 Courtney O. Kelson1
Courtney O. Kelson1 Yekaterina Y. Zaytseva1,2*
Yekaterina Y. Zaytseva1,2*- 1Department of Toxicology and Cancer Biology, University of Kentucky, Lexington, KY, United States
- 2Markey Cancer Center, University of Kentucky, Lexington, KY, United States
Altered lipid metabolism is a well-recognized feature of solid cancers, including colorectal cancer. In colorectal cancer, upregulation of lipid metabolism contributes to initiation, progression, and metastasis; thus, aberrant lipid metabolism contributes to a poor patient outcome. The inactivating mutation of APC, a vital tumor suppressor in the Wnt signaling pathway, is a key event that occurs early in the majority of colorectal cancer cases. The potential crosstalk between lipid metabolism and APC-driven colorectal cancer is poorly understood. This review collectively highlights and summarizes the limited understanding between mutations in APC and the upregulation of Wnt/beta-catenin signaling and lipid metabolism. The interconnection between APC inactivation and aberrant lipid metabolism activates Wnt/beta-catenin signaling which causes transcriptome, epigenetic, and microbiome changes to promote colorectal cancer initiation and progression. Furthermore, the downstream effects of this collaborative effort between aberrant Wnt/beta-catenin signaling and lipid metabolism are enhanced stemness, cellular proliferation, prooncogenic signaling, and survival. Understanding the mechanistic link between APC inactivation and alterations in lipid metabolism may foster identification of new therapeutic targets to enable development of more efficacious strategies for prevention and/or treatment of colorectal cancer.
1 Introduction
Colorectal cancer (CRC) is second only to lung cancer in regard to global cancer mortality for men and women combined (1). Colorectal carcinomas develop through a series of histopathological changes caused by mutations in oncogenes and tumor suppressor genes (2). Epigenetic changes, particularly aberrant DNA methylation, also contribute to pathogenesis and heterogeneity of colorectal cancer (3, 4). Mutation-driven epigenetic abnormalities of DNA methylation at the promoter CpG islands (CGIs) of genes controlling stemness, differentiation, and senescence pathways are important drivers of oncogenic transformation (5). In addition, the epigenome can influence the accumulation of somatic mutations which further promotes development of cancer (4).
The adenomatous polyposis coli (APC) gene is a crucial tumor suppressor and loss of APC function triggers the cascade of events that eventually leads to malignant transformation (6). APC mutations contribute to 80–85% of initiating events in sporadic CRC (7). These mutagenic modifications in APC can present as either germline or somatic mutations. Germline mutations in the APC gene result in familial adenomatous polyposis (FAP), an autosomal dominant inherited condition leading to development of multiple intestinal polyps and CRC (7, 8). Somatic APC mutations occur in 80% of sporadic CRCs and loss of heterozygosity (LOH) of chromosome 5q occurs in 30% to 40% of cases (7, 9). CRC appears to develop via several pathways such as chromosome instability (CIN), microsatellite instability (MSI), and serrated neoplasia pathways (2). The CIN pathway is observed in 65-70% of sporadic CRC and is associated with high APC mutations (approximately 80% of cases). MSI is observed in 12-15% of sporadic CRC and usually has high levels of methylation at the CGIs, but low frequency of APC mutations (2, 10). APC mutation is also less common in the formation of serrated adenocarcinomas via the serrated pathway, which accounts for 10% of all CRCs (11). In the absence of mutations in APC, activation of the Wnt pathway occurs via mutation of other genes or DNA methylation changes of the upstream Wnt antagonists (12).
APC mutations play an important role in activation of the Wnt/β-catenin signaling complex (13). Notably, more than 90% of CRCs have mutations that activate the Wnt pathway and over 80% comprise mutations in APC (14). The Wnt/β-catenin pathway regulates at least 80 target genes (15); is essential for cancer cell proliferation and survival; contributes to cancer cell polarity, stemness maintenance, migration, and invasion (16); supports a cancer-favorable tumor microenvironment; and dampens antitumor immune responses (16, 17).
As a common characteristic of cancer, altered lipid metabolism has been actively investigated as a potential target for therapeutic interventions (18). Dysregulated lipid metabolism presents a targetable metabolic vulnerability in CRC. Lipids are distributed throughout the cell and perform a variety of functions in signal transduction and membrane trafficking as well as a significant role in energy production and storage (19). Alterations in lipid metabolism can contribute to tumor initiation, progression, and metastasis at multiple levels by promoting malignant transformation, cell stemness, enhancing tumorigenic properties of cancer cells, and creating a permissive tumor environment for cancer growth and metastasis (20–22). Lipids can be synthesized de novo or obtained from diet (18). Both de novo lipid synthesis and dietary fatty acid uptake have been actively investigated as potential targets for therapeutic intervention in CRC. However, there is limited understanding of the crosstalk between the APC/Wnt signaling pathway, its downstream effectors, and lipid metabolism as well as how this crosstalk can contribute to CRC initiation, progression, and metastasis. This review summarizes and discusses the current understanding of the link between the mutations in APC gene/activation of Wnt signaling and upregulation of lipid metabolism (Figure 1) and how the interaction between these pathways can be targeted to improve the outcome of CRC (Figure 2; Table 1).
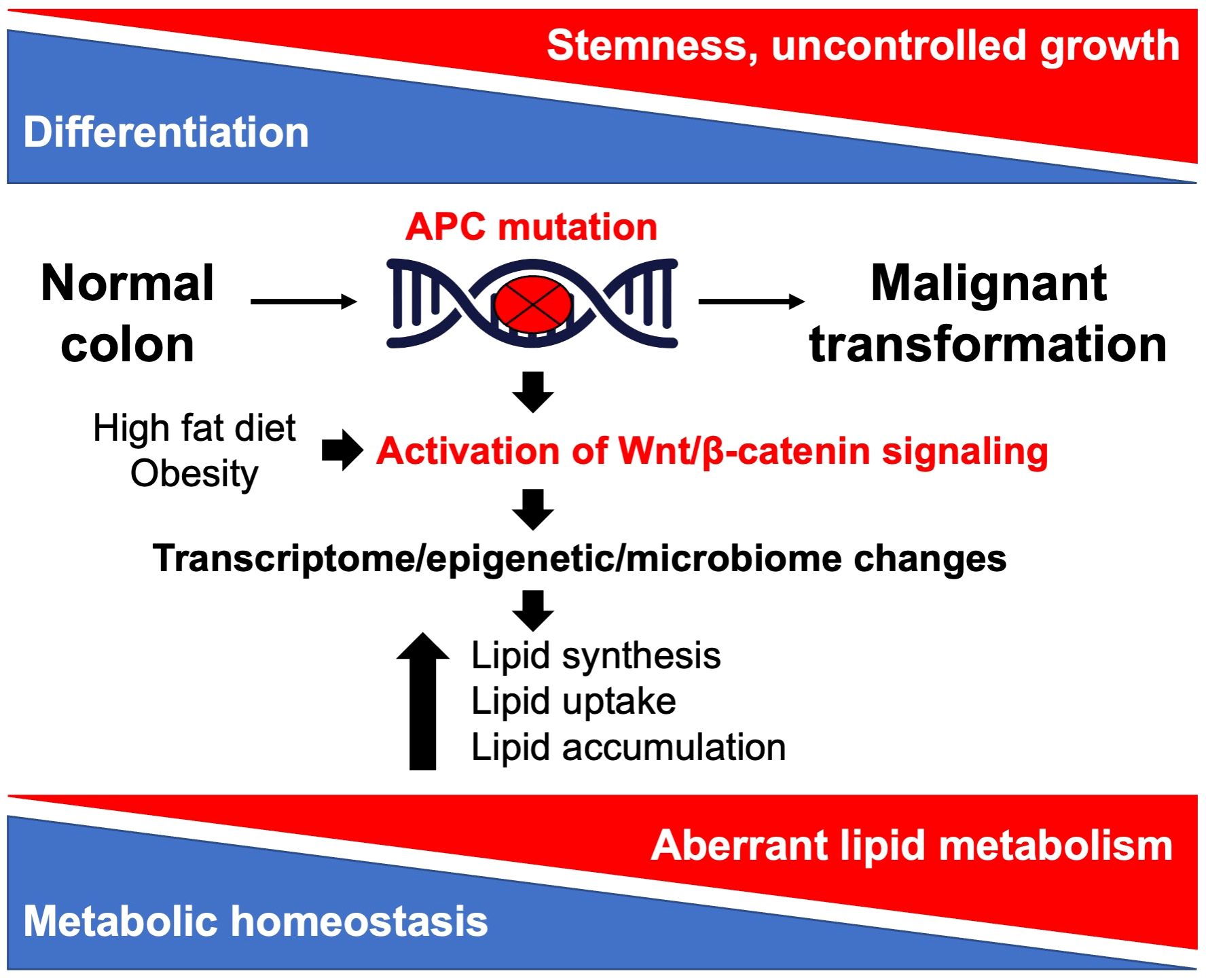
Figure 1 Illustration of the crosstalk between Apc/Wnt signaling and lipid metabolism in colorectal cancer.
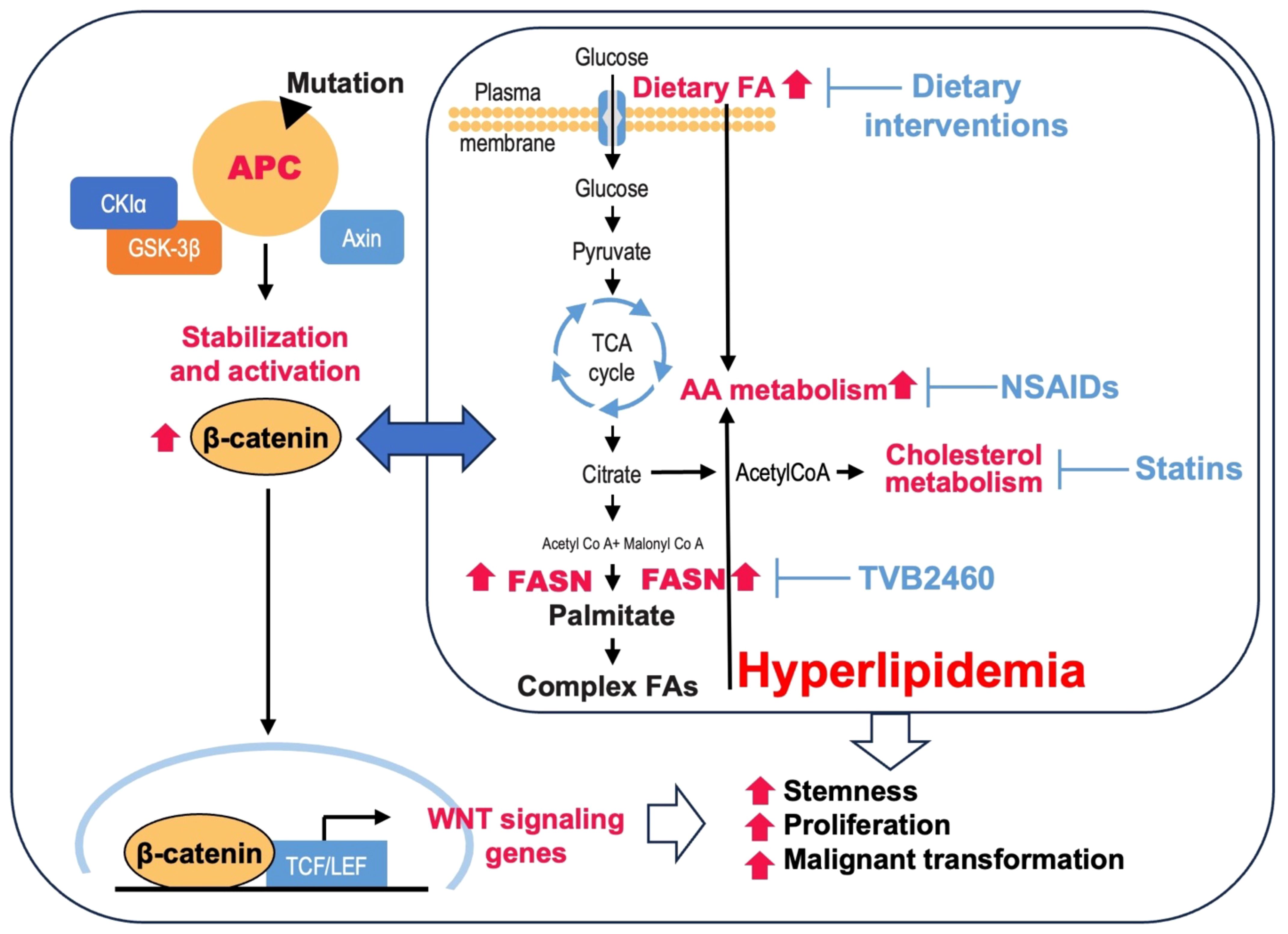
Figure 2 Potential lipid metabolism targets in APC-driven cancer. Loss of APC function due to mutations promotes translocation of β-catenin to the nucleus and increases transcription of Wnt target genes. Mutations in APC and an increase in Wnt signaling are associated with hyperlipidemia via upregulation of de novo lipid synthesis and an increase in dietary fatty acids (FA) in adenomas. Hyperlipidemia further promotes activation of Wnt signaling. Inhibition of de novo lipid synthesis using a novel FASN inhibitor TVB2640, cholesterol metabolism using statins, AA metabolism using NSAIDs, and dietary interventions are potential therapeutic approaches in APC-driven CRC.
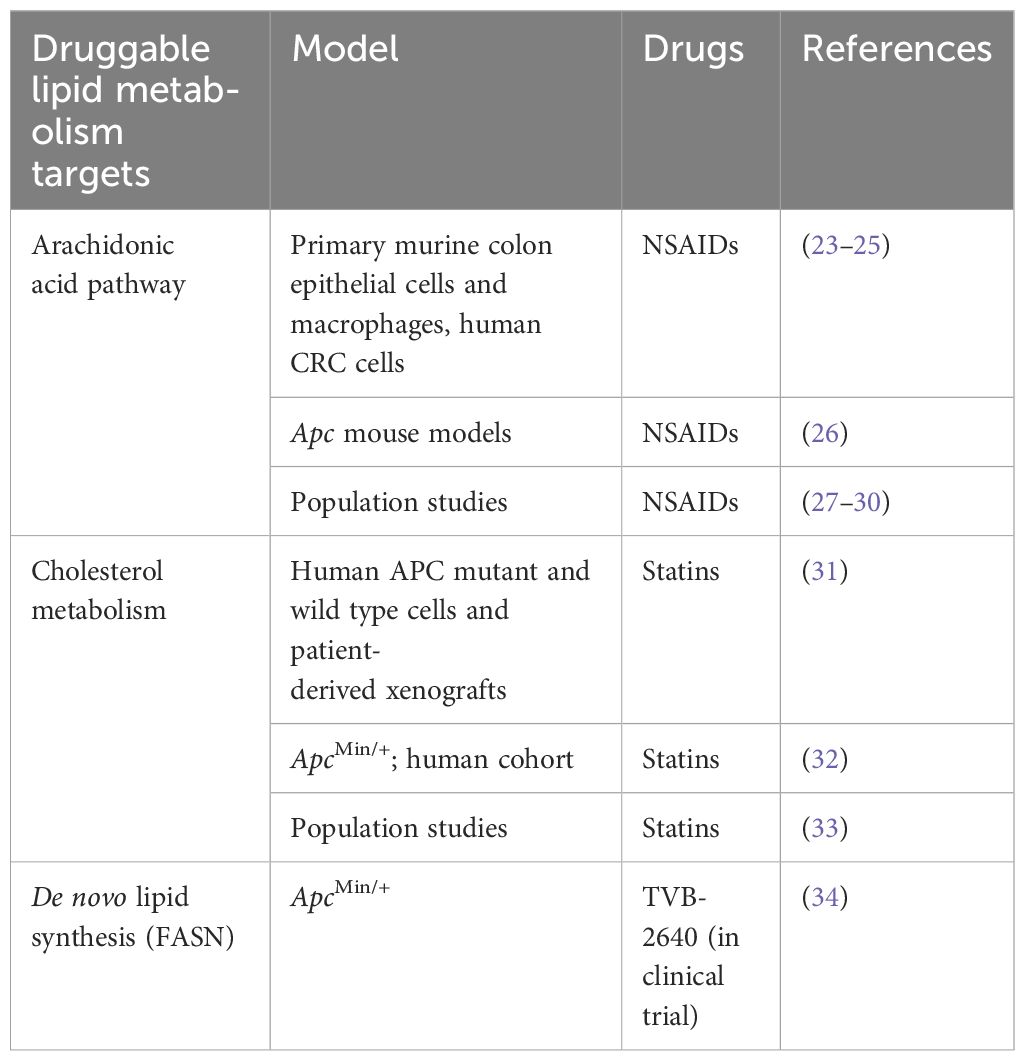
Table 1 The potential lipid metabolism targets for therapeutic intervention in APC-driven colorectal cancer.
2 The APC/Wnt signaling pathway in CRC initiation and progression
The APC locus was initially identified based on its association with FAP (35–38). The molecular characterization of human CRC led to the understanding that mutations of the APC gene play a major role early in the development of CRC (39). Individuals with FAP inherit mutations in the APC gene that lead to formation of thousands of adenomatous polyps, resulting in 100% onset of CRC, typically before the age of 40 if left untreated (40). Furthermore, these individuals acquire biallelic APC mutations, with loss of function in one allele followed by an acquired mutation in the second allele of APC in the adenomas and adenocarcinomas that develop (41). These acquired biallelic APC mutations are recognized as an early and rate limiting step in most sporadic colorectal tumors and all FAP-associated colorectal tumors (13). The germline mutation type and location determine the severity of disease including colonic polyp burden and the presence of extra-colonic FAP manifestations (42). Subtle nonsense, deletion, or nucleotide insertion mutations contribute to biallelic mutations in APC (43). The “top–bottom” and “bottom–top” models are the proposed hypotheses pertaining to the cell of origin within the crypt–villus axis to initiate CRC. Preston et al. found that stem cells at the base of the intestinal crypts induce the initiation event of colorectal adenomas of both sporadic and FAP adenomas by stimulating initial growth via crypt fixation which spreads into adjacent crypts as a secondary event, thus supporting the bottom–top model (44). In contrast, abnormal patterns of proliferation and dysplastic epithelium observed at the tops of crypts in differentiated colonocytes in sporadic tumors suggest that the top–bottom model is responsible for CRC initiation (45, 46). Interestingly, Wnt activation, subsequent of APC or CTNNBL1 mutation, has been identified as an important contributor to malignant transformation in both models (47).
APC-mediated activation of the Wnt signaling pathway is one of the essential early events in the development of CRC (48). The large multi-domain of APC contains various subunits which allow it to bind and interact with various proteins to perform numerous processes like cell migration, cell adhesion, proliferation, differentiation, chromosome segregation, and negative regulation of Wnt/β-catenin signaling (7). The most conserved domain is the armadillo repeat-domain (7). Studies have shown this domain contributes to cell migration and cell adhesion via its binding interactions with cellular molecules including IQ-motif-containing GTPase activation protein 1 (IQGAP1), PP2A, Asef, and KAP3 (49–52). Subsequently, the 15–20-residue domain and SAMP repeats play crucial roles in negative regulation of the Wnt/β-catenin pathway by facilitating the proteasomal degradation of β-catenin (7). Moreover, mutations in the APC gene usually create truncated gene products which lead to the loss of various β-catenin binding sites (20R), Axin interaction sites (SAMP), nuclear localization sequences, and a C-terminal basic region which mediates cytoskeletal interactions (14).
The Wnt signaling pathway plays a crucial role in homeostatic regulation of the epithelium in the intestine (13). Furthermore, this pathway is responsible for regulating differentiation along the crypt–villus axis (53). A major function of the APC gene is tumor suppression of the Wnt signaling pathway via controlling and destabilizing β-catenin turnover. In the absence of a Wnt signal, β-catenin is first phosphorylated by the destruction complex comprised of APC, glycogen synthase kinase 3β (GSK3β), Axin, and casein kinase 1 (CK1) (7). Subsequently, β-catenin is ubiquitinated by β-TrCP200, and finally targeted for proteasomal degradation. In this scenario, the T cell factor/lymphoid enhancer factor (TCF/LEF) in the nucleus binds to the co-repressor protein, Groucho, to suppress transcriptional activity of Wnt targeted genes (7). When a palmitoylated Wnt ligand binds to Frizzled (Fzd) and low-density lipoprotein receptor related protein (LRP) co-receptor, the downstream effects allow β-catenin to evade phosphorylation and enter the nucleus. Specifically, the receptors form a complex to activate the Dishevelled (Dsh) protein. Dsh and Axin bind together to facilitate the formation of Wnt/Fzd/LRP5/6 complex, which prevents formation of the destruction complex (7, 16). Consequently, β-catenin remains stabilized and accumulates in the cytoplasm before it translocates in the nucleus to bind to TCF/LEF. This binding interaction stimulates the transcription of Wnt target genes, such as cyclin D1, c-Myc, Lgr5, promoting proliferation, cell cycle, and stem-like properties, respectively (7, 16). Interestingly, a recent publication demonstrated that intestinal APC mutants outcompeted their wild type counterparts by secreting Notum, a Wnt antagonist/inhibitor, to decrease their stemness and promote differentiation (54). Furthermore, Notum has been identified as a cancer stem cell marker in CRC (55). In gastric cancer cells, Notum enhances stem-like properties by upregulating Sox2 via the PI3K/AKT pathway (56).
Physiological factors within the intestinal environment stimulate epithelial cells to move from the crypt to the villus. In the villus, these epithelial cells differentiate from immature progenitor cells to mature enterocytes in which they lose their ability to proliferate and divide. The optimum activity of the Wnt signaling pathway occurs at the crypts where the progenitor and stem cells are located, and their proliferative activity is the highest, thus, contributing to the initiation and progression of CRC (13).
3 Lipid metabolism in Apc-deficient mouse models
To investigate the significance of APC inactivation in initiation of CRC in animals, Moser et al. created ApcMin/+, the first germline mutant mouse model of intestinal neoplasia. ApcMin/+ carries an autosomal dominant loss of function mutation of the Apc gene generated by exposure to N-ethyl-N-nitrosourea, a highly potent mutagen (57). In this model, a nonsense mutation (TTG to TAG) at the 850th codon of the Apc gene leads to a truncated APC protein and makes these mice prone to intestinal adenomas. ApcMin/+ mice spontaneously develop more than 50 adenomas, on average, throughout the intestinal tract and rarely survive beyond 150 days of age due to anemia and intestinal obstruction (58). The severity of this phenotype decreases the progression of adenoma to invasive carcinoma. While ApcMin/+mice have been used as a model of human APC mutation, these types of APC truncations are not common in inherited and sporadic human CRC (59). Human CRC mutations in APC typically result in truncation of the C-terminal half of the protein without affecting the first 20-amino acid repeat (60). To better recapitulate human disease, Apc1309 and Apc1322T mouse models were developed to express truncated APC that retains the first 20-amino acid repeat. Both of these models develop polyps in the small intestine, but the polyps are more proximal than those in ApcMin/+ mice (59). Interestingly, the hyperlipidemic state has been observed in both ApcMin/+ and Apc1309 mice, suggesting that Apc-driven carcinogenesis is associated with upregulation of lipid metabolism (61). This study shows that both Apc1309 and wild-type (WT) mice have equally low serum levels of triglyceride (TG) and cholesterol at 6 weeks of age, but by 12 weeks of age the TG level increases 10-fold in Apc1309 mice but not in their wild-type counterparts. Moreover, this study demonstrates that aged Apc1309 mice have not only a high cholesterol level but also significantly increased centrilobular-restricted steatosis in the liver. Similar findings were observed in ApcMin/+ mice at 15 weeks of age (61). Interestingly, another study shows that TG and total cholesterol uptake are inhibited in the intestine of ApcMin/+ mice with age, and downregulation of β-oxidation, oxidative stress responses, and cholesterol absorption via the intestinal peroxisome proliferator-activated receptor α (PPARα) are associated with cachexia (62). Consistently, intracellular lipid changes induced by Apc gene mutations were shown in intestinal organoids established from either WT or mice with Apc mutations (Lgr5-EGFP-IRES-CreERT2Apcfl/fl). Using a reversed-phase ultra-high-performance liquid chromatography mass spectrometry-based lipid profiling method, this study showed that the levels of phospholipids (e.g., PC(16:0/16:0), PC(18:1/20:0), PC(38:0), PC(18:1/22:1)), ceramides (e.g., Cer(d18:0/22:0), Cer(d42:0), Cer(d18:1/24:1)) and hexosylceramides (e.g., HexCer(d18:1/16:0), HexCer(d18:1/22:0)) were higher in Apcfl/fl organoids as compared to WT organoids. In contrast, the levels of sphingomyelins (e.g., SM(d18:1/14:0), SM(d18:1/16:0)) were lower in Apcfl/fl organoids as compared to WT mice. These results suggest that the loss of Apc in the organoids leads to alterations in lipid metabolism associated with an increase in intracellular PC, Cer, and HexCer levels (63).
In many human CRCs, Wnt signaling is hyperactivated due to genetic alterations of APC. The level of Wnt activation and its downstream signaling is dosage-dependent based on the number of β-catenin binding repeats that remain in the truncated APC protein (64). An interesting observation made in a recent study is that the level of oncogenic Wnt hyperactivation impacts the metabolic homeostasis, and the metabolism of Wnt‐high tumors in general relies more on mitochondrial metabolism than that of Wnt‐low tumors (65). This study highlights the complexity of the APC/Wnt/β-catenin pathway in different cell types in a context‐dependent manner.
In adipocytes, the canonical Wnt/β-catenin pathway regulates de novo lipogenesis and fatty acid monounsaturation (66). Further, it has been shown that β-catenin mediates the effects of Wnt signaling on lipid metabolism in part by regulating the transcription factors such as MLX interacting protein like (MLXIPL) and sterol regulatory element binding transcription factor 1 (SREBF1). This study found that β-catenin deletion in mice promotes resistance to diet-induced obesity and it occurs via decrease in adipocyte hypertrophy and subsequent protection from metabolic dysfunction (66). Consistently, hepatocyte-associated canonical Wnt signaling is essential for the development of diet-induced fatty liver and obesity (67). β-catenin has also been shown to bind transcription factors involved in regulation of lipid metabolism including peroxisome proliferator-activated receptor γ (PPARγ), retinoid X receptor α (RXRα), and retinoic acid receptor (RAR) (68–70). Dysregulated Wnt signaling due to Apc mutation can also alter bile acid profiles and increase levels of tauro-β-muricholic acid and deoxycholic acid to drive malignant transformations in Lgr5-expressing (Lgr5+) cancer stem cells and promote a progression from adenoma to adenocarcinoma (71). Loss of phospholipid-remodeling enzyme lysophosphatidylcholine acyltransferase 3 (LPCAT3) or driving endogenous cholesterol synthesis through activation of sterol regulatory element-binding protein 2 (SREBP-2) in ApcMin/+ mice markedly promotes intestinal tumor formation, suggesting the crucial importance of cholesterol synthesis and availability in regulation of APC-driven intestinal tumorigenesis (72). Importantly, mutations in Apc promote membrane rigidity and lipid raft stability via an increase in plasma membrane cholesterol, an essential component of the ordered membrane domains. The global transcriptome analyses suggest that the oncogenic APC-induced alterations in cholesterol homeostasis are partly mediated by changes in gene expression associated with cholesterol uptake, efflux, synthesis, intracellular trafficking and metabolism. Changes in plasma membrane lipid homeostasis and in organization of Wnt signaling plasma membrane domains driven by Apc loss and aberrant Wnt signaling lead to vital changes in stimulation of interactions between Wnt receptor and their effectors, thus, further promote oncogenic Wnt signaling and tumorigenesis (73). Besides the role of lipids in remodeling of cellular membranes to promote Wnt signaling, altered lipid metabolism may enhance Wnt signaling by lipidation of Wnt proteins (74). For example, protein-serine O-palmitoleoyltransferase, a key regulator of the Wnt signaling, mediates lipidation of Wnt proteins which is essential for their secretion and signaling activities (75). However, there is a limited data on characterization of Wnt protein lipidation and further studies are needed to understand it’s impact in early intestinal carcinogenesis.
Consistent with other studies showing the impact of Apc loss on lipid metabolism, our group has shown that development of adenomas in an Apc/VillinCre mouse model is driven by de novo lipid synthesis and heterozygous deletion of FASN, a key enzyme of lipid biosynthesis that significantly increases mouse survival and decreases the number of adenomas formed (34). High FASN expression during Apc-driven colon carcinogenesis is associated with enrichment of genes associated with Wnt pathway, FA metabolism, and steroid biosynthesis. Interestingly, heterozygous deletion of FASN in intestinal epithelial cells leads to a significant decrease in the levels of several diglyceride species (C16:1:20:0-DAG; Di-C14-DAG; C14:0:18:0-DAG; C16:1:18:0-DAG; C18:0:18:1-DAG) in mouse adenoma tissues but does not affect the total levels of free fatty acid and sphingolipid species (34). Interestingly, pharmacological inhibition of FASN and lipid biosynthesis disrupts lipid rafts architecture, decreases protein lipidation, and thus, inhibits Wnt/β-catenin signal transduction (76), suggesting these might be the potential mechanisms behind FASN knockout on polyp formation in transgenic animals.
In summary, Apc inactivation leads to significant alteration in intestinal lipid metabolism including upregulation of de novo lipid synthesis and cholesterol synthesis, thus, contributing to stemness, cellular proliferation, survival, lipid raft stability, and support of prooncogenic signaling.
4 Contribution of dietary fatty acids and obesity to Apc-driven carcinogenesis in vivo
It has been shown that a high-fat diet and obesity accelerate the development of intestinal adenomas in mice with mutation or deletion of the Apc gene (77). This study shows that high-fat-diet-induced inflammation from specific dietary fatty acids, rather than obesity or metabolic status, is associated with increased Apc-driven intestinal polyposis (77). Consistently, another study found that when a missense mutation that results in premature termination of the intracellular domain of the long form of the leptin receptor (Leprdb/db)—which results in the phenotype of obesity and diabetes mellitus—is introduced into the Apc1638N/+ background, it induces the formation of not only intestinal but also gastric and colonic tumors (78). The potential mechanisms that drive adenoma formation in C57BL/KsJ-db/db-ApcMin/+ mice are increased expression of insulin-like growth factors (IGFs), as well as hyperlipidemia and hyperinsulinemia (79). Another proposed mechanism is that diet-induced obesity increases the number and function of Lgr5+ ISCs, while also promoting stemness and tumorigenicity of progenitor cells after inactivation of Apc (80). Furthermore, it has been shown that a high-fat diet increases oxidative stress and alters the expression of intestinal gap junction proteins, thereby accelerating membrane permeability and endotoxemia, increasing inflammation, and thus, promoting intestinal tumorigenesis in ApcMin/+ mice (81). Using ChIP-seq and RNA-seq approaches in ApcMin/+ mice, authors of a recent publication demonstrated that keeping mice on a high-fat diet for only three days resulted in epigenetic and transcriptomic changes in lipid metabolic pathways and accelerated tumor growth as compared to mice kept on a low-fat diet. In addition, a high-fat diet has been shown to suppress the activity of pathways related to the immune system, suggesting that an increase in lipid uptake and metabolism provides a more permissive immunologic environment that favors tumor progression (82).
In addition to the effects of high-fat diet on transcriptome, epigenome, and metabolism, it promotes CRC development dependent on gut microbiota (83). To support this finding, paired microbiome and metabolome analyses show that high-fat diet is a dominant determinant of cecal microbiome and metabolome in ApcMin/+ mice and implicates microbially conjugated bile acids as potential drivers of polyposis and CRC progression (84).
One of the potential mechanisms of how high-fat diet and obesity promote intestinal carcinogenesis is through increased activity of the pro-tumorigenic Wnt signaling. The associations of obesity, microbiome, inflammation, and Wnt signaling were examined in Apc+/1638N mice whose obesity was induced by either diet-modified or genetically modified obesity. Studies of Apc+/1638NLepr+/+ fed a low-fat diet (10% kcal fat), Apc+/1638NLepr+/+ fed a high-fat diet (60% kcal fat), and Apc+/1638N Leprdb/db fed a low-fat diet show that the elevated inflammation and changes in microbiome were associated with altered expression of genes associated with oncogenic Wnt signaling and enhanced tumorigenesis (85).
Another mechanism of how lipids can promote colorectal tumorigenesis is through loss of free fatty acid receptor 2 (FFAR2), which is expressed in colon and can be activated by short-chain fatty acids. Loss of FFAR2 promotes the development of colon adenoma in ApcMin/+ mice and enhances long-chain fatty acid β-oxidation and bile acid metabolism (86). These changes in lipid metabolism are associated with significant changes in gut microbiota and an increase in relative abundance of Flavobacteriaceae and Verrucomicrobiaceae in the ApcMin/+-Ffar2-/- mice compared to the ApcMin/+ mice, which may contribute to colon adenoma development in Ffar -deficient mice (86). The crucial role of Apc inactivation and dysregulation of Wnt/β-catenin signaling in obesity is shown in a study demonstrating that inactivation of Pten in Lgr5+ ISCs, whether alone or in combination with obesity, is insufficient to drive adenoma formation in mice, but combination of Pten deletion with Apc loss increases tumor burden and worsens survival (87).
In summary, both high-fat diet and obesity promote Apc-driven carcinogenesis via multiple mechanisms involving changes in transcriptome, metabolome, and microbiome, thus promoting inflammation, stemness, and altering immune responses.
5 Mutant APC and lipid metabolism in human studies
Lipid metabolic pathways that are affected in CRC cells include lipid uptake, lipid synthesis, desaturation, elongation, and mitochondrial oxidation of fatty acids (88). Human lipid profiles depend on many factors including polyp/tumor heterogenicity, diet, and environmental exposures. Therefore, currently available data on CRC lipidome are broad and fraught with inconsistencies. Here, we review several studies performed on FAP/polyp tissues to discuss alterations in lipid metabolism occurring due to APC mutations.
Similar to data reported from the Apc-driven mouse models, studies in FAP show that intestinal polyps exhibit a hyperlipidemic state. Analysis of 28 FAP patients from the National Cancer Center Hospital in Japan shows that the overall prevalence of hyperlipidemia in the patients was 57.7% (15/26) and the prevalence of hypercholesterolemia (220–296 mg/dl) was 53.3% (8/15) (89). Analysis of serum metabolites from 30 FAP patients and 34 healthy individuals detected and quantified using ultra performance liquid chromatography and tandem mass spectrometry (UPLC−MS/MS) demonstrated an increase in metabolites associated with fatty acid metabolism, cholesterol metabolism, bile acid biosynthesis, and bile secretion. In particular, the level of arachidonic acid (AA) was significantly higher in the serum of FAP patients compared with healthy controls (90). Another study reported the fatty acid composition in serum of 38 colectomized FAP patients and 160 healthy individuals. It showed that the levels of linoleic and α-linolenic acid were higher in healthy subjects (difference: 3.96, 95% CI: 2.87–5.04 and 0.06, 95% CI: 0.04–0.08, respectively), and the AA and docosahexaenoic acid levels were lower in healthy individuals (difference: −3.70, 95% CI: −4.35 to −3.06 and −5.26, 95% CI: −6.25 to −4.28, respectively) as compared to the FAP patients (all p ≤ 0.0001) (91). Consistent with previous reports, a study using multi-omics profiling of 135 normal mucosal, benign, and dysplastic polyps and adenocarcinoma samples from six FAP patients showed AA pathway upregulation in benign polyps relative to FAP mucosa. Moreover, this study showed depletion of TG stores suggesting that this lipid class is a key energy source for polyp formation and growth (92). Utilizing human induced pluripotent stem cells (iPSC) from healthy individuals or from patients with FAP, a study showed that APC heterozygosity is responsible for major changes in the transcriptional profile of the cells and key signaling pathways associated with pro-tumorigenic signaling, including alterations in lipid metabolism and PPAR signaling pathway (93). Together, these studies show that hyperlipidemia and altered lipid metabolism are frequently observed in FAP, suggesting its possible link to development of CRC.
To further support the role of lipid metabolism in malignant transformation and CRC, another study investigated CRC lipidome for putative tumor-specific alterations through analysis of three independent retrospective patient cohorts to show that signatures of sphingomyelin and TG species are significantly different in cancerous versus non-diseased tissue. The results of this study demonstrated an increase in the levels of sphingomyelin species with 32–34 carbons and a decrease in the levels of sphingomyelin species with more than 34 carbons in tumor samples. In contrast, the levels of sphingosine-based Cer with longer chains (C24:0–C26:0) were increased and the levels of sphingosine-based Cer with shorter (<C22:0) chains were decreased in tumor samples. For TGs, species with <53 carbons were decreased, while polyunsaturated TGs with 56 carbons were increased in tumor samples. Moreover, the expression of lipogenic enzymes was significantly upregulated in tumor tissue, and FASN gene expression is prognostically detrimental in CRC (94). Even though this study did not assess lipidomic changes in human tumor tissues according to the APC status and staging, the additional quantitative lipidome analysis was performed on adenomas from Apc1638N mice to test whether deregulation of TG species is conserved in mice. Consistent with human data, TG containing poly-unsaturated acyl chains (TG 56:4, TG 56:5, TG 56:6) had a significantly higher abundance in adenomas, whereas shorter and less unsaturated TGs were lower (94). Similarly, FASN was significantly upregulated in mouse adenomas (94), which is consistent with data from our group and suggest that upregulation of FASN is an early event in Apc-driven CRC (34).
In summary, the majority of studies using human tissues demonstrate upregulation of lipid metabolism including de novo lipogenesis and AA metabolism in polyps driven by mutations in APC, suggesting that these pathways can be the major contributors to CRC development and metabolic vulnerabilities during CRC initiation.
6 Discussion: clinical implications and therapeutic strategies based on current knowledge of crosstalk between loss of APC and lipid metabolism
Research by Hans Clevers and colleagues shows that Apc restoration triggers differentiation and restores crypt homeostasis which drive tumor regression in CRC, thus validating the Wnt pathway as an effective therapeutic target in CRC (95). Multiple strategies that target Wnt signaling have been developed, from small molecules to blocking antibodies, and peptide agonists and antagonists (96). However, one of the major challenges of targeting Wnt signaling downstream of mutant APC is significant toxicity since this pathway plays a crucial role in normal development, stem cell maintenance, tissue regeneration, and adult tissue homeostasis (95, 96). In addition, the crosstalk of Wnt signaling with other signaling pathways further challenges the development of effective therapeutics.
Multiple studies suggest that lipid metabolism may be a metabolic vulnerability in CRC, including APC-driven CRC (18, 34, 88, 94). The preclinical studies in Apc mouse models demonstrate that inactivation of Apc is associated with upregulation of fatty acid metabolism and contributes to proliferation of adenomas (34, 61, 63). More importantly, analysis of lipid metabolism in human polyps and tumor tissues may provide better insight into metabolic disturbances that contribute to carcinogenesis and provide new therapeutic strategies for identification, prevention, and therapy of this disease. Upregulation of the AA pathway occurs early in hyperplasia (90, 91) and can activate tumor-related gene expression and signaling pathways (97). This pathway is targeted by non-steroidal anti-inflammatory drugs (NSAIDs), a common preventative treatment for FAP patients. Cyclooxygeneses (COX-1 and COX-2), key enzymes involved in synthesis of prostaglandins from arachidonic acid, are targets of common NSAIDs. Several preclinical studies support the role of the AA pathway in malignant transformation. An increase in production of 4-hydroxy-2-nonenal (4-HNE), a DNA mutagen and an autochthonous mitotic spindle poison, produced by peroxidation of ω6 polyunsaturated fatty acids, and upregulation of COX-2 in primary colon cells by commensal infected microphages have been reported as CRC initiation contributing factors (23–25). Inactivation of COX-1 and COX-2 have been shown to significantly reduce polyp numbers in multiple animal models (26). Furthermore, knockout mutation for prostaglandin E2 receptor EP2 introduced into ApcΔ716 mice results in a reduced number and size of intestinal polyps (98). Importantly, the clinical trial with celecoxib, a selective COX-2 inhibitor, in patients with FAP showed a significant reduction in the number of colorectal polyps (99). Furthermore, a prospective cohort study by Tangrea et al. found a significant reduction in adenoma recurrence in NSAID users, particularly in advanced polyps (28, 30). Interestingly, multiple studies report differences in the gut microbiome and metabolome in patients using NSAIDs as compared to those not using NSAIDs, suggesting that protective anti-inflammatory effects of NSAIDs on polyp formation are potentially mediated by intestinal flora and microbe-derived metabolites (27, 29).
The recent study using a meta-analysis to assess the impact of NSAIDs on colorectal polyp burden in patients with FAP concluded that a short-term use of NSAIDs reduces polyp number and polyp size but with low to very low certainty of evidence (100). Furthermore, evidence supports an association between aspirin use and reduced CRC-related mortality in subpopulations, including tumors overexpressing COX-2 and proximal CRCs (30). Therefore, additional studies are needed to better elucidate the association between use of non-aspirin NSAIDs and CRC prevention low-risk, elderly, and other populations.
There is strong evidence that dietary factors can affect the risk and development of CRC. Several preclinical and human observational studies show the benefits of fish oil, which is rich in two omega-3 fatty acids (EPA and DHA) (101). Results of the Seafood Polyp Prevention Trial, in which 2 g EPA-free fatty acid (FFA) per day (either as FFA or triglyceride), 300 mg aspirin per day, both, or placebo were administered for 12 months, showed no effect on polyp formation; however, it showed a significant decrease in the recurrence of some subtypes of adenomas with both EPA and aspirin (102). In another study, daily supplementation with marine ω-3 fatty acids (1 g) did not reduce the risk of CRC development in the general US population; however, potential benefits were observed in individuals with low plasma levels of ω-3 and in African Americans (103). These studies demonstrate that further research, including research in different racial and ethnics groups, is needed regarding potential therapeutic and dietary intervention to prevent and target APC-driven CRC.
Upregulation of FASN and lipid synthesis is critically important in Apc-driven CRC initiation as it orchestrates changes in the transcriptome, proteome, and metabolic pathways consistent with increased cellular proliferation, ATP production, anabolism, and adenoma formation (34). Therefore, inhibition of FASN should be further investigated as a potential preventive strategy or early-stage treatment for CRC. Currently, a novel FASN inhibitor, TVB-2640, is being tested in several Phase I-II oncology clinical trials (104, 105); thus, providing the opportunity to test inhibition of de novo lipogenesis as a preventive therapeutic approach for CRC.
Targeting cholesterol synthesis may be another potential strategy for CRC. Statin use is associated with a lower risk of incident CRC, CRC-related mortality, and all-cause mortality (33). Several studies have demonstrated that statin use exhibits antitumor effects in CRC. Mechanisms that may be responsible for the antitumor effect of statins include induction of apoptosis and inhibition of cell proliferation, angiogenesis, immune response, and metastatic capacity (106). Interestingly, a recent study shows that treatment with statins leads to a significantly greater inhibition of cell viability and expression of Wnt target in the APC mutant cell lines and patient-derived xenograft models as compared to wild-type APC cells, supporting the use of statins in APC-driven CRC (31). Recent evidence also suggests that the protective effects of statins can be associated with reduced dysbiosis in the gut microbiome. It has been shown that atorvastatin treatment leads to an increase in Lactobacillus reuteri and microbial tryptophan availability, thus eliciting anti-tumorigenic effects via the tryptophan catabolite, indole-3-lactic acid (ILA). The study suggests that interventions with Lactobacillus reuteri or ILA could complement current preventive strategies for CRC (32).
In summary, lipid metabolism is an attractive therapeutic target for CRC initiation, but more studies are needed to better elucidate the mechanistic crosstalk between activation of oncogenic pathways such as APC/Wnt/β-catenin and lipids. Finally, consideration of racial, gender, and dietary differences is important to develop efficacious therapeutic approaches to target lipid metabolism in CRC.
Author contributions
CK: Writing – original draft, Writing – review & editing, Conceptualization. YZ: Supervision, Funding acquisition, Writing – review & editing, Writing – original draft, Conceptualization, Validation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by National Cancer Institute training grant T32 CA165990 (COK) and R01 CA249734 (YYZ), and National Institute of Environmental Health Sciences grant P42 ES007380 (University of Kentucky Superfund Research Center, Project 1-YYZ).
Acknowledgments
The Markey Cancer Center’s Research Communications Office assisted with manuscript preparation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. (2023) 73:233–54. doi: 10.3322/caac.21772
2. Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology. (2020) 158:291–302. doi: 10.1053/j.gastro.2019.08.059
3. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. (2011) 8:686–700. doi: 10.1038/nrgastro.2011.173
4. Heide T, Househam J, Cresswell GD, Spiteri I, Lynn C, Mossner M, et al. The co-evolution of the genome and epigenome in colorectal cancer. Nature. (2022) 611:733–43. doi: 10.1038/s41586-022-05202-1
5. Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O'Brien G, et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and braf(V600E)-induced tumorigenesis. Cancer Cell. (2019) 35:315–328.e6. doi: 10.1016/j.ccell.2019.01.005
6. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. (2001) 3:433–8. doi: 10.1038/35070129
7. Zhang L, Shay JW. Multiple roles of APC and its therapeutic implications in colorectal cancer. J Natl Cancer Inst. (2017) 109. doi: 10.1093/jnci/djw332
8. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. (2001) 10:721–33. doi: 10.1093/hmg/10.7.721
9. Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A, et al. APC mutations in sporadic colorectal tumors: A mutational "hotspot" and interdependence of the "two hits". Proc Natl Acad Sci U.S.A. (2000) 97:3352–7. doi: 10.1073/pnas.97.7.3352
10. Yamada A, Yamamoto Y, Minamiguchi S, Kamada M, Sunami T, Ohashi S, et al. Clinicopathological and molecular characterization of deficient mismatch repair colorectal cancer. Hum Pathol. (2022) 130:1–9. doi: 10.1016/j.humpath.2022.09.005
11. Yamane L, Scapulatempo-Neto C, Reis RM, Guimaraes DP. Serrated pathway in colorectal carcinogenesis. World J Gastroenterol. (2014) 20:2634–40. doi: 10.3748/wjg.v20.i10.2634
12. Satorres C, Garcia-Campos M, Bustamante-Balen M. Molecular features of the serrated pathway to colorectal cancer: Current knowledge and future directions. Gut Liver. (2021) 15:31–43. doi: 10.5009/gnl19402
13. Hankey W, Frankel WL, Groden J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: implications for therapeutic targeting. Cancer Metastasis Rev. (2018) 37:159–72. doi: 10.1007/s10555-017-9725-6
14. Parker TW, Neufeld KL. APC controls Wnt-induced beta-catenin destruction complex recruitment in human colonocytes. Sci Rep. (2020) 10:2957. doi: 10.1038/s41598-020-59899-z
15. Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J. (2010) 427:1–17. doi: 10.1042/BJ20091866
16. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. (2017) 36:1461–73. doi: 10.1038/onc.2016.304
17. Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. (2019) 29:44–65. doi: 10.1016/j.tcb.2018.08.005
18. Zaytseva Y. Lipid metabolism as a targetable metabolic vulnerability in colorectal cancer. Cancers (Basel). (2021) 13(2):301. doi: 10.3390/cancers13020301
19. Fahy E, Cotter D, Sud M, Subramaniam S. Lipid classification, structures and tools. Biochim Biophys Acta. (2011) 1811:637–47. doi: 10.1016/j.bbalip.2011.06.009
20. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C, et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm. (2020) 2:27–59. doi: 10.1002/mco2.27
21. Visweswaran M, Arfuso F, Warrier S, Dharmarajan A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells. (2020) 38:6–14. doi: 10.1002/stem.3101
22. Zhou J, Zhao J, Su C. Role of aberrant lipid metabolism of cancer stem cells in cancer progression. Curr Cancer Drug Targets. (2021) 21:631–9. doi: 10.2174/1568009619666210316112333
23. Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut. (2015) 64:459–68. doi: 10.1136/gutjnl-2014-307213
24. Wang X, Yang Y, Huycke MM. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget. (2017) 8:102176–90. doi: 10.18632/oncotarget.v8i60
25. Wang X, Yang Y, Moore DR, Nimmo SL, Lightfoot SA, Huycke MM. 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology. (2012) 142:543–551.e7. doi: 10.1053/j.gastro.2011.11.020
26. Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. (2009) 136:780–98. doi: 10.1053/j.gastro.2008.12.049
27. Ferrara CR, Bai JDK, McNally EM, Putzel GG, Zhou XK, Wang H, et al. Microbes contribute to chemopreventive efficacy, intestinal tumorigenesis, and the metabolome. Cancer Prev Res (Phila). (2022) 15:803–14. doi: 10.1158/1940-6207.CAPR-22-0244
28. Tangrea JA, Albert PS, Lanza E, Woodson K, Corle D, Hasson M, et al. Non-steroidal anti-inflammatory drug use is associated with reduction in recurrence of advanced and non-advanced colorectal adenomas (United States). Cancer Causes Control. (2003) 14:403–11. doi: 10.1023/A:1024990617158
29. Wu J, Xia C, Liu C, Zhang Q, Xia C. The role of gut microbiota and drug interactions in the development of colorectal cancer. Front Pharmacol. (2023) 14:1265136. doi: 10.3389/fphar.2023.1265136
30. Zaman FY, Orchard SG, Haydon A, Zalcberg JR. Non-aspirin non-steroidal anti-inflammatory drugs in colorectal cancer: A review of clinical studies. Br J Cancer. (2022) 127:1735–43. doi: 10.1038/s41416-022-01882-8
31. Shailes H, Tse WY, Freitas MO, Silver A, Martin SA. Statin treatment as a targeted therapy for APC-mutated colorectal cancer. Front Oncol. (2022) 12:880552. doi: 10.3389/fonc.2022.880552
32. Han JX, Tao ZH, Wang JL, Zhang L, Yu CY, Kang ZR, et al. Microbiota-derived tryptophan catabolites mediate the chemopreventive effects of statins on colorectal cancer. Nat Microbiol. (2023) 8:919–33. doi: 10.1038/s41564-023-01363-5
33. Sun J, Halfvarson J, Bergman D, Ebrahimi F, Roelstraete B, Lochhead P, et al. Statin use and risk of colorectal cancer in patients with inflammatory bowel disease. EClinicalMedicine. (2023) 63:102182. doi: 10.1016/j.eclinm.2023.102182
34. Drury J, Young LEA, Scott TL, Kelson CO, He D, Liu J, et al. Tissue-specific downregulation of fatty acid synthase suppresses intestinal adenoma formation via coordinated reprograming of transcriptome and metabolism in the mouse model of apc-driven colorectal cancer. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23126510
35. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. (1991) 66:589–600. doi: 10.1016/0092-8674(81)90021-0
36. Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. (1991) 66:601–13. doi: 10.1016/0092-8674(81)90022-2
37. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, et al. Identification of FAP locus genes from chromosome 5q21. Science. (1991) 253:661–5. doi: 10.1126/science.1651562
38. Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. (1991) 253:665–9. doi: 10.1126/science.1651563
39. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature. (1992) 359:235–7. doi: 10.1038/359235a0
40. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. (2009) 4:22. doi: 10.1186/1750-1172-4-22
41. Ichii S, Takeda S, Horii A, Nakatsuru S, Miyoshi Y, Emi M, et al. Detailed analysis of genetic alterations in colorectal tumors from patients with and without familial adenomatous polyposis (FAP). Oncogene. (1993) 8:2399–405.
42. Preisler L, Habib A, Shapira G, Kuznitsov-Yanovsky L, Mayshar Y, Carmel-Gross I, et al. Heterozygous APC germline mutations impart predisposition to colorectal cancer. Sci Rep. (2021) 11:5113. doi: 10.1038/s41598-021-84564-4
43. Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. (1994) 54:5953–8.
44. Preston SL, Wong WM, Chan AO, Poulsom R, Jeffery R, Goodlad RA, et al. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. (2003) 63:3819–25.
45. Cole JW, McKalen A. Studies on the morphogenesis of adenomatous polyps in the human colon. Cancer. (1963) 16:998–1002. doi: 10.1002/(ISSN)1097-0142
46. Shih IM, Wang TL, Traverso G, Romans K, Hamilton SR, Ben-Sasson S, et al. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U.S.A. (2001) 98:2640–5. doi: 10.1073/pnas.051629398
47. Basu S, Haase G, Ben-Ze'ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Res 5. (2016). doi: 10.12688/f1000research
48. Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am J Physiol Cell Physiol. (2015) 309:C511–21. doi: 10.1152/ajpcell.00117.2015
49. Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K, et al. Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat Cell Biol. (2002) 4:323–7. doi: 10.1038/ncb779
50. Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. (2000) 289:1194–7. doi: 10.1126/science.289.5482.1194
51. Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. (1999) 283:2089–91. doi: 10.1126/science.283.5410.2089
52. Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. (2004) 7:871–83. doi: 10.1016/j.devcel.2004.10.017
53. Sawa M, Masuda M, Yamada T. Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin Ther Targets. (2016) 20:419–29. doi: 10.1517/14728222.2016.1098619
54. Flanagan DJ, Pentinmikko N, Luopajarvi K, Willis NJ, Gilroy K, Raven AP, et al. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature. (2021) 594:430–5. doi: 10.1038/s41586-021-03525-z
55. Wang H, Gong P, Chen T, Gao S, Wu Z, Wang X, et al. Colorectal cancer stem cell states uncovered by simultaneous single-cell analysis of transcriptome and telomeres. Adv Sci (Weinh). (2021) 8:2004320. doi: 10.1002/advs.202004320
56. Liu Y, Chen H, Xiao L, Dong P, Ma Y, Zhou Y, et al. Notum enhances gastric cancer stem-like cell properties through upregulation of Sox2 by PI3K/AKT signaling pathway. Cell Oncol (Dordr). (2023). doi: 10.1007/s13402-023-00875-w
57. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. (1990) 247:322–4. doi: 10.1126/science.2296722
58. Shoemaker AR, Gould KA, Luongo C, Moser AR, Dove WF. Studies of neoplasia in the Min mouse. Biochim Biophys Acta. (1997) 1332:F25–48. doi: 10.1016/S0304-419X(96)00041-8
59. Zeineldin M, Neufeld KL. More than two decades of Apc modeling in rodents. Biochim Biophys Acta. (2013) 1836:80–9. doi: 10.1016/j.bbcan.2013.01.001
60. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The 'just-right' signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. (2002) 11:1549–60. doi: 10.1093/hmg/11.13.1549
61. Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. (2003) 63:6090–5.
62. Yu B, Peng XH, Wang LY, Wang AB, Su YY, Chen JH, et al. Abnormality of intestinal cholesterol absorption in Apc(Min/+) mice with colon cancer cachexia. Int J Clin Exp Pathol. (2019) 12:759–67.
63. Jukes Z, Freier A, Glymenaki M, Brown R, Parry L, Want E, et al. Lipid profiling of mouse intestinal organoids for studying APC mutations. Biosci Rep. (2021) 41. doi: 10.1042/BSR20202915
64. Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. (2013) 4:2610. doi: 10.1038/ncomms3610
65. Imkeller K, Ambrosi G, Klemm N, Claveras Cabezudo A, Henkel L, Huber W, et al. Metabolic balance in colorectal cancer is maintained by optimal Wnt signaling levels. Mol Syst Biol. (2022) 18:e10874. doi: 10.15252/msb.202110874
66. Bagchi DP, Nishii A, Li Z, DelProposto JB, Corsa CA, Mori H, et al. Wnt/beta-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Mol Metab. (2020) 42:101078. doi: 10.1016/j.molmet.2020.101078
67. Behari J, Li H, Liu S, Stefanovic-Racic M, Alonso L, O'Donnell CP, et al. beta-catenin links hepatic metabolic zonation with lipid metabolism and diet-induced obesity in mice. Am J Pathol. (2014) 184:3284–98. doi: 10.1016/j.ajpath.2014.08.022
68. Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. (2006) 26:5827–37. doi: 10.1128/MCB.00441-06
69. Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, et al. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. (2003) 278:29954–62. doi: 10.1074/jbc.M304761200
70. Easwaran V, Pishvaian M, Salimuddin, Byers S. Cross-regulation of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol. (1999) 9:1415–8. doi: 10.1016/S0960-9822(00)80088-3
71. Sedlak JC, Yilmaz OH, Roper J. Metabolism and colorectal cancer. Annu Rev Pathol. (2023) 18:467–92. doi: 10.1146/annurev-pathmechdis-031521-041113
72. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. (2018) 22:206–220 e4. doi: 10.1016/j.stem.2017.12.017
73. Erazo-Oliveras A, Munoz-Vega M, Mlih M, Thiriveedi V, Salinas ML, Rivera-Rodriguez JM, et al. Mutant APC reshapes Wnt signaling plasma membrane nanodomains by altering cholesterol levels via oncogenic beta-catenin. Nat Commun. (2023) 14:4342. doi: 10.1038/s41467-023-39640-w
74. Kaemmerer E, Gassler N. Wnt lipidation and modifiers in intestinal carcinogenesis and cancer. Cancers (Basel). (2016) 8. doi: 10.3390/cancers8070069
75. Herr P, Basler K. Porcupine-mediated lipidation is required for Wnt recognition by Wls. Dev Biol. (2012) 361:392–402. doi: 10.1016/j.ydbio.2011.11.003
76. Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, et al. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine. (2015) 2:806–22. doi: 10.1016/j.ebiom.2015.06.020
77. Doerner SK, Reis ES, Leung ES, Ko JS, Heaney JD, Berger NA, et al. High-Fat Diet-Induced Complement Activation Mediates Intestinal Inflammation and Neoplasia, Independent of obesity. Mol Cancer Res. (2016) 14:953–65. doi: 10.1158/1541-7786.MCR-16-0153
78. Gravaghi C, Bo J, Laperle KM, Quimby F, Kucherlapati R, Edelmann W, et al. Obesity enhances gastrointestinal tumorigenesis in Apc-mutant mice. Int J Obes (Lond). (2008) 32:1716–9. doi: 10.1038/ijo.2008.149
79. Hata K, Kubota M, Shimizu M, Moriwaki H, Kuno T, Tanaka T, et al. C57BL/KsJ-db/db-Apc mice exhibit an increased incidence of intestinal neoplasms. Int J Mol Sci. (2011) 12:8133–45. doi: 10.3390/ijms12118133
80. Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature. (2016) 531:53–8. doi: 10.1038/nature17173
81. Park MY, Kim MY, Seo YR, Kim JS, Sung MK. High-fat diet accelerates intestinal tumorigenesis through disrupting intestinal cell membrane integrity. J Cancer Prev. (2016) 21:95–103. doi: 10.15430/JCP.2016.21.2.95
82. Qu DC, Neu D, Khawaja ZQ, Wang R, Bartels CF, Lovrenert K, et al. Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-Apc (Min/+) mice. J Transl Genet Genom. (2023) 7:3–16.
83. Yang J, Wei H, Zhou Y, Szeto CH, Li C, Lin Y, et al. High-fat diet promotes colorectal tumorigenesis through modulating gut microbiota and metabolites. Gastroenterology. (2022) 162:135–149.e2. doi: 10.1053/j.gastro.2021.08.041
84. Fu T, Huan T, Rahman G, Zhi H, Xu Z, Oh TG, et al. Paired microbiome and metabolome analyses associate bile acid changes with colorectal cancer progression. Cell Rep. (2023) 42:112997. doi: 10.1016/j.celrep.2023.112997
85. Liu W, Crott JW, Lyu L, Pfalzer AC, Li J, Choi SW, et al. Diet- and genetically-induced obesity produces alterations in the microbiome, inflammation and wnt pathway in the intestine of apc(+/1638N) mice: Comparisons and contrasts. J Cancer. (2016) 7:1780–90. doi: 10.7150/jca.15792
86. Huang YW, Lin CW, Pan P, Echeveste CE, Dong A, Oshima K, et al. Dysregulated free fatty acid receptor 2 exacerbates colonic adenoma formation in apc (Min/+) mice: Relation to metabolism and gut microbiota composition. J Cancer Prev. (2021) 26:32–40. doi: 10.15430/JCP.2021.26.1.32
87. Tabrizian T, Wang D, Guan F, Hu Z, Beck AP, Delahaye F, et al. Apc inactivation, but not obesity, synergizes with Pten deficiency to drive intestinal stem cell-derived tumorigenesis. Endocr Relat Cancer. (2017) 24:253–65. doi: 10.1530/ERC-16-0536
88. Pakiet A, Kobiela J, Stepnowski P, Sledzinski T, Mika A. Changes in lipids composition and metabolism in colorectal cancer: a review. Lipids Health Dis. (2019) 18:29. doi: 10.1186/s12944-019-0977-8
89. Mutoh M, Akasu T, Takahashi M, Niho N, Yoshida T, Sugimura T, et al. Possible involvement of hyperlipidemia in increasing risk of colorectal tumor development in human familial adenomatous polyposis. Jpn J Clin Oncol. (2006) 36:166–71. doi: 10.1093/jjco/hyi233
90. Sun L, Kang Q, Pan Y, Li N, Wang X, He Y, et al. Serum metabolite profiling of familial adenomatous polyposis using ultra performance liquid chromatography and tandem mass spectrometry. Cancer Biol Ther. (2019) 20:1017–28. doi: 10.1080/15384047.2019.1595277
91. Almendingen K, Hostmark AT, Fausa O, Mosdol A, Aabakken L, Vatn MH. Familial adenomatous polyposis patients have high levels of arachidonic acid and docosahexaenoic acid and low levels of linoleic acid and alpha-linolenic acid in serum phospholipids. Int J Cancer. (2007) 120:632–7. doi: 10.1002/ijc.22337
92. Snyder M, Horning A, Esplin E, Wu S, Hanson C, Bararpour N, et al. Multi-omic analysis of familial adenomatous polyposis reveals molecular pathways and polyclonal spreading associated with early tumorigenesis. Res Square. (2021). doi: 10.21203/rs.3.rs-515393/v1
93. Sommer CA, Capilla A, Molina-Estevez FJ, Gianotti-Sommer A, Skvir N, Caballero I, et al. Modeling APC mutagenesis and familial adenomatous polyposis using human iPS cells. PLoS One. (2018) 13:e0200657. doi: 10.1371/journal.pone.0200657
94. Ecker J, Benedetti E, Kindt ASD, Horing M, Perl M, Machmuller AC, et al. The colorectal cancer lipidome: Identification of a robust tumor-specific lipid species signature. Gastroenterology. (2021) 161:910–23.e19.
95. Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, et al. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell. (2015) 161:1539–52. doi: 10.1016/j.cell.2015.05.033
96. Park WJ, Kim MJ. A new wave of targeting 'Undruggable' Wnt signaling for cancer therapy: Challenges and opportunities. Cells. (2023) 12. doi: 10.3390/cells12081110
97. Zhang Y, Liu Y, Sun J, Zhang W, Guo Z, Ma Q. Arachidonic acid metabolism in health and disease. MedComm (2020). (2023) 4:e363. doi: 10.1002/mco2.363
98. Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. (2001) 7:1048–51. doi: 10.1038/nm0901-1048
99. Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. (2000) 342:1946–52. doi: 10.1056/NEJM200006293422603
100. Farooq U, El Alayli A, Duvvuri A, Mansour R, Pasam RT, Malireddy S, et al. Nonsteroidal anti-inflammatory drugs for chemoprevention in patients with familial adenomatous polyposis: A systematic review and meta-analysis. Gastro Hep Adv. (2023) 2:1005–13. doi: 10.1016/j.gastha.2023.05.009
101. Volpato M, Hull MA. Omega-3 polyunsaturated fatty acids as adjuvant therapy of colorectal cancer. Cancer Metastasis Rev. (2018) 37:545–55. doi: 10.1007/s10555-018-9744-y
102. Hull MA, Sprange K, Hepburn T, Tan W, Shafayat A, Rees CJ, et al. Eicosapentaenoic acid and aspirin, alone and in combination, for the prevention of colorectal adenomas (seAFOod Polyp Prevention trial): A multicentre, randomised, double-blind, placebo-controlled, 2 x 2 factorial trial. Lancet. (2018) 392:2583–94. doi: 10.1016/S0140-6736(18)31775-6
103. Song M, Lee IM, Manson JE, Buring JE, Dushkes R, Gordon D, et al. Effect of supplementation with marine omega-3 fatty acid on risk of colorectal adenomas and serrated polyps in the US general population: A prespecified ancillary study of a randomized clinical trial. JAMA Oncol. (2020) 6:108–15. doi: 10.1001/jamaoncol.2019.4587
104. Falchook G, Infante J, Arkenau HT, Patel MR, Dean E, Borazanci E, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine. (2021) 34:100797. doi: 10.1016/j.eclinm.2021.100797
105. Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, et al. FASN inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and FASN inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine. (2017) 16:51–62. doi: 10.1016/j.ebiom.2016.12.012
Keywords: APC gene, APC-mediated signaling, colorectal cancer, lipids, lipid metabolism
Citation: Kelson CO and Zaytseva YY (2024) Altered lipid metabolism in APC-driven colorectal cancer: the potential for therapeutic intervention. Front. Oncol. 14:1343061. doi: 10.3389/fonc.2024.1343061
Received: 22 November 2023; Accepted: 12 March 2024;
Published: 25 March 2024.
Edited by:
Qingfei Zheng, The Ohio State University, United StatesReviewed by:
Mark M. Huycke, University of Oklahoma, United StatesYoutao Lu, University of Pennsylvania, United States
Copyright © 2024 Kelson and Zaytseva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yekaterina Y. Zaytseva, eXl6YXl0MkB1a3kuZWR1