 Yu-jia Jiang
Yu-jia Jiang Yun Xia
Yun Xia Zhuo-jun Han
Zhuo-jun Han Yi-xuan Hu
Yi-xuan Hu Tao Huang
Tao Huang- 1Department of Breast and Thyroid Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China
- 3West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu, China
Familial non-medullary thyroid carcinoma (FNMTC) is a type of thyroid cancer characterized by genetic susceptibility, representing approximately 5% of all non-medullary thyroid carcinomas. While some cases of FNMTC are associated with familial multi-organ tumor predisposition syndromes, the majority occur independently. The genetic mechanisms underlying non-syndromic FNMTC remain unclear. Initial studies utilized SNP linkage analysis to identify susceptibility loci, including the 1q21 locus, 2q21 locus, and 4q32 locus, among others. Subsequent research employed more advanced techniques such as Genome-wide Association Study and Whole Exome Sequencing, leading to the discovery of genes such as IMMP2L, GALNTL4, WDR11-AS1, DUOX2, NOP53, MAP2K5, and others. But FNMTC exhibits strong genetic heterogeneity, with each family having its own pathogenic genes. This is the first article to provide a chromosomal landscape map of susceptibility genes associated with non-syndromic FNMTC and analyze their potential associations. It also presents a detailed summary of variant loci, characteristics, research methodologies, and validation results from different countries.
1 Introduction
Familial non-medullary thyroid carcinoma (FNMTC) refers to a form of thyroid cancer that occurs in families, where there are at least two cases of thyroid cancer of follicular epithelial cell origin in first-degree relatives, excluding individuals with a history of exposure to known thyroid-cancer-causing factors (1, 2). It accounts for approximately 5% of all non-medullary thyroid carcinoma (1, 2). Epidemiological studies have shown that first-degree relatives of FNMTC patients have a 4 to 10 times higher risk of developing the disease compared to the general population. If two individuals in a family are affected, the probability of it being hereditary is estimated at 31% to 38%. If three or more individuals are affected, the probability of it being hereditary is over 94% (3).
Apart from a subset of FNMTC cases (approximately 5%) that are associated with syndromes FNMTC, the majority of FNMTC cases (approximately 95%) occur independently (4). This subset is also known as non-syndromic FNMTC, and the genetic mechanisms underlying this condition remain unclear. The initial studies utilized SNP linkage analysis to identify susceptibility genes for FNMTC, such as the 1q21 fPTC/PRN locus (5), 2q21 NMTC1 locus (6), and 4q32 locus (7). Subsequently, the research progressed to using Genome-wide association study (GWAS), identifying genes like IMMP2L at 7q31.1 (8), GALNTL4 at 11p15.4 (9), WDR11-AS1 at 10q26.12 (10). Alternatively, Whole exome sequencing (WES) was employed to investigate FNMTC susceptibility genes by focusing on one or several pedigrees, resulting in the discovery of genes like DUOX2 at 15q21.1 (11), NOP53 at 19q13.33 (12), and MAP2K5 at 15q23 (13).
However, it is important to note that the identified susceptibility genes, such as MAP2K5 (15q23) (13–15), HABP2 (10q25.3) (16–25), and the 19q13.2 locus (TCO) (26–30), primarily exist within specific families and may not be universally applicable across different families. FNMTC exhibits strong genetic heterogeneity, with each family having its specific pathogenic genes. It is the combination of these genes that may constitute the true susceptibility gene pool for FNMTC.
Therefore, this article provides a comprehensive overview of the chromosomal landscape of susceptibility genes associated with non-syndromic FNMTC (Figure 1). Additionally, it offers a detailed summary of research findings, including information on variant loci, variant characteristics, research methodologies employed, and validation results from various countries.
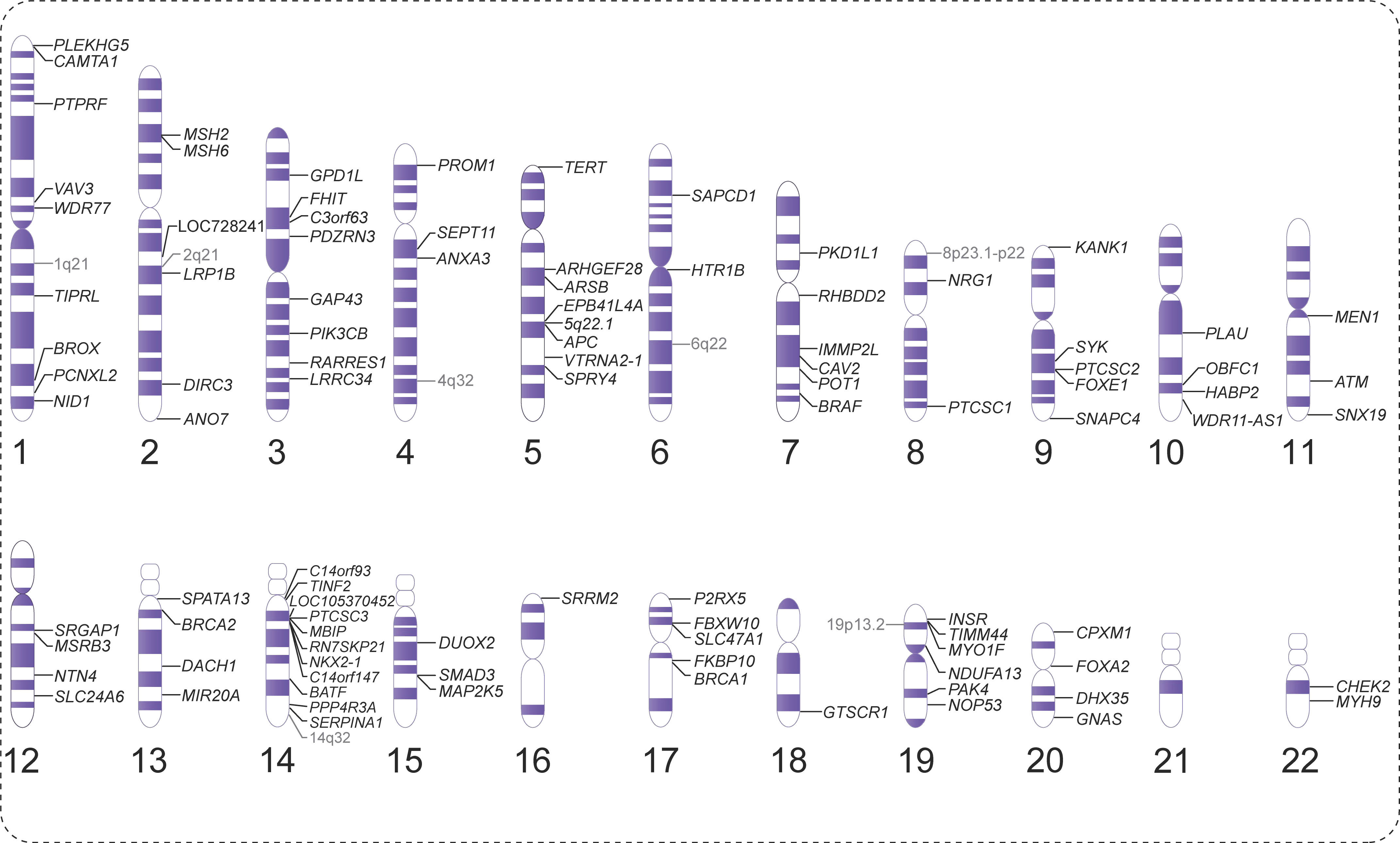
Figure 1 Overview of the chromosomal landscape of susceptibility genes associated with non-syndromic non-medullary thyroid carcinoma (NMTC).
2 Susceptibility chromosomal loci associated with risk of non-syndromic FNMTC
Initially, studies predominantly used SNP linkage analysis to identify susceptibility loci associated with FNMTC, but without specific genes. These loci are mostly related to specific types of NMTC. For instance, the discovery of the 1q21 locus is linked to papillary renal neoplasia (PRN) (31), the 2q21 locus is associated with follicular variant papillary thyroid carcinoma (fvPTC) (6), 4q32 locus was identified in 11 PTC patients and 2 families with undifferentiated thyroid cancer (7), 14q32 locus may be related to multinodular goiter (MNG) (32), and the 19p13.2 microdeletion may be associated with thyroid cancer with oncocytic (TCO) features (26), and so on. However, except for specific subtypes, the aforementioned susceptibility loci have been rarely validated in other NMTC (7, 26, 30, 30, 33, 34). For example, Suh (5) conducted a whole-genome SNP array analysis on 38 families. A significant correlation with chromosome locus 6q22 (the same chromosomal region as the PRN1 locus) and FNMTC was found, with an LOD score of 3.30. Similarly, Cavaco (33) performed SNP linkage analysis on 11 affected family members of Portuguese descent. The highest LOD score obtained was +4.41 at the FTEN locus on chromosome 8p23.1-p22. This suggests that the association between the chromosomal locus and FNMTC may not be universally applicable and could vary among different populations or families.
Significantly, the chromosomal locus at 19p13.2 has been implicated in numerous subsequent studies, exhibiting associations with various genes such as INSR (35), TIMM44 (36), NDUFA13/GRIM-19 (37) and MYO1F (38). Several studies have reported eosinophilic alterations in cells associated with TCO features (27–29). These alterations include abnormal mitochondrial proliferation in pro-oxidant or cancer cells. It is also the locus with the highest number of associated genes for FNMTC identified to date. This provides valuable insights for the fine mapping of susceptibility genes related to specific types of thyroid cancer. Table 1 summarizes the chromosomal loci associated with FNMTC.
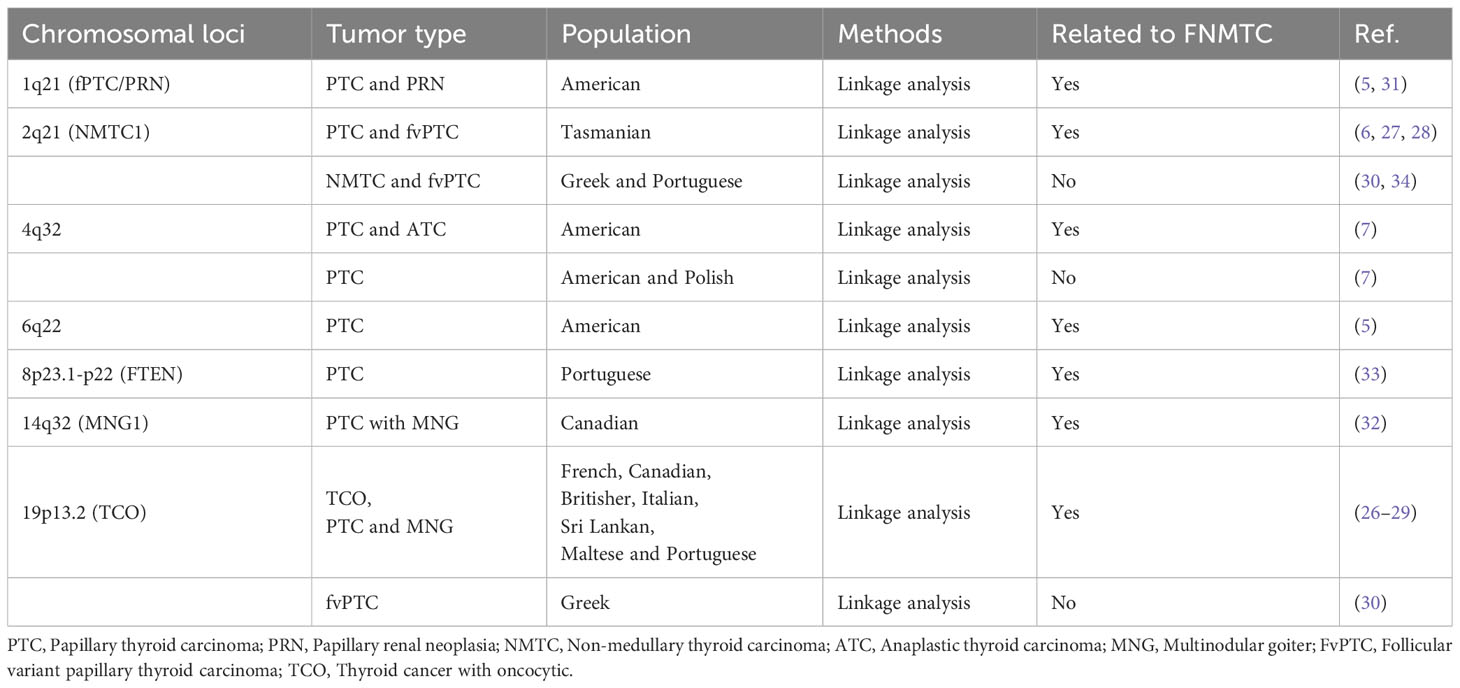
Table 1 Susceptibility chromosomal loci associated with risk of non-syndromic FNMTC.
3 RNA-associated susceptibility genes of FNMTC
RNA-associated susceptibility genes refer to genes that play important roles in the synthesis, modification, and regulation of RNA, and are associated with disease susceptibility. Currently, several RNA and genes associated with FNMTC have been identified, as shown in Table 2.
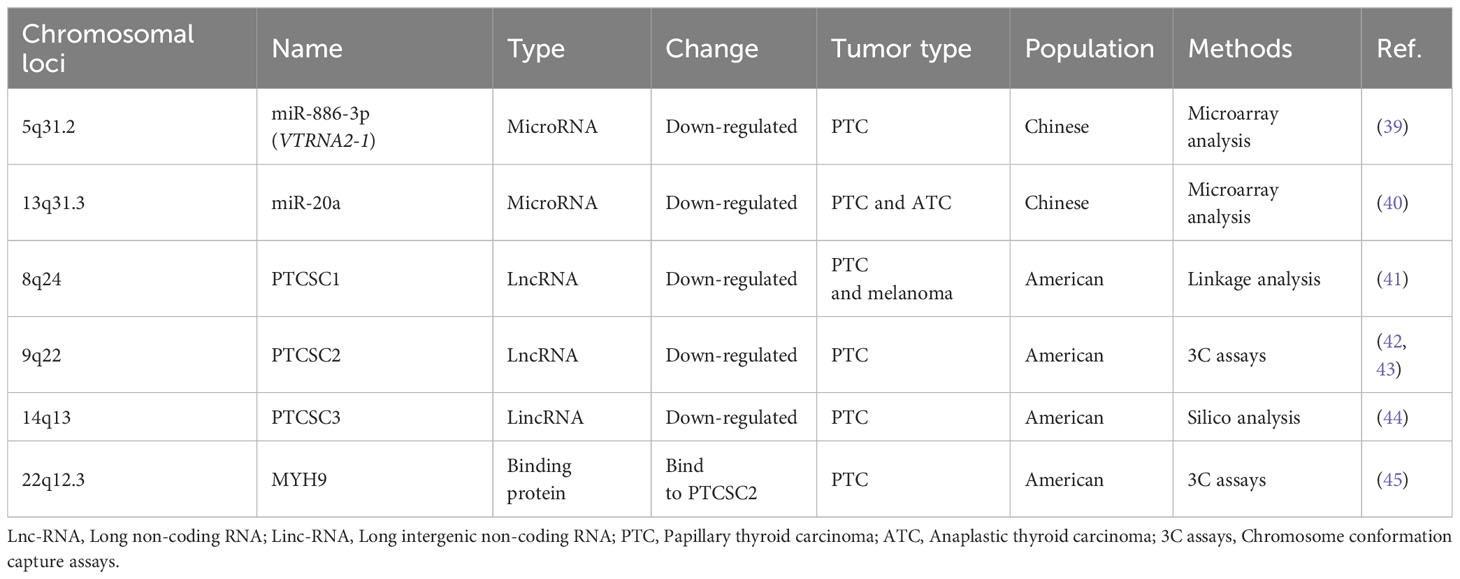
Table 2 RNA-associated susceptibility genes.
Firstly, the researchers found that miR-886-3p was down-regulated by 3-fold in FNMTC compared to SNMTC, while miR-20a was down-regulated by 4-fold in FNMTC compared to SNMTC. Overexpression of miR-886-3p was found to inhibit the expression of DNA replication gene CDC6 and focal adhesion genes PIP5K1C, PXN, and ZYX. This overexpression of miR-886-3p resulted in the inhibition of cell proliferation and invasion, keeping the cells in the S phase. On the other hand, miR-20a is predominantly found in undifferentiated thyroid carcinoma and was shown to down-regulate the expression of LIMK1, a gene associated with reduced cell invasion (39, 40).
Papillary thyroid carcinoma susceptibility candidate (PTCSC) gene families belong to long non-coding RNA (lnc-RNA) that have been implicated in the development of papillary thyroid carcinoma (41). Currently, studies have shown that PTCSC1, PTCSC2, and PTCSC3 are associated with the occurrence of FNMTC.
PTCSC1, also known as AK023948, is a gene that has been found to interact with DHX9 and the PI3K regulatory subunit p85-β (PIK3R2). The activation of AK023948 through this interaction leads to the phosphorylation and activation of protein kinase AKT. However, it is important to note that this specific association between AK023948 and breast cancer cell growth has not been explained or explored in the context of other studies on FNMTC (46).
An interaction between rs965513, a susceptibility SNP for FNMTC, PTCSC2 has been observed. The SNP rs965513 in the 9q22 region play a crucial role in the genetic predisposition to PTC. And the [AA] risk genotype of rs965513 is associated with decreased expression of FOXE1, PTCSC2 unspliced transcript in unaffected thyroid tissue. The decrease in gene abundance is associated with dedifferentiation, which promotes malignant transformation (42, 43). Furthermore, MYH9 has been found to bind to the lncRNA PTCSC2, regulating the p53 signaling pathway by inhibiting FOXE1 expression (45).
PTCSC3, located downstream of SNP rs944289 at a distance of approximately 3.2 kb, exhibits characteristics of tumor suppressor genes (44). Rogounovitch et al. (47) confirmed the association of the T allele of SNP rs944289 with the risk of both PTC and benign thyroid nodules in the Japanese population.
4 Candidate genes identified by WES and susceptibility genes identified by GWAS
GWAS is a method used to detect genetic variations and polymorphisms across the whole genome in multiple individuals. GWAS aims to identify genetic variations that are most likely to influence a specific trait by examining the genotype-phenotype relationship. So far, GWAS studies from various populations have revealed several susceptibility genes for FNMTC research (8–10, 35, 48, 49) (Tables 3, 4).
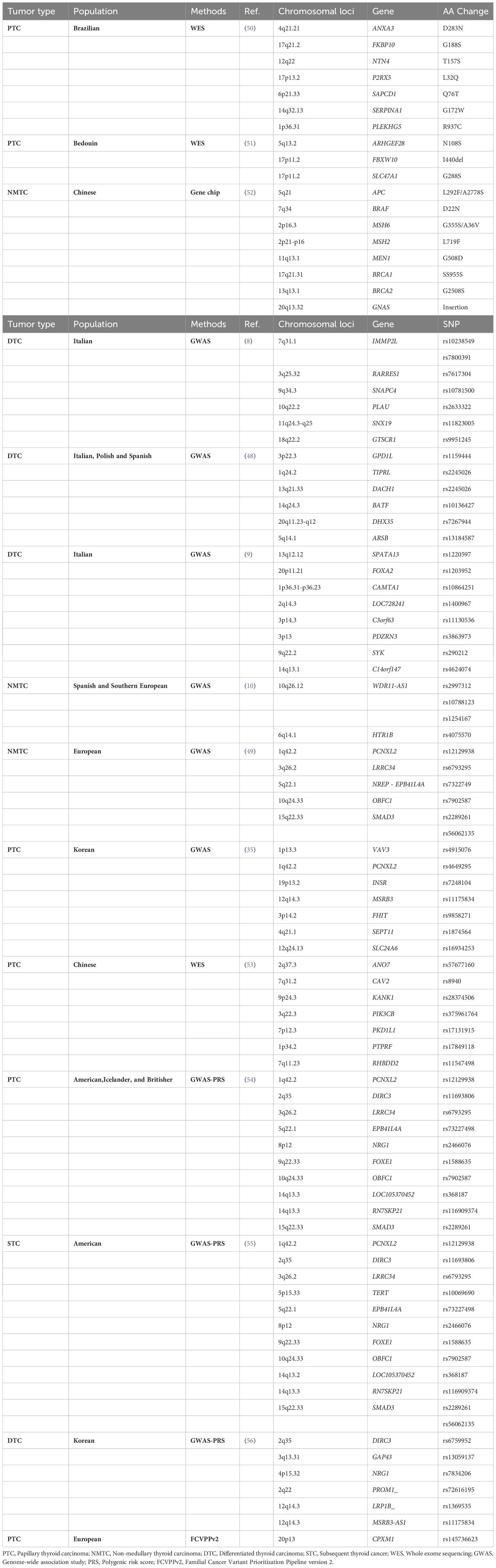
Table 3 Candidate genes identified by whole exome sequencing (WES) and susceptibility genes identified by genome-wide association studies (GWAS).
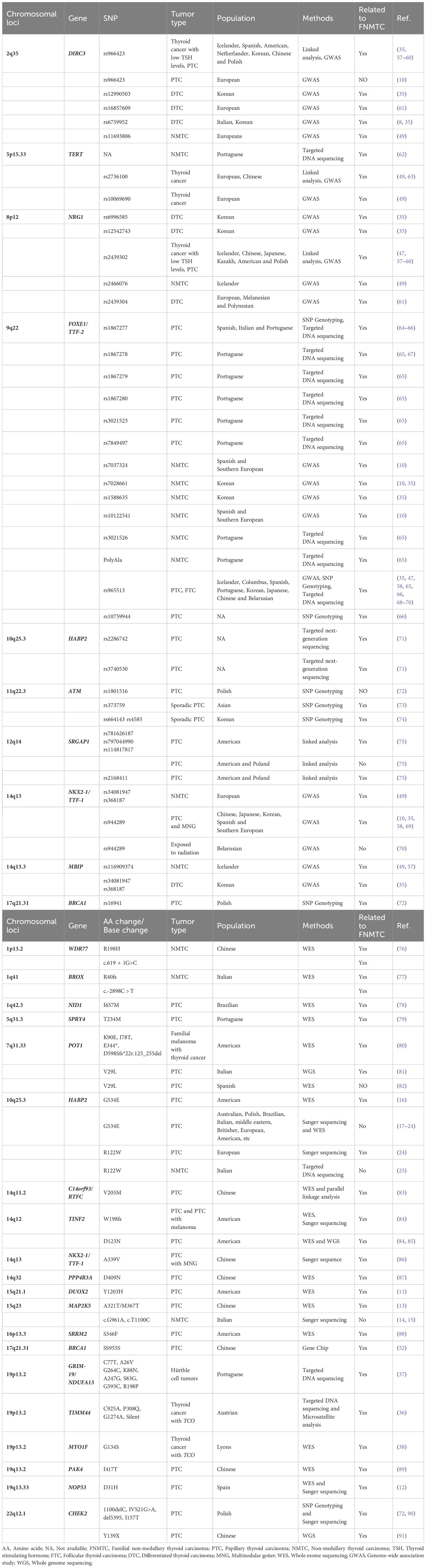
Table 4 Identified susceptibility genes associated with Familial non-medullary thyroid carcinoma.
Although GWAS studies have identified numerous risk SNPs, FNMTC cannot currently be explained by single-gene variants. Therefore, researchers have started using polygenic risk scores (PRS) to assess the risk of developing thyroid cancer.
For example, a study utilizing GWAS data from the United States, Iceland, and the United Kingdom proposed a 10-SNP PRS assessment model (rs12129938, rs11693806, rs6793295, rs73227498, rs2466076, rs1588635, rs7902587, rs368187, rs116909374, and rs2289261), which could be applied for personalized assessment of thyroid cancer susceptibility (54). A study analyzed the PRS of 12 thyroid cancer-associated SNPs (rs11693806, rs2466076, rs1588635, rs368187, rs116909374, rs12129938, rs6793295, rs73227498, rs7902587, rs2289261, and rs56062135) in childhood cancer survivors of European population. The findings showed that for each one standard deviation increase in PRS (1.57) (55). Furthermore, another study compared the high PRS tertile with the low PRS tertile and found that the PRS of six SNPs (rs6759952, rs13059137, rs7834206, rs72616195, rs1369535, rs11175834) increased the risk of thyroid cancer by 3.9 times (56). These studies suggest that utilizing PRS can better assess an individual’s risk of developing thyroid cancer. However, it is important to note that PRS is still evolving, and further validation and refinement are necessary before widespread clinical implementation. (Table 3).
In addition to the aforementioned PRS method, Asta et al. (92) introduced the familial cancer variant prioritization pipeline (FCVPP), which is a comprehensive approach for analyzing germline genomes in Mendelian cancer pedigrees. This pipeline involves variant calling, quality control, frequency screening, segregation analysis, and bioinformatics assessment. By considering these steps and ensuring accurate pedigree information, researchers can enhance the likelihood of identifying novel genes associated with Mendelian types of cancer. Building upon this, Abhishek et al. (93) introduced FCVPPv2, an enhanced pipeline for prioritizing variants in pedigrees that incorporates multiple tools to assess variant deleteriousness and intolerance scores. The pipeline also encompasses the evaluation of non-coding regions using various datasets and tools. Application of the pipeline on a papillary thyroid cancer family revealed one variant in the CPXM1 gene (c.G1717A:p.G573R), suggesting its potential involvement in tumorigenesis (Table 3). However, further functional characterization is required to validate its role in cancer predisposition. Overall, FCVPPv2 provides a comprehensive approach for predicting high-risk cancer predisposing variants in familial cancer pedigrees.
WES is also widely used for the screening of candidate genes. However, most of these loci have not been mentioned or functionally validated in other studies, as indicated in Table 3.
For example, Yang et al. (52), identified 10 germline mutations were found in 8 genes, including APC (L292F/A2778S), BRAF (D22N), MSH6 (G355S/A36V), MSH2 (L719F), MEN1 (G508D), BRCA1 (SS955S), BRCA2 (G2508S), and GNAS insertions. The mutation rate of these eight genes was found to be 53.2% in the FNMTC group and 31.3% in the SNMTC group. However, it is important to note that the sample size might have contributed to the lack of statistical significance in distinguishing FNMTC from SNMTC, with an odds ratio of 2.46 and a P-value of 0.16. Furthermore, Majdalani et al. (51) suggested that ARHGEF28 is a promising candidate due to its high expression in the thyroid and potential associations with other genes. Protein-protein interactions suggest that ARHGEF28 may predispose individuals to PTC through associations with SQSTM1-TP53 or PTCSC2-FOXE1-TP53. Zhu et al. (53) identified Seven novel candidate FNMTC pathogenic genes (ANO7, CAV2, KANK1, PIK3CB, PKD1L1, PTPRF, and RHBDD2). Notably, three of these genes (PIK3CB, CAV2, and KANK1) have been linked to tumorigenesis through the PI3K/Akt signaling pathway. Sarquis et al. (50) performed WES on three FNMTC pedigrees in Brazil and identified seven potential FNMTC susceptibility genes. The three FNMTC pedigrees had a total of 11 patients with an average age of diagnosis at 41.5 years.
Although this class only involves the analysis of candidate genes at the level of bioinformatics, it may also have certain significance for expanding the FNMTC gene library. In the future, more verification is needed to confirm the contribution of these genes.
5 Identified susceptibility genes associated with FNMTC
In the FNMTC susceptibility gene map, most of the genes have been validated through other pedigrees or functional studies, but there is controversy surrounding many genes. Some of them may be susceptibility genes for specific subtypes of NMTC, while others may only belong to specific certain populations. This once again highlights that the genetic susceptibility of FNMTC is not determined by a single gene. Therefore, the following text and Table 4 provide detailed information.
5.1 1p13.2 WDR77
WD repeat domain 77 (WDR77), also known as methyltransferase-like protein 50 and androgen receptor-associated protein p44, was studied by Zhao et al. through WES in two unrelated Chinese pedigrees with FNMTC. In one pedigree, a missense mutation (p.R198H) was found in exon 6 of WDR77, while in the other pedigree, a splice site mutation (c.619 + 1G>c) was identified at the 5’ end of intron 6 (76).
WDR77 forms a complex with protein arginine methyltransferase 5 (PRMT5) to regulate the formation of histone H4 arginine 3 dimethylation (H4R3me2) in cells. The functional activity of WDR77 protein is dependent on its subcellular localization. For example, in early developing prostate epithelial cells, WDR77 is located in the cytoplasm and plays a crucial role in cell proliferation (94, 95). Functional studies have shown that mutations in WDR77 disrupt the formation of the PRMT5/WDR77 complex, resulting in reduced H4R3me2 levels within cells. Knockdown of the WDR77 gene leads to an increased proliferation rate in thyroid cancer cells (76).
5.2 q41 BROX
Pasquali et al. conducted WES analysis on five FNMTC pedigrees and identified two pedigrees with loss-of-function mutations in the BROX gene. In one pedigree, a frameshift deletion mutation (chr1:222892283 NM_001288579:c.119delG:p.Arg40fs) occurred in the BROX gene, while in another pedigree, a missense mutation (chr1:222886144 NM_144695:c.2898C>T) was observed (77). However, there is currently no direct evidence of a functional effect associated with the second variant.
BROX encodes human Brox, which contains a Bro1 domain-like sequence involved in the endosomal sorting of cargo proteins and degradation processes in lysosomes, such as integrin and EGFR (96–98). The researchers hypothesized that a BROX mutation is sufficient to alter the degradation of EGFR, leading to the accumulation of EGFR and subsequently abnormal cell growth (77).
5.3 1q42.3 NID1
WES was performed on a Brazilian family with a history of PTC. Previously reported FNMTC susceptibility genes were examined, and novel candidate genes were identified using PhenoDB. No variants in known FNMTC susceptibility genes co-segregated with the disease. However, a missense variant (c.1971T>G:p.Ile657Met) in the NID1 gene co-segregated with the disease and had a low allele frequency. In silico analysis suggested its deleteriousness, and NID1 expression was observed in PTC cells but not normal thyroid tissue. TCGA data showed higher NID1 expression associated with the risk of recurrence and lateral neck lymph node metastasis in PTCs. The NID1 variant in this study shows potential as a novel FNMTC predisposing gene (78).
5.4 2q35 DIRC3
DIRC3 (Disrupted in Renal Cancer 3) was already known to associate hereditary renal cancer and was believed to have tumor suppressor activity (99). In a GWAS study involving 561 Icelandic thyroid cancer cases and 40,013 controls, rs966423 variant of the DIRC3 was found to be associated with thyroid cancer risk and levels of thyroid-stimulating hormone for the first time (57). Multiple reports have shown the prognostic significance of the rs966423 variant and its pathogenic effects in cases of DTC (35, 57–60). Subsequent replication studies identified additional variants, rs6759952 (8, 35) and rs12990503 (35). However, Mankickova et al. (10) were unable to establish an association between rs966423 and thyroid cancer in European populations. More recently, GWAS analysis discovered novel variants, 11693806 (49) located near DIRC3, and rs16857609 (61), that are associated with European ancestry.
5.5 5p15.33 TERT
Ge et al. (63) conducted a study on thyroid cancer in Chinese populations. They found that TERT rs2736100 was significantly associated with thyroid cancer in the Chinese cohort. Other study in populations of European ancestry confirmed this association (rs2736100). Additionally, they identified rs10069690 as the variant with the strongest association in this region and suggested that these two variants likely represent the same underlying association signal, with rs10069690 providing a slight refinement in the European ancestry study groups compared to the Chinese population (49).
In an article that examined 75 FNMTC families, the promoter region of the TERT gene was sequenced. Somatic mutations in the TERT promoter, as well as RAS and BRAF mutations, were evaluated, along with the entire EIF1AX gene in 54 familial cases of thyroid tumors. The study found that FNMTC is associated with short telomeres, an increased copy number of the TERT gene, and elevated telomerase activity in both germline and somatic cells.
Additionally, the study suggested that TERT promoter and EIF1AX mutations are infrequently involved in the development of FNMTC. However, TERT mutations were more frequently observed in late-onset FNMTC cases compared to BRAF mutations, indicating a potential correlation between TERT mutations and the late onset of FNMTC (62).
5.6 5q31.3 SPRY4
Marques et al. (79) performed WES on leukocyte DNA from six affected members of FNMTC family. Shared genetic variants were identified through bioinformatic analysis and validated using Sanger sequencing. After filtering, a specific variant (c.701C>T, p.Thr234Met) in the SPRY4 gene was identified as the most promising. In vitro experiments showed that this variant increased cell viability and colony formation, indicating enhanced proliferation and clonogenic capacity. Analysis suggested that the SPRY4 variant acted through the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway, with increased sensitivity to a MEK inhibitor in thyroid cancer cells.
5.7 7q31.33 POT1
POT1, also known as GLM9, CMM10, and HPOT1, is involved in telomere maintenance. Mutations in POT1 were first reported in familial melanoma, where some family members also developed thyroid cancer (80, 100). Capezzone et al. found that peripheral blood samples from familial PTC patients exhibited an imbalance in the telomere-telomerase complex, characterized by shortened telomeres and TERT gene amplification (101). Richard et al. used functional variant approaches to confirm the association between low-frequency intronic regulatory POT1 variants in survivors of childhood cancer and subsequent development of malignant thyroid tumors. Intronic variations in POT1 may affect key proteins involved in telomere maintenance and genomic integrity (102).
Recently, Srivastava et al. (81) reported a pedigree with FNMTC, in which a novel mutation (p.Val29Leu) was identified in the POT1 gene among family members. In vitro experiments showed that the p.Val29Leu mutation in the POT1 gene exhibited increased telomere length, indicating loss of function caused by the mutation and resulting in telomere dysfunction. However, Orois et al. (82) did not find potential pathogenic mutations in the POT1 gene through WES in four FNMTC pedigrees from Spain.
5.8 8p12 NRG1
The NRG1 gene encodes neuregulin 1, a signaling protein involved in the development of both malignant and benign thyroid tumors. Additionally, it plays a role in protecting against oxidative damage, and influencing cell proliferation (57, 59, 103, 104).
Studies identified strong associations in the NRG1 gene, specifically with rs6996585, rs12542743 and rs2439302 (35, 47, 57–60). Guibon et al. (61) conducted fine mapping of the 8p12 (NRG1) locus, identifying rs2439304 as being associated with DTC. Furthermore, another variant at the NRG1 locus (rs2466076) was found to be associated with 3001 NMTC cases and 287,550 controls from Iceland (49).
5.9 9q22 FOXE1/TTF-2
The FOXE1 gene, located on chromosome 9q22.33, encodes the FOXE1 transcription factor (also known as thyroid transcription factor 2, TTF-2). This gene is involved in regulating the expression of genes such as thyroglobulin and thyroid peroxidase, and it plays an essential role in the formation and development of the thyroid gland.
Regarding the association of FOXE1 with FNMTC, several studies have been conducted to investigate its potential role. In one study (64), the SNP rs1867277 variation at the 5’UTR was found to regulate FOXE1 transcription. Another study (65, 67) tested the FOXE1 gene in 60 Portuguese FNMTC families and 80 SNMTC cases and identified ten germline variants in the promoter and coding sequence of the gene. One variant, c.743C > G (p.A248G), showed gene-disease co-segregation in one FNMTC family and was detected in a disseminated PTC case.
In another study (66), a total of 672 patients from 133 FNMTC progeny were tested at 11 known candidate loci, including the chromosomal locus 9q22.33 near FOXE1. The SNPs rs965513 and rs10759944 at 9q22.33 showed the most consistent association with FNMTC. However, Bonora et al. (66) reported that among the previously mentioned rs944289 (68) and rs1867277 (64) SNPs, only rs1867277 was consistently associated with FNMTC. Furthermore, validation from 95 FNMTC cases carrying three risk alleles of related SNPs (rs965513, rs10759944, rs1867277) at the 9q22.33 locus did not find any deleterious missense mutations in the entire coding sequence of FOXE1. Therefore, the aforementioned FOXE1-associated SNPs were not consistently shown to be associated with FNMTC.
A study conducted on the Icelandic population and people of European ancestry (67) showed that two simultaneous polymorphic changes (rs944289 and rs965513) near the FOXE1 gene increased the risk of PTC and follicular thyroid carcinoma (FTC) development. Overall, it should be noted that not all literature supports a direct association between the FOXE1 gene and FNMTC.
5.10 10q25.3 HABP2
Gara et al. (16) performed exome sequencing on seven FNMTC patients from one family, in which the HABP2 G534E mutation was found to promote tumorigenesis, growth, invasion, and loss of tumor suppression. Weeks et al. (17) conducted Sanger sequencing on 37 Australian families with FNMTC to validate the presence of the HABP2 G534E mutation. Additionally, WES was performed on 59 participants from 20 families to validate the HABP2 mutation. However, no explainable HABP2 G534E mutation was detected in the Australian family. Furthermore, studies conducted in regions including Europe and the Middle East have also failed to replicate this data (17–24).
In another study by Kern et al. (24), Sanger sequencing was performed on 20 patients with familial PTC from 11 European families. They identified c.1601G > A (p.Gly534Glu) mutations in two patients at the heterozygous level and c.364C > T (p.Arg122Trp) mutations in three patients. The minor allele frequencies (MAF) for these mutations were 5.0% and 7.5%, respectively. Therefore, these findings do not support the pathogenicity of the HABP2 c.1601G > A (p.Gly534Glu) variant. However, the study highlighted the presence of a novel mutation, c.364C > T (p.Arg122Trp), which was not found in another study (25) involving 32 Italian FNMTC families that did not identify the HABP2 R122W variant.
Finally, additional HABP2 variants (rs2286742 and rs3740530) were identified, which have the potential to increase the risk of PTC in a recessive model, respectively (71).
Therefore, considering potential ethnic variations across different regions, further investigation is warranted in a broader cohort of patients with familial PTC to thoroughly evaluate its pathogenicity.
5.11 11q22.3ATM 17q21.31 BRCA1 22q12.1 CHEK2
Ionizing radiation has been documented to directly increase the risk of thyroid and breast cancer by causing DNA double-strand breaks, thereby promoting carcinogenesis. The process of DNA damage repair is influenced by genetic polymorphisms and mutations in the ATM-BRCA1-CHEK2 pathway. In mammalian cells, DNA double-strand breaks activate the ATM kinase, which phosphorylates and activates CHEK2. This activation leads to the phosphorylation of BRCA1, initiating DNA repair. If the repair fails, apoptosis may occur (72).
Wojcicka et al. conducted a study and found that certain genetic variations, such as ATM D1853N, BRCA1 E1038G, and CHEK2 I157T, were associated with an increased susceptibility to PTC. They also discovered that specific single-nucleotide polymorphisms (SNPs), namely rs17879961 in CHEK2 and rs16941 in BRCA1, were significantly associated with PTC susceptibility. However, they did not find an association between ATM rs1801516 and PTC (72).
In another study, it was suggested that polymorphic changes in the ATM gene increased the risk of sporadic PTC and DTC in Caucasians (105, 106). Gu et al. (73) investigated the relationship between polymorphic changes in ATM and the increased risk of PTC in an Asian population. Among the four studied polymorphisms (rs664677, rs373759, rs4988099, rs189037), only rs373759 was found to be associated with an increased risk of PTC development. Strong linkage disequilibrium was observed among three ATM SNPs (rs373759, rs664143, and rs4585). The presence of the ATM haplotype (C-G-T) +/- was associated with a lower risk of PTC compared to the absence of this haplotype (C-G-T) -/-, suggesting the potential role of ATM genetic polymorphisms in thyroid cancer development in the Korean population (74).
Siołek et al. (90) demonstrated a significant correlation between carrying CHEK2 mutations (such as 1100delC, IVS21G > A, del5395, and I157T) and an increased risk of PTC. To further investigate this relationship, they genotyped 468 PTC patients and 468 age- and sex-matched cancer-free controls. Among the PTC cases, 73 out of 468 (15.6%) carried CHEK2 mutations, while only 28 out of 460 (6.0%) of the controls had these mutations. Zhao et al. (91) conducted a study involving whole genome sequencing of from two PTC patients in the same family. As a result, a new heterozygous germline mutation in CHEK2 (c.417C→A) was identified in all affected members of the family.
Functional analysis revealed that the CHEK2 c.417C > A variant introduces a premature termination codon (Y139X), resulting in the production of a truncated protein. This loss-of-function variant led to reduced p53 phosphorylation and decreased abundance of the p53 protein. In addition to the Y139X variant, two rare missense variants (R180C and H371Y) were also detected in the CHEK2 gene.
5.12 12q14 SRGAP1
An SNP-linked analysis of 38 families suggested that three variants (rs781626187, rs797044990 and rs114817817) in the SRGAP1 gene might serve as potential candidate genes for FNMTC (75). These variants could potentially lead to the loss of gene function, resulting in the inactivation of the CDC42 protein. This, in turn, can impact the regulation of intracellular signaling networks, influencing various signaling pathways and promoting tumorigenesis (107). Additionally, the identified variants were not found in population studies conducted in Ohio or Poland. However, a different SNP (rs2168411) of the SRGAP1 gene demonstrated an association with PTC in both the Ohio and Poland populations (75).
5.13 14q11.2 C14orf93/RTFC
Liu et al. (83) conducted a study involving one Chinese family with FNMTC using WES and linkage analysis and validated their findings in 14 additional FNMTC families. Through this analysis, they identified six candidate genes: C14orf93/RTFC (chr14:23465462), PYGL (chr14:51383415), BMP4 (chr14:54418885), PLCB4 (chr20: 9440331), CCDC54 (chr3:107097080), and EIF2AK4 (chr15:40268998). Further screening based on allele frequency and linkage analysis revealed that C14orf93/RTFC was the only remaining candidate gene among the six identified. The other five genes were excluded from further consideration. In particular, the V205M RTFC mutant was found to be the only oncogenic mutation that promoted cell survival, cell migration, and colony-forming capacity under starvation conditions.
5.14 14q12 TINF2
The study involved genome sequencing analysis of a large family with cases of PTC and melanoma. Screening for coding variants in the shelterin genes was conducted in 24 families using WES. A frameshift mutation in the TINF2 gene (TINF2 p.Trp198fs) was identified, showing complete co-segregation with PTC and melanoma in the key family. This mutation was unique to this family. Reduced TINF2 expression and disrupted binding to TERF1 were observed in individuals with the mutation. These individuals also exhibited significantly longer telomeres compared to unaffected family members and healthy controls. In addition, rare missense and synonymous variants in TINF2 and ACD were found in some families (84, 85).
5.15 14q13 NKX2-1/TTF-1,MBIP
The TTF-1 gene is located on chromosome 14q13 and encodes the thyroid transcription factor-1 (TTF-1). This protein plays a role in activating the transcription of thyroglobulin, thyroid peroxidase, and thyroid stimulating hormone receptors.
Ngan et al. (86) conducted targeted DNA sequencing of the SRGAP1 gene in 20 patients with PTC who had a history of MNG, as well as in 284 patients with PTC without a history of MNG. Among the latter group, four patients with PTC were found to have the NKX2-1/TTF-1 mutation pattern.
Functional experiments have indicated that mutations in this gene, such as the TTF-1 A339V mutation, can stimulate thyroid cell proliferation independently of thyroid-stimulating hormone. These mutations have also been shown to activate STAT3 and Akt signaling pathways and increase the expression of cyclin D2. These findings suggest that the TTF-1 A339V mutation may be associated with the development of PTC. However, these results have not been consistently confirmed in other experiments (108).
Gudmundsson et al. (47, 68) discovered that downstream of the NKX2-1 gene, there is a risk allele T for the SNP rs944289. Liao et al. (109) conducted genetic testing on a Chinese family and found that the T allele of rs944289 is a risk allele for members of this family to develop MNG and PTC. This association was also confirmed by other countries (10, 35, 58, 69). Another genome-wide study conducted by Takahashi et al. (70) focused on Belarusians who were exposed to radiation from the Chernobyl incident at 18 years of age or younger. However, this study did not find an association between radiation-associated PTC and the SNP rs944289 located at the 14q13.3 locus. Other relevant variants, such as rs34081947 and rs368187, were initially identified in a GWAS conducted on European populations (49).
In addition to rs944289, Gudmundsson also proposed that the variants (rs116909374 on 14q13.3) were significantly associated with thyroid cancer. On chromosome 14q13.3, the nearby gene MBIP and NKX2-1/TTF-1 are important candidates to consider for their roles in thyroid development (57).
5.16 15q21.1 DUOX2
Ohye et al. (11) utilized WES to identify a novel germline mutation in the DUOX2 gene associated with FNMTC. This mutation, known as DUOX2 Y1203H, affects the function of DUOX2, which is responsible for hydrogen peroxide (H2O2) production in the thyroid gland. H2O2 is essential for iodide tyrosine residue oxidation and the synthesis of thyroxine and triiodothyronine by thyroid peroxidase.
The study found that DUOX2 Y1203H is a functionally active mutation, and it was observed to be highly expressed in normal thyroid tissue, potentially increasing the risk of tumorigenesis. Notably, the expression of DUOX2 was found to be elevated in patients harboring the rs965513 variant, suggesting a potential link between dysregulated H2O2 metabolism mediated by DUOX2 and hereditary thyroid carcinoma with either high or low penetrance. However, a study on 33 Italian FNMTC pedigrees did not find these mutations (110).
5.17 15q23 MAP2K5
Ye et al. (13) conducted a study utilizing WES in 33 patients with FNMTC and further validated their results in an additional 44 intra-family patients. Through this analysis, they identified five candidate genes: MAP2K5, ZNF500, MUC6, IGSF3, and FRG1.
An interesting aspect to consider is that the pathogenesis of FNMTC diverges from the classical MAP2K1/2-ERK1/2 signaling pathway typically associated with sporadic cases. In FNMTC, there is a distinct activation of the MAP2K5-ERK5 pathway, which subsequently causes changes in gene expression downstream. These alterations ultimately drive the malignant transformation of thyroid epithelial cells.
Additionally, a separate study targeting MAP2K5 DNA assays in Italian families did not yield any positive results (14, 15).
5.18 16p13.3 SRRM2
This study (88) involved a sample of 6 FNMTC patients from a single family, which was then validated by including an additional 138 FNMTC families. The study also screened for the presence of the FNMTC susceptibility gene SRRM2 in 1170 sporadic PTC cases from Ohio, as well as 1404 healthy controls.
The data obtained from this study suggest that the S346F mutation in the SRRM2 gene may play a role in PTC development. This mutation affects the splicing process and leads to variable splicing of downstream target genes. A total of 7 SRRM2 mutations were identified in 7 PTC cases, while no mutations were found in the 1404 controls. However, it is worth noting that these mutations were not detected in the 138 FNMTC families analyzed.
5.19 19p13.2 GRIM-19/NDUFA13
There is a cluster of genes involved in mitochondrial metabolism, including GRIM-19. In a study of 52 thyroid tumors, researchers searched for GRIM-19 mutations. They found somatic missense mutations in three sporadic Hurthle cell carcinomas and a germline mutation in a Hurthle cell papillary carcinoma. No mutations were detected in non-Hurthle cell carcinomas or blood donor samples. One of the sporadic Hurthle cell papillary carcinomas with a GRIM-19 mutation also had a RET/PTC-1 rearrangement. Interestingly, no GRIM-19 mutations were identified in familial Hurthle cell tumors, suggesting that GRIM-19 may not be the TCO gene. The GRIM-19 mutations may contribute to the development of sporadic or familial Hurthle cell tumors by affecting GRIM-19’s role in mitochondrial metabolism and cell death (37).
5.20 19p13.2 TIMM44
This particular study (36) focused on 8 families affected by oncocytic thyroid carcinomas. The researchers conducted a systematic screening of 14 candidate genes and observed their localization in regions associated with affected members of the families with oncocytic thyroid carcinomas.
Through this screening, the study identified two new variants in TIMM44. These variants were located in exon 9 and exon 13 of the gene, specifically at 19p13.2. TIMM44 is known as a mitochondrial endosomal transporter involved in the import of nuclear-encoded proteins into mitochondria. Interestingly, these TIMM44 variants were found to co-occur with the oncocytic thyroid carcinomas phenotype within the affected families.
5.21 19p13.2 MYO1F
A studies (38) revealed a novel heterozygous mutation in exon 5 of the MYO1F gene (c.400G > A, NM: 012335; p.Gly134Ser) that maps to the linkage site. In addition to this mutation, another variant was found in exon 7 among the 192 FNMTC families studied.
This variant leads to an increase in mitochondrial mass and significantly elevates reactive oxygen species levels. Furthermore, it confers a significant advantage in terms of colony formation, invasion, and anchorage-independent growth. Moreover, there is another variant in exon 7 that causes exon skipping, which is predicted to alter the ATP-binding domain in MYO1F.
5.22 19q13.33 NOP53
A study (12) conducted WES of one family and analyzed an additional 44 FNMTC families using Sanger sequencing. In this study, a shared germline variant in NOP53, known as Asp31His (rs78530808, with a minor allele frequency of 1.8%), was identified in three non-syndromic FNMTC families.
Functional studies of NOP53 in thyroid cancer cell lines have demonstrated its oncogenic function. However, considering the relatively high frequency of this variant in the general population, it suggests that NOP53 may not be a causative gene but rather a low penetrance gene associated with FNMTC.
7 Pathway and process enrichment analysis
We performed pathway and process enrichment analysis on the given gene list above using the following ontology source from Metascape (version v3.5.20230501, https://metascape.org/): KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, CORUM, WikiPathways, and PANTHER Pathway. All genes in the genome have been used as the enrichment background. Terms with a p-value < 0.01, a minimum count of 3, and an enrichment factor > 1.5 are collected and grouped into clusters based on their membership similarities. The most statistically significant term within a cluster is chosen to represent the cluster.
According to Figure 2A, we have found that the currently reported genes are mainly involved in key processes related to carcinogenesis, such as negative regulation of DNA metabolic process, endocrine system development, cell activation, and regulation of cell-substrate adhesion in GO Biological Processes. Additionally, key pathways associated with thyroid cancer, such as DNA damage response (only ATM dependent) and EGF/EGFR signaling pathway, were identified in WikiPathways.
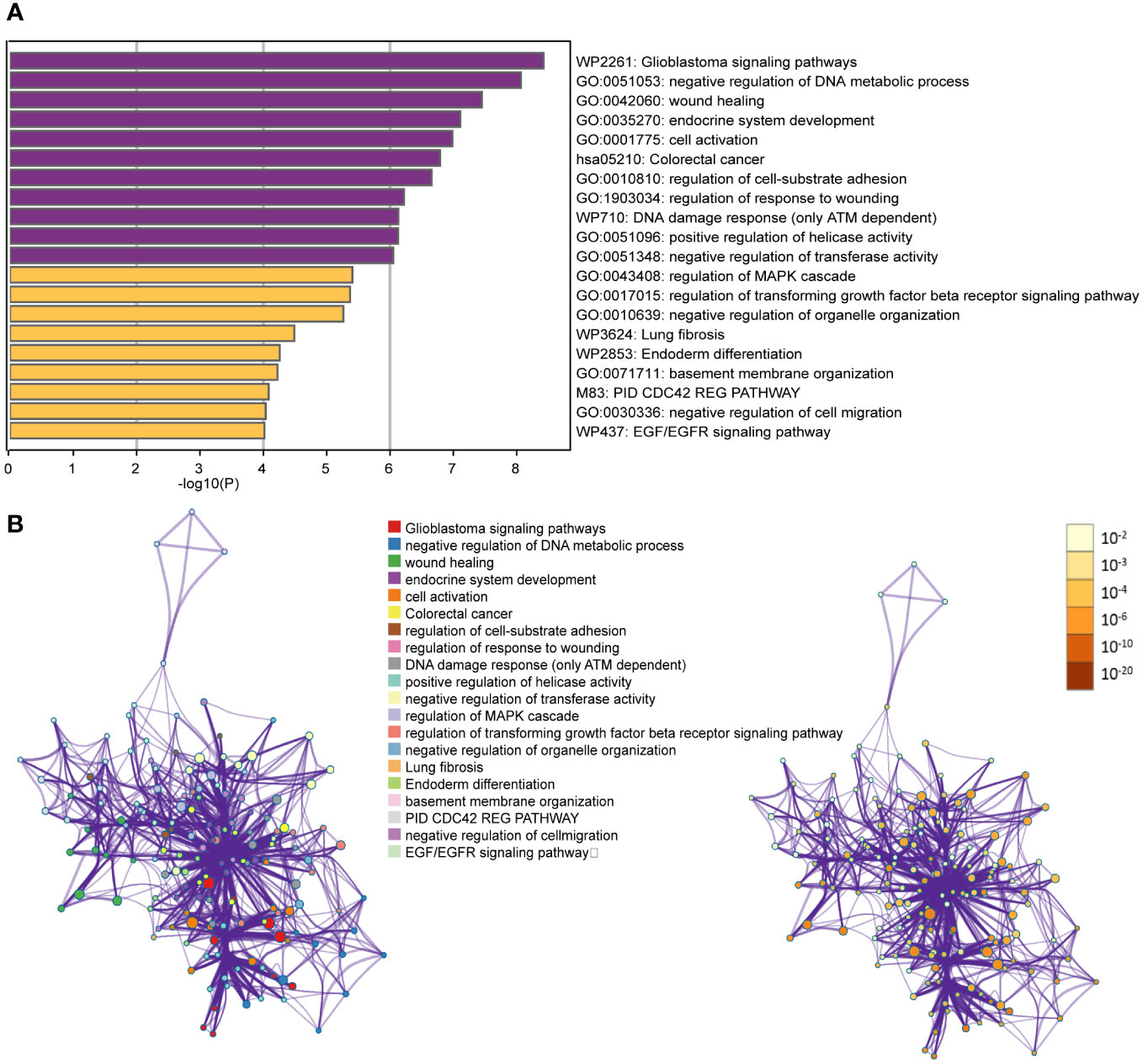
Figure 2 (A) Bar graph of enriched terms across input gene lists, colored by p-values; (B) Network of enriched terms: colored by cluster ID, where nodes that share the same cluster ID are typically close to each other; colored by p-value, where terms containing more genes tend to have a more significant p-value.
To enhance the representation of term relationships, we have created a network plot using a subset of enriched terms. In this network, terms with a similarity greater than 0.3 are connected by edges. Our selection process involved choosing terms with the best p-values from each of the 20 clusters. We ensured that each cluster had no more than 15 terms and that the total number of terms did not exceed 250. The resulting network is visualized using Cytoscape (111), where each node represents an enriched term. Nodes are initially colored based on their cluster ID and then further differentiated by their p-value (Figure 2B).
8 Protein-protein interaction enrichment analysis
For each given gene list, protein-protein interaction enrichment analysis has been carried out with the following databases: STRING6, BioGrid7, OmniPath8, InWeb_IM9.Only physical interactions in STRING (physical score > 0.132) and BioGrid are used. MCODE_1 shows the three best-scoring terms (GO:0051053, negative regulation of DNA metabolic process, Log10(P)= -11.6; GO:0032205, negative regulation of telomere maintenance, Log10(P)= -9.5; GO:0051096, positive regulation of helicase activity, Log10(P)= -9.0) by p-value have been retained as the functional description of the corresponding components, shown in the tables underneath corresponding network plots within Figure 3. VAV3, SYK, PIK3CB, ATM, MSH2, MSH6, TINF2, STN1, and POT1 potentially have protein-protein interaction relationships among them.
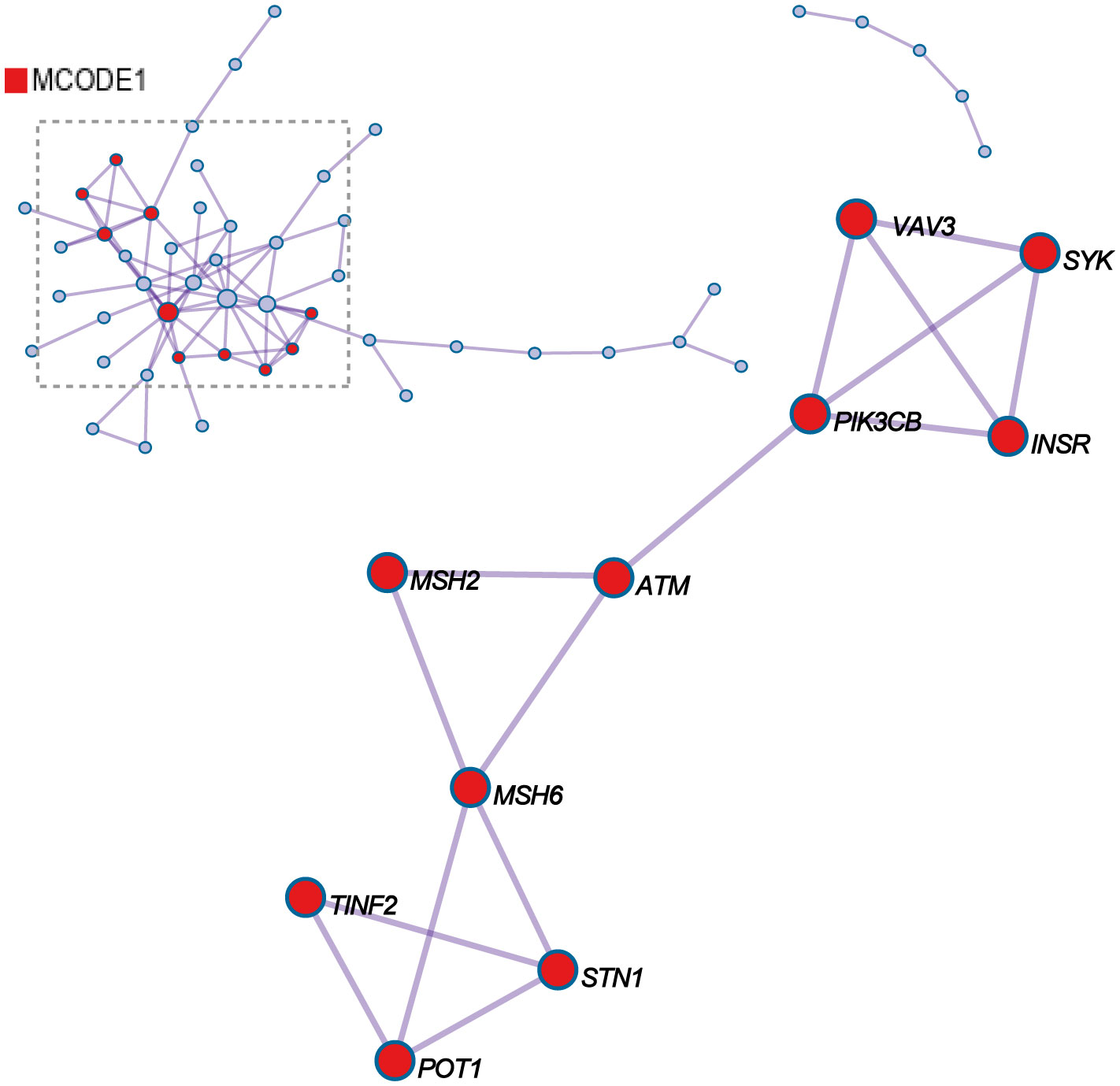
Figure 3 Protein-protein interaction network and MCODE components identified in the gene lists.
9 Discussion
Currently, no significant loci have been successfully identified in the research on FNMTC, indicating that the genetic causal model of FNMTC may be complex and mostly validated only in individual pedigrees, without genetic replication in other pedigrees. This may be related to various factors, with racial differences being one possible reason. Another important factor is the genetic heterogeneity of inherited diseases, where different gene variations can lead to the same disease, or in other words, a single factor does not determine a particular disease.
The strength of this article lies in its first-time summary of the chromosomal landscape map, which facilitates researchers to visually identify gene clustering loci. We also provide a summary of the localization and methodologies of susceptibility genes from different countries. The integration presented in this study may provide new insights for further research.
However, the limitation of this study is that FNMTC susceptibility genes are family-specific pathogenic genes, and each family may have its unique mechanisms of disease development. Currently, there is no precise localization of the causative genes for this condition.
It is indeed worth discussing the potential avenues for future research in the field of FNMTC, such as the development of new assessment methods like PRS and FCVPP. Researchers may uncover novel insights into the molecular mechanisms underlying FNMTC by investigating gene expression patterns, studying individual cells, and examining copy number variations in the future. These investigations could potentially lead to the identification of biomarkers for early detection, as well as the development of targeted therapeutic approaches.
10 Conclusion
This review provides an overview of the recent advancements in non-syndrome FNMTC susceptibility gene research, offering comprehensive theoretical support for subsequent investigations in this field. To explore the pathogenesis of FNMTC, more FNMTC pedigrees need to be included to expand and improve the FNMTC gene database. For non-syndromic FNMTC patients, early detection, diagnosis, and treatment may be an effective way to prevent disease progression.
Author contributions
YJ-J: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft. YX: Conceptualization, Data curation, Formal Analysis, Methodology, Writing – original draft. Z-JH: Data curation, Writing – original draft. Y-XH: Writing – original draft, Writing – review & editing. TH: Conceptualization, Methodology, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
FNMTC, Familial non-medullary thyroid carcinoma; NMTC, Non-medullary thyroid carcinoma; PTC, Papillary thyroid carcinoma; ATC, Anaplastic thyroid carcinoma; DTC, Differentiated thyroid carcinoma; FTC, Follicular thyroid carcinoma; WES, Whole exome sequencing; GWAS, Genome-wide association study; WGS, Whole genome sequencing; PRN, Papillary renal neoplasia; FvPTC, Follicular variant papillary thyroid carcinoma; TCO, Thyroid cancer with oncocytic; TSH, Thyroid stimulating hormone; MNG, Multinodular goiter; Lnc-RNA, Long non-coding RNA; Linc-RNA, Long intergenic non-coding RNA; 3C assays, Chromosome conformation capture assays; PTCSC, Papillary thyroid carcinoma susceptibility candidate; PRS, Polygenic risk scores; FCVPP, Familial Cancer Variant Prioritization Pipeline; PRMT5, Protein arginine methyltransferase 5; H4R3me2, Histone H4 arginine 3 dimethylation; EGFR, Epidermal growth factor receptor; DIRC3, Disrupted in Renal Cancer 3; TTF, Thyroid transcription factor; MAF, Minor allele frequencies; SNPs, Single-nucleotide polymorphisms; H2O2, Hydrogen peroxide.
References
1. Khan A, Smellie J, Nutting C, Harrington K, Newbold K. Familial nonmedullary thyroid cancer: A review of the genetics. Thyroid. (2010) 20:795–801. doi: 10.1089/thy.2009.0216
2. Vriens MR, Suh I, Moses W, Kebebew E. Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid. (2009) 19:1343–9. doi: 10.1089/thy.2009.1607
3. Charkes ND. On the prevalence of familial nonmedullary thyroid cancer in multiply affected kindreds. Thyroid. (2006) 16:181–6. doi: 10.1089/thy.2006.16.181
4. Peiling YS, Ngeow J. Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr Relat Cancer. (2016) 23:R577–R95. doi: 10.1530/ERC-16-0067
5. Suh I, Filetti S, Vriens MR, Guerrero MA, Tumino S, Wong M, et al. Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: a SNP array-based linkage analysis of 38 families. Surgery. (2009) 146:1073–80. doi: 10.1016/j.surg.2009.09.012
6. McKay JD, Lesueur F, Jonard L, Pastore A, Williamson J, Hoffman L, et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am J Hum Genet. (2001) 69:440–6. doi: 10.1086/321979
7. He HL, Li W, Wu DY, Nagy R, Liyanarachchi S, Akagi K, et al. Ultra-rare mutation in long-range enhancer predisposes to thyroid carcinoma with high penetrance. PloS One. (2013) 8:e61920. doi: 10.1371/journal.pone.0061920
8. Kohler A, Chen B, Gemignani F, Elisei R, Romei C, Figlioli G, et al. Genome-wide association study on differentiated thyroid cancer. J Clin Endocrinol Metab. (2013) 98:E1674–81. doi: 10.1210/jc.2013-1941
9. Figlioli G, Chen B, Elisei R, Romei C, Campo C, Cipollini M, et al. Novel genetic variants in differentiated thyroid cancer and assessment of the cumulative risk. Sci Rep. (2015) 5:8922. doi: 10.1038/srep08922
10. Mancikova V, Cruz R, Inglada-Perez L, Fernandez-Rozadilla C, Landa I, Cameselle-Teijeiro J, et al. Thyroid cancer GWAS identifies 10q26.12 and 6q14.1 as novel susceptibility loci and reveals genetic heterogeneity among populations. Int J Cancer. (2015) 137:1870–8. doi: 10.1002/ijc.29557
11. Bann DV, Jin Q, Sheldon KE, Houser KR, Nguyen L, Warrick JI, et al. Genetic variants implicate dual oxidase-2 in familial and sporadic nonmedullary thyroid cancer. Cancer Res. (2019) 79:5490–9. doi: 10.1158/0008-5472.CAN-19-0721
12. Orois A, Gara SK, Mora M, Halperin I, Martinez S, Alfayate R, et al. NOP53 as A candidate modifier locus for familial non-medullary thyroid cancer. Genes-Basel. (2019) 10:899. doi: 10.3390/genes10110899
13. Ye F, Gao H, Xiao L, Zuo Z, Liu Y, Zhao Q, et al. Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int J Cancer. (2019) 144:1321–30. doi: 10.1002/ijc.31825
14. Ye F, Gao H, Xiao L, Ding H, Huang Z, Jiang Y, et al. Authors' reply to: Absence of the MAP2K5 germline variants c.G961A and c.T1100C in a wide series of familial non-medullary thyroid carcinoma Italian families. International Journal of Cancer 2019; in press. Int J Cancer. (2019) 145:601–2. doi: 10.1002/ijc.32243
15. Cirello V, Colombo C, Persani L, Fugazzola L. Absence of the MAP2K5 germline variants c.G961A and c.T1100C in a wide series of familial nonmedullary thyroid carcinoma Italian families. Int J Cancer. (2019) 145:600. doi: 10.1002/ijc.32244
16. Gara SK, Jia L, Merino MJ, Agarwal SK, Zhang L, Cam M, et al. Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med. (2015) 373:448–55. doi: 10.1056/NEJMoa1502449
17. Weeks AL, Wilson SG, Ward L, Goldblatt J, Hui J, Walsh JP. HABP2 germline variants are uncommon in familial nonmedullary thyroid cancer. BMC Med Genet. (2016) 17:60. doi: 10.1186/s12881-016-0323-1
18. Kowalik A, Gasior-Perczak D, Gromek M, Siolek M, Walczyk A, Palyga I, et al. The p.G534E variant of HABP2 is not associated with sporadic papillary thyroid carcinoma in a Polish population. Oncotarget. (2017) 8:58304–8. doi: 10.18632/oncotarget.16870
19. de Mello LEB, Araujo AN, Alves CX, de Paiva FJP, Brandao-Neto J, Cerutti JM. The G534E variant in HABP2 is not associated with increased risk of familial nonmedullary thyroid cancer in Brazilian Kindreds. Clin Endocrinol (Oxf). (2017) 87:113–4. doi: 10.1111/cen.13352
20. Cantara S, Marzocchi C, Castagna MG, Pacini F. HABP2 G534E variation in familial non-medullary thyroid cancer: an Italian series. J Endocrinol Invest. (2017) 40:557–60. doi: 10.1007/s40618-016-0583-9
21. Ruiz-Ferrer M, Fernandez RM, Navarro E, Antinolo G, Borrego S. G534E variant in HABP2 and nonmedullary thyroid cancer. Thyroid. (2016) 26:987–8. doi: 10.1089/thy.2016.0193
22. Alzahrani AS, Murugan AK, Qasem E, Al-Hindi H. HABP2 gene mutations do not cause familial or sporadic non-medullary thyroid cancer in a highly inbred Middle Eastern population. Thyroid. (2016) 26:667–71. doi: 10.1089/thy.2015.0537
23. Sahasrabudhe R, Stultz J, Williamson J, Lott P, Estrada A, Bohorquez M, et al. The HABP2 G534E variant is an unlikely cause of familial non-medullary thyroid cancer. J Clin Endocrinol Metab. (2016) 10:1098–103. doi: 10.1210/jc.2015-3928
24. Kern B, Coppin L, Romanet P, Crepin M, Szuster I, Renaud F, et al. Multiple HABP2 variants in familial papillary thyroid carcinoma: Contribution of a group of "thyroid-checked" controls. Eur J Med Genet. (2017) 60:178–84. doi: 10.1016/j.ejmg.2017.01.001
25. Colombo C, Fugazzola L, Muzza M, Proverbio MC, Cirello V. Letter regarding the article: "Multiple HABP2 variants in familial papillary thyroid carcinoma: Contribution of a group of "thyroid-checked" controls" by Kern et al. Eur J Med Genet. (2018) 61:104–5. doi: 10.1016/j.ejmg.2017.07.012
26. Bevan S, Pal T, Greenberg CR, Green H, Wixey J, Bignell G, et al. A comprehensive analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1. J Clin Endocrinol Metab. (2001) 86:3701–4. doi: 10.1210/jcem.86.8.7725
27. McKay JD, Thompson D, Lesueur F, Stankov K, Pastore A, Watfah C, et al. Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer. J Med Genet. (2004) 41:407–12. doi: 10.1136/jmg.2003.017350
28. Prazeres HJ, Rodrigues F, Soares P, Naidenov P, Figueiredo P, Campos B, et al. Loss of heterozygosity at 19p13.2 and 2q21 in tumours from familial clusters of non-medullary thyroid carcinoma. Fam Cancer. (2008) 7:141–9. doi: 10.1007/s10689-007-9160-x
29. Canzian F, Amati P, Harach HR, Kraimps JL, Lesueur F, Barbier J, et al. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet. (1998) 63:1743–8. doi: 10.1086/302164
30. Tsilchorozidou T, Vafiadou E, Yovos JG, Romeo G, McKay J, Lesueur F, et al. A Greek family with a follicular variant of familial papillary thyroid carcinoma: TCO, MNG1, fPTC/PRN, and NMTC1 excluded as susceptibility loci. Thyroid. (2005) 15:1349–54. doi: 10.1089/thy.2005.15.1349
31. Malchoff CD, Sarfarazi M, Tendler B, Forouhar F, Whalen G, Joshi V, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab. (2000) 85:1758–64. doi: 10.1210/jcem.85.5.6557
32. Bignell GR, Canzian F, Shayeghi M, Stark M, Shugart YY, Biggs P, et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am J Hum Genet. (1997) 61:1123–30. doi: 10.1086/301610
33. Cavaco BM, Batista PF, Sobrinho LG, Leite V. Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density single-nucleotide polymorphism genome-wide linkage analysis. J Clin Endocrinol Metab. (2008) 93:4426–30. doi: 10.1210/jc.2008-0449
34. Cavaco BM, Batista PF, Martins C, Banito A, Do RF, Limbert E, et al. Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr Relat Cancer. (2008) 15:207–15. doi: 10.1677/erc-07-0214
35. Son HY, Hwangbo Y, Yoo SK, Im SW, Yang SD, Kwak SJ, et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat Commun. (2017) 8:15966. doi: 10.1038/ncomms15966
36. Bonora E, Evangelisti C, Bonichon F, Tallini G, Romeo G. Novel germline variants identified in the inner mitochondrial membrane transporter TIMM44 and their role in predisposition to oncocytic thyroid carcinomas. Br J Cancer. (2006) 95:1529–36. doi: 10.1038/sj.bjc.6603455
37. Máximo V, Botelho T, Capela J, Soares P, Lima J, Taveira A, et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. Virchows Arch. (2005) 447:138–9. doi: 10.1038/sj.bjc.6602547
38. Diquigiovanni C, Bergamini C, Evangelisti C, Isidori F, Vettori A, Tiso N, et al. Mutant MYO1F alters the mitochondrial network and induces tumor proliferation in thyroid cancer. Int J Cancer. (2018) 143:1706–19. doi: 10.1002/ijc.31548
39. Xiong Y, Zhang L, Holloway AK, Wu X, Su L, Kebebew E. MiR-886-3p regulates cell proliferation and migration, and is dysregulated in familial non-medullary thyroid cancer. PloS One. (2011) 6:e24717. doi: 10.1371/journal.pone.0024717
40. Xiong Y, Zhang L, Kebebew E. MiR-20a is upregulated in anaplastic thyroid cancer and targets LIMK1. PloS One. (2014) 9:e96103. doi: 10.1371/journal.pone.0096103
41. He H, Nagy R, Liyanarachchi S, Jiao H, Li W, Suster S, et al. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. (2009) 69:625–31. doi: 10.1158/0008-5472.Can-08-1071
42. He H, Li W, Liyanarachchi S, Jendrzejewski J, Srinivas M, Davuluri RV, et al. Genetic predisposition to papillary thyroid carcinoma: involvement of FOXE1, TSHR, and a novel lincRNA gene, PTCSC2. J Clin Endocrinol Metab. (2015) 100:E164–72. doi: 10.1210/jc.2014-2147
43. He H, Li W, Liyanarachchi S, Srinivas M, Wang Y, Akagi K, et al. Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc Natl Acad Sci U S A. (2015) 112:6128–33. doi: 10.1073/pnas.1506255112
44. Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, et al. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc Natl Acad Sci U S A. (2012) 109:8646–51. doi: 10.1073/pnas.1205654109
45. Wang Y, He H, Li W, Phay J, Shen R, Yu L, et al. MYH9 binds to lncRNA gene PTCSC2 and regulates FOXE1 in the 9q22 thyroid cancer risk locus. Proc Natl Acad Sci U S A. (2017) 114:474–9. doi: 10.1073/pnas.1619917114
46. Koirala P, Huang J, Ho TT, Wu F, Ding X, Mo YY. LncRNA AK023948 is a positive regulator of AKT. Nat Commun. (2017) 8:14422. doi: 10.1038/ncomms14422
47. Rogounovitch TI, Bychkov A, Takahashi M, Mitsutake N, Nakashima M, Nikitski AV, et al. The common genetic variant rs944289 on chromosome 14q13.3 associates with risk of both Malignant and benign thyroid tumors in the Japanese population. Thyroid. (2015) 25:333–40. doi: 10.1089/thy.2014.0431
48. Figlioli G, Kohler A, Chen B, Elisei R, Romei C, Cipollini M, et al. Novel genome-wide association study-based candidate loci for differentiated thyroid cancer risk. J Clin Endocrinol Metab. (2014) 99:E2084–92. doi: 10.1210/jc.2014-1734
49. Gudmundsson J, Thorleifsson G, Sigurdsson JK, Stefansdottir L, Jonasson JG, Gudjonsson SA, et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat Commun. (2017) 8:14517. doi: 10.1038/ncomms14517
50. Sarquis M, Moraes DC, Bastos-Rodrigues L, Azevedo PG, Ramos AV, Reis FV, et al. Germline mutations in familial papillary thyroid cancer. Endocr Pathol. (2020) 31:14–20. doi: 10.1007/s12022-020-09607-4
51. Majdalani P, Yoel U, Nasasra T, Fraenkel M, Haim A, Loewenthal N, et al. Novel susceptibility genes drive familial non-medullary thyroid cancer in a large consanguineous kindred. Int J Mol Sci. (2023) 24:8233. doi: 10.3390/ijms24098233
52. Yu Y, Dong L, Li D, Chuai S, Wu Z, Zheng X, et al. Targeted DNA sequencing detects mutations related to susceptibility among familial non-medullary thyroid cancer. Sci Rep. (2015) 5:16129. doi: 10.1038/srep16129
53. Zhu JW, Wu KL, Lin ZY, Bai SW, Wu J, Li PK, et al. Identification of susceptibility gene mutations associated with the pathogenesis of familial nonmedullary thyroid cancer. Mol Genet Genom Med. (2019) 7:e1015. doi: 10.1002/mgg3.1015
54. Liyanarachchi S, Gudmundsson J, Ferkingstad E, He H, Jonasson JG, Tragante V, et al. Assessing thyroid cancer risk using polygenic risk scores. Proc Natl Acad Sci U S A. (2020) 117:5997–6002. doi: 10.1073/pnas.1919976117
55. Song N, Liu Q, Wilson CL, Sapkota Y, Ehrhardt MJ, Gibson TM, et al. Polygenic risk score improves risk stratification and prediction of subsequent thyroid cancer after childhood cancer. Cancer Epidemiol Biomarkers Prev. (2021) 30:2096–104. doi: 10.1158/1055-9965.EPI-21-0448
56. Song SS, Huang S, Park S. Association of polygenetic risk scores related to cell differentiation and inflammation with thyroid cancer risk and genetic interaction with dietary intake. Cancers (Basel). (2021) 13:1510. doi: 10.3390/cancers13071510
57. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Masson G, He HL, et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat Genet. (2012) 44:319–U126. doi: 10.1038/ng.1046
58. Wang YL, Feng SH, Guo SC, Wei WJ, Li DS, Wang Y, et al. Confirmation of papillary thyroid cancer susceptibility loci identified by genome-wide association studies of chromosomes 14q13, 9q22, 2q35 and 8p12 in a Chinese population. J Med Genet. (2013) 50:689–95. doi: 10.1136/jmedgenet-2013-101687
59. Liyanarachchi S, Wojcicka A, Li W, Czetwertynska M, Stachlewska E, Nagy R, et al. Cumulative risk impact of five genetic variants associated with papillary thyroid carcinoma. Thyroid. (2013) 23:1532–40. doi: 10.1089/thy.2013.0102
60. Mussazhanova Z, Rogounovitch TI, Saenko VA, Krykpayeva A, Espenbetova M, Azizov B, et al. The contribution of genetic variants to the risk of papillary thyroid carcinoma in the Kazakh population: study of common single nucleotide polymorphisms and their clinicopathological correlations. Front Endocrinol (Lausanne). (2020) 11:543500. doi: 10.3389/fendo.2020.543500
61. Guibon J, Sugier PE, Kulkarni O, Karimi M, Bacq-Daian D, Besse C, et al. Fine-mapping of two differentiated thyroid carcinoma susceptibility loci at 2q35 and 8p12 in Europeans, Melanesians and Polynesians. Oncotarget. (2021) 12:493–506. doi: 10.18632/oncotarget.27888
62. Marques IJ, Moura MM, Cabrera R, Pinto AE, Simoes-Pereira J, Santos C, et al. Identification of somatic TERT promoter mutations in familial nonmedullary thyroid carcinomas. Clin Endocrinol. (2017) 87:394–9. doi: 10.1111/cen.13375
63. Ge MH, Shi M, An CM, Yang WJ, Nie XL, Zhang J, et al. Functional evaluation of TERT-CLPTM1L genetic variants associated with susceptibility of papillary thyroid carcinoma. Sci Rep-Uk. (2016) 6:26037. doi: 10.1038/srep26037
64. Landa I, Ruiz-Llorente S, Montero-Conde C, Inglada-Perez L, Schiavi F, Leskela S, et al. The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PloS Genet. (2009) 5:e1000637. doi: 10.1371/journal.pgen.1000637
65. Tomaz RA, Sousa I, Silva JG, Santos C, Teixeira MR, Leite V, et al. FOXE1 polymorphisms are associated with familial and sporadic nonmedullary thyroid cancer susceptibility. Clin Endocrinol (Oxf). (2012) 77:926–33. doi: 10.1111/j.1365-2265.2012.04505.x
66. Bonora E, Rizzato C, Diquigiovanni C, Oudot-Mellakh T, Campa D, Vargiolu M, et al. The FOXE1 locus is a major genetic determinant for familial nonmedullary thyroid carcinoma. Int J Cancer. (2014) 134:2098–107. doi: 10.1002/ijc.28543
67. Pereira JS, Da SJ, Tomaz RA, Pinto AE, Bugalho MJ, Leite V, et al. Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine. (2015) 49:204–14. doi: 10.1007/s12020-014-0470-0
68. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet. (2009) 41:460–4. doi: 10.1038/ng.339
69. Matsuse M, Takahashi M, Mitsutake N, Nishihara E, Hirokawa M, Kawaguchi T, et al. The FOXE1 and NKX2-1 loci are associated with susceptibility to papillary thyroid carcinoma in the Japanese population. J Med Genet. (2011) 48:645–8. doi: 10.1136/jmedgenet-2011-100063
70. Takahashi M, Saenko VA, Rogounovitch TI, Kawaguchi T, Drozd VM, Takigawa-Imamura H, et al. The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum Mol Genet. (2010) 19:2516–23. doi: 10.1093/hmg/ddq123
71. Shen CT, Zhang GQ, Qiu ZL, Song HJ, Sun ZK, Luo QY. Targeted next-generation sequencing in papillary thyroid carcinoma patients looking for germline variants predisposing to the disease. Endocrine. (2019) 64:622–31. doi: 10.1007/s12020-019-01878-0
72. Wojcicka A, Czetwertynska M, Swierniak M, Dlugosinska J, Maciag M, Czajka A, et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer. (2014) 53:516–23. doi: 10.1002/gcc.22162
73. Gu Y, Yu Y, Ai L, Shi J, Liu X, Sun H, et al. Association of the ATM gene polymorphisms with papillary thyroid cancer. Endocrine. (2014) 45:454–61. doi: 10.1007/s12020-013-0020-1
74. Song CM, Kwon TK, Park BL, Ji YB, Tae K. Single nucleotide polymorphisms of ataxia telangiectasia mutated and the risk of papillary thyroid carcinoma. Environ Mol Mutagen. (2015) 56:70–6. doi: 10.1002/em.21898
75. He HL, Bronisz A, Liyanarachchi S, Nagy R, Li W, Huang YG, et al. SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J Clin Endocr Metab. (2013) 98:E973–E80. doi: 10.1210/jc.2012-3823
76. Zhao Y, Yu T, Sun J, Wang F, Cheng C, He S, et al. Germ-line mutations in WDR77 predispose to familial papillary thyroid cancer. Proc Natl Acad Sci U.S.A. (2021) 118:e2026327118. doi: 10.1073/pnas.2026327118
77. Pasquali D, Torella A, Accardo G, Esposito D, Del Vecchio Blanco F, Salvatore D, et al. BROX haploinsufficiency in familial nonmedullary thyroid cancer. J Endocrinol Invest. (2021) 44:165–71. doi: 10.1007/s40618-020-01286-6
78. de Mello LEB, Carneiro TNR, Araujo AN, Alves CX, Galante PAF, Buzatto VC, et al. Identification of NID1 as a novel candidate susceptibility gene for familial non-medullary thyroid carcinoma using whole-exome sequencing. Endocr Connect. (2022) 11:e210406. doi: 10.1530/EC-21-0406
79. Marques IJ, Gomes I, Pojo M, Pires C, Moura MM, Cabrera R, et al. Identification of SPRY4 as a novel candidate susceptibility gene for familial nonmedullary thyroid cancer. Thyroid. (2021) 31:1366–75. doi: 10.1089/thy.2020.0290
80. Wilson TL, Hattangady N, Lerario AM, Williams C, Koeppe E, Quinonez S, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. (2017) 16:561–6. doi: 10.1007/s10689-017-9984-y
81. Srivastava A, Miao B, Skopelitou D, Kumar V, Kumar A, Paramasivam N, et al. A germline mutation in the POT1 gene is a candidate for familial non-medullary thyroid cancer. Cancers (Basel). (2020) 12:1441. doi: 10.3390/cancers12061441
82. Orois A, Badenas C, Reverter JL, López V, Potrony M, Mora M, et al. Lack of mutations in POT1 gene in selected families with familial non-medullary thyroid cancer. Hormones cancer. (2020) 11:111–6. doi: 10.1007/s12672-020-00383-5
83. Liu C, Yu Y, Yin G, Zhang J, Wen W, Ruan X, et al. C14orf93 ( RTFC ) is identified as a novel susceptibility gene for familial nonmedullary thyroid cancer. Biochem Biophys Res Commun. (2017) 482:590–6. doi: 10.1016/j.bbrc.2016.11.078
84. He HL, Li W, Comiskey DF, Liyanarachchi S, Nieminen TT, Wang YQ, et al. A truncating germline mutation of TINF2 in individuals with thyroid cancer or melanoma results in longer telomeres. Thyroid. (2020) 30:204–13. doi: 10.1089/thy.2019.0156
85. Wang Y, Liyanarachchi S, Miller KE, Nieminen TT, Comiskey DF Jr., Li W, et al. Identification of rare variants predisposing to thyroid cancer. Thyroid. (2019) 29:946–55. doi: 10.1089/thy.2018.0736
86. Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst. (2009) 101:162–75. doi: 10.1093/jnci/djn471
87. Hu Y, Han Z, Guo H, Zhang N, Shen N, Jiang Y, et al. Identification of a Novel Germline PPP4R3A Missense Mutation Asp409Asn on Familial Non-Medullary Thyroid Carcinoma. Biomedicines. (2024) 12:244. doi: 10.3390/biomedicines12010244
88. Tomsic J, He H, Akagi K, Liyanarachchi S, Pan Q, Bertani B, et al. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep. (2015) 5:10566. doi: 10.1038/srep10566
89. Jiang Y, Xia Y, Hu Y, Han Z, Guo A, Huang T. Identification of P21 (CDKN1A) Activated Kinase 4 as a Susceptibility Gene for Familial Non-medullary Thyroid Carcinoma. Thyroid. [in press]. doi: 10.1089/thy.2023.0564
90. Siolek M, Cybulski C, Gasior-Perczak D, Kowalik A, Kozak-Klonowska B, Kowalska A, et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int J Cancer. (2015) 137:548–52. doi: 10.1002/ijc.29426
91. Zhao YY, Yu T, Chen L, Xie DH, Wang FL, Fu LP, et al. A germline CHEK2 mutation in a family with papillary thyroid cancer. Thyroid. (2020) 30:924–30. doi: 10.1089/thy.2019.0774
92. Försti A, Kumar A, Paramasivam N, Schlesner M, Catalano C, Dymerska D, et al. Pedigree based DNA sequencing pipeline for germline genomes of cancer families. Hered Cancer Clin Pr. (2016) 14:16. doi: 10.1186/s13053-016-0058-1
93. Kumar A, Bandapalli OR, Paramasivam N, Giangiobbe S, Diquigiovanni C, Bonora E, et al. Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci Rep-Uk. (2018) 8:11635. doi: 10.1038/s41598-018-29952-z
94. Altayyar MA, Sheng X, Wang Z. WD repeat domain 77 protein regulates translation of E2F1 and E2F3 mRNA. Mol Cell Biol. (2020) 40:e00302–20. doi: 10.1128/mcb.00302-20
95. O'Bryant D, Wang Z. The essential role of WD repeat domain 77 in prostate tumor initiation induced by Pten loss. Oncogene. (2018) 37:4151–63. doi: 10.1038/s41388-018-0254-8
96. McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad Sci U S A. (2008) 105:7687–91. doi: 10.1073/pnas.0801567105
97. Pradhan-Sundd T, Verheyen EM. The role of Bro1-domain-containing protein Myopic in endosomal trafficking of Wnt/Wingless. Dev Biol. (2014) 392:93–107. doi: 10.1016/j.ydbio.2014.04.019
98. Mu RL, Dussupt V, Jiang JS, Sette P, Rudd V, Chuenchor W, et al. Two distinct binding modes define the interaction of brox with the C-terminal tails of CHMP5 and CHMP4B. Structure. (2012) 20:887–98. doi: 10.1016/j.str.2012.03.001
99. Bodmer D, Schepens M, Eleveld MJ, Schoenmakers EF, Geurts van Kessel A. Disruption of a novel gene, DIRC3, and expression of DIRC3-HSPBAP1 fusion transcripts in a case of familial renal cell cancer and t (2,3)(q35;q21). Genes Chromosomes Cancer. (2003) 38:107–16. doi: 10.1002/gcc.10243
100. Potrony M, Puig-Butille JA, Ribera-Sola M, Iyer V, Robles-Espinoza CD, Aguilera P, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. (2019) 181:105–13. doi: 10.1111/bjd.17443
101. Capezzone M, Cantara S, Marchisotta S, Filetti S, De Santi MM, Rossi B, et al. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J Clin Endocrinol Metab. (2008) 93:3950–7. doi: 10.1210/jc.2008-0372
102. Richard MA, Lupo PJ, Morton LM, Yasui YA, Sapkota YA, Arnold MA, et al. Genetic variation in POT1 and risk of thyroid subsequent Malignant neoplasm: A report from the Childhood Cancer Survivor Study. PloS One. (2020) 15:e0228887. doi: 10.1371/journal.pone.0228887
103. He H, Li W, Liyanarachchi S, Wang Y, Yu L, Genutis LK, et al. The role of NRG1 in the predisposition to papillary thyroid carcinoma. J Clin Endocrinol Metab. (2018) 103:1369–79. doi: 10.1210/jc.2017-01798
104. Zhang TT, Qu N, Sun GH, Zhang L, Wang YJ, Mu XM, et al. NRG1 regulates redox homeostasis via NRF2 in papillary thyroid cancer. Int J Oncol. (2018) 53:685–93. doi: 10.3892/ijo.2018.4426
105. Dombernowsky SL, Weischer M, Allin KH, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. Risk of cancer by ATM missense mutations in the general population. J Clin Oncol. (2008) 26:3057–62. doi: 10.1200/jco.2007.14.6613
106. Akulevich NM, Saenko VA, Rogounovitch TI, Drozd VM, Lushnikov EF, Ivanov VK, et al. Polymorphisms of DNA damage response genes in radiation-related and sporadic papillary thyroid carcinoma. Endocr Relat Cancer. (2009) 16:491–503. doi: 10.1677/erc-08-0336
107. Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci. (2004) 117:1291–300. doi: 10.1242/jcs.01115
108. Cantara S, Capuano S, Formichi C, Pisu M, Capezzone M, Pacini F. Lack of germline A339V mutation in thyroid transcription factor-1 (TITF-1/NKX2.1) gene in familial papillary thyroid cancer. Thyroid Res. (2010) 3:4. doi: 10.1186/1756-6614-3-4
109. Liao S, Song W, Liu Y, Deng S, Liang Y, Tang Z, et al. Familial multinodular goiter syndrome with papillary thyroid carcinomas: mutational analysis of the associated genes in 5 cases from 1 Chinese family. BMC endocrine Disord. (2013) 13:48. doi: 10.1186/1472-6823-13-48
110. Cirello V, Colombo C, Karapanou O, Pogliaghi G, Persani L, Fugazzola L. Clinical and genetic features of a large monocentric series of familial non-medullary thyroid cancers. Front Endocrinol. (2021) 11:589340. doi: 10.3389/fendo.2020.589340
Keywords: thyroid carcinoma, familial non-medullary thyroid carcinoma, chromosomal locus, GWAS, WES, genetic mutation
Citation: Jiang Y-j, Xia Y, Han Z-j, Hu Y-x and Huang T (2024) Chromosomal localization of mutated genes in non-syndromic familial thyroid cancer. Front. Oncol. 14:1286426. doi: 10.3389/fonc.2024.1286426
Received: 31 August 2023; Accepted: 29 February 2024;
Published: 20 March 2024.
Edited by:
Valentina Cirello, University of Milan, ItalyReviewed by:
Ali Alzahrani, King Faisal Specialist Hospital and Research Centre, Saudi ArabiaA. Kumar, University of Kiel, Germany
Copyright © 2024 Jiang, Xia, Han, Hu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Huang, aHVhbmd0YW93aEAxNjMuY29t; Yi-xuan Hu, ZW1lcnkxMDMwQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship