li-fraumeni syndrome: mutation of TP53 is a biomarker of hereditary predisposition to tumor: new insights and advances in the treatment. Cancers (Basel). (2022) 14(15):3664. doi: 10.3390/cancers14153664
10. Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. (1999) 286:2528–31. doi: 10.1126/science.286.5449.2528
11. Vahteristo P, Tamminen A, Karvinen P, Eerola H, Eklund C, Aaltonen LA, et al. p53, CHK2, and CHK1 genes in finnish families with li-fraumeni syndrome: further evidence of CHK2 in inherited cancer predisposition1. Cancer Res. (2001) 61:5718–22.
12. Ning L, Wang H, San D, Sang J. C-terminal domain of Chk1 regulates its subcellular location and kinase activity for DNA repair. Chin Sci Bull. (2011) 56:3138–47. doi: 10.1007/s11434-011-4538-4
13. Emptage RP, Schoenberger MJ, Ferguson KM, Marmorstein R. Intramolecular autoinhibition of checkpoint kinase 1 is mediated by conserved basic motifs of the C-terminal kinase-associated 1 domain. J Biol Chem. (2017) 292:19024–33. doi: 10.1074/jbc.M117.811265
14. Kosoy A, O'Connell MJ. Regulation of Chk1 by its C-terminal domain. Mol Biol Cell. (2008) 19:4546–53. doi: 10.1091/mbc.e08-04-0444
15. Smits VAJ, Gillespie DA. DNA damage control: regulation and functions of checkpoint kinase 1. FEBS J. (2015) 282:3681–92. doi: 10.1111/febs.13387
16. Han X, Tang J, Wang J, Ren F, Zheng J, Gragg M, et al. Conformational change of human checkpoint kinase 1 (Chk1) induced by DNA damage. J Biol Chem. (2016) 291:12951–9. doi: 10.1074/jbc.M115.713248
17. Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. (2000) 100:681–92. doi: 10.1016/S0092-8674(00)80704-7
18. Lopes JL, Chaudhry S, Lopes GS, Levin NK, Tainsky MA. FANCM, RAD1, CHEK1 and TP53I3 act as BRCA-like tumor suppressors and are mutated in hereditary ovarian cancer. Cancer Genet. (2019) 235-236:57–64. doi: 10.1016/j.cancergen.2019.04.061
19. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. (2000) 14:1448–59. doi: 10.1101/gad.14.12.1448
20. Tho LM, Libertini S, Rampling R, Sansom O, Gillespie DA. Chk1 is essential for chemical carcinogen-induced mouse skin tumorigenesis. Oncogene. (2012) 31:1366–75. doi: 10.1038/onc.2011.326
21. Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, Vassilopoulos A, et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene. (2010) 29:4007–17. doi: 10.1038/onc.2010.163
22. Niida H, Murata K, Shimada M, Ogawa K, Ohta K, Suzuki K, et al. Cooperative functions of Chk1 and Chk2 reduce tumour susceptibility in vivo. EMBO J. (2010) 29:3558–70. doi: 10.1038/emboj.2010.218
23. Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res. (2007) 67:6574–81. doi: 10.1158/0008-5472.CAN-06-3545
24. Fadaka AO, Bakare OO, Sibuyi NRS, Klein A. Gene expression alterations and molecular analysis of CHEK1 in solid tumors. Cancers (Basel). (2020) 12(3):662. doi: 10.3390/cancers12030662
25. Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. (2014) 134:1013–23. doi: 10.1002/ijc.28226
26. Neizer-Ashun F, Bhattacharya R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. (2021) 497:202–11. doi: 10.1016/j.canlet.2020.09.016
27. Wang W-J, Wu S-P, Liu J-B, Shi Y-S, Huang X, Zhang Q-B, et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. (2013) 73:1219–31. doi: 10.1158/0008-5472.CAN-12-1408
28. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. (2014) 16:864–75. doi: 10.1038/ncb3013
29. David L, Fernandez-Vidal A, Bertoli S, Grgurevic S, Lepage B, Deshaies D, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signaling. (2016) 9:ra90–ra. doi: 10.1126/scisignal.aac9704
30. Menoyo A, Alazzouzi H, Espín E, Armengol M, Yamamoto H, Schwartz JR. S. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability1. Cancer Res. (2001) 61:7727–30.
31. Bertoni F, Codegoni AM, Furlan D, Tibiletti MG, Capella C, Broggini M. CHK1 frameshift mutations in genetically unstable colorectal and endometrial cancers. Genes Chromosomes Cancer. (1999) 26:176–80. doi: 10.1002/(ISSN)1098-2264
32. Kim CJ, Lee JH, Song JW, Cho YG, Kim SY, Nam SW, et al. Chk1 frameshift mutation in sporadic and hereditary non-polyposis colorectal cancers with microsatellite instability. Eur J Surg Oncol (EJSO). (2007) 33:580–5. doi: 10.1016/j.ejso.2007.02.007
33. Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability1. Cancer Res. (2002) 62:4095–9.
34. Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. (2010) 34:433–43. doi: 10.1097/PAS.0b013e3181cf3d79
35. Lynch HT, Casey MJ, Snyder CL, Bewtra C, Lynch JF, Butts M, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol. (2009) 3:97–137. doi: 10.1016/j.molonc.2009.02.004
36. Shaw PA, McLaughlin JR, Zweemer RP, Narod SA, Risch H, Verheijen RH, et al. Histopathologic features of genetically determined ovarian cancer. Int J Gynecol Pathol. (2002) 21:407–11. doi: 10.1097/00004347-200210000-00011
37. Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. (2003) 97:2187–95. doi: 10.1002/cncr.11310
38. Maehle L, Apold J, Paulsen T, Hagen B, Løvslett K, Fiane B, et al. High risk for ovarian cancer in a prospective series is restricted to BRCA1/2 mutation carriers. Clin Cancer Res. (2008) 14:7569–73. doi: 10.1158/1078-0432.CCR-08-0112
39. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. (2012) 30:2654–63. doi: 10.1200/JCO.2011.39.8545
40. Okamoto A, Glasspool RM, Mabuchi S, Matsumura N, Nomura H, Itamochi H, et al. Gynecologic Cancer InterGroup (GCIG) consensus review for clear cell carcinoma of the ovary. Int J Gynecol Cancer. (2014) 24:S20–5. doi: 10.1097/IGC.0000000000000289
41. Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol. (2012) 13:385–94. doi: 10.1016/S1470-2045(11)70404-1
42. Zhu C, Xu Z, Zhang T, Qian L, Xiao W, Wei H, et al. Updates of pathogenesis, diagnostic and therapeutic perspectives for ovarian clear cell carcinoma. J Cancer. (2021) 12:2295–316. doi: 10.7150/jca.53395
43. Xiao W, Awadallah A, Xin W. Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int J Clin Exp Pathol. (2012) 5:642–50.
44. Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. (2012) 25:615–24. doi: 10.1038/modpathol.2011.189
45. King CM, Barbara C, Prentice A, Brenton JD, Charnock-Jones DS. Models of endometriosis and their utility in studying progression to ovarian clear cell carcinoma. J Pathol. (2016) 238:185–96. doi: 10.1002/path.4657
46. Iida Y, Okamoto A, Hollis RL, Gourley C, Herrington CS. Clear cell carcinoma of the ovary: a clinical and molecular perspective. Int J Gynecol Cancer. (2021) 31:605–16. doi: 10.1136/ijgc-2020-001656
47. Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun. (2015) 6:6118. doi: 10.1038/ncomms7118
48. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discovery. (2015) 5:752–67. doi: 10.1158/2159-8290.CD-14-0849
49. Yap TA, Stadler ZK, Stout LA, Schneider BP. Aligning germline cancer predisposition with tumor-based next-generation sequencing for modern oncology diagnosis, interception, and therapeutic development. Am Soc Clin Oncol Educ Book. (2023) 43:e390738. doi: 10.1200/EDBK_390738
50. Stafford JL, Dyson G, Levin NK, Chaudhry S, Rosati R, Kalpage H, et al. Reanalysis of BRCA1/2 negative high risk ovarian cancer patients reveals novel germline risk loci and insights into missing heritability. PloS One. (2017) 12:e0178450. doi: 10.1371/journal.pone.0178450
51. Wu J, Wei Y, Pan J, Jin S, Gu W, Gan H, et al. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int J Cancer. (2021) 148:673–81. doi: 10.1002/ijc.33324
52. Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. (2000) 14:1439–47. doi: 10.1101/gad.14.12.1439
53. Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. (2004) 6:45–59. doi: 10.1016/j.ccr.2004.06.015
54. Poehlmann A, Roessner A. Importance of DNA damage checkpoints in the pathogenesis of human cancers. Pathol Res Practice. (2010) 206:591–601. doi: 10.1016/j.prp.2010.06.006
55. Boles NC, Peddibhotla S, Chen AJ, Goodell MA, Rosen JM. Chk1 haploinsufficiency results in anemia and defective erythropoiesis. PloS One. (2010) 5:e8581. doi: 10.1371/journal.pone.0008581
56. Schuler F, Afreen S, Manzl C, Häcker G, Erlacher M, Villunger A. Checkpoint kinase 1 is essential for fetal and adult hematopoiesis. EMBO Rep. (2019) 20:e47026. doi: 10.15252/embr.201847026
57. Zhang H, Chen T, Wu K, Hou Z, Zhao S, Zhang C, et al. Dominant mutations in CHK1 cause pronuclear fusion failure and zygote arrest that can be rescued by CHK1 inhibitor. Cell Res. (2021) 31:814–7. doi: 10.1038/s41422-021-00507-8
58. Chen B, Guo J, Wang T, Lee Q, Ming J, Ding F, et al. Maternal heterozygous mutation in CHEK1 leads to mitotic arrest in human zygotes. Protein Cell. (2022) 13:148–54. doi: 10.1007/s13238-021-00844-9
Keywords: CHEK1, germline mutation, splicing error, ovarian cancer, inherited cancer
Citation: Qian J, Peng M, Li Y, Liu W, Zou X, Chen H, Zhou S, Xiao S and Zhou J (2024) Case report: A germline CHEK1 c.613 + 2T>C leads to a splicing error in a family with multiple cancer patients. Front. Oncol. 14:1380093. doi: 10.3389/fonc.2024.1380093
Received: 01 February 2024; Accepted: 21 March 2024;
Published: 15 April 2024.
Edited by:
Thierry André Magnaldo, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Veenu Tripathi, National Cancer Institute (NIH), United StatesJiabi Qian, Guangzhou Medical University, China
Copyright © 2024 Qian, Peng, Li, Liu, Zou, Chen, Zhou, Xiao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhua Zhou, Znl5empoQHN1ZGEuZWR1LmNu; Sheng Xiao, c3hpYW9AcGFydG5lcnMub3Jn
†These authors have contributed equally to this work and share first authorship