
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 01 March 2024
Sec. Pediatric Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1320541
This article is part of the Research TopicGlobal Approaches to Molecular Diagnostics for Pediatric CancerView all 8 articles
 Francesco Pellegrino1*
Francesco Pellegrino1* Elisa Tirtei2,3Federico Divincenzo3Anna Campello2,3
Elisa Tirtei2,3Federico Divincenzo3Anna Campello2,3 Carlotta Rubino1Elisabetta Augustoni1Alessandra Linari4
Carlotta Rubino1Elisabetta Augustoni1Alessandra Linari4 Sebastian Dorin Asaftei2,3
Sebastian Dorin Asaftei2,3 Franca Fagioli2,3
Franca Fagioli2,3Introduction: Malignant ectomesenchymoma (MEM) is a soft tissue tumour, consisting of both malignant neuroectodermal elements and one or more mesenchymal elements.
Case presentation and review of the literature: Here we describe the case of a 6-months-old male, previously treated in another hospital for abdominal rhabdomyosarcoma (RMS). Histological re-examination demonstrated that the tumour had mesenchymal and neuroectodermal elements components, with a new diagnosis of abdominal-pelvic MEM. A Next-Generation Sequencing (NGS) analysis was performed on a surgical tumour specimen and revealed the presence of a somatic mutation, already reported in MEM cases. We carried out a review of the literature and we found 33 new cases of MEM since the last review. We reported the clinic-pathologic features of new cases of MEM, highlighting the role of molecular studies in supporting the diagnosis of this ambiguous tumours.
Conclusion: We promote the importance of a diagnosis based on an integrative morpho-molecular approach, that routinely include molecular analysis and the use of bioinformatic mutation detection tools, to support diagnostic and therapeutical queries and to highlight tumour biology and behaviour.
Malignant ectomesenchymoma (MEM) is an uncommon and rapidly progressing soft tissue tumour, composed of both malignant mesenchymal and neuroectodermal elements (1–3). The predominant mesenchymal elements are usually embryonal rhabdomyosarcoma (ERMS), while the neuroectodermal component may present with multiple degrees of differentiation, emerging as neuroblastoma (NB), ganglioneuroma (GN), ganglioneuroblastoma (GNB) or ganglion cells (GC). Most commonly the neural component occurs as clustered GC and, rarely, as primitive neuroblastic elements. Sporadic cases of alveolar rhabdomyosarcoma (ARMS), malignant peripheral nerve sheath tumour, and primary peripheral neuroectodermal tumour have been reported (4).
MEMs are presumed to originate from the remnants of migratory cells of the neuronal crest, which constitute the ectomesenchyme (5). These pluripotent cells are widespread throughout the body; thus MEMs may potentially develop at any site within the soft tissue or in the central nervous system. The most common primary sites include pelvic and retroperitoneal region and urogenital sites, whereas a less often they arise in the head and neck region or mediastinum (3). The exact aetiology is unknown.
It is commonly assumed that MEM patients should be treated according to RMS protocols, as the risk factors, treatment and outcomes of these tumours are comparable to other highly malignant paediatric soft tissue tumours, such as ERMS (6, 7).
More than 60 cases of MEM have been reported in the literature, predominantly concerning young children and adolescents. Here we expand the knowledge of this rare tumour, by describing the clinicopathologic spectrum of the 22 cases that have not been previously reported as well as reviewing the previously reported cases, highlighting the supportive role of immunohistochemistry and molecular analysis for diagnosis.
The patient was a 6-month-old male infant, treated in another hospital for abdominal rhabdomyosarcoma (RMS). The patient was treated according to EpSSG (European paediatric Soft tissue sarcoma Study Group) protocol, with 4 IVADo (Ifosfamide, Vincristine, Actinomycin, Doxorubicin) cycles (8). In the post-chemotherapy Magnetic Resonance Imaging (MRI), the patient showed a stable disease according to Response Evaluation Criteria in Solid Tumours (RECIST v1.1) (9).
Two more chemotherapy cycles were performed according to the VIT (Vincristine, Irinotecan and Temozolomide) regimen (10), and the subsequent radiological evaluation showed a stable disease (SD) according to RECIST 1.1 with a mild tumour volume reduction, inferior to 20%. Then he was referred to our hospital, where radical excision of the tumour was performed without surgical complication. Histological examination demonstrated that the tumour presented two different elements. The main component was composed by mesenchymal and neuroectodermal elements. The first element consisted of spindle cells, similar to rhabdomyosarcomatous elements. Immunoistochemical staining showed positivity for desmin and myogenin. Thus, ERMS was considered. The tumour’s neuroectodermal element showed ganglion-like cells, positive for synaptophysin, so ganglioneuroma was considered. Based on the pathological findings, a malignant ectomesenchymoma suspected (Figure 1).
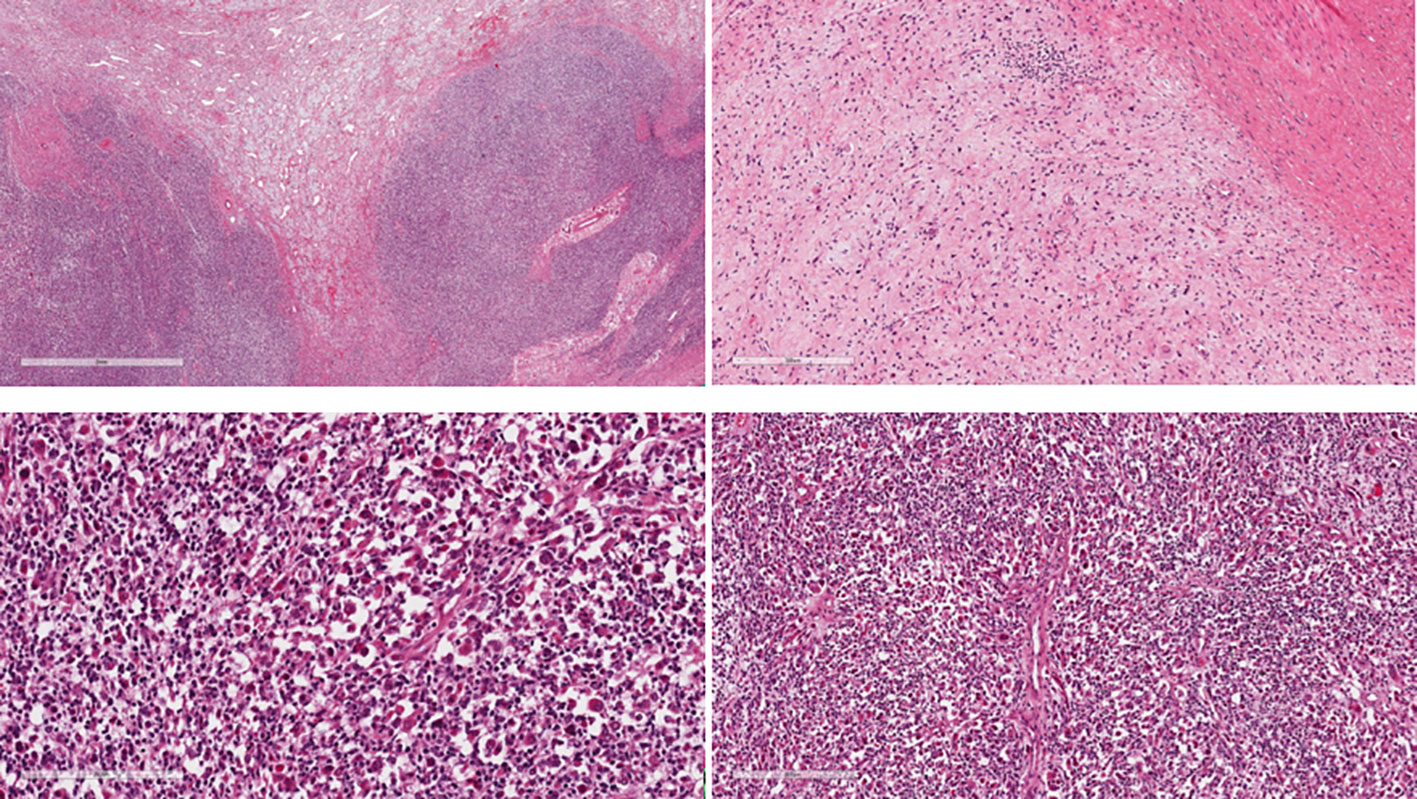
Figure 1 The tumour shows biphasic atypical cells composed of mesenchymal and neuroectodermal elements predominantly intermingled but also with distinct borders at places.
A Whole Exome Sequencing (WES) analysis was performed on a surgical tumour specimen with Illumina® DNA platform to detect DNA tumour mutations. The analysis revealed the presence of somatic mutation HRAS : NM_176795:exon2:c.G37C:p.G13R (Variant Allele Frequency (VAF):72%), already reported in MEM cases (3). No mutations of FOX1 were detected.
HRAS G13R is hotspot mutation that lies within the Guanosine-5’-triphosphate (GTP) binding domain of Harvey Rat Sarcoma Virus Oncogene (HRAS) protein, and results in activation of Mapk and Pi3k signalling (11). G13R mutation predicts a loss of HRAS protein function (11).
The patient received two more VIT regimen cycles as postoperative chemotherapy. Then, a computer-tomography (CT) was performed, which showed a stable complete remission. Therefore, the patient was discharged and prescribed maintenance chemotherapy with Cyclophosphamide and Vinorelbine for 12 months (12).
A first radiological evaluation with CT was performed 3 months after starting maintenance, and a complete remission was confirmed. However, the radiological evaluation performed 6 months after the beginning of maintenance treatment, showed a rectal tumour mass at the scar site of the previous surgery. A complete staging was performed with MRI and a whole-body Positron Emission Tomography (PET)-CT scan to exclude any distant metastasis. Due to the location of the tumour, which infiltrated nearby organs (rectum, bladder, and prostate), surgeons performed a mass debulking because a complete resection was not feasible. Then, two chemotherapy courses following a Topotecan and Cyclophosphamide regimen were administered (13). Approximately 2 months after tumour recurrence, the patient’s clinical condition worsened, and he passed away.
The mesenchymal component shows intersecting fascicles of pleomorphic spindle cells similar to rhabdomyosarcomatous elements. The cells present vesicular nuclei and a scant amount of eosinophilic cytoplasm.
The neuroectodermal component is arranged in loose irregularly oriented bundles with isolated ganglion-like cells with copious eosinophilic cytoplasm.
Iimmunohistochemistry: RMS cell: anti-desmin and anti-myogenin positive. GN cells: anti- Synaptophysin and S-100 positive (images not available).
In this retrospective study, we conducted a systematic literature search of MEDLINE, EMBASE and Pubmed databases to identify studies describing cases of MEMs. The search and selection of articles was carried out in accordance with PRISMA guidelines.
No restrictions were imposed on the language and type of studies, including case reports that described patients with MEMs. All human patients with a confirmed histological diagnosis of MEM, with no restrictions on age or other demographics, were included.
The following data were collected from the studies retrieved: first author, year of publication, type of article, number of cases described, sex and age of the patients, past medical history, clinical manifestation at onset, disease site radiographic imaging, histology and immunohistochemistry, type of treatment (surgery, medical therapy, clinical and/or imaging surveillance), follow-up, outcome. The databases research produced a total of around 90 literature articles on MEMs. Applying the filters to select articles subsequent to the last review of Nael et al., and after application of PRISMA guidelines, 12 article were retrieved, for a total of 33 new cases (Figure 2).
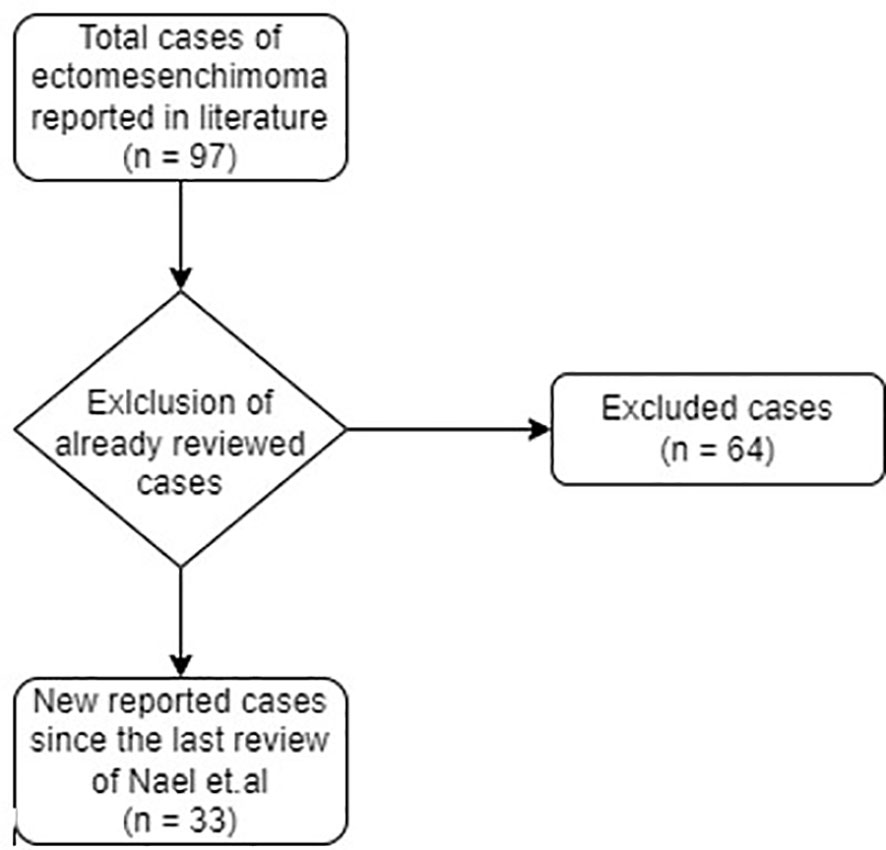
Figure 2 Selection of new MEMs cases, reported in literature since the last review of 2014.
Including our case, 98 MEM cases were described in literature. Freitas et al. initially reported 40 MEM cases from 1946 to 1998 (14), while Nael et al. described a further 24 MEM cases, from 1998 to 2014, with related data regarding gender, age, primary site, histology pattern, treatment, and survival of patients since the time of presentation (15). After reviewing the literature from 2014 to present, we found another 33 MEM cases. The clinic-pathologic features of new reported cases of MEM and our new case are summarised in Table 1.
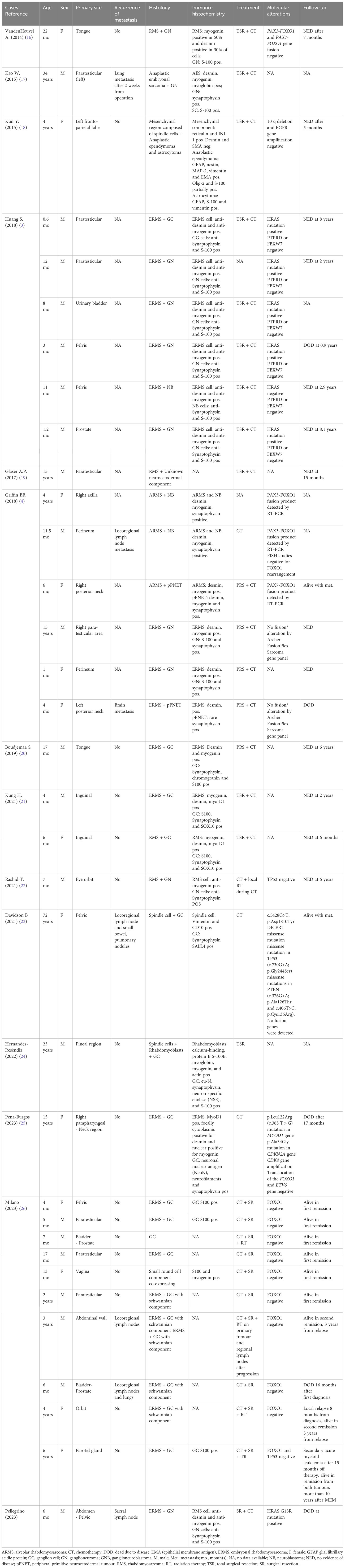
Table 1 Cases of malignant ectomesenchymoma reported in literature after the last Review of Nael et al. in 2014.
Thirteen (38%) of these are females and 21 (62%) are males. The age at diagnosis ranged from 0.6 months to 72 years, with a median age of 12 months. Eighteen patients (53%) developed the tumour in the first year of life (3, 4, 21, 22, 26).
The most common tumour localisation was the genitourinary/pelvic region (23 cases, 67%) (3, 4, 17, 19, 21, 23, 26). ERMS was the prevalent mesenchymal component in the majority (21/34) of tumours (3, 4, 20, 21, 25, 26).
Immunohistochemical analysis revealed diffuse positivity in ERMS-like mesenchymal elements for desmin and myogenin, while neuroectodermal elements (GNB, GN e GC) presented a diffuse positivity for synaptophysin and S-100. Interestingly, in cases of MEMs with ARMS-like and NB elements, both type of cells presented diffuse positivity for desmin, myogenin and synaptophysin.
Seventeen patients received primary total surgical excision of the tumour (3, 16–19, 21, 24), while 11 underwent partial surgical excision (4, 20, 26), in both cases followed and/or preceded by chemotherapy. The chemotherapy regimen was the same used for RMS which included ifosfamide, vincristine, actinomycin-D (IVA) or ifosfamide, vincristine, actinomycin-D, doxorubicin (IVADo) for nine courses, plus surgery and/or radiotherapy according to the risk of local failure (27).
Radiotherapy (RT) was performed in five patients (22, 26).
Locoregional lymph-nodes metastases were reported in five patients (4, 23, 26) and four patients also had distant metastatic disease. The most common site of distant metastasis were lungs (75%). Distant metastasis was always detected at first diagnosis, except for one patient with lung metastasis, in whom they were detected after 2 weeks from surgery. The presence of distant metastasis is more common in patients with abdominal/pelvis MEM and is usually associated with a worst prognosis.
Griffin et al. detected rearrangements of the FOXO1 gene in three cases with alveolar RMS morphology, including two with PAX2-FOXO1 fusion, and one PAX7-FOXO1 translocation (4).
Moreover, Huang et al. studied seven MEMs by RNA sequencing and found HRAS, PTPRD and FBXW7 mutations respectively in six, two and one cases. No fusion genes were reported. They observed oncogenic mutations in RAS signalling pathway also in the control paediatric ERMS. The HRAS mutations detected in the MEMs case described by Huang, were identical to 3 ERMS cases reported in literature (3). Furthermore, Davidson and co-workers sequenced the paraffin-embedded tissue (FFPE) material from the patient’s lymph node metastasis, containing both neoplastic cellular elements, in which they reported the presence of missense mutations in DICER1, TP53 and PTEN (23). However, molecular studies of MEMs are so far restricted to these few reports (3, 4, 23).
Follow-up data are available for 28 patients (82%), of whom 23 were surviving with no evidence of disease (NED) following multimodality treatment approach, while 4 died due to disease (DOD) and 1 was alive with metastasic disease. Follow-up period ranged from 5 to 72 months, with an average of 38.5 months. Five patients were lost to follow-up.
Malignant ectomesenchymoma (MEM) is an extremely rare tumour, with only about 75 cases having been reported to date (14). The last review was conducted by Nael et al. in 2014 (15). Therefore, the aim of this review is to update the epidemiologic and clinical data about this rare disease, to improve our still limited knowledge of biological behaviour, histological characteristics, treatment and prognosis.
MEMs present most commonly in children, primarily involving infants during the first year of life (14). The most common anatomical site is pelvic and abdominal region, followed head and neck, and mediastinum (3). Our review shows a male-to-female ratio of 1.75, confirming a slightly male predominance. MEMs are composed of both neuroectodermal and one or more mesenchymal neoplastic elements (1). Combining data from the Freitas et al. and the Nael et al. studies (14, 15), and our own observations, the most common mesenchymal element is ERMS, and less often other variants. The neuroectodermal elements may cover the entire spectrum of neuroblastic phenotypes, but they were predominantly GN and GC. Only sporadic cases have been reported with malignant peripheral nerve sheath tumour (MPNST) (7, 28).
In most cases, the malignant component consisted of the mesenchymal elements, as in our patient.
MEMs should be distinguished from another biphenotypic neuromuscular tumour, known as “benign triton tumour” or “neuromuscular choristomas”, which also contains neural tissue and skeletal muscle at varying levels of differentiation. The main difference would consist in the absence of malignant degeneration in benign triton tumours. However, VandenHeuvel reported the case of a 35-month-old girl affected by MEM, arising in association with benign triton tumour in the tongue. This finding can suggest that also triton tumours may have malignant potential and may have a possible relationship with MEMs (16).
Previous molecular studies performed on MEMs have revealed chromosomal changes and gene rearrangements. Though quite limited, the data reported in literature demonstrate remarkable overlapping characteristics between MEMs and RMS, including demographic features (male predominance, young age < 2 years), anatomic distribution and immunohistochemistry. In particular the retained H3K27me3 expression, reported in all the 7 cases described by Huang (23) suggest a closer relationship to RMS than MPNST, in which H3K27me3 expression is lost. Unfortunately, the major limitation of this study is the impossibility to perform H3K27me3 expression in our patient.
In the recent years, also cytogenetic abnormalities suggest that MEM might potentially constitute a variant of RMS (3, 6, 7, 29). For example, the DICER1 mutation reported by Davidson and previously not described in MEM, has been described in ERMS as well as in RMS and in anaplastic sarcoma, consolidating the link between MEM and ERMS evidenced in the Huang report (23).
The latest edition of World Health Organization classification of soft tissue and bone tumours categorised MEM under “Skeletal Muscle Tumor” (30), while in the 2013 edition, they were classified as nerve sheath tumours. All these data suggest a stronger link to RMS than MPNST, as previously suggested by Kleinschimidt-DeMasters et al, who found that intracranial MEMs showed gene expression pattern similar to MPNST (28).
Nevertheless, due to the singularity of these tumours, the diagnostic criteria of MEM are not well defined and may be ambiguous; it is also challenging to determine prognostic factors and outcomes. Some MEMs have initially been diagnosed as pure RMS because the biphasic histological pattern characteristic of the MEM can be hardly detected in small biopsies (31).
Moreover, most cases showed a wide range of growth patterns, including myxoid-cellular areas, fascicular spindle cell and compact round cells, and immunomarkers of multiple lineages of differentiation, which complicate the diagnosis. Also in our case, the first diagnosis was ERMS, and the patient was treated according to the RMS protocols. However, after examining a larger amount of tissue obtained from the surgery, our final diagnosis was MEM.
This, along with other cases of MEMs, has taught us to be prudent in the histological diagnosis of MEM because biphasic, heterologous tumours may not be correctly identified when only small specimens are examined on routine hematoxylin and eosin-stained sections; ancillary studies may be required, such as immunohistochemistry, electron microscopy, or molecular analysis. It is supposed that the more undifferentiated the tumour is, the higher the probability of expressing markers of various differentiation lineages. Molecular data can even provide diagnostic accuracy in cases that are tricky to interpret.
It is well-known that, regarding soft tissue pathologies, molecular characterisation can contribute untangle the equivocal morphologic overlap between subtypes of sarcomas, especially when they arise in non-canonical anatomical sites (32).
Nevertheless, the results of molecular studies must always be contextualised in the context of an accurate histopathologic evaluation. In fact, a detected genetic mutation is not in itself a diagnosis, but merely a partial indicator.
Therefore, we think that the future diagnostic approach should routinely include molecular analysis and the use of bioinformatic mutation detection tools, in order to find peculiar gene rearrangements and mutations of MEMs. This will be integrative and supportive for the pathologist and fundamental in providing a better understanding of the disease aetiology of MEMs and in the search for new therapeutic targets and biomarkers.
General consensus for the management and therapeutic regimen for MEM seems to be a multimodal treatment strategy, including a combination of surgery and chemotherapy (7, 20). When the tumour is unresectable or the disease has metastasised in other sites, the prognosis is worse (15, 17). Considering the 5 patients lost at follow-up, our review revealed MEMs to have the same prognosis as other paediatric chemotherapy sensitive soft tissue sarcomas, with 82% (14/17) of children affected by MEM surviving with no evidence of disease (NED) following multimodality treatment approach.
MEMs have different and peculiar characteristics that distinguish them from other soft tissue sarcomas and an increased risk of delay in diagnosis. Despite the paucity of reported data, we emphasise the importance of an integrative morpho-molecular approach to support the diagnosis and understanding of the biology and behaviour of this rare and insidious tumour.
FP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. ET: Conceptualization, Data curation, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. FD: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. AC: Data curation, Investigation, Methodology, Software, Supervision, Validation, Writing – review & editing. CR: Data curation, Investigation, Validation, Writing – review & editing. EA: Data curation, Investigation, Validation, Writing – review & editing. AL: Formal analysis, Investigation, Methodology, Supervision, Writing – review & editing. SA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – review & editing. FF: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank our patient and previous investigators who have documented their findings.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MEM, Malignant Ectomesenchymoma; RMS, Rhabdomyosarcoma; NGS, Next-Generation Sequencing; ERMS, Embryonal rhabdomyosarcoma; NB, Neuroblastoma; GN, Ganglioneuroma;
GNB, Ganglioneuroblastoma; GC, Ganglion cells; ARMS, Alveolar rhabdomyosarcoma; EpSSG, European paediatric Soft tissue sarcoma Study Group; IVADo, Ifosfamide, Vincristine, Actinomycin, Doxorubicin; MRI, Magnetic Resonance Imaging; VIT, Vincristine, Irinotecan and Temozolomide; SD, Stable disease; GTP, Guanosine-5’-triphosphate; HRAS, Harvey Rat Sarcoma Virus Oncogene; CT, Computer Tomography; RT, Radiotherapy; NED, No evidence of disease; DOD, Died of disease; MPNST, Malignant peripheral nerve sheath tumour.
1. Pediatric Treatment Editorial Board PDQ. Childhood Soft Tissue Sarcoma Treatment (PDQ®): Health Professional Version. 2022 Mar 2. In: PDQ Cancer Information Summaries. National Cancer Institute (US, Bethesda (MD (2002).
2. Mouton SC, Rosenberg HS, Cohen MC, Drut R, Emms M, Kaschula RO. Malignant ectomesenchymoma in childhood. Pediatr Pathol Lab Med. (1996) 16:607–24.
3. Huang SC, Alaggio R, Sung YS, Chen CL, Zhang L, Kao YC, et al. Frequent HRAS mutations in Malignant ectomesenchymoma: overlapping genetic abnormalities with embryonal rhabdomyosarcoma. Am J Surg Pathol. (2016) 40:876–85. doi: 10.1097/PAS.0000000000000612
4. Griffin BB, Chou PM, George D, Jennings LJ, Arva NC. Malignant ectomesenchymoma: series analysis of a histologically and genetically heterogeneous tumor. Int J Surg Pathol. (2018) 26:200–12. doi: 10.1177/1066896917734915
5. Karcioglu Z, Someren A, Mathes SJ. Ectomesenchymoma. A Malignant tumor of migratory neural crest (ectomesenchyme) remnants showing ganglionic, schwannian, melanocytic and rhabdomyoblastic differentiation. Cancer. (1977) 39:2486–96.
6. Floris G, Debiec-Rychter M, Wozniak A, Magrini E, Manfioletti G, De Wever I, et al. Malignant ectomesenchymoma: genetic profile reflects rhabdomyosarcomatous differentiation. Diagn Mol Pathol. (2007) 16:243–8. doi: 10.1097/PDM.0b013e3180645105
7. Dantonello TM, Leuschner I, Vokuhl C, Gfroerer S, Schuck A, Kube S, et al. Malignant ectomesenchymoma in children and adolescents: report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer. (2013) 60:224–9. doi: 10.1002/pbc.24174
8. Bisogno G, Ferrari A, Bergeron C, Scagnellato A, Prete A, Alaggio R, et al. The IVADo regimen–a pilot study with ifosfamide, vincristine, actinomycin D, and doxorubicin in children with metastatic soft tissue sarcoma: a pilot study of behalf of the European pediatric Soft tissue sarcoma Study Group. Cancer. (2005) 103:1719–24. doi: 10.1002/cncr.20928
9. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. (2009) 45:228–47. doi: 10.1016/j.ejca.2008.10.026
10. Raciborska A, Bilska K, Drabko K, Chaber R, Pogorzala M, Wyrobek E, et al. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer. (2013) 60:1621–5. doi: 10.1002/pbc.24621
11. Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. (2012) 44:783–7. doi: 10.1038/ng.2316
12. Casanova M, Ferrari A, Bisogno G, Merks JH, De Salvo GL, Meazza C, et al. Vinorelbine and low-dose cyclophosphamide in the treatment of pediatric sarcomas: pilot study for the upcoming European Rhabdomyosarcoma Protocol. Cancer. (2004) 101:1664–71. doi: 10.1002/cncr.20544
13. Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. (2006) 47:795–800. doi: 10.1002/pbc.20719
14. Freitas AB, Aguiar PH, Miura FK, Yasuda A, Soglia J, Soglia F, et al. Malignant ectomesenchymoma. Case report and review of the literature. Pediatr Neurosurgery. (1999) 30:320–30. doi: 10.1159/000028818
15. Nael A, Siaghani P, Wu WW, Nael K, Shane L, Romansky SG. Metastatic Malignant ectomesenchymoma initially presenting as a pelvic mass: report of a case and review of literature. Case Rep Pediatr. (2014) 2014:792925. doi: 10.1155/2014/792925
16. VandenHeuvel KA, Carpentieri DF, Chen J, Fung KM, Parham DM. Ectomesenchymoma with embryonal rhabdomyosarcoma and ganglioneuroma, arising in association with benign triton tumor of the tongue. Pediatr Dev Pathol. (2014) 17:226–30. doi: 10.2350/14-01-1433-CR.1
17. Kao WT, Chiang YT, Tzou KY. An adult paratesticular Malignant ectomesenchymoma with post-operative flare-up of lung metastasis. Urol Case Rep. (2015) 3:164–6. doi: 10.1016/j.eucr.2015.06.010
18. Kun Y, Duan Z, Mei X, Xu Y, Li J, Li S, et al. A rare case of Malignant pediatric ectomesenchymoma arising from the cerebrum. Int J Clin Exp Pathol. (2015) 8:8545–50.
19. Glaser AP, Bowen DK, Lindgren BW, Meeks JJ. Robot-assisted retroperitoneal lymph node dissection (RA-RPLND) in the adolescent population. J Pediatr Urol. (2017) 13:223–4. doi: 10.1016/j.jpurol.2017.01.007
20. Boudjemaa S, Petit A. Malignant ectomesenchymoma: A potential pitfall of diagnosis in the spectrum of pediatric small blue round cell tumors. Appl Immunohistochem Mol Morphol. (2019) 27:e63–4. doi: 10.1097/PAI.0000000000000584
21. Kung HC, Hsiao YT, Huang HY, Hsiao CC. Infant Malignant ectomesenchymoma masquerading as inguinal hernia in two patients. Pediatr Neonatol. (2021) 62:324–6. doi: 10.1016/j.pedneo.2021.01.003
22. Rashid T, Bagatell R, Pawel B, Bentley RC, Kreissman SG, Deel MD. More than meets the eye? A cautionary tale of Malignant ectomesenchymoma treated as low-risk orbital rhabdomyosarcoma. J Pediatr Hematol Oncol. (2021) 43:e854–8. doi: 10.1097/MPH.0000000000001901
23. Davidson B, Kleinberg L, Børresen IM, Slettevoll F, Fangberget A, Hindosh D, et al. Primary uterine ectomesenchymoma harboring a DICER1 mutation: case report with molecular analysis. Virchows Arch. (2021) 479:419–24. doi: 10.1007/s00428-021-03057-x
24. Hernández-Reséndiz R, Villanueva-Castro E, Chávez-Macías L, Gómez-Apo E, Ortiz-Plata A, Salinas-Lara C, et al. Teratoma with Malignant ectomesenchymoma in the pineal region: A case report. Cureus. (2022) 14:e27711. doi: 10.7759/cureus.27711
25. Pena-Burgos EM, De Sabando DP, Utrilla C, Pozo-Kreilinger JJ, Sastre A, Rubio P, et al. First reported case of Malignant ectomesenchymoma with p.Leu122Arg mutation in MYOD1 gene: extensive intra- and extracranial tumor in a 15-year-old female. Head Neck Pathol. (2023) 17:855–63. doi: 10.1007/s12105-023-01542-0
26. Milano GM, Orbach D, Casanova M, Berlanga P, Schoot RA, Corradini N, et al. Malignant ectomesenchymoma in children: The European pediatric Soft tissue sarcoma Study Group experience. Pediatr Blood Cancer. (2023) 70:e30116. doi: 10.1002/pbc.30116
27. Bisogno G, Jenney M, Bergeron C, Melcón SG, Ferrari A, De Salvo GL, et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. (2018) 19:1061–1071. doi: 10.1016/S1470-2045(18)30337-1
28. Kleinschmidt-DeMasters BK, Lovell MA, Donson AM, Wilkinson CC, Madden JR, Addo-Yobo SO, et al. Molecular array analyses of 51 pediatric tumors shows overlap between Malignant intracranial ectomesenchymoma and MPNST but not medulloblastoma or atypical teratoid rhabdoid tumor. Acta Neuropathol. (2007) 113:695–703. doi: 10.1007/s00401-007-0210-0
29. Boué DR, Parham DM, Webber B, Crist WM, Qualman SJ. Clinicopathologic study of ectomesenchymomas from Intergroup Rhabdomyosarcoma Study Groups III and IV. Pediatr Dev Pathol. (2000) 3:290–300. doi: 10.1007/s100249910039
30. Choi, Hyuk J, Ro JY. The 2020 WHO classification of tumors of soft tissue: selected changes and new entities. Adv In Anatomic Pathol. (2021) 28:44–58. doi: 10.1097/PAP.0000000000000284
31. Howley S, Stack D, Morris T, McDermott M, Capra M, Betts D, et al. Ectomesenchymoma with t(1;12)(p32;p13) evolving from embryonal rhabdomyosarcoma shows no rearrangement of ETV6. Hum Pathol. (2012) 43:299–302. doi: 10.1016/j.humpath.2011.03.010
Keywords: malignant ectomesenchymoma, soft tissue tumor, morpho-molecular analysis, pediatric oncology, next generation sequencing
Citation: Pellegrino F, Tirtei E, Divincenzo F, Campello A, Rubino C, Augustoni E, Linari A, Asaftei SD and Fagioli F (2024) An integrative morpho-molecular approach in malignant ectomesenchymoma diagnosis: report of a new paediatric case and a review of the literature. Front. Oncol. 14:1320541. doi: 10.3389/fonc.2024.1320541
Received: 12 October 2023; Accepted: 08 February 2024;
Published: 01 March 2024.
Edited by:
Jeremy Wang, University of North Carolina at Chapel Hill, United StatesReviewed by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2024 Pellegrino, Tirtei, Divincenzo, Campello, Rubino, Augustoni, Linari, Asaftei and Fagioli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Pellegrino, Zi5wZWxsZWdyaW5vQHVuaXRvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.