 Kamilla JA. Bigos1*Conrado G. Quiles1Sapna Lunj1Danielle J. Smith1Mechthild Krause2,3,4,5,6,7,8,9,10Esther GC. Troost4,6,7,8,9,10,11,12Catharine M. West13Peter Hoskin1,14Ananya Choudhury1,15
Kamilla JA. Bigos1*Conrado G. Quiles1Sapna Lunj1Danielle J. Smith1Mechthild Krause2,3,4,5,6,7,8,9,10Esther GC. Troost4,6,7,8,9,10,11,12Catharine M. West13Peter Hoskin1,14Ananya Choudhury1,15- 1Division of Cancer Sciences, University of Manchester, Manchester, United Kingdom
- 2German Cancer Consortium (DKTK), partner site Dresden and German Cancer Research Center (DKFZ), Heidelberg, Germany
- 3Department of Radiation Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 4OncoRay – National Center for Radiation Research in Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Helmholtz-Zentrum Dresden - Rossendorf, Dresden, Germany
- 5Translational Radiooncology and Clinical Radiotherapy, Helmholtz-Zentrum Dresden - Rossendorf, Dresden, Germany
- 6Translational Radiation Oncology, National Center for Tumor Diseases (NCT), Partner Site Dresden, Dresden, Germany
- 7Translational Radiooncology and Clinical Radiotherapy and Image-guided High Precision Radiotherapy, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 8Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 9Translational Radiooncology and Clinical Radiotherapy and Image-guided High Precision Radiotherapy, Helmholtz Association / Helmholtz-Zentrum Dresden - Rossendorf (HZDR), Dresden, Germany
- 10School of Medicine, Technische Universitat Dresden, Dresden, Germany
- 11Department of Radiotherapy and Radiation Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 12Institute of Radiooncology – OncoRay, Helmholtz-Zentrum Dresden-Rossendorf, Rossendorf, Germany
- 13Division of Cancer Sciences, University of Manchester, Manchester Academic Health Science Centre, Christie Hospital, Manchester, United Kingdom
- 14Mount Vernon Cancer Centre, Northwood, United Kingdom
- 15Christie Hospital NHS Foundation Trust, Manchester, Germany
Hypoxia is a common feature of solid tumours affecting their biology and response to therapy. One of the main transcription factors activated by hypoxia is hypoxia-inducible factor (HIF), which regulates the expression of genes involved in various aspects of tumourigenesis including proliferative capacity, angiogenesis, immune evasion, metabolic reprogramming, extracellular matrix (ECM) remodelling, and cell migration. This can negatively impact patient outcomes by inducing therapeutic resistance. The importance of hypoxia is clearly demonstrated by continued research into finding clinically relevant hypoxia biomarkers, and hypoxia-targeting therapies. One of the problems is the lack of clinically applicable methods of hypoxia detection, and lack of standardisation. Additionally, a lot of the methods of detecting hypoxia do not take into consideration the complexity of the hypoxic tumour microenvironment (TME). Therefore, this needs further elucidation as approximately 50% of solid tumours are hypoxic. The ECM is important component of the hypoxic TME, and is developed by both cancer associated fibroblasts (CAFs) and tumour cells. However, it is important to distinguish the different roles to develop both biomarkers and novel compounds. Fibronectin (FN), collagen (COL) and hyaluronic acid (HA) are important components of the ECM that create ECM fibres. These fibres are crosslinked by specific enzymes including lysyl oxidase (LOX) which regulates the stiffness of tumours and induces fibrosis. This is partially regulated by HIFs. The review highlights the importance of understanding the role of matrix stiffness in different solid tumours as current data shows contradictory results on the impact on therapeutic resistance. The review also indicates that further research is needed into identifying different CAF subtypes and their exact roles; with some showing pro-tumorigenic capacity and others having anti-tumorigenic roles. This has made it difficult to fully elucidate the role of CAFs within the TME. However, it is clear that this is an important area of research that requires unravelling as current strategies to target CAFs have resulted in worsened prognosis. The role of immune cells within the tumour microenvironment is also discussed as hypoxia has been associated with modulating immune cells to create an anti-tumorigenic environment. Which has led to the development of immunotherapies including PD-L1. These hypoxia-induced changes can confer resistance to conventional therapies, such as chemotherapy, radiotherapy, and immunotherapy. This review summarizes the current knowledge on the impact of hypoxia on the TME and its implications for therapy resistance. It also discusses the potential of hypoxia biomarkers as prognostic and predictive indictors of treatment response, as well as the challenges and opportunities of targeting hypoxia in clinical trials.
Hypoxia
Hypoxia is a state in which there is a lack of sufficient oxygen supply to tissues and organs. This has detrimental effects on cells as they require oxygen for their function. The physiological response to hypoxia is to induce cell death. In the context of cancer, overconsumption of oxygen leads to low levels of oxygen. However, tumour cells find mechanisms to adapt to these harsh conditions, enabling tumour cell survival. Hypoxia modulates tumour growth, invasion, and resistance to therapy induced by rapid tumour cell proliferation, abnormal tumour vasculature, high interstitial pressure, or low oxygen delivery. These combined features can enhance a tumours ability to metastasise (1, 2). Hypoxia can be described as chronic, acute or cycling within solid tumours. Cycling hypoxia is defined by tumours undergoing periodic exposure to hypoxia followed by reoxygenation, which is associated with enhancing common hallmarks of cancer (3, 4). This has a significant role in promoting resistance to both radiotherapy and chemotherapy (2). Schwarz et al., were the first to propose that hypoxia drives resistance to radiotherapy (5). Thomlinson and Gray also observed hypoxia in bronchial carcinomas. These tumours were found to grow in solid cords surrounded by stroma, cords larger than 180 microns, were associated with a necrotic centre due to insufficient oxygen supply (1). These landmark studies led researchers to explore the impact of hypoxia on anti-tumour response.
The hypoxic tumour microenvironment (TME) has been defined as a condition where the partial pressure of oxygen (pO2) is below 10 mmHg (6). Physoxia describes the maintenance of physiological levels of oxygen in tumours. It can differ dependent on tissue type and location, highlighting why hypoxia also varies between different tumour types (Table 1).
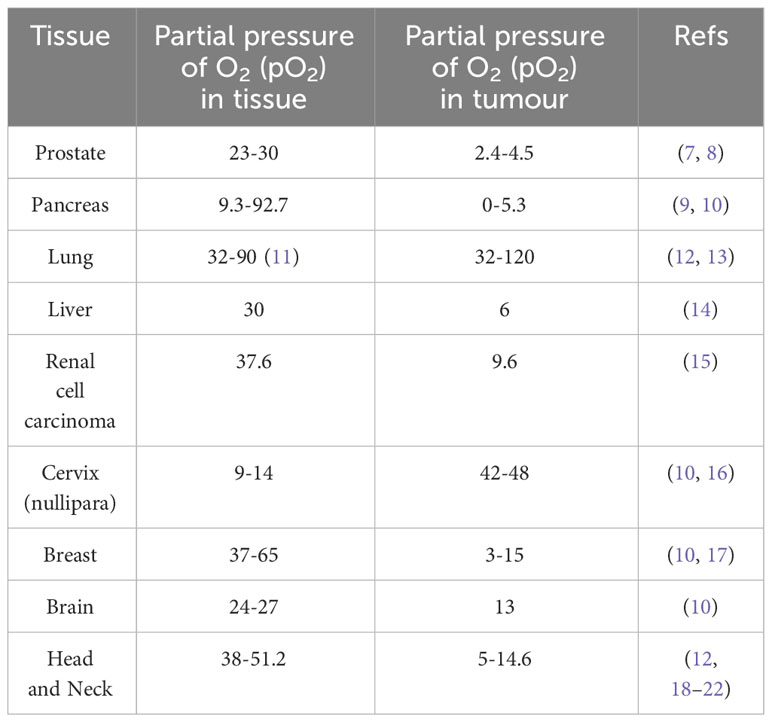
Table 1 Tissue and tumour physoxia and hypoxia expressed as partial pressure of O2 (pO2).
Hypoxia- inducible factor activation by hypoxia
HIF, a transcription factor responding to low oxygen levels, regulates gene expression to adapt tumour cell behaviour for survival (Table 2). It activates genes supporting hypoxia adaptation, like those linked to angiogenesis, erythropoiesis, glucose uptake, and anaerobic metabolism, while suppressing non-essential genes for survival (46).
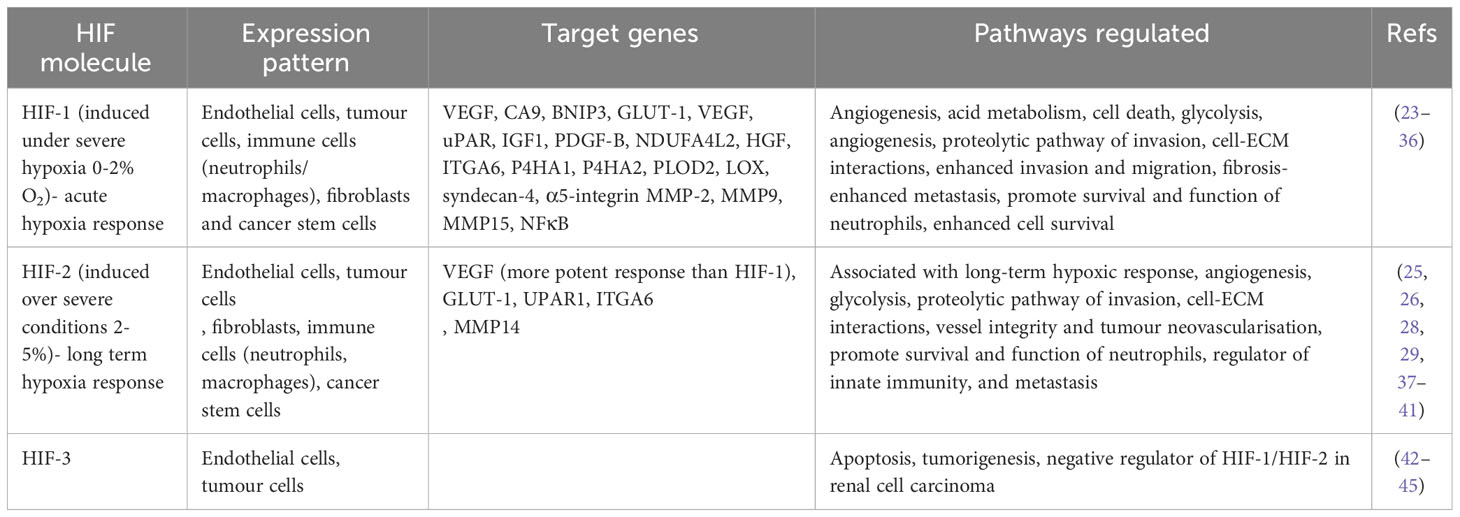
Table 2 HIF molecule expression patterns and the pathways they target.
Cellular response to hypoxia induces intracellular signalling pathways regulated by HIF (2). HIF-1 is a heterodimer composed of an oxygen-sensitive subunit, HIF-1α, and HIF-1β which is constitutively expressed (47). Under physoxia, HIF-1α is degraded however, under hypoxia, HIF-1α is stabilised and forms a heterodimer with HIF-1β. This complex acts as a transcriptional activator that binds to DNA sequences called hypoxia response elements (HRE) in the promoters of target genes (14). HIF-2 is also a heterodimer with and oxygen-sensitive HIF-2α subunit, whilst sharing HIF-1β with HIF-1α as a constitutively expressed subunit (48). However, HIF-1 and HIF-2 have distinctive roles and this is why both are considered as separate therapeutic targets (Table 2). HIF-3 is the least characterised HIF and has multiple isoforms which are variants of HIF-3α. These isoforms have been identified with different tissue distribution and functional properties. HIF-3 does not have a transactivation domain like HIF-1/HIF-2 but instead contains a polypeptide that represses HRE-responsive gene expression (49).
Hypoxia as a biomarker
The amount of hypoxia varies in solid tumours, with around 50% of tumours having high levels of hypoxia (50). Numerous studies show that most hypoxic tumours have the worse prognosis (51). Therefore, the importance of hypoxia as an adverse prognostic factor has led to interest in developing methods for its measurement and targeting (51).
In the 1960s, studies were published on the use of oxygen electrodes to measure hypoxia in cervical cancers (CC) (52). Oxygen electrodes (Eppendorf probes) or fibre optic probes (OxyLite) measure oxygen tension directly. The approach is based on inserting an electrode into an accessible tumour and measuring oxygen at multiple points within several tracks (51). Although this is considered the gold standard, it is invasive, operator-dependent and cannot account for heterogeneity (53). The result of heterogeneity is the overestimation of viable hypoxic cells as both areas with healthy cells and necrotic cells are measured (51, 54, 55). The key differences in the two methods are summarised by Griffiths & Robinson (53). The output of these methods is pO2 (Table 1). Fyles et al. (56) showed that oxygenation measured by oxygen electrodes was able to predict radiation response and survival in patients with CC. Nordsmark et al. (57) show that in advanced head and neck squamous cell carcinoma (HNSCC), patients with pO2 value of ≤2.5mmHg had a stronger predictive ability for radiation response. However, despite the ability to predict radiation response using this method, due to the limitations of the electrode systems, this led to their discontinuation (Table 3).
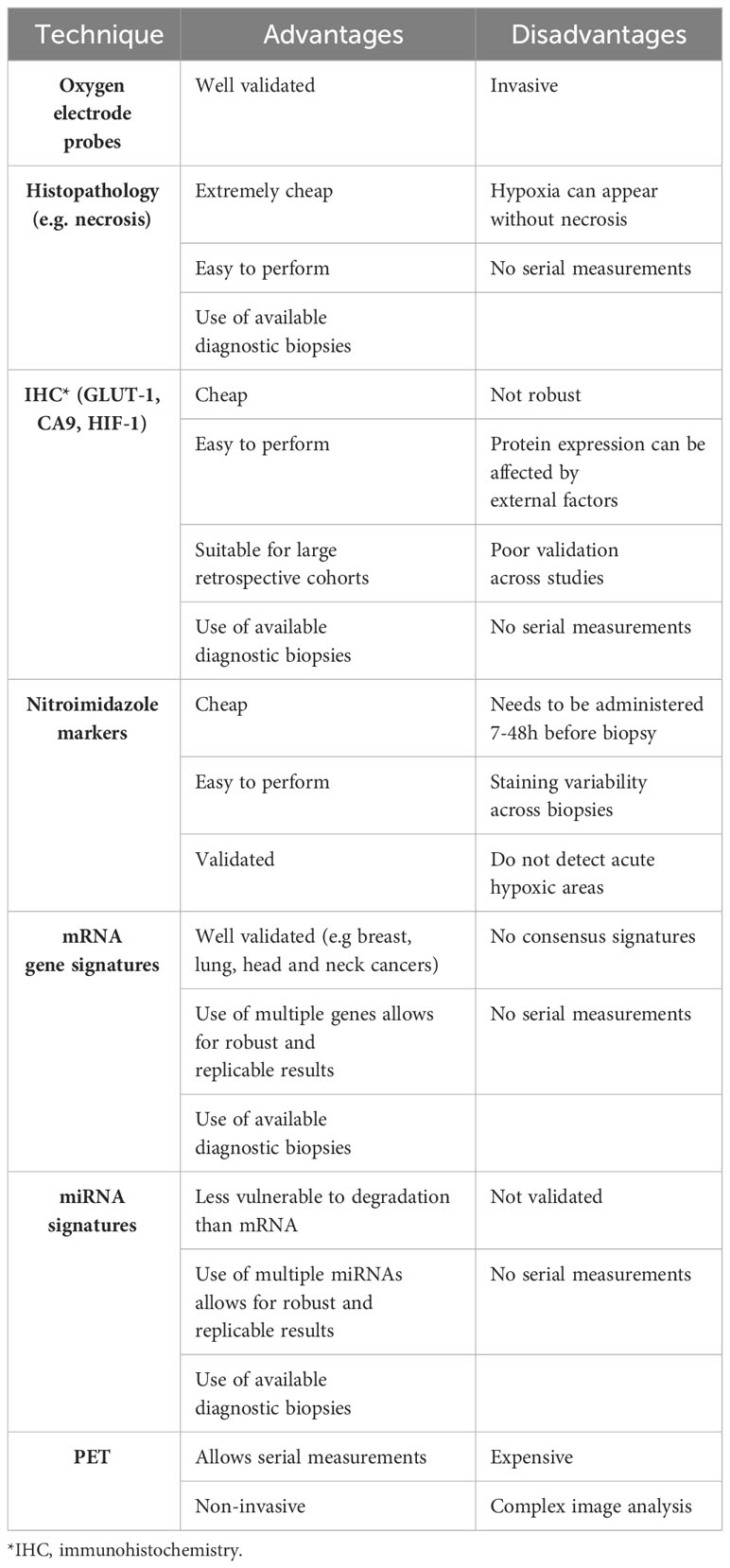
Table 3 Advantages and disadvantages of hypoxia detection methods.
Despite the limitations, the studies in oxygen measurement show that there is a clinical benefit to measuring hypoxia, which has led to continued research in the field including hypoxia targeted therapies (73). The methods include increasing oxygen delivery to tumours by breathing hyperbaric oxygen or carbogen; increasing vascular perfusion (e.g., nicotinamide); giving oxygen-mimetic radiosensitisers with radiotherapy (e.g., nimorazole); hypoxia-activated prodrugs (e.g., tirapazamine, evofosfamide); and small molecule inhibitors of hypoxia-relevant molecular targets (e.g. belzutifan, SLC-0111) (74). Overall, there is currently a clinical need to measure tumour hypoxia.
Currently, the most widely studied approach for assessing hypoxia in cancer patients involves endogenous markers, which has the advantage that large retrospective studies can be carried out (51, 54, 75). The approach uses immunohistochemistry on pre-treatment diagnostic biopsies (76). The markers used are hypoxia inducible in vitro, e.g., HIF-1α, CA9 and GLUT1 (76, 77). Many reports link high tumour HIF-1α expression with a poor prognosis including meta-analyses of studies in brain (n=1,422) (78), breast [BCa] (n=6,201) (79), digestive system (n=5,964) (80), hepatocellular [HCC] (n=3,570) (81), and oral cavity (n=1,471) (82) cancers. HIF-1α expression has been shown to be increased independently of hypoxia in clear cell renal carcinoma (ccRCC) due to a mutation in the von Hippel-Lindau (VHL) gene. The mutation inactivates normoxic proteasomal degradation of HIF-1α inducing elevated levels of HIF (83). Therefore, expression of hypoxia-related endogenous probes may not necessarily correlate with hypoxia and, are not a good standalone hypoxic marker. Meta-analyses have also shown high tumour expression of CA9 is an adverse prognostic factor in multiple cancers: renal cell carcinoma [RCC] (n=2,611) (84), oral squamous cell carcinoma [OSSC] (n=1,616) (85), and head and neck [H&N] cancer (n=1,470) (86). Several meta-analyses also show GLUT1 is an adverse prognostic factor in several cancers: lung (n=1,423 (87) and n=1,665 (88)), BCa (n=1,861) (89), colorectal cancer [CRC] (n=2,077) (90), and OSCC (n=1,301) (91). Furthermore, two meta-analyses in mixed cancer types showed high GLUT1 expression in tumours is associated with a poor prognosis (n=4,079 (92), and n=4,794 (93)). Although some of these markers have shown promise in hypoxia detection and stratifying patient therapy, this method relies on standardisation and establishment of guidelines across different laboratories, a common limitation in tissue-specific biomarkers (94). More focus is needed on systems that can be implemented into clinic (11).
Alternative measurements of hypoxia have also been developed including the indirect measurement of hypoxia using exogenous probes such as 2-nitroimidazole compounds (pimonidazole and EF5). These probes diffuse passively across the cell membrane and the nitro-group is enzymatically reduced to a reactive species inside the cell. Under hypoxic conditions, the nitro species undergoes further reduction and forms covalent bonds with central macromolecules which results in the accumulation of 2-nitroimidazole in hypoxic cells. However, within normoxic cells, the nitro species is re-oxidised and can diffuse out the cells (95, 96). Tumour biopsies are collected and hypoxia is detected using monoclonal antibodies. Multiple studies have shown the prognostic significance of these markers in H&N cancers, PCa, sarcomas, laryngeal cancer (LC), and glioblastoma (GBM). These markers are associated with locoregional control (LRC) (97), aggressive phenotype (98), metastases (99), poor outcome (100) and short time to recurrence (101).
Imaging-based markers for hypoxia are undergoing development such as 18F-MISO PET which uses a radioactive tracer, fluoromisonidazole (FMISO), to measure the levels of oxygen within tumours alongside positron emission tomography (PET) imaging. This is associated with survival outcomes and treatment response. In prospective hypothesis- generating and validation cohorts, patients with H&N cancer received 18F-MISO PET at different timepoints before or during chemoradiotherapy (CRT). Tumour hypoxia after 2 weeks of CRT correlated with a low LRC, whereas patients with oxic tumours had a good prognosis (102, 103). Carles et al. (104) prospectively analysed 35 HNSCC patients evaluated with 18F-MISO PET during CRT, correlating the changes in size and location of hypoxic areas within the tumour by a new classification parameter. The classification parameter distinguished between patients who had early or late disease progression, and how their hypoxic regions changed during CRT. Some of the radiomic features, particularly the low grey-level zone emphasis was able to predict local recurrence with high accuracy. Therefore, it was concluded that 18F-MISO PET hypoxia scanning has potential to be useful for personalised treatment plans and outcome prediction in HNSCC patients. However, 18F-MISO can bind to non-hypoxic cells in conditions of inflammation, infection or oxidative stress (105). Moreover, 18F-MISO has a slow blood clearance and tissue uptake therefore, it takes a long time to reach stable distribution in tumours (106). Although in a meta-analysis study, it has been shown that PET measured hypoxia is a robust parameter with a strong impact on outcome of HNSCC and that the most commonly investigated tracers 18F-MISO and FAZA (18F-Fluoroazomycin-arabinosid) can probably be used equivalently in multicentre trials (107). Dynamic contrast enhanced-MRI (DCE-MRI) has been shown to reflect heterogenous tumour perfusion and subtle tumour volume change during radiation/chemotherapy in prospective analysis of 62 CC patients. DCE-MRI is associated with tissue oxygenation and therefore, can be a good parameter for assessing hypoxia. In CC patients, they showed that there are independent and better predictors of tumour recurrence and death than clinical prognostic factors. Combining clinical prognostic factors and MRI parameters improves early prediction of treatment failure. This offers the potential of altering the treatment plan for patients (108).
Each of the techniques described have individual advantages and disadvantages (Table 3). The ability to measure hypoxia would aid clinical decision-making, with a robust predictive biomarker being the holy grail. However, a robust biomarker is not sufficient for its implementation into clinic, as shown by the oxygen electrode studies. Feasibility, economic costs, and undesirable effects in patients also need to be considered.
Hypoxia and the tumour microenvironment
The TME is a complex environment formed around tumours involving the interplay of several cell types and molecules. In solid tumours, the TME consists of cancer cells, surrounding blood vessels, immune cells, cancer-associated fibroblasts (CAFs), signalling molecules and the extracellular matrix (ECM) (Figure 1). The tumour type, stage of cancer and location can influence the nature of the TME. Tumour cells can change the composition of the microenvironment by releasing extracellular signals that promote angiogenesis or induce peripheral immune tolerance to evade immune detection (109). Features including hypoxia, the metabolic microenvironment, acidic niche and mechanical environment also play important roles in the phenotype of the TME (110). The hypoxic TME can help the tumour grow, spread or become resistant to treatments.
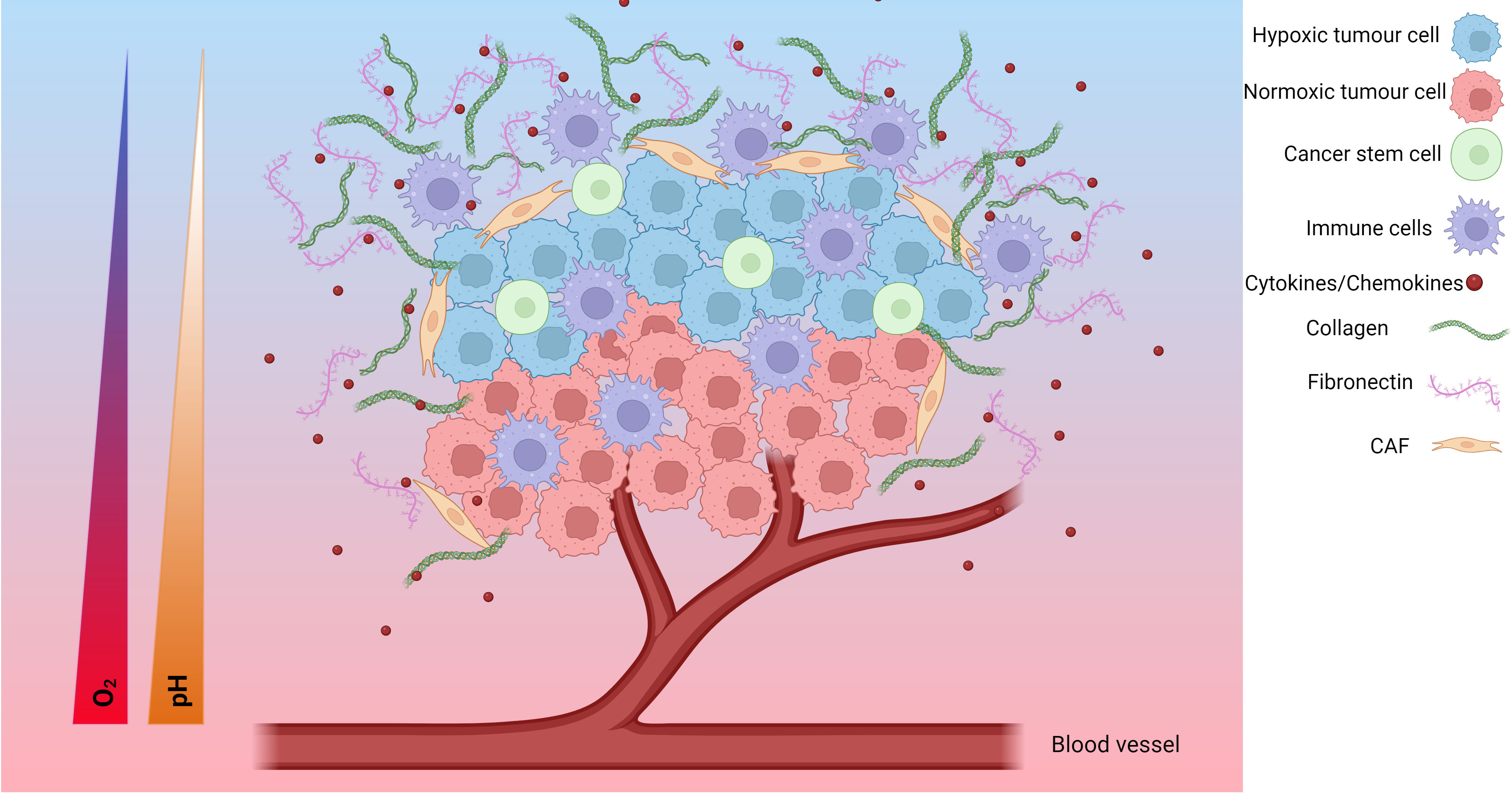
Figure 1 The hypoxic tumour microenvironment (TME). Hypoxia plays an important role in the development of the TME. The TME is composed of hypoxic tumour cells, cancer stem cells, tumour cells, immune cells, cytokines/chemokines, collagen, fibronectin, cancer associated fibroblasts (CAFs), endothelial cells and, blood vessels. As the tumour cells grow, the tumour cells further away from the blood supply have limited access to oxygen and become hypoxic tumour cells. Additionally, the TME undergoes a metabolic switch to meet the demands of the TME which involves increased glucose uptake and production of lactate resulting in an acidic TME characterised by a decreasing pH. These changes result in a change in cytokine/chemokine release, a change in immune cell phenotype, modification of the extracellular matrix, and activation of CAFs. Together, these form a more pro-tumorigenic environment prone to increased invasive and metastatic potential, as well as increased resistance to chemoradiotherapy. Created with BioRender.com.
Metabolic switch within the TME is essential for cancer cell growth and metabolism, and adaptation to the hypoxic microenvironment. This phenomenon is described as the Warburg effect and is characterised by cell metabolism favouring glycolysis to meet the demand of the cancer cells for survival. The Warburg effect involves increased rate of glucose uptake and preferential production of lactate. This induces acidification of TME from accumulation of large amounts of metabolic waste products including lactic acid, carbon dioxide and bicarbonate protons (111). These products lower the pH of the extracellular space and create a gradient between the intracellular and extracellular pH. This has an impact on multiple hallmarks of cancer including suppression of immune response, increasing tumour invasion and metastasis, and modulating proliferation (112). Hypoxia also enhances upregulation of glucose transporters including GLUT1 and enzymes of the glycolytic pathway (e.g. CA9) feeding back into the glycolysis cycle (113, 114). A further consequence of acidification is the enhanced activity of matrix metalloproteinases (MMPs) which have a role in degrading the ECM and basement membrane (115). Therefore, the invasive and metastatic potential of tumours under hypoxia is enhanced. Acidification can also enhance resistance to chemotherapy and radiotherapy by reducing the uptake of drugs and activating DNA repair mechanisms (83).
This review will focus on the ECM, CAFs and immune cells within the hypoxic TME, both as individual components and how they interplay to create a more pro-tumorigenic microenvironment that can be manipulated for better patient outcome.
Hypoxia and the extracellular matrix in cancer
The ECM is a complex network of biomolecules in the extracellular region that provides structural and mechanical support to surrounding cells. Several reports show its importance in the development of cancer (116). The ECM comprises a wide range of molecules that include structural proteins (e.g. fibronectin [FN], collagen [COL]), signalling molecules including cytokines and growth factors, (e.g. endothelial growth factor [EGF], transforming growth factor β [TGF-β]) and enzymes (e.g. MMPs, lysyl oxidase [LOX], prolyl 4-hydroxylase [P4HA]). The main protein components of the ECM are FN, COL, elastin and laminin proteins (117–121).
(116) CAFs are located in the tumour stroma and are the main producers of ECM (122–124). However, cancer cells also produce ECM and are important in determining the tumour ECM composition (125). In cancer development, the normal ECM phenotype shifts towards a cancerous ECM phenotype (126, 127). During this process, the ECM undergoes increased remodelling and deregulation of the levels of growth factors and enzymes (e.g. MMPs and TGF-β). Because of this remodelling process, a more fibrotic and stiffened ECM develops in cancer (116, 127). The transformation of the ECM towards a cancerous phenotype is a key process for tumour development that promotes cell growth and survival, metastasis and recruitment of cancer-associated cells (e.g. CAFs and tumour-associated macrophages [TAMs]), and modulates immune responses (4, 126, 128).
Hypoxia enhances ECM remodelling as a driver of tumour desmoplasia, a complex process that includes ECM degradation, composition and structural changes, generating a fibrotic and stiffer ECM (129). Degradation of the basement ECM membrane is enhanced in hypoxia as it drives neoplasia and tumour re-oxygenation (130). As reviewed by Chang and Chaudhuri (131), degradation of the basement membrane is a key for tumour progression that promotes local tissue invasion, and tumour metastasis (131). BCa patients with no basement membrane degradation have five-year overall survival (OS) rate of 99%. However, the percentage drops down to 85% for patients with local invasion of the basement membrane, and 27% once metastatic (132). Hypoxia increases MMPs secretion to induce basement ECM membrane degradation through HIF signalling (133, 134). For example, HIF-1 activity drives MMP2, MMP9, MMP14 and MMP15 overexpression (129, 134, 135). MMP2, MMP9, and MMP14 target and promote COLIV degradation, an activity associated with the destruction of basement membranes in BCa (136). MMP1 is also overexpressed due to HIF-1 activity in bladder cancer (BLCA), leading to increased migratory capacity in the presence of reactive oxygen species (ROS) (137).
The changes in the composition and structure of the ECM under hypoxia are driven by increased deposition and crosslinking of COL, FN and hyaluronic acid (HA) (138). Higher numbers and crosslinking of ECM fibres increase ECM stiffness and induces fibrosis. Fibrosis is associated with metastasis and poor cancer prognosis (139, 140). For example, fibrosis reduces the expected survival time of patients with non-small cell lung carcinoma (NSCLC) by 60% (141). Mechanistically, fibrosis promotes cancer development and spread through enhancing integrin mechanosensory pathways that activate EMT transition (e.g. FAK/Rho/ROCK signalling) (139, 140) leading to high ECM stiffness, fibrosis, and increased metastasis (129). Fibrosis induction is mostly mediated through increased COL deposition and enhanced expression of ECM remodelling enzymes (e.g. P4HA1, P4HA2, PLOD2) (129). Hypoxic induction of COL1 deposition has been known for 40 years and confirmed in several studies (142–145). Other studies also show HIF-mediated deposition and/or gene expression of fibrillar COLs (COL3 (146), COL5 (147–150), COL11 (149, 151) and COL27 (149)), basement membrane COLs (COL4 (150, 152, 153), COL7 (154), COL10 (151, 155) and COL18 (153, 156)), filament-forming COLs (COL6 (146)), fibril-associated COLs (COL9 (150, 151), COL14 (157)) and transmembrane COLs (COL13 (150)). Recent studies show hypoxia can also enhance fibrosis through increasing FN expression (158, 159). Increase in HA under hypoxia was first reported by Gao et al. (160). The findings were recently validated by Chen et al., who associated the increase in HA to higher invasive capacity of GBM cells (161). However, the role of HA in hypoxia-induced fibrosis has not been widely explored, and requires further research.
Enhanced deposition of ECM proteins (e.g. COL, FN, HA) is not sufficient to induce fibrosis. ECM crosslinking enzymes such as LOX are required to stiffen the ECM (129, 162). Increased expression of the LOX family members under hypoxia has been reported in several cancer studies (138, 163, 164), stromal cell, and endothelial cells (165). Similarly, hypoxia has been reported to promote the expression of other COL crosslinking enzymes through HIF signalling (PLOD1 (166), PLOD2 (167), P4HA1 (149, 168–170) and P4HA2 (149, 169)).
We can infer that higher expression of crosslinking enzymes under hypoxia enhances COL and FN fibrogenesis, leading to a fibrotic and desmoplastic ECM in cancer, but it is currently believed that hypoxic fibrosis generates migratory tracks to promote cancer cell migration and metastasis (Figure 2). This model is supported by reports showing that the expression of ECM-crosslinking enzymes is necessary for metastasis in in vivo models, and is associated with worse patient outcome (170, 171). Hypoxia also increases the expression of integrin receptors, further enhancing the mechanosensory pathways driving EMT transition. Hongo et al., have shown hypoxia enhances cancer cell migration through upregulation of α2, α5 and β1 integrins (172). Ju et al., showed upregulation of integrins α1, α5, α11 and β1, with α5 and β1 integrin also increased cancer cell migration (173). As the integrins described above are COL and FN receptors, it is possible that their upregulation under hypoxia provides a migratory advantage in a context where hypoxia is inducing fibrosis due to increased COL and FN deposition and crosslinking. However, there is evidence in the literature contradicting the current model. Kakkad et al., and Goggins et al., have shown hypoxia reduces COL fibre density through HIF-1 signalling using in vivo BCa and PCa xenograft models (174, 175). Furthermore, Kuchnio et al., demonstrated that low prolyl-hydroxylase 2 (PHD2), a protein that induces HIF-1 hydroxylation and degradation, impairs ECM deposition, fibrogenesis, and metastasis (176). These findings were validated by Madsen et al., who showed PHD2 inactivation impairs CAF activation, ECM deposition, and fibrosis (177).
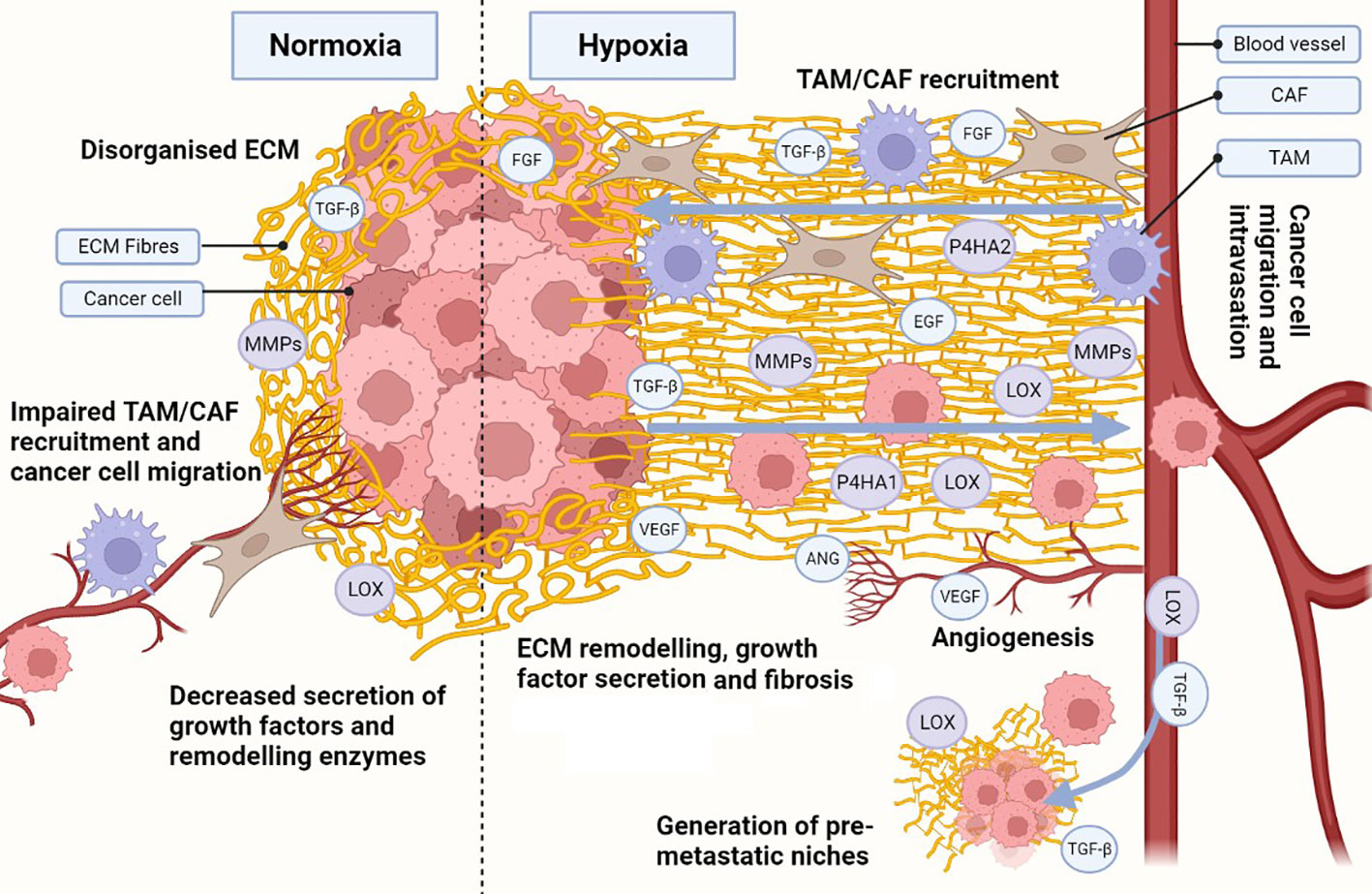
Figure 2 Hypoxia influences the development of a cancerous ECM. Hypoxia in the ECM increases collagen (COL) and fibronectin (FN) deposition, as well as secretion of metalloproteinases (MMPs), lysyl oxidases (LOX) and prolyl 4-hydroxylase subunit alpha (P4HA) 1 and 2. Increased MMPs, LOX and P4HA1/2 promote the generation of organised aligned COL and FN fibre tracks in the ECM, enhancing cell migration. Hypoxia also induces the secretion of growth factors and cytokines, which are also released due to ECM remodelling, establishing a synergistic effect. Release of growth factors (e.g. transforming growth factor β [TGF-β], endothelial growth factor [EGF], fibroblast growth factor [FGF]) enhance not only cancer cell growth and survival, but also recruitment of cancer-associated fibroblasts (CAFs) and tumour associated macrophages (TAMs). TAMs and CAFs participate in the secretion of growth factors, ECM remodelling and COL/FN deposition, increasing the synergistic effect. Under hypoxic stress, angiogenesis is activated through secretion of angiogenic growth factors (e.g. vascular endothelial growth factor [VEGF], angiopoietin [ANG]). The angiogenic process allows for the development of new blood vessels, enhancing ECM remodelling during the process. Additionally, an organised hypoxic ECM provides migratory tracks directing cells towards blood vessels and enhancing intravasation. Pro-tumorigenic growth factors and enzymes (e.g. TGF-β, LOX) can intravasate and travel to distant healthy tissues, generating pre-metastatic niches through ECM remodelling. The same migratory tracks enhance cancerous cell migration and intravasation, allowing them to circulate and eventually colonise the pre-metastatic niches and seed new tumour cells. Created with BioRender.com.
There are several possible explanations to the contradictory findings. Most studies addressing hypoxia in the ECM have been in fibroblasts, as they are considered the main drivers of fibrosis in cancer (139, 140). However, Tian et al., have shown cancer cells also change the ECM composition and remodelling in the development of fibrosis (178). Furthermore, the same study highlights fibroblasts and cancer cells have distinct contributions to the ECM during the development of fibrosis (178). Therefore, it is possible hypoxia drives distinct ECM remodelling process in fibroblasts and cancer cells.
Regarding the hypoxia mechanism driving ECM remodelling, most studies have focused on the role of HIF-1/2 (110). However, the role of HIF-3, the unfolded protein response (UPR) and the DNA damage response (DDR) pathways in the induction of fibrosis is poorly understood. Distinct levels of oxygen deprivation (179), and the length of exposure to hypoxia (3, 180) can change the signalling of the hypoxia pathways. Therefore, variability across experimental settings can also explain the differences reported in literature. In addition, to the current data no study has mechanistically proven hypoxias ability to enhance ECM fibrogenesis. Therefore, further clarification of the mechanisms driving hypoxia ECM remodelling, and its links with fibrosis and metastasis in hypoxic tumours is needed.
Hypoxia and cancer associated fibroblasts
CAFs are a type of stromal cell within the TME characterised by an elongated morphology. They can be derived from different cell types including resident fibroblasts, mesenchymal stem cells (MSCs), pericytes, smooth muscle cells, endothelial cells, epithelial cells, fibrocytes, stellate cells and adipocytes (Figure 3). CAFs are characterised by a lack of protein expression for epithelial, endothelial, or hematopoietic cells however, do express mesenchymal biomarkers including vimentin, α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP) and platelet-derived growth factor receptor-alpha (PDGFR-α) (183). They can be regulated by both HIF-dependent and HIF-independent mechanisms (110).
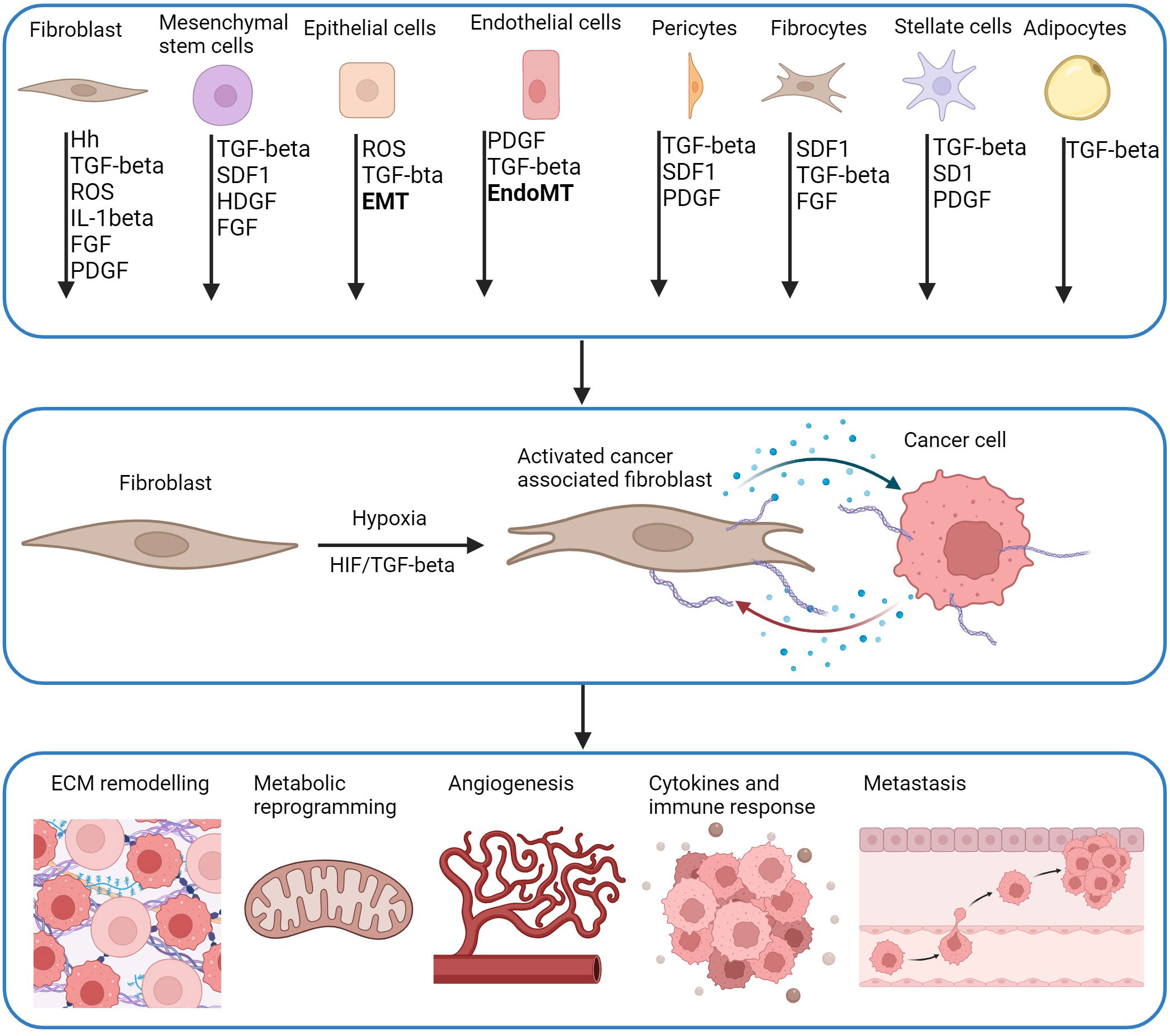
Figure 3 CAF activation pathways and downstream effects. CAFs are derived from multiple cell types and activated by multiple molecules including hedgehog (Hh), transforming growth factor-β (TGF-β), reactive oxygen species (ROS), interleukin-1β (IL-1β), fibroblast growth factor (FGF), platelet-derived growth factor (PDGR), stromal cell-derived factor 1 (SDF-1), heparin binding growth factor (HBGF) which can be driven by hypoxia. Precursor cells including mesenchymal stem cells (MSCs) are a source of CAFs activated by CXCL-12 and TGF-β derived from tumour cells. Pericytes, fibrocytes, stellate cells and adipocytes are also recruited by tumours by CXCL-12 and TGF-β, and are activated by TGF-β and PDGF. CAFs can derive from mature epithelial cells that differentiate into functional CAFs by TGF-β mediated epithelial-mesenchymal transition (EMT). Endothelial cells undergo EndoMT to differentiate into CAFs through TGF-β and SMAD signalling. HIF and TGF-β have a role in the function of CAFS. Genes associated with changes in ECM remodelling, metabolic reprogramming, angiogenesis, immune response and metastasis are direct transcriptional targets of HIF in CAFs or cancer cells. This creates bi-directional communication between CAFs and cancer cells through release of cytokines and chemokines (blue dots) which promotes proliferation of both cells, and further enhancement of pro-tumorigenic pathways (Adapted from (181, 182)). Created with BioRender.com.
CAFs interact with multiple TME cells and have a significant impact on tumour biology including angiogenesis, invasion, immune evasion, metastasis and drug resistance (183) (Figure 3). Hypoxia can alter the phenotype and function of CAFs, modulating the crosstalk between different cells of the TME (110). Due to their diverse role within the TME, tumour-promoting CAFs have been considered potential therapeutic targets in cancer (110, 183, 184). More recently, different CAF subtypes have been identified with either pro-tumorigenic or anti-tumorigenic effects (185). Although research is emerging within this field, the focus on hypoxia is limited. Therefore, it is important to differentiate between subtypes and cancer types, to deliver a more targeted and personalised therapy for patients.
CAF activation by hypoxia
Fibroblasts are activated in response to multiple signalling molecules including TGF-β, interleukin-1β (IL-1β), PDGF, ROS, stromal cell-derived factor 1 (SDF1), sonic hedgehog protein (SHH), hepatoma derived growth factor (HDGF) and FGF. These can be secreted by both cancer cells and stromal cells (Figure 2). CAF activation can mediate cancer progression under hypoxic conditions (Figure 3), HIF/TGF-β activation regulate ECM remodelling, immune response, metabolic reprogramming, angiogenesis and metastasis (181).
Hypoxia activates resident fibroblasts specifically by ROS and the activation of the HIF-1α pathway which are both driven by hypoxia. Resident fibroblasts are stimulated and mobilised by TGF-β1 to activate CAFs (Figure 3). PDGF, FGF, SHH and IL-1β secreted by tumours, also play a role in the activation of resident fibroblasts into CAFs activating ERK, Shh/Smo and NFκB pathways. Pericytes, fibrocytes, stellate cells and adipocyte-derived CAFs are recruited by tumours by TGF-β and SDF-1, and are activated by TGF-β or PDGF. CAFs also originate from precursor cells that are recruited by tumour cells. These are activated by the multiple signalling molecules described. MSC-derived CAFs are activated by SDF-1,TGF-β, HDGF and FGF. Epithelial cells can differentiate into CAFs through TGF-β and ROS mediated EMT (181). Endothelial cells undergo a similar process known as EndoMT which is driven by TGF-β and PDGF (Figure 3) (186).
CAFs respond much like cancer cells to hypoxia and undergo metabolic reprogramming to adapt to the TME and to support glycolysis. CAFs also provide essential metabolites for tumour growth. They have a bi-directional role with cancer cells, and enhance tumour proliferation. The metabolic switch is important in transforming fibroblasts to CAFs (23). Both melanoma tumour cells and CRC CAFs undergo glycolysis under hypoxic conditions however, CAF proliferation decreases relative to primary foreskin fibroblasts. It is proposed that the increase in glycolysis is to aid proliferation of tumour cells (23). Becker et al. (187) show that hypoxia induces epigenetic reprogramming of normal fibroblasts which results in a pro-glycolytic CAF-like phenotype. This induces the metabolism of BCa cells and promotes tumour growth. Together, these data highlight the role of CAFs in promoting tumour growth, and how this can be driven by HIFs.
CAF subtypes
Hypoxia induces oncogenic signals that can change both CAF phenotype and function reviewed by Han et al. (188). Rhim et al. (189) and Özdemir et al. (190) highlight that the lack of clinical success in targeting CAFs could be attributed to poor understanding of their heterogeneity. Therefore, further elucidation of this could help develop more targeted therapies.
These studies demonstrated that there is no common consensus for defining CAFs within literature. Without a standardised naming convention, it will be difficult to stratify patients who may benefit from CAF-targeting therapeutics. CAFs have a significant role in regulating multiple pro-tumorigenic pathways by the hypoxic TME (Figure 3). Although, it is important to fully understand the CAF/cancer cell axis, a deeper insight into CAF subtypes is needed to fully elucidate these pathways.
CAFs and hypoxia
Contrary to data often presented on the role of HIF-1α as a tumour-promoting factor, Kim et al. (191) have shown that targeted deletion of HIF-1α in stromal cells enhances tumour growth. In mammary stromal cells, HIF-1α is a negative regulator of tumour development. HIF-1α null and VEGF-A-null mammary tumours were associated with reduced hypoxia and decreased permeability and density of tumours (191). TAM infiltration was reduced in both null mammary tumours. In contrast, Chiavarina et al. (192), showed that fibroblasts expressing HIF-1α increased xenograft MDA-MB-231 breast tumour volume by ~3-fold. This was associated with a reduction in caveolin-1 expression and an increase in aerobic glycolysis shown by loss of mitochondrial activity and increased lactate production. Direct activation of HIF-1α in MDA-MB-231 cells in a xenograft model, showed a 3-fold reduction in tumour volume. These data suggest that HIF-1α effects are cell type dependent. The fibroblasts analysed by Kim et al. (191), were derived from a mouse mammary model, compared to Chiavarna et al. (192) who used a human immortalised cell line, hTERT-BJ1.
In androgen-deprived prostate tumours, hypoxia is common and associated with activation of HIF-1 and induction of TGF-β expression. This results in differentiation of fibroblasts to myofibroblasts and production of CXCL13 which drives B-cell recruitment and a more aggressive phenotype. This was highlighted in a transgenic adenocarcinoma mouse model, where the emergence of castration-resistant PCa was inhibited by inhibition of TGF-β or blocking phosphodiesterase-5 which prevents activation of myofibroblasts and CXCL13 (193). These studies used different animal models, cancer models and methods which could cause discrepancies within the data. The genetic homogeneity within these species means that there is lack of genetic variation that is more representative of the human population. Most murine cancers are derived from mesenchymal origin whilst human tumours mainly arise from epithelial tumours (194). Additionally, the origin, differentiation and malignancy can change the nature of the interaction between HIF-1α and CAFs.
Despite the evidence that CAFs have a tumour-promoting role, there are studies that highlight tumour-supressing properties. It is unknown whether fibroblast subtypes are normal fibroblasts that are resistant to differentiating into CAFs, or a subtype of anti-tumour CAFs. Hu et al. (195) emphasise the challenge of personalising treatments due to our lack of understanding CAFs functional distinctions within patient tumours. However, they were able to identify 3 CAF subtypes from patients with NSCLC, the different subtypes correlated with clinical response to targeted therapies and tumour immune microenvironment. They showed that NSCLC cells with mutant EGFR which accounts for 20% of NSCLC resistance to tyrosine kinase inhibitors (TKIs) had differing responses to EGFR TKI, Osimertinib, dependent on CAF subtype.
Meflin is a protein expressed in CAFs within the pancreatic TME. Low Meflin expression correlated with straight collagen fibre alignment and a more aggressive pancreatic cancer (PaCa) phenotype (196). Lida et al., showed that Meflin positive CAFs have a tumour suppressive role in a PDAC mouse model. Am80 was identified as a reagent that induces Meflin expression in CAFs and increased PDAC sensitivity to chemotherapeutics. Furthermore, tumour vessel area and intra-tumoral drug delivery was enhanced following treatment with Am80. Meflin suppressed tissue stiffening by interacting with LOX to inhibit its COL crosslinking activity (197). CAFs are largely associated with creating an immunosuppressive TME, reducing the efficacy of immune checkpoint blockade. However, CAFs expressing Meflin in patients with NSCLC, showed enhanced survival and favourable therapeutic response to immune checkpoint blockade. Higher prevalence of Meflin-positive CAFs is positively correlated with CD4-positive T-cell infiltration and vascularisation within NSCLC tumour. The anti-tumorigenic role of Meflin positive CAFs has also been shown in CRC models (198). Takahashi et al., propose that hypoxia depletes expression of Meflin on CAFs resulting in cancer-promoting CAFs that induce chemoresistance (199).
Brechbuhl et al., show that estrogen-receptor (ER+) BCa have two CAF subtypes defined by their CD146 expression. CD146neg CAFs supress expression of ER and are less sensitive to tamoxifen. Whilst CD146pos CAFs maintain ER expression in BCa cells and sustain estrogen-dependent proliferation and sensitivity to tamoxifen. This is also shown in PaCa (200), blocking CD146 in CAFs significantly enhanced tumour cell migration and invasion in co-cultures with PaCa cells (201).
There has been limited research on the role of hypoxia in regulating CAF phenotype. Madsen et al., show that chronic hypoxia and HIF-1α stabilisation deactivate CAFs in an orthotopic BCa model. The loss of PHD2 activity following chronic hypoxia prevents both CAF-induced ECM remodelling and BCa metastasis (177). This suggests that hypoxia may have a positive interaction with CAFs, and that the influence of hypoxia differs between different cell types. Despite the evidence of both pro-tumorigenic and anti-tumorigenic roles of CAFs, there is limited research on the role of hypoxia in regulating CAF expression and role within the TME. All these data imply that targeting HIF-1α and its signalling pathways in CAFs may provide different outcomes between different patients and cancers. Therefore, it is essential that specific characteristics of individual patient tumours and CAFs is taken into consideration rather than a one-size-fits-all approach.
Hypoxia and immune cell regulation
Several infiltrated immune cells can be found within the TME including CD8 T cells, CD4 T cells, regulatory T cells (Tregs), natural killer cells (NK), TAMs and dendritic cells (DCs) (202). Under physiological conditions, a tightly regulated balance between immune activation and immune suppression is needed to maintain homeostasis (203). External factors including hypoxia can alter this balance (204). Data supports the role of hypoxia in dampening the anti-tumour immune response by modulating key processes including immune cell activation, infiltration and function (205). Hypoxia-induced immune modulation can occur via direct and indirect mechanisms including changes in cytokines and growth factors, expression of immune checkpoint molecules and metabolic activity (206–209). The role of hypoxia in modulating immune cell activity is complex and may be tumour-dependent or person-dependent. In addition, hypoxia can have different effects on the distinct immune cells present within the TME.
Regulation of T cells
T cells (CD8+ and CD4+) are an important component of the adaptive immune system and play a key role in eliminating tumour cells via various cytotoxic activities (210, 211). The anti-tumour function of T cells can be suppressed in several ways. This includes up-regulation of inhibitory checkpoint molecules and the presence of other immune cells such as Tregs, TAMs and myeloid-derived suppressor cells (MDSCs) (210). Evidence suggests that hypoxia plays an important role in inducing T cell dysfunction by modulating expression of checkpoint molecules and immune cell infiltrate via changes in growth factors, cytokines, chemokines and intra-tumoural pH (212–214).
The presence of hypoxia in tumours causes up-regulation of inhibitory immune checkpoint molecules and growth factors that reduce the cytotoxic and survival potential of T cells (212–222). Hypoxia has been shown to up-regulate immune checkpoint molecules on T cells (TIGIT, TIM3 and VISTA) in pre-clinical models of melanoma, CRC and lung cancer. They are associated with reduced anti-tumour T cell activity (6, 159, 213, 215–222).
VEGF is a hypoxia-responsive growth factor that has a role in immune regulation. In vivo studies investigating the effects of inhibiting VEGF on immune regulation suggest that blocking VEGF improves the cytotoxic potential of CD8+ T cells by increasing production of IFNγ and TGF-β (223, 224). However, careful consideration of the anti-VEGF approach needs to be highlighted as although inhibiting VEGF improved anti-tumour immune cell phenotype, this was accompanied by increased hypoxia. Using combinational therapies may help eliminate the risk of negative side effects. In addition, targeting hypoxia via VEGF or HIF-1α may need a deeper understanding of the cell-type or tumour expression profile. Especially since Palazon et al. (224) show that deletion of either HIF-1α or VEGF in CD8+ T cells reduces their ability to infiltrate and kill BCa tumours.
Single-cell analysis of tumours from HCC patients demonstrates that regions of high hypoxia have increased Treg infiltration and decreased granzyme B positive T cells compared with low hypoxic regions (214). In vitro analysis suggested that hypoxia increases secretion of CCL28 which increases Treg migration (212). In addition, the hypoxic TME is more acidic and glucose deprived which leads to changes in immune cell metabolism (i.e. activation of glycolysis, increased lactic acid levels and amino acid metabolism) (24, 207–209, 225–231). These metabolic changes provide Tregs with a survival advantage, as they are resistant to high levels of lactate. Meanwhile, other immune cells including NK cells, DCs, CD8+ and CD4+ T cells cytotoxicity and maturation is inhibited, causing increased immune suppression (207–209, 226, 231–233). In vitro co-culture analysis using HCC cell lines suggests that hypoxia increases expression of indoleamine 2, 3-dioxygenase 1 (IDO1) in monocyte-derived macrophages and Tregs leading to reduced CD8+ T cell proliferation and cytotoxic effects and expansion of Tregs (227, 229). The effects of hypoxia on IDO1 are potentially cell-type specific as expression of IDO1 in tumour cells (i.e. OC cells) is reduced under hypoxic conditions (229). This along with the differences observed in targeting VEGF and HIF-1α in different cell types highlights the complexity associated with drug-targeting. Suggesting that a greater understanding of the wide-spread expression profile of targets of interests is needed to improve drug efficacy and reduce toxicity.
Regulation of NK cells
NK cells form part of the innate immune response that can induce rapid and strong anti-tumour activity without the need for priming by antigen-presenting cells (211). However, under hypoxic conditions expression of PD-L1 on NK cells is increased, leading to reduced CD8+ T cell proliferation (234, 235). In addition, hypoxia reduces NK cell activation in response to tumour cells by reducing the expression of NK cell surface receptors (i.e. NKp46, NKp30, NKp44 and NKG2D) (236). Although hypoxia reduces expression of receptors associated with NK cell activation, the surface density and function of the Fc-γ receptor CD16 remains unaffected under hypoxia (236), suggesting that hypoxia does not affect the cytotoxic potential of NK cells. Similar to observations in T cells, hypoxia affects NK cell activity to different extents and may be tumour-dependent, person-dependent or dependent on the severity of hypoxia (236–239). Balsomo et al. (236) found that even in the presence of hypoxia, NK cells were able to efficiently kill melanoma cells but the same was not observed for HCC and BCa cell lines (237, 238). In vitro analysis of HCC cell lines demonstrated that HCC cells and NK cells exposed to 6%, oxygen significantly inhibited NK cell toxicity against the HCC tumour cells (238). However, at oxygen concentrations <6% there was no significant decline in NK cell activity, suggesting differences in downstream pathways activated at different oxygen concentrations. This is an important observation as it suggests that the extent of hypoxia can influence the resulting NK cell response either positively or negatively. Therefore, biomarkers that allow for gradients of tumour hypoxia to be identified would be advantageous in providing personalised treatment approach to targeting immune cells.
Regulation of myeloid-derived suppressor cells
MDSCs are immune cells derived from the myeloid linage that have potent immunosuppressive activity even under physiological conditions (240). Hypoxia increases intra-tumoural lactate, promoting production of MDSCs which reduces T cell and NK cell proliferation and cytotoxic potential of T cells. Noman et al. (241) showed that hypoxia upregulates the expression of PD-L1 but not PD-L2, PD-1, CTLA-4, CD80 or CD86 on splenic MDSCs. Subsequent inhibition of PD-L1 increased the cytotoxic capability of CD4+ and CD8+ T cells by restoring the production of IFNγ. Down-regulation of IL6 and IL10 is the proposed mechanism for improved T cell cytotoxicity by PD-L1 blockage (241).
Regulation of monocyte-derived tumour-associated macrophages
TAMs are innate immune cells that are recruited into the tumour and differentiate from monocytes. Recruitment and differentiation of TAMs is orchestrated by chemotactic signals from soluble factors such as CCL2, CCL5, colony-stimulating factor 1 (CSF1), VEGF, semaphorin 3A (SEMA3A) endothelial cell monocyte-activating polypeptide-II (EMAP-II), endothelin, stromal cell-derived factor 1α (SDF1α) and oncosatin M (211, 242). Within the tumour, TAMs are found in abundance in both vascular and avascular stromal areas. TAMs can exist as M1 or M2 phenotype, with M2 TAMs associated with a pro-tumorigenic potential. Hypoxia modulates TAM recruitment, phenotype and function to induce an immunosuppressive TME.
Hypoxia inhibits the mobility of TAMs, resulting in accumulation of TAMs within hypoxic areas. Sica et al. (243) showed that defective expression of CCR2, a monocyte chemotactic protein in TAMs derived from patients with OC induces monocyte migration towards CCR2. Chemokines and cytokines produced by TAMs are key modulators of angiogenesis and metastasis, promoting a pro-tumorigenic microenvironment. TNF-α and IL-1 are secreted by macrophages and blood monocytes (244, 245). Both cytokines are associated with stimulating VEGF, and therefore may drive angiogenesis (246, 247).
Increased lactic acid in regions of hypoxia induces an M2 TAM phenotype which is associated with poor prognosis in BCa, CC, PCa and BLCA (231, 248–250). Exosomes secreted from hypoxic tumours contain elevated levels of cytokines and chemokines that drive macrophage recruitment and M2 polarisation. Park et al. (251) showed that exosomal protein secretion in hypoxic melanoma, squamous skin carcinoma and lung cancer cells was 3-4 fold higher than in normoxic cells. The hypoxic exosomes were associated with secretion of immunosuppressive mediators including TGF-β1and chemo-attractants including, CSF-1 associated with M2-like macrophage polarisation. The role of exosomes in promoting M2 polarisation has been demonstrated further by Shou et al. (252) in oesophageal cancer. Downregulation of PTEN is alsoshown by Zhu et al. (253) via exosomal miR223 derived from hypoxic TAMs co-cultured with OC cells. This resulted in decreased apoptosis, increased cell viability and enhanced drug resistance. In cancer, an M2-phenotype macrophage can enhance immunosuppression, promote angiogenesis and drug resistance. Polarisation of TAMs to an M2-like phenotype induces secretion of IL-10, TGF-β or VEGF which inhibit T cell function, induce Tregs and promote angiogenesis.
iNOS expression is increased in the presence of HIF-1α in macrophages under hypoxic conditions (24). Doedens et al. (24) show that hypoxia mediates suppression of T cell infiltration in vitro dependent on macrophage expression of HIF-1α in BCa models. IDO expression has been shown to increase monocyte-derived macrophages in a CCL20 dependent manner when co-cultured with hypoxic HCC cells. This correlated with increased HIF-1α expression. Additionally, the monocyte-derived macrophages were shown to supress T cell function and induce Tregs, generating an immunosuppressive microenvironment (227).
Regulation of monocyte-derived dendritic cells
DCs are specialised antigen presenting cells that differentiate from circulating monocytes and link the adaptive and innate immune response (211). Low oxygen tension has been associated with the differentiation and function of DCs.
Hypoxia reduces the ability of DCs to uptake tumour antigen and downregulates the expression of DC differentiation and activation markers including CD40, CD80 and MHCII via increased production of factors including IL-10, iNOS and VEGF. This affects the ability of DCs to process and present tumour antigens, reducing T cell priming and ultimately the induction of treatment induced immunogenic cell death (254). In addition, hypoxia induces increased secretion of osteopontin by DCs, a factor associated with enhanced migration of tumour cells (255).
Regulation of immune cells by hypoxia via other cells in the TME
There is strong evidence for the role of hypoxia in inducing direct effects on immune cells. However, hypoxia can also modulate immune cells indirectly via actions of tumour cells, endothelial cells and CAFs.
In terms of immune regulation by tumour cells, hypoxia has been shown to modulate epithelial mesenchymal plasticity. In lung adenocarcinoma, hypoxia-induced a mesenchymal phenotype in some hypoxic tumour cells. These hypoxic subclones demonstrated increased resistance to CD8+ T cell and NK cell-mediated lysis via TGF-β signalling (256). In vitro data using human cell lines derived from metastatic PCa and BCa tumours (DU145 and MDA-MB-231) demonstrates that hypoxia (0.5% O2) increases tumour cell expression of PD-L1 in a HIF-1α dependent manner leading to increased T cell apoptosis (257). In vivo models of melanoma suggest that increased expression of NANOG under hypoxic conditions upregulates the expression and secretion of TGF-β, and promotes the infiltration of immunosuppressive cells (258). In vivo models of BCa suggest that hypoxic mammary tumours secrete a variety of cytokines and growth factors (CCL2, G-CSF, TNF-α, VEGF, TIMP-1 and MMP-9) that increase infiltration of MDSCs (CD11b+/Ly6Cmed/Ly6G+). This increase in myeloid cell infiltration is inversely correlated with NK cell suppression, which enabled a microenvironment primed for metastatic growth of disseminated tumour cells within the lung. HIF-1α expression has been shown to negatively correlate with major histocompatibility complex (MHC) class I chain-associated genes which are essential for tumour antigen presentation and immune recognition. In vitro, analysis of PaCa demonstrated that hypoxia reduces the expression of MHC on the surface of PaCa cells. In addition, hypoxia induced the shedding of membrane bound MHC into extracellular space forming a soluble MHC which acts as a decoy and reduces the capacity of antigen presenting immune cells to present antigens to CD8+ T cells, thereby reducing T cells priming and T-cell mediated killing.
Hypoxia can induce changes to endothelial cells via increased expression of VEGF and FGF, altering interstitial pressure, intra-tumoural perfusion and expression of adhesion molecules. Changes in adhesion molecules (i.e. ICAM1, P-selectin, E-selectin, MAdCAM-1 and VCAM) on endothelial cells can lead to reduced immune cell infiltration and shift the balance of the infiltrated immune cell profile towards a more immune suppressive phenotype.
Hypoxia also modulates chemokine and cytokine production by CAFs, which increases immune cells associated with an immunosuppressive microenvironment. Data demonstrates that hypoxia increases CAF secretion of CXCL13 which promotes B cell recruitment in PCa. Increased B cell tumour infiltration is associated with progression to castration-resistant disease and neuroendocrine differentiation, both of which are associated with poor prognosis (193). In BCa, hypoxia-induced CAF secretion of CXCL12 suppresses the anti-tumour activity of T cells, DCs, NK cells and enhances the pro-tumour activity of Tregs, MDSCs and TAMs. Hypoxia up-regulates TGF-β secretion which up-regulates expression of PD-L1 and PD-L2 on CAFs. This promotes T cell exhaustion and immune evasion (259).
The tumour microenvironment and resistance to therapy
The ECM, CAFs and immune cells are all shown to be impacted by hypoxia, having an effect on cancer cell resistance to chemotherapy, radiotherapy and immunotherapy. One way in which hypoxia induces resistance is by modulating the TME. This phenomenon has attracted research interest for a long time, yet it still remains unsolved (260, 261).
Our understanding to date includes knowledge of the hypoxic TME inducing cell quiescence and causing resistance of tumours to cell-cycle specific drugs including alkylating agents (e.g cisplatin), antimetabolites (e.g gemcitabine), mitotic inhibitors (e.g paclitaxel) and cyclin-dependent kinase (CDK) inhibitors (e.g Palbociclib) (262–266). Hypoxia can also activate survival pathways including PI3K/AKT, MAPK and NFκB which induces resistance to apoptosis and DNA damage summarised by Rohwer and Cramer (267). Hypoxia is also a well reported factor contributing to poor response to radiotherapy (1, 268, 269). and suppression of immune response by activation of expression of immune checkpoint inhibitors. It also alters the composition and function of immune cells within the TME (213, 270).
Resistance to chemotherapy
Resistance to chemotherapy is controlled by multiple mechanisms, and these effects are often enhanced by hypoxia. Biomechanical and biophysical properties of the ECM of solid tumours often induce resistance to chemotherapy. Hydroxylation of collagen by P4HA1, P4HA2, PLOD1 and PLOD-2 mediates tissue stiffness and is regulated by HIF-1 (110). Hayashi et al. (271) highlight that increased tissue stiffness is associated with a poorer clinical complete response in BCa patients, relative to patients with a low tissue stiffness score (10% versus 38% respectively). This has also been shown in PaCa by Rice et al. (272), where matrix stiffness was correlated with chemoresistance to paclitaxel. Matrix stiffness has been proposed to enhance chemoresistance by inducing cell cycle arrest in the G0 phase. Physical signals induced by matrix stiffness are transduced to tumour cells via integrin receptors which have a role in mechano-transduction. This alters cell morphology, proliferative capacity and invasive ability of tumour cells. However, there is conflicting data on the exact role of matrix stiffness in therapeutic efficacy dependent on the tumour type. In metastatic CRC, increased ECM stiffness was regulated by highly activated metastases-associated fibroblasts which reduced the efficacy of bevacizumab. Addition of anti-RAS, a hypertension drug, to the regime enhanced response to bevacizumab by reducing stiffness of the ECM (273). Qin et al., highlight how intermediate matrix stiffness (38 kPa) induces resistance to doxorubicin measured by cell death rates (29.6% cell death) in BCa cells (MDA-MB-231), compared to low matrix stiffness (10 kPa- 48.5% cell death) and high matrix stiffness (57 kPa- 55.2% cell death). It is proposed that high expression of integrin-linked kinase (ILK) and translational coactivator, Yes-associated protein (YAP), within the BCa matrix is associated with resistance to doxorubicin (274). In contrast, MCF-7 BCa cells exposed to a rigid matrix stiffness (2710 kPa), the IC50 of cisplatin and taxol decreased significantly (p < 0.01), compared to the soft matrix stiffness (5.3 kPa) which showed more resistance. OC cells, SKOV3, were also shown to have enhanced survival within the soft matrix (0.5 kPa), compared to a stiffer substrate (25 kPa) following treatment with 1 μM cisplatin. This correlated with overexpression of multi-drug resistance proteins, ABCB1 and ABCB4, in cells grown on the soft gel matrix (275). Furthermore, in osteosarcoma cell lines, the IC50 value and viability of cells was significantly higher at 7kPa compared to the 55 kPa matrix (276). Bordeleau et al., have correlated increased matrix stiffness with disruption of vessel architecture and integrity and promotion of tumour-like vascular phenotype. Increased stiffness of collagen resulted in increased outgrowth of angiogenic sprouts from spheroids, and a 1.5-fold increase in branching, compared to the softer collagen gels. Furthermore, they demonstrated that MMPs played a key role in promoting increased angiogenesis which was found in stiffer matrices. This provides a potential explanation for reduced chemotherapeutic efficacy following increased matrix stiffness (277). Overall, these data demonstrate the contradictions in data, and that it is important to consider multiple components of the matrix including stiffness and its effects on vasculature. These data have all been demonstrated in a normoxic context however, we know hypoxia has an essential role in regulating matrix stiffness (110), and therefore is likely to modulate response to chemotherapy.
Another consequence of a hypoxic TME is its acidification, this is detrimental to resistance to therapies through a mechanism described as ‘ion trapping’ (278). The phenomenon of ‘ion trapping’ is described as weak bases isolated in acidic compartments, and weak acids sequestering into alkaline compartments. This has consequences for weak base drugs including anthracyclines and vinca alkaloids (279). Furthermore, Wachsberger et al. (280) showed that chronic exposure to acidic conditions activates heat-shock protein, HSP-27, inducing cisplatin resistance. Vukovic & Tannock (281)., observed that intracellular acidification arrested cells in G1 phase, making the cells more resistant to mitoxantrone, paclitaxel and topotecan in murine mammary carcinoma cells and urothelial cancer cell lines. These chemotherapeutics are all weak bases therefore, acidic conditions reduce the cytotoxicity of these drugs by inhibiting their uptake due to a larger proportion of the drug molecules becoming protonated, limiting diffusion to cells.
The cell cycle plays an important role in regulating cancer cell proliferation and apoptosis. The hypoxic TME induces cell cycle arrest or quiescence in cancer cells (260). This is particularly prevalent in the G1/S and G2/M checkpoints, which protect cells from DNA damage and genomic instability. Cells become “stuck” in these cell cycle phases making them less sensitive to chemotherapies that target rapidly dividing cells. Chronic hypoxia is associated with the induction of a quiescent state in cancer cells, where the cells are temporarily and reversibly arrested in G0 phase. This is associated with a more aggressive tumour phenotype (282). The G0 phase of the cell cycle is where cells are not actively dividing. These cells remain dormant until the cells are exposed to favourable conditions (283). Druker et al., reviewed the role of hypoxia and its control of the cell cycle (284) highlighting that identifying G0-arrested cells within tumours remains a challenge as there is a lack of easily measurable markers to measure this state (283). BCa cells exposed to chronic hypoxia (1% O2 for up to 7 days) were shown to enter G0/G1 cell cycle phase (282), and it is established that this phase of the cell cycle is resistant to cytotoxic chemotherapies (285).
p16INK4A has been related to inducing cell senescence, and inhibition of pRb phosphorylation through cyclinD/CDK4 (286). Box et al. (287) measured cell cycle arrest genes in multiple cancer cell lines and primary fibroblasts. They found that different cell types had different cell cycle arrest profiles in response to hypoxia. BCa cells (HTB-30), CC cells (HeLa) and human mammary epithelial cells all showed induction of G1/S arrest after initial exposure to hypoxia. However, a HCC cell line (Hep3B) lacked observable G1/S arrest in hypoxia conditions. Although all cell lines showed reduced proliferation at 24 h. Loss of multiple cyclin-dependent kinase inhibitors (CDKI) was found in the cell lines including p16, p21 and p27. Methylation of P16 has been associated with reduced sensitivity to paclitaxel in patients with advanced NSCLC (288). Therefore, regulation of CDKI by hypoxia plays a role in resistance to chemotherapeutics. Yano et al. (285) highlighted that 90% of cancer cells within the centre of tumours are in G0/G1 phase following implantation of MKN45 metastatic stomach adenocarcinoma cells in nude mice. Furthermore, 75% of cancer cells located >100 µm from tumour blood vessels are also in this phase. Therefore, most drugs currently used in clinics are ineffective in solid tumours as they target cancer cells in S/G2/M phases.
Dysregulation of DNA repair pathways is associated with initiation and progression of cancers. Hypoxia increases DDR proteins, decreasing homologous directed repair by downregulating BRCA1, BRCA2 and RAD51, decreasing mismatch repair proteins MLH1, MSH2, MSH3 (289) and downregulating base excision repair factors including, APE1, OGG1 and MYH (290). With these mechanisms active within hypoxic tumours treated with chemotherapies, it can be difficult to effectively treat solid tumours. PARP inhibitors have shown promise in overcoming dysregulation of DNA repair pathways (291), Shelton et al. (292) showed in vitro and in vivo enhancement of the effects of 5-FU, irinotecan or oxaliplatin and radiation with PARP inhibitor, ABT-888 in CRC cells. However, hypoxia has been associated with reduced efficacy of PARP inhibitors in multiple cancer cell lines (293). Dysregulation of DNA repair pathways can have significant implications for patients, which leads to genomic instabilities and accumulation of mutations. Understanding DNA repair deficiencies in tumours can aid in treatment decisions including the use of PARP inhibitors. Other targets such as DNA-dependent protein kinase inhibitors and Rad51 inhibitors are currently being explored preclinically. Prognostic biomarkers can indicate whether a patient is suitable for these treatments by considering mutations in BRCA1/BRCA2 in BCa which indicates a more aggressive phenotype. Identifying these mutations can inform clinicians that patients will be suitable for treatment with PARP inhibitors (294). Weil et al., show that dependent on the DNA repair pathway defects, breast and ovarian tumours can be more sensitive with platinum-based drugs or PARP inhibitors (295). This demonstrates that identifying patients with DNA repair defects can guide treatment decisions. In BRCA deficient cells, PARP inhibition is 3 times more effective than cisplatin. Lou et al., highlight the benefits of profiling patients with DDR pathway profiling. Patients with a low DDR score did not benefit from adjuvant chemotherapy with anti-PD1 (296).
Many chemotherapies induce apoptosis of cells however, resistance to chemotherapy is characterised by a reduction in apoptosis. The B-cell lymphoma-2 protein (Bcl-2) family of proteins are one of the main regulators of the intrinsic apoptosis pathway, and in chemoresistance upregulation of Bcl-2 proteins is observed which offsets the pro-apoptotic proteins. Hypoxia has been indicated as a key regulator Bcl-2 proteins, downregulating pro-apoptotic proteins and upregulating anti-apoptotic proteins in HCC and lung cancer cell lines (297). Treatment of OC cell lines with cisplatin was associated with upregulation of Bcl-xL and subsequent chemoresistance (298). Additionally, dysregulation of inhibitors of apoptosis (IAPs) have been correlated with chemoresistance in multiple cancers. IAP overexpression of cIAP1 in oesophageal squamous carcinoma correlated with resistance to cisplatin and camptothecin (299). Overexpression of ML-IAP (Livin) correlated with resistance to etoposide, vincristine, 5-FU in CRC cells (300). In primary cells derived from melanoma patients, Nachmias et al. (301) also showed resistance to etoposide correlating with increased expression of the gene. These data were associated with patient clinical response.
Our knowledge on the dysregulation of apoptosis in solid tumours has led to the exploration of novel drug compounds that increase apoptosis (Table 4). There are multiple molecules within the apoptotic pathway that can be targeted; agonists to TRAIL and SMAC mimetics have been developed. Birinapant has been tested in patient-derived xenograft models of OC and CRC and melanoma showing growth inhibition following intraperitoneal administration (30mg/kg) (302). Tolinapant (ASTX660) is another SMAC mimetic and IAP antagonist which has shown success preclinically in HNSCC, BLCA and CRC (303). In H&N cancer, tolinapant enhanced radiation-induced immunogenic cell death in syngeneic mouse models. An in vitro BLCA study showed that tolinapant induced necroptosis. It was proposed that this mechanism could help overcome resistance to cisplatin in BLCA (303). Crawford et al. (304) show that there may be clinical benefit of combining tolinapant with FOLFOX chemotherapy in microsatellite stable CRC with elevated cIAP1 and cIAP2.
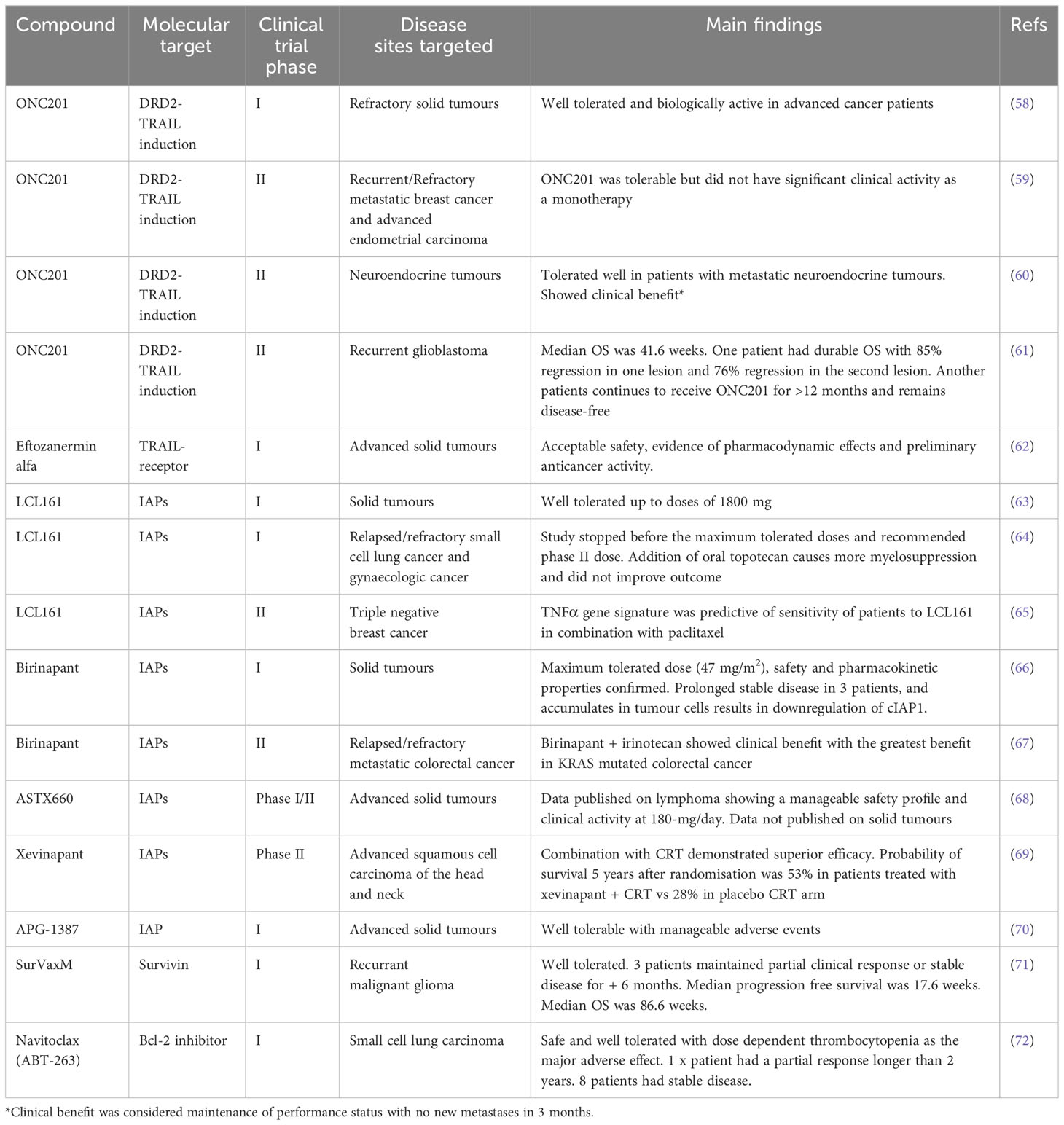
Table 4 Clinical trials targeting different apoptotic pathways within solid tumours.
Myeloid cell leukemia-1 (Mcl-1) is a member of the anti-apoptotic Bcl-2 family, and has been identified as an apoptotic survival factor in TNBC. Mcl-1 is commonly amplified in 56% of TNBC tumours and its overexpression associated with poor clinical prognosis (305). This was one the studies that led to the investigation of Mcl-1 as a target for patients with poor prognosis. Pre-clinical models in PaCa, BCa, lung cancer and OC cell lines showed Mcl-1 inhibition in vivo and in vitro (306–308). Both preclinical studies and clinical trials show that modulation of apoptosis is a valuable target in tumours with dysregulated apoptosis, and offers a strategy for overcoming chemoresistance.
Hypoxia upregulates expression of drug efflux pumps and confers with resistance to chemotherapy. Multidrug resistance protein 1 (MRP1), multidrug resistance-associated protein 1 (MRAP1) and breast cancer resistance protein (BRCP) are all regulated by HIF-1α in CRC (309) and HIF-2α in OC (310). Additionally, reduced drug uptake due to poor blood supply is characteristic of hypoxic tumours (311). There are limited studies that correlate clinical data with chemotherapy response. One of the proposed reasons for limited knowledge on MDR1/P-gp expression is poor sensitivity and specificity and difficulty in quantifying levels of protein by immunohistochemistry, and normal tissue contamination (312). Furthermore, clinical trials targeting drug efflux pumps show little promise of improving patient outcome. Despite attempts to target efflux pumps that are upregulated in cancer cell lines, there is limited efficacy in the drugs developed. Mohelnikova-Duchonova et al. (313) show that in PaCa, multiple ABC transporters are upregulated compared to normal tissue. This highlights that tumours are likely to adapt to alternative mechanisms of resistance if single efflux pumps are targeted (313). In contrast, A phase I trial combining tyrosine kinase inhibitor, pazopanib, with topotecan which is an approved treatment for SCLC, CC and metastatic OC showed a 1.7-fold increase in patient exposure compared to topotecan treatment alone. Pazopanib was trialled due to its mild affinity for Pgp/ABCB1 and high affinity for BCRP/ABCG2 (314).
Resistance to radiotherapy
Approximately 50% of all cancer patients undergo radiotherapy as part of their treatment, 60% with curative intent (315). Radiotherapy uses high-energy radiation to target and destroy cancer cells. There are two main ways of delivering radiotherapy to patients; external beam radiation which uses an external source of radiation to target tumours, or internal radiation therapy including brachytherapy which uses radioactive sources including seeds, wires or pellets that are placed directly inside or near the tumour (316). However, it was discovered by Gray et al. (317) that tumour cells are less damaged by a dose of X- or γ-radiation than oxygenated cells versus anoxia at the time of irradiation.
Other forms of radiotherapy include protons, electrons and carbon ions. Proton beam therapy delivers a higher dose of radiation than conventional radiation using photons whilst sparing the surrounding tissue. However, it is only used in a subset of patients, particularly children and patients with complex brain cancer, H&N cancers and sarcomas in the UK. Treatment is limited to these tumour types as proton beam therapy requires a high degree of accuracy and precision which means that tumours that change shape or move due to close proximity to organs of the autonomic system, are unsuitable for this therapy. It is beneficial to use proton beam therapy in children as its localised effects reduce late-stage toxicity (318). Dose painting can be used to target more hypoxic regions of tumours with a higher dose of protons, to try and overcome the potential of radio-resistance and induce permanent damage to DNA of cancer cells (319).
Oxygen is an essential factor that influences response to radiotherapy in solid tumours. Gray et al. (317) showed that when partial pressure of oxygen is below 20mmHg at the time of irradiation, the cells become resistant to radiation damage. When tumours are targeted with radiation, they absorb the radiation and produce highly reactive free radicals either directly or indirectly. The radicals produced are unstable and highly reactive with oxygen, which induces damage to the target tissue. Radiation induces DNA damage by producing ROS and inducing apoptosis. Therefore, presence of oxygen in tumours enhances the effect of radiation by increasing ROS production and inhibiting DNA repair. To produce the same effect in hypoxic conditions, 2.5-3.0-fold increase in radiation is required (320). Horsman et al. (75) review and summarise the clinical impact of hypoxia in patient outcome following radiotherapy (321–323). As previously described, solid tumours have varying degrees of tissue oxygenation that fluctuates overtime. Therefore, when radiotherapy is delivered during periods of reduced oxygen, its effectiveness is reduced (324). Thiruthaneeswaran et al. (325) summarise the role of hypoxia on radiotherapy and the challenges that remain within the field that need to be overcome to offer patients the best clinical outcome.
Understanding the hypoxic TME is crucial for optimizing treatment outcomes. Hypoxia alters radiation effectiveness, necessitating higher doses for hypoxic tumours, yet this can lead to increased side effects (326). Approaches including altered fractionation, dose escalation, and high linear energy transfer (high-LET) radiation help combat hypoxia’s challenges. To improve efficacy, radio-sensitizing agents like nitroimidazoles or carbogen can be employed. Wang et al. stress the need for precise dose guidelines in clinical practice. Overall, tailoring treatment plans to individual tumour biology remains key for achieving optimal outcomes (327).
When discussing radiotherapy, we often refer to the 6R’s of radiotherapy which are of mechanisms that are important in determining the response of biological tissue to multiple doses of radiation. These include repair, redistribution, repopulation, reoxygenation, radiosensitivity and reactivation of the immune system (328–330). Hypoxia plays a role in dysregulating the 6R’s and impacting patient response to treatment.
Disruption of the 6 R’s by hypoxia has been discussed by Rakotomalala et al. (50). Wozny et al. (331) show that hypoxia (1% O2) increases non-homologous end-joining (NHEJ) and increases radio-resistance in HNSCC. Hypoxia disrupts the capacity of cells to repair radiation-induced DNA making hypoxic cells less susceptible to damage by radiation. Primary human fibroblasts exposed to chronic hypoxia (0.2% O2) were shown to have defective repair of DNA double-strand breaks (DSBs) following irradiation, compared to cells grown in normoxic conditions. It was proposed that unrepaired DNA-DSBs drive genetic rearrangement and genomic instability under hypoxic conditions. Genomic instability can increase metastatic capacity of tumours. The authors propose that understanding the DNA-DSB repair defects regulated by hypoxia in tumours could improve the treatment modalities used for cancer (332). Exposing H1299 lung carcinoma cells to chronic hypoxia (72 h 0.2% O2) downregulated homologous recombination (HR) proteins which increased sensitivity to DNA cross-linking agents and increased radiosensitivity compared to acutely hypoxic cells (6h 0.2% O2) and anoxic cells. Therefore, chronically hypoxic cells that are repair deficient may be a novel target to selectively kill hypoxic cells (333). BCa cells cultured in 3% O2, showed reduced expression of RAD50, RAD51, BRCA1 and BRCA2 which are essential for HRR. The same genes were downregulated in PCa with the addition of RAD54, and NHEJ genes (Ku70, LIG4 and XRCC4) when cultured in 0.2% O2 (334, 335).
Redistribution describes alterations to the cell cycle following radiotherapy. Hypoxic cells enter arrest in G1/S and G2/M phases making cells less sensitive to radiation, hindering the redistribution of cells to more sensitive cell cycle phases. Enhanced expression of HIF-1α correlates with upregulation of p21 and CDKI-1 phosphorylation in PCa. Luo et al. (336) showed that this correlates with radioresistance both in vitro and in vivo which maintains PCa cells in G0/G1 or S phase.
Repopulation following radiotherapy describes the proliferation of cancer cells that have survived irradiation. Hypoxic conditions often drive rapid proliferation and therefore the accelerated growth reduces the efficacy of radiotherapy. Cancer stem cells play an important role in recurrence of tumours following radiotherapy. Luo et al. (336) showed that WNT/β-catenin signalling is responsible for progression of PCa tumours following radiotherapy which is driven by HIF-1α and is responsible for repopulation. In gastric cancer, hypoxia has been shown to increase KDM4B (lysine demethylase) which induces cyclin A1 expression following irradiation. This enhances cancer cell proliferation following treatment (337).
Reoxygenation of tumour cells occurs between radiotherapy fractions. Reoxygenation describes when death of oxygenated cells within normoxic regions decreases the oxygen consumption within these areas which enables molecular oxygen to diffuse to hypoxic regions that are located 70-100 micrometres from functional blood vessels. More hypoxic tumours with disrupted vasculature struggle to reoxygenate and therefore make the cells more resistant to radiotherapy (Figure 4). Adding treatments to a patients plan which enhances oxygen delivery can help overcome this mechanism. Moeller et al. (338) show that through a poorly understood mechanism, tumours exposed to radiation release cytokines that inhibit apoptosis of endothelial cells. This is regulated by HIF-1, and enhances oxygen delivery to tumour cells that survive initial irradiation. In a CC model, tumour cells rapidly repopulated following irradiation as the decreased tumour bulk caused by the initial dose, reduced tumour mass and created a favourable growth environment. This was mediated by Akt/mTOR dependent mechanisms, which activated HIF-1 intra-tumoural activity (339). HIF-1 stabilisation in these tumours further enhances radioresistance. Increased lactate levels induced by metabolic switch in HNSCC and CC are associated with reduced sensitivity to radiotherapy (340, 341). Autophagy has been attributed to a cytoprotective role, protecting cells from damage including radiation. This has been shown in osteosarcoma, BCa and rectal cancers (342, 343).
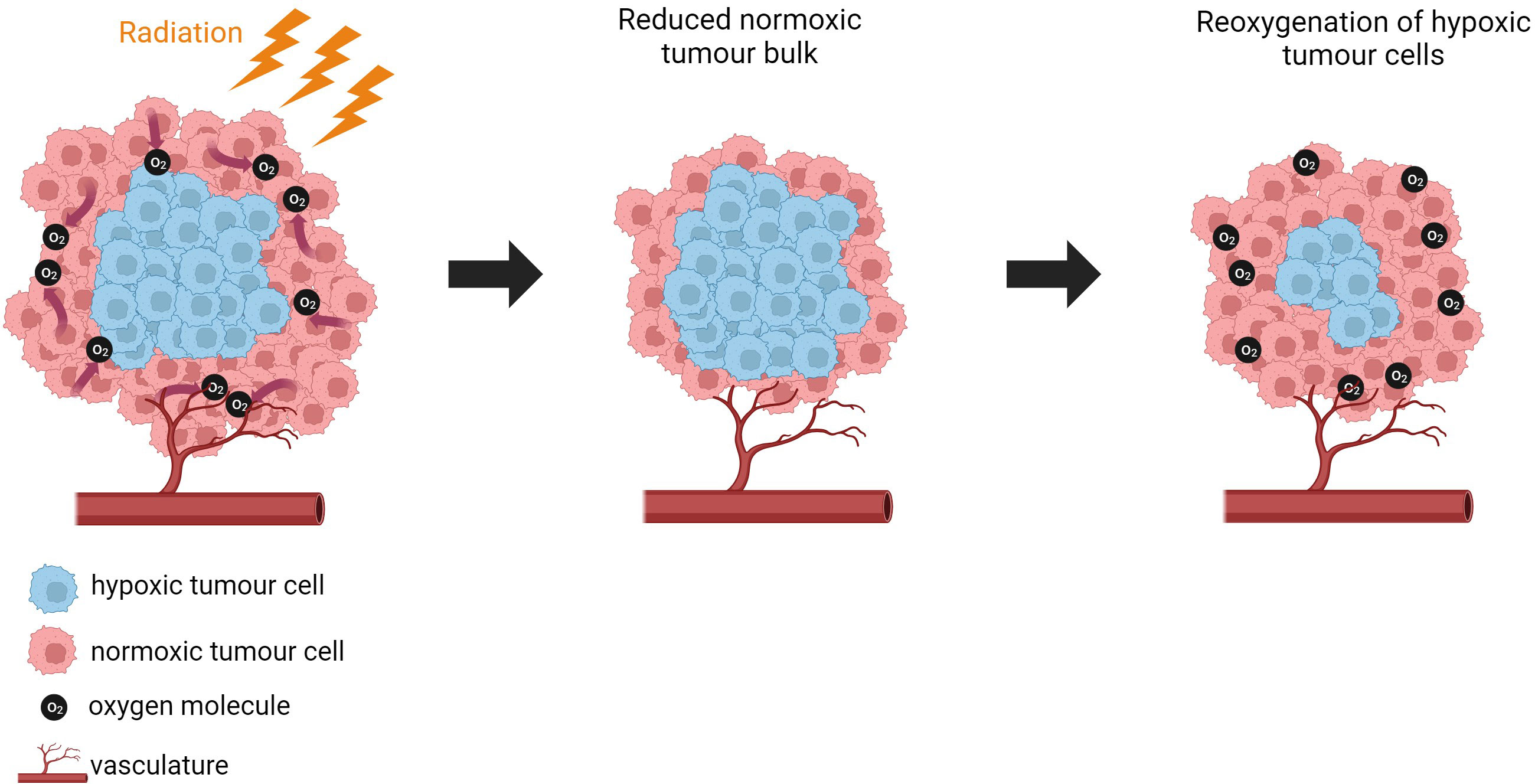
Figure 4 Tumour reoxygenation. Normoxic tumour cells are killed by irradiation which induces a reduction in tumour bulk. Oxygen consumption is reduced by normoxic cells which enables oxygen to diffuse to hypoxic regions. This induces vessel regrowth and reoxygenation of hypoxic cells. The reoxygenated cells are more sensitive for reirradiation. Created with BioRender.com.
The mechanisms of apoptosis activated during cell death can impede cancer cell radiosensitivity. Hypoxia directly modulates the cancer cell apoptotic response, which reduces cancer cells radiosensitivity. Cuisnier et al. (344) showed that chronic hypoxia (3% or 5% O2) led to overexpression of Bcl-2 in CC cells (KB-3-1). Overexpression of Bcl-2 inhibited radiation induced apoptosis by inhibiting ROS production. Furthermore, expression of the pro-apoptotic molecule, Bax, was reduced with no translocation of the gene in the mitochondria. Decreased Bax expression mediated by HIF-1 was also shown by Bamodu et al. (345) in HCC which correlated with a radiation-resistant phenotype. This mechanism driven by PDK1 which is a HIF-1α target gene which drives PI3K/AKT/mTOR pathway, inducing radiotherapy resistance. Rakotomalala et al. (50) summarise further mechanisms of resistance to cell death.
Hypoxia and immune cell regulation play a role in the anti-tumour microenvironment therefore, the ‘6th R’ was identified by Boustani et al. (328). It describes the reactivation of the anti-tumour immune response following irradiation. Radiotherapy induced tumour cell death releases tumour-associated antigens, activates DCs and produces cytokines and chemokines that stimulate the priming and infiltration of T cells into the tumour. However, within a hypoxic TME, the immunogenicity of tumour cells reduces by increasing immunosuppressive cells. There are currently ongoing trials combing radiotherapy with immunotherapy (328). More recently, a ‘7th R of Radiobiology’ has been described by Taghizadeh-Hesery (346) as ‘reinforcement’ by the TME. This describes that radiation cancer cell response can be altered by both cellular and noncellular components that surround the tumour, and this can be regulated by hypoxia (346).
Resistance to immunotherapy
As researchers have gained more insight into the crucial role of anti-tumour immunity and hypoxia in patient prognosis, immunotherapies have been introduced as a therapeutic intervention for some solid tumours (270, 347). HIF-1α upregulates PD-L1 on both tumour and stromal cells, whilst its receptors PD-1, CTLA-4 and LAG3 are all expressed on immune cells. Overexpression of PD-L1 is associated with resistance to immunotherapies in hypoxic melanoma, CRC and glioma. Combining metformin, which reduces oxygen consumption with anti-PD-1 therapy in melanoma and CRC showed improved efficacy in hypoxic tumours. Inhibiting HIF-1α in glioma reduced PD-L1 expression and enhanced immunotherapy efficacy (348, 349). Furthermore, retrospective analysis of HNSCC patient samples treated with immunotherapy either as front-line therapy or following platinum failure found that patients, with higher %CA9/mean intensity (%CA9/I) associated with more hypoxic tumours, had reduced efficacy of anti-PD-1 therapy. Metastatic/recurrent patients with a lower % CA9 were associated with improved OS, low CA9/I was associated with a 12-month OS rate of 51.3% versus 14.1% in patients with high CA9/I. Patients with low hypoxia and high CD8 had better efficacy with anti-PD-1 (350). Upregulation of HIF-1α/CXCL12 correlates with higher PD-L1 expression in HCC, and therefore worse prognosis in these patients (351).
Pseudohypoxia and cancer
Oxygen-dependent HIF activation has been extensively covered in this review, but it is important to consider oxygen-independent HIF activation leading to pseudohypoxia. Interest is growing in this concept as it may explain the persistence of the Warburg effect in cancers where even non-hypoxic cells preferentially use glycolysis for ATP production (271). A number of gene mutations have been associated with this phenomenon. These gene mutations have led to the development of compounds for clinical trials and approval of antagonists for multiple solid tumours, as one of the implications of pseudohypoxia is resistance to conventional therapies (352, 353).
Von Hippel-Lindau
Mutations in the VHL gene result in the loss of the Von Hippel-Lindau tumour suppressor protein (pVHL), which is responsible for targeting HIF for degradation. Without functional pVHL, HIF remains stabilized, contributing to the pseudohypoxic state and the development of various tumours (354). In the case of ccRCC, this results in a highly angiogenic tumour due to overproduction of hypoxia inducible VEGFA mRNA. Initial VHL inactivation in RCC induces expression and accumulation of both HIF-1α and HIF-2α however, HIF-2α expression becomes dominantly expressed in chronic hypoxia and supresses HIF-1α protein (355). This promotes oncogenic potential by driving tumour progression and metastasis through activation of hypoxia-sensitive signalling pathways and overexpression of HIF-2α target genes (352). The understanding of the biology underpinning this phenomenon has produced early positive results using a HIF-2α antagonist to treat ccRCC, resulting in approval by the U.S. Food and Drug Administration (FDA) (353).
Mouse double minute 2 homolog (MDM2)
Data from Retinoblastoma and Myelodysplastic syndromes (MDS) implicate MDM2 in pseudohypoxia. Zhang et al., have shown that MDM2 promotes cell survival in retinoblastoma through regulating both pVHL and HIF-1α resulting in HIF stabilisation (356). Studies in MDS suggest that TP53 loss of function mutations can affect oxygen-independent HIF degradation by MDM2. This results in accumulation of HIF-1α and pseudohypoxia (357). These data suggest that novel agents such as MDM2 inhibitors could have a role in the treatment of cancers with a number of clinical trials under way (358).
Other genetic factors
Pseudohypoxia can also be driven by genetic mutations or alterations in other genes involved in the oxygen-sensing and response pathways (359). Mutations in genes associated with angiogenesis such HIF-dependent neovascularisation through VEGF or through anti-apoptotic proteins such as IAP-2 can result in activation of hypoxia-associated pathways (360). Understanding the molecular basis of oxygen-independent pseudohypoxia will lead to novel target and biomarker discovery and potential for improved patient outcomes.
Clinical trials
This review has defined the biological importance of tumour hypoxia with its ubiquitous effects. The clinical importance of hypoxic regions within solid tumors has been known since the early 20th century. Data from numerous studies reveal the prevalence of hypoxia in various types of human tumours, though there is significant variability among individual cases.
Over the past four decades, controlled clinical trials have demonstrated that radiation resistance due to tumour hypoxia can be mitigated by interventions like normobaric or hyperbaric oxygen therapy and the use of nitroimidazoles as hypoxic radiation sensitizers. More recently, hypoxic cytotoxins, drugs that selectively target cells in hypoxic environments, have gained attention.
As far back as 2008, a systematic review involving 10,108 patients across 86 randomized trials aimed to modify tumour hypoxia in patients receiving primary radiation therapy found that hypoxic modification significantly improved the efficacy of radiotherapy (361). Hypoxic modification resulted in better LRC and an associated improvement in OS. However, the incidence of distant metastases and radiation-related complications did not show significant changes. There are only two solid tumours in which hypoxia modification is standard of care. HNSCC where the ARCON Trial and DAHANCA 5 trial confirmed that hypoxia modification improved LRC (97, 323). In the ARCON trial, LRC improved by 80% in patients with T3 and T4 laryngeal cancer which aided in organ preservation (97). Within the DAHANCA-5 trial, LRC increased to 49% with addition of nimarazole versus 33% in the placebo group (323). Since DAHANCA-5 randomised patients between radiotherapy alone and radiotherapy with nimorazole, nimorazole is used as a standard of care in Scandanavia. The BCON trial confirmed that radiotherapy with carbogen and nicotinamide was superior to radiotherapy alone for muscle-invasive BLCA with a 13% absolute improvement in OS (362). Updated 10-year outcomes show that benefit is maintained especially if patients are stratified by hypoxia biomarkers such as necrosis or a 24-gene transcriptomic signature (363).
More recently, Bourigault et al., have shown that the antimalarial drug atovaquone can reduce tumour hypoxia detected using hypoxia PET in NSCLC patients. This is a small promising phase II study which poses the challenge of how investigators pursue clinical trials without industrial support (364).
Pseudohypoxia may prove to be an area of greater interest with industry already pursuing clinical trials with a novel molecular agent targeting HIF-2α approved by the FDA based on data from a small phase II study while the phase III trial is awaited (353).
Despite the substantial evidence supporting the benefits of hypoxic modification, its implementation in clinical practice remains limited. There are many reasons why this is including prejudice, lack of familiarity and funding structures (365).
Conclusions and future perspectives
Hypoxia is a key regulator of the TME and plays a role in regulating hallmarks of cancer (366), mediating chemotherapy, radiotherapy and immunotherapy resistance. This review has highlighted how hypoxia regulates multiple pro-tumorigenic pathways through HIF to induce changes in the ECM, activate CAFs to enhance tumorigenesis and promote an immunosuppressive microenvironment. Additionally, the role of hypoxia in inducing changes to tumour cell metabolism was also discussed showing its role in resistance to therapies due to acidification of the TME (83).
Whilst there is strong evidence supporting the role of hypoxia in enhancing tumorigenesis and its role in complicating treatment in a clinical setting, there are still gaps in knowledge. It is important to be able to decipher the complexity of the TME including clarifying the contradictions in ECM data, and how hypoxia interacts with CAFs and their specific influence on both the ECM and TME. Many in vitro models used to study hypoxia often rely on 2D models of a single cell type which do not take into consideration the whole TME (367). Therefore, using new models able to recapitulate the hypoxic TME in 3D with multiple cell types may drive research further. This can provide novel insights into how the mechanosensory interactions influence resistance to therapies for example through matrix stiffness, and the specific cell types that can be targeted to evade this mechanism of resistance (354, 368). Representation of cycling hypoxia which is more physiologically relevant than acute or chronic hypoxia may be beneficial (3). Understanding the specific roles of CAFs within the TME is critical as there are multiple subtypes with differing roles. Until this is fully clarified, it will be difficult to develop targeted strategies
Further elucidation of the role of HIF-3 may provide important insights into HIF biology, and new mechanisms of therapeutic targets (369). Additionally, continued research into pseudohypoxia could provide a novel strategy to target tumours.
Hypoxia research has enabled the development of hypoxia-specific treatments that have shown some clinical success (97, 323, 362). As summarised by Hoskin (365), the problem lies with the lack of clinical implementation despite significant evidence supporting benefits of hypoxia modification. Development of hypoxic biomarkers is still of interest and perhaps indicates that there is a future for hypoxia-targeted therapies in clinics with better methods of stratifying patients. Some limitations with current biomarkers that have been developed is that they use platforms that are not clinically applicable. Furthermore, many of the biomarkers are not validated in prospective clinical trials. Understanding the best format for analysing hypoxia gene/protein signatures in tissue (fresh frozen/FFPE), and using platforms that are clinically available and affordable could help overcome this.
The complex hypoxic TME provides an opportunity for identifying novel therapeutic targets. However, as we have learnt from treatments already implemented in clinic, for example anti-PD-1 therapies, there are often mechanisms that induce resistance (351). Understanding the complex interactions between the different cell types within the hypoxic TME, and the differing expression patterns regulated by hypoxia could help develop more targeted treatments. Developing biomarkers using platforms that we know are clinically accessible can overcome the challenge of clinical implementation.
Author contributions
KB: Writing – original draft, Writing – review & editing. CQ: Writing – review & editing. SL: Writing – review & editing. DS: Writing – review & editing. MK: Writing – review & editing. ET: Writing – review & editing. CW: Writing – review & editing. PH: Writing – review & editing. AC: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. KB was supported by Cancer Research UK RadNet Manchester [C1994/A28701]. CQ was supported by Prostate Cancer UK [MA-CT21-005]. SL was supported by the Cancer Research UK Manchester Centre award [CTRQQR-2021\100010]. CW, PH and AC was supported by NIHR Manchester Biomedical Research Centre.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer (1955) 9(4):539–49. doi: 10.1038/bjc.1955.55
2. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83. doi: 10.2147/HP.S93413
3. Saxena K, Jolly MK. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules (2019) 9(8):339. doi: 10.3390/biom9080339
4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
5. Schwarz G. Uber Desensibilisierung gegen Rontgen- llnd Radiumstrahlen. Munchener Medizinische Wochenschrift (1909) 56:1217–8.
6. Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: New opportunities and challenges. Cells. (2019) 8(9):1083. doi: 10.3390/cells8091083
7. Movsas B, Chapman JD, Hanlon AL, Horwitz EM, Pinover WH, Greenberg RE, et al. Hypoxia in human prostate carcinoma: an Eppendorf PO2 study. Am J Clin Oncol (2001) 24(5):458–61. doi: 10.1097/00000421-200110000-00009
8. Movsas B, Chapman JD, Horwitz EM, Pinover WH, Greenberg RE, Hanlon AL, et al. Hypoxic regions exist in human prostate carcinoma. Urology (1999) 53(1):11–8. doi: 10.1016/S0090-4295(98)00500-7
9. Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat OncologyBiologyPhysics (2000) 48(4):919–22. doi: 10.1016/S0360-3016(00)00803-8
10. Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal (2007) 9(8):1221–36. doi: 10.1089/ars.2007.1628
11. Cuppen E, Elemento O, Rosenquist R, Nikic S, IJzerman M, Zaleski ID, et al. Implementation of whole-genome and transcriptome sequencing into clinical cancer care. JCO Precis Oncol (2022) 6:1–11. doi: 10.1200/PO.22.00245
12. Falk SJ, Ward R, Bleehen NM. The influence of carbogen breathing on tumour tissue oxygenation in man evaluated by computerised p02 histography. Br J Cancer (1992) 66(5):919–24. doi: 10.1038/bjc.1992.386
13. Le QT, Chen E, Salim A, Cao H, Kong CS, Whyte R, et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non–small cell lung cancers. Clin Cancer Res (2006) 12(5):1507–14. doi: 10.1158/1078-0432.CCR-05-2049
14. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br J Radiol (2014) 87(1035):20130676. doi: 10.1259/bjr.20130676
15. Lawrentschuk N, Poon AMT, Foo SS, Putra LGJ, Murone C, Davis ID, et al. Assessing regional hypoxia in human renal tumours using 18F-fluoromisonidazole positron emission tomography. BJU Int (2005) 96(4):540–6. doi: 10.1111/j.1464-410X.2005.05681.x
16. Höckel M, Schlenger K, Knoop C, Vaupel P. Oxygenation of carcinomas of the uterine cervix: Evaluation by computerized O2 tension measurements. Cancer Res (1991) 51(22):6098–102.
17. Vaupel P, Mayer A, Briest S, Höckel M. Oxygenation gain factor: A novel parameter characterizing the association between hemoglobin level and the oxygenation status of breast cancers. Cancer Res (2003) 63(22):7634–7.
18. Lartigau E, Le Ridant AM, Lambin P, Weeger P, Martin L, Sigal R, et al. Oxygenation of head and neck tumors. Cancer (1993) 71(7):2319–25. doi: 10.1002/1097-0142(19930401)71:7<2319::AID-CNCR2820710724>3.0.CO;2-C
19. Lartigau E, Lusinchi A, Weeger P, Wibault P, Luboinski B, Eschwege F, et al. Variations in tumour oxygen tension (pO2) during accelerated radiotherapy of head and neck carcinoma. Eur J Cancer (1998) 34(6):856–61. doi: 10.1016/S0959-8049(97)10172-1
20. Nordsmark M, Bentzen SM, Overgaard J. Measurement of Human Tumour Oxygenation Status by a Polarographic Needle Electrode: An analysis of inter- and intratumour heterogeneity. Acta Oncol (Madr). (1994) 33(4):383–9. doi: 10.3109/02841869409098433
21. Becker A, Hänsgen G, Bloching M, Weigel C, Lautenschläger C, Dunst J. Oxygenation of squamous cell carcinoma of the head and neck: comparison of primary tumors, neck node metastases, and normal tissue. Int J Radiat OncologyBiologyPhysics. (1998) 42(1):35–41. doi: 10.1016/S0360-3016(98)00182-5
22. Le QT, Kovacs MS, Dorie MJ, Koong A, Terris DJ, Pinto HA, et al. Comparison of the comet assay and the oxygen microelectrode for measuring tumor oxygenation in head-and-neck cancer patients. Int J Radiat OncologyBiologyPhysics. (2003) 56(2):375–83. doi: 10.1016/S0360-3016(02)04503-0
23. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep (2015) 10(8):1335–48. doi: 10.1016/j.celrep.2015.02.006
24. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi: 10.1158/0008-5472.CAN-10-1439
25. Sowter HM, Raval R, Moore J, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1α versus hif-2α in regulation of the transcriptional response to hypoxia1. Cancer Res (2003) 63(19):6130–4.
26. Carroll VA, Ashcroft M. Role of Hypoxia-Inducible Factor (HIF)-1α versus HIF-2α in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-Like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res (2006) 66(12):6264–70. doi: 10.1158/0008-5472.CAN-05-2519
27. Schito L, Rey S, Tafani M, Zhang H, Wong CCL, Russo A, et al. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor β promotes lymphatic metastasis of hypoxic breast cancer cells. Proc Natl Acad Sci (2012) 109(40):E2707–16. doi: 10.1073/pnas.1214019109
28. Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W, et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol (2021) 14(1):14. doi: 10.1186/s13045-020-01030-w
29. Brooks DLP, Schwab LP, Krutilina R, Parke DN, Sethuraman A, Hoogewijs D, et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol Cancer (2016) 15(1):26. doi: 10.1186/s12943-016-0510-x
30. Koike T, Kimura N, Miyazaki K, Yabuta T, Kumamoto K, Takenoshita S, et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc Natl Acad Sci (2004) 101(21):8132–7. doi: 10.1073/pnas.0402088101
31. Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res (2003) 63(5):1138–43.
32. Choi SB, Park JB, Song TJ, Choi SY. Molecular mechanism of HIF-1-independent VEGF expression in a hepatocellular carcinoma cell line. Int J Mol Med (2011) 28(3):449–54. doi: 10.3892/ijmm.2011.719
33. Zhu G, Tang Y, Geng N, Zheng M, Jiang J, Li L, et al. HIF-α/MIF and NF-κB/IL-6 axes contribute to the recruitment of CD11b+Gr-1+ Myeloid cells in hypoxic microenvironment of HNSCC. Neoplasia (2014) 16(2):168–W21. doi: 10.1593/neo.132034
34. Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, et al. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell (2004) 6(5):485–95. doi: 10.1016/j.ccr.2004.09.026
35. Walmsley SR, Cadwallader KA, Chilvers ER. The role of HIF-1α in myeloid cell inflammation. Trends Immunol (2005) 26(8):434–9. doi: 10.1016/j.it.2005.06.007
36. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J Exp Med (2005) 201(1):105–15. doi: 10.1084/jem.20040624
37. Petrella BL, Lohi J, Brinckerhoff CE. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel–Lindau renal cell carcinoma. Oncogene (2005) 24(6):1043–52. doi: 10.1038/sj.onc.1208305
38. Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, et al. Endothelial deletion of hypoxia-inducible factor–2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood (2009) 114(2):469–77. doi: 10.1182/blood-2008-12-193581
39. Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1α and HIF-2α Differentially regulate metastatic success. Cancer Cell (2012) 21(1):52–65. doi: 10.1016/j.ccr.2011.11.017
40. Thompson AAR, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood (2014) 123(3):366–76. doi: 10.1182/blood-2013-05-500207
41. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest (2010) 120(8):2699–714. doi: 10.1172/JCI39506
42. Jaskiewicz M, Moszynska A, Serocki M, Króliczewski J, Bartoszewska S, Collawn JF, et al. Hypoxia-inducible factor (HIF)-3a2 serves as an endothelial cell fate executor during chronic hypoxia. EXCLI J (2022) 21:454–69. doi: 10.17179/excli2021-4622
43. Tanaka T, Wiesener M, Bernhardt W, Eckardt KU, Warnecke C. The human HIF (hypoxia-inducible factor)-3alpha gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem J (2009) 424(1):143–51. doi: 10.1042/BJ20090120
44. Zhou X, Guo X, Chen M, Xie C, Jiang J. HIF-3α promotes metastatic phenotypes in pancreatic cancer by transcriptional regulation of the RhoC-ROCK1 signaling pathway. Mol Cancer Res (2018) 16(1):124–34. doi: 10.1158/1541-7786.MCR-17-0256
45. Maynard MA, Evans AJ, Hosomi T, Hara S, Jewett MA, Ohh M. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB journal : Off Publ Fed Am Societies Exp Biol (2005) 19(11):1396–406. doi: 10.1096/fj.05-3788com
46. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol (2020) 21(5):268–83. doi: 10.1038/s41580-020-0227-y
47. Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the α Subunit *. J Biol Chem (1997) 272(17):11205–14. doi: 10.1074/jbc.272.17.11205
48. Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors — Similar but not identical. Mol Cells (2010) 29(5):435–42. doi: 10.1007/s10059-010-0067-2
49. Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ (2008) 15(4):678–85. doi: 10.1038/cdd.2008.21
50. Rakotomalala A, Escande A, Furlan A, Meignan S, Lartigau E. Hypoxia in solid tumors: How low oxygenation impacts the “Six Rs” of radiotherapy. Front Endocrinol (Lausanne) (2021) 12. doi: 10.3389/fendo.2021.742215
51. Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid Redox Signal (2014) 21(10):1516–54. doi: 10.1089/ars.2013.5378
52. Kolstad P. Oxygen tension and radiocurability in cancer of the cervix. Acta Obstet Gynecol Scand (1964) 43(S7):100–2. doi: 10.3109/00016346409155842
53. Griffiths JR, Robinson SP. The OxyLite: A fibre-optic oxygen sensor. Br J Radiol (1999) 72(859):627–30. doi: 10.1259/bjr.72.859.10624317
55. Harris BHL, Barberis A, West CML, Buffa FM. Gene expression signatures as biomarkers of tumour hypoxia. Clin Oncol (2015) 27(10):547–60. doi: 10.1016/j.clon.2015.07.004
56. Fyles AW, Milosevic M, Pintilie M, Hill RP. Cervix cancer oxygenation measured following external radiation therapy. Int J Radiat OncologyBiologyPhysics (1998) 42(4):751–3. doi: 10.1016/S0360-3016(98)00307-1
57. Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiotherapy Oncol [Internet]. (1996) 41(1):31–9. doi: 10.1016/S0167-8140(96)91811-3
58. Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, et al. First-in-human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res (2017) 23(15):4163–9. doi: 10.1158/1078-0432.CCR-16-2658
59. Atkins SLP, Greer YE, Jenkins S, Gatti-Mays ME, Houston N, Lee S, et al. A single-arm, open-label phase II study of ONC201 in recurrent/refractory metastatic breast cancer and advanced endometrial carcinoma. Oncologist. (2023) 28(10):919–e972. doi: 10.1093/oncolo/oyad164
60. Anderson PM, Trucco MM, Tarapore RS, Zahler S, Thomas S, Gortz J, et al. Phase II study of ONC201 in neuroendocrine tumors including pheochromocytoma-paraganglioma and desmoplastic small round cell tumor. Clin Cancer Res (2022) 28(9):1773–82. doi: 10.1158/1078-0432.CCR-21-4030
61. Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget. (2017) 8(45):79298–304. doi: 10.18632/oncotarget.17837
62. LoRusso P, Ratain MJ, Doi T, Rasco DW, de Jonge MJA, Moreno V, et al. Eftozanermin alfa (ABBV-621) monotherapy in patients with previously treated solid tumors: findings of a phase 1, first-in-human study. Invest New Drugs (2022) 40(4):762–72. doi: 10.1007/s10637-022-01247-1
63. Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol (2014) 32(28):3103–10. doi: 10.1200/JCO.2013.52.3993
64. Johnson ML, Patel MR, Aljumaily R, Jones SF, Burris Iii HA, Spigel DR. A phase ib dose-escalation study of LCL161 plus oral topotecan for patients with relapsed/refractory small cell lung cancer and select gynecologic Malignancies. Oncologist. (2023) 28(7):640–e559. doi: 10.1093/oncolo/oyad029
65. Bardia A, Parton M, Kümmel S, Estévez LG, Huang CS, Cortés J, et al. Paclitaxel with inhibitor of apoptosis antagonist, LCL161, for localized triple-negative breast cancer, prospectively stratified by gene signature in a biomarker-driven neoadjuvant trial. J Clin Oncol (2018) 36:JCO2017748392. doi: 10.1200/JCO.2017.74.8392
66. Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, et al. A phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther (2015) 14(11):2569–75. doi: 10.1158/1535-7163.MCT-15-0475
67. Senzer NN, LoRusso P, Martin LP, Schilder RJ, Amaravadi RK, Papadopoulos KP, et al. Phase II clinical activity and tolerability of the SMAC-mimetic birinapant (TL32711) plus irinotecan in irinotecan in relapsed/refractory metastatic colorectal cancer. J Clin Oncol (2013) 31:3261. doi: 10.1200/jco.2013.31.15_suppl.3621
68. Mita MM, LoRusso PM, Papadopoulos KP, Gordon MS, Mita AC, Ferraldeschi R, et al. A phase I study of ASTX660, an antagonist of inhibitors of apoptosis proteins, in adults with advanced cancers or lymphoma. Clin Cancer Res (2020) 26(12):2819–26. doi: 10.1158/1078-0432.CCR-19-1430
69. Tao Y, Sun XS, Pointreau Y, Le Tourneau C, Sire C, Kaminsky MC, et al. Extended follow-up of a phase 2 trial of xevinapant plus chemoradiotherapy in high-risk locally advanced squamous cell carcinoma of the head and neck: a randomised clinical trial. Eur J Cancer. (2023) 183:24–37. doi: 10.1016/j.ejca.2022.12.015
70. Rasco DW, Li Y, Tang Y, Men L, Wang H, Ji J, et al. A phase I study of a novel IAP inhibitor APG-1387 as a monotherapy or in combination with pembrolizumab in treatments of patients with advanced solid tumors. ASCO Annu meeting (2019) 38:508. doi: 10.1200/JCO.2019.37.15_suppl.3125
71. Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent Malignant glioma. Cancer Immunology Immunother (2016) 65(11):1339–52. doi: 10.1007/s00262-016-1890-x
72. Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol (2011) 29(7):909–16. doi: 10.1200/JCO.2010.31.6208
73. Li Y, Zhao L, Li XF. Targeting hypoxia: Hypoxia-activated prodrugs in cancer therapy. Front Oncol (2021) 11:11. doi: 10.3389/fonc.2021.700407
74. Tharmalingham H, Hoskin P. Clinical trials targeting hypoxia. Br J Radiol (2019) 92(1093):1–11. doi: 10.1259/bjr.20170966
75. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol (2012) 9(12):674–87. doi: 10.1038/nrclinonc.2012.171
76. Le QT, Courter D. Clinical biomarkers for hypoxia targeting. Cancer Metastasis Rev (2008) 27(3):351–62. doi: 10.1007/s10555-008-9144-9
77. Kim J, Choi KU, Lee IS, Choi YJ, Kim WT, Shin DH, et al. Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol Lett (2015) 9(4):1699–706. doi: 10.3892/ol.2015.2914
78. Liu Q, Cao P. Clinical and prognostic significance of HIF-1α in glioma patients: a meta-analysis. Int J Clin Exp Med (2015) 8(12):22073.
79. Shamis SAK, McMillan DC, Edwards J. The relationship between hypoxia-inducible factor 1α (HIF-1α) and patient survival in breast cancer: Systematic review and meta-analysis. Crit Rev Oncol Hematol (2021) 159:1–11. doi: 10.1016/j.critrevonc.2021.103231
80. Arjmand MH, Moradi A, Rahimi HR, Es-Haghi A, Akbari A, Hadipanah MR, et al. Prognostic value of HIF-1α in digestive system Malignancies: evidence from a systematic review and meta-analysis. Gastroenterol Hepatol Bed Bench. (2022) 15(2):108–19.
81. Méndez-Blanco C, Fernández-Palanca P, Fondevila F, González-Gallego J, Mauriz JL. Prognostic and clinicopathological significance of hypoxia-inducible factors 1α and 2α in hepatocellular carcinoma: A systematic review with meta-analysis. Ther Adv Med Oncol (2021) 13:1–28. doi: 10.1177/1758835920987071
82. Zhou J, Huang S, Wang L, Yuan X, Dong Q, Zhang D, et al. Clinical and prognostic significance of HIF-1α overexpression in oral squamous cell carcinoma: A meta-analysis. World J Surg Oncol (2017) 15(1):1–8. doi: 10.1186/s12957-017-1163-y
83. Omran Z, Scaife P, Stewart S, Rauch C. Physical and biological characteristics of multi drug resistance (MDR): An integral approach considering pH and drug resistance in cancer. Semin Cancer Biol Epub. (2017) 43:42–8. doi: 10.1016/j.semcancer.2017.01.002
84. Zhao Z, Liao G, Li Y, Zhou S, Zou H, Ferno S. Prognostic value of carbonic anhydrase IX immunohistochemical expression in renal cell carcinoma: a meta-analysis of the literature. PloS One (2014) 9(11):1–16. doi: 10.1371/journal.pone.0114096
85. Lorenzo-Pouso AI, Gallas-Torreira M, Pérez-Sayáns M, Chamorro-Petronacci CM, Alvarez-Calderon O, Takkouche B, et al. Prognostic value of CAIX expression in oral squamous cell carcinoma: A systematic review and meta-analysis. J Enzyme Inhib Med Chem (2020) 35(1):1258. doi: 10.1080/14756366.2020.1772250
86. Peridis S, Pilgrim G, Athanasopoulos I, Parpounas K. Carbonic anhydrase-9 expression in head and neck cancer: A meta-analysis. Eur Arch Otorhinolaryngol (2011) 268(5):661–70. doi: 10.1007/s00405-011-1488-z
87. Zhang B, Xie Z, Li B. The clinicopathologic impacts and prognostic significance of GLUT1 expression in patients with lung cancer: A meta-analysis. Gene. (2019) 689:76–83. doi: 10.1016/j.gene.2018.12.006
88. Tan Z, Yang C, Zhang X, Zheng P, Shen W. Expression of glucose transporter 1 and prognosis in non-small cell lung cancer: A pooled analysis of 1665 patients. Oncotarget. (2017) 8(37):60954–61. doi: 10.18632/oncotarget.17604
89. Deng Y, Zou J, Deng T, Liu J. Clinicopathological and prognostic significance of GLUT1 in breast cancer: A meta-analysis. Medicine (2018) 97(48):1–6. doi: 10.1097/MD.0000000000012961
90. Yang J, Wen J, Tian T, Lu Z, Wang Y, Wang Z, et al. GLUT-1 overexpression as an unfavorable prognostic biomarker in patients with colorectal cancer. Oncotarget. (2017) 8(7):11788–96. doi: 10.18632/oncotarget.14352
91. Li CX, Sun JL, Gong ZC, Lin ZQ, Liu H. Prognostic value of GLUT-1 expression in oral squamous cell carcinoma: A prisma-compliant meta-analysis. Medicine. (2016) 95(45):1–7. doi: 10.1097/MD.0000000000005324
92. Yu M, Yongzhi H, Chen S, Luo X, Lin Y, Zhou Y, et al. The prognostic value of GLUT1 in cancers: a systematic review and meta-analysis. Oncotarget. (2017) 8(26):43356–67. doi: 10.18632/oncotarget.17445
93. Zhao ZX, Lu LW, Qiu J, Li QP, Xu F, Liu BJ, et al. Glucose transporter-1 as an independent prognostic marker for cancer: a meta-analysis. Oncotarget. (2017) 9(2):2728–38. doi: 10.18632/oncotarget.18964
94. Speirs V. Quality considerations when using tissue samples for biomarker studies in cancer research. biomark Insights (2021) 16:117727192110095. doi: 10.1177/11772719211009513
95. Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer. (1995) 61(4):567–73. doi: 10.1002/ijc.2910610422
96. Wardman P. Nitroimidazoles as hypoxic cell radiosensitizers and hypoxia probes: misonidazole, myths and mistakes. Br J Radiol (2019) 92(1093):1–14. doi: 10.1259/bjr.20170915
97. Kaanders JHAM, Pop LAM, Marres HAM, Bruaset I, van den Hoogen FJA, Merkx MAW, et al. ARCON: Experience in 215 patients with advanced head-and-neck cancer. Int J Radiat OncologyBiologyPhysics (2002) 52(3):769–78. doi: 10.1016/S0360-3016(01)02678-5
98. Ragnum HB, Vlatkovic L, Lie AK, Axcrona K, Julin CH, Frikstad KM, et al. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br J Cancer (2015) 112(2):382–90. doi: 10.1038/bjc.2014.604
99. Evans SM, Fraker D, Hahn SM, Gleason K, Jenkins WT, Jenkins K, et al. EF5 binding and clinical outcome in human soft tissue sarcomas. Int J Radiat OncologyBiologyPhysics. (2006) 64(3):922–7. doi: 10.1016/j.ijrobp.2005.05.068
100. Rademakers SE, Hoogsteen IJ, Rijken PF, Oosterwijk E, Terhaard CH, Doornaert PA, et al. Pattern of CAIX expression is prognostic for outcome and predicts response to ARCON in patients with laryngeal cancer treated in a phase III randomized trial. Radiotherapy Oncol (2013) 108(3):517–22. doi: 10.1016/j.radonc.2013.04.022
101. Evans SM, Jenkins KW, Chen HI, Jenkins WT, Judy KD, Hwang WT, et al. The relationship among hypoxia, proliferation, and outcome in patients with de novo glioblastoma: A pilot study. Transl Oncol (2010) 3(3):160–9. doi: 10.1593/tlo.09265
102. Zips D, Zöphel K, Abolmaali N, Perrin R, Abramyuk A, Haase R, et al. Exploratory prospective trial of hypoxia-specific PET imaging during radiochemotherapy in patients with locally advanced head-and-neck cancer. Radiother Oncol (2012) 105(1):21–8. doi: 10.1016/j.radonc.2012.08.019
103. Löck S, Perrin R, Seidlitz A, Bandurska-Luque A, Zschaeck S, Zöphel K, et al. Residual tumour hypoxia in head-and-neck cancer patients undergoing primary radiochemotherapy, final results of a prospective trial on repeat FMISO-PET imaging. Radiother Oncol (2017) 124(3):533–40. doi: 10.1016/j.radonc.2017.08.010
104. Carles M, Fechter T, Grosu AL, Sörensen A, Thomann B, Stoian RG, et al. 18F-FMISO-PET hypoxia monitoring for head-and-neck cancer patients: Radiomics analyses predict the outcome of chemo-radiotherapy. Cancers (Basel) (2021) 13(14):1–16. doi: 10.3390/cancers13143449
105. Reeves KM, Song PN, Angermeier A, Della Manna D, Li Y, Wang J, et al. 18F-FMISO PET imaging identifies hypoxia and immunosuppressive tumor microenvironments and guides targeted evofosfamide therapy in tumors refractory to PD-1 and CTLA-4 inhibition. Clin Cancer Res (2022) 28(2):327–37. doi: 10.1158/1078-0432.CCR-21-2394
106. Challapalli A, Carroll L, Aboagye EO. Molecular mechanisms of hypoxia in cancer. Clin Transl Imaging (2017) 5(3):225–53. doi: 10.1007/s40336-017-0231-1
107. Zschaeck S, Löck S, Hofheinz F, Zips D, Saksø Mortensen L, Zöphel K, et al. Individual patient data meta-analysis of FMISO and FAZA hypoxia PET scans from head and neck cancer patients undergoing definitive radio-chemotherapy. Radiotherapy Oncol (2020) 149:189–96. doi: 10.1016/j.radonc.2020.05.022
108. Mayr NA, Yuh WTC, Jajoura D, Wang JZ, Lo SS, Montebello JF, et al. Ultra-early predictive assay for treatment failure using functional magnetic resonance imaging and clinical prognostic parameters in cervical cancer. Cancer. (2010) 116(4):903–12. doi: 10.1002/cncr.24822
109. Tian X, Shen H, Li Z, Wang T, Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol (2019) 12(1):84. doi: 10.1186/s13045-019-0772-z
110. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. (2014) 14(6):430–9. doi: 10.1038/nrc3726
111. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
112. Bogdanov A, Bogdanov A, Chubenko V, Volkov N, Moiseenko F, Moiseyenko V. Tumor acidity: From hallmark of cancer to target of treatment. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.979154
113. McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology. (2019) 157(3):823–37. doi: 10.1053/j.gastro.2019.05.004
114. Zhang JZ, Behrooz A, Ismail-Beigi F. Regulation of glucose transport by hypoxia. Am J Kidney Diseases. (1999) 34(1):189–202. doi: 10.1016/S0272-6386(99)70131-9
115. Ji K, Mayernik L, Moin K, Sloane BF. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev (2019) 38(1–2):103–12. doi: 10.1007/s10555-019-09796-3
116. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep (2014) 15(12):1243–53. doi: 10.15252/embr.201439246
117. Hynes RO. Extracellular matrix: not just prety fibrils. Sci (1979). (2009) 326(5957):1216–9. doi: 10.1126/science.1176009
118. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol (2014) 15(12):802–12. doi: 10.1038/nrm3896
119. Hansen NUB, Genovese F, Leeming DJ, Karsdal MA. The importance of extracellular matrix for cell function and in vivo likeness. Exp Mol Pathol (2015) 98(2):286–94. doi: 10.1016/j.yexmp.2015.01.006
120. Mouw JK, Ou G, Weaver VM, Regeneration T, Francisco S, Francisco S, et al. Extracellular matrix assembly: A multiscale deconstruction. Nat Rev Mol Cell Biol (2015) 15(12):771–85. doi: 10.1038/nrm3902
121. Cox TR. The matrix in cancer. Nat Rev Cancer (2021) 21(4):217–38. doi: 10.1038/s41568-020-00329-7
122. da Cunha BR, Domingos C, Buzzo Stefanini AC, Henrique T, Polachini GM, Castelo-Branco P, et al. Cellular interactions in the tumor microenvironment: The role of secretome. J Cancer. (2019) 10(19):4574. doi: 10.7150/jca.21780
123. Cirri P, Chiarugi P. Cancer associated fibroblasts : the dark side of the coin. Am J Cancer Res (2011) 1(4):482–97.
124. Belhabib I, Zaghdoudi S, Lac C, Bousquet C, Jean C. Extracellular matrices and cancer-associated fibroblasts: Targets for cancer diagnosis and therapy? Cancers (2021) 13:1–38. doi: 10.3390/cancers13143466
125. Xiong GF, Xu R. Function of cancer cell-derived extracellular matrix in tumor progression. J Cancer Metastasis Treat (2016) 2(9):357–64. doi: 10.20517/2394-4722.2016.08
126. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol (2016) 33(4):230–6. doi: 10.1016/j.tibtech.2015.01.004
127. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
128. Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol (2012) 196(4):395–406. doi: 10.1083/jcb.201102147
129. Gilkes DM, Wirtz D, Semenza GL. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. (2015) 14(6):430–9. doi: 10.1038/nrc3726
130. Yu X, Ye F. Role of angiopoietins in development of cancer and neoplasia associated with viral infection. Cells. (2020) 9(2):1–17. doi: 10.3390/cells9020457
131. Chang J, Chaudhuri O. Beyond proteases: Basement membrane mechanics and cancer invasion. J Cell Biol (2019) 218(8):2456–69. doi: 10.1083/jcb.201903066
132. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
133. Hapke RY, Haake SM. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett (2020) 487:10–20. doi: 10.1016/j.canlet.2020.05.012
134. Schito L, Semenza GL. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer. (2016) 2(12):758–70. doi: 10.1016/j.trecan.2016.10.016
135. Zhu SK, Zhou Y, Cheng C, Zhong S, Wu HQ, Wang B, et al. Overexpression of membrane-type 2 matrix metalloproteinase induced by hypoxia-inducible factor-1α in pancreatic cancer: Implications for tumor progression and prognosis. Mol Clin Oncol (2014) 2(6):973–81. doi: 10.3892/mco.2014.357
136. Nyante SJ, Wang T, Tan X, Ozdowski EF, Lawton TJ. Quantitative expression of MMPs 2, 9, 14, and collagen IV in LCIS and paired normal breast tissue. Sci Rep (2019) 9(1):1–9. doi: 10.1038/s41598-019-48602-6
137. Shin DH, Dier U, Melendez JA, Hempel N. Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim Biophys Acta (2015) 1852(12):2593–602. doi: 10.1016/j.bbadis.2015.09.001
138. Ji F, Wang Y, Qiu L, Li S, Zhu J, Liang Z, et al. Hypoxia inducible factor 1α-mediated LOX expression correlates with migration and invasion in epithelial ovarian cancer. Int J Oncol (2013) 42(5):1578–88. doi: 10.3892/ijo.2013.1878
139. Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer. (2020) 1873(2):1–31. doi: 10.1016/j.bbcan.2020.188356
140. Landolt L, Spagnoli GC, Hertig A, Brocheriou I, Marti HP. Fibrosis and cancer: shared features and mechanisms suggest common targeted therapeutic approaches. Nephrol Dial Transplant. (2022) 37(6):1024–32. doi: 10.1093/ndt/gfaa301
141. Whittaker Brown SA, Padilla M, Mhango G, Taioli E, Powell C, Wisnivesky J. Outcomes of older patients with pulmonary fibrosis and non-small cell lung cancer. Ann Am Thorac Soc (2019) 16(8):1034–40. doi: 10.1513/AnnalsATS.201808-510OC
142. Buechler C, Krautbauer S, Eisinger K. Adipose tissue fibrosis. World J Diabetes. (2015) 6(4):548. doi: 10.4239/wjd.v6.i4.548
143. Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. (2002) 35(5):1010–21. doi: 10.1053/jhep.2002.32524
144. Levene CI, Kapoor R, Heale G. The effect of hypoxia on the synthesis of collagen and glycosaminoglycans by cultured pig aortic endothelium. Atherosclerosis. (1982) 44(3):327–37. doi: 10.1016/0021-9150(82)90007-7
145. Kang Y, Roh MR, Rajadurai S, Rajadurai A, Kumar R, Njauw CN, et al. Hypoxia and HIF-1α Regulate collagen production in keloids. J Invest Dermatol (2020) 140(11):2157–65. doi: 10.1016/j.jid.2020.01.036
146. Novotny T, Eckhardt A, Doubkova M, Knitlova J, Vondrasek D, Vanaskova E, et al. The possible role of hypoxia in the affected tissue of relapsed clubfoot. Sci Rep (2022) 12(1):1–10 doi: 10.1038/s41598-022-08519-z
147. Braun RK, Broytman O, Braun FM, Brinkman JA, Clithero A, Modi D, et al. Chronic intermittent hypoxia worsens bleomycin-induced lung fibrosis in rats. Respir Physiol Neurobiol (2018) 256:97. doi: 10.1016/j.resp.2017.04.010
148. Jiang M, Ren L, Chen Y, Wang H, Wu H, Cheng S, et al. Identification of a hypoxia-related signature for predicting prognosis and the immune microenvironment in bladder cancer. Front Mol Biosci (2021) 8:380. doi: 10.3389/fmolb.2021.613359
149. Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res (2013) 73(11):3285–96. doi: 10.1158/0008-5472.CAN-12-3963
150. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. (2005) 105(2):659–69. doi: 10.1182/blood-2004-07-2958
151. Khan WS, Adesida AB, Hardingham TE. Hypoxic conditions increase hypoxia-inducible transcription factor 2alpha and enhance chondrogenesis in stem cells from the infrapatellar fat pad of osteoarthritis patients. Arthritis Res Ther (2007) 9(3):1–9. doi: 10.1186/ar2211
152. Tajima R, Kawaguchi N, Horino Y, Takahashi Y, Toriyama K, Inou K, et al. Hypoxic enhancement of type IV collagen secretion accelerates adipose conversion of 3T3-L1 fibroblasts. Biochim Biophys Acta (2001) 1540(3):179–87. doi: 10.1016/S0167-4889(01)00114-8
153. Bignon M, Pichol-Thievend C, Hardouin J, Malbouyres M, Bréchot N, Nasciutti L, et al. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood. (2011) 118(14):3979–89. doi: 10.1182/blood-2010-10-313296
154. Li H, Tong L, Tao H, Liu Z. Genome-wide analysis of the hypoxia-related DNA methylation-driven genes in lung adenocarcinoma progression. Biosci Rep (2020) 40(2):1–14. doi: 10.1042/BSR20194200
155. Ishizuka S, Sakai T, Hiraiwa H, Hamada T, Knudson W, Omachi T, et al. Hypoxia-inducible factor-2α induces expression of type X collagen and matrix metalloproteinases 13 in osteoarthritic meniscal cells. Inflammation Res (2016) 65(6):439–48. doi: 10.1007/s00011-016-0926-1
156. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. (2007) 117(12):3810–20. doi: 10.1172/JCI30487
157. Copple BL, Bai S, Burgoon LD, Moon JO. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int (2011) 31(2):230–44. doi: 10.1111/j.1478-3231.2010.02347.x
158. Takahashi M, Okada K, Ouch R, Konno T, Usui K, Suzuki H, et al. Fibronectin plays a major role in hypoxia-induced lenvatinib resistance in hepatocellular carcinoma PLC/PRF/5 cells. Pharmazie. (2021) 76(12):594–601. doi: 10.1691/ph.2021.1854
159. Kuo YL, Jou IM, Jeng SF, Chu CH, Huang JS, Hsu TI, et al. Hypoxia-induced epithelial-mesenchymal transition and fibrosis for the development of breast capsular contracture. Sci Rep (2019) 9(1):1–6. doi: 10.1038/s41598-019-46439-7
160. Gao F, Okunieff P, Han Z, Ding I, Wang L, Liu W, et al. Hypoxia-induced alterations in hyaluronan and hyaluronidase. Adv Exp Med Biol (2005) 566:249–56. doi: 10.1007/0-387-26206-7_33
161. Chen JWE, Lumibao J, Blazek A, Gaskins HR, Harley B. Hypoxia activates enhanced invasive potential and endogenous hyaluronic acid production by glioblastoma cells. Biomater Sci (2018) 6(4):854–62. doi: 10.1039/C7BM01195D
162. Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol (2022) 15(1):1–15. doi: 10.1186/s13045-022-01252-0
163. Wong CCL, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci U.S.A. (2011) 108(39):16369–74. doi: 10.1073/pnas.1113483108
164. Kasashima H, Yashiro M, Kinoshita H, Fukuoka T, Morisaki T, Masuda G, et al. Lysyl oxidase is associated with the epithelial-mesenchymal transition of gastric cancer cells in hypoxia. Gastric Cancer (2016) 19(2):431–42. doi: 10.1007/s10120-015-0510-3
165. Xie Q, Xie J, Tian T, Ma Q, Zhang Q, Zhu B, et al. Hypoxia triggers angiogenesis by increasing expression of LOX genes in 3-D culture of ASCs and ECs. Exp Cell Res (2017) 352(1):157–63. doi: 10.1016/j.yexcr.2017.02.011
166. Wang Z, Shi Y, Ying C, Jiang Y, Hu J. Hypoxia-induced PLOD1 overexpression contributes to the Malignant phenotype of glioblastoma via NF-κB signaling. Oncogene (2021) 40(8):1458–75. doi: 10.1038/s41388-020-01635-y
167. Rosell-García T, Palomo-Álvarez O, Rodríguez-Pascual F. A hierarchical network of hypoxia-inducible factor and SMAD proteins governs procollagen lysyl hydroxylase 2 induction by hypoxia and transforming growth factor β1. J Biol Chem (2019) 294(39):14308–18. doi: 10.1074/jbc.RA119.007674
168. Takahashi Y, Takahashi S, Shiga Y, Yoshimi T, Miura T. Hypoxic induction of prolyl 4-hydroxylase alpha (I) in cultured cells. J Biol Chem (2000) 275(19):14139–46. doi: 10.1074/jbc.275.19.14139
169. Hofbauer KH, Gess B, Lohaus C, Meyer HE, Katschinski D, Kurtz A. Oxygen tension regulates the expression of a group of procollagen hydroxylases. Eur J Biochem (2003) 270(22):4515–22. doi: 10.1046/j.1432-1033.2003.03846.x
170. Zhu X, Liu S, Yang X, Wang W, Shao W, Ji T. P4HA1 as an unfavorable prognostic marker promotes cell migration and invasion of glioblastoma via inducing EMT process under hypoxia microenvironment. Am J Cancer Res (2021) 11(2):590–617.
171. Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med (2015) 7(8):1063–76. doi: 10.15252/emmm.201404827
172. Hongo K, Tsuno NH, Kawai K, Sasaki K, Kaneko M, Hiyoshi M, et al. Hypoxia enhances colon cancer migration and invasion through promotion of epithelial-mesenchymal transition. J Surg Res (2013) 182(1):75–84. doi: 10.1016/j.jss.2012.08.034
173. Ju JA, Godet I, Ye IC, Byun J, Jayatilaka H, Lee SJ, et al. Hypoxia Selectively Enhances Integrin α5β1 receptor expression in breast cancer to promote metastasis. Mol Cancer Res (2017) 15(6):723–34. doi: 10.1158/1541-7786.MCR-16-0338
174. Kakkad SM, Solaiyappan M, O’Rourke B, Stasinopoulos I, Ackerstaff E, Raman V, et al. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia. (2010) 12(8):608–17. doi: 10.1593/neo.10344
175. Goggins E, Kakkad S, Mironchik Y, Jacob D, Wildes F, Krishnamachary B, et al. Hypoxia inducible factors modify collagen I fibers in MDA-MB-231 triple negative breast cancer xenografts. Neoplasia. (2018) 20(2):131–9. doi: 10.1016/j.neo.2017.11.010
176. Kuchnio A, Moens S, Bruning U, Kuchnio K, Cruys B, Thienpont B, et al. The cancer cell oxygen sensor PHD2 promotes metastasis via activation of cancer-associated fibroblasts. Cell Rep (2015) 12(6):992–1005. doi: 10.1016/j.celrep.2015.07.010
177. Madsen CD, Pedersen JT, Venning FA, Singh LB, Moeendarbary E, Charras G, et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep (2015) 16(10):1394–408. doi: 10.15252/embr.201540107
178. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci U S A. (2019) 116(39):19609–18. doi: 10.1073/pnas.1908626116
179. Luo Z, Tian M, Yang G, Tan Q, Chen Y, Li G, et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct Target Ther (2022) 7(1):1–30. doi: 10.1038/s41392-022-01080-1
180. Bolland H, Ma TS, Ramlee S, Ramadan K, Hammond EM. Links between the unfolded protein response and the DNA damage response in hypoxia: a systematic review. Biochem Soc Trans (2021) 49(3):1251–63. doi: 10.1042/BST20200861
181. Kim I, Choi S, Yoo S, Lee M, Kim IS. Cancer-associated fibroblasts in the hypoxic tumor microenvironment. Cancers (Basel) (2022) 14(14):3321. doi: 10.3390/cancers14143321
182. Louault K, Li RR, DeClerck YA. Cancer-associated fibroblasts: understanding their heterogeneity. Cancers (Basel) (2020) 12(11):3108. doi: 10.3390/cancers12113108
183. Glabman RA, Choyke PL, Sato N. Cancer-associated fibroblasts: Tumorigenicity and targeting for cancer therapy. Cancers (Basel) (2022) 14(16):3906. doi: 10.3390/cancers14163906
184. Yang D, Liu J, Qian H, Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med (2023) 55(7):1322–32. doi: 10.1038/s12276-023-01013-0
185. Gieniec KA, Butler LM, Worthley DL, Woods SL. Cancer-associated fibroblasts-heroes or villains? Br J Cancer (2019) 121(4):293–302. doi: 10.1038/s41416-019-0509-3
186. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol (2012) 13(10):616–30. doi: 10.1038/nrm3434
187. Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (2020) 31(9):107701. doi: 10.1016/j.celrep.2020.107701
188. Han C, Liu T, Yin R. Biomarkers for cancer-associated fibroblasts. biomark Res (2020) 8(1):64. doi: 10.1186/s40364-020-00245-w
189. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
190. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
191. Kim JW, Evans C, Weidemann A, Takeda N, Lee YS, Stockmann C, et al. Loss of fibroblast HIF-1α accelerates tumorigenesis. Cancer Res (2012) 72(13):3187–95. doi: 10.1158/0008-5472.CAN-12-0534
192. Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle (2010) 9(17):3534–51. doi: 10.4161/cc.9.17.12908
193. Ammirante M, Shalapour S, Kang Y, Jamieson CAM, Karin M. Tissue injury and hypoxia promote Malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc Natl Acad Sci (2014) 111(41):14776–81. doi: 10.1073/pnas.1416498111
194. Perlman RL. Mouse models of human disease: An evolutionary perspective. Evol Med Public Health (2016) 2016(1):170–6. doi: 10.1093/emph/eow014
195. Hu H, Piotrowska Z, Hare PJ, Chen H, Mulvey HE, Mayfield A, et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell (2021) 39(11):1531–47. doi: 10.1016/j.ccell.2021.09.003
196. Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res (2019) 79(20):5367–81. doi: 10.1158/0008-5472.CAN-19-0454
197. Iida T, Mizutani Y, Esaki N, Ponik SM, Burkel BM, Weng L, et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene. (2022) 41(19):2764–77. doi: 10.1038/s41388-022-02288-9
198. Kobayashi H, Gieniec KA, Wright JA, Wang T, Asai N, Mizutani Y, et al. The balance of stromal BMP signaling mediated by GREM1 and ISLR drives colorectal carcinogenesis. Gastroenterology. (2021) 160(4):1224–1239.e30. doi: 10.1053/j.gastro.2020.11.011
199. Takahashi M, Kobayashi H, Mizutani Y, Hara A, Iida T, Miyai Y, et al. Roles of the mesenchymal stromal/stem cell marker meflin/islr in cancer fibrosis. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.749924
200. Zheng B, Ohuchida K, Chijiiwa Y, Zhao M, Mizuuchi Y, Cui L, et al. CD146 attenuation in cancer-associated fibroblasts promotes pancreatic cancer progression. Mol Carcinog. (2016) 55(11):1560–72. doi: 10.1002/mc.22409
201. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res (2017) 23(7):1710–21. doi: 10.1158/1078-0432.CCR-15-2851
202. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
203. Crimeen-Irwin B, Scalzo K, Gloster S, Mottram PL, Plebanski M. Failure of immune homeostasis – the consequences of under and over reactivity. Curr Drug Targets Immune Endocr Metabol Disord (2005) 5(4):413–22. doi: 10.2174/156800805774912980
204. Khouzam RA, Zaarour RF, Brodaczewska K, Azakir B, Venkatesh GH, Thiery J. The effect of hypoxia and hypoxia-associated pathways in the regulation of antitumor response: Friends or foes? Front Immunol (2022) 13(828875). doi: 10.3389/fimmu.2022.828875
205. Semenza GL. Intratumoral hypoxia and mechanisms of immune evasion mediated by hypoxia-inducible factors. Physiology (2021) 36(2):73–83. doi: 10.1152/physiol.00034.2020
206. Chen Y, Gaber T. Hypoxia/HIF modulates immune responses. Biomedicines (2021) 9(3):260. doi: 10.3390/biomedicines9030260
207. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
208. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene (2019) 38(19):3681–95. doi: 10.1038/s41388-019-0688-7
209. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
210. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
211. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev (2018) 32(19–20):1267–84. doi: 10.1101/gad.314617.118
212. Ren L, Yu Y, Wang L, Zhu Z, Lu R, Yao Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget. (2016) 7(46):75763–73. doi: 10.18632/oncotarget.12409
213. Hu M, Li Y, Lu Y, Wang M, Li Y, Wang C, et al. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ. (2021) 9:e11306. doi: 10.7717/peerj.11306
214. Suthen S, Lim CJ, Nguyen PHD, Dutertre CA, Lai HLH, Wasser M, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology. (2022) 76(5):1329–44. doi: 10.1002/hep.32419
215. Wang B, Zhao Q, Zhang Y, Liu Z, Zheng Z, Liu S, et al. Targeting hypoxia in the tumor microenvironment: a potential strategy to improve cancer immunotherapy. J Exp Clin Cancer Res (2021) 40(1):24. doi: 10.1186/s13046-020-01820-7
216. Ohta A. Oxygen-dependent regulation of immune checkpoint mechanisms. Int Immunol (2018) 30(8):335–43. doi: 10.1093/intimm/dxy038
217. Lequeux A, Noman MZ, Xiao M, Van Moer K, Hasmim M, Benoit A, et al. Targeting HIF-1 alpha transcriptional activity drives cytotoxic immune effector cells into melanoma and improves combination immunotherapy. Oncogene. (2021) 40(28):4725–35. doi: 10.1038/s41388-021-01846-x
218. Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. (2017) 6(1):e1249561. doi: 10.1080/2162402X.2016.1249561
219. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. (2015) 6(29):27359–77. doi: 10.18632/oncotarget.4751
220. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Invest (2015) 125(5):2046–58. doi: 10.1172/JCI80445
221. Hosseinkhani N, Shadbad MA, Asghari Jafarabadi M, Karim Ahangar N, Asadzadeh Z, Mohammadi SM, et al. A systematic review and meta-analysis on the significance of TIGIT in solid cancers: Dual TIGIT/PD-1 blockade to overcome immune-resistance in solid cancers. Int J Mol Sci (2021) 22(19):10389. doi: 10.3390/ijms221910389
222. Sun F, Guo ZS, Gregory AD, Shapiro SD, Xiao G, Qu Z. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J Immunother Cancer (2020) 8(1):1–12. doi: 10.1136/jitc-2019-000294
223. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi: 10.1038/nm863
224. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell (2017) 683(e5):669–83. doi: 10.1016/j.ccell.2017.10.003
225. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17(4):364–8. doi: 10.1038/ni.3415
226. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
227. Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res (2016) 76(4):818–30. doi: 10.1158/0008-5472.CAN-15-0977
228. Song X, Zhang Y, Zhang L, Song W, Shi L. Hypoxia enhances indoleamine 2,3-Dioxygenase production in dendritic cells. Oncotarget. (2018) 9(14):11572–80. doi: 10.18632/oncotarget.24098
229. Liu J, Zhang H, Jia L, Sun H. Effects of Treg cells and IDO on human epithelial ovarian cancer cells under hypoxic conditions. Mol Med Rep (2015) 11(3):1708–14. doi: 10.3892/mmr.2014.2893
230. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
231. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
232. Husain Z, Seth P, Sukhatme VP. Tumor-derived lactate and myeloid-derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology. (2013) 2(11):e26383. doi: 10.4161/onci.26383
233. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal (2015) 8(396):1–12. doi: 10.1126/scisignal.aab2610
234. Sierra JM, Secchiari F, Nuñez SY, Iraolagoitia XLR, Ziblat A, Friedrich AD, et al. Tumor-experienced human NK cells express high levels of PD-L1 and inhibit CD8+ T cell proliferation. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.745939
235. Shurin MR, Umansky V. Cross-talk between HIF and PD-1/PD-L1 pathways in carcinogenesis and therapy. J Clin Invest (2022) 132(9):1–4. doi: 10.1172/JCI159473
236. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol (2013) 43(10):2756–64. doi: 10.1002/eji.201343448
237. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, Van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci (2013) 110(43):17450–5. doi: 10.1073/pnas.1304790110
238. Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand J Immunol (2003) 58:607–12. doi: 10.1111/j.1365-3083.2003.01347.x
239. Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res (2014) 74(23):6820–32. doi: 10.1158/0008-5472.CAN-14-0303
240. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
241. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
242. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by sema3A/nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24(6):695–709. doi: 10.1016/j.ccr.2013.11.007
243. Sica A, Saccani A, Bottazzi B, Bernasconi S, Allavena P, Gaetano B, et al. Defective expression of the monocyte chemotactic protein-1 receptor CCR2 in macrophages associated with human ovarian carcinoma. J Immunol (2000) 164(2):733–8. doi: 10.4049/jimmunol.164.2.733
244. Hempel SL, Monick MM, Hunninghake GW. Effect of hypoxia on release of IL-1 and TNF by human alveolar macrophages. Am J Respir Cell Mol Biol (1996) 14(2):170–6. doi: 10.1165/ajrcmb.14.2.8630267
245. Scannell G, Waxman K, Kaml GJ, Ioli G, Gatanaga T, Yamamoto R, et al. Hypoxia induces a human macrophage cell line to release tumor necrosis factor-alpha and its soluble receptors in vitro. J Surg Res (1993) 54(4):281–5. doi: 10.1006/jsre.1993.1044
246. Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol (1997) 17(7):4015–23. doi: 10.1128/MCB.17.7.4015
247. Salven P, Hattori K, Heissig B, Rafii S. Interleukin-1alpha promotes angiogenesis in vivo via VEGFR-2 pathway by inducing inflammatory cell VEGF synthesis and secretion. FASEB journal : Off Publ Fed Am Societies Exp Biol (2002) 16(11):1471–3. doi: 10.1096/fj.02-0134fje
248. Tiainen S, Tumelius R, Rilla K, Hämäläinen K, Tammi M, Tammi R, et al. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology (2015) 66(6):873–83. doi: 10.1111/his.12607
249. Petrillo M, Zannoni GF, Martinelli E, Anchora LP, Ferrandina G, Tropeano G, et al. Polarisation of tumor-associated macrophages toward M2 phenotype correlates with poor response to chemoradiation and reduced survival in patients with locally advanced cervical cancer. PloS One (2015) 10(9):e0136654. doi: 10.1371/journal.pone.0136654
250. Lanciotti M, Masieri L, Raspollini MR, Minervini A, Mari A, Comito G, et al. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. BioMed Res Int (2014) 2014:486798. doi: 10.1155/2014/486798
251. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. (2019) 38(26):5158–73. doi: 10.1038/s41388-019-0782-x
252. Shou Y, Wang X, Chen C, Liang Y, Yang C, Xiao Q, et al. Exosomal miR-301a-3p from esophageal squamous cell carcinoma cells promotes angiogenesis by inducing M2 polarization of macrophages via the PTEN/PI3K/AKT signaling pathway. Cancer Cell Int (2022) 22(1):153. doi: 10.1186/s12935-022-02570-6
253. Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q, et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res (2019) 38(1):81. doi: 10.1186/s13046-019-1095-1
254. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2(10):1096–103. doi: 10.1038/nm1096-1096
255. Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology (2009) 128:237–49. doi: 10.1111/j.1365-2567.2008.02954.x
256. Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology. (2017) 6(2):1–10. doi: 10.1080/2162402X.2016.1271858
257. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
258. Hasmim M, Noman MZ, Messai Y, Bordereaux D, Gros G, Baud V. Cutting edge: Hypoxia-induced nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J Immunol (2013) 191(12):5802–6. doi: 10.4049/jimmunol.1302140
259. Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, et al. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. (2023) 22(1):29. doi: 10.1186/s12943-023-01731-z
260. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. (2019) 18(1):157. doi: 10.1186/s12943-019-1089-9
261. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res (1998) 58(7):1408–16.
262. Yano S, Takehara K, Tazawa H, Kishimoto H, Urata Y, Kagawa S, et al. Cell-cycle-dependent drug-resistant quiescent cancer cells induce tumor angiogenesis after chemotherapy as visualized by real-time FUCCI imaging. Cell Cycle (2017) 16(5):406–14. doi: 10.1080/15384101.2016.1220461
263. Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci (2016) 107(1):5–11. doi: 10.1111/cas.12817
264. Wu FH, Mu L, Li XL, Hu YB, Liu H, Han LT, et al. Characterization and functional analysis of a slow-cycling subpopulation in colorectal cancer enriched by cell cycle inducer combined chemotherapy. Oncotarget (2017) 8(45):78466–79. doi: 10.18632/oncotarget.19638
265. Ewton DZ, Hu J, Vilenchik M, Deng X, Luk KC, Polonskaia A, et al. Inactivation of mirk/dyrk1b kinase targets quiescent pancreatic cancer cells. Mol Cancer Ther (2011) 10(11):2104–14. doi: 10.1158/1535-7163.MCT-11-0498
266. Menegakis A, Klompmaker R, Vennin C, Arbusà A, Damen M, Broek B, et al. Resistance of hypoxic cells to ionizing radiation is mediated in part via hypoxia-induced quiescence. Cells (2021) 10(3):610. doi: 10.3390/cells10030610
267. Rohwer N, Cramer T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resistance Updates (2011) 14(3):191–201. doi: 10.1016/j.drup.2011.03.001
268. Wiechec E, Matic N, Ali A, Roberg K. Hypoxia induces radioresistance, epithelialmesenchymal transition, cancer stem celllike phenotype and changes in genes possessing multiple biological functions in head and neck squamous cell carcinoma. Oncol Rep (2022) 47(3):58. doi: 10.3892/or.2022.8269
269. Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, et al. Expression of hypoxia-inducible factor-1α: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer1. Cancer Res (2001) 61(7):2911–6.
270. Lequeux A, Noman MZ, Xiao M, Sauvage D, Van Moer K, Viry E, et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett (2019) 458:13–20. doi: 10.1016/j.canlet.2019.05.021
271. Hayashi M, Yamamoto Y, Ibusuki M. Evaluation of tumor stiffness by elastography is predictive for pathologic complete response to neoadjuvant chemotherapy in patients with breast cancer. Ann Surg Oncol (2012) 19:3042–9. doi: 10.1245/s10434-012-2343-1
272. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis (2017) 6(7):e352. doi: 10.1038/oncsis.2017.54
273. Shen Y, Wang X, Lu J, Salfenmoser M, Wirsik NM, Schleussner N, et al. Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell (2020) 37(6):800–17. doi: 10.1016/j.ccell.2020.05.005
274. Qin X, Lv X, Li P, Yang R, Xia Q, Chen Y, et al. Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2020) 1866(3):165625. doi: 10.1016/j.bbadis.2019.165625
275. Fan Y, Sun Q, Li X, Feng J, Ao Z, Li X, et al. Substrate stiffness modulates the growth, phenotype, and chemoresistance of ovarian cancer cells. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.718834
276. Li S, Bai H, Chen X, Gong S, Xiao J, Li D, et al. Soft substrate promotes osteosarcoma cell self-renewal, differentiation, and drug resistance through miR-29b and its target protein spin 1. ACS Biomater Sci Eng. (2020) 6(10):5588–98. doi: 10.1021/acsbiomaterials.0c00816
277. Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci U S A. (2017) 114(3):492–7. doi: 10.1073/pnas.1613855114
278. Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm (2011) 8(6):2032–8. doi: 10.1021/mp200292c
279. Raghunand N, Gillies RJ. pH and drug resistance in tumors. Drug Resist Updat. (2000) 3(1):39–47. doi: 10.1054/drup.2000.0119
280. Wachsberger PR, Landry J, Storck C, Davis K, O’Hara MD, Owen CS, et al. Mammalian cells adapted to growth at pH 6.7 have elevated HSP27 levels and are resistant to cisplatin. Int J Hyperthermia (1997) 13(3):251–5. doi: 10.3109/02656739709023533
281. Vukovic V, Tannock IF. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. Br J Cancer. (1997) 75(8):1167–72. doi: 10.1038/bjc.1997.201
282. Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J Cell Biochem (2017) 118(10):3237–48. doi: 10.1002/jcb.25972
283. Wiecek AJ, Cutty SJ, Kornai D, Parreno-Centeno M, Gourmet LE, Tagliazucchi GM, et al. Genomic hallmarks and therapeutic implications of G0 cell cycle arrest in cancer. Genome Biol (2023) 24(1):128. doi: 10.1186/s13059-023-02963-4
284. Druker J, Wilson JW, Child F, Shakir D, Fasanya T, Rocha S. Role of hypoxia in the control of the cell cycle. Int J Mol Sci (2021) 22(9):1–15. doi: 10.3390/ijms22094874
285. Yano S, Miwa S, Mii S, Hiroshima Y, Uehara F, Yamamoto M, et al. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle (2014) 13(6):953–60. doi: 10.4161/cc.27818
286. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. (1993) 366(6456):704–7. doi: 10.1038/366704a0
287. Box AH, Demetrick AJ. Cell cycle kinase inhibitor expression and hypoxia-induced cell cycle arrest in human cancer cell lines. Carcinogenesis. (2004) 25(12):2325–35. doi: 10.1093/carcin/bgh274
288. Liu Z, Lin H, Gan Y, Cui C, Zhang B, Gu L, et al. P16 methylation leads to paclitaxel resistance of advanced non-small cell lung cancer. J Cancer. (2019) 10(7):1726–33. doi: 10.7150/jca.26482
289. Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol (2003) 23(9):3265–73. doi: 10.1128/MCB.23.9.3265-3273.2003
290. Chan N, Ali M, McCallum GP, Kumareswaran R, Koritzinsky M, Wouters BG, et al. Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol Cancer Res (2014) 12(10):1407–15. doi: 10.1158/1541-7786.MCR-14-0246
291. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery (2017) 7(1):20–37. doi: 10.1158/2159-8290.CD-16-0860
292. Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, et al. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys (2013) 86(3):469–76. doi: 10.1016/j.ijrobp.2013.02.015
293. Mehibel M, Xu Y, Li CG, Moon EJ, Thakkar KN, Diep AN, et al. Eliminating hypoxic tumor cells improves response to PARP inhibitors in homologous recombination-deficient cancer models. J Clin Invest (2021) 131(11):e146256. doi: 10.1172/JCI146256
294. Tufail M. DNA repair pathways in breast cancer: from mechanisms to clinical applications. Breast Cancer Res Treat (2023) 200(3):305–21. doi: 10.1007/s10549-023-06995-z
295. Weil MK, Chen AP. PARP inhibitor treatment in ovarian and breast cancer. Curr Probl Cancer. (2011) 35(1):7–50. doi: 10.1016/j.currproblcancer.2010.12.002
296. Lou S, Wang Y, Zhang J, Yin X, Zhang Y, Wang Y, et al. Patient-level DNA damage repair pathway profiles and anti-tumor immunity for gastric cancer. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.806324
297. Sermeus A, Genin M, Maincent A, Fransolet M, Notte A, Leclere L, et al. Hypoxia-induced modulation of apoptosis and BCL-2 family proteins in different cancer cell types. PloS One (2012) 7(11):e47519. doi: 10.1371/journal.pone.0047519
298. Brotin E, Meryet-Figuière M, Simonin K, Duval RE, Villedieu M, Leroy-Dudal J, et al. Bcl-XL and MCL-1 constitute pertinent targets in ovarian carcinoma and their concomitant inhibition is sufficient to induce apoptosis. Int J Cancer (2010) 126(4):885–95. doi: 10.1002/ijc.24787
299. Imoto I, Yang ZQ, Pimkhaokham A, Tsuda H, Shimada Y, Imamura M, et al. Identification of cIAP1 As a Candidate Target Gene within an Amplicon at 11q22 in Esophageal Squamous Cell Carcinomas1. Cancer Res (2001) 61(18):6629–34.
300. Oh BY, Kim KH, Chung SS, Lee RA. Silencing the livin gene enhances the cytotoxic effects of anticancer drugs on colon cancer cells. Ann Surg Treat Res (2016) 91(6):273–7. doi: 10.4174/astr.2016.91.6.273
301. Nachmias B, Ashhab Y, Bucholtz V, Drize O, Kadouri L, Lotem M, et al. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: implications for drug-resistant melanoma. Cancer Res (2003) 63(19):6340–9.
302. Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol Cancer Ther (2014) 13(4):867–79. doi: 10.1158/1535-7163.MCT-13-0798
303. Wang L, Hu C, Zhao Y, Hu X. Novel smac mimetic ASTX660 (Tolinapant) and TNF-α synergistically induce necroptosis in bladder cancer cells in vitro upon apoptosis inhibition. Biochem Biophys Res Commun (2022) 602:8–14. doi: 10.1016/j.bbrc.2022.02.053
304. Crawford N, Stott KJ, Sessler T, McCann C, McDaid W, Lees A, et al. Clinical positioning of the IAP antagonist tolinapant (ASTX660) in colorectal cancer. Mol Cancer Ther (2021) 20(9):1627–39. doi: 10.1158/1535-7163.MCT-20-1050
305. Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3β Activity and associates with poor prognosis in human breast cancer. Cancer Res (2007) 67(10):4564–71. doi: 10.1158/0008-5472.CAN-06-1788
306. Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady ASA, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther (2014) 13(3):565–75. doi: 10.1158/1535-7163.MCT-12-0767
307. Mitchell C, Yacoub A, Hossein H, Pandya Martin A, Bareford MD, Eulitt PJ, et al. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther (2010) 10(9):903–17. doi: 10.4161/cbt.10.9.13273
308. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis (2015) 6(1):e1590. doi: 10.1038/cddis.2014.561
309. Lv Y, Zhao S, Han J, Zheng L, Yang Z, Zhao L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. Onco Targets Ther (2015) 8:1941–8. doi: 10.2147/OTT.S82835
310. He M, Wu H, Jiang Q, Liu Y, Han L, Yan Y, et al. Hypoxia-inducible factor-2α directly promotes BCRP expression and mediates the resistance of ovarian cancer stem cells to adriamycin. Mol Oncol (2019) 13(2):403–21. doi: 10.1002/1878-0261.12419
311. Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer (2017) 17(12):738–50. doi: 10.1038/nrc.2017.93
312. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. (2003) 8(5):411–24. doi: 10.1634/theoncologist.8-5-411
313. Mohelnikova-Duchonova B, Brynychova V, Oliverius M, Honsova E, Kala Z, Muckova K, et al. Differences in transcript levels of ABC transporters between pancreatic adenocarcinoma and nonneoplastic tissues. Pancreas. (2013) 42(4):707–16. doi: 10.1097/MPA.0b013e318279b861
314. Kerklaan BM, Lolkema MPJ, Devriese LA, Voest EE, Nol-Boekel A, Mergui-Roelvink M, et al. Phase I and pharmacological study of pazopanib in combination with oral topotecan in patients with advanced solid tumours. Br J Cancer. (2015) 113(5):706–15. doi: 10.1038/bjc.2015.257
315. Barnett GC, West CML, Dunning AM, Elliott RM, Coles CE, Pharoah PDP, et al. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer. (2009) 9(2):134–42. doi: 10.1038/nrc2587
316. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: Current advances and future directions. Int J Med Sci (2012) 9(3):193–9. doi: 10.7150/ijms.3635
317. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol (1953) 26(312):638–48. doi: 10.1259/0007-1285-26-312-638
318. Burnet NG, Mackay RI, Smith E, Chadwick AL, Whitfield GA, Thomson DJ, et al. Proton beam therapy: perspectives on the National Health Service England clinical service and research programme. Br J Radiol (2020) 93(1107):20190873. doi: 10.1259/bjr.20190873
319. Flynn RT, Bowen SR, Bentzen SM, Rockwell Mackie T, Jeraj R. Intensity-modulated x-ray (IMXT) versus proton (IMPT) therapy for theragnostic hypoxia-based dose painting. Phys Med Biol (2008) 53(15):4153–67. doi: 10.1088/0031-9155/53/15/010
320. Horsman MR, Wouters BG, Joiner MC, Overgaard J. The oxygen effect and fractionated radiotherapy. 4th ed. London: CRC press (2009).
321. Toustrup K, Sørensen BS, Nordsmark M, Busk M, Wiuf C, Alsner J, et al. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res (2011) 71(17):5923–31. doi: 10.1158/0008-5472.CAN-11-1182
322. Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, et al. Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol (2006) 24(13):2098–104. doi: 10.1200/JCO.2005.05.2878
323. Overgaard J, Hansen HS, Overgaard M, Bastholt L, Berthelsen A, Specht L, et al. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother Oncol (1998) 46(2):135–46. doi: 10.1016/s0167-8140(97)00220-x
324. Telarovic I, Wenger RH, Pruschy M. Interfering with tumor hypoxia for radiotherapy optimization. J Exp Clin Cancer Res (2021) 40(1):197. doi: 10.1186/s13046-021-02000-x
325. Thiruthaneeswaran N, Bibby BAS, Yang L, Hoskin PJ, Bristow RG, Choudhury A, et al. Lost in application: Measuring hypoxia for radiotherapy optimisation. Eur J Cancer. (2021) 148:260–76. doi: 10.1016/j.ejca.2021.01.039
326. Wenzl T, Wilkens JJ. Theoretical analysis of the dose dependence of the oxygen enhancement ratio and its relevance for clinical applications. Radiat Oncol (2011) 6(1):171. doi: 10.1186/1748-717X-6-171
327. Wang JZ, Li XA, Mayr NA. Dose escalation to combat hypoxia in prostate cancer: a radiobiological study on clinical data. Br J Radiol (2006) 79(947):905–11. doi: 10.1259/bjr/18700614
328. Boustani J, Grapin M, Laurent PA, Apetoh L, Mirjolet C. The 6th R of radiobiology: Reactivation of anti-tumor immune response. Cancers (Basel) (2019) 11:1–16. doi: 10.3390/cancers11060860
329. Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. Int J Radiat Biol (1989) 56(6):1045–8. doi: 10.1080/09553008914552491
330. Withers HR. The four R’s of radiotherapy. In: Lett JT, Adler H, editors. Advances in radiation biology, vol. 5. United States: Elsevier (1975). p. 241–71. Available at: https://www.sciencedirect.com/science/article/pii/B9780120354054500128.
331. Wozny AS, Alphonse G, Cassard A, Malésys C, Louati S, Beuve M, et al. Impact of hypoxia on the double-strand break repair after photon and carbon ion irradiation of radioresistant HNSCC cells. Sci Rep (2020) 10(1):21357. doi: 10.1038/s41598-020-78354-7
332. Kumareswaran R, Ludkovski O, Meng A, Sykes J, Pintilie M, Bristow RG. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J Cell Sci (2012) 125(Pt 1):189–99. doi: 10.1242/jcs.092262
333. Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res (2008) 68(2):605–14. doi: 10.1158/0008-5472.CAN-07-5472
334. Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, et al. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiotherapy Oncol (2005) 76(2):168–76. doi: 10.1016/j.radonc.2005.06.025
335. Fanale D, Bazan V, Caruso S, Castiglia M, Bronte G, Rolfo C, et al. Hypoxia and human genome stability: downregulation of BRCA2 expression in breast cancer cell lines. BioMed Res Int (2013) 2013:746858. doi: 10.1155/2013/746858
336. Luo Y, Li M, Zuo X, Basourakos SP, Zhang J, Zhao J, et al. βcatenin nuclear translocation induced by HIF1α overexpression leads to the radioresistance of prostate cancer. Int J Oncol (2018) 52(6):1827–40. doi: 10.3892/ijo.2018.4368
337. Kim JG, Yi JM, Park SJ, Kim JS, Son TG, Yang K, et al. Histone demethylase JMJD2B-mediated cell proliferation regulated by hypoxia and radiation in gastric cancer cell. Biochim Biophys Acta (2012) 1819(11-12):1200–7). doi: 10.1016/j.bbagrm.2012.10.001
338. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell (2004) 5(5):429–41. doi: 10.1016/S1535-6108(04)00115-1
339. Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, et al. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J Biol Chem (2009) 284(8):5332–42. doi: 10.1074/jbc.M806653200
340. Quennet V, Yaromina A, Zips D, Rosner A, Walenta S, Baumann M, et al. Tumor lactate content predicts for response to fractionated irradiation of human squamous cell carcinomas in nude mice. Radiother Oncol (2006) 81(2):130–5. doi: 10.1016/j.radonc.2006.08.012
341. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60(4):916–21.
342. Feng H, Wang J, Chen W, Shan B, Guo Y, Xu J, et al. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J Bone Oncol (2016) 5(2):67–73. doi: 10.1016/j.jbo.2016.03.001
343. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res (2008) 68(5):1485–94. doi: 10.1158/0008-5472.CAN-07-0562
344. Cuisnier O, Serduc R, Lavieille JP, Longuet M, Reyt E, Riva C. Chronic hypoxia protects against gamma-irradiation-induced apoptosis by inducing bcl-2 up-regulation and inhibiting mitochondrial translocation and conformational change of bax protein. Int J Oncol (2003) 23(4):1033–41.
345. Bamodu OA, Chang HL, Ong JR, Lee WH, Yeh CT, Tsai JT. Elevated PDK1 expression drives PI3K/AKT/MTOR signaling promotes radiation-resistant and dedifferentiated phenotype of hepatocellular carcinoma. Cells (2020) 9(3):746. doi: 10.3390/cells9030746
346. Taghizadeh-Hesary F. “Reinforcement” by tumor microenvironment: The seventh “R” of radiobiology. Int J Radiat OncologyBiologyPhysics (2023) 000:1–7. doi: 10.1016/j.ijrobp.2023.09.027
347. Diesendruck Y, Benhar I. Novel immune check point inhibiting antibodies in cancer therapy—Opportunities and challenges. Drug Resistance Updates (2017) 30:39–47. doi: 10.1016/j.drup.2017.02.001
348. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
349. Ding X, Wang L, Zhang X, Xu J, Li P, Liang H, et al. The relationship between expression of PD-L1 and HIF-1α in glioma cells under hypoxia. J Hematol Oncol (2021) 14(1):92. doi: 10.1200/JCO.2021.39.15_suppl.e14043
350. Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J Immunother Cancer (2021) 9(5):e002088. doi: 10.1136/jitc-2020-002088
351. Semaan A, Dietrich D, Bergheim D, Dietrich J, Kalff JC, Branchi V, et al. CXCL12 expression and PD-L1 expression serve as prognostic biomarkers in HCC and are induced by hypoxia. Virchows Arch (2017) 470(2):185–96. doi: 10.1007/s00428-016-2051-5
352. Mazumder S, Higgins PJ, Samarakoon R. Downstream targets of VHL/HIF-α Signaling in renal clear cell carcinoma progression: Mechanisms and therapeutic relevance. Cancers (Basel). (2023) 15(4):1316. doi: 10.3390/cancers15041316
353. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von hippel-lindau disease. N Engl J Med (2021) 385(22):2036–46. doi: 10.1056/NEJMoa2103425
354. Ohh M, Taber CC, Ferens FG, Tarade D. Hypoxia-inducible factor underlies von Hippel-Lindau disease stigmata. Elife (2022) 11:1–18. doi: 10.7554/eLife.80774
355. Raval RR, Lau KW, Tran MGB, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol (2005) 25(13):5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005
356. Zhang S, Xu H, Li W, Ji J, Jin Q, Chen L, et al. MDM2 promotes cancer cell survival through regulating the expression of HIF-1α and pVHL in retinoblastoma. Pathol Oncol Res (2023) 29:1610801. doi: 10.3389/pore.2023.1610801
357. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci (2019) 110(5):1510–7. doi: 10.1111/cas.13990
358. Wang S, Chen FE. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur J Med Chem (2022) 236:114334. doi: 10.1016/j.ejmech.2022.114334
359. Kluckova K, Tennant DA. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res (2018) 372(2):367–78. doi: 10.1007/s00441-018-2801-6
360. Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol (2004) 57(10):1009–14. doi: 10.1136/jcp.2003.015032
361. Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol (2007) 25(26):4066–74. doi: 10.1200/JCO.2007.12.7878
362. Hoskin P, Rojas A, Bentzen S, Saunders M. Radiotherapy with concurrent carbogen and nicotinamide in bladder carcinoma. J Clin Oncol (2010) 28:4912–8. doi: 10.1200/JCO.2010.28.4950
363. Song YP, Mistry H, Irlam J, Valentine H, Yang L, Lane B, et al. Long-term outcomes of radical radiation therapy with hypoxia modification with biomarker discovery for stratification: 10-Year update of the BCON (bladder carbogen nicotinamide) phase 3 randomized trial (ISRCTN45938399. Int J Radiat Oncol Biol Phys (2021) 110(5):1407–15. doi: 10.1016/j.ijrobp.2021.03.001
364. Bourigault P, Skwarski M, Macpherson RE, Higgins GS, McGowan DR. Investigation of atovaquone-induced spatial changes in tumour hypoxia assessed by hypoxia PET/CT in non-small cell lung cancer patients. EJNMMI Res (2021) 11(1):130. doi: 10.1186/s13550-021-00871-x
365. Hoskin P. Evidence-based practice in oncology, when it suits us? BMJ Oncol (2023) 2(1):e000061. doi: 10.1136/bmjonc-2023-000061
366. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
367. Fontana F, Marzagalli M, Sommariva M, Gagliano N, Limonta P. In vitro 3D cultures to model the tumor microenvironment. Cancers (Basel) (2021) 13(12):2970. doi: 10.3390/cancers13122970
368. Pavlacky J, Polak J. Technical feasibility and physiological relevance of hypoxic cell culture models. Front Endocrinol (Lausanne). (2020) 11:57. doi: 10.3389/fendo.2020.00057
Keywords: hypoxia, tumour microenvironment, extracellular matrix, immune cells, cancer associated fibroblasts
Citation: Bigos KJA, Quiles CG, Lunj S, Smith DJ, Krause M, Troost EGC, West CM, Hoskin P and Choudhury A (2024) Tumour response to hypoxia: understanding the hypoxic tumour microenvironment to improve treatment outcome in solid tumours. Front. Oncol. 14:1331355. doi: 10.3389/fonc.2024.1331355
Received: 01 November 2023; Accepted: 08 January 2024;
Published: 30 January 2024.
Edited by:
Katarzyna Leszczyńska, Polish Academy of Sciences, PolandCopyright © 2024 Bigos, Quiles, Lunj, Smith, Krause, Troost, West, Hoskin and Choudhury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamilla JA. Bigos, kamilla.bigos@manchester.ac.uk