 Rachel R. Tindall
Rachel R. Tindall Jennifer M. Bailey-Lundberg
Jennifer M. Bailey-Lundberg Yanna Cao
Yanna Cao Tien C. Ko1*
Tien C. Ko1*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 04 March 2024
Sec. Cancer Molecular Targets and Therapeutics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1362247
This article is part of the Research Topic Emerging Anti-cancer Compounds and Immunomodulators for Pancreatic Cancer Treatment View all 10 articles
The transforming growth factor (TGF)-β superfamily has important physiologic roles and is dysregulated in many pathologic processes, including pancreatic cancer. Pancreatic cancer is one of the most lethal cancer diagnoses, and current therapies are largely ineffective due to tumor resistance and late-stage diagnosis with poor prognosis. Recent efforts are focused on the potential of immunotherapies in improving therapeutic results for patients with pancreatic cancer, among which TGF-β has been identified as a promising target. This review focuses on the role of TGF-β in the diseased pancreas and pancreatic cancer. It also aims to summarize the current status of therapies targeting the TGF-β superfamily and postulate potential future directions in targeting the TGF-β signaling pathways.
Cytokines mediate the body’s natural response to injury at a systemic level (1). These cytokines can be subcategorized into the transforming growth factor (TGF)-β superfamily, interleukins, interferons, chemokines, and the tumor necrosis factor (TNF) superfamily (2–5). The TGF-β superfamily, one of the major groups, was first described as a family of growth factors released by fibroblasts that stimulated cell growth (6). In 1981, further investigation into these growth factors led to the purification of TGF-β, the first named member of the TGF-β superfamily (7, 8). While studying the purification techniques of this protein, isoforms of TGF-β were discovered, which were called TGF-β1, TGF-β2, and TGF-β3 (9–11). The role of these proteins was further elucidated with the discovery of other members, including the bone morphogenic proteins (BMP)s (12). Currently, 33 proteins are recognized in this superfamily, with subtypes including TGF-βs, BMPs, growth differentiation factors (GDF)s, inhibins, and activins (Table 1) (13–16). This mini-review focuses on the role of the TGF-β superfamily in pancreatic diseases, including pancreatic ductal adenocarcinoma (PDAC), and the current therapeutics targeting these pathways.
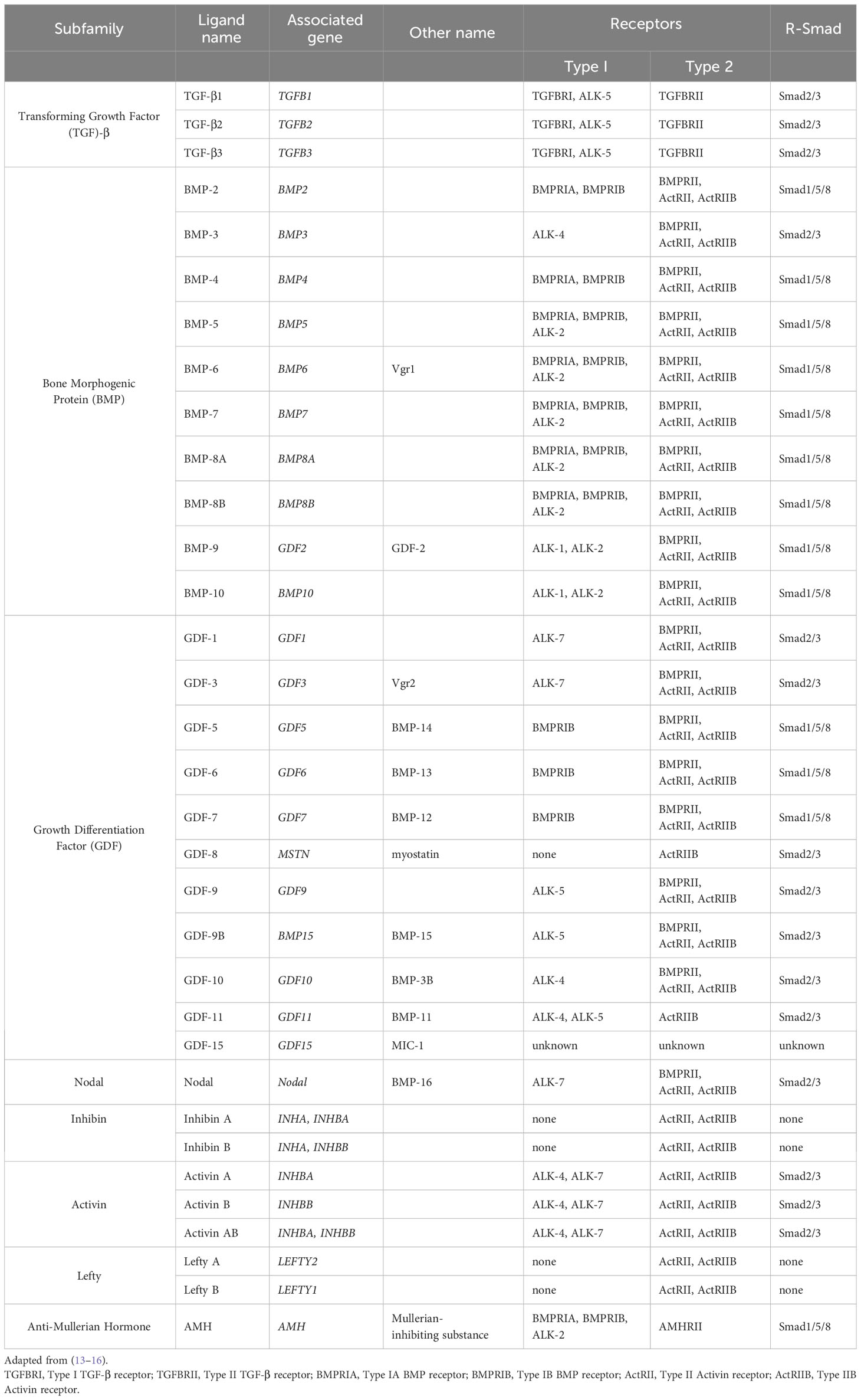
Table 1 Members of the TGF-β superfamily.
TGF-β is produced in a latent form. Activation of the latent form is initiated by regulatory T cells with a transmembrane protein, glycoprotein A repetitions predominant (GARP), which binds and cleaves pro-TGF-β to produce latent TGF-β. The latent form is activated by integrins (Figure 1A) (17, 18). BMPs, GDFs, and Lefty A and B are produced and processed similarly, with inactive precursors being cleaved and activated by proteases (Table 1) (19–21). The activins and inhibins are composed of common subunits and are formed by cleavage of dimerized subunits; inhibins are αβ heterodimers, and activins are ββ homodimers (Table 1) (16). The TGF-β superfamily is essential in physiologic functions, including tissue development and differentiation, regulation of immunologic responses, and tissue healing (13). These members activate physiologic activities through canonical and non-canonical signaling (22). Canonical signaling occurs through the SMAD pathway, where receptor-activated (R) SMAD1/5/8 and SMAD2/3 are phosphorylated by receptors following ligand binding (Figure 1B). These phosphorylated SMADs then complex with SMAD4 and translocate to the nucleus to regulate the expression of the target genes (Figure 1C) (13). Non-canonical pathways can be activated upon the ligand binding to the receptors, such as Erk, involved in epithelial-mesenchymal transition (EMT), and JNK/p38, involved in EMT and apoptosis (23).
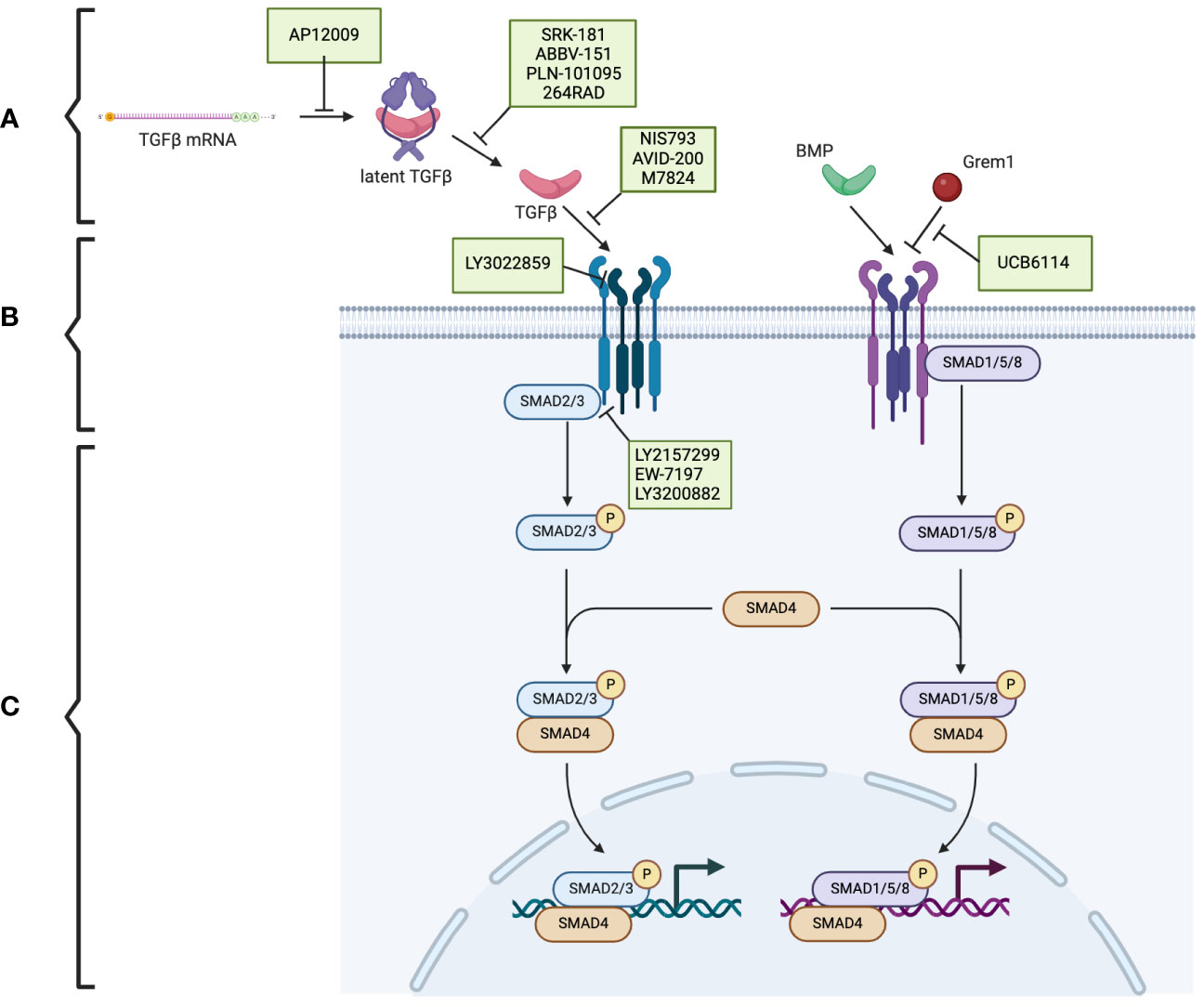
Figure 1 The TGF-β superfamily signaling pathway and the potential therapeutic targets in pancreatic cancer. (A) The TGF-β superfamily members. (B) TGF-β receptors and the phosphorylation/activation of the intracellular mediators of SMAD2/3 and SMAD1/5/8. (C) Downstream of the phosphorylated SMAD2/3 and SMAD1/5/8. Therapeutics (indicated in green boxes) targeting specific steps in the TGF-β superfamily signaling pathway are currently under investigation. Created using BioRender.com.
These signaling pathways play an essential role in proliferation and in controlling the growth of specific cell types, including epithelial cells, endothelial cells, immune cells, and neuronal cells, through growth inhibition and induction of apoptosis (24). BMPs are specifically involved in developing and maintaining skeletal tissues and are regulated extracellularly by antagonists, including Noggin, Chordin, and Gremlin1 (Grem1) (Figure 1A) (14). Additionally, activin A is a critical mesoderm-inducing factor (25), and GDFs are primarily involved in developmental processes (26). Inhibins antagonize activin signaling, and lefty inhibits phosphorylation of SMAD2 and subsequently regulates downstream signaling (Table 1) (16, 27).
TGF-β also plays a role in immunoregulation by inhibiting T-lymphocyte proliferation and promoting T-cell differentiation (28–30). Additionally, these proteins play an essential role in fibroblast activation and are involved in routine wound healing; TGF-β1 is secreted by the platelets forming the hemostatic plug and is a chemoattractant for monocytes and fibroblasts essential to tissue repair (31–34).
In addition to the essential role of TGF-β in physiologic mechanisms, aberrantly increased TGF-β has been shown to contribute to excess fibrosis (35). Administration of exogenous TGF-β leads to fibrosis in subcutaneous tissues, lung parenchyma, and hepatic tissue (36–38). Furthermore, dysregulation of the TGF-β signaling pathway contributes to carcinogenesis (39). For example, tumor cells have been shown to evade the growth regulation of TGF-β through mutations in the TGF-β receptors and SMAD family (40, 41).
PDAC is currently the third leading cause of cancer-related death (42). Poor outcomes can be attributed to both late diagnosis and a fibrotic tumor microenvironment that surrounds the cancer cells, creating a chemo-resistant barrier. Further understanding of the role of the TGF-β superfamily may elucidate potential targets for novel therapies that could improve patient outcomes.
TGF-β activity is paradoxical in pancreatic diseases, promoting or suppressing disease progression. The TGF-β superfamily modulates acute pancreatitis (AP) by regulating inflammation and apoptosis through canonical or non-canonical signaling. The TGF-β superfamily also plays distinct roles in the progression of chronic pancreatitis (CP) and PDAC through effects on pancreatic stellate cells (PSCs) and the extracellular matrix (ECM) production.
AP results from injury to the pancreatic acinar cells, leading to premature activation of pancreatic enzymes and causing pancreatic autodigestion and tissue inflammation (43). Apoptosis and suppression of proliferation have been reported to limit the inflammatory cascade in response to the insult in AP (44). TGF-β is released by various cells at the site of injury and induces apoptosis and suppress the proliferation of pancreatic acinar cells (45). However, this was contradicted by a study showing the induction of apoptosis upon suppressing the TGF-β signaling pathway in pancreatic epithelial cells (46). Additionally, our group has demonstrated that BMP signaling is upregulated in AP and causes dysregulation of autophagic processes. Administration of a BMP antagonist Noggin in vivo in a mouse model attenuated AP inflammation, suggesting a proinflammatory role of BMP signaling in AP (47).
In addition to involvement with apoptosis, TGF-β mediates the inflammatory response in AP through T-cell activation. Specifically, TGF-β induces the differentiation of both Th9 and Th17 cells, which are proinflammatory (48, 49). Th17 cells are known to secrete IL-17, associated with increased inflammatory markers and severity of AP (50).
CP results from repeated injury to the pancreas from recurrent bouts of AP, which leads to the replacement of normal pancreatic tissue with fibrotic scarring (51). This is primarily facilitated by the activation of PSCs, which secrete growth factors and chemokines such as TGF-β and produce excess ECM (52). The TGF-β secreted by activated PSCs is directly related to the characteristic fibrosis of CP (53). This fibrosis results from increased ECM production by PSCs (54) and inhibition of matrix metalloproteinases, which are involved in ECM degradation (55). Our group has shown that BMPs oppose the fibrogenic function of TGF-β on PSCs in CP by activating the SMAD1/5/8 pathway, which inhibits SMAD2 (56, 57).
Other modulators of TGF-β superfamily pathways are also involved in CP pathophysiology. Our group has shown that Grem1, an endogenous BMP antagonist, is pro-fibrogenic in a CP mouse model (58). Additionally, SMAD7, a known inhibitory SMAD, suppressed TGF-β signaling and modulated CP fibrosis through decreased ECM deposition and decreased inflammatory cell response in an in vivo mouse model (59).
The TGF-β superfamily plays dual roles in PDAC, promoting tumorigenesis in some capacities while acting as an inhibitor in others (60). In the early stages of PDAC, TGF-β has been shown to suppress tumor progression by promoting apoptosis and regulation of the cell cycle and promoting the stroma’s development by activating PSCs and increasing stromal production (61). Additionally, BMP2 expression is increased in pancreatic cancer and has variable mitogenic effects on pancreatic cancer cell lines, with a greater capacity to stimulate growth in cell lines with SMAD mutations (62). BMP signaling has also been shown to play a role in EMT through canonical BMP signaling, mediated by Grem1 inhibitory feedback, resulting in a maintenance of heterogeneity (63). Our group has also shown that activated fibroblasts express Grem1 and that increased expression is associated with a more severe tumor stage (64).
PDAC is associated with several common mutations in oncogenes and tumor suppressor genes, including mutations in KRAS, TP53, CDKN2A, and SMAD4. These mutations can affect TGF-β signaling pathways at various points, including the intracellular signaling molecules and receptors (65).
SMAD4 mutations are common in PDAC and are identified in approximately 60% of cases (66). Interestingly, an isolated SMAD4 mutation does not independently cause cancer; it must be paired with another mutation, such as KRAS (67). SMAD4 is part of the intracellular signaling pathway that complexes with activated and phosphorylated SMAD2/3 and SMAD1/5/8 in response to TGF-β and BMP binding their respective receptors (68). Mutation of SMAD4 results in loss of the tumor suppressor function of canonical TGF-β signaling (69). Loss of SMAD4 results in decreased T-cell recruitment and a suppressed immune response (70). Additionally, knockout of Smad4 in a PDAC mouse model has increased tumor sensitivity to host immune control and induced DNA damage (71).
Mutations in TGF-β receptors have been identified as disruptions of TGF-β signaling pathways that result in the loss of TGF-β suppressive effects. Studies have shown that mutations in TGFBR1, which encodes TGF-β type I receptor (TGFBRI), occur in approximately 1% of cases, and mutations in TGFBR2, which encodes TGF-β Type II receptor (TGFBRII), appear in approximately 4% of patients (72). Type III TGF-β receptor mutations also occur and result in increased EMT-associated increased motility and invasiveness (73).
Disruption in the expression of other receptors has also been reported. Deletion of ACVR1B, which encodes the ALK-4 receptor for activin A, is associated with a more aggressive cancer phenotype (74). Interestingly, mutations in BMPRI are described in patients with hereditary juvenile polyposis. BMPRI and BMPRII mRNA levels are upregulated in pancreatic cancers, and cells with higher levels have been shown to have more significant metastatic potential (75, 76).
KRAS is frequently mutated in human carcinomas and approximately 85% of PDAC cases (77). KRAS mutations are often detectable early in disease progression (78). GREM1 is upregulated in hereditary mixed polyposis syndrome, where duplications of the gene result in increased antagonism of BMP signaling (79). Similar mutations are observed in sporadic intestinal polyps (80). However, mutations in GREM1 have not been reported in cases of PDAC.
Management options for pancreatic cancer depend primarily on the stage of the cancer when it is diagnosed. Distant metastasis, retroperitoneal invasion, and invasion of the mesenteric root are contraindications to surgical resection. Chemotherapy is the standard of care for metastatic pancreatic cancer. Gemcitabine was considered the first line for a couple of decades following a randomized control trial showing more favorable outcomes than fluorouracil. However, survival for patients treated with gemcitabine was still dismal, with a median survival of 5.65 months (81). This regimen has been improved following the PRODIGE and MPACT studies, which evaluated FOLFIRINOX and albumin-bound paclitaxel plus gemcitabine, showing significant improvement in survival time compared to gemcitabine alone (82, 83). Despite these improved regimens, outcomes remain poor, leading to a focus on the potential of other treatment modalities, including immunotherapy.
Interestingly, chemotherapy has been shown to alter the tumor microenvironment through reprogramming and increased synthesis of chemokines, including TGF-β (84). Thus, TGF-β appears to be involved in the resistance to chemotherapy. Inhibiting TGF-β has become a focus of therapeutic intervention and shows promising results in treating PDAC.
Because of the complexity of the TGF-β signaling pathway, numerous potential targets are under investigation (Figure 1). Therapeutic strategies include antisense oligonucleotides, neutralizing antibodies, ligand traps, and small molecule kinase inhibitors. Many of these therapies are being investigated in several cancers, including pancreatic cancer.
Trabedersen (AP12009), specific for TGFB2 mRNA, reduced TGFB2 expression in human pancreatic cancer cell lines, resulted in decreased proliferation and migration, and reversed immunosuppressive effects (Figure 1A) (85). A phase 2 clinical study showed a good safety profile, with the only identified adverse effect being transient thrombocytopenia and a mean survival of 13.4 months for 61 patients with pancreatic cancer (86). Further clinical trials have yet to be published.
In preclinical studies, SRK-181, which specifically targets latent TGF-β1, countered TGF-β-mediated resistance to cancer checkpoint blockade therapy (Figure 1A) (87). It is currently under investigation in the DRAGON trial (NCT04291079), a phase 1 clinical trial investigating it as a monotherapy or in combination with anti-PD-L1 in patients with solid tumors, including pancreatic tumors, which has shown no dose-limiting toxicity and adverse effects limited to fatigue, anorexia, and nausea. One patient with pancreatic cancer who was treated with SRK-181 as a monotherapy showed stable disease (88).
Livmoniplimab (ABBV-151) targets GARP-TGF-β1 and prevents the release of active TGF-β1 (Figure 1A). It is currently under investigation in a phase 1 trial (NCT03821935), investigating it as a single agent or combined with Budigalimab in patients with locally advanced or metastatic solid tumors. This clinical trial is still in the recruiting phase, and preliminary results are not yet available (89).
PLN-101095 targets integrin αvβ8 and αvβ1 and prevents activation of TGF-β (Figure 1A). It has shown enhanced response to standard chemotherapy regimens in preclinical studies (90) and is currently in a phase 1 clinical trial (91). Additionally, 264RAD inhibits integrin αvβ6 and has shown promising results in preclinical trials (92); however, further clinical trials have not been pursued.
NIS793 binds and neutralizes active TGF-β with high affinity and has been shown to decrease fibroblasts and enhance tumor cell chemosensitivity (Figure 1B) (93). In a phase 1b trial (NCT02947165), 120 patients, of which ten had pancreatic cancer, were treated with NIS793 as a monotherapy or in combination with spartalizumab. Partial response was observed in 2.5% of patients, and stable disease was observed in 24.2% of patients. While no dose-limiting toxicity was observed, nearly half experienced an adverse event, most commonly rash (94). A phase 2 trial (NCT04390763) and phase 3 trial (NCT04935359) are ongoing to evaluate the drug’s effect in patients with metastatic pancreatic ductal adenocarcinoma.
LY3022859 targets the type II TGF-β receptor and inhibits signaling activation (Figure 1B). A phase 1 trial (NCT01646203) was discontinued due to patients developing uncontrollable cytokine release syndrome (95).
AVID-200 is explicitly designed to resemble the receptor ectodomain for TGF-β1 and TGF-β3 and has been shown to enhance the efficacy of immune checkpoint inhibitors in preclinical trials (Figure 1B) (96, 97). It recently underwent a phase 1 clinical trial (NCT03834662) for solid tumors, including PDAC (98).
Activated T-cells present PD-1 on the surface, which can be exploited by tumor cells expressing PD-L1. PD-L1 binding to PD-1 inactivates the T-cells and prevents the T-cell-regulated destruction of the tumor cells (99). Thus, the PD-1 signaling pathway has been identified as a promising target for cancer immunotherapy (100). Specific interest has arisen in dual inhibition of the PD-1 and TGF-β signaling pathways, which is hypothesized to enhance the anti-tumor activity (101). Bintrafusp alfa (M7824) is a bifunctional fusion protein with a type II TGF-β receptor fused to an antibody against PD-L1 (Figure 1B) (102), which has undergone a phase 1 clinical trial (NCT02517398) that included five patients with pancreatic cancer. Three patients had a response of stable disease, one of partial response, and one of progressive disease (103).
Galunisertib (LY2157299) is an oral drug that inhibits the type I TGF-β receptor kinase and down-regulates the phosphorylation of SMAD2 (Figure 1B) (104). In phase 1 and 2 clinical trials, a combination of galunisertib and gemcitabine resulted in an improved survival time of 8.9 months compared to 7.1 months in patients treated with just galunisertib with minimal increase in toxicity in patients with locally advanced or metastatic pancreatic adenocarcinoma (105).
Vactosertib (EW-7197), a type I TGF-β receptor inhibitor, has been shown to augment gemcitabine and decrease the expression of ECM components, improving the sensitivity of pancreatic cancer cells to gemcitabine (Figure 1B) (106). It also has synergistic effects when combined with T1-44, an inhibitor of PRMT5 methyltransferase (107). It has been investigated in a phase 1b clinical trial in combination with FOLFOX in sixteen patients with pancreatic ductal adenocarcinoma; three patients had a partial response, and five had stable disease (108).
LY3200882, an oral type I TGF-β receptor inhibitor, was investigated in a phase 1 clinical trial (Figure 1B). LY3200882 was used in combination with gemcitabine and nab-paclitaxel in twelve patients with pancreatic cancer. Six of the twelve patients had partial responses, and all but one demonstrated decreased tumor size (109).
In addition to TGF-β, other members of the TGF-β superfamily are promising targets for cancer therapeutics. Interestingly, ginisortamab (UCB6114), an antibody that neutralizes Grem1 and blocks its antagonistic effects on BMP signaling, has been shown to restore BMP signaling pathways in human colorectal cancer cell lines and fibroblasts (Figure 1B) (110). It is currently being evaluated by a phase 1/2 clinical trial (NCT04393298) in advanced solid tumors, including pancreatic adenocarcinomas.
GDF-15 has also been identified as a potential target. While the exact signaling pathway has yet to be elucidated, recent studies have identified a unique GDF-15 receptor glial cell-derived neurotrophic factor family receptor α-like (GFRAL) and have shown that GDF-15 inhibits leukocyte integrin activation and T cell migration, which is reversed with neutralization of GDF-15 (111, 112). Visugromab (CTL-002), a neutralizing antibody of GDF-15, is currently under evaluation in phase 2a of the GDFATHER trial (NCT04725474) and is showing promising results in combination with nivolumab in advanced non-small cell lung cancer and urothelial cancer.
TGF-β and associated proteins undoubtedly play a role in the development of pancreatic disease and disease progression from AP to CP to PDAC. However, the heterogeneous nature of pancreatic tissue and the dynamic role of the TGF-β superfamily and associated signaling pathways result in nuanced implications for therapeutics that target these pathways.
Systemic therapies such as chemotherapy have been the standard of care for patients with pancreatic cancer following trials such as PRODIGE 4/ACCORD 11 and MPACT; however, outcomes remain very poor with short survival times (83, 113). This has led to an interest in targeted therapies such as immunotherapy, which modulate a patient’s immune system response. Such therapies have survival benefits in several types of solid tumors, including upper gastrointestinal tumors and colorectal cancers (114, 115). However, similar benefits from these immunotherapies have not been seen in pancreatic cancer, mainly because pancreatic tumors are immunologically cold due to the unique immunosuppressive tumor microenvironment with limited immune cells (116). Thus, current strategies seek to target components of the microenvironment that contribute to this immunosuppression to improve the responsiveness of tumors to immunotherapy (117). Subsequently, TGF-β signaling pathways became targets of interest, given the role of TGF-β in immunosuppression and ECM production.
Several therapeutics that target TGF-β and associated pathway molecules are currently under investigation in early clinical trials for solid tumors, including pancreatic tumors. These targets include modulating the TGF-β signaling pathway directly and targeting other proteins in the superfamily, such as Grem1, which inhibits BMP signaling. Additionally, antagonism of BMP has been suggested as a potential target to block and reduce pancreatic cancer invasiveness (118). However, due to the context-dependent manner of BMP signaling, the efficacy of such therapeutics varies greatly, and further investigation into the subtleties of BMP signaling in both oncogenic and tumor-suppressive functions is warranted (119).
Because the TGF-β superfamily has heterogenous roles in pancreatic tumor development, the effectiveness of these therapies has yet to be fully elucidated. TGF-β signaling pathways are involved in immunosuppression and ECM production, but TGF-β pathways also regulate cell cycle progression. Regardless, data from the recent clinical trials suggest hopeful results. Side effect profiles were essentially minimal, but the potential adverse effects of TGF-β targeting drugs when delivered systemically should be a point of investigation in future studies, as blockage of the signaling pathways has previously been shown to have contradictory effects depending on cell type (120). Targeted drug delivery to the pancreatic tumor microenvironment may help mitigate such effects.
Ultimately, definitive management of pancreatic disease will likely require a multifaceted treatment plan due to the heterogeneous nature of the disease processes. TGF-β has been shown to augment the microenvironment, which likely contributes to the characteristic resistance and poor outcomes of PDAC. Inhibition of these signaling pathways shows promising results in boosting the effects of traditional therapeutics. Pairing modulators of TGF-β signaling pathways with conventional systemic treatments such as chemotherapy and other immune modulators such as PD-1 inhibitors will address the mechanisms of resistance that have contributed to the poor outcomes of pancreatic cancer and allow for a more comprehensive treatment regimen.
RT: Conceptualization, Validation, Writing – original draft, Writing – review & editing, Funding acquisition, Investigation, Visualization. JB-L: Conceptualization, Writing – review & editing, Funding acquisition, Investigation, Visualization. YC: Conceptualization, Supervision, Validation, Writing – review & editing, Investigation, Project administration, Visualization. TK: Conceptualization, Supervision, Writing – review & editing, Funding acquisition, Investigation, Project administration.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partially supported by the NIH R21 AA027014 and Jack H Mayfield M.D. Distinguished Professorship in Surgery (TK), Dean’s fund for Summer Research Program at UTHealth MMS (RT). JB-L is supported by NIH R01 CA277161.
Figure 1 was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. (2009) 9:480–90. doi: 10.1038/nri2580
2. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
3. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
4. Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. (2000) 12:121–7. doi: 10.1016/S1074-7613(00)80165-X
5. Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. (1999) 17:331–67. doi: 10.1146/annurev.immunol.17.1.331
6. de Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci U S A. (1978) 75:4001–5. doi: 10.1073/pnas.75.8.4001
7. Moses HL, Branum EL, Proper JA, Robinson RA. Transforming growth factor production by chemically transformed cells. Cancer Res. (1981) 41:2842–8.
8. Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. (1981) 78:5339–43. doi: 10.1073/pnas.78.9.5339
9. Cheifetz S, Weatherbee JA, Tsang ML, Anderson JK, Mole JE, Lucas R, et al. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell. (1987) 48:409–15. doi: 10.1016/0092-8674(87)90192-9
10. Derynck R, Lindquist PB, Lee A, Wen D, Tamm J, Graycar JL, et al. A new type of transforming growth factor-beta, TGF-beta 3. EMBO J. (1988) 7:3737–43. doi: 10.1002/embj.1988.7.issue-12
11. ten Dijke P, Hansen P, Iwata KK, Pieler C, Foulkes JG. Identification of another member of the transforming growth factor type beta gene family. Proc Natl Acad Sci U S A. (1988) 85:4715–9. doi: 10.1073/pnas.85.13.4715
12. Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, et al. Novel regulators of bone formation: molecular clones and activities. Science. (1988) 242:1528–34. doi: 10.1126/science.3201241
13. Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. (2016) 8. doi: 10.1101/cshperspect.a021873
14. Katagiri T, Watabe T. Bone morphogenetic proteins. Cold Spring Harb Perspect Biol. (2016) 8. doi: 10.1101/cshperspect.a021899
15. Low EL, Baker AH, Bradshaw AC. TGFbeta, smooth muscle cells and coronary artery disease: a review. Cell Signal. (2019) 53:90–101. doi: 10.1016/j.cellsig.2018.09.004
16. Namwanje M, Brown CW. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol. (2016) 8. doi: 10.1101/cshperspect.a021881
17. Lienart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, et al. Structural basis of latent TGF-beta1 presentation and activation by GARP on human regulatory T cells. Science. (2018) 362:952–6. doi: 10.1126/science.aau2909
18. Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. (2008) 87:601–15. doi: 10.1016/j.ejcb.2008.01.012
19. Nelsen SM, Christian JL. Site-specific cleavage of BMP4 by furin, PC6, and PC7. J Biol Chem. (2009) 284:27157–66. doi: 10.1074/jbc.M109.028506
20. Bauskin AR, Brown DA, Junankar S, Rasiah KK, Eggleton S, Hunter M, et al. The propeptide mediates formation of stromal stores of PROMIC-1: role in determining prostate cancer outcome. Cancer Res. (2005) 65:2330–6. doi: 10.1158/0008-5472.CAN-04-3827
21. Ulloa L, Creemers JW, Roy S, Liu S, Mason J, Tabibzadeh S. Lefty proteins exhibit unique processing and activate the MAPK pathway. J Biol Chem. (2001) 276:21387–96. doi: 10.1074/jbc.M006933200
22. Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-beta - an excellent servant but a bad master. J Transl Med. (2012) 10:183. doi: 10.1186/1479-5876-10-183
23. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. (2009) 19:128–39. doi: 10.1038/cr.2008.328
24. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. (2003) 3:807–21. doi: 10.1038/nrc1208
25. Asashima M, Nakano H, Shimada K, Kinoshita K, Ishii K, Shibai H, et al. Mesodermal induction in early amphibian embryos by activin A (erythroid differentiation factor). Rouxs Arch Dev Biol. (1990) 198:330–5. doi: 10.1007/BF00383771
26. Wischhusen J, Melero I, Fridman WH. Growth/differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Front Immunol. (2020) 11:951. doi: 10.3389/fimmu.2020.00951
27. Ulloa L, Tabibzadeh S. Lefty inhibits receptor-regulated Smad phosphorylation induced by the activated transforming growth factor-beta receptor. J Biol Chem. (2001) 276:21397–404. doi: 10.1074/jbc.M010783200
28. Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. (1986) 163:1037–50. doi: 10.1084/jem.163.5.1037
29. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
30. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. (2008) 9:632–40. doi: 10.1038/ni.1607
31. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. (1983) 258:7155–60. doi: 10.1016/S0021-9258(18)32345-7
32. Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. (1987) 84:5788–92. doi: 10.1073/pnas.84.16.5788
33. Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J Exp Med. (1987) 165:251–6. doi: 10.1084/jem.165.1.251
34. Lodyga M, Hinz B. TGF-beta1 - A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol. (2020) 101:123–39. doi: 10.1016/j.semcdb.2019.12.010
35. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol Cancer. (2022) 21:104. doi: 10.1186/s12943-022-01569-x
36. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. (1986) 83:4167–71. doi: 10.1073/pnas.83.12.4167
37. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. (1997) 100:768–76. doi: 10.1172/JCI119590
38. Dooley S, ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res. (2012) 347:245–56. doi: 10.1007/s00441-011-1246-y
39. Korkut A, Zaidi S, Kanchi RS, Rao S, Gough NR, Schultz A, et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-beta superfamily. Cell Syst. (2018) 7:422–37.e7. doi: 10.1016/j.cels.2018.08.010
40. Bharathy S, Xie W, Yingling JM, Reiss M. Cancer-associated transforming growth factor beta type II receptor gene mutant causes activation of bone morphogenic protein-Smads and invasive phenotype. Cancer Res. (2008) 68:1656–66. doi: 10.1158/0008-5472.CAN-07-5089
41. Shi Y, Hata A, Lo RS, Massague J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. (1997) 388:87–93. doi: 10.1038/40431
42. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
43. Bhatia M, Wong FL, Cao Y, Lau HY, Huang J, Puneet P, et al. Pathophysiology of acute pancreatitis. Pancreatology. (2005) 5:132–44. doi: 10.1159/000085265
44. Bhatia M. Apoptosis of pancreatic acinar cells in acute pancreatitis: is it good or bad? J Cell Mol Med. (2004) 8:402–9. doi: 10.1111/j.1582-4934.2004.tb00330.x
45. Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, et al. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. (1997) 99:2365–74. doi: 10.1172/JCI119418
46. Liu X, Yu M, Chen Y, Zhang J. Galunisertib (LY2157299), a transforming growth factor-beta receptor I kinase inhibitor, attenuates acute pancreatitis in rats. Braz J Med Biol Res. (2016) 49:e5388. doi: 10.1590/1414-431X20165388
47. Cao Y, Yang W, Tyler MA, Gao X, Duan C, Kim SO, et al. Noggin attenuates cerulein-induced acute pancreatitis and impaired autophagy. Pancreas. (2013) 42:301–7. doi: 10.1097/MPA.0b013e31825b9f2c
48. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
49. Jabeen R, Kaplan MH. The symphony of the ninth: the development and function of Th9 cells. Curr Opin Immunol. (2012) 24:303–7. doi: 10.1016/j.coi.2012.02.001
50. Jia R, Tang M, Qiu L, Sun R, Cheng L, Ma X, et al. Increased interleukin-23/17 axis and C-reactive protein are associated with severity of acute pancreatitis in patients. Pancreas. (2015) 44:321–5. doi: 10.1097/MPA.0000000000000284
51. Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. (2007) 132:1557–73. doi: 10.1053/j.gastro.2007.03.001
52. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol. (2012) 3:344. doi: 10.3389/fphys.2012.00344
53. Yoo BM, Yeo M, Oh TY, Choi JH, Kim WW, Kim JH, et al. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas. (2005) 30:e71–9. doi: 10.1097/01.mpa.0000157388.54016.0a
54. Vogelmann R, Ruf D, Wagner M, Adler G, Menke A. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-beta1 transgenic mouse model. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G164–72. doi: 10.1152/ajpgi.2001.280.1.G164
55. Shek FW, Benyon RC, Walker FM, McCrudden PR, Pender SL, Williams EJ, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. (2002) 160:1787–98. doi: 10.1016/S0002-9440(10)61125-X
56. Gao X, Cao Y, Yang W, Duan C, Aronson JF, Rastellini C, et al. BMP2 inhibits TGF-beta-induced pancreatic stellate cell activation and extracellular matrix formation. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G804–13. doi: 10.1152/ajpgi.00306.2012
57. Gao X, Cao Y, Staloch DA, Gonzales MA, Aronson JF, Chao C, et al. Bone morphogenetic protein signaling protects against cerulein-induced pancreatic fibrosis. PLoS One. (2014) 9:e89114. doi: 10.1371/journal.pone.0089114
58. Staloch D, Gao X, Liu K, Xu M, Feng X, Aronson JF, et al. Gremlin is a key pro-fibrogenic factor in chronic pancreatitis. J Mol Med (Berl). (2015) 93:1085–93. doi: 10.1007/s00109-015-1308-9
59. Li X, Nania S, Fejzibegovic N, Moro CF, Klopp-Schulze L, Verbeke C, et al. Cerulein-induced pancreatic fibrosis is modulated by Smad7, the major negative regulator of transforming growth factor-beta signaling. Biochim Biophys Acta. (2016) 1862:1839–46. doi: 10.1016/j.bbadis.2016.06.017
60. Akhurst RJ, Derynck R. TGF-beta signaling in cancer–a double-edged sword. Trends Cell Biol. (2001) 11:S44–51. doi: 10.1016/s0962-8924(01)02130-4
61. Principe DR, Timbers KE, Atia LG, Koch RM, Rana A. TGFbeta signaling in the pancreatic tumor microenvironment. Cancers (Basel). (2021) 13:5086. doi: 10.3390/cancers13205086
62. Kleeff J, Maruyama H, Ishiwata T, Sawhney H, Friess H, Buchler MW, et al. Bone morphogenetic protein 2 exerts diverse effects on cell growth in vitro and is expressed in human pancreatic cancer in vivo. Gastroenterology. (1999) 116:1202–16. doi: 10.1016/s0016-5085(99)70024-7
63. Lan L, Evan T, Li H, Hussain A, Ruiz EJ, Zaw Thin M, et al. GREM1 is required to maintain cellular heterogeneity in pancreatic cancer. Nature. (2022) 607:163–8. doi: 10.1038/s41586-022-04888-7
64. Davis JM, Cheng B, Drake MM, Yu Q, Yang B, Li J, et al. Pancreatic stromal Gremlin 1 expression during pancreatic tumorigenesis. Genes Dis. (2022) 9:108–15. doi: 10.1016/j.gendis.2020.05.001
65. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. (2008) 3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305
66. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. (1996) 271:350–3. doi: 10.1126/science.271.5247.350
67. Leung L, Radulovich N, Zhu CQ, Wang D, To C, Ibrahimov E, et al. Loss of canonical Smad4 signaling promotes KRAS driven Malignant transformation of human pancreatic duct epithelial cells and metastasis. PloS One. (2013) 8:e84366. doi: 10.1371/journal.pone.0084366
68. Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. (2002) 296:1646–7. doi: 10.1126/science.1071809
69. Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor beta (TGF-beta) target genes and distinguishes TGF-beta-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. (2005) 25:8108–25. doi: 10.1128/MCB.25.18.8108-8125.2005
70. Principe DR, Underwood PW, Kumar S, Timbers KE, Koch RM, Trevino JG, et al. Loss of SMAD4 is associated with poor tumor immunogenicity and reduced PD-L1 expression in pancreatic cancer. Front Oncol. (2022) 12:806963. doi: 10.3389/fonc.2022.806963
71. Xiong W, He W, Wang T, He S, Xu F, Wang Z, et al. Smad4 deficiency promotes pancreatic cancer immunogenicity by activating the cancer-autonomous DNA-sensing signaling axis. Adv Sci (Weinh). (2022) 9:e2103029. doi: 10.1002/advs.202103029
72. Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. (1998) 58:5329–32.
73. Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis. (2008) 29:252–62. doi: 10.1093/carcin/bgm249
74. Togashi Y, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, Fujita Y, et al. Homozygous deletion of the activin A receptor, type IB gene is associated with an aggressive cancer phenotype in pancreatic cancer. Mol Cancer. (2014) 13:126. doi: 10.1186/1476-4598-13-126
75. Zhou XP, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M, Hampel H, et al. Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet. (2001) 69:704–11. doi: 10.1086/323703
76. Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, et al. Gene expression profiles in normal and cancer cells. Science. (1997) 276:1268–72. doi: 10.1126/science.276.5316.1268
77. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discovery. (2014) 13:828–51. doi: 10.1038/nrd4389
78. Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. (2005) 7:17–23. doi: 10.1593/neo.04445
79. Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. (2012) 44:699–703. doi: 10.1038/ng.2263
80. Davis H, Irshad S, Bansal M, Rafferty H, Boitsova T, Bardella C, et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med. (2015) 21:62–70. doi: 10.1038/nm.3750
81. Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. (1997) 15:2403–13. doi: 10.1200/JCO.1997.15.6.2403
82. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
83. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. (2013) 369:1691–703. doi: 10.1056/NEJMoa1304369
84. Principe DR, Narbutis M, Kumar S, Park A, Viswakarma N, Dorman MJ, et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. (2020) 80:3101–15. doi: 10.1158/0008-5472.CAN-19-2959
85. Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. (2011) 102:1193–200. doi: 10.1111/j.1349-7006.2011.01917.x
86. Oettle H, Seufferlein T, Luger T, Schmid RM, v. Wichert G, Endlicher E, et al. Final results of a phase I/II study in patients with pancreatic cancer, Malignant melanoma, and colorectal carcinoma with trabedersen. J Clin Oncol. (2012) 30:4034–4. doi: 10.1200/jco.2012.30.15_suppl.4034
87. Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. (2020) 12:eaay8456. doi: 10.1126/scitranslmed.aay8456
88. Yap T, Gainor J, McKean M, Johnson M, Bockornv B, Barve M, et al 780 SRK-181, a latent TGFβ1 inhibitor: safety, efficacy, and biomarker results from the dose escalation portion of a phase I trial (DRAGON trial) in patients with advanced solid tumors. J ImmunoTherapy Cancer. (2022) 10:A812–2. doi: 10.1136/jitc-2022-SITC2022.0780
89. Powderly J, Spira A, Kondo S, Doi T, Luke JJ, Rasco D, et al. Model informed dosing regimen and phase I results of the anti-PD-1 antibody budigalimab (ABBV-181). Clin Transl Sci. (2021) 14:277–87. doi: 10.1111/cts.12855
90. Fajardo DC, Moore CL, Porazinski S, McLean B, Borbilas E, Australian Pancreatic, et al 1467 Selective targeting of integrins αVβ8 and αVβ1 within the dynamic ecosystem of pancreatic cancer to improve the overall anti-tumor response. J ImmunoTherapy Cancer. (2023) 11:A1631–1. doi: 10.1136/jitc-2023-SITC2023.1467
91. Daud A, Barnes CN, Owen S, Turner SM, Sznol M, Lefebvre EA, et al 714 Phase 1a trial of PLN-101095, an integrin αvβ8 and αvβ1 inhibitor, as monotherapy and in combination with pembrolizumab, in treatment-resistant patients with advanced or metastatic solid tumors. J ImmunoTherapy Cancer. (2023) 11:A809–9. doi: 10.1136/jitc-2023-SITC2023.0714
92. Reader CS, Vallath S, Steele CW, Haider S, Brentnall A, Desai A, et al. The integrin alphavbeta6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J Pathol. (2019) 249:332–42. doi: 10.1002/path.5320
93. Qiang L, Hoffman MT, Ali LR, Castillo JI, Kageler L, Temesgen A, et al. Transforming growth factor-beta blockade in pancreatic cancer enhances sensitivity to combination chemotherapy. Gastroenterology. (2023) 165:874–90.e10. doi: 10.1053/j.gastro.2023.05.038
94. Bauer TM, Santoro A, Lin CC, Garrido-Laguna I, Joerger M, Greil R, et al Phase I/Ib, open-label, multicenter, dose-escalation study of the anti-TGF-β monoclonal antibody, NIS793, in combination with spartalizumab in adult patients with advanced tumors. J ImmunoTherapy Cancer. (2023) 11:e007353. doi: 10.1136/jitc-2023-007353
95. Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2017) 79:673–80. doi: 10.1007/s00280-017-3245-5
96. O'Connor-McCourt MD, Tremblay G, Lenferink A, Sulea T, Zwaagstra J, Koropatnick J. Abstract 1759: AVID200, a highly potent TGF-beta trap, exhibits optimal isoform selectivity for enhancing anti-tumor T-cell activity, without promoting metastasis or cardiotoxicity. Cancer Res. (2018) 78:1759–9. doi: 10.1158/1538-7445.Am2018-1759
97. Tremblay G, Gruosso T, Denis J-F, Figueredo R, Koropatnick J, O'Connor-McCourt M. Abstract 6710: AVID200, a first-in-class selective TGF-beta 1 and -beta 3 inhibitor, sensitizes tumors to immune checkpoint blockade therapies. Cancer Res. (2020) 80:6710–0. doi: 10.1158/1538-7445.Am2020-6710
98. Yap TA, Lakhani NJ, Araujo DV, Ahnert JR, Chandana SR, Sharma M, et al. AVID200, first-in-class TGF-beta 1 and 3 selective and potent inhibitor: Safety and biomarker results of a phase I monotherapy dose-escalation study in patients with advanced solid tumors. J Clin Oncol. (2020) 38:3587–7. doi: 10.1200/JCO.2020.38.15_suppl.3587
99. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. (2017) 214:895–904. doi: 10.1084/jem.20160801
100. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J BioMed Sci. (2017) 24:26. doi: 10.1186/s12929-017-0329-9
101. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, et al. Dual inhibition of TGF-beta and PD-L1: a novel approach to cancer treatment. Mol Oncol. (2022) 16:2117–34. doi: 10.1002/1878-0261.13146
102. Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFbeta Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. (2018) 7:e1426519. doi: 10.1080/2162402X.2018.1426519
103. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin Cancer Res. (2018) 24:1287–95. doi: 10.1158/1078-0432.CCR-17-2653
104. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. (2015) 9:4479–99. doi: 10.2147/DDDT.S86621
105. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. (2018) 119:1208–14. doi: 10.1038/s41416-018-0246-z
106. Lee JE, Lee P, Yoon YC, Han BS, Ko S, Park MS, et al. Vactosertib, TGF-beta receptor I inhibitor, augments the sensitization of the anti-cancer activity of gemcitabine in pancreatic cancer. BioMed Pharmacother. (2023) 162:114716. doi: 10.1016/j.biopha.2023.114716
107. Hong E, Barczak W, Park S, Heo JS, Ooshima A, Munro S, et al. Combination treatment of T1-44, a PRMT5 inhibitor with Vactosertib, an inhibitor of TGF-beta signaling, inhibits invasion and prolongs survival in a mouse model of pancreatic tumors. Cell Death Dis. (2023) 14:93. doi: 10.1038/s41419-023-05630-5
108. Kim ST, Hong JY, Park YS, Kim S-J, Park JO. Phase 1b study of vactosertib in combination with oxaliplatin with 5FU/LV (FOLFOX) in patients with metastatic pancreatic cancer who have failed first-line gemcitabine/nab-paclitaxel. J Clin Oncol. (2022) 40:e16299–9. doi: 10.1200/JCO.2022.40.16_suppl.e16299
109. Yap TA, Vieito M, Baldini C, Sepulveda-Sanchez JM, Kondo S, Simonelli M, et al. First-in-human phase I study of a next-generation, oral, TGFbeta receptor 1 inhibitor, LY3200882, in patients with advanced cancer. Clin Cancer Res. (2021) 27:6666–76. doi: 10.1158/1078-0432.CCR-21-1504
110. Davies GCG, Dedi N, Jones PS, Kevorkian L, McMillan D, Ottone C, et al. Discovery of ginisortamab, a potent and novel anti-gremlin-1 antibody in clinical development for the treatment of cancer. MAbs. (2023) 15:2289681. doi: 10.1080/19420862.2023.2289681
111. Haake M, Haack B, Schafer T, Harter PN, Mattavelli G, Eiring P, et al. Tumor-derived GDF-15 blocks LFA-1 dependent T cell recruitment and suppresses responses to anti-PD-1 treatment. Nat Commun. (2023) 14:4253. doi: 10.1038/s41467-023-39817-3
112. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. (2017) 23:1158–66. doi: 10.1038/nm.4394
113. Gourgou-Bourgade S, Bascoul-Mollevi C, Desseigne F, Ychou M, Bouche O, Guimbaud R, et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: results from the PRODIGE 4/ACCORD 11 randomized trial. J Clin Oncol. (2013) 31:23–9. doi: 10.1200/JCO.2012.44.4869
114. Kelly RJ, Ajani JA, Kuzdzal J, Zander T, Van Cutsem E, Piessen G, et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med. (2021) 384:1191–203. doi: 10.1056/NEJMoa2032125
115. André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
116. Ullman NA, Burchard PR, Dunne RF, Linehan DC. Immunologic strategies in pancreatic cancer: making cold tumors hot. J Clin Oncol. (2022) 40:2789–805. doi: 10.1200/JCO.21.02616
117. Zabransky DJ, Yarchoan M, Jaffee EM. Strategies for heating up cold tumors to boost immunotherapies. Annu Rev Cancer Biol. (2023) 7:149–70. doi: 10.1146/annurev-cancerbio-061421-040258
118. Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis. (2009) 30:238–48. doi: 10.1093/carcin/bgn274
119. Ehata S, Yokoyama Y, Takahashi K, Miyazono K. Bi-directional roles of bone morphogenetic proteins in cancer: another molecular Jekyll and Hyde? Pathol Int. (2013) 63:287–96. doi: 10.1111/pin.12067
Keywords: TGF-β, acute pancreatitis, chronic pancreatitis, pancreatic ductal adenocarcinoma, pancreatic stellate cells
Citation: Tindall RR, Bailey-Lundberg JM, Cao Y and Ko TC (2024) The TGF-β superfamily as potential therapeutic targets in pancreatic cancer. Front. Oncol. 14:1362247. doi: 10.3389/fonc.2024.1362247
Received: 28 December 2023; Accepted: 15 February 2024;
Published: 04 March 2024.
Edited by:
Yinan Zhang, Nanjing University of Chinese Medicine, ChinaReviewed by:
Jingjing Zhuang, Shandong University, ChinaCopyright © 2024 Tindall, Bailey-Lundberg, Cao and Ko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanna Cao, WWFubmEuQ2FvQHV0aC50bWMuZWR1; Tien C. Ko, VGllbi5DLktvQHV0aC50bWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.