 Anna Maria Cacic1,2*
Anna Maria Cacic1,2* Felicitas Isabel Schulz1,2
Felicitas Isabel Schulz1,2 Ulrich Germing1,2Sascha Dietrich1,2
Ulrich Germing1,2Sascha Dietrich1,2 Norbert Gattermann1,2
Norbert Gattermann1,2- 1Department of Hematology, Oncology and Clinical Immunology, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine Universität Düsseldorf, Düsseldorf, Germany
- 2Center for Integrated Oncology Aachen Bonn Cologne Düsseldorf (CIO ABCD), Düsseldorf, Germany
Clonal hematopoiesis of indeterminate potential (CHIP) has fascinated the medical community for some time. Discovered about a decade ago, this phenomenon links age-related alterations in hematopoiesis not only to the later development of hematological malignancies but also to an increased risk of early-onset cardiovascular disease and some other disorders. CHIP is detected in the blood and is characterized by clonally expanded somatic mutations in cancer-associated genes, predisposing to the development of hematologic neoplasms such as MDS and AML. CHIP-associated mutations often involve DNA damage repair genes and are frequently observed following prior cytotoxic cancer therapy. Genetic predisposition seems to be a contributing factor. It came as a surprise that CHIP significantly elevates the risk of myocardial infarction and stroke, and also contributes to heart failure and pulmonary hypertension. Meanwhile, evidence of mutant clonal macrophages in vessel walls and organ parenchyma helps to explain the pathophysiology. Besides aging, there are some risk factors promoting the appearance of CHIP, such as smoking, chronic inflammation, chronic sleep deprivation, and high birth weight. This article describes fundamental aspects of CHIP and explains its association with hematologic malignancies, cardiovascular disorders, and other medical conditions, while also exploring potential progress in the clinical management of affected individuals. While it is important to diagnose conditions that can lead to adverse, but potentially preventable, effects, it is equally important not to stress patients by confronting them with disconcerting findings that cannot be remedied. Individuals with diagnosed or suspected CHIP should receive counseling in a specialized outpatient clinic, where professionals from relevant medical specialties may help them to avoid the development of CHIP-related health problems. Unfortunately, useful treatments and clinical guidelines for managing CHIP are still largely lacking. However, there are some promising approaches regarding the management of cardiovascular disease risk. In the future, strategies aimed at restoration of gene function or inhibition of inflammatory mediators may become an option.
Introduction
Clonal hematopoiesis (CH) is characterized by the presence of a population of blood cells derived from a mutated multipotent stem/progenitor cell that has gained an abnormal growth advantage.
Specifically, clonal hematopoiesis of indeterminate potential (CHIP), as defined by the new WHO classification of hematolymphoid tumors, refers to CH with somatic mutations in myeloid malignancy-associated genes detected in the blood or bone marrow at a variant allele frequency (VAF) of ≥2% in individuals without diagnosed hematologic disorder or unexplained cytopenia (1–3). It should be emphasized that CHIP is not yet a disease, since the accepted definition excludes persistent (≥4 months) cytopenia and an overt pathology associated with the somatic lesion (3, 4). Using a VAF of ≥2% as cut-off, up to 20% of individuals above the age of 70 have detectable CH (5–7). Considering the demography in Germany, it is estimated that 2.75 million people are affected (8). Regarding the individual person with CH, intraindividual heterogeneity has been reported, with disparities observed between affected cell lineages, tissue compartments (peripheral blood vs. bone marrow), and anatomic locations (9). CHIP mutations can arise in both the myeloid lineage (referred to as M-CHIP) and the lymphoid lineage (known as L-CHIP) of the hematopoietic system (10). Both myeloid and lymphoid somatic gene mutations, as well as myeloid and lymphoid mosaic chromosomal alterations (mCAs), are associated with the risk of lineage-specific hematologic malignancies (10). In the case of M-CHIP, the majority of mutations are found in three genes, namely DNMT3A, TET2, and ASXL1 (10). Somatic mutations in myeloid-biased CH are correlated with an elevated risk of myeloid malignancy, cardiovascular disease (CVD), and all-cause mortality (11). L-CHIP appears to be distributed more evenly across a larger number of genes, resembling the distribution of variants in the remaining M-CHIP genes (10). Somatic mutations in lymphoid-biased CH are associated with the development of lymphoid malignancies and late-onset manifestations of autoimmunity and immune deficiency (11).
The CHIP clones identified so far exhibit a range of large-scale mCAs, including deletions, duplications, and copy-neutral loss of heterozygosity (CN-LOH) on all chromosomes (12–16). mCA-driven CH primarily predisposes to lymphoid malignancies such as chronic lymphocytic leukemia (CLL) and is associated with a twofold increase in all-cause mortality (16, 17). Notably, events involving JAK2-mutated CN-LOH have been demonstrated to significantly elevate cardiovascular risk (17). Additionally, mCA-associated CH may lead to impaired immune function, making individuals more susceptible to infections (18).
Again, it is important to note that the term CHIP should only be applied to individuals with normal blood counts (19). In cases where CHIP is identified alongside one or more persistent cytopenias that are otherwise unexplained by hematologic or non-hematologic conditions and that do not meet diagnostic criteria for defined myeloid neoplasms (MNs), the condition is referred to as CCUS (clonal cytopenia of undetermined significance) (1). Both CCUS and CHIP become more prevalent with age and are relatively common among elderly individuals (2). Patients with CHIP have an elevated risk of developing a MN compared to controls without CHIP (3, 5, 6, 20–23), and they also have an increased risk of developing progressive atherosclerosis and related cardiovascular disorders (5, 24). However, it should be noted that in the majority of patients with CHIP, neither malignancy nor severe CVD manifests (19).
Understanding the intricate interplay between genetic predisposition, somatic mutations, and environmental influences is crucial for unraveling the complexities of CHIP and its implications for health. Genetic predisposition seems to play a significant role, contributing to the accumulation of somatic mutations in hematopoietic stem cells (HSCs). Some of these mutations are key drivers of CHIP. Acquisition of CH mutations occurs through natural mechanisms during aging, such as point mutations due to spontaneous deamination of 5-methylcytosine to thymine (25), or less commonly through errors during repair of double-stranded DNA breaks, creating small insertions/deletions (indels), or through replication errors produced by DNA polymerase (26).
CH is associated with inflammation that promotes HSC expansion, increasing the risk of hematologic and cardiologic diseases. This relationship is believed to create a “CH expansion-inflammation cycle” (27, 28). When a stem cell acquires a CH-associated gene mutation, it gains enhanced fitness in an inflammatory microenvironment, leading to CH-mutant hematopoietic stem and progenitor cell (HSPC) expansion. In turn, this results in increased and chronic expression of proinflammatory cytokines and other mediators that alter the hematopoietic milieu, creating an environment that suppresses normal HSPCs and establishes a vicious cycle of enhanced CH cell fitness and inflammation. Targeting inflammatory mechanisms may disrupt this cycle, reducing the risk of cancer and comorbid inflammatory diseases.
CHIP and hematologic risks
The accumulation of somatic cancer-associated mutations in HSCs, as observed in CHIP, enhances the risk of developing a hematologic neoplasm such as myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). Presumably, this leukemic transformation is driven by exaggerated stimulation of inflammatory processes in the bone marrow, coupled with inflammation associated with aging, ultimately resulting in the functional impairment of HSCs (29). Usually, the development of myeloid malignancies involves progression through certain precursor stages (CH, CHIP, CCUS).
Early-stage CH is characterized by the initial acquisition of a somatic mutation in a leukemia-associated gene, resulting in improved fitness and clonal expansion of the affected stem cell (30). The most frequent gene mutations associated with CHIP are DNMT3A, TET2, and ASXL1 (DTA genes). Individuals affected by these mutations in the context of CHIP exhibit no evidence of hematologic malignancy or cytopenia (31). Such mutations are detectable in approximately 5% of individuals aged 60-69 years, and about 10% of those aged 70-79 years (5, 32). Driver mutations associated with CHIP exhibit distinct patterns of clonal expansion over time. Previous findings indicate that DNMT3A-mutant clones are more likely to expand earlier in life (33). While clones with mutations in DNMT3A and TP53 display the slowest annual growth rate at 5%, clonal lineages with mutations in TET2, ASXL1, PPM1D and SF3B1 expand at approximately twice the rate, and those with mutations in splicing factors such as SRSF2, PTPN11, and U2AF1 grow at an average annual rate of 15-20% (33). The rapid growth of the latter set of mutations may contribute to the observed worse prognoses and increased disease severity associated with CHIP mutations in splicing factor genes (34). The risk of hematologic malignancy appears to be determined by the size of the clone as well as the type and number of mutated genes. The presence of mutations in spliceosome genes, IDH1/2 mutations, TP53 mutations, multiple mutations and a high VAF are associated with an increased risk of myeloid malignancies, whereas the risk appears to be lowest in patients with single DNMT3A or TET2 mutations (35). Advanced precursor states preceding the development of MDS and AML include CCUS and ICUS. As expected, CCUS, which carries a detectable clonal marker, has a significantly poorer prognosis than ICUS (31), with a considerable risk of transformation to MDS/AML (36). As an alternative to extensive mutation screening based on next-generation DNA sequencing, HUMARA (human androgen receptor gene-based assay) can be harnessed to assess clonality and thus differentiate between CCUS/MDS and ICUS (37).
The risk of malignant transformation is relatively low for CHIP, with a yearly progression rate of 0.5-1.0% (5, 24). ICUS carries a 9% probability of malignant clonal evolution at both 5 years and 10 years, while CCUS shows a probability of 82% at 5 years and 95% at 10 years (36). The outcome in CCUS is influenced by several clone metrics, including the number of mutations and specific genes affected (31). Epigenetic modifier genes DNMT3A, TET2, and ASXL1 are commonly mutated in CCUS but confer a lower risk, while spliceosome genes U2AF1, SRSF2, and SF3B1 are associated with the highest risk of progression (31, 36). A VAF >10% is associated with a 50-fold increase in the likelihood of progression (5). CCUS with a VAF of more than 20%, combined with mutations in spliceosome genes and DTA mutations, is considered “high-risk CCUS” and carries a 5-year cumulative probability of progression of 95% (36). Further research is needed to elucidate the mechanisms of disease progression and identify predictive markers that can guide prognostication and treatment decisions in individuals with CH, CHIP, CCUS, and ICUS.
An increased prevalence of CHIP has been observed following cytotoxic therapies such as chemotherapy, radiotherapy, and PARP inhibitor therapy (38–41), suggesting that these treatments contribute to the emergence and expansion of mutated cells in the hematopoietic system, thereby fostering the development of therapy-related myeloid neoplasms (t-MNs). In particular, cancer therapy involving radiation, platinum, and topoisomerase II inhibitors shows a preference for selecting mutations in DNA damage response (DDR) genes such as TP53, PPM1D and CHEK2 (38). Screening for CHIP in individuals who have undergone genotoxic treatment may help to identify those who have an elevated risk of developing a hematologic malignancy.
In addition to single-nucleotide variants (SNVs) and small indels, which contribute to the risk of malignancy in the context of CHIP, large structural changes to chromosomes, namely mCAs, can also play a role in driving clonal expansion (12, 13). Recent research has revealed that the presence of either SNVs/indels alone or mCAs alone is associated with a relatively low risk or cancer transformation (10, 42). However, when both forms of CH exist simultaneously, there is a greater risk of cancer transformation (10, 42). Specific forms of mCAs may predispose individuals to L-CHIP, while other forms are more likely to result in M-CHIP (10).
CHIP and cardiovascular risks
Myeloid precursor conditions, especially CHIP, have been linked to increased all-cause mortality (5, 6, 16, 32), predominantly attributed to cardiovascular causes, including coronary artery disease and stroke (5, 16, 43). CHIP mutations in genes such as DNMT3A, TET2, ASXL1, TP53, JAK2, and SF3B1 have been identified that are particularly associated with an increased risk of developing cardiovascular diseases (44). Table 1 presents the major cardiovascular conditions and their corresponding CHIP-associated mutations, according to current literature.
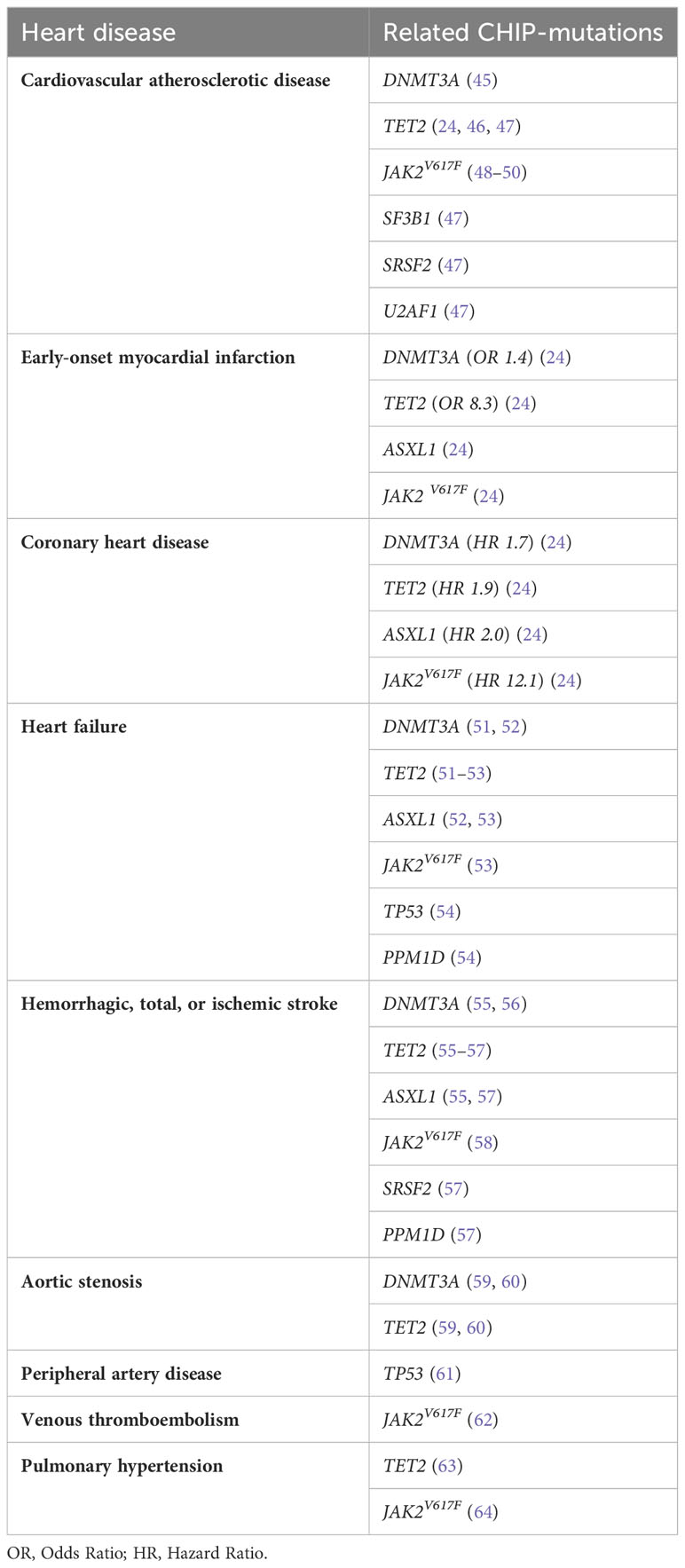
Table 1 CHIP and diverse cardiovascular conditions.
CHIP has been associated with cardiovascular atherosclerotic disease (CAD). Notably, the TET2 gene mutation has been strongly linked to increased cardiovascular morbidity and mortality due to CAD (24, 46, 47). Mutations in TET2 were reported to accelerate atherosclerosis through altered function of the NOD-like receptor 3 (NLRP3)/interleukin-1ß (IL-1ß) inflammasome in mutant monocytes/macrophages residing in the blood vessel wall (46, 65). Such dysregulation of macrophage gene expression leads to increased activation of inflammatory pathways, including enhanced IL-1ß production (65). The ensuing chronic inflammatory state promotes the formation of atherosclerotic plaques, thus increasing the risk of myocardial infarction (MI) and stroke (66). CHIP was also found to be independently associated with adverse outcomes in individuals with established CAD, particularly in cases with TET2 and spliceosome (SF3B1/SRSF2/U2AF1) mutations (47). The JAK2V617F mutation, known for promoting the proliferation of mature myeloid cells (48), has also been linked to increased AIM2 inflammasome activation and IL-1ß production (49), and with increased neutrophil cellular trap formation, thereby reportedly increasing the risk of atherosclerotic thrombosis in myeloproliferative neoplasms (MPNs) (62). An elevated burden of inflammatory macrophages within atherosclerotic lesions is associated with necrotic core formation and potential plaque instability (49). Inhibition of IL-1ß has shown promise in mitigating these effects (49, 50). Recently, Rauch and colleagues demonstrated that the loss of DNMT3A mimics the effects observed in the absence of TET2 (45). This was evidenced by a consistent macrophage phenotype characterized by significant upregulation of CXCL1, CXCL2, CXCL3, and IL-6, ultimately contributing to the promotion of accelerated atherosclerosis (45).
The presence of CHIP has been associated with a 1.9-fold increase of coronary heart disease (CHD) (24). The risk of CHD is significantly increased in carriers of DNMT3A, TET2, and ASXL1 CHIP mutations (1.7 to 2.0-fold increase), while JAK2V617 CHIP carriers face an even higher risk of 12.1-fold elevation (24). DNMT3A is linked to elevated coronary-artery calcification (CAC) (24), highlighting its role in atherosclerotic burden. Notably, individuals with CHIP and atherosclerosis or CHD often lack traditional risk factors such as hypercholesterolemia, type 2 diabetes (T2D), hypertension, or smoking (67, 68). This suggests that the association between CHIP and CHD is primarily driven by an increased atherosclerotic burden, rather than other factors that may cause MI (24). This is in line with the finding that atherosclerosis is mainly driven by interactions between clonal monocytes-macrophages and the endothelium but also increased expression of pro-inflammatory genes such as IL-1ß and IL-6 (24, 46, 69, 70). Both atherosclerosis and coronary events follow a dose-response pattern with increasing size of the mutated clone (24). Mouse models using animals prone to hypercholesterolemia and carrying TET2 mutations showed a marked increase in atherosclerotic lesions in the aortic root and aorta (24). These mice exhibited increased expression of several genes of the CXC chemokine gene cluster, including CXCL1, CXCL2, CXCL3, and Pf4, and classic proinflammatory cytokine genes such as IL-1ß and IL-6, which contribute to atherosclerosis (24). Additionally, these TET2-deficient mice displayed significantly larger plaque size (24, 46), increased atherosclerotic lesion area (24), xanthoma development in the spleen and middle ear (24), foam-cell accumulation and glomerulosclerosis in the kidney (24), and inflammatory infiltrates in the liver and lung (24).
CHIP is associated with a 4-fold increased risk of early-onset (before the age of 50 years) MI, combined with significant enrichment of mutations in DNMT3A, TET2, JAK2, and ASXL1 (24). In patients with both ischemic and non-ischemic heart failure (HF), the presence of CHIP is linked to rapid disease progression and unfavorable overall survival (51, 71). The risk of cardiac death related to CHIP is dependent on the size of the clone, with larger clonal amplification (VAF >10%) correlating with a higher risk of HF (53). CHIP is generally associated with HF risk factors and biomarkers and is specifically associated with HF in individuals below 65 years of age (72). Mutations in ASXL1, TET2, and JAK2 have emerged as new risk factors for HF, and are significantly associated with rapid HF progression, with odds ratios of 1.58x, 1.59x and 2.5x, respectively (52, 53). Also, DDR genes like TP53 and PPM1D seem to be risk factors for HF (54). In studies of patients with chronic HF, approximately 18.5% exhibited 47 mutations with a VAF ≥ 2%, most commonly DNMT3A and TET2 mutations, which were associated with worse long-term clinical outcomes, including death and rehospitalization (51, 52). Importantly, CHIP increased the risk of HF by 25%, independent of traditional CVD risk factors (53). Hematopoietic/myeloid dysfunction has been shown to contribute to pathological cardiac remodeling in HF mouse models with gene mutations in TET2 and DNMT3A (65, 73), suggesting that TET2-CH as well as DNMT3A-CH may generally play a role in altered heart remodeling and HF. Furthermore, DNMT3A and JAK2 mutations, now known to promote atherosclerosis and HF, drive the proliferation of bone marrow cells with proinflammatory characteristics (24, 46, 53, 65, 73, 74). Recently, Abplanalp and colleagues conducted an in-depth exploration of the cell-intrinsic effects of CHIP in HF (75). Their findings revealed that DNMT3A-mutant monocytes exhibited elevated expression of genes associated with inflammation (CCR2) and phagocytosis (CALR, CYBA) (75). Additionally, CD4+ T cells and NK cells displayed heightened activation signatures and effector functions (75). CD4+ T cells demonstrated upregulation of markers indicative of T cell effector and killing function, such as LYZ and CXCL8, along with increased expression of drivers of proliferation (SELENOF) and markers of T cell differentiation (SLAMF6) (75). Similarly, CD4+ NK cells exhibited increased expression of the important lysozyme molecule, LYZ (75). Importantly, they demonstrated that DNMT3A-mutant macrophages can indirectly promote pro-hypertrophic activity in wild-type macrophages through paracrine signaling (75).
Individuals with CHIP have an increased risk of hemorrhagic or ischemic stroke (55, 57). The analysis of clonal dynamics following an ischemic stroke revealed that clones with a VAF of 1% have an estimated doubling time of approximately 8 years on average (57). Notably, SRSF2-mutated clones displayed the most robust expansion characteristics, doubling in size every 2.8 years (57). Therefore, individuals harboring SRSF2-mutated clones are at the greatest risk of developing a hematologic malignancy following an ischemic stroke. Furthermore, TET2 mutations are highly enriched in patients with ischemic stroke (57), suggesting a central role of TET2-driven CH in stroke pathogenesis (57). Other studies have identified DNMT3A (30.0%) and TET2 (11.4%) as the most common mutations in individuals with CHIP who experienced a first-ever acute ischemic stroke, and CHIP was specifically associated with recurrent stroke in patients with hyperinflammation (56). TET2 showed the strongest association with total and ischemic strokes, while both DNMT3A and TET2 were associated with an increased risk of hemorrhagic stroke (55). Moreover, mutations in ASXL1 and PPM1D have been associated with stroke, too (55, 57). More recently, the JAK2V617F mutation has also been associated with ischemic stroke, with a prevalence of 11.3% (58). Patients who experienced an ischemic stroke and had CH mutations displayed elevated levels of Interferon-γ (IFN-γ) (57). In addition, the presence of TET2 mutations was associated with elevated IL-1ß levels (57). Furthermore, there was a notable increase in white matter lesion load, which showed correlations with patient age and clone size (57).
CHIP has also been implicated in poor outcomes following transcatheter aortic valve implantation (TAVI) in patients with calcific aortic stenosis (76). In such patients with severe degenerative aortic valve stenosis, DNMT3A and TET2 mutations again played a significant role in promoting higher monocyte expression of inflammatory mediators such as IL-1ß, IL-6, and NLRP3 (59, 60, 77, 78). Furthermore, CHIP has been associated with peripheral artery disease (TP53) (61), venous thromboembolism (JAK2V617F) (62) and pulmonary hypertension (TET2, JAK2V617F) (63, 64).
Etiology of CHIP and associated diseases
A variety of factors have been reported to contribute to the development of CHIP and the diseases that are promoted or aggravated by CHIP. Such risk factors are listed in Table 2.
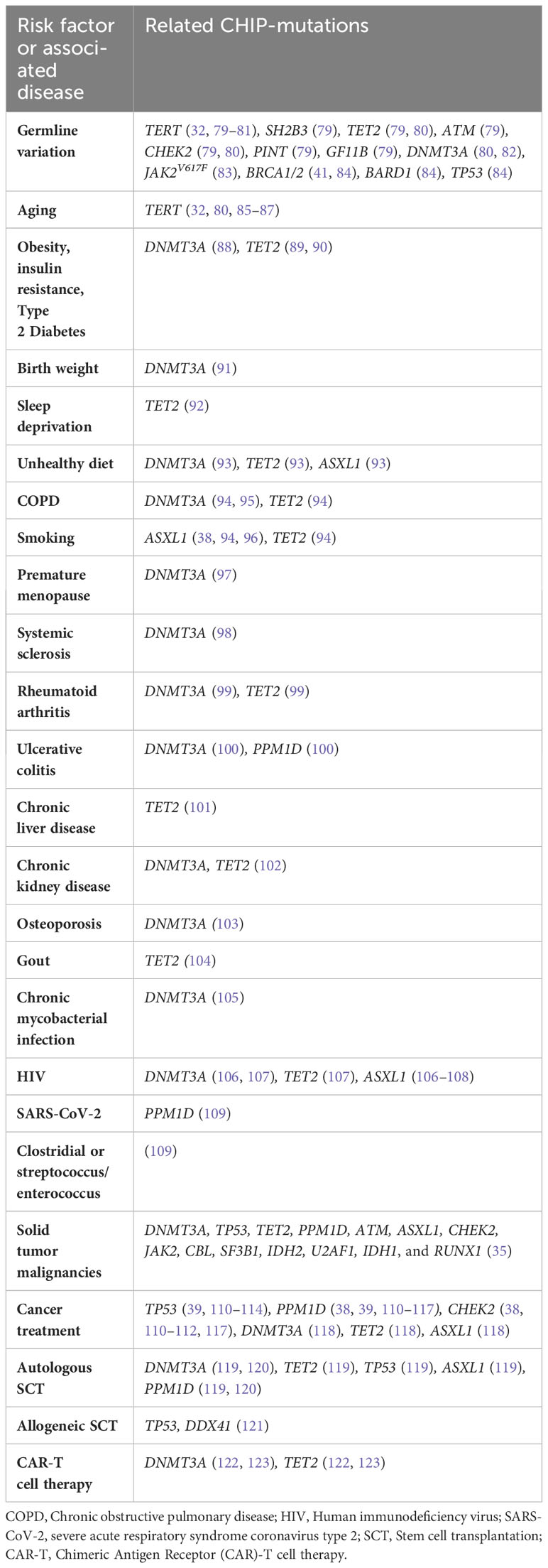
Table 2 CHIP, risk factors and associated diseases.
The development of CHIP and its association with various diseases is influenced by several risk factors. Germline variation in genes such as TERT, SH2B3, TET2, ATM, CHEK2, PINT, and GF11B plays a significant role in predisposing individuals to the emergence of CHIP (79). The TERT locus, in particular, harbors the most important risk allele for CHIP (80, 81), since germline deletion of intron 3 results in decreased leukocyte telomere length (LTL) (32). Altogether, four gene loci have been identified that are variably associated with an increased susceptibility to TET2-CHIP (80), namely TERT, CHEK, TET2, and the TCL1A locus, which also results in an elevated risk of DNMT3A-CHIP (80). In contrast, missense variants in PARP1 and LY75 are associated with lower CHIP incidence (81). The driver mutation JAK2V617 can be acquired as early as in utero or childhood, followed by lifelong clonal evolution eventually leading to MPNs (83). Acquisition of a DNMT3A mutation has also been detected to occur in utero or childhood (82). Another study identified cancer susceptibility germline mutations in the DDR genes BRCA1, BRCA2, BARD1 and TP53 in patients with t-MNs (84). The reported association of gBRCA1/2 mutation status and increased t-MN incidence is probably indirect, owing to more extensive platinum-based chemotherapy, which increased the likelihood of accumulating CH-associated gene mutations in PPM1D and TP53 (41). Studies of CHIP prevalence have revealed interesting patterns regarding race, ethnicity, and ancestry. There is a modestly lower prevalence of CHIP in Hispanics and East Asians compared to other populations (24, 124). Similarly, previous studies showed that the prevalence of AML is lower in East Asian populations than in Caucasian populations (125). CHIP may thus be more common in the Western population. However, a particular germline variant in TET2, rs144418061, which is associated with an increased risk of CHIP, is found exclusively in individuals of African descent (80), highlighting the importance of considering ancestral backgrounds in understanding CHIP prevalence.
Aging is a major factor in the accumulation of somatic mutations in HSCs due to cumulative DNA damage (126), and patients with inherited telomeropathies have a higher prevalence of CHIP (85), with common variants at the TERT gene (80) (telomere brink hypothesis (32, 86, 87, 127)). Recently, familial CH was reported in a long telomere syndrome (128). The authors suggested that delayed telomere attrition or even telomere lengthening over time allowed for the expansion of CHIP clones by enabling multiple cell divisions while avoiding cellular senescence (128). Hotspot mutations DNMT3A and TET2 were most common (128).
CHIP is furthermore associated with obesity, insulin resistance, and T2D. Obesity-related insulin resistance is associated with a higher incidence of TET2-CHIP (89). In general, diabetic patients exhibit 1.3-fold increased odds of CHIP (5). In mice, TET2-driven CHIP leads to an increased expression of IL-1ß in white adipose tissue, exacerbating age- and obesity-related insulin-resistance and hyperglycemia (89). This sets off a positive feedback loop (89, 90), promoting HSC self-renewal and inflammation progression, ultimately resulting in the loss of ß-cell mass in T2D (89, 129) which, in the end, would be responsible for the pathogenesis of diabetes and atherosclerosis (90). DNMT3A mutations are also more frequent in adipose tissue-derived macrophages in mice fed a high-fat diet (88). Moreover, a recent study demonstrated that birth weight is associated with CHIP and cardiovascular outcomes in adulthood (91). A 1-kg increase in birth weight was associated with a 3% increased risk of CHIP, mainly driven by a stronger association observed between birth weight and DNMT3A-CHIP (91). Sleep deprivation enhances the incidence of CVD, diabetes, obesity, and cancer (130). Sleep-deprived mice have increased atherosclerotic lesions, along with systemic monocytosis and neutrophilia (130). When sleep is fragmented, Tet2-/- Ldlr-/- mice show a 1.6-fold increase in the emergence of CHIP (92). Addiction and psychiatric diseases are also associated with CHIP, potentially linked to the critical component of sleep fragmentation (32). Unhealthy diet, characterized by highly processed food, red meat, added salt, and low intake of fruits and vegetables, is associated with a higher incidence of CHIP, CVD, and mortality (93). DNMT3A, TET2, and ASXL1 mutations were associated with unhealthy diet, although no statistically significant differences were observed between the distributions of CHIP-associated genes across dietary classes (93). However, some studies show no direct associations between diet and CHIP (131), possibly attributable to variations in study populations, measurement errors, and other factors that require further investigation (132).
Smoking consistently associates with CHIP (6, 32, 96), and individuals who never smoked have a decreased risk of developing CHIP (131). Current and past smokers show a higher frequency of ASXL1 mutations (38, 94, 96). Tobacco smoke has also been found to promote the expansion of TET2-mutated blood cells, potentially acting as a contributing factor to chronic obstructive pulmonary disease (COPD) (94). COPD is associated with CHIP (94), with DNMT3A mutations most commonly involved (95). The risk factors for COPD, such as smoking and aging, are also important risk factors for CHIP (94). Individuals with CHIP have a 1.6-fold and 2.2-fold increased risk of moderate and very severe COPD, respectively (94). In mouse models, the absence of TET2 has been linked to increased lung infiltration of immune cells, elevated IFN-γ levels, reduced Transforming growth factor ß (TGF-ß) signaling in immune cells, and accelerated development of emphysema in the lungs (94). In COPD patients, defects in DNMT3A in macrophages have been associated with increased production of IL-6 and tumor necrosis factor (TNF)-α, as well as global hypomethylation in the genome (95). To date, it is uncertain in which causal relationship CHIP and COPD are related to each other, however, based on previous findings, it is likely that the presence of somatic mutations such as CHIP contributes to the development and exacerbation of COPD (94).
Premature menopause (PM), with an odds ratio of 1.40, is linked to an increased risk of CHIP (97). PM is furthermore associated with a higher incidence of CAD (HR 1.36), with a stronger association observed in cases of natural PM (97). DNMT3A is the only candidate driver mutation associated with PM-related CHIP (97).
CHIP is also associated with various chronic inflammatory conditions. Patients with systemic sclerosis (SS) have an elevated prevalence of CHIP, with 25% of individuals affected compared to only 4% in the control cohort without SS (98). Notably, among the CHIP cases in SS, DNMT3A was the most commonly mutated gene (98). Similarly, rheumatoid arthritis (RA) patients have a 17% higher prevalence of CHIP, which increases to 25% in individuals aged 70-79 years, with DNMT3A and TET2 mutations being the most frequent (99). Patients with ulcerative colitis (UC), an inflammatory bowel disease (IBD) characterized by T-cell infiltration in the colon and overproduction of TNF-α and IFN-γ, also have an association with CHIP (100). These patients are at an increased risk of ischemic heart disease (133–135), with DNMT3A and PPM1D mutations being the most common alterations (100, 136). Since patients with IBD are a high-risk population for developing CH, there is concern that clonal expansion induced by inflammation could potentially make IBD patients with CHIP more prone to develop MNs (136). Furthermore, IBD carriers carry elevated levels of Th17 cells and Th17 related cytokines which are important in the pathogenesis of mucosal damage in IBD (137), displaying a similar pathogenesis to DNMT3A-CHIP carriers with severe degenerative aortic stenosis (60). More recently, CHIP was associated with an increased prevalence and incidence of chronic liver disease (OR = 2.01), with CHIP individuals being more likely to show liver inflammation and fibrosis compared to those without CHIP (101). Similarly, mice transplanted with TET2-deficient HSCs had more severe liver inflammation and fibrosis (101). Another chronic disease associated with CHIP is chronic kidney disease, with gene mutations most commonly found in DNMT3A and TET2 (102). CHIP is linked to kidney function decline and end-stage kidney disease in the general population, although the effect sizes were relatively modest (138). Moreover, it has been observed that impaired kidney function associated with CHIP is correlated with anemia in patients with advanced chronic kidney disease (102). Furthermore, research has demonstrated the association of DNMT3A-CHIP with an elevated occurrence of osteoporosis diagnoses and reduced bone marrow density (103). This phenomenon is driven by the expression of proinflammatory IL-20 from DNMT3A-mutant macrophages (103). Further, TET2-mutant CHIP is linked to an increased risk of incident gout. This association is supported by the observation of heightened NLRP3-independent IL-1ß secretion from TET2 knockout macrophages in response to monosodium urate crystals in vitro (104), indicating a pathogenesis similar to that of gout. CHIP is associated with a broad range of chronic infections. Mice models have shown that chronic mycobacterial infection leads to DNMT3A-CHIP clone expansion through an IFN-γ-dependent mechanism (105). Moreover, HIV patients have a twofold higher prevalence of CHIP (108), and HIV is associated with a higher risk of MDS (106). The Swiss HIV cohort study found a greater prevalence of ASXL1 mutations (108), while the ARCHIVE study revealed a higher prevalence of DNMT3A (48.5%), TET2 (20.5%), and ASXL1 (11.4%) mutations in HIV patients (107). ASXL1 and DNMT3A mutations are frequently detected in HIV patients with MDS (106). Given the association of CHIP with increased cardiac risk, it is worth exploring whether CHIP could explain the elevated risk of CVD in HIV patients (139). Moreover, PPM1D-CHIP carriers have been observed to experience a more severe course of disease following SARS-CoV-2 infection (109), and CHIP is associated with an increased risk of clostridial or streptococcal/enterococcal infections (109).
CHIP is common among patients with solid tumors, as summarized by Marshall and colleagues (35). Associations exist between CHIP and several types of cancer, including bladder tumors (38, 140, 141), breast cancer (38, 140–143), colorectal cancer (38, 140, 142, 144, 145), endometrial cancer (38, 140), esophagogastric cancer (38, 140, 143), head and neck cancer (38, 140, 143), kidney cancer (38, 140, 141), lung cancer (38, 140–143), melanoma (38, 140, 141), ovarian cancer (38, 41, 140, 143), pancreatic cancer (38, 140–143), prostate cancer (38, 140–142, 146), and thyroid cancer (38, 140, 141, 143). Across those malignancies, mutations in the genes DNMT3A, TP53, TET2, PPM1D, ATM, ASXL1, CHEK2, JAK2, CBL, SF3B1, IDH2, U2AF1, IDH1, and RUNX1 were the most common (35). The presence of CHIP in these patients suggests a potential link between CH and the development or progression of solid tumors. CHIP-associated mutations may play a contributory role in the initiation and progression of certain cancers by promoting increased inflammatory activity, DNA damage, and other pertinent cellular alterations.
Finally, cancer treatment is strongly associated with CHIP. There is compelling evidence that CH mutations may drive the transformation into t-MNs (38). Concerning the progression of CH to t-MNs, it has been observed that mutations are differentially selected based on various drug exposures. Therefore, it is essential to identify patients who exhibit risk factors associated with the development of t-MN (38). The highest CHIP prevalence is observed in patients who have undergone external beam radiation therapy, cytotoxic chemotherapy (particularly with topoisomerase II inhibitors, anthracyclines, or carboplatin), and radionuclide therapy (38). Furthermore, ambient exposure to radiation is associated with CHIP, as exemplified by the NASA Twin Study where two astronauts exhibited CHIP nearly two decades earlier than the mean age and displayed more significant shifts in clone size compared to age-matched controls or radiotherapy patients (147). It has been shown that ionizing radiation, topoisomerase II inhibitors, and cisplatin select for mutations in DDR genes TP53, PPM1D, and CHEK2 (38, 110–112). Particularly the chemotherapy agents cisplatin, etoposide, and doxorubicin selectively promote the growth of TP53- and PPM1D-mutated CHIP clones that are resistant to chemotherapeutic agents (110). Those mutations are more commonly detected in therapy-related acute myeloid leukemia (t-AML) than de novo AML (110). Similarly, CHEK2-mutant blood cells can expand under therapy with topoisomerase inhibitors, leading to secondary MN, such as t-AML or secondary MDS (38). Clones with lower VAF can be detected in the blood even before t-AML diagnosis and prior to chemotherapy (112, 148). PPM1D mutations are most frequently observed in secondary therapy-related myelodysplastic syndromes (t-MDS) (110, 115) and in patients with bronchial (116) and ovarian carcinoma (113) after chemotherapy. In patients with ovarian carcinoma, TP53 mutations may also be found after chemotherapy (113). Presumably, mutations in DDR genes confer a selective advantage to HSCs under the selective pressure exerted on the hematopoietic system, with certain cytotoxic classes including platinum, radionuclide therapy, and topoisomerase I/II inhibitors creating a particular advantage for DDR-mutant HSCs (38, 117). While TP53 mutations provide a selective advantage to cells under cytotoxic stress through a dominant-negative effect, mutations in exon 6 of PPM1D lead to a truncated gene product lacking a C-terminal degradation domain, which prolongs the lifespan of the protein (114). PPM1D-mutated HSCs exhibit reduced apoptosis and increased resistance to cytotoxic chemotherapy, allowing the clones to expand under chemotherapy (110, 114). In contrast to DDR mutations, mutated DTA genes tend to foster proliferation and clonal expansion through mechanisms involving inflammation (118). A few cycles of chemotherapy are sufficient to cause significant expansion of a CH clone (149). Several risk factors have been identified for the development of t-MNs, including the number of mutations, clone size, presence of TP53 and PPM1D mutations, spliceosome mutations, and exposure to PARP inhibitor therapy (38, 39). Using PARP inhibitors in tumors with homologous recombination deficiency due to germline mutations in BRCA1/2 can provide a selective advantage for pre-existing TP53- and PPM1D-mutated clones, resulting in increased t-MN risk (39–41). This advantage is mainly attributed to the increased exposure to platinum-containing cytostatic drugs and PARP-inhibitors, such as rucaparib, used in high-grade ovarian cancer treatment (40, 41). CH-related alterations in TP53 (most common), ATM, GNAS, and JAK2 have been identified in cell-free DNA from patients with invasive glioma who underwent treatment with temozolomide, a mutagenic alkylating agent, in combination with radiation therapy (150). It is believed that in deep remission from the primary disease, a CHIP clone may persist and accumulate additional mutations, potentially leading to the development of a myeloid neoplasia (151).
In the context of cell therapies, such as autologous stem cell transplantation (SCT), allogeneic SCT, and Chimeric Antigen Receptor (CAR)-T cell therapy, an increased prevalence of CHIP has been observed. For example, SCT for Non-Hodgkin lymphoma is associated with a 30% incidence of CHIP (152). In autologous SCT, the prior exposure to cytotoxic agents used in conventional chemotherapeutic regimens can promote the occurrence of de novo mutations and also favor the selection and expansion of existing clones, with most common mutations observed in DNMT3A, TET2, TP53, ASXL1, and PPM1D (119). Patients with single and comutated DNMT3A- and PPM1D-CH were found to have lower stem cell yield and delayed recovery of platelet count after autologous SCT (120), thus suggesting investigation for CHIP prior to autologous SCT may be plausible. In allogeneic SCT, which involves the transfer of HSCs from a foreign donor to the recipient, a rare complication known as donor cell leukemia (DCL) or secondary myeloid neoplasia can occur. The complication seems to result from the transfer of pre-existing donor CHIP to the recipient (153). It has been estimated that there are approximately 0.8% DCLs in Europe, with 28% of them being CHIP-related (153). However, it has also been demonstrated that the presence of donor DNMT3A-CH is associated with enhanced recipient survival due to a decreased risk of relapse and an expanded network of inflammatory cytokines in recipients (121). The risk of DCL in allogeneic hematopoietic cell transplantation is primarily driven by somatic mutations associated with MDS, particularly in genes like TP53 or splicing factor mutations (121). In addition, there is a risk of DCL associated with germline predisposition in donors, particularly when mutations in the DDX41 gene are present (121).
Although it appears that CHIP is generally associated with unfavorable outcomes, there are at least two exceptions. In the context of CAR-T cell therapy, which exclusively utilizes autologous preparations, the presence of CHIP can influence the treatment outcome beneficially. CHIP can contribute to increased inflammatory reactions and altered interactions between immune cells, potentially affecting the efficacy and behavior of the transfected immune cells (24, 124, 154). Mutations in genes such as TET2 and DNMT3A, which are commonly associated with CHIP, may thus have implications for CAR-T cell therapy by enhancing antitumor activity (122, 123). Overall, there is a consistently high prevalence of CHIP prior to CAR-T cell therapy (ranging from 34% to 48%) (155). This high prevalence may be attributed to the intensive pretreatment received by these patients, contributing to frequent clonal progression in the long term after CAR-T cell therapy, which is relevant to the development of t-MNs (155). Recently, CHIP was shown to attenuate the risk of Alzheimer’s Disease (AD). A higher VAF was significantly associated with protection against AD when modeled as a continuous variable (156). CHIP-associated mutations were found in brain samples (156, 157), presumably due to infiltration of the brain by marrow-derived mutant myeloid cells that have adopted a microglial-like phenotype (156). These mutant cells residing in the brain may ameliorate neurodegenerative disorders due to their superior phagocytic activity.
Table 3 summarizes CHIP-associated genes, presumed mechanisms of action, and related medical conditions. The listed mechanisms reflect our current knowledge rather than a comprehensive understanding of each gene’s function and its impact on CHIP-associated outcomes.
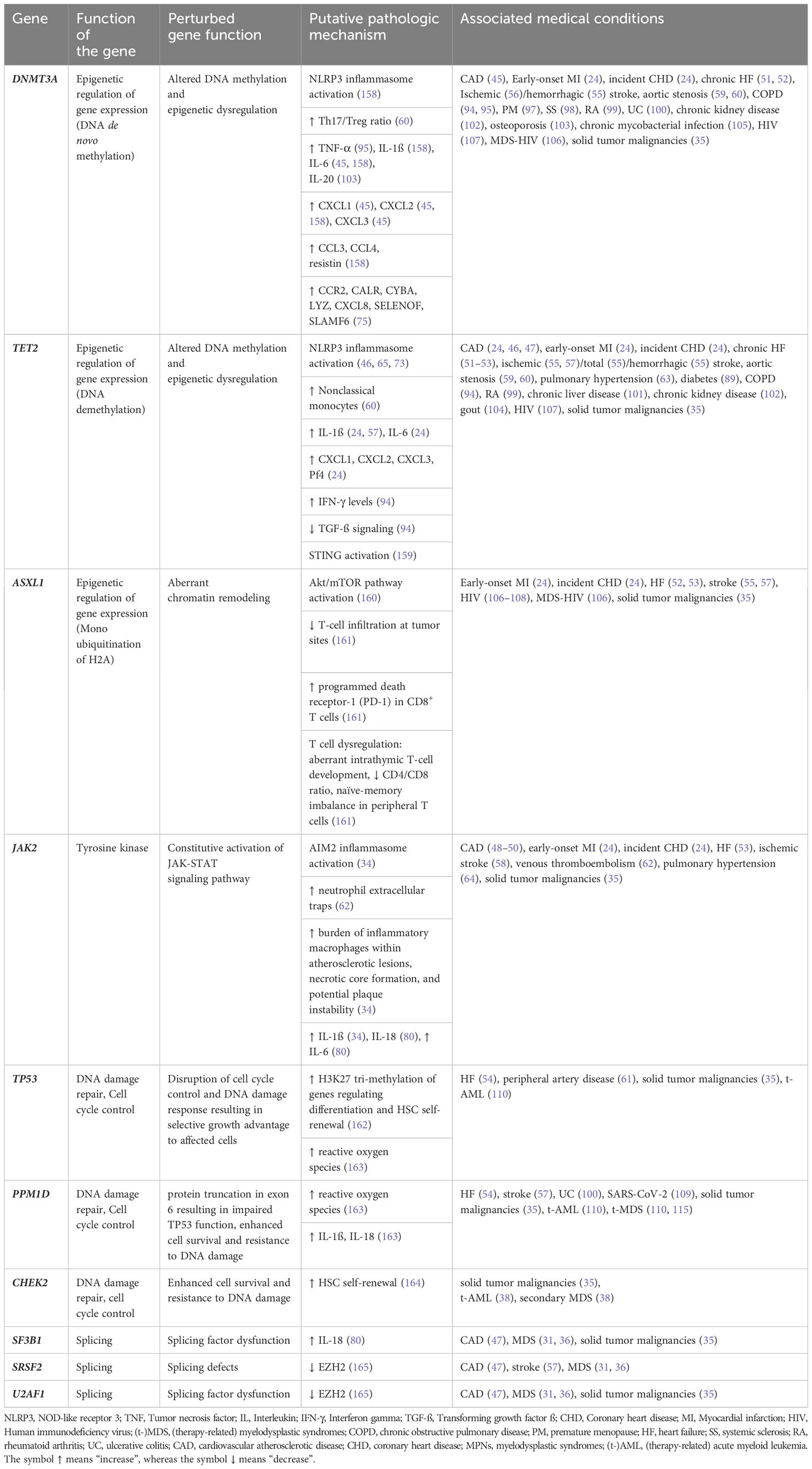
Table 3 Frequently mutated CHIP genes and associated mechanisms.
Future perspectives and clinical implications
Research on CHIP has already yielded significant insights, but many questions remain. A better knowledge of the impact of genetic mutations on cellular function could help to develop new diagnostic and therapeutic approaches. Identifying individuals with CHIP is expected to enable more precise risk assessment for hematologic disorders and cardiovascular complications, potentially leading to improved prevention and treatment strategies. Several studies have explored therapeutic approaches for cardiovascular risks related to CHIP (s. Table 4).
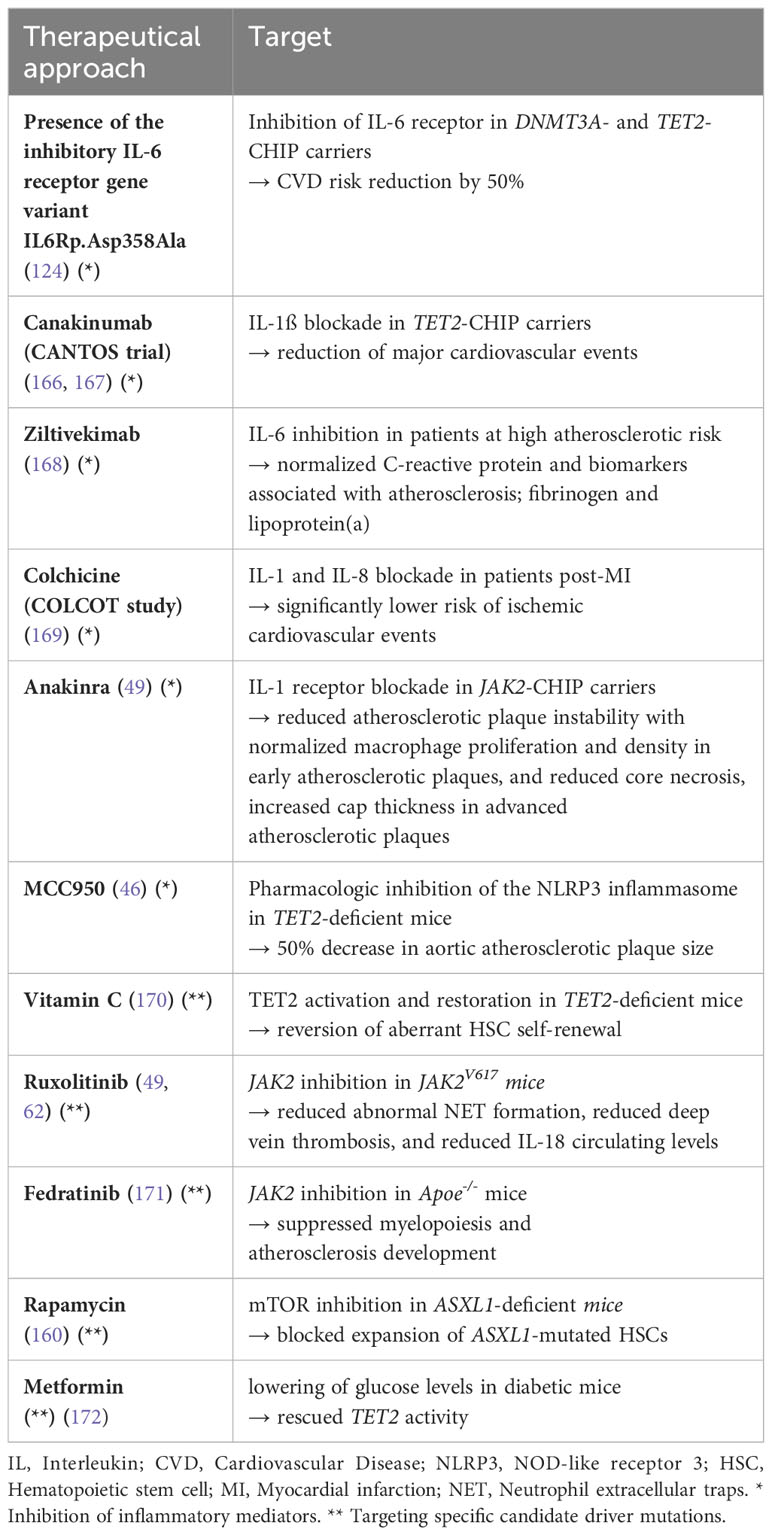
Table 4 Current therapeutical approaches for CHIP.
Inhibition of inflammatory mediators has been a focus in these studies. For example, the CANTOS clinical trial demonstrated that blocking IL-1ß (an upstream mediator of IL-6) with canakinumab resulted in reduced major adverse cardiovascular events in high-risk atherosclerosis patients (167). The reduction in events was more pronounced in TET2-CHIP patients (166). A phase-2 study is currently being conducted in Leipzig, Germany, testing the efficacy and safety of canakinumab for the treatment of anemia in low-risk MDS patients (ClinicalTrials.gov Identifier: NCT05237713). Another study used ziltivekimab for IL-6 inhibition in patients at high atherosclerotic risk and achieved normalized expression of C-reactive protein and biomarkers associated with atherosclerosis, such as fibrinogen and lipoprotein(a) (168). Interestingly, the presence of the inhibitory IL-6 receptor gene variant IL6R p.Asp.358 Ala reduced CVD risk by 50% in DNMT3A- and TET2-CHIP carriers (124). Non-selective IL-1 receptor blockade with anakinra and targeted IL-1ß blockade in a JAK2-CHIP model resulted in decreased atherosclerotic plaque instability, normalized macrophage proliferation and density in early plaques, and reduced core necrosis and increased cap thickness in advanced plaques (49). The COLCOT study explored modulating IL-1 and IL-8 using colchicine in patients recruited within 30 days post-MI, thereby significantly lowering risk of ischemic cardiovascular events (169). Pharmacologic inhibition of the NLRP3 inflammasome with MCC950 in TET2-deficient mice led to a 50% decrease in aortic atherosclerotic plaque size, which was greater than the nonsignificant reduction observed in wildtype mice (46). Targeted inhibition of the AIM2 inflammasome, which is overactive in JAK2-mutant CHIP, may offer similar benefits (49).
Further investigations have focused on targeting specific driver mutations in CHIP. For example, vitamin C metabolites that activate TET2 and mimic the restoration of TET2 via elevated 5-hydroxymethycytosine formation in TET2-deficient mice showed potential as a therapy for TET2-CHIP carriers (170). Treatment with the JAK2 inhibitor ruxolitinib reduced abnormal NET formation (62), deep vein thrombosis (62), and circulating levels of IL-18 (49) in JAK2V617 mice. JAK2 inhibition with fedratinib in Apoe-/- mice suppressed myelopoiesis and the development of atherosclerosis (171). Rapamycin treatment has been shown to block expansion of ASXL1-mutated HSCs through mTOR inhibition (160). The heightened self-renewal and neoplastic transformation observed in TET2-mutated HSPCs has recently been demonstrated to result from the activation of the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-stimulator of interferon genes (STING) pathway (159). Hence, targeting the STING pathway could represent a promising intervention strategy for relevant hematopoietic diseases. Finally, TET2 activity could be restored in diabetic mice by lowering blood glucose levels with metformin (172), suggesting that this may also hold promise for mitigating CVD risk in patients with TET2-driven CHIP.
Overall, some therapeutic approaches are emerging that mainly focus on inhibition of inflammatory pathways and restoration of gene function. However, further research is needed to confirm the efficacy and safety of such treatments in individuals with CHIP.
CHIP detection and risk management
The prevalence of CHIP depends on the age of the study cohort and the sensitivity of the DNA sequencing method (26). Currently, the threshold for CHIP is a VAF ≥2%, which means that a heterozygous somatic mutation in a leukemia-associated gene is present in ≥4% of all blood cells (5). Since there is generally no screening for CHIP in routine clinical practice, CHIP is often an incidental finding during tumor diagnostics, for example, when DNA from blood cells serves as a normal control, or in the context of genetic workup for other disease (149). Regarding molecular tumor diagnostics, it is important to note that, as long as a normal wildtype finding in the blood is not available, CH-related driver mutations detected in tumor tissue should not be interpreted as tumor mutations (149). It has been shown that mutations in CH-associated genes were detectable in 65% of solid tumors, of which 8% were CH mutations instead of tumor mutations, with DNMT3A most common (173). This interference can be resolved by parallel sequencing of tumor material and DNA from peripheral blood (149).
The most sensitive technique, i.e. ‘error corrected sequencing’ (ECS) can identify point mutations and small indels from adult blood and marrow samples at a VAF as low as 0.01% (26, 174). ‘Targeted sequencing’ looks at a specific selection of genes, mostly through gene panel sequencing using next generation sequencing (NGS), detecting clones with a VAF ≥0.5% (26, 175). Targeted sequencing provides greater sequencing depth and sensitivity, and therefore detects more people with CH (26), than whole exome (WES) (5, 6) or whole genome (WGS) sequencing (32), which typically have a much lower depth of coverage. WGS covers the entire genome but is relatively insensitive to smaller clones, detecting clones with a VAF ≥7% (26). WES covers only the protein-coding regions of the genome but is somewhat more sensitive to smaller clones, detecting clones with a VAF around 3% or higher (26). Currently, the biological significance of very small clones is unclear (175–177).
Regarding the future management of people with CHIP, setting up ‘CHIP clinics’ is an important initiative. Here, individuals with certain risk factors for CHIP will be offered adequate screening, and individuals who are already diagnosed with CHIP will receive counseling and, if possible, a plan for intervention, considering individual lifestyle and environmental influences (178). For that purpose, ideally, hematologists and cardiologists should cooperate with hepatologists, geneticists, psychologists, and nutritionists in an interdisciplinary consultative clinic (178). Comprehensive screening for CHIP mutations and identification of potential correlations with clinical patterns will facilitate consultation tailored to patients’ individual needs and will also support further research into CHIP.
However, the absence of established guidelines for “CHIP consultation” suggests that the way forward will not be easy. There is pronounced variability regarding the health effects of CHIP mutations, depending on genes affected, VAFs, dynamics of clonal expansion, and interaction between multiple genetic aberrations. This precludes simple algorithms for counseling.
In terms of hematological risk management, CHIP consultation is particularly relevant for patients carrying mutations linked to an increased risk of hematological malignancy, such as driver mutations for MNs, especially IDH1/IDH2, TP53, or spliceosome genes (179). The Clonal Hematopoiesis Risk Score (CHRS) is a prognostic tool to assess the risk of malignancy in individuals with CHIP or CCUS. It helps to identify a minority of high-risk CHIP/CCUS cases among the majority of cases with minimal risk of progression to MN (180). Critical predictors of the 10-year probability of developing MN include high risk mutations, single DNMT3A mutations, presence of 2 or more mutations, age ≥65 years, diagnosis of CCUS or CHIP, maximum VAF ≥0.2, mean corpuscular volume (MCV) ≥100 fl, and red cell distribution width (RDW) ≥15% (180). For patients who are estimated to be at high risk of progression to MN, it is advisable to initiate regular monitoring of blood counts. If abnormalities are detected, the threshold for performing bone marrow biopsy should be low, and repeated deep sequencing should be considered for monitoring the clonal dynamics of CHIP. If a mutation has a VAF of ≥10%, or if there are ≥2 mutations present, it is recommended to conduct differential blood counts every 3 months in the first year and perhaps annually from the second year onward (181). If there is no evidence of progression, regular health check-ups are still advisable (181). Individuals with CHIP should also be evaluated for potential participation in a clinical trial aimed at preventing progression. Such trials are important to corroborate potential treatment options, such as administering high-dose vitamin C to restore gene function in TET2-mutated individuals (170). Since we do not yet have convincing therapies, clinical guidelines are also lacking.
Regarding cardiovascular risk management, the presence of a JAK2V617F mutation and events involving JAK2-mutated CN-LOH, for instance, are strongly associated with an increased risk of CVD (179). Somatic mutations in the three most frequently mutated CHIP genes, DNMT3A, TET2 and ASXL1, have also been linked to a variety of cardiovascular problems. Certain CHIP mutations may be associated with lifestyle habits. For example, mutations in ASXL1 seem to be promoted by smoking (38, 94, 96). Therefore, it should be helpful to counsel high-risk patients regarding lifestyle factors, in order to ameliorate the development of cardiovascular complications. Another approach is to suppress certain inflammatory pathways, in order to reduce the risk of CVD in patients with a high pro-inflammatory profile. Examples for that approach are the CANTOS (166, 167) and COLCOT (169) studies, using canakinumab and colchicine, respectively. In the future, such treatments may become available in the context of a CHIP clinic.
Counseling for CHIP becomes even more complex when germline mutations are incidentally detected along with somatic CHIP mutations. The potential impact on family members is considerable. According to ethical guidelines, it is ultimately the affected individual’s decision whether or not they prefer to be informed about such findings. They also retain the autonomy to decide whether or not their healthcare provider should convey the information to their relatives, who might carry the same germline mutation.
It has already been shown that interest in CHIP testing is high in certain patient groups. A study involving over 500 young breast cancer survivors gauged their perspective on testing for CHIP mutations. Being presented with a case vignette, 78.6% of respondents opted for CHIP testing (182). The interest in testing increased to 85.8% if the possibility of consultation in a specialized CHIP clinic was introduced, and further rose to 92.7% if the scenario included the availability of a hypothetical CHIP treatment (182). Patients expressed a strong desire to receive information about CHIP through printed material (62.7%) or website-based resources (60.2%) (182). Above all, in-person discussions, particularly with oncologists or cancer specialists, were the preferred mode of communication (88.1%) (182). Upon receiving a CHIP diagnosis, patients indicated their interest in periodic communications, lifestyle modifications, annual blood tests, and possible treatment (182). The preferred setting for CHIP follow-up care was predominantly a specialized breast oncology clinic (39.6%) or a CHIP-focused clinic (32.8%) (182). The levels of anxiety when learning about CHIP varied. Specifically, 26.9% reported no anxiety, 37.9% experienced mild anxiety, 24.4% had moderate anxiety, 2.5% indicated severe anxiety, and only 1.3% reported very severe anxiety (182). In summary, patients showed a keen interest in obtaining information about CHIP from specialized providers, along with receiving individualized risk assessment and health recommendations (182).
Conclusion and outlook
CHIP is a fascinating field of research that expands our understanding of the development of hematologic disorders and cardiovascular risks. Several studies have demonstrated that CHIP can be a precursor to MDS and AML, in particular after cytotoxic therapy, suggesting that such treatments promote the emergence and expansion of mutated cells, thereby increasing the risk of leukemic transformation. However, only 0.5-1% of people affected by CHIP will develop a hematological malignancy. Interestingly, CHIP turned out to be associated with diverse cardiovascular diseases. Abnormal interaction between mutant monocytes-macrophages and the endothelium, along with increased expression of pro-inflammatory molecules like IL-1ß and IL-6, substantially promotes atherosclerotic development and also affects the myocardium, resulting in MI and/or HF. Since people with premature CAD do not exhibit traditional risk factors, they should be investigated for CHIP as a potential explanation for their cardiovascular problem. This requires deep sequencing of peripheral blood samples.
A better understanding of the impact of specific mutations, their interactions with environmental factors, and their role in disease progression will help to identify individuals at high risk of hematological and/or cardiovascular complications and will foster the development of potential therapeutic and preventive strategies. Recurring patterns have already been identified in various CHIP-associated conditions (Figure 1).
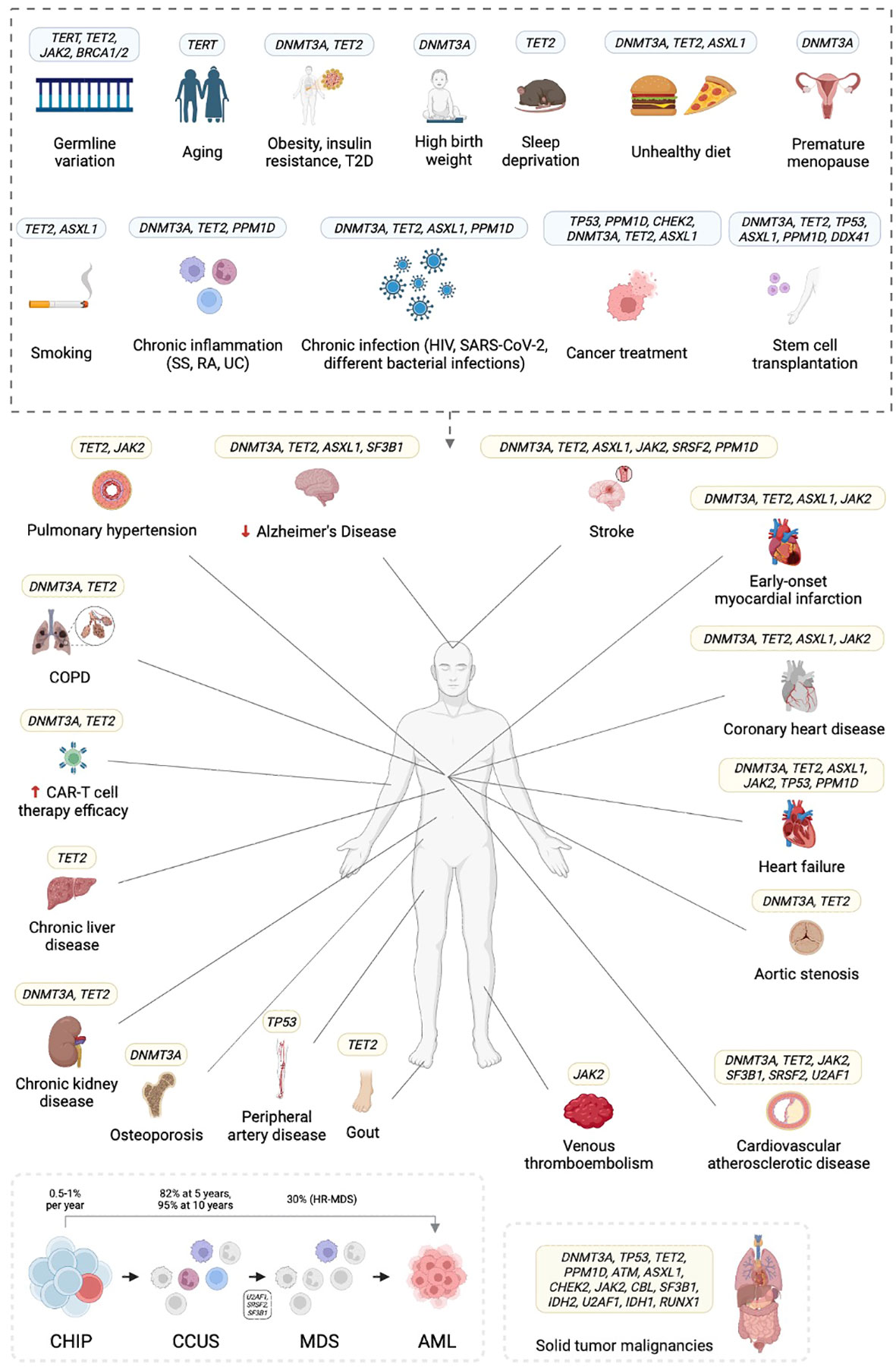
Figure 1 Clonal hematopoiesis of indeterminate potential (CHIP) is associated with several risk factors and confers hematologic, cardiovascular, and other risks. T2D, type 2 diabetes; COPD, chronic obstructive pulmonary disease; SS, systemic sclerosis; RA, rheumatoid arthritis; UC, ulcerative colitis; HIV, human immunodeficiency virus; SARS-CoV-2, severe acute respiratory syndrome coronavirus type 2; CAR-T, chimeric antigen receptor (CAR)-T cell therapy; CCUS, clonal cytopenia of undetermined significance; (HR-)MDS, (high risk) myelodysplastic syndrome; AML, acute myeloid leukemia. Created in BioRender.com.
Currently, treatment strategies for CHIP beyond the modification of traditional risk factors are limited. By targeting specific molecular pathways and inflammatory processes, it may be possible to mitigate the increased risk of cardiovascular events associated with the clonal expansion of abnormal myeloid cells. Besides neutralizing inflammatory mediators such as IL-1ß or IL-6, targeted inhibition of specific candidate driver mutations in CHIP, such as TET2 or JAK2, might be pursued. However, further research is needed to validate these approaches.
Individuals with CHIP should receive proper counseling, which may best be provided in a specialized CHIP outpatient clinic. We and others are moving in that direction. Understanding the mutational landscape and associated clinical implications of CHIP variants is crucial for risk stratification and personalized management, taking into consideration individual lifestyle factors. We are going to focus our scientific interest on patients with early-onset cardiovascular diseases, hematological patients with suspected but not confirmed MDS, and breast cancer patients with BRCA1/2 germline mutations who have received chemotherapy and/or PARP inhibitor therapy. Such patients will be screened for CHIP-related mutations, and those identified with CHIP will receive counseling in our CH outpatient clinic according to the current state of knowledge.
Author contributions
AC: Writing – original draft, Writing – review & editing. FS: Writing – review & editing. UG: Supervision, Writing – review & editing. SD: Writing – review & editing. NG: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research has received funding by Deutsche Krebshilfe e.V. which sponsors the doctoral research program ‘Cancer Prevention -Graduate School’. The title of the corresponding research project is ‘Mutational analysis for hematologic and cardiologic risk management in patients with clonal hematopoiesis of indeterminate potential (CHIP)’.
Acknowledgments
The authors thank the following cooperation partners for their contribution to the project: Prof. Dr. Dagmar Wieczorek, Dr. rer. nat. Beate Betz and Dr. rer. nat. Harald Surowy (Institute for Human Genetics, University Hospital Düsseldorf), Kerstin Becker (West German Genome Center Cologne), PD Dr. Florian Bönner, PD Dr. Amin Polzin and Dr. rer. nat. Daniel Oehler (Department of Cardiology, Pulmonology and Angiology, University Hospital Düsseldorf), Prof. Dr. Rita Schmutzler and PD Dr. rer. nat. Eric Hahnen (Center for Familial Breast and Ovarian Cancer, University Hospital Cologne), Prof. Dr. Tanja Fehm, Dr. med. Bernadette Jäger and Dr. rer. nat. Dieter Niederacher (Department of Gynecology and Obstetrics, University Hospital Düsseldorf), Prof. Dr. Andreas Schuppert (Institute of Computational Biomedicine, RWTH Aachen), Dr. rer. nat. Ute Fischer (Department of Pediatric Oncology, Hematology and Clinical Immunology, University Hospital Düsseldorf), and Prof. Dr. Gesine Kögler (Institute for Transplantation Diagnostics and Cellular Therapies, University Hospital Düsseldorf).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
2. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka H-M, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: Integrating morphologic, clinical, and genomic data. Blood (2022) 140:1200–28. doi: 10.1182/blood.2022015850
3. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
4. Valent P, Orazi A, Steensma DP, Ebert BJ, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget (2017) 8:73483–500. doi: 10.18632/oncotarget.19008
5. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
6. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
7. Xie M, Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and Malignancies. Nat Med (2014) 20:1472–8. doi: 10.1038/nm.3733
8. Heuser M, Thol F, Ganser A. Clonal hematopoiesis of indeterminate potential. Dtsch Ärztebl Int (2016) 113:317–22. doi: 10.3238/arztebl.2016.0317
9. Hartmann L, Hecker JS, Rothenberg-Thurley M, Rivière J, Jentzsch M, Ksienzyk B, et al. Compartment-specific mutational landscape of clonal hematopoiesis. Leukemia (2022) 36:2647–55. doi: 10.1038/s41375-022-01700-3
10. Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med (2021) 27:1921–7. doi: 10.1038/s41591-021-01521-4
11. Von Beck K, Von Beck T, Ferrell PB, Bick AG, Kishtagari A. Lymphoid clonal hematopoiesis: Implications for Malignancy, immunity, and treatment. Blood Cancer J (2023) 13:5. doi: 10.1038/s41408-022-00773-8
12. Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet (2012) 44:651–8. doi: 10.1038/ng.2270
13. Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet (2012) 44:642–50. doi: 10.1038/ng.2271
14. Machiela MJ, Zhou W, Sampson JN, Dean MC, Jacobs KB, Black A, et al. Characterization of large structural genetic mosaicism in human autosomes. Am J Hum Genet (2015) 96:487–97. doi: 10.1016/j.ajhg.2015.01.011
15. Vattathil S, Scheet P. Extensive hidden genomic mosaicism revealed in normal tissue. Am J Hum Genet (2016) 98:571–8. doi: 10.1016/j.ajhg.2016.02.003
16. Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature (2018) 559:350–5. doi: 10.1038/s41586-018-0321-x
17. Loh P-R, Genovese G, McCarroll SA. Monogenic and polygenic inheritance become instruments for clonal selection. Nature (2020) 584:136–41. doi: 10.1038/s41586-020-2430-6
18. Zekavat SM, Lin S-H, Bick AG, Liu A, Paruchuri K, Uddin MM, et al. Hematopoietic mosaic chromosomal alterations and risk for infection among 767,891 individuals without blood cancer. medRxiv (2020) 2020.11.12.20230821. doi: 10.1101/2020.11.12.20230821
19. Valent P, Kern W, Hoermann G, Milosevic Feenstra JD, Sotlar K, Pfeilstöcker M, et al. Clonal hematopoiesis with oncogenic potential (CHOP): Separation from CHIP and roads to AML. Int J Mol Sci (2019) 20:789. doi: 10.3390/ijms20030789
20. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet (2012) 44:1179–81. doi: 10.1038/ng.2413
21. Malcovati L, Cazzola M. The shadowlands of MDS: Idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology (2015) 2015:299–307. doi: 10.1182/asheducation-2015.1.299
22. Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer (2017) 17:5–19. doi: 10.1038/nrc.2016.112
23. Valent P. ICUS, IDUS CHIP. and CCUS: Diagnostic criteria, separation from MDS and clinical implications. Pathobiology (2019) 86:30–8. doi: 10.1159/000489042
24. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
25. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
26. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science (2019) 366:eaan4673. doi: 10.1126/science.aan4673
27. Caiado F, Kovtonyuk LV, Gonullu NG, Fullin J, Boettcher S, Manz MG. Aging drives TET2+/– clonal hematopoiesis via IL-1 signaling. Blood (2023) 141:886–903. doi: 10.1182/blood.2022016835
28. Rauh MJ. Breaking the CH inflammation-expansion cycle. Blood (2023) 141:815–6. doi: 10.1182/blood.2022019104
29. Kouroukli O, Symeonidis A, Foukas P, Maragkou M-K, Kourea EP. Bone marrow immune microenvironment in myelodysplastic syndromes. Cancers (2022) 14:5656. doi: 10.3390/cancers14225656
30. Fey MF, Liechti-Gallati S, von Rohr A, Borisch B, Theilkas L, Schneider V, et al. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe [see comments]. Blood (1994) 83:931–8. doi: 10.1182/blood.V83.4.931.931
31. Osman AEWG. When are idiopathic and clonal cytopenias of unknown significance (ICUS or CCUS)? Hematology (2021) 2021:399–404. doi: 10.1182/hematology.2021000272
32. Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood (2017) 130:742–52. doi: 10.1182/blood-2017-02-769869
33. Fabre MA, Guilherme de Almeida J, Fiorillo E, Mitchell E, Damaskou A, Rak J, et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature (2022) 606:335–42. doi: 10.1038/s41586-022-04785-z
34. Hou H-A, Liu C-Y, Kuo Y-Y, Chou W-C, Tsai C-H, Lin C-C, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget (2016) 7:9084–101. doi: 10.18632/oncotarget.7000
35. Marshall CH, Gondek LP, Luo J, Antonarakis ES. Clonal hematopoiesis of indeterminate potential in patients with solid tumor Malignancies. Cancer Res (2022) 82:4107–13. doi: 10.1158/0008-5472.CAN-22-0985
36. Malcovati L, Gallì A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood (2017) 129:3371–8. doi: 10.1182/blood-2017-01-763425
37. Schroeder T, Ruf L, Bernhardt B, Aivado M, Aul C, Gattermann N, et al. Distinguishing myelodysplastic syndromes (MDS) from idiopathic cytopenia of undetermined significance (ICUS): HUMARA unravels clonality in a subgroup of patients. Ann Oncol (2010) 21:2267–71. doi: 10.1093/annonc/mdq233
38. Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet (2020) 52:1219–26. doi: 10.1038/s41588-020-00710-0
39. Martin JE, Khalife-Hachem S, Grinda T, Kfoury M, Garciaz S, Pasquier F, et al. Therapy-related myeloid neoplasms following treatment with PARP inhibitors: New molecular insights. Ann Oncol (2021) 32:1046–8. doi: 10.1016/j.annonc.2021.04.015
40. Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, Aghajanian C, et al. Preexisting TP53-variant clonal hematopoiesis and risk of secondary myeloid neoplasms in patients with high-grade ovarian cancer treated with rucaparib. JAMA Oncol (2021) 7:1772. doi: 10.1001/jamaoncol.2021.4664
41. Weber-Lassalle K, Ernst C, Reuss A, Möllenhoff K, Baumann K, Jackisch C, et al. Clonal hematopoiesis–associated gene mutations in a clinical cohort of 448 patients with ovarian cancer. JNCI J Natl Cancer Inst (2022) 114:565–70. doi: 10.1093/jnci/djab231
42. Saiki R, Momozawa Y, Nannya Y, Nakagawa MM, Ochi Y, Yoshizato T, et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat Med (2021) 27:1239–49. doi: 10.1038/s41591-021-01411-9
43. Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell (2022) 185:1630–45. doi: 10.1016/j.cell.2022.04.004
44. Senguttuvan NB, Subramanian V, Venkatesan V, Muralidharan TR, Sankaranarayanan K. Clonal hematopoiesis of indeterminate potential (CHIP) and cardiovascular diseases—an updated systematic review. J Genet Eng Biotechnol (2021) 19:105. doi: 10.1186/s43141-021-00205-3
45. Rauch PJ, Gopakumar J, Silver AJ, Nachun D, Ahmad H, McConkey M, et al. Loss-of-function mutations in DNMT3A and TET2 lead to accelerated atherosclerosis and concordant macrophage phenotypes. Nat Cardiovasc Res (2023) 2:805–18. doi: 10.1038/s44161-023-00326-7
46. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (2017) 355:842–7. doi: 10.1126/science.aag1381
47. Gumuser ED, Schuermans A, Mi Jemma Cho S, Sporn ZA, Uddin M, Paruchuri K, et al. Clonal hematopoiesis of indeterminate potential predicts adverse outcomes in patients with atherosclerotic cardiovascular disease. J Am Coll Cardiol (2023) 81:1996–2009. doi: 10.1016/j.jacc.2023.03.401
48. James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434:1144–8. doi: 10.1038/nature03546
49. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
50. Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2 V617F mice. Circ Res (2018) 123(11):e35–47. doi: 10.1161/CIRCRESAHA.118.313283
51. Pascual-Figal DA, Bayes-Genis A, Díez-Díez M, Hernández-Vicente Á, Vásquez-Andrés D, de la Barrera J, et al. Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J Am Coll Cardiol (2021) 77:1747–59. doi: 10.1016/j.jacc.2021.02.028
52. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol (2019) 4:25. doi: 10.1001/jamacardio.2018.3965
53. Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, et al. Supplemental association of clonal hematopoiesis with incident heart failure. J Am Coll Cardiol (2021) 78:42–52. doi: 10.1016/j.jacc.2021.04.085
54. Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight (2021) 6:e146076. doi: 10.1172/jci.insight.146076
55. Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke (2022) 53:788–97. doi: 10.1161/STROKEAHA.121.037388
56. Qiu X, Dai Y, Cheng S, Gu HQ, Jiang Y, Meng X, et al. Somatic mutation contributing to clonal haematopoiesis is a risk factor of recurrent stroke in first-ever acute ischaemic stroke: A prospective cohort study. Stroke Vasc Neurol (2023) 8:103–10. doi: 10.1136/svn-2022-001756
57. Arends CM, Liman TG, Strzelecka PM, Kufner A, Löwe P, Huo S, et al. Associations of clonal hematopoiesis with recurrent vascular events and death in patients with incident ischemic stroke. Blood (2023) 141:787–99. doi: 10.1182/blood.2022017661
58. Kristiansen MH, Kjær L, Skov V, Larsen MK, Ellervik C, Hasselbalch HC, et al. JAK2V617F mutation is highly prevalent in patients with ischemic stroke: A case-control study. Blood Adv (2023) 7(19):5825–34. doi: 10.1182/bloodadvances.2023010588
59. Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol (2020) 5:1170. doi: 10.1001/jamacardio.2020.2468
60. Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J (2020) 41:933–9. doi: 10.1093/eurheartj/ehz591
61. Zekavat SM, Viana-Huete V, Zuriaga MA, Uddin M, Paruchuri K, et al. TP53- mediated clonal hematopoiesis confers increased risk for incident peripheral artery disease. medRxiv (2021) 2021.08.22.21262430. doi: 10.1101/2021.08.22.21262430
62. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med (2018) 10:eaan8292. doi: 10.1126/scitranslmed.aan8292
63. Potus F, Pauciulo MW, Cook EK, Zhu N, Hsieh A, Welch CL, et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation (2020) 141:1986–2000. doi: 10.1161/CIRCULATIONAHA.119.044320
64. Kimishima Y, Misaka T, Yokokawa T, Wada K, Ueda K, Sugimoto K, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun (2021) 12:6177. doi: 10.1038/s41467-021-26435-0
65. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. TET2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037
66. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res (2018) 122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362
67. Baber U, Mehran R, Sartori S, Schoos MM, Sillesen H, Muntendam P, et al. Prevalence, impact, and predictive value of detecting subclinical coronary and carotid atherosclerosis in asymptomatic adults: the BioImage study. J Am Coll Cardiol (2015) 65:1065–74. doi: 10.1016/j.jacc.2015.01.017
68. Wilkins JT, Ning H, Berry J, Zhao L, Dyer AR, Lloyd-Jones DM. Lifetime risk and years lived free of total cardiovascular disease. JAMA (2012) 308:1795. doi: 10.1001/jama.2012.14312
69. Libby P, Sidlow R, Lin AE, Gupta D, Jones LW, Moslehi J, et al. Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC review topic of the week. J Am Coll Cardiol (2019) 74:567–77. doi: 10.1016/j.jacc.2019.06.007
70. Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv (2018) 2:3404–10. doi: 10.1182/bloodadvances.2018020222
71. Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur Heart J (2021) 42:257–65. doi: 10.1093/eurheartj/ehaa845
72. Shi C, Aboumsallem JP, Suthahar N, de Graaf AO, Jansen JH, van Zeventer IA, et al. Clonal haematopoiesis of indeterminate potential: Associations with heart failure incidence, clinical parameters and biomarkers. Eur J Heart Fail (2023) 25:4–13. doi: 10.1002/ejhf.2715
73. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-mediated gene editing to assess the roles of TET2 and DNMT3A in clonal hematopoiesis and cardiovascular disease. Circ Res (2018) 123:335–41. doi: 10.1161/CIRCRESAHA.118.313225
74. Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, et al. JAK2-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci (2019) 4:684–97. doi: 10.1016/j.jacbts.2019.05.013
75. Abplanalp WT, Schuhmacher B, Cremer S, Merten M, Shumliakivska M, Macinkovic I, et al. Cell-intrinsic effects of clonal hematopoiesis in heart failure. Nat Cardiovasc Res (2023) 2:819–34. doi: 10.1038/s44161-023-00322-x
76. Heimlich JB, Bick AG. Somatic mutations in cardiovascular disease. Circ Res (2022) 130:149–61. doi: 10.1161/CIRCRESAHA.121.319809
77. El Husseini D, Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, et al. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J Mol Cell Cardiol (2014) 72:146–56. doi: 10.1016/j.yjmcc.2014.02.014
78. Mathieu P, Bouchareb R, Boulanger M-C. Innate and adaptive immunity in calcific aortic valve disease. J Immunol Res (2015) 2015:1–11. doi: 10.1155/2015/851945
79. Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood (2016) 128:1121–8. doi: 10.1182/blood-2015-06-652941
80. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature (2020) 586:763–8. doi: 10.1038/s41586-020-2819-2
81. Kessler MD, Damask A, O'Keeffe S, Banerjee N, Li D, Watanabe K, et al. Exome sequencing of 628,388 individuals identifies common and rare variant associations with clonal hematopoiesis phenotypes. medRxiv (2022) 2021.12.29.21268342. doi: 10.1101/2021.12.29.21268342
82. Williams N, Lee J, Moore L, Baxter JE, Hewinson J, Dawson KJ, et al. Phylogenetic reconstruction of myeloproliferative neoplasm reveals very early origins and lifelong evolution. bioRxiv (2020) 2020:11.09.374710. doi: 10.1101/2020.11.09.374710
83. Williams N, Lee J, Moore L, Baxter JE, Hewinson J, Dawson KJ, et al. Driver mutation acquisition in utero and childhood followed by lifelong clonal evolution underlie myeloproliferative neoplasms. Blood (2020) 136:LBA–1. doi: 10.1182/blood-2020-143813
84. Schulz E, Valentin A, Ulz P, Beham-Schmid C, Lind K, Rupp V, et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J Med Genet (2012) 49:422–8. doi: 10.1136/jmedgenet-2011-100674
85. Perdigones N, Perin JC, Schiano I, Nicholas P, Biegel JA, Mason PJ, et al. Clonal hematopoiesis in patients with dyskeratosis congenita. Am J Hematol (2016) 91:1227–33. doi: 10.1002/ajh.24552
86. Nakao T, Bick AG, Taub MA, Zekavat SM, Uddin MM, Niroula A, et al. Bidirectional Mendelian randomization supports bidirectional causality between telomere length and clonal hematopoiesis of intermediate potential. medRxiv (2021) 2021:2.26.21252199. doi: 10.1101/2021.02.26.21252199
87. Codd V, Wang Q, Allara E, Musicha C, Kaptoge S, Stoma S, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat Genet (2021) 53:1425–33. doi: 10.1038/s41588-021-00944-6
88. You D, Nilsson E, Tenen DE, Lyubetskaya A, Lo JC, Jiang R, et al. DNMT3A is an epigenetic mediator of adipose insulin resistance. eLife (2017) 6:e30766. doi: 10.7554/eLife.30766
89. Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, et al. TET2-loss-of-function-driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep (2020) 33:108326. doi: 10.1016/j.celrep.2020.108326
90. Asada S, Kitamura T. Clonal hematopoiesis and associated diseases: A review of recent findings. Cancer Sci (2021) 112:3962–71. doi: 10.1111/cas.15094
91. Schuermans A, Nakao T, Ruan Y, Koyama S, Yu Z, Uddin MM, et al. Birth weight is associated with clonal hematopoiesis of indeterminate potential and cardiovascular outcomes in adulthood. J Am Heart Assoc (2023) 12(13):e030220. doi: 10.1161/JAHA.123.030220
92. Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell (2021) 184:1348–1361.e22. doi: 10.1016/j.cell.2021.01.049
93. Bhattacharya R, Zekavat SM, Uddin MM, Pirruccello J, Niroula A, Gibson C, et al. Association of diet quality with prevalence of clonal hematopoiesis and adverse cardiovascular events. JAMA Cardiol (2021) 6:1069–77. doi: 10.1001/jamacardio.2021.1678
94. Miller PG, Qiao D, Rojas-Quintero J, Honigberg MC, Sperling AS, Gibson CJ, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood (2022) 139:357–68. doi: 10.1182/blood.2021013531
95. Kuhnert S, Mansouri S, Rieger MA, Savai R, Avci E, Díaz-Piña G, et al. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with COPD. Cells (2022) 11:2121. doi: 10.3390/cells11132121
96. Dawoud AAZ, Tapper WJ, Cross NCP. Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia (2020) 34:2660–72. doi: 10.1038/s41375-020-0896-8
97. Honigberg MC, Zekavat SM, Niroula A, Griffin GK, Bick AG, Pirruccello JP, et al. Premature menopause, clonal hematopoiesis, and coronary artery disease in postmenopausal women. Circulation (2021) 143:410–23. doi: 10.1161/CIRCULATIONAHA.120.051775
98. Ricard L, Hirsch P, Largeaud L, Deswarte C, Jachiet V, Mohty M, et al. Clonal haematopoiesis is increased in early onset in systemic sclerosis. Rheumatol Oxf Engl (2020) 59:3499–504. doi: 10.1093/rheumatology/keaa282
99. Savola P, Lundgren S, Keränen MAI, Almusa H, Ellonen P, Leirisalo-Repo M, et al. Clonal hematopoiesis in patients with rheumatoid arthritis. Blood Cancer J (2018) 8:69. doi: 10.1038/s41408-018-0107-2
100. Zhang CRC, Nix D, Gregory M, Ciorba MA, Ostrander EL, Newberry RD, et al. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp Hematol (2019) 80:36–41.e3. doi: 10.1016/j.exphem.2019.11.008
101. Wong WJ, Emdin C, Bick AG, Zekavat SM, Niroula A, Pirruccello JP, et al. Clonal haematopoiesis and risk of chronic liver disease. Nature (2023) 616:747–54. doi: 10.1038/s41586-023-05857-4
102. Vlasschaert C, McNaughton AJM, Chong M, Cook EK, Hopman W, Kestenbaum B, et al. Association of clonal hematopoiesis of indeterminate potential with worse kidney function and anemia in two cohorts of patients with advanced chronic kidney disease. J Am Soc Nephrol (2022) 33:985–95. doi: 10.1681/ASN.2021060774
103. Kim PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Słabicki M, et al. DNMT3A-mutated clonal hematopoiesis promotes osteoporosis. J Exp Med (2021) 218:e20211872. doi: 10.1084/jem.20211872
104. Agrawal M, Niroula A, Cunin P, McConkey M, Shkolnik V, Kim PG, et al. TET2 -mutant clonal hematopoiesis and risk of gout. Blood (2022) 140:1094–103. doi: 10.1182/blood.2022015384
105. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives DNMT3A-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell (2021) 28:1428–1442.e6. doi: 10.1016/j.stem.2021.03.002
106. Kaner JD, Thibaud S, Jasra S, Wang Y, Janakiram M, Sharma A, et al. HIV portends a poor prognosis in myelodysplastic syndromes. Leuk Lymphoma (2019) 60:3529–35. doi: 10.1080/10428194.2019.1633631
107. Dharan NJ, Yeh P, Bloch M, Yeung MM, Baker D, Guinto J, et al. HIV is associated with an increased risk of age-related clonal hematopoiesis among older adults. Nat Med (2021) 27:1006–11. doi: 10.1038/s41591-021-01357-y
108. Bick AG, Popadin K, Thorball CW, Uddin MM, Zanni MV, Yu B, et al. Increased prevalence of clonal hematopoiesis of indeterminate potential amongst people living with HIV. Sci Rep (2022) 12:577. doi: 10.1038/s41598-021-04308-2
109. Bolton KL, Koh Y, Foote MB, Im H, Jee J, Sun CH, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat Commun (2021) 12:5975. doi: 10.1038/s41467-021-26138-6
110. Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell (2018) 23:700–713.e6. doi: 10.1016/j.stem.2018.10.004
111. Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell (2017) 21:374–382.e4. doi: 10.1016/j.stem.2017.07.010
112. Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid Malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol (2017) 18:112–21. doi: 10.1016/S1470-2045(16)30627-1
113. Swisher EM, Harrell MI, Norquist BM, Walsh T, Brady M, Lee M, et al. Somatic mosaic mutations in PPM1D and TP53 in the blood of women with ovarian carcinoma. JAMA Oncol (2016) 2:370. doi: 10.1001/jamaoncol.2015.6053
114. Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, Gibson C, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood (2018) 132:1095–105. doi: 10.1182/blood-2018-05-850339
115. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med (2017) 376:536–47. doi: 10.1056/NEJMoa1611604
116. Zajkowicz A, Butkiewicz D, Drosik A, Giglok M, Suwiński R, Rusin M. Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients. Br J Cancer (2015) 112:1114–20. doi: 10.1038/bjc.2015.79
117. Chan ICC, Wiley BJ, Bolton KL. What clonal hematopoiesis can teach us about MDS. Front Oncol (2022) 12:794021. doi: 10.3389/fonc.2022.794021
118. Avagyan S, Henninger JE, Mannherz WP, Mistry M, Yoon J, Yang S, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science (2021) 374:768–72. doi: 10.1126/science.aba9304
119. Mouhieddine TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun (2020) 11:2996. doi: 10.1038/s41467-020-16805-5
120. Stelmach P, Richter S, Sauer S, Fabre MA, Gu M, Rohde C, et al. Clonal hematopoiesis with DNMT3A and PPM1D mutations impairs regeneration in autologous stem cell transplant recipients. Haematologica (2023) 108(12). doi: 10.3324/haematol.2023.282992
121. Gibson CJ, Kim HT, Zhao L, Murdock HM, Hambley B, Ogata A, et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J Clin Oncol (2022) 40:189–201. doi: 10.1200/JCO.21.02286
122. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
123. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med (2021) 13:eabh0272. doi: 10.1126/scitranslmed.abh0272
124. Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation (2020) 141:124–31. doi: 10.1161/CIRCULATIONAHA.119.044362
125. Wei H, Wang Y, Zhou C, Lin D, Liu B, Liu K, et al. Distinct genetic alteration profiles of acute myeloid leukemia between Caucasian and Eastern Asian population. J Hematol Oncol J Hematol Oncol (2018) 11:18. doi: 10.1186/s13045-018-0566-8
126. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell (2012) 150:264–78. doi: 10.1016/j.cell.2012.06.023
127. Aviv A, Levy D. Hemothelium, clonal hematopoiesis of indeterminate potential, and atherosclerosis. Circulation (2019) 139:7–9. doi: 10.1161/CIRCULATIONAHA.118.038434
128. DeBoy EA, Tassia MG, Schratz KE, Yan SM, Cosner ZL, McNally EJ, et al. Familial clonal hematopoiesis in a long telomere syndrome. N Engl J Med (2023) 388:2422–33. doi: 10.1056/NEJMoa2300503
129. Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes (2010) 17:314–21. doi: 10.1097/MED.0b013e32833bf6dc
130. McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature (2019) 566:383–7. doi: 10.1038/s41586-019-0948-2
131. Haring B, Reiner AP, Liu J, Tobias DK, Whitsel E, Berger JS, et al. Healthy lifestyle and clonal hematopoiesis of indeterminate potential: Results from the women’s health initiative. J Am Heart Assoc (2021) 10:e018789. doi: 10.1161/JAHA.120.018789
132. Haring B, Wissel S, Manson JE. Somatic mutations and clonal hematopoiesis as drivers of age-related cardiovascular risk. Curr Cardiol Rep (2022) 24:1049–58. doi: 10.1007/s11886-022-01724-2
133. Card TR, Langan SM, Chu TPC. Extra-gastrointestinal manifestations of inflammatory bowel disease may be less common than previously reported. Dig Dis Sci (2016) 61:2619–26. doi: 10.1007/s10620-016-4195-1
134. Kristensen SL, Ahlehoff O, Lindhardsen J, Erichsen R, Jensen GV, Torp-Pedersen C, et al. Disease activity in inflammatory bowel disease is associated with increased risk of myocardial infarction, stroke and cardiovascular death–a Danish nationwide cohort study. PloS One (2013) 8:e56944. doi: 10.1371/journal.pone.0056944
135. Singh S, Nagpal SJ, Murad MH, Yadav S, Kane SV, Pardi DS, et al. Inflammatory bowel disease is associated with an increased risk of melanoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc (2014) 12:210–8. doi: 10.1016/j.cgh.2013.04.033
136. Nathan D, Selvan ME, Guisado D, Collatuzzo G, Boffetta P, Cohen L, et al. MDS-472 Clonal hematopoiesis in inflammatory bowel diseases. Clin Lymphoma Myeloma Leuk (2023) 23:S368. doi: 10.1016/S2152-2650(23)01192-8
137. Jiang W, Su J, Zhang X, Cheng X, Zhou J, Shi R, et al. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm Res Off J Eur Histamine Res Soc Al (2014) 63:943–50. doi: 10.1007/s00011-014-0768-7
138. Huang Z, Vlasschaert C, Robinson-Cohen C, Pan Y, Sun X, Lash JP, et al. Emerging evidence on the role of clonal hematopoiesis of indeterminate potential in chronic kidney disease. Transl Res (2023) 256:87–94. doi: 10.1016/j.trsl.2022.12.009
139. Rieger MA. Einfluss der klonalen Hämatopoese auf nicht-hämatologische Erkrankungen und Alterungsprozesse. Inn Med (2022) 63:1115–25. doi: 10.1007/s00108-022-01409-6
140. Zhang Y, Yao Y, Xu Y, Li L, Gong Y, Zhang K, et al. Author correction: Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat Commun (2021) 12:1048. doi: 10.1038/s41467-021-21285-2
141. Severson EA, Riedlinger GM, Connelly CF, Vergilio JA, Goldfinger M, Ramkissoon S, et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood (2018) 131:2501–5. doi: 10.1182/blood-2018-03-840629
142. Antonarakis ES, Kuang Z, Tukachinsky H, Parachoniak C, Kelly AD, Gjoerup O, et al. Prevalence of inferred clonal hematopoiesis (CH) detected on comprehensive genomic profiling (CGP) of solid tumor tissue or circulating tumor DNA (ctDNA). J Clin Oncol (2021) 39:3009–9. doi: 10.1200/JCO.2021.39.15_suppl.3009
143. Li Z, Huang W, Yin JC, Na C, Wu X, Shao Y, et al. Comprehensive next-generation profiling of clonal hematopoiesis in cancer patients using paired tumor-blood sequencing for guiding personalized therapies. Clin Transl Med (2020) 10:e222. doi: 10.1364/MICROSCOPY.2020.MW2A.2
144. Chan HT, Nagayama S, Chin YM, Otaki M, Hayashi R, Kiyotani K, et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol (2020) 14:1719–30. doi: 10.1002/1878-0261.12727
145. Arends CM, Dimitriou S, Stahler A, Hablesreiter R, Strzelecka PM, Stein CM, et al. Clonal hematopoiesis is associated with improved survival in patients with metastatic colorectal cancer from the FIRE-3 trial. Blood (2022) 139:1593–7. doi: 10.1182/blood.2021014108
146. Marshall CH, Holler AE, Tsai H-L, Gondek L, Luo J, Antonarakis ES. Clonal hematopoiesis in prostate cancer inferred from somatic tumor profiling. J Clin Oncol (2021) 39:e17001–1. doi: 10.1200/JCO.2021.39.15_suppl.e17001
147. Mencia-Trinchant N, MacKay MJ, Chin C, Afshinnekoo E, Foox J, Meydan C, et al. Clonal hematopoiesis before, during, and after human spaceflight. Cell Rep (2020) 33:108458. doi: 10.1016/j.celrep.2020.108458
148. Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature (2015) 518:552–5. doi: 10.1038/nature13968
149. Arends CM, Damm F. Klonale Hämatopoese und solide Neoplasien. Inn Med (2022) 63:1133–40. doi: 10.1007/s00108-022-01404-x
150. Okamura R, Piccioni DE, Boichard A, Lee S, Jimenez RE, Sicklick JK, et al, et al. High prevalence of clonal hematopoiesis-type genomic abnormalities in cell-free DNA in invasive gliomas after treatment. Int J Cancer (2021) 148:2839–47. doi: 10.1002/ijc.33481
151. Papadopoulou V, Schoumans J, Basset V, Solly F, Pasquier J, Blum S, et al. Single-center, observational study of AML/MDS-EB with IDH1/2 mutations: Genetic profile, immunophenotypes, mutational kinetics and outcomes. Hematology (2023) 28:2180704. doi: 10.1080/16078454.2023.2180704
152. Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35:1598–605. doi: 10.1200/JCO.2016.71.6712
153. Engel N, Rovo A, Badoglio M, Labopin M, Basak GW, Beguin Y, et al. European experience and risk factor analysis of donor cell-derived leukaemias/MDS following haematopoietic cell transplantation. Leukemia (2019) 33:508–17. doi: 10.1038/s41375-018-0218-6
154. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
155. Teipel R, Kroschinsky F, Kramer M, Kretschmann T, Egger-Heidrich K, Krüger T, et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Adv (2022) 6:1941–6. doi: 10.1182/bloodadvances.2021005747
156. Bouzid H, Belk JA, Jan M, Qi Y, Sarnowski C, Wirth S, et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nat Med (2023) 29:1662–70. doi: 10.1038/s41591-023-02397-2
157. Keogh MJ, Wei W, Aryaman J, Walker L, van den Ameele J, Coxhead J, et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat Commun (2018) 9:4257. doi: 10.1038/s41467-018-06331-w
158. Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, et al. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ Res (2021) 128:216–28. doi: 10.1161/CIRCRESAHA.120.317104
159. Xie J, Sheng M, Rong S, Zhou D, Wang C, Wu W, et al. STING activation in TET2-mutated hematopoietic stem/progenitor cells contributes to the increased self-renewal and neoplastic transformation. Leukemia (2023) 37:2457–67. doi: 10.1038/s41375-023-02055-z
160. Fujino T, Goyama S, Sugiura Y, Inoue D, Asada S, Yamasaki S, et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat Commun (2021) 12:1826. doi: 10.1038/s41467-021-22053-y
161. Liu X, Sato N, Shimosato Y, Wang TW, Denda T, Chang YH, et al. CHIP-associated mutant ASXL1 in blood cells promotes solid tumor progression. Cancer Sci (2022) 113:1182–94. doi: 10.1111/cas.15294
162. Chen S, Wang Q, Yu H, Capitano ML, Vemula S, Nabinger SC, et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun (2019) 10:5649. doi: 10.1038/s41467-019-13542-2
163. Yura Y, Miura-Yura E, Katanasaka Y, Min KD, Chavkin N, Polizio AH, et al. The cancer therapy-related clonal hematopoiesis driver gene PPM1D promotes inflammation and non-ischemic heart failure in mice. Circ Res (2021) 129:684–98. doi: 10.1161/CIRCRESAHA.121.319314
164. Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature (2020) 586:769–75. doi: 10.1038/s41586-020-2786-7
165. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet (2010) 42:722–6. doi: 10.1038/ng.621
166. Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, et al. TET2-Driven clonal hematopoiesis and response to canakinumab: An exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol (2022) 7:521–8. doi: 10.1001/jamacardio.2022.0386
167. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
168. Wada Y, Jensen C, Meyer ASP, Zonoozi AAM, Honda H. Efficacy and safety of interleukin-6 inhibition with ziltivekimab in patients at high risk of atherosclerotic events in Japan (RESCUE-2): A randomized, double-blind, placebo-controlled, phase 2 trial. J Cardiol (2023) 82(4):279–85. doi: 10.1016/j.jjcc.2023.05.006
169. Tardif J-C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
170. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell (2017) 170:1079–1095.e20. doi: 10.1016/j.cell.2017.07.032
171. Tang Y, Liu W, Wang W, Fidler T, Woods B, Levine RL, et al. Inhibition of JAK2 suppresses myelopoiesis and atherosclerosis in Apoe-/- mice. Cardiovasc Drugs Ther (2020) 34:145–52. doi: 10.1007/s10557-020-06943-9
172. Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature (2018) 559:637–41. doi: 10.1038/s41586-018-0350-5
173. Coombs CC, Gillis NK, Tan X, Berg JS, Ball M, Balasis ME, et al. Identification of clonal hematopoiesis mutations in solid tumor patients undergoing unpaired next-generation sequencing assays. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:5918–24. doi: 10.1158/1078-0432.CCR-18-1201
174. Wong WH, Tong S, Druley TE. Error-corrected sequencing of cord bloods identifies pediatric AML-associated clonal hematopoiesis. Blood (2017) 130:2687–7. doi: 10.1182/blood.V130.Suppl_1.2687.2687
175. Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun (2016) 7:12484. doi: 10.1038/ncomms12484
176. McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep (2015) 10:1239–45. doi: 10.1016/j.celrep.2015.02.005
177. Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney LALM, et al. Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am J Hum Genet (2017) 101:50–64. doi: 10.1016/j.ajhg.2017.05.013
178. Götze KS, Lengerke C. Bedeutung der klonalen Hämatopoese für hämatologische Neoplasien. Inn Med (2022) 63:1107–14. doi: 10.1007/s00108-022-01401-0
179. Steensma DP, Bolton KL. What to tell your patient with clonal hematopoiesis and why: Insights from 2 specialized clinics. Blood (2020) 136:1623–31. doi: 10.1182/blood.2019004291
180. Weeks LD, Niroula A, Neuberg D, Wong W, Lindsley RC, Luskin M, et al. Prediction of risk for myeloid Malignancy in clonal hematopoiesis. NEJM Evid (2023) 2(5). doi: 10.1056/EVIDoa2200310
181. Haferlach C, Heuser M. Diagnostik bei unklaren Zytopenien – wie und wann suchen wir nach klonaler Hämatopoese? Inn Med (2022) 63:1141–7. doi: 10.1007/s00108-022-01402-z
Keywords: clonal hematopoiesis of indeterminate potential (CHIP), preleukemia, myelodysplastic syndromes, germline mutations, PARP inhibitors, mutation analysis, cardiovascular risk factors, genetic counseling
Citation: Cacic AM, Schulz FI, Germing U, Dietrich S and Gattermann N (2023) Molecular and clinical aspects relevant for counseling individuals with clonal hematopoiesis of indeterminate potential. Front. Oncol. 13:1303785. doi: 10.3389/fonc.2023.1303785
Received: 28 September 2023; Accepted: 28 November 2023;
Published: 15 December 2023.
Edited by:
Eduardo Anguita, San Carlos University Clinical Hospital, SpainReviewed by:
Francesco Tarantini, University of Bari Aldo Moro, ItalySergei Vatolin, Case Western Reserve University, United States
Abhishek Niroula, University of Gothenburg, Sweden
Copyright © 2023 Cacic, Schulz, Germing, Dietrich and Gattermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Maria Cacic, QW5uYU1hcmlhLkNhY2ljQG1lZC51bmktZHVlc3NlbGRvcmYuZGU=