 Jian Li
Jian Li Jialu Wu1
Jialu Wu1 Xijie Yu
Xijie Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 March 2024
Sec. Cancer Metabolism
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1360471
This article is part of the Research Topic Metabolic Crosstalk between Cancer Cells and Immune Cells in the Tumor Microenvironment: Cellular and Molecular Insights, and their Therapeutic Implications View all 11 articles
Bone is a common site of metastasis for lung cancer. The “seed and soil” hypothesis suggests that the bone marrow microenvironment (“soil”) may provide a conducive survival environment for metastasizing tumor cells (“seeds”). The bone marrow microenvironment, comprising a complex array of cells, includes bone marrow adipocytes (BMAs), which constitute about 70% of the adult bone marrow volume and may play a significant role in tumor bone metastasis. BMAs can directly provide energy for tumor cells, promoting their proliferation and migration. Furthermore, BMAs participate in the tumor microenvironment’s osteogenesis regulation, osteoclast(OC) regulation, and immune response through the secretion of adipokines, cytokines, and inflammatory factors. However, the precise mechanisms of BMAs in lung cancer bone metastasis remain largely unclear. This review primarily explores the role of BMAs and their secreted adipokines (leptin, adiponectin, Nesfatin-1, Resistin, chemerin, visfatin) in lung cancer bone metastasis, aiming to provide new insights into the mechanisms and clinical treatment of lung cancer bone metastasis.
According to data released by the Global Burden of Disease Study in 2020, an estimated 2.2 million people worldwide are afflicted with lung cancer, with approximately 1.8 million deaths. As the cancer with the highest incidence and mortality rate globally, lung cancer is the leading cause of cancer death among men and the second leading cause among women. Despite substantial regional variations in incidence and mortality rates between men and women, statistics show that rates in men are approximately twice those in women (1). Compared to other malignancies, lung cancer continues to have a low survival rate, with a 5-year survival rate of only 5% for advanced stages (2). One of the main characteristics of malignant tumors is their ability to metastasize, and over 90% of lung cancer patients die from complications related to metastasis. Bone is among the most common metastasis sites for lung cancer, with an incidence rate of 30-40%. Clinical data indicate that approximately 40-48% of patients with advanced lung cancer exhibit bone metastasis at initial diagnosis (3). Once bone metastasis occurs, the median survival time of patients significantly reduces to only five months (3).Depending on radiological features, lung cancer bone metastasis can be classified into osteolytic, osteoblastic, and mixed types. Studies have shown that approximately 70% of lung cancer bone metastases are osteolytic, with osteoblastic types being less common (4). Following osteolytic bone metastasis in lung cancer, about 50% of patients will experience skeletal-related events (SREs), including intractable bone pain, pathological fractures, spinal cord compression, and hypercalcemia, which accelerate disease deterioration, diminish quality of life, decrease physical abilities, increase medical expenses, and elevate mortality rates. Given that current treatments for lung cancer bone metastasis are limited to symptom relief and cannot effectively slow the progression or fundamentally alter the pathological process, elucidating the pathogenic mechanisms of lung cancer bone metastasis and exploring effective early diagnosis and treatment strategies are crucial.
The occurrence of lung cancer bone metastasis results from a series of complex pathological processes, which can generally be divided into tumor invasion, tumor cell migration, and bone tissue invasion stages. The initiation phase of bone metastasis involves tumor cells escaping the primary site and entering the circulation, forming disseminated tumor cells (DTCs) (5). The epithelial-mesenchymal transition (EMT) plays a key role in this process, where epithelial cells lose polarity and cell-cell adhesion, acquiring mesenchymal characteristics (6). Studies have demonstrated that abnormal activation of the Wnt/β-catenin pathway can induce the onset and progression of tumor EMT (7). Both in vitro and in vivo studies have observed an increase in β-catenin expression under hypoxic conditions, promoting EMT progression, affecting lung cancer cell migration capabilities, and inducing morphological changes (8). Lung cancer cells can also secrete E-cadherin and matrix metalloproteinases(MMPs), degrading the extracellular matrix, reducing cell adhesion and cross-linking, facilitating tumor cell detachment from the tumor matrix into the circulation as circulating tumor cells (CTCs), and ultimately forming DTCs that migrate to the bone marrow microenvironment (9). Upon reaching the bone marrow microenvironment, tumor cells undergo stages of settlement, survival, and dormancy, eventually reactivating and forming proliferative metastatic foci (5) (Figure 1).
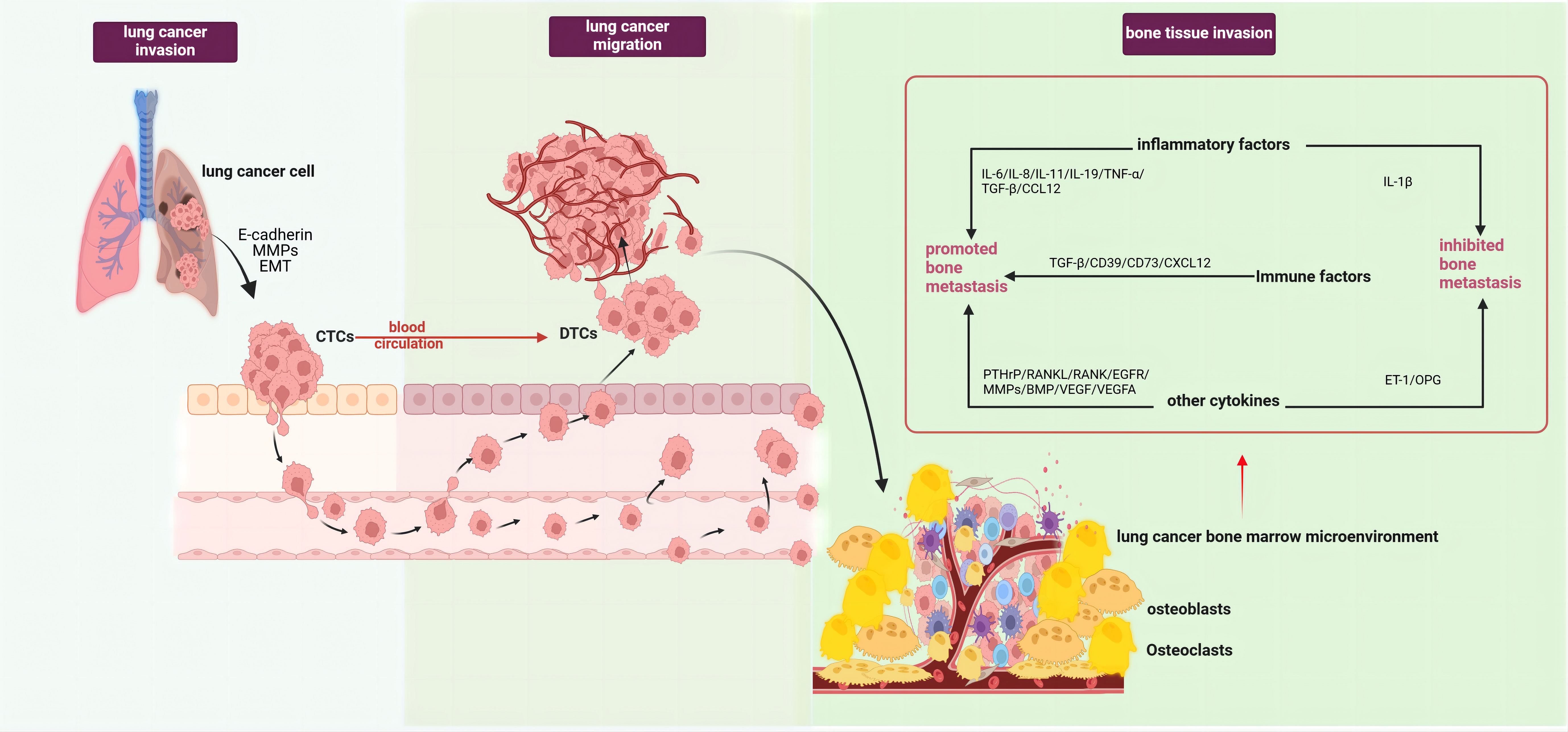
Figure 1 Pathological processes of lung cancer bone metastasis (created with BioRender.com.). The occurrence of bone metastasis in lung cancer could be divided into three stages, tumor invasion, tumor cell migration, and bone tissue invasion. In the first stage, lung cancer cells secreted E-cadherin and MMPs, along with EMT of lung cancer cells, enabling their entry into circulation and formation of DTCs. In the second stage, lung cancer cells detached from the tumor matrix and entered circulation to become CTCs. In the third stage, after lung cancer cells migrated to the bone marrow microenvironment, under the combined effects of various interactions between different cell types and factors within the tumor microenvironment, they became reactivated and formed proliferative metastatic lesions. This stage mainly involved the roles of various factors in the tumor microenvironment in bone metastasis,such as pro-metastatic inflammatory factors (IL-6, IL-8, IL-11, IL-19, TNF-α, CCL12), immune factors (TGF-β, CD39, CD73, CXCL12), and other factors (PTHrP, MMPs, RANKL, RANK, BMP, EGFR/VEGF, VEGFA). Factors that inhibited lung cancer bone metastasis included IL-1β, OPG, and ET-1. EMT, epithelial-mesenchymal transition; MMPs, matrix metalloproteinases; DTCs, Disseminated tumor cells; CTCs, Circulating tumor cells; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; IL-8, Interleukin 8; IL-11, Interleukin 11, IL-19, Interleukin 19; CCL12, C-C motif chemokine ligand 12; TNF-α, Tumor necrosis factor alpha; TGF-β, Transforming growth factor beta; CD39, Cluster of differentiation 39; CD73, Cluster of differentiation 73; CXCL12, C-X-C motif chemokine ligand 12; PTHrP, Parathyroid hormone-related protein; MMPs, Matrix metalloproteinases; RANKL, Receptor activator of nuclear factor kappa B ligand; RANK, Receptor activator of nuclear factor kappa B; BMP, Bone morphogenetic protein; OPG, Osteoprotegerin; ET-1, Endothelin 1.
The “seed and soil” hypothesis suggests that during the process of bone metastasis, the bone marrow microenvironment (“soil”) may provide an “ecological niche” suitable for metastasis, laying the foundation for the seeding, invasion, and proliferation of tumor cells (“seeds”) (10). A conducive “soil” may determine whether the “seeds” will germinate, as research has found that less than 0.01% of circulating tumor cells ultimately form distal metastases (11). The activation process of tumor cells within the bone marrow microenvironment (the germination process of “seeds” in the “soil”) is the result of bilateral interaction.
On one hand, tumor cells interact with various cells in the bone microenvironment, such as bone marrow stromal cells (BMSCs), osteoclast (OCs), osteoblasts (OBs), endothelial cells (ECs), BMAs, immune cells, etc., causing adaptive changes in the bone marrow microenvironment and providing favorable conditions for tumor cell invasion and growth (12). For instance, in vivo studies have found that BMSCs can be chemotactically guided to tumor cells, participating in the construction of the tumor microenvironment (13). Lung cancer cells can also activate OCs, causing bone matrix dissolution and creating conditions for their adhesion and settlement (14). ECs in the microenvironment can enhance tumor metastasis through angiogenesis, providing an energy source and new pathways for tumor cell invasion and migration (15).
On the other hand, various growth factors secreted by lung cancer cells, such as EGF-like domain multiple 6, bone morphogenetic protein-7 (BMP-7), transforming growth factor-beta (TGF-β), endothelin-1 (ET-1), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGF), etc., can directly affect the composition of the tumor microenvironment (16–19). For instance, EGF-like domain multiple 6 secreted by lung adenocarcinoma cells can enhance the EMT process, activate the Wnt/β-catenin and PI3K/AKT/mTOR pathways, promoting lung adenocarcinoma cell proliferation, migration, and invasion capabilities. Overexpression of this factor in a nude mouse model can enhance tumor growth and exacerbate bone resorption. In vitro studies have also found that it can increase OC differentiation of mouse bone marrow mononuclear macrophages via the NF-κB and c-Fos/NFATc1 signaling pathways (16). In vitro cell studies have shown that downregulating BMP-7 expression can significantly inhibit the invasiveness of lung adenocarcinoma SPC-A1 cells, while upregulating BMP-7 notably promotes the migration ability of A549 cells (17). Clinical studies have also found BMP-7 expression in the tumor cell membrane and cytoplasm of non-small cell lung cancer (NSCLC), with high cytoplasmic BMP-7 expression associated with tumor progression and adverse prognosis. These results all demonstrate that BMP-7 secreted by lung cancer cells, through affecting cell invasiveness and migration capability, promotes its growth and spread in bone tissues (18). In vitro cell studies indicate that TGF-β secreted by lung cancer cells can not only promote tumor microenvironment angiogenesis to facilitate lung cancer cell proliferation and migration but also regulate T cell activity to inhibit the immune system’s recognition and attack on tumor cells, helping the tumor evade immune surveillance (19). Additionally, TGF-β can increase the invasive and migratory capabilities of lung cancer cells, thereby promoting bone metastasis (20). Both in vivo and in vitro studies have confirmed that MMPs and urokinase plasminogen activator secreted by lung cancer cells can specifically degrade bone matrix components (such as collagen and trabeculae), leading to bone tissue destruction and dissolution, facilitating tumor cell invasion into bone tissues (21). A study on microRNA-328 (miR-328) secreted by lung adenocarcinoma A549 cells discovered that miR-328, potentially through the downregulation of neuropilin-2 (Nrp-2) expression in A549-derived extracellular vesicles (A549-Exos) in vitro, enhanced OC formation and bone resorption. Meanwhile, the in vivo administration of a miR-328 inhibitor in A549-Exos significantly inhibited bone resorption (22). Lastly, the unique invasiveness, infiltrative capability, and rapid growth and migration ability of tumor cells also affect the transformation of the metastatic foci’s microenvironment (23). Tumor cells can also engage in inflammatory reactions and a series of complex interactions with immune cells in the microenvironment, enabling tumor cells to evade immune surveillance, thereby promoting their proliferation and migration (24). Additionally, the metabolic decomposition of BMAs can produce a large amount of lipids, providing energy for tumor cells, promoting their proliferation, migration, and invasion. BMAs can also secrete certain adipokines, such as leptin, by activating signaling pathways such as PI3K, HIF, and MAPK, enhancing the proliferation, migration, and invasion capabilities of lung cancer cells (25, 26) (Figure 1).
The osteolytic lesions in lung cancer bone metastasis are primarily caused by the activation of OCs. Lung cancer cells can directly influence OCs or indirectly upregulate their function through secreted active factors, thereby promoting lung cancer bone metastasis (27). For instance, lung cancer cells secrete extracellular vesicles containing amphiregulin (AREG), which induces abnormal activation of the EGFR signaling in OCs, upregulating MMP-9 and thus triggering osteolytic metastasis (28). Lung adenocarcinoma secreted miR-21 suppresses programmed cell death 4 (PDCD4), promoting the generation of OCs and hence facilitating osteolytic lesions in lung cancer (29). Additionally, experimental studies with lung cancer cells have found that they secrete parathyroid hormone-related protein (PTHrP), which binds to the PTH/PTHrP receptor, enhances the expression of the receptor activator of nuclear factor-kappa B ligand (RANKL), and inhibits the synthesis of osteoprotegerin (OPG) (30). RANKL binds to the receptor activator of nuclear factor κB (RANK) on the surface of OC precursor cells, inducing OC aggregation and activation, enhancing their activity, and causing osteolytic destruction (31). The release of insulin-like growth factor-1 (IGF-1) and TGF-β from bone matrix, in turn, acts on tumor cells: IGF-1 promotes tumor cell proliferation and inhibits apoptosis, while TGF-β induces the tumor to secrete more PTHrP, activating more OCs, and dissolving the bone matrix again (32), forming a “vicious cycle” that promotes the occurrence and development of tumor bone metastasis. Furthermore, in vivo and in vitro studies have discovered that lung adenocarcinoma A549 cells induce OCs to secrete the ligand IL-19 for IL-20RB, activating the downstream JAK1/STAT3 signaling pathway, and promoting lung cancer proliferation in the bone microenvironment (33). Importantly, blocking IL-20RB with neutralizing antibodies can significantly inhibit lung cancer bone metastasis. These data demonstrate the important role played by activated OCs in osteolytic bone metastasis of lung cancer (Figure 2).
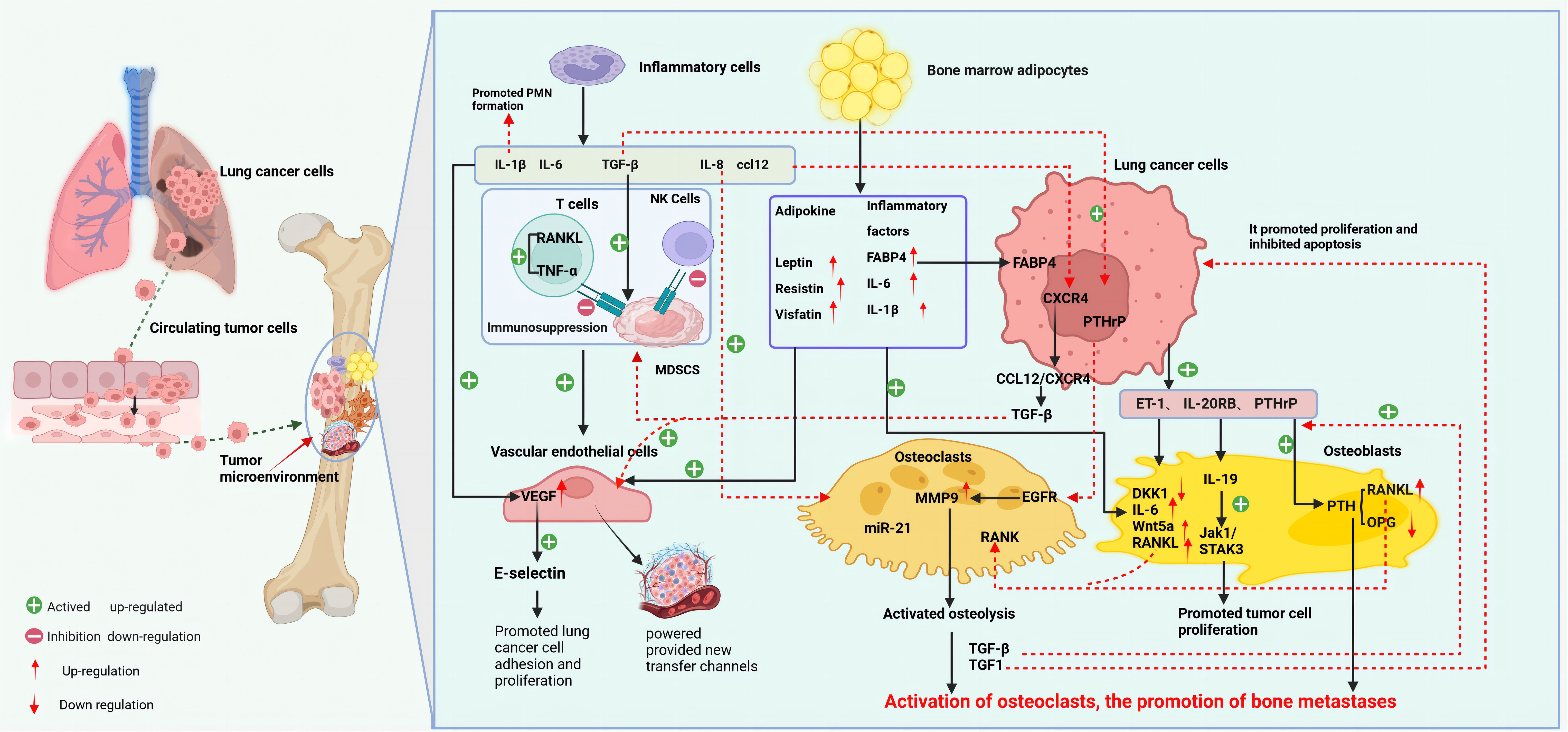
Figure 2 Mechanism of bone metastasis in lung cancer (created with BioRender.com.). The main mechanism of bone metastasis in lung cancer is the interaction between various cells (OC, OB, EC, BMA, inflammatory cells, etc.) in the bone microenvironment and lung cancer cells, which leads to adaptive changes in the bone marrow microenvironment for tumor cell invasion and growth. At the same time, various factors secreted by lung cancer cells also participate in and affect the composition of the tumor microenvironment. Bone metastasis in lung cancer mainly manifests as osteolytic lesions. Lung cancer cells can directly or indirectly activate OC, initiating a vicious cycle of bone resorption, promoting bone matrix dissolution and lung cancer bone metastasis. OB metastasis is relatively rare in lung cancer, mainly through the downregulation of DKK1 in OB by ET-1 secreted by lung cancer cells, upregulation of osteogenic genes such as IL-6 and RANKL, and promotion of OB metastasis. Lung cancer cells, inflammatory cells, and BMA in the bone tumor microenvironment release inflammatory factors, participate in the construction of an immune-suppressive and tumor angiogenesis microenvironment, and provide conditions for lung cancer cells to evade immune surveillance, proliferate,and migrate. BMA, bone marrow adipocytes; ccl12, chemokine 12; CXCR4, CXC chemokine receptor 4; DKK1, Dickkopf-1; ET-1, endothelin-1; EC, endothelial cells; EGFR, epidermal growth factor receptor; IL-19, Interleukin-19; IL-20R, interleukin 20 receptor; MDSCs, myeloid-derived suppressor cells; MMP9, matrix metalloproteinase 9; OB, osteoblasts; OC, osteoclasts; PTHrP, parathyroid hormone-related protein; PTH, parathyroid hormone; RANK, receptor activator of nuclear factor κ B; RANKL, RANK ligand; sVCAM1, soluble vascular cell adhesion molecule-1.
Compared to osteolytic metastasis, osteoblastic (bone-forming) metastasis of lung cancer is less common (34). Currently, most studies on osteoblastic metastasis suggest that under various influences, OBs participate in tumor bone metastasis through secreting molecules to affect OC formation and tumor progression. Factors promoting osteogenic differentiation of OBs include BMP, ET-1, semaphorin 3A (Sema3A), vascular endothelial growth factor (VEGF), monocyte chemoattractant protein-1 (MCP-1), and interleukin-6 (IL-6), etc. (34–36). For example, Wnt/β-catenin signaling and DKK1 (a Wnt signaling pathway inhibitor) can promote lung cancer metastasis, particularly bone metastasis (37). In vitro cell studies discovered that ET-1 significantly downregulates DKK1 in OBs, while also upregulates osteogenic genes (such as IL-6, Wnt5a, RANKL, etc.) (38). Thus, ET-1, as a key tumor factor, can not only upregulate genes promoting osteogenesis but also downregulate negative regulators of osteogenesis, participating in tumor osteoblastic metastasis. Additionally, miR-139-5p was found to positively regulate the osteogenic differentiation of mesenchymal stem cells (MSCs) (39). However, Feng et al. found that miR-139-5p inhibits osteogenic differentiation. In vivo model studies showed that upregulation of miR-139-5p reduced cell proliferation and osteogenic differentiation in MSCs by targeting NOTCH1 and inhibiting the Wnt/β-catenin signaling pathway (40). In vitro cell studies also found that hypoxia-inducible factor-1α (HIF-1α) can inhibit osteogenic differentiation by upregulating Sema4D, thus participating in lung cancer bone metastasis (41). Engblom et al. found that OBs also have a distal regulatory role in lung cancer progression (42). However, the specific mechanisms of OBs in lung cancer bone metastasis are still not fully understood and require further in-depth research (Figure 2).
It is well-documented that inflammation plays a critical role in tumor progression. Both lung cancer cells and inflammatory cells secrete pro-inflammatory cytokines, which not only directly participate in the formation of the pre-metastatic niche (PMN) but also engage in the activation, proliferation, and migration of tumor cells in the microenvironment, thereby facilitating bone metastasis (43). Evidence suggests that cytokines such as interleukin-1β (IL-1β), IL-6, interleukin-7 (IL-7), interleukin-8 (IL-8), interleukin-11 (IL-11), tumor necrosis factor-alpha (TNF-α), TGF-β, and C-C motif chemokine ligand 12 (CCL12) play significant roles in the bone metastasis of lung cancer (43–46). For instance, studies have shown that blocking IL-1β in K-ras mutant lung adenocarcinoma (KM-LUAD) mice, which express high levels of IL-1β in the lungs, significantly reduces tumor load when IL-1β monoclonal antibodies are administered at 6 and 14 weeks of age (47). Clinical research also confirms that inhibiting IL-1β significantly reduces the incidence rate of lung cancer in a dose-dependent manner (48). Lung cancer cells secrete IL-6 and IL-11, which can activate various signaling pathways such as PI3K/Akt and MAPK, promoting cell proliferation and migration (49–52). In vitro studies reveal that IL-11 can stimulate OC formation and activation, accelerate bone resorption, and release growth factors such as TGF-β in the bone matrix, thereby promoting the growth and metastasis of lung cancer cells (53). Additionally, IL-8 has been found to increase the invasive capabilities of tumor cells and promote OC maturation and activation, leading to the release of key enzymes such as acid phosphatase and MMPs that degrade the bone matrix and further enhance bone dissolution (54). IL-8 also induces BMSC to secrete RANKL while inhibiting OPG secretion, leading to RANKL/OPG dysregulation, thus promoting OC activation and maturation and subsequent bone resorption (55). Moreover, IL-1, IL-6, IL-8, TNF-α, and TGF-β secreted by lung cancer cells can activate downstream signaling pathways to increase VEGF expression, thereby promoting angiogenesis in the bone tumor microenvironment and indirectly facilitating bone metastasis (56). These inflammatory cytokines can also recruit and induce immune cells, notably myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, and neutrophils, creating an immunosuppressive local milieu that promotes tumor cell survival and thereby indirectly facilitating bone metastasis (57). In vitro cell studies have shown that activation of the KRAS signaling pathway can upregulate tumor cell secretion of CCL12, which promotes tumor cell recruitment to target organs through binding to its receptor CXCR4 (46). Research has confirmed that upregulation of CXCL12 promotes cancer cell metastasis and growth in bone metastases of breast and prostate cancers (58, 59). The expression of CXCR4 is elevated in bone destruction areas of NSCLC bone metastasis patients (60). Furthermore, clinical studies have found TGF-β promoting CD39 and CD73 expression on MDSCs, which can suppress T cell and NK cell activity, thus contributing to the formation of an immunosuppressive microenvironment and enabling tumor cells to evade immune surveillance (61). These findings underscore the pivotal role of inflammatory cells and their secreted cytokines in lung cancer bone metastasis, though the complex inflammatory response mechanisms involved in lung cancer bone metastasis require further research (Figure 2).
The RANK/RANKL signaling axis, crucial for maintaining bone homeostasis, plays a key role in tumor bone metastasis. RANKL, a critical regulator of OC differentiation, is chiefly secreted by OBs, osteocytes, and activated T cells. Binding of RANKL to RANK activates signaling pathways in OC precursors, promoting OC formation and enhancing bone resorption (62). OPG, a competitive receptor for RANKL, inhibits the RANKL-RANK interaction, thereby reducing OC formation and activity (63).
The RANKL/RANK signaling pathway directly affects the functionality of lung cancer cells and OCs, playing a role in bone metastasis of lung cancer. In vitro studies have revealed that RANKL, by activating the RANK receptor, can promote lung cancer cell proliferation and growth while inhibiting apoptosis (64). Other research has shown that activation of the RANKL/RANK signaling pathway enhances OC functionality within the bone marrow microenvironment, aiding bone resorption and consequently facilitating bone metastasis (65).
The indirect mechanisms in bone metastasis include: 1) Activation of NF-κB, MAPK, and other signaling pathways to promote lung cancer proliferation and growth (66). 2) Release of inflammatory cytokines and other cellular factors, altering the composition of the tumor microenvironment and indirectly facilitating bone metastasis (62). 3) Regulation of the activation and function of immune cells within the tumor microenvironment. In certain cases, activation of the RANKL/RANK signaling pathway induces immunosuppressive molecules (e.g., TGF-β from Treg cells), regulating the immune response balance, allowing lung cancer cells to evade immune surveillance and promote bone metastasis (67). Moreover, RANKL can stimulate the maturation and activation of dendritic cells, macrophages, and other immune cells, enhancing their cytokine and chemokine production, thus boosting antigen presentation and T cell activation capabilities (65, 68). Additionally, RANKL/RANK signaling activation can regulate the NF-κB signaling pathway to inhibit B cell apoptosis, promoting B cell survival and proliferation capacity, providing a protective environment for lung cancer cells to escape immune surveillance, thereby inhibiting lung cancer cell apoptosis and promoting bone metastasis (69). In summary, RANKL/RANK maintains immune homeostasis through various immune regulation mechanisms, indirectly participating in bone metastasis. 4) Regulation of VEGF receptor activation and tumor angiogenesis. It also promotes EC growth, migration, and lumen formation, contributing to tumor microenvironment angiogenesis (70, 71). The RANK/RANKL signaling axis plays a complex role in lung cancer bone metastasis, with the specific mechanisms requiring further in-depth study.
Apoptosis functions as a critical protective mechanism within organisms, which eliminates aberrant cells and prevents tumor genesis. When apoptosis of tumor cells is inhibited, cells may evade immune surveillance, thereby enhancing their survival and propagation (72). In vitro studies have revealed that lung cancer cells can suppress the initiation of apoptosis by upregulating the anti-apoptotic protein Bcl-2, which blocks the release of apoptogenic cytochrome c; they can also inhibit apoptosis by downregulating pro-apoptotic proteins, such as Bax and Bak, thus promoting their survival within the bone marrow microenvironment (73, 74). Furthermore, lung cancer cells can interact with other cells in the bone microenvironment (such as OBs, OCs, and BMAs), not only augmenting the anti-apoptotic capabilities of lung cancer cells but also enhancing the function of OCs (73). For instance, proteins of the Bcl-2 family can inhibit the apoptosis of lung cancer cells and promote the differentiation and function of OCs. Cytokines released during the apoptosis of lung cancer cells, such as TNF-α, TGF-β, and VEGF, can indirectly promote bone metastasis by fostering inflammation and angiogenesis within the tumor microenvironment (75). Additionally, the release of lactate and ATP post-apoptosis can upregulate the metabolic state of bone cells and increase acidification of the tumor microenvironment, indirectly facilitating bone resorption in the context of lung cancer bone metastasis (76).
During tumor metastasis, lung cancer cells and ECs release VEGF to promote the formation of new blood vessels, supplying the tumor with additional nutrients and growth factors, and providing new physical pathways for tumor metastasis. For example, MDSCs, as critical molecules in PMN formation, can produce VEGFA, which upregulates E-selectin, thereby enhancing the adhesion of tumor cells, and facilitating the homing and proliferation of circulating tumor cells (77). Furthermore, MDSCs can secrete MMP9 to regulate the function of VEGF to promote angiogenesis and tumor cell extravasation and migration (78, 79). Increasing research indicates that adipokines (such as leptin, resistin, visfatin, etc.) and inflammatory factors (such as IL-1β, IL-6, chemokines, FABP4, etc.) secreted by BMA can also regulate angiogenesis, indirectly facilitating the progression of tumor bone metastasis (80–83). Occupying 70% of the bone marrow cavity volume, BMAs constitute a major component of the bone marrow microenvironment, and their role in tumor bone metastasis is gaining increasing attention, particularly the regulatory effects of secreted adipokines on energy metabolism, endocrine functions, and inflammatory responses in influencing tumor growth and migration (81, 83). Despite numerous studies on tumor bone metastasis and the bone marrow microenvironment, the mechanisms linking BMAs and lung cancer bone metastasis remain largely unexplored. This text will next focus on elucidating the mechanisms underlying the role of BMAs and their secreted adipokines in lung cancer bone metastasis (Figure 2).
BMAs originate from a distinct cell population within the bone marrow, comprising MSCs and preadipocytes of the marrow adipogenic lineage (84). While historically regarded as an inert adipose tissue, recent studies have identified unique characteristics and functions of BMAs, distinguishing them from white, brown, and beige adipose tissues (85, 86). In humans, BMAs are primarily located in the marrow of long bones, especially within the trabecular bone at the epiphyses and metaphyses, and near the endosteal surface of the bone shaft (87). The abundance of BMAs in the bone marrow increases with aging, obesity, the application of peroxisome proliferator-activated receptor γ (PPARγ), and radiation exposure (85, 88). Studies in ovariectomized mouse models have shown that estrogen deficiency leads to an increase in BMAs, which can be reversed by estrogen supplementation (89). Clinical research on osteoporosis has revealed that the age-related increase in BMAs is associated with bone loss, suggesting BMAs as negative regulators of bone mass (90). However, this correlation is not uniformly observed across studies. In C57BL/6 mice (with the lowest trabecular and cortical bone density among all mouse strains), BMAs are scarce, whereas they are abundant in C3H/He mice (a unique strain with higher bone density) (91, 92), implying genetic regulation of BMA distribution and the need for further comprehensive analysis (Figure 2).
BMAs can secrete factors like RANKL, IL-6, and TNF-α, which activate OCs to promote bone resorption within the bone microenvironment (93, 94). In vitro studies have shown that BMAs can upregulate the RANK expression on OCs, leading to increased OC formation (95). Research on breast cancer bone metastasis demonstrated that activated OCs release acid phosphatase, acidifying the bone microenvironment (96, 97). This acidic environment upregulates matrix MMPs, and BMAs further contribute to bone matrix degradation by upregulating expression of OC-specific genes such as cathepsin K, facilitating tumor cell growth (98). IL-1β and IL-6 can induce EMT in breast cancer cells via the STAT3 pathway and promote angiogenesis, which suggests that BMAs may similarly exacerbate bone destruction in lung cancer bone metastasis (Figure 2).
Studies have shown that enhanced adipogenic differentiation of bone marrow MSCs in the bone marrow microenvironment leads to a decrease in their osteogenic potential (99). BMAs contribute to the regulation of OB function by secreting inflammatory factors such as IL-6, IL-1β, and TNF-α (100–102). Both tumor cells and BMAs can produce IL-6, which promotes tumor cell proliferation, induces OC activation, and downregulates OB activity (102). Additionally, IL-6 promotes adipogenic differentiation of MSCs while inhibiting osteogenesis (103). An increase in palmitic acid and arachidonic acid, associated with increased BMAs, heightens the cytotoxic effects on OBs. In vitro studies indicate that palmitate-induced lipotoxicity in OBs and osteocytes is mediated by autophagy dysregulation, leading to OB apoptosis (104). Given that lung cancer cells also secrete IL-6 and TNF-α, it is speculated that lung cancer cells and BMAs, by regulating OB function and inhibiting osteogenic differentiation, indirectly disrupt bone remodeling and promote lung cancer bone metastasis (Figure 2).
BMAs can regulate EC function through both direct and indirect mechanisms. Directly, they secrete cytokines and metabolic products influencing EC growth, proliferation, and migration (105–107). In vitro studies have found that adiponectin secreted by BMAs affects EC growth and migration and influences the expression of EC adhesion molecules, facilitating closer proximity and invasion by tumor cells into bone (107, 108). Indirectly, cytokines secreted by BMAs activate multiple signaling pathways affecting ECs. Studies on breast cancer bone metastasis showed that BMAs-secreted IL-1β activates the p38-MAPK pathway, increasing EC permeability and vasculogenesis (108). Additionally, the impact of BMAs-secreted IL-6 on HIF-1α and VEGF levels participates in the regulation of angiogenesis (109). The presence of adiponectin and IL-6 in the bone marrow microenvironment of lung cancer bone metastasis suggests that BMAs may promote EC growth and new blood vessel formation, providing additional nutrients and pathways for cancer metastasis (Figure 2).
BMAs are capable of secreting pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and leptin, and they induce BMSCs to participate in the inflammatory immune response by regulating B cell responses and lymphocyte production (110). For instance, the inflammatory cytokine IL-1β, secreted by BMAs, can upregulate the expression of leptin (111). A clinical study involving 116 lung cancer patients with bone metastasis found significantly higher levels of leptin and its receptor in patients with bone metastases compared to those without, suggesting that the formation of lung adenocarcinoma bone metastatic lesions is closely related to leptin (112). Leptin can inhibit the activity of macrophages and natural killer cells, reducing their ability to kill lung cancer cells. It can also regulate the expression of immune checkpoint molecules on the surface of lung cancer cells, such as programmed death-ligand 1 (PD-L1), to inhibit the immune cells’ ability to kill tumor cells. PD-L1, in conjunction with its receptor PD-1, forms an immune checkpoint that suppresses the activation and proliferation of effector T cells and promotes the increase and upregulation of regulatory T cells (Tregs) (113, 114). Ultimately, lung cancer cells can escape immune surveillance by regulating the leptin signaling pathway, thereby promoting the occurrence and development of lung cancer bone metastases. Additionally, IL-6, TNF-α, CXCL12, and leptin are considered to significantly promote tumor cell migration and proliferation, as well as inhibit apoptosis and activate autophagy, facilitating the development of tumor bone metastasis (115, 116) (Figure 2).
Current research posits that BMAs, through their metabolic processes, produce a substantial quantity of lipids, including fatty acids, triglycerides, phospholipids, and cholesterol (117, 118). These lipids serve as an effective energy source for tumor cells, promoting their proliferation, migration, and invasion (118). Lipids are not only essential components of tumor cell membranes but also serve as energy sources during high metabolic demands (117). Fatty acids, fundamental components of lipids, have been found to be released from triacylglycerol in BMAs through lipolysis. These fatty acids can supply energy for tumor cell growth and metabolism via the microcirculation (119). Furthermore, fatty acid-binding protein 4 (FABP4) secreted by BMAs can increase the stability of fatty acids. FABP4 facilitates lipid transport and the transfer of free fatty acids to tumor cells, playing a role in the process of tumor bone metastasis (120, 121). Studies utilizing BMA-enriched mouse models found upregulated levels of FABP4 in Prostate Cancer(PCa) cells directly in contact with BMAs, suggesting a bidirectional interaction between FABP4 and the PPARγ pathway may enhance the invasiveness of tumor cells in bone metastasis (121). Additionally, co-culture studies inducing lipolysis in adipocytes and beta-oxidation in cancer cells have demonstrated that adipocytes can act as an energy source for cancer cells (122). In PCa, in vitro studies showed that adipocytes could enhance PCa cell migration. Breast cancer research found that adipocytes near invasive cancer cells promoted the migration and growth of breast tumor cells (119). These studies collectively underscore the significant role of BMAs as an energy source in the process of tumor bone metastasis (123). Given the metabolic secretion of a large amount of lipids by BMAs in the bone marrow microenvironment of lung cancer bone metastasis, it is speculated that BMAs, as an energy source, facilitate the development of lung cancer bone metastasis.
Furthermore, certain immunomodulatory adipokines secreted by BMAs may participate in the process of tumor bone metastasis through interactions with adipocytes, immune cells, and tumor cells (110, 124). The following sections will discuss the mechanisms of action of leptin, adiponectin, Nesfatin-1, Resistin, chemerin, and visfatin in lung cancer bone metastasis.
Leptin is a protein composed of 146 amino acids encoded by the ob gene, acting as a neuromodulatory, immunoregulatory, and endocrine hormone with multifunctional roles across various organs (125). By binding to specific leptin receptors, leptin activates intracellular signaling pathways, regulating the transcription of target genes to exert biological effects (125). The interaction of leptin with central and peripheral receptors yields divergent, sometimes opposing effects; while binding to peripheral receptors may increase bone mass, interaction with central receptors can induce bone loss (126).
The binding of leptin to its receptor can activate several signaling pathways associated with tumor progression, including TGF-β, JAK/STAT, PI3K, HIF, and MAPK pathways (127). In vitro studies on lung cancer bone metastasis have shown that leptin promotes metastasis of the A549 human lung cancer cell line through a TGF-β-dependent induction of EMT (128). Leptin can also block the endoplasmic reticulum stress-related pathway, preventing apoptosis and promoting proliferation in lung adenocarcinoma A549 cells (129). Inhibition of the leptin-related pathway significantly induces apoptosis in these cells. Ieptin can regulate apoptosis-related factors, such as members of the Bcl-2 family and caspases, to inhibit apoptosis in lung cancer cells (130). This evidence confirms that leptin enhances lung cancer cell growth and migration through suppressing apoptosis (Table 1).
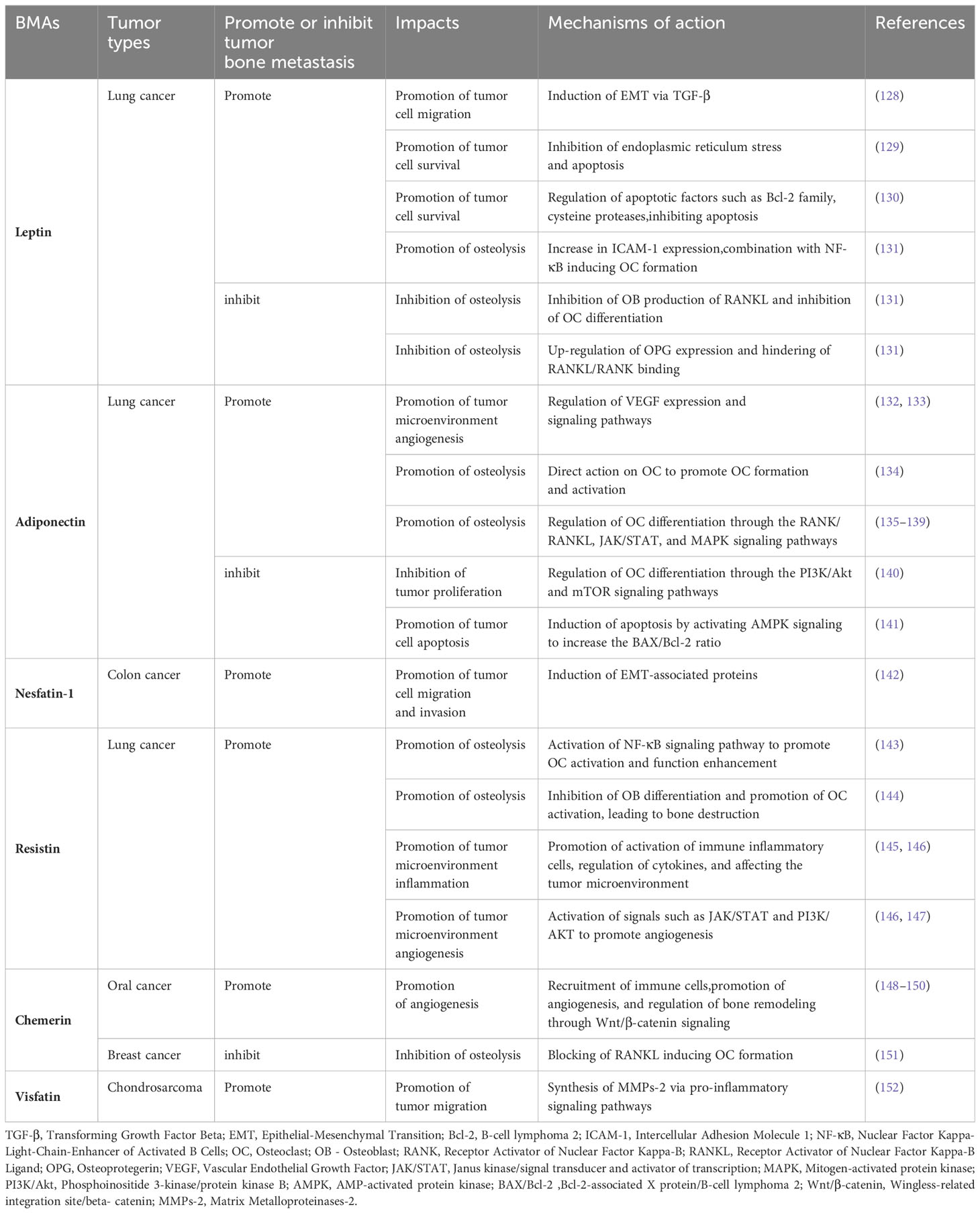
Table 1 Leptin, Adiponectin, Nesfatin-1, Resistin, Chemerin, Visfatin: Mechanisms of action in tumor bone metastasis - Literature table.
Leptin’s indirect effects in lung cancer bone metastasis mainly involve regulation of OCs and OBs. Leptin inhibits the expression of RANKL in OBs, thereby suppressing OC differentiation; it also increases the expression of OPG, preventing the RANKL/RANK binding and indirectly inhibiting bone resorption (131). Leptin can regulate other factors and pathways affecting bone resorption, such as matrix MMP2 and MMP9, which are involved in extracellular matrix remodeling, tumor progression, and bone absorption (131, 153). Clinical studies on lung cancer bone metastasis have found that overactivation of MMP2/MMP9 promotes osteolytic metastasis and bone destruction in advanced cancer (154). In vitro studies showed that leptin enhanced production of soluble intercellular adhesion molecule-1 (ICAM-1) in lung cancer cells through triggering a signaling cascade involving JAK1/2, STAT3, FAK, ERK, and GSK3αβ (154). Leptin-stimulated production of soluble ICAM-1, in coordination with RANKL activation, synergistically induces OC formation, suggesting that leptin indirectly promotes tumor-induced bone resorption. Animal experiments have shown that leptin promotes proliferation and differentiation of OBs and enhances bone matrix synthesis and secretion, including collagen, alkaline phosphatase, and osteocalcin (155, 156). Further research is needed to determine whether this effect is similarly active in lung cancer bone metastasis (Table 1).
Leptin directly influences the proliferation, differentiation, and activity of various immune cells (such as monocytes, T cells, B cells, and macrophages) and interacts with other cytokines in the tumor microenvironment (157). Leptin indirectly regulates immune function in the lung cancer bone metastasis microenvironment by affecting the function of other cells (158). Cell studies have demonstrated that leptin promotes monocyte proliferation, induces macrophage phagocytosis and pro-inflammatory cytokine secretion, and acts as a nutritional factor to prevent apoptosis, playing a role in adaptive immunity by regulating T and B cell populations (159). Leptin dose-dependently promotes naïve CD4+ T cell proliferation and polarizes CD4+ T cells towards a Th1 phenotype (160), which in turn facilitates lung cancer secretion of inflammatory cytokines such as TNF-α and IL-6, promoting lung cancer bone metastasis. Beyond its effects on T cells, leptin maintains the homeostasis of murine B cells by inducing Bcl-2 and cyclin D1, promoting cell cycle entry and preventing apoptosis, thereby promoting lung cancer cell proliferation (161). Leptin interacts with inflammatory cytokines, such as VEGF and TGF-β, to establish and maintain an inflammatory immune state within the tumor microenvironment (162). Leptin can also upregulate the function of MDSCs, which suppress T cell activation and proliferation through the release of immunosuppressive factors and direct interaction with T cells (163), allowing lung cancer cells to evade immune surveillance (Table 1).
It is crucial to note that the specific mechanisms of leptin in lung cancer bone metastasis require further investigation due to the complexity of tumor metastasis and individual variability, which may lead to diverse leptin response patterns. Additionally, factors such as lung cancer type (e.g., adenocarcinoma, squamous cell carcinoma), molecular subtypes, and the microenvironment may also influence the effects of leptin.
While previous studies suggested adiponectin was primarily secreted by white adipose tissue (WAT), recent research shows that BMAs produce more adiponectin than WAT, especially in cancer patients. Adiponectin’s role in the tumor microenvironment contrasts with that of leptin, sparking debate over its impact on cancer (164). Although earlier results pointed towards anti-tumor effects, recent studies have highlighted adiponectin’s significant role in promoting tumor metastasis (165).
Adiponectin primarily exerts its influence by inhibiting the proliferation of lung cancer cells and inducing their apoptosis. An in vitro study on lung cancer revealed that adiponectin can suppress the proliferation of lung cancer cells by inhibiting the PI3K/Akt signaling pathway and the phosphorylation of mTOR (mammalian target of rapamycin) (140). Additionally, it induces cell apoptosis by activating the AMPK signaling pathway and increasing the BAX/Bcl-2 ratio (140, 141) (Table 1). Animal experimental studies have also discovered adiponectin’s role in prohibiting lung cancer cell proliferation through inhibiting the Wnt/β-catenin signaling pathway (166).
Adiponectin indirectly participates in lung cancer bone metastasis by regulating angiogenesis, influencing OC activities, and impacting immune functions. Research indicates that adiponectin affects vessel formation by regulating the expression of VEGF and its signaling pathways (165). Adiponectin can enhance the expression of VEGFs (including VEGF-A, VEGF-B, VEGF-C, and VEGF-D) in ECs. VEGFs operate on receptors on ECs, such as VEGFR-1 and VEGFR-2, promoting EC proliferation and tubule formation through activating the VEGFR-2 signaling pathway. Moreover, adiponectin promotes angiogenesis by inhibiting angiogenesis inhibitors, such as Angiopoietin-1 (132, 133).
Adiponectin directly and indirectly partakes in the development and maturation of OCs, influencing bone metastasis in lung cancer. On one side, in vitro experiments have shown that adiponectin can directly influence OCs, promoting their formation, activation, and increasing the release of lysosomal enzymes, thereby enhancing bone resorption (134). On the other hand, it indirectly contributes to the formation and maturation of OCs mainly through binding to receptors on cell types such as OBs and OCs (167). Receptors include Ob-Rb (long-form receptor primarily distributed in MSCs); upon binding, adiponectin can activate multiple signaling pathways and regulate the expression of related genes, contributing to the differentiation of hematopoietic stem cells (HSCs) into OCs (166). Pathways implicated include RANK/RANKL, JAK/STAT, and MAPK, promoting bone metastasis by influencing OC formation and maturation (135–139) (Table 1). In summary, adiponectin exerts an indirect influence on osteolytic metastasis in lung cancer primarily through modulating angiogenesis within the tumor microenvironment, participating in the regulation of OC activity, and affecting the equilibrium of bone remodeling.
Adiponectin might play a role in lung cancer bone metastasis through regulating immune cell functions. In vitro studies indicate that adiponectin can increase the quantity and function of regulatory T cells (Tregs) while reducing the activity of natural killer (NK) cells, diminishing attacks on cancer cells, thus creating a favorable environment for lung cancer bone metastasis (168). However, differing studies suggest adiponectin possesses anti-inflammatory properties, inhibiting the NF-κB signaling pathway and cytokine release, thereby reducing the production of inflammatory mediators. Consequently, it suppresses the inflammatory response surrounding lung cancer cells, reducing their survival and proliferation capabilities (169, 170).
Discovered in 2006, Nesfatin-1 is an anorexigenic neuropeptide initially associated with food intake and energy regulation, hence being considered as a hormone regulating body weight and appetite. Beyond the central system, Nesfatin-1 is also present in various organs and tissues, such as the stomach, intestines, spleen, and adipose tissue (171). However, research on Nesfatin-1 secreted by BMAs in lung cancer bone metastasis is lacking. Recent studies have predominantly focused on Nesfatin-1 secreted by white adipose tissue, observing elevated expression levels of Nesfatin-1 in lung cancer, breast cancer, and other tumors. It has been found that Nesfatin-1 is associated with tumor invasion, metastasis, and prognosis (172, 173).
Nesfatin-1 secreted by adipose tissue in the tumor microenvironment might directly influence tumor bone metastasis by regulating the expression and function of EMT-related proteins like E-cadherin, N-cadherin, and Vimentin (174). Some studies have demonstrated that Nesfatin-1/Nucleobindin-2 can suppress the expression of E-cadherin and increase the expression of N-cadherin and Vimentin, thereby inducing migration, invasion, and EMT of colon cancer cells (142) (Table 1). Since both peripheral and bone marrow fat can secrete Nesfatin-1, it can be inferred that in the bone marrow microenvironment of lung cancer bone metastasis, Nesfatin-1 secreted by BMAs might promote the migration and invasion of lung cancer cells by inducing EMT.
Nesfatin-1 might indirectly affect the invasion and metastasis of tumor cells in bone tissue by influencing the activity of immune cells and the production of inflammatory cytokines (175). Clinical research has shown that Nesfatin-1 can induce an increase in CCL2 expression in human synovial tissues, favoring M1 macrophage polarization, thereby increasing the expression of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α, and promoting the progression of inflammatory responses (176). This, consequently, indirectly affects the invasion and metastasis of tumor cells in bone tissue. Additionally, studies in rats have found that Nesfatin-1 can inhibit cell apoptosis induced by IL-1β inflammation and promote angiogenesis by inhibiting neutrophil recruitment, cell apoptosis, and activating VEGF, mechanisms that can indirectly promote tumor bone metastasis (177, 178). There is currently no direct evidence regarding Nesfatin-1’s mechanisms in lung cancer bone metastasis, further research to clarify these mechanisms is required.
Resistin, a peptide hormone, is primarily secreted by white adipose tissue but also produced by other tissues such as bone marrow adipose, liver, and muscle (179). Initial studies suggested its association with insulin resistance and type 2 diabetes (180). Recent research, however, has linked Resistin to bone marrow fat and bone metabolism, suggesting it may influence OC and osteoblast OB functions, participate in inflammatory responses and immune regulation, and affect the tumor microenvironment in tumor bone metastasis.
Resistin may directly promote lung cancer bone metastasis by activating OC function. In vitro studies have revealed that resistin can activate the NF-κB signaling pathway, leading to the release of inflammatory cytokines like TNF-α and IL-6, which further enhance OC activation and functionality, accelerating bone tissue destruction and bone metastasis (143). Additionally, resistin has been found to inhibit OB differentiation and bone-forming functions while promoting OC activation and functionality, resulting in bone tissue destruction and remodeling (144) (Table 1).
By regulating the production of inflammatory cytokines and the activity of immune cells, as well as activating tumor-associated signaling pathways like JAK/STAT and PI3K/AKT, resistin can indirectly promote cellular growth, angiogenesis, and immune response in the tumor microenvironment, facilitating lung cancer bone metastasis (145). Resistin can promote the production and activation of inflammatory cells like macrophages and lymphocytes and regulate the inflammatory response by modulating cytokine release (146) (Table 1). These immune cells play an essential role in lung cancer bone metastasis, including promoting inflammatory responses, regulating immune responses, and affecting tumor cell invasion and migration. Moreover, animal models of lung cancer have shown that mice treated with anti-resistin antibodies exhibited reduced rates of lung cancer development and metastasis (181), further proving that resistin, by modulating inflammatory responses in the tumor microenvironment, indirectly facilitates lung cancer bone metastasis.
Resistin can activate signaling pathways such as JAK/STAT and PI3K/AKT, thereby influencing cell growth and angiogenesis within the tumor microenvironment, as well as suppressing the body’s anti-tumor immune response, indirectly facilitating the occurrence and progression of tumor bone metastasis. Studies have found that resistin activates the PI3K and Akt signaling pathways, while inhibitors of PI3K and Akt, or siRNA, can reduce the expression of VEGF-A induced by resistin (146). Concurrently, both in vitro and in vivo studies have demonstrated that resistin promotes the expression of VEGF-A and angiogenesis within the tumor microenvironment by inhibiting the expression of miR-5-3p through the PI3K/Akt signaling cascade, thus affecting tumor bone metastasis (146, 147) (Table 1).
Chemerin is a small peptide hormone predominantly secreted by adipose tissue, with subsequent production noted in the liver, kidneys, and other tissues. Initially identified for its roles in inflammation and immune regulation, recent clinical studies have unveiled Chemerin’s capability to inhibit osteogenic differentiation in favor of adipogenesis (182). The Wnt pathway plays a crucial role in bone biology and the regulation of bone tumors, especially the classic Wnt/β-catenin signaling pathway is closely related to tumor bone metastasis. Relevant studies on mouse bone structure have shown that inhibiting the Wnt/β-catenin signaling pathway and activating the RANK signal promote bone resorption. Simultaneously, it has been demonstrated that Chemerin participates in the progression of bone tumors (151, 183). However, the role of Chemerin in cancer remains contentious, with a majority of studies highlighting its anticancer effects, while a minority suggest its protumorigenic capabilities. These effects derive from its regulation of angiogenesis and modulation of the immune-inflammatory response in the bone marrow microenvironment, indirectly contributing to the osteolytic metastasis of lung cancer.
Antitumor activities of Chemerin have been identified in a murine model of breast cancer, where it binds to its receptor ChemR23/CMKLR1 and inhibits neovascularization, thereby suppressing growth and invasion of breast cancer cells (148). Moreover, in Chemerin-treated media derived from metastatic breast cancer cells, an increase in the RANKL/OPG ratio and a reduction in the secretion of MMPs (e.g., MMP-2, MMP-9) and Cathepsin K were observed (148) (Table 1), thus inhibiting RANKL-induced OC formation and consequentially suppressing bone dissolution and tumor bone metastasis.
On the contrary, protumorigenic activity is evidenced through the recruitment of innate immune defenses and activation of endothelial vasculogenesis, as well as the suppression of Wnt/β-catenin signaling, reducing OB differentiation and stimulating OC differentiation and proliferation through RANK signaling activation (149). This mechanism plays a crucial role in tumor bone metastasis. Furthermore, studies in oral squamous cell carcinoma (OSCC) have shown that Chemerin enhances the formation of human umbilical vein ECs (HUVECs), promoting angiogenesis and subsequently tumor growth and migration. Additionally, Chemerin can upregulate pro-angiogenic factors such as VEGF-A, MMP-9, MMP-2, and S100A9 in neutrophils through the activation of the MEK/ERK signaling pathway (150, 184) (Table 1), thereby facilitating tumor vascularization and bone metastasis in lung cancer. Hence, the balance between Chemerin’s anticancer and protumorigenic effects ultimately dictates tumor progression, underscoring the need for further research into Chemerin’s role within the lung cancer bone metastasis.
Visfatin, an inflammatory adipokine also known as pre-B cell colony-enhancing factor (PBEF), plays a pivotal role in NAD+ biosynthesis as the rate-limiting enzyme in nicotinamide metabolism, implicating its involvement in B-cell development, apoptosis, and glucose metabolism. Initially discovered in visceral adipose tissue, Visfatin has been widely recognized for its compensatory response in obesity-induced insulin resistance (185). Recent studies have expanded our understanding of Visfatin’s functionality, uncovering its significant role in the genesis and development of tumors as well as bone metabolism. Elevated levels of Visfatin expression have been associated with certain tumor types, correlating with aggressiveness, migration, and prognosis (152). By promoting the synthesis of MMP-2 and activation of the AP-1 transcription factor through the ERK, p38, and JNK signaling pathways, Visfatin enhances tumor cell migration (186) (Table 1). Additionally, overexpression of Visfatin has been linked to increased pulmonary metastasis in a murine model of chondrosarcoma. In the mouse bone marrow, it disrupts the balance between bone resorption and formation, tilting towards a pro-inflammatory phenotype in MSCs differentiating towards BMAs and OBs (144). As an adipokine, Visfatin’s exact action in bone regulation and immune modulation remains to be fully elucidated. However, its capacity to induce pro-inflammatory transcription factors, such as NF-kB, and modulate pathways including MAPK, PI3K, and ROS (187), suggests that Visfatin might act as a potential tumor-promoting factor in lung cancer bone metastasis through upregulation of MMP-2, enhanced differentiation of OBs, and the promotion of inflammatory responses in MSCs, thereby indirectly facilitating osteolysis and the progression of pulmonary cancer bone metastasis.
This review primarily explored how BMAs directly or indirectly interact with various cells within the bone microenvironment of lung cancer, as well as its pathological role in modulating the tumor microenvironment and thus influencing bone metastasis in lung cancer. The contributions of BMAs are summarized through three main aspects: (1) BMAs provide direct energy supply to lung cancer cells through lipid metabolites and FABP4, with lipids also serving as building blocks for the cancer cell membrane; (2) Direct effects of BMAs in bone metastasis of lung cancer include secretion of pro-inflammatory cytokines like IL-1β that participate in PMN formation and CCL12 which directly interacts with CXCR4 on lung cancer cells, promoting cell proliferation and invasion. Secreted adipokines like leptin activate TGF-β, PI3K/Akt, and MAPK signaling pathways, upregulating MMP2/MMP9 secretion and facilitating EMT, thereby promoting bone metastasis; (3) Indirect effects of BMAs are mediated through interactions with OCs, OBs, ECs, and MSCs, indirectly aiding lung cancer bone metastasis. For instance, leptin secreted by BMAs may regulate osteogenesis and osteolysis by inhibiting OC activity, suppressing RANKL production and enhancing OPG secretion. Adiponectin influences OC activity, increases regulatory T cells, and inhibits NK cell activity, contributing to immune evasion by cancer cells. Resistin could activate the NF-κB pathway, releasing inflammatory cytokines like TNF-α and IL-6, thus promoting OC activation and inhibiting OB differentiation, essential for lung cancer bone metastasis.
Despite the existence of both metastasis-promoting and inhibitory factors secreted by BMAs, they predominantly exert a pro-metastatic role in lung cancer bone metastasis. Reducing BMAs in the bone microenvironment could represent a novel approach to inhibit lung cancer bone metastasis. Considering the shared origin and mutual regulation during differentiation between BMAs and OBs, promoting osteogenic differentiation to decrease BMAs might suppress lung cancer bone metastasis. Z-DNA binding protein 1 (ZBP1) has been identified as a novel regulator of osteogenic and adipogenic differentiation through the Wnt/β-catenin signaling pathway (188). Therefore, ZBP1 could serve as a novel therapeutic target for treating lung cancer bone metastasis. Additionally, inhibiting the actions of BMA-secreted factors could indirectly repress lung cancer bone metastasis. For instance, clinical research on metastatic renal cell carcinoma (RCC) patients undergoing tyrosine kinase inhibitor (TKI) therapy revealed that the adiponectin-AdipoR1 axis inhibits tumor cell migration and invasion by blocking the GSK3β/β-Catenin pathway (189). This raises the possibility that adiponectin-AdipoR1 might exhibit similar effects in lung cancer bone metastasis, potentially serving as a therapeutic target for further verification.
In summary, BMAs and their secreted adipokines present effective therapeutic targets for lung cancer bone metastasis, paving new directions for future research in this field.
JL: Writing – original draft, Writing – review & editing. JW: Writing – original draft. YN: Writing – review & editing. XY: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Fund program: The National Natural Science Foundation of China (Grant Number 82273294).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Qin N, Ma H, Jin G. Annual progress in epidemiological research on lung cancer in 2022. Chin Med J. (2023) 103:1068–73. doi: 10.3760/cma.j.cn112137-20221213-02640
3. Riihimaki M, Hemminki A, Fallah M. Metastatic sites and survival in lung cancer. Lung Cancer. (2014) 86:78–84. doi: 10.1016/j.lungcan.2014.07.020
4. Katakami N, Kunikane H, Takeda K. Prospective study on the incidence of bone metastasis (BM) and skeletal-related events (SREs) in patients (pts) with stage IIIB and IV lung cancer-CSP-HOR 13. J Thorac Oncol. (2014) 9:231–8. doi: 10.1097/JTO.0000000000000051
5. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nature reviews. Clin Oncol. (2017) 14:155–67. doi: 10.1038/nrclinonc.2016.144
6. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
7. Tang Q, Chen J, Di Z, Yuan W, Zhou Z, Liu Z, et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/β-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. (2020) 39:232. doi: 10.1186/s13046-020-01690-z
8. Wang S, Cheng Y, Gao Y, He Z, Zhou W, Chang R, et al. SH2B1 promotes epithelial-mesenchymal transition through the IRS1/β-catenin signaling axis in lung adenocarcinoma. Mol carcinogenesis. (2018) 57:640–52. doi: 10.1002/mc.22788
9. Hamilton G, Rath B. Circulating tumor cell interactions with macrophages: implications for biology and treatment. Trans Lung Cancer Res. (2017) 6:418–30. doi: 10.21037/tlcr.2017.07.04
10. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature reviews. Cancer. (2003) 06:453–8. doi: 10.1038/nrc1098
11. Rucci N, Teti A. Osteomimicry: how the seed grows in the soil. Calcified Tissue Int. (2018) 102:131–40. doi: 10.1007/s00223-017-0365-1
12. Zhang X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun (London England). (2019) 39:76. doi: 10.1186/s40880-019-0425-1
13. Zhang W, Xu Z, Hao X, He T, Li J, Shen Y, et al. Bone metastasis initiation is coupled with bone remodeling through osteogenic differentiation of NG2+ Cells. Cancer Discovery. (2023) 13:474–95. doi: 10.1158/2159-8290.CD-22-0220
14. Zhao M-N, Zhang L-F, Sun Z, Qiao L-H, Yang T, Ren Y-Z, et al. A novel microRNA-182/Interleukin-8 regulatory axis controls osteolytic bone metastasis of lung cancer. Cell Death Dis. (2023) 14:298. doi: 10.1038/s41419-023-05819-8
15. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
16. Yang M, Sun Yi, Sun J, Wang Z, Zhou Y, Yao G, et al. Differentially expressed and survival-related proteins of lung adenocarcinoma with bone metastasis. Cancer Med. (2018) 7:1081–92. doi: 10.1002/cam4.1363
17. Song X, Cheng Xu, Jin X, Ruan S, Xu X, Lu F, et al. EGFL6 promotes bone metastasis of lung adenocarcinoma by increasing cancer cell Malignancy and bone resorption. Clin Exp metastasis. (2023) 40:357–71. doi: 10.1007/s10585-023-10219-5
18. Liu Y, Chen J, Yang Y, Zhang L, Jiang WG. Molecular impact of bone morphogenetic protein 7, on lung cancer cells and its clinical significance. Int J Mol Med. (2012) 29:1016–24. doi: 10.3892/ijmm.2012.948
19. Aoki M, Umehara T, Kamimura Go, Imamura N, Morizono S, Nonaka Y, et al. Expression of bone morphogenetic protein-7 significantly correlates with non-small cell lung cancer progression and prognosis: A retrospective cohort study. Clinical medicine insights. Oncology. (2019) 13:1179554919852087. doi: 10.1177/1179554919852087
20. Juárez P, Guise TA. TGF-β in cancer and bone: implications for treatment of bone metastases. Bone. (2011), 48:23–9. doi: 10.1016/j.bone.2010.08.004
21. Liu L, Chen X, Wang Y, Qu Z, Lu Q, Zhao J, et al. Notch3 is important for TGF-β-induced epithelial-mesenchymal transition in non-small cell lung cancer bone metastasis by regulating ZEB-1. Cancer Gene Ther. (2014) 21:364–72. doi: 10.1038/cgt.2014.39
22. Wu M, Kong D, Zhang Y. SPON2 promotes the bone metastasis of lung adenocarcinoma via activation of the NF-κB signaling pathway. Bone. (2023) 167:116630. doi: 10.1016/j.bone.2022.116630
23. Zhang C, Qin J, Yang Lu, Zhu Z, Yang J, Su W, et al. Exosomal miR-328 originated from pulmonary adenocarcinoma cells enhances osteoclastogenesis via downregulating Nrp-2 expression. Cell Death Discovery. (2022) 8:405. doi: 10.1038/s41420-022-01194-z
24. Kang J, Manna FLa, Bonollo F, Sampson N, Alberts IL, Mingels C, et al. Tumor microenvironment mechanisms and bone metastatic disease progression of prostate cancer. Cancer Lett. (2022) 530:156–69. doi: 10.1016/j.canlet.2022.01.015
25. Yuan Z, Li Y, Zhang S, Wang X, Dou He, Yu Xi, et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. (2023) 22:48. doi: 10.1186/s12943-023-01744-8
26. Shin E, Koo JaS. The role of adipokines and bone marrow adipocytes in breast cancer bone metastasis. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21144967
27. Wu S, Pan Y, Mao Y, Chen Yu, He Y. Current progress and mechanisms of bone metastasis in lung cancer: a narrative review. Trans Lung Cancer Res. (2021) 10:439–51. doi: 10.21037/tlcr-20-835
28. Taverna S, Pucci M, Giallombardo M, Bella MADi, Santarpia M, Reclusa P, et al. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci Rep. (2017) 7:3170. doi: 10.1038/s41598-017-03460-y
29. Xu Z, Liu X, Wang H, Li J, Dai L, Li J, et al. Lung adenocarcinoma cell-derived exosomal miR-21 facilitates osteoclastogenesis. Gene. (2018) 666:116–22. doi: 10.1016/j.gene.2018.05.008
30. Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. (2016) 23:315–23. doi: 10.1016/j.cmet.2015.11.003
31. Dou C, Li J, Kang F, Cao Z, Yang X, Jiang H, et al. Dual effect of cyanidin on RANKL-induced differentiation and fusion of osteoclasts. J Cell Physiol. (2016) 231:558–67. doi: 10.1002/jcp.24916
32. Chappard D, Bouvard B, Baslé M-F, Legrand E, Audran M. Bone metastasis: histological changes and pathophysiological mechanisms in osteolytic or osteosclerotic localizations. A review. Morphol: Bull l’Association Des anatomistes. (2011) 95:65–75. doi: 10.1016/j.morpho.2011.02.004
33. He Y, Luo W, Liu Y, Wang Y, Ma C, Wu Q, et al. IL-20RB mediates tumoral response to osteoclastic niches and promotes bone metastasis of lung cancer. J Clin Invest. (2022), 132:20. doi: 10.1172/JCI157917
34. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. (2018) 6:16. doi: 10.1038/s41413-018-0019-6
35. Rahim F, Hajizamani S, Mortaz E, Ahmadzadeh A, Shahjahani M, Shahrabi S, et al. Molecular regulation of bone marrow metastasis in prostate and breast cancer. Bone Marrow Res. (2014), 405920. doi: 10.1155/2014/405920
36. Jeong HM, Cho SW, Park SI. Osteoblasts are the centerpiece of the metastatic bone microenvironment. Endocrinol Metab (Seoul). (2016) 31:485–92. doi: 10.3803/EnM.2016.31.4.485
37. Pang H, Ma N, Jiao Mi, Shen W, Xin Bo, Wang T, et al. The biological effects of dickkopf1 on small cell lung cancer cells and bone metastasis. Oncol Res. (2017) 25:35–42. doi: 10.3727/096504016X14719078133249
38. Clines GA, Mohammad KS, Bao Y, Stephens OW, Suva LJ, Shaughnessy JD, et al. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol (Baltimore Md.). (2007) 21:486–98. doi: 10.1210/me.2006-0346
39. Xu S, Yang F, Liu R, Li X, Fan H, Liu J, et al. Serum microRNA-139-5p is downregulated in lung cancer patients with lytic bone metastasis. Oncol Rep. (2018) 39:2376–84. doi: 10.3892/or.2018.6316
40. Feng Y, Wan P, Yin L, Lou X. The inhibition of microRNA-139-5p promoted osteoporosis of bone marrow-derived mesenchymal stem cells by targeting wnt/beta-catenin signaling pathway by NOTCH1. J Microbiol Biotechnol. (2020) 30:448–58. doi: 10.4014/jmb.1908.08036
41. Chen WG, Sun J, Shen WW, Yang SZ, Zhang Y, Hu X, et al. Sema4D expression and secretion are increased by HIF-1alpha and inhibit osteogenesis in bone metastases of lung cancer. Clin Exp Metastasis. (2019) 36:39–56. doi: 10.1007/s10585-018-9951-5
42. Engblom C, Pfirschke C, Zilionis R, Martins JDaS, Bos SA, Courties G, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. (2017) 358:6367. doi: 10.1126/science.aal5081
43. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. (2019) 133:2159–67. doi: 10.1182/blood-2018-11-844548
44. Zhou J, Lu X, Zhu H, Ding N, Zhang Y, Xu X, et al. Resistance to immune checkpoint inhibitors in advanced lung cancer: Clinical characteristics, potential prognostic factors and next strategy. Front Immunol. (2023) 14:1089026. doi: 10.3389/fimmu.2023.1089026
45. Renaud S, Falcoz PE, Schaeffer M. Prognostic value of the KRAS G12V mutation in 841 surgically resected Caucasian lung adenocarcinoma cases. Br J Cancer. (2015) 113:1206–15. doi: 10.1038/bjc.2015.327
46. D’Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J Bone Oncol. (2019) 15:004–4. doi: 10.1016/j.jbo.2018.10.004
47. Yuan Bo, Clowers MJ, Velasco WV, Peng S, Peng Q, Shi Y, et al. Targeting IL-1β as an immunopreventive and therapeutic modality for K-ras-mutant lung cancer. JCI Insight. (2022) 7:11. doi: 10.1172/jci.insight.157788
48. Pretre V, Papadopoulos D, Regard J, Pelletier M, Woo J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine. (2022) 153:155s850. doi: 10.1016/j.cyto.2022.155850
49. Maimon A, Levi-Yahid V, Ben-Meir K, Halpern A, Talmi Z, Priya S, et al. Myeloid cell-derived PROS1 inhibits tumor metastasis by regulating inflammatory and immune responses via IL-10. J Clin Invest. (2021) 131:10. doi: 10.1172/JCI126089
50. Zhao X, Lin Y, Jiang B, Yin J, Lu C, Wang J, et al. Icaritin inhibits lung cancer-induced osteoclastogenesis by suppressing the expression of IL-6 and TNF-a and through AMPK/mTOR signaling pathway. Anti-canc Drugs. (2020) 31:1004–11. doi: 10.1097/CAD.0000000000000976
51. Gross AC, Cam H, Phelps DA, Saraf AJ, Bid HK, Cam M, et al. IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to metastasis. JCI Insight. (2018) 3:16. doi: 10.1172/jci.insight.99791
52. Liu N-N, Yi C-X, Wei L-Q, Zhou J-A, Jiang T, Hu C-C, et al. The intratumor mycobiome promotes lung cancer progression via myeloid-derived suppressor cells. Cancer Cell. (2023) 41:1927–1944.e9. doi: 10.1016/j.ccell.2023.08.012
53. Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J. Regulation of osteoclast differentiation by cytokine networks. Immune Net. (2018) 18:e8. doi: 10.4110/in.2018.18.e8
54. Raimondo S, Saieva L, Vicario E, Pucci M, Toscani D, Manno M, et al. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol. (2019) 12:2. doi: 10.1186/s13045-018-0689-y
55. Bendre MS, Margulies AG, Walser B, Akel NS, Bhattacharrya S, Skinner RA, et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. (2005) 65:11001–9. doi: 10.1158/0008-5472.CAN-05-2630
56. Tung Y-T, Huang P-W, Chou Y-C, Lai C-W, Wang H-P, Ho H-C, et al. Lung tumorigenesis induced by human vascular endothelial growth factor (hVEGF)-A165 overexpression in transgenic mice and amelioration of tumor formation by miR-16. Oncotarget. (2015) 12:10222–38. doi: 10.18632/oncotarget.3390
57. Lee H-M, Lee H-J, Chang J-E. Inflammatory cytokine.An attractive target for cancer treatment. Biomedicines. (2022) 10:9. doi: 10.3390/biomedicines10092116
58. Gandhi S, Opyrchal M, Grimm MJ, Slomba RT, Kokolus KM, Witkiewicz A, et al. Systemic infusion of TLR3-ligand and IFN-α in patients with breast cancer reprograms local tumor microenvironments for selective CTL influx. J Immunother Cancer. (2023) 11:11. doi: 10.1136/jitc-2023-007381
59. Heidegger I, Fotakis G, Offermann A, Goveia J, Daum S, Salcher S, et al. Comprehensive characterization of the prostate tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in prostate cancer. Mol Cancer. (2022) 21:132. doi: 10.1186/s12943-022-01597-7
60. Liao T, Chen W, Sun J, Zhang Y, Hu X, Yang S, et al. CXCR4 accelerates osteoclastogenesis induced by non-small cell lung carcinoma cells through self-potentiation and VCAM1 secretion. Cell Physiol Biochem. (2018) 50:1084–99. doi: 10.1159/000494533
61. Li J, Wang L, Chen X, Li L, Li Yu, Ping Yu, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
62. Yasuda H. Discovery of the RANKL/RANK/OPG system. J Bone mineral Metab. (2021) 39:2–11. doi: 10.1007/s00774-020-01175-1
63. Yao Z, Getting SJ, Locke IC. Regulation of TNF-induced osteoclast differentiation. Cells. (2021) 11:1. doi: 10.3390/cells11010132
64. Brouns AJWM, Hendriks LEL, Berge IJR-vd, Driessen AJHM, Roemen GMJM, van Herpen BLJ, et al. Association of RANKL and EGFR gene expression with bone metastases in patients with metastatic non-small cell lung cancer. Front Oncol. (2023) 13:1145001. doi: 10.3389/fonc.2023.1145001
65. Ono T, Hayashi M, Sasaki F, Nakashima T. RANKL biology: bone metabolism, the immune system, and beyond. Inflammation Regenerat. (2020) 40:2. doi: 10.1186/s41232-019-0111-3
66. Zhang Y, Liang J, Liu P, Wang Q, Liu L, Zhao H. The RANK/RANKL/OPG system and tumor bone metastasis: Potential mechanisms and therapeutic strategies. Front Endocrinol. (2022) 13:1063815. doi: 10.3389/fendo.2022.1063815
67. Leon-Oliva DDe, Barrena-Blázquez S, Jiménez-Álvarez L, Fraile-Martinez O, García-Montero C, López-González L, et al. The RANK-RANKL-OPG system: A multifaceted regulator of homeostasis, immunity, and cancer. Med (Kaunas Lithuania). (2023) 59:10. doi: 10.3390/medicina59101752
68. Deng J, Lu C, Zhao Q, Chen K, Ma S, Li Z. The Th17/Treg cell balance: crosstalk among the immune system, bone and microbes in periodontitis. J periodontal Res. (2022) 57:246–55. doi: 10.1111/jre.12958
69. Titanji K. Beyond antibodies: B cells and the OPG/RANK-RANKL pathway in health, non-HIV disease and HIV-induced bone loss. Front Immunol. (2017) 8:1851. doi: 10.3389/fimmu.2017.01851
70. Rochette L, Meloux A, Rigal E, Zeller M, Cottin Y, Vergely C. The role of osteoprotegerin and its ligands in vascular function. Int J Mol Sci. (2019) 20:3. doi: 10.3390/ijms20030705
71. Romeo SG, Alawi KM, Rodrigues J, Singh A, Kusumbe AP, Ramasamy SK. Endothelial proteolytic activity and interaction with non-resorbing osteoclasts mediate bone elongation. Nat Cell Biol. (2019) 21:430–41. doi: 10.1038/s41556-019-0304-7
72. Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Macciò A. EGFR-mutated non-small cell lung cancer and resistance to immunotherapy. Role of the tumor microenvironment. Int J Mol Sci. (2022) 23:12. doi: 10.3390/ijms23126489
73. Chen J, Zhao L, Xu M-F, Huang Di, Sun X-L, Zhang Y-X. Novel isobavachalcone derivatives induce apoptosis and necroptosis in human non-small cell lung cancer H1975 cells. J Enzyme inhibition Medic Chem. (2024) 39:2292006. doi: 10.1080/14756366.2023.2292006
74. Liu J-H, Li C, Cao L, Zhang C-H, Zhang Z-H. Cucurbitacin B regulates lung cancer cell proliferation and apoptosis via inhibiting the IL-6/STAT3 pathway through the lncRNA XIST/miR-let-7c axis. Pharm Biol. (2022) 60:154–62. doi: 10.1080/13880209.2021.2016866
75. Su L, Chen Y, Huang C, Wu S, Wang X, Zhao X, et al. Targeting Src reactivates pyroptosis to reverse chemoresistance in lung and pancreatic cancer models. Sci Trans Med. (2023) 15:678:eabl7895. doi: 10.1126/scitranslmed.abl7895
76. Pennington Z, Goodwin ML, Westbroek EM, Cottrill E, Ahmed AK, Sciubba DM. Lactate and cancer: spinal metastases and potential therapeutic targets (part 2). Ann Trans Med. (2019) 10:221. doi: 10.21037/atm.2019.01.85
77. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U.S.A. (2011) 108:3725–30. doi: 10.1073/pnas.1100446108
78. Gyamfi J, Eom M, Koo JS, Choi J. Multifaceted roles of interleukin-6 in adipocyte–breast cancer cell interaction. Trans Oncol. (2018) 11:275–85. doi: 10.1016/j.tranon.2017.12.009
79. Tacchio MDi, Macas J, Weissenberger J, Sommer K, Bähr O, Steinbach JP, et al. Tumor vessel normalization, immunostimulatory reprogramming, and improved survival in glioblastoma with combined inhibition of PD-1, angiopoietin-2, and VEGF. Cancer Immunol Res. (2019) 7:1910–27. doi: 10.1158/2326-6066.CIR-18-0865
80. Rami AZA, Hamid AA, Anuar NNM, Aminuddin A, Ugusman A. Exploring the relationship of perivascular adipose tissue inflammation and the development of vascular pathologies. Mediators Inflammation. (2022) 2022:2734321. doi: 10.1155/2022/2734321
81. Li Y, Cao S, Gaculenko A, Zhan Y, Bozec A, Chen X. Distinct metabolism of bone marrow adipocytes and their role in bone metastasis. Front Endocrinol. (2022) 13:90 2033. doi: 10.3389/fendo.2022.902033
82. Galley JC, Singh S, Awata WMC, Alves JV, Bruder-Nascimento T. Adipokines. Deciphering the cardiovascular signature of adipose tissue. Biochem Pharmacol. (2022) 206:115324. doi: 10.1016/j.bcp.2022.115324
83. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Bone marrow fat: linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. (2014) 33:527–43. doi: 10.1007/s10555-013-9484-y
84. Rendina-Ruedy E, Rosen CJ. Lipids in the bone marrow: an evolving perspective. Cell Metab. (2020) 31:219–31. doi: 10.1016/j.cmet.2019.09.015
85. Wang H, Leng Y, Gong Y. Bone marrow fat and hematopoiesis. Front Endocrinol. (2018) 9:694. doi: 10.3389/fendo.2018.00694
86. Horowitz MC, Berry R, Holtrup B, Sebo Z, Nelson T, Fretz JA, et al. Bone marrow adipocytes. Adipocyte. (2017) 6:193–204. doi: 10.1080/21623945.2017.1367881
87. Li Q, Wu Y, Kang N. Marrow adipose tissue: its origin, function, and regulation in bone remodeling and regeneration. Stem Cells Int. (2018) 2018:7098456. doi: 10.1155/2018/7098456
88. Hawkes CP, Mostoufi-Moab S. Fat-bone interaction within the bone marrow milieu: Impact on hematopoiesis and systemic energy metabolism. Bone. (2019) 119:57–64. doi: 10.1016/j.bone.2018.03.012
89. Ali D, Figeac F, Caci A, Ditzel N, Schmal C, Kerckhofs G, et al. High-fat diet-induced obesity augments the deleterious effects of estrogen deficiency on bone: Evidence from ovariectomized mice. Aging Cell. (2022) 21:e13726. doi: 10.1111/acel.13726
90. Beekman KM, Duque G, Corsi A, Tencerova M, Bisschop PH, Paccou J. Osteoporosis and bone marrow adipose tissue. Curr osteoporosis Rep. (2023) 21:45–55. doi: 10.1007/s11914-022-00768-1
91. Gawronska-Kozak B, Staszkiewicz J, Gimble JM, Kirk-Ballard H. Recruitment of fat cell precursors during high fat diet in C57BL/6J mice is fat depot specific. Obesity. (2014) 22:1091–102. doi: 10.1002/oby.20671
92. Hardouin P, Pansini V, Cortet B. Bone marrow fat. Joint Bone Spine. (2014) 81:313–9. doi: 10.1016/j.jbspin.2014.02.013
93. Oliveira MC, Vullings J, van de Loo FAJ. Osteoporosis and osteoarthritis are two sides of the same coin paid for obesity. Nutrition. (2020) 70:110486. doi: 10.1016/j.nut.2019.04.001
94. Histing T, Andonyan A, Klein M, Scheuer C, Stenger D, Holstein JH. N T Veith,et al.Obesity does not affect the healing of femur fractures in mice. Injury. (2016) 47:1435–44. doi: 10.1016/j.injury.2016.04.030
95. Xu F, Du Yu. Shilong Hang, Anmin Chen, Fengjin Guo, Tao Xu.Adipocytes regulate the bone marrow microenvironment in a mouse model of obesity. Mol Med Rep. (2013) 8:823–8. doi: 10.3892/mmr.2013.1572
96. Gall CélineLe, Bellahcène A, Bonnelye E, Gasser JürgA, Castronovo V, Green J, et al. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. (2007) 67:9894–902. doi: 10.1158/0008-5472.CAN-06-3940
97. Duong L, Wesolowski GA, Leung P, Oballa R, Pickarski M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol Cancer Ther. (2014) 13:2898–909. doi: 10.1158/1535-7163.MCT-14-0253
98. Cheng M, Liu P, Xu LX. Iron promotes breast cancer cell migration via IL-6/JAK2/STAT3 signaling pathways in a paracrine or autocrine IL-6-rich inflammatory environment. J inorganic Biochem. (2020) 210:111159. doi: 10.1016/j.jinorgbio.2020.111159
99. Lin Z, He H, Wang M, Liang J. MicroRNA-130a controls bone marrow mesenchymal stem cell differentiation towards the osteoblastic and adipogenic fate. Cell proliferation. (2019) 52:e12688. doi: 10.1111/cpr.12688
100. Liu Yu, Almeida M, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. Skeletal inflammation and attenuation of Wnt signaling, Wnt ligand expression, and bone formation in atherosclerotic ApoE-null mice. American journal of physiology. Endocrinol Metab. (2016) 310:E762–73. doi: 10.1152/ajpendo.00501.2015
101. Huang M, Liu H, Zhu L, Li X, Li J, Yang S, et al. Mechanical loading attenuates breast cancer-associated bone metastasis in obese mice by regulating the bone marrow microenvironment. J Cell Physiol. (2021) 236:6391–406. doi: 10.1002/jcp.30314
102. Xie C, Chen Q. Adipokines: new therapeutic target for osteoarthritis? Curr Rheumatol Rep. (2019) 21:71. doi: 10.1007/s11926-019-0868-z
103. Singh L, Tyagi S, Myers D, Duque G. Good, bad, or ugly: the biological roles of bone marrow fat. Curr Osteoporos Rep. (2018) 16:130–7. doi: 10.1007/s11914-018-0427-y
104. Gunaratnam K, Vidal C, Boadle R, Thekkedam C, Duque G. Mechanisms of palmitate-induced cell death in human osteoblasts. Biol Open. (2013) 2:1382–9. doi: 10.1242/bio.20136700
105. Favaretto F, Bettini S, Busetto L, Milan G, Vettor R. Adipogenic progenitors in different organs: Pathophysiological implications. Rev endocrine Metab Disord. (2022) 23:71–85. doi: 10.1007/s11154-021-09686-6
106. Archivum Immunologiae et. Therapiae Experimentalis. Annual report for 1964. Arch Immunol Ther Exp (Warsz). (1965) 13:Suppl:1–15.
107. Lu Y, Gao X, Wang R, Sun J, Guo B, Wei R, et al. Adiponectin inhibits proliferation of vascular endothelial cells induced by Ox-LDL by promoting dephosphorylation of Caveolin-1 and depolymerization of eNOS and up-regulating release of NO. Int Immunopharmacol. (2019) 73:424–34. doi: 10.1016/j.intimp.2019.05.017
108. Tulotta C, Ottewell P. The role of IL-1B in breast cancer bone metastasis. Endocr Relat Cancer. (2018) 25:R421–34. doi: 10.1530/ERC-17-0309
109. Gyamfi J, Eom M, Koo JS, Choi J. Multifaceted roles of interleukin-6 in adipocyte-breast cancer cell interaction. Transl Oncol. (2018) 11:275–85. doi: 10.1016/j.tranon.2017.12.009
110. Aaron N, Costa S, Rosen CJ, Qiang Li. The implications of bone marrow adipose tissue on inflammaging. Front Endocrinol. (2022) 13:853765. doi: 10.3389/fendo.2022.853765
111. Evangelista GCM, Salvador PA, Soares SMA, Barros LRC, Xavier FHdaC, Abdo LM, et al. 4T1 mammary carcinoma colonization of metastatic niches is accelerated by obesity. Front Oncol. (2019) 9:685. doi: 10.3389/fonc.2019.00685
112. Feng HL, Guo P, Wang J, Liu QY, Xu JF, Yang HC, et al. Association of the expression of leptin and leptin receptor with bone metastasis in pulmonary adenocarcinoma. Chin J Oncol. (2016) 38:840–4. doi: 10.3760/cma.j.issn.0253-3766.2016.11.008
113. Oyanagi J, Koh Y, Sato K, Mori K, Teraoka S, Akamatsu H, et al. Predictive value of serum protein levels in patients with advanced non-small cell lung cancer treated with nivolumab. Lung Cancer. (2019) 6:107–13. doi: 10.1016/j.lungcan.2019.03.020
114. Olea-Flores M, Juárez-Cruz JC, Zuñiga-Eulogio MD, Acosta E, García-Rodríguez E, Zacapala-Gomez AE, et al. New actors driving the epithelial-mesenchymal transition in cancer the role of leptin. Biomolecules. (2020) 10:12. doi: 10.3390/biom10121676
115. Morris EV, Edwards CM. Adipokines, adiposity, and bone marrow adipocytes: Dangerous accomplices in multiple myeloma. J Cell Physiol. (2018) 233:9159–66. doi: 10.1002/jcp.26884
116. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis. (2015) 32:353–68. doi: 10.1007/s10585-015-9714-5
117. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
118. Samimi A, Ghanavat M, Shahrabi S, Azizidoost S, Saki N. Role of bone marrow adipocytes in leukemia and chemotherapy challenges. Cell Mol Life Sci. (2019) 76:2489–97. doi: 10.1007/s00018-019-03031-6
119. Wang C, Wang J, Chen K, Pang H, Li X, Zhu J, et al. Caprylic acid (C8:0) promotes bone metastasis of prostate cancer by dysregulated adipo-osteogenic balance in bone marrow. Cancer Sci. (2020) 111:3600–12. doi: 10.1111/cas.14606
120. Luis Géraldine, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. (2021) 07:102006. doi: 10.1016/j.redox.2021.102006
121. Herroon MK, Rajagurubandara E, Hardaway AL, Powell K, Turchick A, Feldmann D, et al. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget. (2013) 4:2108–23. doi: 10.18632/oncotarget.1482
122. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. (2019) 129:3006–17. doi: 10.1172/JCI127201
123. Yip RKH, Rimes JS, Capaldo BD, Vaillant François, Mouchemore KA, Pal B, et al. Mammary tumour cells remodel the bone marrow vascular microenvironment to support metastasis. Nat Commun. (2021) 12:6920. doi: 10.1038/s41467-021-26556-6
124. Wang K, Jiang L, Hu A, Sun C, Zhou L, Huang Y, et al. Vertebral-specific activation of the CX3CL1/ICAM-1 signaling network mediates non-small-cell lung cancer spinal metastasis by engaging tumor cell-vertebral bone marrow endothelial cell interactions. Theranostics. (2021) 11:4770–89. doi: 10.7150/thno.54235
125. Zhang Y, Chua S. Leptin function and regulation. Compr Physiol. (2017) 8:351–69. doi: 10.1002/cphy.c160041
126. Sáinz N, Barrenetxe J, Moreno-Aliaga MaríaJ, Martínez JoséA. Leptin resistance and diet-induced obesity: central and peripheral actions of leptin. Metabolism: Clin Exp. (2015) 64:35–46. doi: 10.1016/j.metabol.2014.10.015
127. Münzberg H. Christopher D Morrison.Structure, production and signaling of leptin. Metabolism: Clin Exp. (2015) 64:13–23. doi: 10.1016/j.09.010.metabol.2014
128. Xu M, Cao F-L, Li N, Gao X, Su X, Jiang X. Leptin induces epithelial-to-mesenchymal transition via activation of the ERK signaling pathway in lung cancer cells. Oncol letters. (2018) 16:4782–8. doi: 10.3892/ol.2018.9230
129. Wang W, Yan H, Dou C, Su Y. Human leptin triggers proliferation of A549 cells via blocking endoplasmic reticulum stress-related apoptosis. Biochemistry. (2013) 78:1333–41. doi: 10.1134/S0006297913120031
130. Zheng X-J, Yang Z-X, Dong Y-J, Zhang G-Y, Sun M-F, An X-K, et al. Downregulation of leptin inhibits growth and induces apoptosis of lung cancer cells via the Notch and JAK/STAT3 signaling pathways. Biol Open. (2016) 5:794–800. doi: 10.1242/bio.017798
131. Maroni P. Leptin, adiponectin, and sam68 in bone metastasis from breast cancer. Int J Mol Sci. (2020) 21:3. doi: 10.3390/ijms21031051
132. Boura P, Loukides S, Grapsa D, Achimastos A, Syrigos K. The diverse roles of adiponectin in non-small-cell lung cancer: current data and future perspectives. Future Oncol. (2015) 11:2193–203. doi: 10.2217/fon.15.96
133. Roger Lijnen H, Scroyen I. Effect of vascular endothelial growth factor receptor 2 antagonism on adiposity in obese mice. J Mol endocrinology. (2013) 50:319–24. doi: 10.1530/JME-12-0244
134. Maroni P, Luzzati A, Perrucchini G, Cannavò L, Bendinelli P. Leptin receptor, KHDRBS1 (KH RNA binding domain containing, signal transduction associated 1), and adiponectin in bone metastasis from breast carcinoma. Immunohistochemical Study. Biomedicines. (2020) 8:11. doi: 10.3390/biomedicines8110510
135. Zhang H-Q, Wang L-J, Liu S-H, Li J, Xiao L-G, Yang G-T. Adiponectin regulates bone mass in AIS osteopenia via RANKL/OPG and IL6 pathway. J Trans Med. (2019) 17:64. doi: 10.1186/s12967-019-1805-7
136. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology. (2010) 52:1713–22. doi: 10.1002/hep.23892
137. Akifusa S, Kamio N, Shimazaki Y, Yamaguchi N, Nonaka K, Yamashita Y. Involvement of the JAK-STAT pathway and SOCS3 in the regulation of adiponectin-generated reactive oxygen species in murine macrophage RAW 264 cells. J Cell Biochem. (2010),111, 597–606. doi: 10.1002/jcb.22745
138. Luo X-H, Guo L-J, Xie H, Yuan L-Q, Wu X-P, Zhou H-D, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone mineral research: Off J Am Soc Bone Mineral Res. (2006) 21:1648–56. doi: 10.1359/jbmr.060707
139. Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol. (2018) 8:1031–63. doi: 10.1002/cphy.c170046
140. Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. (2010) 1804:581–91. doi: 10.1016/j.bbapap.2009.09.012
141. Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1-and-R2-mediated AMPK activation. Mol Endocrinol. (2010) 24:1441–52. doi: 10.1210/me.2009-0498
142. Ren Le, Bao D, Wang L, Xu Q, Xu Y, Shi Z. Nucleobindin-2/nesfatin-1 enhances the cell proliferation, migration, invasion and epithelial-mesenchymal transition in gastric carcinoma. J Cell Mol Med. (2022) 26:4986–94. doi: 10.1111/jcmm.17522
143. Sudan SK, Deshmukh SK, Poosarla T, Holliday NP, Dyess DL, Singh AP, et al. Resistin: An inflammatory cytokine with multi-faceted roles in cancer. Biochimica et biophysica acta. Rev cancer. (2020) 1874:188419. doi: 10.1016/j.bbcan.2020.188419
144. Muruganandan S, Ionescu AM, Sinal CJ. At the crossroads of the adipocyte and osteoclast differentiation programs: future therapeutic perspectives. Int J Mol Sci. (2020) 21:7. doi: 10.3390/ijms21072277
145. Ntikoudi E, Kiagia M, Boura P, Syrigos KN. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat Rev. (2014) 40:22–30. doi: 10.1016/j.ctrv.2013.06.005
146. Tzanavari T, Tasoulas J, Vakaki C, Mihailidou C, Tsourouflis G, Theocharis S. The role of adipokines in the establishment and progression of head and neck neoplasms. Curr Medic Chem. (2019) 26:4726–48. doi: 10.2174/0929867325666180713154505
147. Chen S-S, Tang C-H, Chie M-J, Tsai C-H, Fong Y-C, Lu Y-C, et al. Resistin facilitates VEGF-A-dependent angiogenesis by inhibiting miR-16-5p in human chondrosarcoma cells. Cell Death Dis. (2019) 10:31. doi: 10.1038/s41419-018-1241-2
148. Kim H, Lee J-H, Lee SK, Song N-Y, Son SH, Kim KiR, et al. Chemerin treatment inhibits the growth and bone invasion of breast cancer cells. Int J Mol Sci. (2020) 21:8. doi: 10.3390/ijms21082871
149. Ostrowska Z, Morawiecka-Pietrzak Małgorzata, Pluskiewicz W, Świętochowska Elżbieta, Strzelczyk J, Gołąbek K, et al. The relationship between chemerin, bone metabolism, the RANKL/RANK/OPG system, and bone mineral density in girls with anorexia nervosa. Endokrynologia Polska. (2022) 73:26–34. doi: 10.5603/EP.a2021.0103
150. Hu X, Xiang F, Feng Y, Gao F, Ge S, Wang C, et al. Neutrophils promote tumor progression in oral squamous cell carcinoma by regulating EMT and JAK2/STAT3 signaling through chemerin. Front Oncol. (2022) 12:812044. doi: 10.3389/fonc.2022.812044
151. Han L, Zhang Yu, Wan S, Wei Q, Shang W, Huang G, et al. Loss of chemerin triggers bone remodeling in vivo and in vitro. Mol Metab. (2021) 53:101322. doi: 10.1016/j.molmet.2021.101322
152. Dahl TB, Holm S, Aukrust Pål, Halvorsen B. Visfatin/NAMPT: a multifaceted molecule with diverse roles in physiology and pathophysiology. Annu Rev Nutr. (2012) 32:229–43. doi: 10.1146/annurev-nutr-071811-150746
153. Olea-Flores M, Zuñiga-Eulogio M, Tacuba-Saavedra A, Bueno-Salgado M, Sánchez-Carvajal A, Vargas-Santiago Y, et al. Leptin promotes expression of EMT-related transcription factors and invasion in a src and FAK-dependent pathway in MCF10A mammary epithelial cells. Cells. (2019) 8:10. doi: 10.3390/cells8101133
154. Tsai CF, Chen JH, Wu CT, Chang PC, Wang SL, Yeh WL. Induction of osteoclast-like cell formation by leptin-induced soluble intercellular adhesion molecule secreted from cancer cells. Ther Adv Med Oncol(2019). (2019) 14:1758835919846806. doi: 10.1177/1758835919846806
155. Reid IR, Baldock PA, Cornish J. Effects of leptin on the skeleton. Endocrine Rev. (2018) 39:938–59. doi: 10.1210/er.2017-00226
156. Mei L, Li M, Zhang T. MicroRNA miR-874-3p inhibits osteoporosis by targeting leptin (LEP). Bioengineered. (2021) 12:11756–67. doi: 10.1080/21655979.2021.2009618
157. Vita E, Stefani A, Piro G, Mastrantoni L, Cintoni M, Cicchetti G, et al. Leptin-mediated meta-inflammation may provide survival benefit in patients receiving maintenance immunotherapy for extensive-stage small cell lung cancer (ES-SCLC). Cancer immunology Immunother: CII. (2023) 72:3803–12. doi: 10.1007/s00262-023-03533-0
158. Kim J-W, Mahiddine FY, Kim GA. Leptin modulates the metastasis of canine inflammatory mammary adenocarcinoma cells through downregulation of lysosomal protective protein cathepsin A (CTSA). Int J Mol Sci. (2020) 21:23. doi: 10.3390/ijms21238963
159. Sanchez-Margalet V, Martin-Romero C. Human leptin signaling in human peripheral blood mononuclear cells: activation of the JAK-STAT pathway. Cell Immunol. (2001) 211:30–6. doi: 10.1006/cimm.2001.1815
160. Maurya R, Sebastian P, Namdeo M, Devender M, Gertler A. COVID-19 severity in obesity: leptin and inflammatory cytokine interplay in the link between high morbidity and mortality. Front Immunol. (2021) 12:649359. doi: 10.3389/fimmu.2021.649359
161. Lam QLK, Wang S, Ko OKH, Kincade PW, Lu L. Leptin signaling maintains B-cell homeostasis via induction of Bcl-2 and Cyclin D1. Proc Natl Acad Sci United States America. (2010) 107:13812–7. doi: 10.1073/pnas.1004185107
162. Skrypnik D, Skrypnik K, Suliburska J, Bogdański Paweł. Leptin-VEGF crosstalk in excess body mass and related disorders: A systematic review. Obes reviews: an Off J Int Assoc Study Obes. (2023) 24:e13575. doi: 10.1111/obr.13575
163. Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J leukocyte Biol. (2018) 103:395–407. doi: 10.1002/JLB.4HI0517-210R
164. Luo G, He Y, Yu X. Bone marrow adipocyte: an intimate partner with tumor cells in bone metastasis. Front Endocrinol. (2018) 9:339. doi: 10.3389/fendo.2018.00339
165. Man K, Ng KTP, Xu A, Cheng Q, Lo CM, Xiao JW, et al. Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin Cancer research: an Off J Am Assoc Cancer Res. (2010) 16:967–77. doi: 10.1158/1078-0432.CCR-09-1487
166. Wang Y, Zhang X, Shao J, Liu H, Liu X, Luo En. Adiponectin regulates BMSC osteogenic differentiation and osteogenesis through the Wnt/β-catenin pathway. Sci Rep. (2017) 7:3652. doi: 10.1038/s41598-017-03899-z
167. Mukohira H, Hara T, Abe S, Tani-Ichi S, Sehara-Fujisawa A, Nagasawa T, et al. Mesenchymal stromal cells in bone marrow express adiponectin and are efficiently targeted by an adiponectin promoter-driven Cre transgene. Int Immunol. (2019) 131:729–42. doi: 10.1093/intimm/dxz042
168. Illiano M, Nigro E, Sapio L, Caiafa I, Spina A, Scudiero O, et al. Adiponectin down-regulates CREB and inhibits proliferation of A549 lung cancer cells. Pulmonary Pharmacol Ther. (2017) 45:114–20. doi: 10.1016/j.pupt.2017.05.009
169. Tsai J-R, Liu P-L, Chen Y-H, Chou S-H, Cheng Y-J, Hwang J-J, et al. Curcumin inhibits non-small cell lung cancer cells metastasis through the adiponectin/NF-κb/MMPs signaling pathway. PloS One. (2015) 10:e0144462. doi: 10.1371/journal.pone.0144462
170. Jian M, Kwan JS-C, Bunting M, Ng RC-L, Chan KHo. Adiponectin suppresses amyloid-β oligomer (AβO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling pathway. J Neuroinflamm. (2019) 16:110. doi: 10.1186/s12974-019-1492-6
171. Rupp SK, Stengel A. Interactions between nesfatin-1 and the autonomic nervous system-An overview. Peptides. (2022) 149:170719. doi: 10.1016/j.peptides.2021.170719
172. Suzuki S, Takagi K, Miki Y, Onodera Y, Akahira JI, Ebata A, et al. Nucleobindin 2 in human breast carcinoma as a potent prognostic factor. Cancer Sci. (2012) 103:136–43. doi: 10.1111/j.1349-7006.2011.02119.x
173. Altan B, Kaira K, Okada S, Saito T, Yamada E, Bao H, et al. High expression of nucleobindin 2 is associated with poor prognosis in gastric cancer. Tumor Biol. (2017) 39:1–7. doi: 10.1177/1010428317703817
174. Akrida I, Papadaki H. Adipokines and epithelial-mesenchymal transition (EMT) in cancer. Mol Cell Biochem. (2023) 1:30. doi: 10.1007/s11010-023-04670-x
175. Kmiecik AM, Dzięgiel P, Podhorska-Okołów M. Nucleobindin-2/nesfatin-1-A new cancer related molecule? Int J Mol Sci. (2021) 22:15. doi: 10.3390/ijms22158313
176. Chang J-W, Liu S-C, Lin Y-Y, He X-Y, Wu Y-S, Su C-M, et al. Nesfatin-1 stimulates CCL2-dependent monocyte migration and M1 macrophage polarization: implications for rheumatoid arthritis therapy. Int J Biol Sci. (2023) 19:281–93. doi: 10.7150/ijbs.77987
177. Jiang L, Xu K, Li J, Zhou X, Xu L, Wu Z, et al. Nesfatin-1 suppresses interleukin-1β-induced inflammation, apoptosis, and cartilage matrix destruction in chondrocytes and ameliorates osteoarthritis in rats. Aging. (2020) 12:1760–77. doi: 10.18632/aging.102711
178. Solmaz A, Bahadır E, Gülçiçek OB, Yiğitbaş H, Çelik A, Karagöz Ayça, et al. Nesfatin-1 improves oxidative skin injury in normoglycemic or hyperglycemic rats. Peptides. (2016) 78:1–10. doi: 10.1016/j.peptides.2015.12.006
179. Tripathi D, Kant S, Pandey S, Ehtesham NZ. Resistin in metabolism, inflammation, and disease. FEBS J. (2020) 287:3141–9. doi: 10.1111/febs.15322
180. Deepika F, Bathina S, Armamento-Villareal R. Novel adipokines and their role in bone metabolism: A narrative review. Biomedicines. (2023) 11:2. doi: 10.3390/biomedicines11020644
181. Kuo C-H, Chen K-F, Chou S-H, Huang Y-F, Wu C-Y, Cheng D-E, et al. Lung tumor-associated dendritic cell-derived resistin promoted cancer progression by increasing Wolf-Hirschhorn syndrome candidate 1/Twist pathway. Carcinogenesis. (2013) 34:2600–9. doi: 10.1093/carcin/bgt281
182. Wang X, Tang Y, Xiao R. Chemerin contributes to inflammatory responses and suppresses osteogenic differentiation in chronic periodontitis. Oral Dis. (2023) 29:1706–14. doi: 10.1111/odi.14130
183. Wani AK, Prakash A, Sena S, Akhtar N, Singh R, Chopra C, et al. Unraveling molecular signatures in rare bone tumors and navigating the cancer pathway landscapes for targeted therapeutics. Crit Rev oncology/hematology. (2024) 196:104291. doi: 10.1016/j.critrevonc.2024.104291
184. Gao F, Feng Y, Hu X, Zhang X, Li T, Wang Y, et al. Neutrophils regulate tumor angiogenesis in oral squamous cell carcinoma and the role of Chemerin. Int Immunopharmacol. (2023) 121:110540. doi: 10.1016/j.intimp.2023.110540
185. Dakroub A, Nasser SA, Younis N, Bhagani H, Al-Dhaheri Y, Pintus G, et al. Visfatin: A possible role in cardiovasculo-metabolic disorders. Cells. (2020) 9:11. doi: 10.3390/cells9112444
186. Hung S-Y, Lin C-Y, Yu C-C, Chen H-T, Lien M-Y, Huang Y-W, et al. Visfatin promotes the metastatic potential of chondrosarcoma cells by stimulating AP-1-dependent MMP-2 production in the MAPK pathway. Int J Mol Sci. (2021) 22:16. doi: 10.3390/ijms22168642
187. Zhang Z, Xiao Ke, Wang S, Ansari AR, Niu X, Yang W, et al. Visfatin is a multifaceted molecule that exerts regulation effects on inflammation and apoptosis in RAW264.7 cells and mice immune organs. Front Immunol. (2022) 13:1018973. doi: 10.3389/fimmu.2022.1018973
188. Zhao X, Xie L, Wang Z, Wang J, Xu H, Han X, et al. ZBP1 (DAI/DLM-1) promotes osteogenic differentiation while inhibiting adipogenic differentiation in mesenchymal stem cells through a positive feedback loop of Wnt/β-catenin signaling. Bone Res. (2020) 8:12. doi: 10.1038/s41413-020-0085-4
Keywords: bone marrow adipocytes, adipokines, tumor bone metastasis, lung cancer bone metastasis, immune response
Citation: Li J, Wu J, Xie Y and Yu X (2024) Bone marrow adipocytes and lung cancer bone metastasis: unraveling the role of adipokines in the tumor microenvironment. Front. Oncol. 14:1360471. doi: 10.3389/fonc.2024.1360471
Received: 23 December 2023; Accepted: 08 March 2024;
Published: 20 March 2024.
Edited by:
Parmanand Malvi, University of Alabama at Birmingham, United StatesReviewed by:
Amit Kumar, University of Maryland, United StatesCopyright © 2024 Li, Wu, Xie and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijie Yu, eGlqaWV5dUBzY3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.