
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 24 November 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1301179
This article is part of the Research TopicWomen in Neuro-Oncology and Neurosurgical Oncology Vol II: 2023View all 7 articles
 Giulia Cerretti1*†
Giulia Cerretti1*† Federico Pessina2,3
Federico Pessina2,3 Enrico Franceschi4
Enrico Franceschi4 Valeria Barresi5
Valeria Barresi5 Alessandro Salvalaggio6,7
Alessandro Salvalaggio6,7 Marta Padovan1
Marta Padovan1 Renzo Manara8,9Vincenzo Di Nunno4Beatrice Claudia Bono2,3Giovanni Librizzi8
Renzo Manara8,9Vincenzo Di Nunno4Beatrice Claudia Bono2,3Giovanni Librizzi8 Mario Caccese1
Mario Caccese1 Marta Scorsetti10Marta Maccari1
Marta Scorsetti10Marta Maccari1 Giuseppe Minniti11,12Pierina Navarria10
Giuseppe Minniti11,12Pierina Navarria10 Giuseppe Lombardi1
Giuseppe Lombardi1Ependymomas are rare glial tumors with clinical and biological heterogeneity, categorized into supratentorial ependymoma, posterior fossa ependymoma, and spinal cord ependymoma, according to anatomical localization. Spinal ependymoma comprises four different types: spinal ependymoma, spinal ependymoma MYCN-amplified, myxopapillary ependymoma, and subependymoma. The clinical onset largely depends on the spinal location of the tumor. Both non-specific and specific sensory and/or motor symptoms can be present. Owing to diverse features and the low incidence of spinal ependymomas, most of the current clinical management is derived from small retrospective studies, particularly in adults. Treatment involves primarily surgical resection, aiming at maximal safe resection. The use of radiotherapy remains controversial and the optimal dose has not been established; it is usually considered after subtotal resection for WHO grade 2 ependymoma and for WHO grade 3 ependymoma regardless of the extent of resection. There are limited systemic treatments available, with limited durable results and modest improvement in progression-free survival. Thus, chemotherapy is usually reserved for recurrent cases where resection and/or radiation is not feasible. Recently, a combination of temozolomide and lapatinib has shown modest results with a median progression-free survival (PFS) of 7.8 months in recurrent spinal ependymomas. Other studies have explored the use of temozolomide, platinum compounds, etoposide, and bevacizumab, but standard treatment options have not yet been defined. New treatment options with targeted treatments and immunotherapy are being investigated. Neurological and supportive care are crucial, even in the early stages. Post-surgical rehabilitation can improve the consequences of surgery and maintain a good quality of life, especially in young patients with long life expectancy. Here, we focus on the diagnosis and treatment recommendations for adults with spinal ependymoma, and discuss recent molecular advances and new treatment perspectives.
Ependymomas are glial tumors that originate from the ependymal cells within the cerebral ventricles or in the spinal canal.
Primary spinal intradural tumors in adults are extremely rare entities and intramedullary neoplasms are even more rare, representing 5%–10% of all spinal tumors; spinal ependymomas are the most common adult intramedullary tumor, accounting for more than 60% of all intramedullary lesions (1).
The reported incidence of IDSCTs (intradural spinal cord tumors) is approximately 0.3 per 100,000 per year (2).
Men are slightly more affected than women (1.3:1), and there is a higher incidence in White individuals compared to Black individuals (3).
The location of the tumor largely depends on the patient’s age. While 90% of ependymomas are intracranial in children, 50%–60% of adult ependymomas occur in the spine (4). The cervical and lumbar portions of the spinal cord, including the filum terminale, are the most common sites of spinal ependymomas in adults (1).
Currently, no known environmental causes or specific oncogenic drivers have been identified. An increased incidence of ependymomas has been associated with the familial syndrome neurofibromatosis type 2. However, a Danish retrospective study reported that mutations in NF1 and NF2 were associated with ependymoma development in patients under 18 years old, but germline mutations were observed in fewer than 4% of cases (5).
Ependymoma survival rates are more favorable in adults than in children. The most recent population-based epidemiological data indicate a 10-year relative survival rate of 86.7% for adult ependymoma, with patients diagnosed under the age of 14 having a 10-year relative survival rate of 72% (6).
Because of the rare location, it is difficult to retrieve detailed information about epidemiology and survival outcomes in adult spinal ependymoma specifically (7).
Khalid et al. retrospectively assessed survival in 2,126 patients affected by spinal ependymoma; overall survival (OS) at 1 year, 3 years, and 5 years after diagnosis was 97.0%, 94.3%, and 93.3%, respectively (7).
Wostrack et al. demonstrated in a retrospective cohort of 158 patients with resected spinal ependymoma a 5-year progression-free survival (PFS) rate of 80% (1).
Mainly according to retrospective studies, risk factors for early progression of ependymomas include supratentorial location, histological grade 3, and subtotal resection (8). However, also site-specific molecular alterations may influence the prognosis. For this reason, the 2021 World Health Organization (WHO) classification of CNS tumors classifies ependymomas, not only on their histopathological features and anatomical site, but also on their molecular alterations (9). In addition, DNA methylation and gene expression profiling of ependymomas have identified at least nine subgroups characterized by distinct DNA methylation patterns and genetic alterations, which demonstrates the heterogeneity and complexity of these tumors (10, 11). Adult spinal ependymomas can be included in three out of nine of the methylation classes, specifically spinal subependymoma (SP-SE), spinal myxopapillary ependymoma (SP-MPE), and classic ependymoma (SP-EPN) (12).
An extensive thematic bibliographic research on the PubMed database was performed. English, pertinent articles, dating between 1984 and July 2023, have been identified.
The terms used for the PubMed search are: spinal ependymoma, spinal ependymoma in adult, methylation classes, target treatments in ependymoma, spinal symptoms, neurological outcomes, EANO guidelines, chemotherapy, radiotherapy, MYCN, temozolomide (TMZ), lapatinib, and WHO classification.
Only articles regarding the adult population and regarding ependymomas located in the spine have been included in this review.
Given the low incidence of spinal ependymoma in adults, there are few reports on the symptoms and clinical presentation of the tumor (13–17) and studies aiming to distinguish the clinical features from other spine tumors or other diseases are missing.
Early symptoms are non-specific, and the onset is usually slow and progressive. The majority of patients had experienced symptoms for more than 6 months before the diagnosis, usually between 3 and 4 years (13–15, 18). A shorter duration of symptoms (from 2 weeks to 24 months) before diagnosis was observed for anaplastic ependymoma (19).
Sensory symptoms, in particular dysesthesias (numbness/tingling), are the most common (58%), followed by weakness (45%), back pain (35%), and radiating back pain (27%) (14–17). Sensory and motor impairments may determine gait abnormalities. Patients may also report aspecific symptoms as fatigue, drowsiness, or sleep disturbances (16).
Sensory complaints (numbness or tingling) are also the earliest to present in upwards of 70% of patients usually beginning distally at lower limbs with proximal progression. Pain at the level of tumor (rarely with a radicular distribution) may also occur early in the course of the disease progression (18). Sphincteric and sexual dysfunction also tend to occur early while weakness and associated spasticity usually occur late in disease progression and may be asymmetric. Motor involvement indicates that the tumor has significantly thinned the surrounding spinal cord (20). In anaplastic ependymoma, the most common symptom is pain, followed by sensory deficits, limb weakness, and sphincter dysfunction (19).
Tumor location affects the symptoms, with cervical ependymomas associated with upper limb involvement, thoracic ependymomas with symptoms at lower limbs, and lumbar ependymomas often presenting with back and leg pain, including radicular distribution, urogenital, and anorectal dysfunction. Lower limb impairment and sphincteric dysfunction may also be determined by cervical and thoracic ependymoma (18). Conus ependymoma is associated with pain, sexual dysfunction, and sphincteric disturbances as early symptoms, as well as progressive leg (one or two) weakness and/or numbness.
Cervical ependymomas present with sensory (more often) or motor deficit or isolated pain to one or both arms. Mild lower limb symptoms (weakness, spasticity, or sensory impairment) may be found at clinical examination at the moment of the diagnosis even if not reported by the patient. However, complaints at lower limbs may occasionally begin at the same time as upper limbs. Among presenting signs, atrophy of hands or proximal muscles may also be detected at clinical examination. Thoracic ependymomas present with weakness or numbness at lower limbs. Bladder or bowel disturbances may be reported at the diagnosis of both cervical and thoracic ependymomas.
Pain, sexual dysfunctions, and sphincteric disturbances as early symptoms are more common in conus ependymoma. Progressive leg (one or two) weakness and/or numbness are also associated with conus ependymoma. Notably, signs and symptoms at the moment of the diagnosis or in the course of follow-up may be asymmetric. Weakness may progress to para- or even tetraparesis with a subsequent loss of ambulation and a significant reduction of quality of life (17, 21) (Table 1).
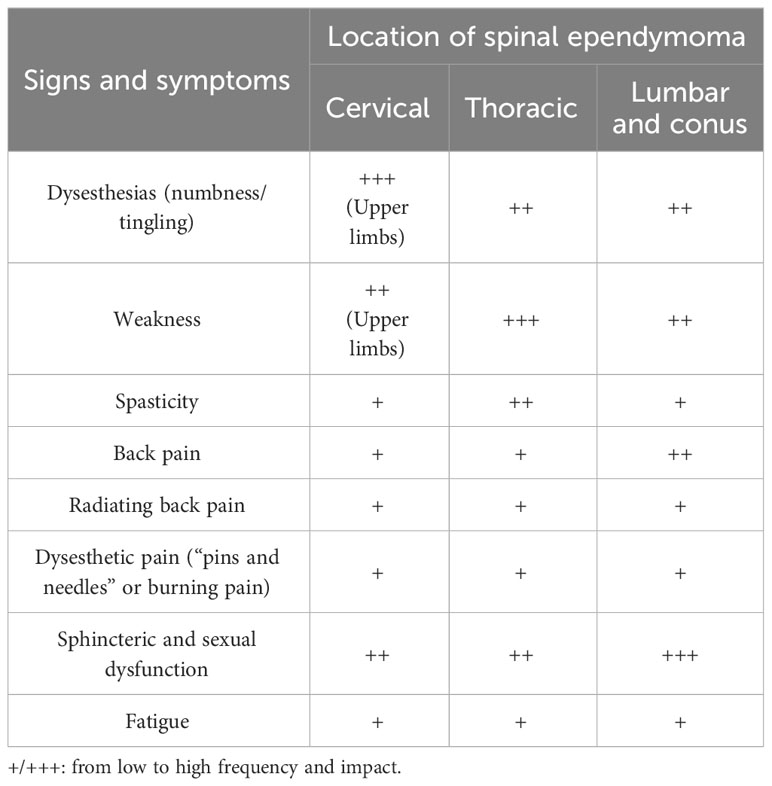
Table 1 Signs and symptoms of spinal ependymoma.
The clinical course is usually slowly progressive with an accelerated progression in the months preceding the diagnosis. Acute worsening of symptoms is usually due to intratumoral hemorrhage, which represents a rare complication.
Neurological deficits are classified according to Modified McCormick grade (from I corresponding to minimal sensory symptoms to V corresponding to paraplegia or tetraplegia) and ASIA score (22). The majority of adult patients with spinal ependymoma have a pre-surgical modified McCormick grade I or II (15, 17, 23) or III in case of anaplastic ependymoma (19).
The most reliable predicting factor of post-surgical neurological outcome is the preoperative neurological state (13, 15, 24). Severe and long-term neurological deficits will not improve significantly after tumor resection, even if a gross total resection is reached (13). In the immediate post-surgery, patients may complain of a worsening of the previous symptoms of the onset of a new deficit, usually sensory, related to the posterior column traumatism caused by midline myelotomy, transient edema, or vascular compromise. In particular, patients may suffer from dysesthetic pain (“pins and needles” or burning pain) scarcely responsive to treatment, but self-limiting in few months (13, 17). After the transient worsening in the months post-surgery, usually patients improve at the level of their pre-operative deficits or may further improve, but some patients showed a worsening of their clinical status (19, 24). A progressive neurological worsening after surgery may underlie the presence of intramedullary cyst, tumor recurrence, or hematoma (15, 18).
A self-limiting orthostatic hypotension may occasionally occur after removal of upper thoracic and cervical ependymoma, thus interfering with the early mobilization, which is encouraged (13).
Because a risk of cerebrospinal fluid (CSF) dissemination exists for all patients with ependymoma, clinical and MRI evaluation should include both cranial and complete spinal districts. This extensive evaluation should be performed at the time of diagnosis with the aim of staging the disease and along the course of follow up. From a clinical point of view, it means that a comprehensive neurological evaluation should be performed including cranial nerves, cognitive functions, motor and sensory systems, gait, and cerebellar function; patients should be asked for new onset or worsening of headache, backpain, or radicular pain and sexual, bowel, bladder, and sphincteric disturbances (25–27). Late relapses may be asymptomatic; therefore, a meticulous neurological evaluation and enhanced MRI should be performed regularly in the follow-up (25, 27).
Considering the young age of these patients (usually in their 30s or 40s) and long life expectation, a satisfactory quality of life represents an important aim. In a study addressing this topic, almost half of the patients reported that the disease interfered at least moderately with work, general activities, walking, or enjoyment of life. Over 40% of patients stopped working or reduced their working hours due to the disease. Pain and weakness (and related accessibility constraints) represented the preeminent symptoms preventing the return to work. One-third of patients required help at home from a caregiver. Two-thirds of patients encountered difficulties when returning to sport activities due to pain, coordination problems, accessibility constraints, and fatigue symptoms (16, 17).
Low-grade ependymoma (specifically subependymoma and myxopapillary ependymoma) is often regarded as a tumor with a benign trajectory; however, many patients continue to have significant deficits and symptoms years after their initial diagnosis, and even in the best cases, ependymoma should be considered a chronic disease associated with symptom burden and high morbidity.
Among pharmacologic medications, patients are often treated for neuropathic pain with gabapentin and antidepressants (13); myorelaxants may be considered in case of spasticity. Early and intensive physical and occupational therapy optimize functional recovery and ultimately quality of life.
In the last decade, molecular studies provide extensive evidence that different tumor types harbor distinct methylation profile, depending on their cell of origin and on the molecular alterations acquired during tumorigenesis (28). Therefore, DNA methylation profile, integrated with morphological and genetic features, was used to classify and diagnose tumors in the central nervous system (29).
In 2015, Pajtler et al., using DNA methylation profiling of 500 tumors, classified ependymal tumors in nine different molecular subgroups, across three anatomical compartments (supratentorial, posterior fossa, and spinal) (10). They showed that, in spite of similar histology, ependymomas arising in the spinal cord have a different DNA methylation profile, genetic alterations, and overall better clinical outcome, compared to those originating in the supratentorial compartment or in the posterior fossa (10). Ependymal tumors in the spinal cord were classified into spinal ependymoma, spinal subependymoma and myxopapillary ependymoma (10). However, a subsequent study demonstrated a distinct DNA methylation profile in a subgroup of spinal ependymomas featuring dismal prognosis in spite of aggressive treatment and displaying MYCN amplification (30). Therefore, the fifth edition of the WHO classification of CNS tumors categorizes spinal ependymal tumors into four different subtypes: spinal ependymoma, spinal ependymoma MYCN-amplified, myxopapillary ependymoma, and subependymoma (31).
Spinal ependymoma is a well-demarcated tumor, histologically composed of monotonous glial cells, embedded in a glial fibrillary matrix, and showing rounded to oval nuclei. By definition, it lacks histopathological features of myxopapillary ependymoma or subependymoma and MYCN amplification (32). Tumor cells have fibrillary processes arranged around blood vessels and forming peri-vascular anucleate zones (so-called peri-vascular pseudorosettes) (Figure 1A). Only rare cases show true ependymal rosettes, consisting in glial cells arranged around empty lumina (32).
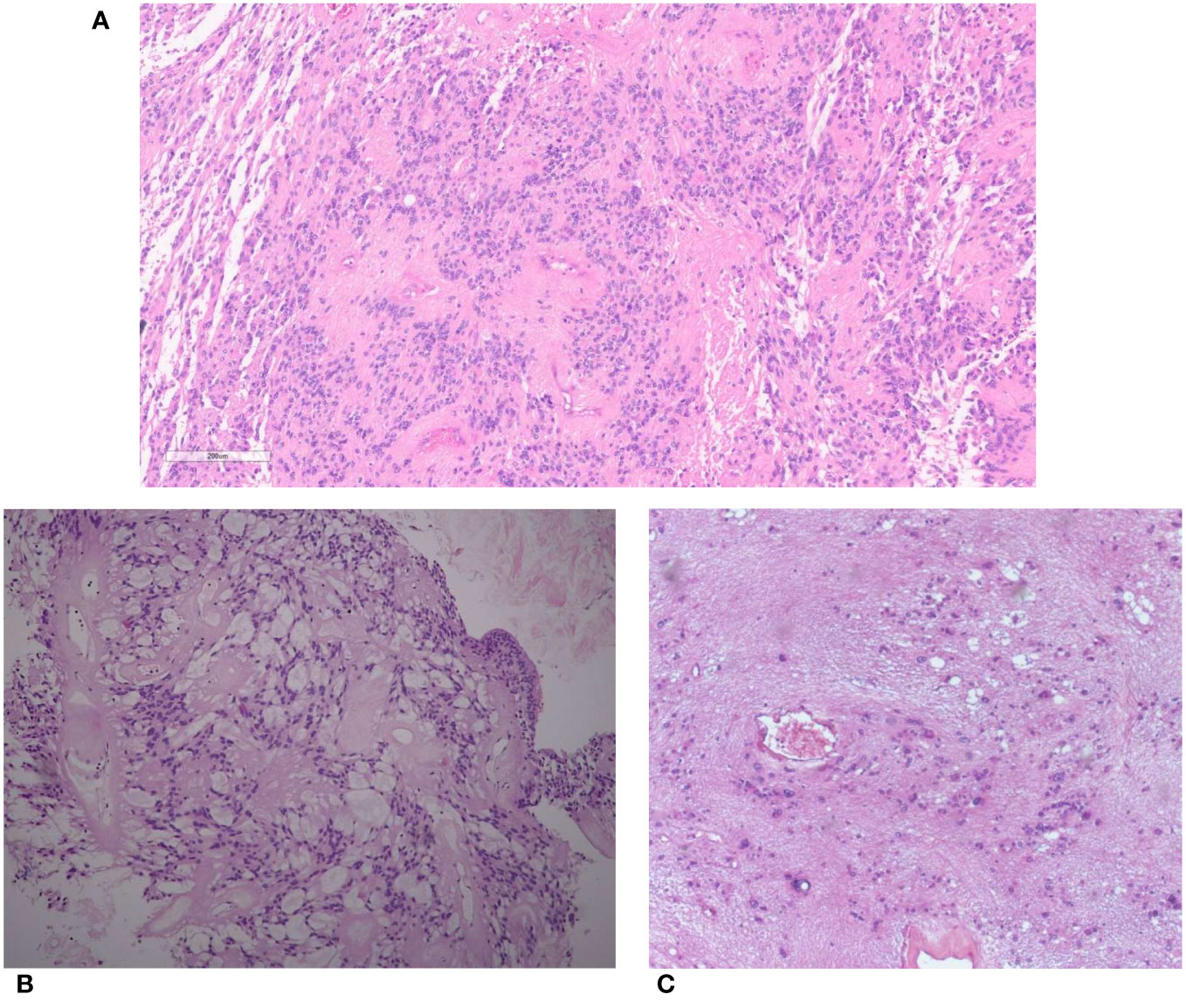
Figure 1 (A) Spinal ependymoma. Tumor cells with rounded to oval nuclei have fibrillary processes arranged around blood vessels (peri-vascular pseudorosettes). (B) Myxopapillary ependymoma. Tumor cells are arranged around blood vessels with interposed myxoid material. (C) Subependymoma. Tumor cells with uniform nuclei clusterized in a fibrillary matrix.
The papillary, clear cell, and tanycytic morphological variants, previously considered as subtypes of ependymoma, are now included as histopathological patterns (31).
Based on its morphological features, spinal ependymoma is graded into CNS WHO grade 2 and 3, the latter showing brisk mitotic activity and high cell density (32). The most frequent genetic alterations in this tumor subtype are chromosome 22q loss and NF2 mutations (10) (Table 2).
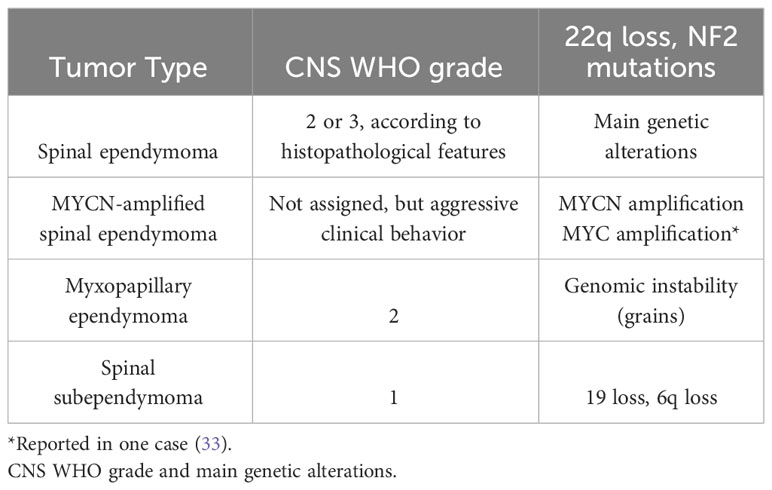
Table 2 Spinal ependymal tumors.
Spinal ependymoma MYCN-amplified harbors a distinct DNA methylation profile and, by definition, MYCN amplification in tumor cells (34). It is a rare, recently defined tumor type with a higher incidence in women (1.7:1) and a median age at a diagnosis of 31 years (30, 35, 36), and a worse outcome compared with other spinal ependymomas. The reported median PFS after initial surgery and OS were 17 and 87 months, respectively, and it also undergoes frequent dissemination (30, 36).
Histologically, it features classical ependymoma morphology with peri-vascular pseudorosettes and true rosettes, but compared to spinal ependymoma, it invariably shows high mitotic activity, necrosis, and microvascular proliferation (34). In spite of its high-grade histological features and its adverse clinical outcome, it has yet to be assigned a definite CNS WHO grade. Spinal ependymoma MYCN-amplified is characterized by strong and intense MYCN immunostaining. Therefore, immunohistochemical staining for MYCN might be used as a screening method to identify this tumor subtype, although gene amplification should be confirmed using other methods, such as FISH analysis. Notably, a recent report described a case of a spinal ependymoma, with poor clinical outcome and DNA methylation profile consistent with ependymoma MYCN-amplified, lacking MYCN amplification and rather harboring MYC amplification (33). This suggests that the molecular portrait of spinal ependymoma MYCN-amplified may also include alterations in genes encoding for other proteins of the MYC family members beyond MYCN (Table 2).
MYCN, a member of the MYC group of oncogenes, codes for a transcription factor involved in the regulation of neuronal embryogenesis. It is involved in the oncogenesis of multiple tumor types, including neuroblastoma, pediatric glioblastoma, and medulloblastoma. The specific mechanisms by which MYCN promotes oncogenesis in spinal ependymomas have not yet been elucidated. Interestingly, multiple schwannomas have been reported in one patient with a MYCN-amplified spinal ependymoma, suggesting a possible relationship with neurofibromatosis type 2 (35, 36).
Myxopapillary ependymoma is a subtype almost exclusively occurring at the conus medullaris or filum terminale (37). It is defined by the presence of spindle or epithelioid neoplastic cells arranged around blood vessels (papillary feature) with interposed myxoid material (myxoid feature) or microcyst formation (38) (Figure 1B). This tumor was traditionally classified as WHO grade 1; however, reported rates of 10-year PFS of 60% and possible metastasization (39, 40) led to its reclassification as CNS WHO grade 2 in the fifth edition of WHO Classification (38) (Table 2).
Rarely, myxopapillary ependymomas can also develop outside of the CNS, usually within the sacrococcygeal or presacral tissues.
They are rare tumors with an incidence of 0.6–1.0 cases per 1 million person-years and a higher frequency in men (36, 41). Myxopapillary ependymomas are more common in adults with a bimodal peak approximately 25–29 years and 45–59 years. Overall prognosis is favorable (10-year survival rates > 90%) and complete resection is important for prognosis, but it can prove challenging (36, 41). CSF cytology is warranted before determining adjuvant treatments to exclude leptomeningeal dissemination (25). Although this is a rare event, distant metastases may also occur (36, 42). Spinal myxopapillary ependymoma is not characterized by specific genetic alterations, but it invariably features a high genomic instability, mainly consisting in copy number gains (10). Since this tumor displays HOXB13 upregulation, the immunohistochemical detection of this protein may be used in routine practice for the distinction from other tumor types (43, 44). A recent study on 185 tumors classified as myxopapillary ependymomas based on DNA methylation suggested that these diseases can be further subdivided into two methylation subtypes, named “A” and “B” (43). Tumors in subtype A were characterized by a higher number of copy number variations, papillary morphology, and worse outcome compared to tumors in subtype B (43). However, since methylation subtyping was not an independent prognostic factor after a multivariate survival analysis including histology, localization, and resection status (43), the utility of this distinction in clinical practice is to be determined.
Spinal subependymoma is a tumor preferentially localized at the cervicothoracic segment (45), and is histologically characterized by clusters of uniform to mildly pleomorphic tumor cell nuclei in an abundant fibrillary matrix (Figure 1C) and classified as CNS WHO grade 1 (46). It may show chromosome 19 and 6q loss, while other copy number alterations are infrequent (10, 12) (Table 2).
Within the spinal cord, spinal ependymoma with and without Myc amplification, myxopapillary ependymoma, and subependymoma are four distinct WHO histologic subtypes. In spite of some imaging overlap, site, demographic, and distinct imaging features might provide useful clues about ependymoma subtype. Even though histologic evaluation remains necessary for differentiating these tumors from other tumor subtypes (e.g., astrocytoma, metastasis, lymphomas, etc.) and for detecting N-Myc amplification, neuroimaging (above all MRI) retains a pivotal role in defining tumor site, recurrence, or dissemination and in surgical planning.
Spinal ependymomas can be found anywhere along the spinal cord but the distribution is not even: nearly half of ependymomas (44%) occur in the cervical cord, approximately 25% occur at the cervicothoracic junction, and 25% occur in the thoracic cord alone (47). As ependymomas arise from the central canal and are typically slowly expansive rather than infiltrative (48), they are characterized by a central spinal epicenter with a clear delimitation from the peripherally displaced normal cord. Vertebral canal widening, posterior vertebral body scalloping, and foraminal enlargement commonly confirm the slow tumor growth. Among neurofibromatosis type 2 patients, spinal ependymomas should be suspected as they represent up to 80% of intradural spinal tumor and affect 33%–53% of patients. Ependymomas usually extend less than astrocytomas (mean extension less than 4 vertebral segments).
Ependymomas are slightly and heterogeneously T2-hyperintense. Sagittal imaging best depicts the tumor features: markedly T2 hyperintense syringohydromyelia above or below the tumor is seen in 9%–50% of cases; peritumoral edema is present in approximately 2/3 of cases and tumoral cysts are present in approximately 1/5 of cases (49). Ependymoma have the tendency to bleed, leading to hemosiderin staining, i.e., a T2 hypointense rim at the caudal or cranial margins (“cap sign”), in 20%–33% of cases (48). This feature is suggestive of but not pathognomonic for ependymoma as it may also be seen in other tumors of the spinal cord (e.g., paragangliomas or hemangioblastomas). Superficial siderosis has also been reported (50, 51).
Most ependymomas are homogeneously T1-iso/hypointense; mixed-signal lesions reveal the occurrence of intratumoral cysts, necrosis, or hemorrhage. Ependymomas show vivid homogeneous enhancement. Calcifications are uncommon. In patients harboring N-Myc mutation, ependymomas show dismal prognosis due to a higher aggressivity and higher rate of tumor dissemination (Figure 2A).
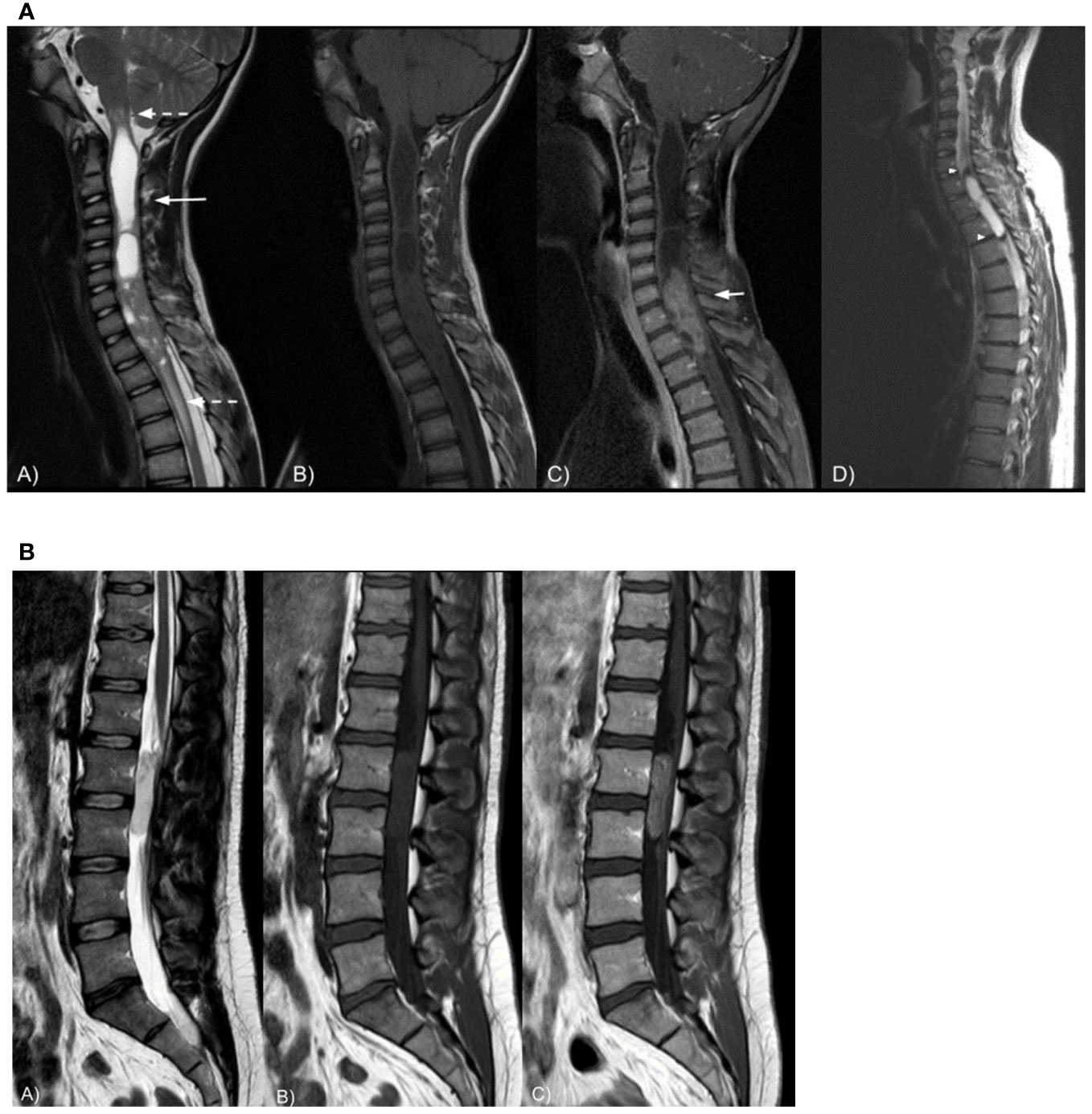
Figure 2 (A) Sagittal spine MRI in a 24-year-old female patient shows a T2-inhomogeneous (A), T1-isointense intramedullary (B) cervical mass with intratumoral cysts and syringomyelia (long arrow). The lesion is surrounded by moderate peritumoral edema (dashed arrows) and demonstrates strong contrast enhancement (C, short arrow). Sagittal T2-weighted spine MRI (D) in a 26-year-old male patient shows a cervico-thoracic intramedullary cystic lesion with marked peripheral hypointensity likely due to hemosiderin staining, i.e., the cap sign (arrowheads). Spinal cord ependymoma was proven at histopathology in both cases. (B) Sagittal spine MRI in a 34-year-old male patient shows a T2-hyperintense (A), T1-isointense (B) extramedullary intradural lumbar mass displacing the nerve roots of cauda equina posteriorly. The lesion demonstrates vivid and slightly inhomogeneous contrast enhancement (C). Myxopapillary ependymoma was proven at histopathology.
Myxopapillary ependymoma is extramedullary and intradural spinal tumor typically located in the lumbo-sacral region due to its origin in the filum terminale. Rarely, this subtype originates in the cervicothoracic region or fourth ventricle (52). Myxopapillary ependymoma can also extend into the neuroforamina; thus, differential diagnosis includes extradural tumors.
In the lumbo-sacral region, they typically appear as well-defined, encapsulated, oval, or sausage-shaped masses, often spanning more than one vertebra, though small oval tumors are also seen. The latter tend to displace the nerve roots of the cauda equina, while larger lesions often compress or encase them. Large, myxopapillary ependymomas may cause scalloping of the vertebral bodies and enlarge the spinal canal. Lesions are usually T1-isointense and T2-hyperintense. However, calcifications, hemorrhages, and cystic degeneration can be encountered (53), giving the mass an inhomogeneous aspect. Mucinous components occasionally result in T1 hyperintensity while calcifications might appear hyper- or hypointense. Hemorrhages often result in hypointense T2-hypointense tumor margins (cap sign) or, rarely, in brain, spinal cord, and nerve roots, surface T2-hypointense siderosis (54). Homogeneous contrast enhancement is common (Figure 2B).
Spinal cord subependymoma is a rare, slow-growing, indolent, benign spinal cord tumor. It is radiologically similar to a spinal cord classic ependymoma as it is intramedullary, in the cervical or cervicothoracic region, typically T1 hypo- to isointense to white matter and T2 hyperintense. T2 signal might be heterogeneous and depends on the degree of cystic changes and associated hemorrhages or calcifications. However, subependymoma typically shows no or just slight enhancement. In addition, differently from ependymomas, the tumor growth is typically eccentric with the normal spinal cord included in the tumor (bamboo leaf sign in 76%) (55).
According to the most recent European guidelines (3, 25), the gold standard of treatment for any suspected spinal ependymoma is surgical resection (25), with the aims of obtaining the tumor’s histomolecular characterization, achieving complete resection (3), whenever it is possible, and preserving/improving the patient’s functional status (56). Indeed, in both pediatric and adult spinal ependymomas, tumor grading and the extent of resection seem to be, to date, the only independent factors capable of carrying a significant impact on patient prognosis (3, 56–59).
In spinal ependymomas, the extent of resection depends on several factors, including tumor location and volume, the presence of a capsule, and histomolecular grading (56–60).
Thanks to advances in modern microsurgical techniques, a gross total resection while maintaining/improving the patient’s functional outcome is possible in most cases (84%–93%) also because these tumors only rarely infiltrate the spinal cord (56, 57, 61).
Intraoperative neuromonitoring, including motor evoked potentials (MEPs) and somatosensory evoked potentials (SEPs), is of utmost importance to prevent and reduce the risk of postoperative neurological deficit related to surgical maneuvers (62). The D-wave monitoring, whose electrodes can be placed either subdural or epidural, allows recording the corticospinal activity without any peripheral conductivity-related artifacts, and it is the most specific predictor of postoperative transient/permanent motor deficit (63).
A standard posterior midline approach is usually tailored according to the tumor extension. A posterior laminectomy is usually performed including one level above and one level below the tumoral mass, sparing the articular facets to avoid any long-term segmental instability. In this regard, intraoperative neuronavigation (intraoperative ultrasound, CT-MRI-based neuronavigation) can be employed during the initial phases of the procedure to precisely locate the tumor and tailor its exposure.
Under microscopic view, a midline durotomy is performed and the dura is suspended laterally with the aid of suture stitches. The arachnoid is then sharply and widely opened to fully expose the posterior spinal cord surface. At this point, the tumor is usually visible and well-distinguishable from the normal tissues. Neuronavigation could be employed, again, in cases of non-superficial tumors.
Whenever possible, en bloc resection must be preferred over piecemeal resection (which is usually performed with the aid of an ultrasonic aspirator) to minimize the risks of CSF dissemination (63). A midline posterior myelotomy is performed under neuromonitoring guidance. Tumor dissection from the normal tissue is then performed both sharply and bluntly with the aid of micro scissors, micro forceps, and micro dissectors. The dissection plane must always be preserved and carefully maintained.
Tumors with more distinct capsules are associated with fewer post-operative neurologic deficits, likely because they are more easily separated from normal tissue.
Careful hemostasis is performed at the end of the resection with constant irrigation, which should be preferred over bipolar coagulation to avoid any inadvertent ischemic injury to the nearby eloquent structures. A watertight dural closure is performed to minimize the risk of postoperative CSF leakage. Autologous fat tissue and/or synthetic materials can also be used to strengthen the dural seal.
The association between extent of resection (EOR) and PFS in patients with spinal cord ependymoma has been diffusely recorded in retrospective studies (64, 65) (Table 3). However, evidence of definitive relationships with PFS or OS are lacking. In a large review, Oh et al. analyzed 175 patients with SCE across 43 different studies, focusing on the association between tumor grading and outcome. For the entire cohort, maximal resection was associated with better outcome even after controlling for adjuvant radiation therapy. In the group of patients with grade II ependymomas, those with gross total resection (GTR) had a significantly lower recurrence rate compared to patients with subtotal resection (STR), while patients with myxopapillary ependymoma showed a similar recurrence rate in case of GTR and STR (12.1% vs. 26.1%), respectively (60).
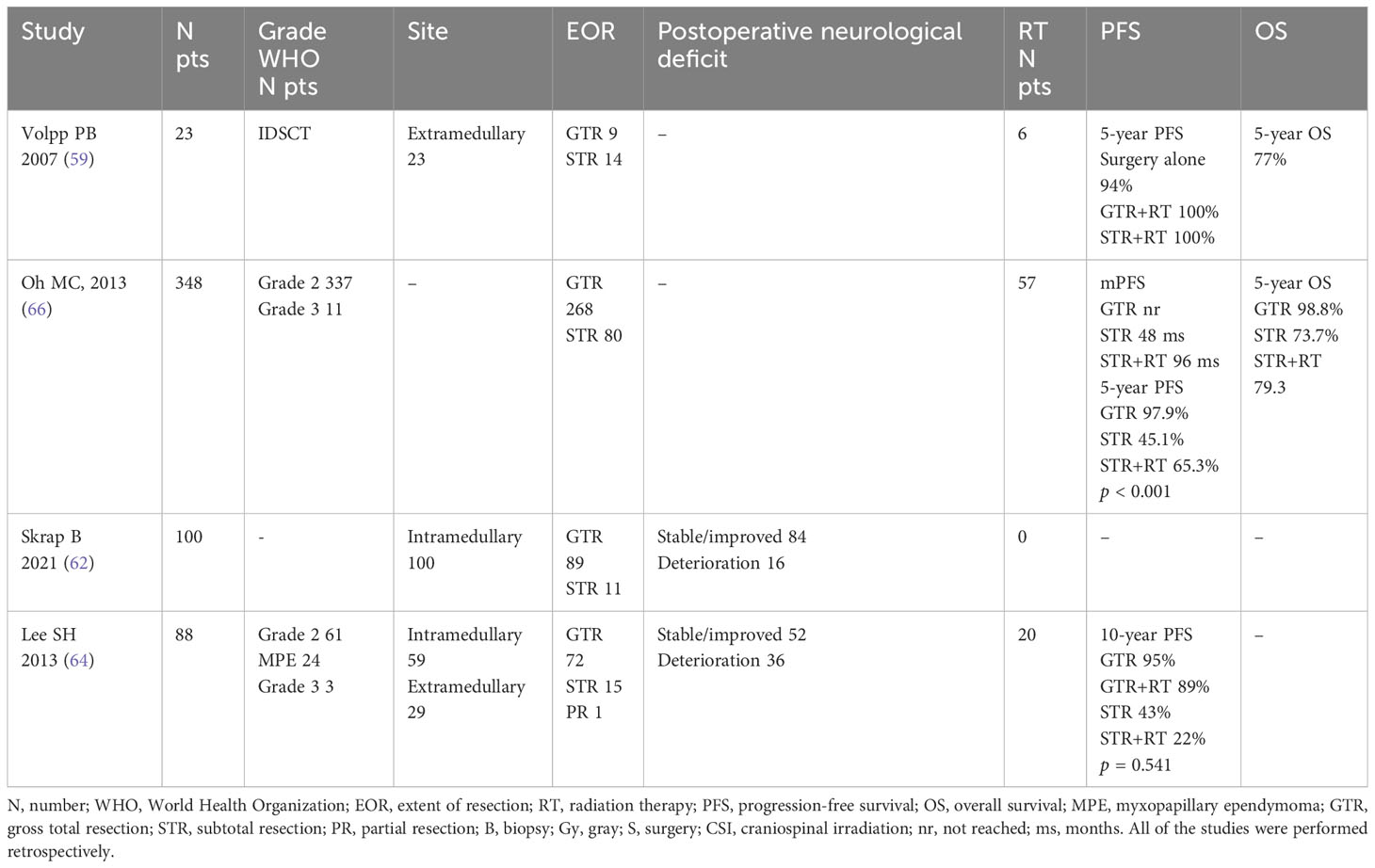
Table 3 Studies analyzing surgery in spinal ependymoma.
For the grade III ependymoma group, GTR strongly impacted outcome (no recurrences among patients underwent maximal resection compared to 80% of patients with STR). The small population in the study represents a relevant limit for this results interpretation (60).
Lee et al. showed that GTR alone is a good treatment strategy for spinal ependymomas. Early diagnosis and surgery, before severe paralysis, are important to obtain good functional outcomes. Subtotal resection with radiation therapy for intramedullary lesions appears to offer no advantages over gross total removal (64).
Despite efforts to preserve normal tissue, post-operative neurologic deficits are unfortunately possible, with risk best predicted by the patient’s baseline neurologic function. Compared to patients with deficits at baseline, patients with good neurologic function before surgery showed a lower risk of post-operative neurologic impairment (67). Thoracic tumor location, related to the region’s relatively limited blood supply, are more likely to worsen post-operative status (57, 65). Interestingly, extent of surgical resection has not been found to be related to post-operative neurologic function (67).
N, number; WHO, World Health Organization; EOR, extent of resection; RT, radiation therapy; PFS, progression-free survival; OS, overall survival; MPE, myxopapillary ependymoma; GTR, gross total resection; STR, subtotal resection; PR, partial resection; B, biopsy; Gy, gray; S, surgery; CSI, craniospinal irradiation; nr, not reached; ms, months. All of the studies were performed retrospectively.
The role of radiation therapy in spinal ependymoma is still a topic under investigation due to the lack of results provided by prospective or randomized trials (Table 4). The large part of published studies are retrospective, with a limited number of patients included, grade 2 and 3 together, in which a small percentage of patients received adjuvant RT. Notwithstanding, RT has proven to be an effective local treatment in spinal ependymoma; above all, in cases of incomplete surgical resection, available data related to the benefit on survival are discordant.
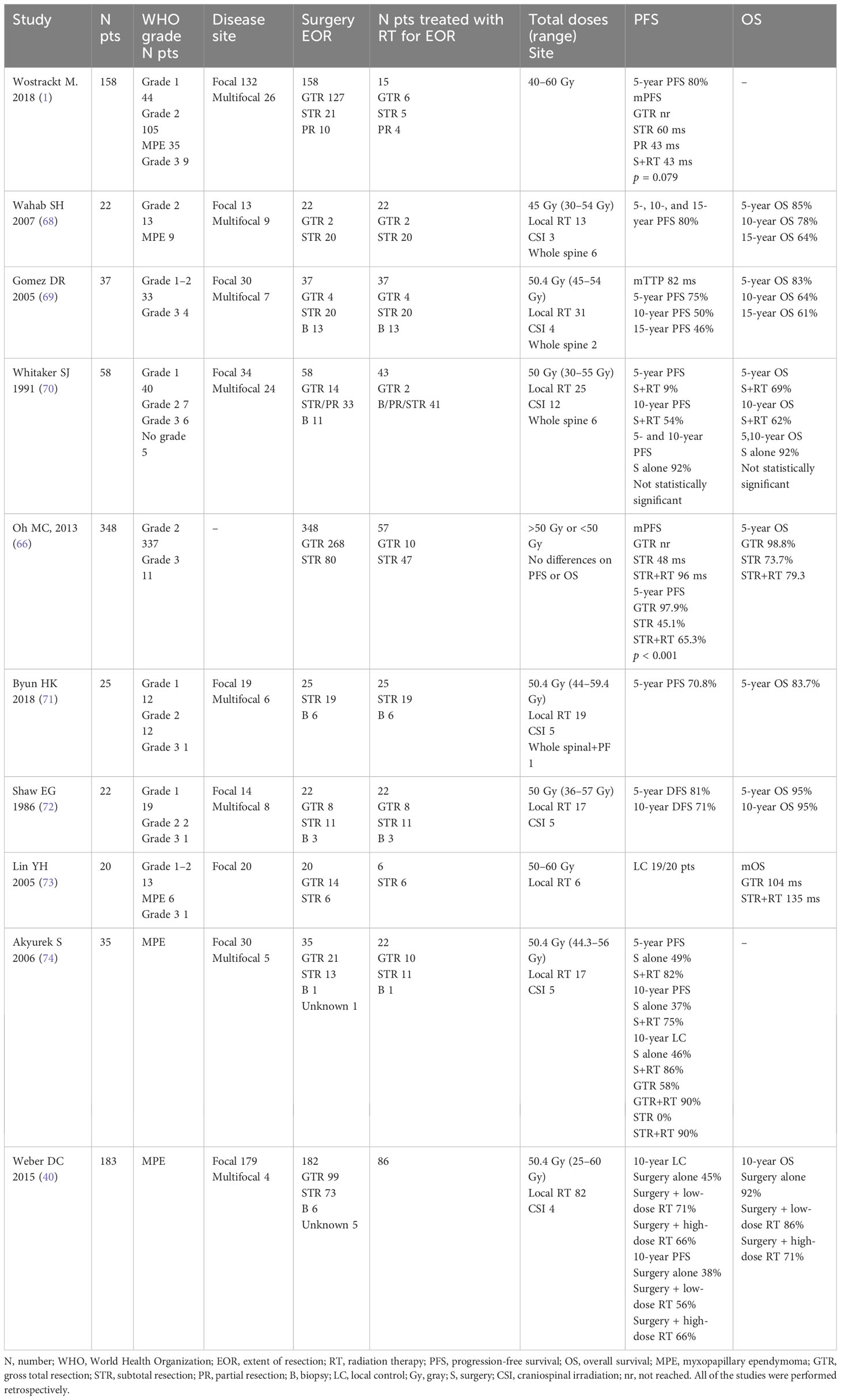
Table 4 Studies analyzing radiation therapy in spinal cord ependymoma.
The results of the multicenter retrospective study led by Wostrack et al. that evaluated 158 patients with spinal ependymoma did not show an improve survival in patients receiving adjuvant RT following subtotal resection, although only 15 cases received RT (1). Data from the SEER database in which a deep learning algorithm was applied to >2,200 patients with spinal ependymoma, assessing predictive factors of OS, identified RT as an independent predictor (75). To date, suggestions regarding the use of adjuvant RT and total doses to deliver widely varied in relation to histological subtype, grade, and extent of surgical resection (66, 68–70, 76).
For grade 1 spinal subependymoma, RT is not indicated given the possibility of obtaining complete surgical resection in almost all patients, the favorable patients prognosis, and the low incidence of local recurrence.
In grade 2 ependymoma patients receiving gross total resection (GTR), adjuvant RT does not improve PFS and OS, and accordingly is not recommended. On the other hand, when GTR is not feasible because of infiltration of spinal cord or nerve roots, the recurrence rate reaches up to 50%–70% without adjuvant therapy, with a 5-year survival rate of 73.7%. A comprehensive review of the literature on 348 WHO grade 2 and 3 classic spinal cord ependymoma patients has been conducted aiming to evaluate whether adjuvant RT improves tumor control in patients who underwent surgical resection (66). Patients who received GTR were 77%, and 33% received STR; among these, adjuvant RT has been administered in 58.8% of the cases. On univariate and multivariate analysis, the use of adjuvant RT after STR significantly affected PFS [p < 0.001; hazard ratio (HR) =2.26, p = 0.047]. By contrast, improved OS was only associated with GTR (GTR versus STR + RT group; HR = 0.07, p = 0.001) and grade 2 ependymomas (HR = 0.16, p = 0.001). No correlation between RT doses employed (≤50 Gy vs. >50 Gy) on PFS or OS has been recorded. This high recurrence rate has promoted the use of adjuvant RT for patients receiving incomplete surgical resection (72).
The role of radiotherapy for Myxopapillary ependymoma (MPE) in adults, reclassified by the 2021 WHO classification of CNS tumors as grade 2, is controversial too. Unlike the other grade 2 ependymomas, MPE typically located in the conus medullaris, cauda equina, and filum terminal region has a tendency towards a leptomeningeal spread occurring in up to 10% of the initially localized disease. Owing to the close adhesion to the nerve roots and the production of a myxoid matrix, a safe GTR of MPE may be challenging with a GTR ranging from 53% to 75% (77, 78). In this context, the role of RT is crucial. Mixed results provided by retrospective series are available, some did not demonstrate a benefit in the recurrence rates adding adjuvant RT, and others underlined the superiority of a combined local treatment approach, consisting of surgery followed by RT over surgery resection alone (40, 73, 74, 79). The experience of the MD Anderson Cancer Center showed that the addition of postoperative radiotherapy to surgery was associated with significantly longer 10-year PFS rates (75% for the combination vs. 37% for surgery alone) (79). Among 11 patients who received GTR alone, 5 (45%) had recurrence. A total of 12 (34%) patients had disease recurrence, all in the neural axis; 8 of them had treatment failure at the primary site only, 3 in the distant neural axis only, and 1 at the primary site and in the distant neural axis. Patient age (>35 years; p = 0.002) and adjuvant RT (p = 0.04) significantly affected PFS. The larger published series evaluating the outcome of 183 MPE patients underlined the use of adjuvant RT as the factor that allowed an increase of the 10-year PFS from 40% to 70% in patients receiving a combined treatment with respect to those treated with surgery alone. This would justify a more liberal use of adjuvant RT, especially in those patients with subtotal resection and piecemeal resection, or questionable GTR in patients with spinal MPE (40). In summary, although adjuvant RT may not ultimately affect OS, decreasing recurrence can appreciably benefit patient outcomes by avoiding repeated surgeries, which are associated with significant morbidities.
Grade 3 spinal ependymomas are rare entities, accounting for between 2% and 8% of all spinal ependymomas, and the patient prognosis is poor (71). Postoperative radiotherapy is indicated regardless of the extent of resection as suggested by international guidelines (25).
The optimal dose of radiation for spinal cord ependymomas also remains to be determined. Most authors currently recommend doses of 45–54 Gy with long-term follow-up because recurrence can occur many years after initial treatment. High doses (≥50 Gy) have been proven to increase the local control and PFS (10-year PFS from <40% to 70%) with good tolerance and without substantial late toxicity (40, 66, 79). In cases of cranial and spinal dissemination, craniospinal irradiation (CSI) of 36 Gy is recommended with a boost up to 45–54 Gy on local lesions (25). However, the recommended dose of radiation has not been clearly defined because high doses of radiation may be associated with increased risk of radiation myelopathy (40, 71, 79).
Ependymal tumors of the spinal cord are more common in adults than in children and have a better prognosis as compared to spinal cord astrocytoma (25, 80, 81). Owing to the rarity of these tumors, there are very little data investigating the clinical impact of systemic therapy (82) (Table 5).
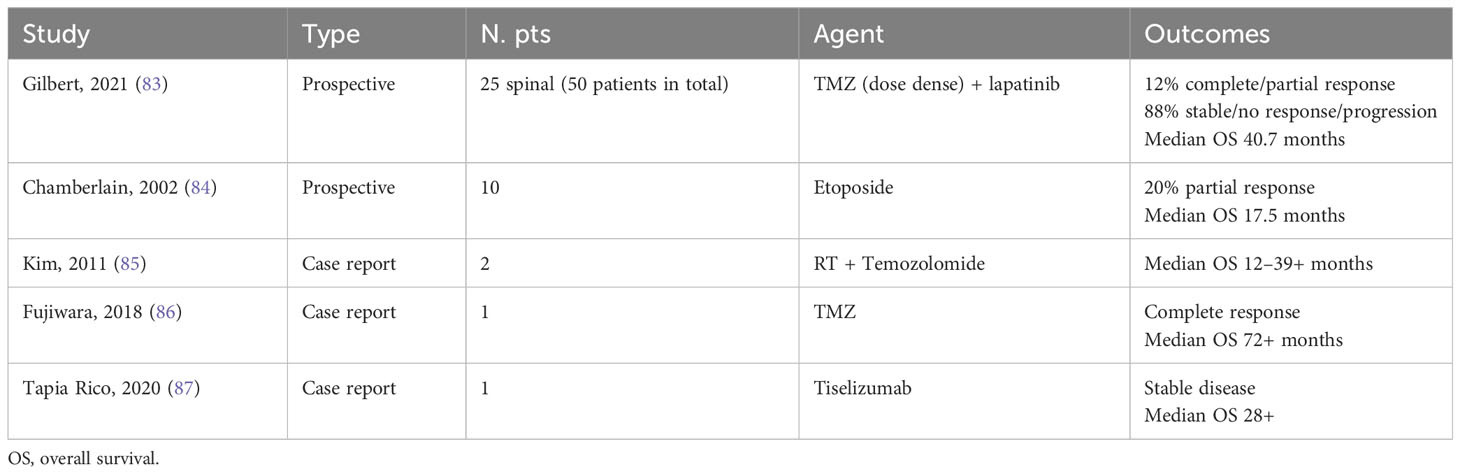
Table 5 Studies evaluating systemic therapy in patients with spinal cord ependymoma.
To date, no prospective studies investigated the role of adjuvant systemic treatments. The only experience of concurrent radiation and TMZ chemotherapy is represented by a small case series. In one of these studies, a single patient with anaplastic spinal cord ependymoma had an OS of 39 months (88). A similar OS was observed in another distinct series (89). Thus, the only setting where systemic treatments have been investigated is the advanced disease refractory to loco-regional treatments. In 2020, Gilbert MR et al. published the results of a prospective phase II study investigating the combination between dose-dense TMZ and lapatinib within adult patients with recurrent ependymoma (83).
In this single-arm study, 50 patients received TMZ at a dose of 125 mg/m (2) as a single daily dose on days 1–7 and 15–21 of a 28-day cycle in combination with a single daily dose of lapatinib 1,250 mg orally.
Between the 50 patients enrolled, 25 (50%) had a spinal cord ependymoma with a variable tumor grade. In particular, 7 patients had anaplastic grade 3 tumors, while 16 patients had grade 2 (n = 8) and grade 1 (n = 8) spinal cord ependymomas; and 2 of unknown grade (83). The primary endpoint of the study was the median mPFS. In the overall cohort, after a follow-up of 4.41 years, the mPFS was 7.8 months (95% CI: 5.5-12.2) and median OS was 2.25 years (95% CI: 1.7–3.97 years). Tumor response was observed in eight patients (16%) consisting of two complete and six partial responses (83).
In patients with spinal cord tumors, the median PFS was 0.9 years (95% CI: 0.46–1.84 years). Three patients (12%) showed radiological responses and 22 patients (88%) showed stable disease. One complete response was observed within the eight patients with myxopapillary grade I ependymoma. The remaining responses (six partial responses and one complete response) were observed within patients with more aggressive spine tumors. Of note, the majority of patients with spine tumors experienced a clinical benefit consisting of pain reduction (62%), loss of control of bladder/bowel (73%), and numbness/tingling reduction (57%).
In the overall population, neither tumor grade, nor tumor localization, nor prior systemic treatment received significantly affected the PFS on univariate analyses (83).
Regarding treatment-related toxicity, there was a significant rate of myelotoxicity represented mainly by neutropenia, leukopenia, and thrombocytopenia (7 cases of grade 3/4 thrombocytopenia and 18 cases of grade 3/4 leukopenia/neutropenia) (83).
To date, this study represents the larger prospective series assessing a systemic treatment in patients with spinal cord ependymoma. The foreseeable limits of the study are related to the small number of patients with spinal cord ependymomas and the heterogeneity of this cohort (previously treated/untreated patients and different tumor-grade tumors). Toxicity is another issue that may reduce the use of this combination in clinical practice, even if myelotoxicity was significantly reduced by escalating TMZ dosing after two cycles of treatments.
Another phase II study carried out by Chamberlain MC investigated the role of oral etoposide in 10 patients with recurrent spinal ependymoma. In this study, mPFS and mOS were 15 months (range 2.5–45) and 17.5 months (range 3–45), respectively (84).
Except for these phase II studies, the majority of data investigating systemic treatments came from small retrospective series and single case reports.
Retrospective case series suggested a potential role of TMZ and or other alkylating agents, including cisplatin and cyclophosphamide (86, 88).
A single retrospective study also suggested a clinical benefit with bevacizumab (90).
To date, basket trials are now recruiting adult patients with spinal cord ependymoma and testing brigatinib (a target agent active on tumors cells with EML4-ALK translocation) or neratinib (an EGFR and HER 2 inhibitor) in patients with neurofibromatosis type 2-associated progressive tumors (NCT04374305).
Similarly, selumetinib (a MEK inhibitor) is being tested in a similar population (NCT03095248).
In the immunotherapy era, preclinical studies identified ependymomas as tumors with an immune-suppressive phenotype mainly composed of exhausted T cells (91, 92). The only case report investigating the programmed death 1 (PD1) inhibitor tislelizumab in a metastatic myxo-papillary ependymoma resulted in a durable stable disease with a PFS of 18 months (87).
To date, a phase II study is currently investigating the PD1 inhibitor nivolumab (NCT03173950) in adult patients with rare central nervous system tumors including spinal ependymoma.
Other immunological treatments consist of CAR-T therapy. These engineered T cells have achieved important outcomes in solid and hematologic malignancies and represent a concrete hope also in patients with central nervous system tumors (93–95). The majority of trials investigating CAR T are tailored to pediatric patients. The NCT04661384 is a phase I trial investigating the IL13Ralpha2-CAR T cells within patients with leptomeningeal involvement from ependymoma, glioblastoma, and other malignancies. The accrual is opened also for adult patients.
Adult spinal ependymomas are rare tumors. However, important molecular insights have been gained in recent years. Optimal treatment requires surgery with the goal of gross total resection and radiotherapy when indicated. No standard systemic treatment has been established, although promising results have been achieved with the combination of TMZ + lapatinib. Promising and new treatments are being investigated, such as brigatinib, selumetinib, and neratinib together with immunotherapy strategies, including CAR-Ts.
Dedicated neurological supportive care and a multidisciplinary approach must always be favored to achieve optimal disease management, limit toxicity, and preserve quality of life.
GC: Writing – original draft, Writing – review & editing. FP: Writing – original draft. EF: Writing – original draft. VB: Writing – original draft. AS: Writing – original draft. MP: Writing – original draft. RM: Writing – original draft. VD: Writing – review & editing. BB: Writing – review & editing. GLi: Writing – original draft. MC: Writing – original draft. MS: Writing – review & editing. MM: Writing – review & editing. GM: Writing – original draft. PN: Writing – original draft. GLo: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research received “Ricerca Corrente 2023” funding from the Italian Ministry of Health (CDC 099183) to cover publication costs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wostrack M, Ringel F, Eicker SO, Jägersberg M, Schaller K, Kerschbaumer J, et al. Spinal ependymoma in adults: a multicenter investigation of surgical outcome and progression-free survival. J Neurosurg Spine (2018) 28(6):654–62. doi: 10.3171/2017.9.SPINE17494
2. Segal D, Lidar Z, Corn A, Constantini S. Delay in diagnosis of primary intradural spinal cord tumors. Surg Neurol Int (2012) 3:52. doi: 10.4103/2152-7806.96075
3. Rudà R, Bruno F, Pellerino A, Soffietti R. Ependymoma: Evaluation and management updates. Curr Oncol Rep (2022) 24(8):985–93. doi: 10.1007/s11912-022-01260-w
4. Elsamadicy AA, Koo AB, David WB, Lee V, Zogg CK, Kundishora AJ, et al. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neuro-Oncol Adv (2020) 2(1):vdaa019. doi: 10.1093/noajnl/vdaa019
5. Foss-Skiftesvik J, Stoltze UK, van Overeem Hansen T, Ahlborn LB, Sørensen E, Ostrowski SR, et al. Redefining germline predisposition in children with molecularly characterized ependymoma: a population-based 20-year cohort. Acta Neuropathol Commun (2022) 10(1):123. doi: 10.1186/s40478-022-01429-1
6. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2015–2019. Neuro-Oncol (2022) 24(Supplement_5):v1–v95. doi: 10.1093/neuonc/noac202
7. Khalid SI, Adogwa O, Kelly R, Metha A, Bagley C, Cheng J, et al. Adult spinal ependymomas: An epidemiologic study. World Neurosurg (2018) 111:e53–61. doi: 10.1016/j.wneu.2017.11.165
8. Vera-Bolanos E, Aldape K, Yuan Y, Wu J, Wani K, Necesito-Reyes MJ, et al. Clinical course and progression-free survival of adult intracranial and spinal ependymoma patients. Neuro-Oncol (2015) 17(3):440–7. doi: 10.1093/neuonc/nou162
9. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
10. Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell (2015) 27(5). doi: 10.1016/j.ccell.2015.04.002
11. Träger M, Schweizer L, Pérez E, Schmid S, Hain EG, Dittmayer C, et al. Adult intracranial ependymoma - relevance of DNA methylation profiling for diagnosis, prognosis and treatment. Neuro-Oncol (2023) 25(7):1286–98. doi: 10.1093/neuonc/noad030
12. Witt H, Gramatzki D, Hentschel B, Pajtler KW, Felsberg J, Schackert G, et al. DNA methylation-based classification of ependymomas in adulthood: implications for diagnosis and treatment. Neuro-Oncol (2018) 20(12):1616–24. doi: 10.1093/neuonc/noy118
13. Schwartz TH, McCormick PC. Intramedullary ependymomas: clinical presentation, surgical treatment strategies and prognosis. J Neurooncol (2000) 47(3):211–8. doi: 10.1023/a:1006414405305
14. Armstrong TS, Vera-Bolanos E, Gilbert MR. Clinical course of adult patients with ependymoma: results of the adult ependymoma outcomes project. Cancer (2011) 117(22):5133–41. doi: 10.1002/cncr.26181
15. Kucia EJ, Bambakidis NC, Chang SW, Spetzler RF. Surgical technique and outcomes in the treatment of spinal cord ependymomas, part 1: intramedullary ependymomas. Neurosurgery (2011) 68(1 Suppl Operative):57–63. doi: 10.1227/NEU.0b013e318208f181
16. Walbert T, Mendoza TR, Vera-Bolaños E, Acquaye A, Gilbert MR, Armstrong TS. Symptoms and socio-economic impact of ependymoma on adult patients: results of the adult ependymoma outcomes project 2. J Neurooncol (2015) 121(2):341–8. doi: 10.1007/s11060-014-1638-4
17. Butenschoen VM, Gloßner T, Hostettler IC, Meyer B, Wostrack M. Quality of life and return to work and sports after spinal ependymoma resection. Sci Rep (2022) 12(1):4926. doi: 10.1038/s41598-022-09036-9
18. McCormick PC, Torres R, Post KD, Stein BM. Intramedullary ependymoma of the spinal cord. J Neurosurg (1990) 72(4):523–32. doi: 10.3171/jns.1990.72.4.0523
19. Wu L, Wang L, Zou W, Yang J, Jia W, Xu Y. Primary spinal anaplastic ependymoma: A single-institute retrospective cohort and systematic review. Front Oncol (2023) 13:1083085. doi: 10.3389/fonc.2023.1083085
20. Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg (1993) 79(2):204–9. doi: 10.3171/jns.1993.79.2.0204
21. Alhalabi OT, Heene S, Landré V, Neumann JO, Scherer M, Ishak B, et al. Spinal oncologic paraparesis: Analysis of neurological and surgical outcomes in patients with intramedullary, extramedullary, and extradural tumors. Front Oncol (2022) 12:1003084. doi: 10.3389/fonc.2022.1003084
22. Cofano F, Giambra C, Costa P, Zeppa P, Bianconi A, Mammi M, et al. Management of extramedullary intradural spinal tumors: The impact of clinical status, intraoperative neurophysiological monitoring and surgical approach on outcomes in a 12-year double-center experience. Front Neurol (2020) 11:598619. doi: 10.3389/fneur.2020.598619
23. Myrseth E, Habiba S, Rekand T, Sætran HA, Mørk S, Grønning M. Intramedullary spinal cord and filum tumours-long-term outcome: single institution case series. Acta Neurochir (Wien) (2022) 164(11):3047–56. doi: 10.1007/s00701-022-05350-3
24. Lee SM, Cho YE, Kwon YM. Neurological outcome after surgical treatment of intramedullary spinal cord tumors. Korean J Spine (2014) 11(3):121–6. doi: 10.14245/kjs.2014.11.3.121
25. Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, Santarius T, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro-Oncol (2018). doi: 10.1093/neuonc/nox166
26. Gilbert MR, Ruda R, Soffietti R. Ependymomas in adults. Curr Neurol Neurosci Rep (2010) 10(3):240–7. doi: 10.1007/s11910-010-0109-3
27. Wu J, Ranjan S. Neoplastic myelopathies. Contin Minneap Minn. (2018) 24(2, Spinal Cord Disorders):474–96. doi: 10.1212/CON.0000000000000585
28. Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res (2012) 22(2):407–19. doi: 10.1101/gr.119867.110
29. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature (2018) 555(7697):469–74. doi: 10.1038/nature26000
30. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol (Berl) (2019). doi: 10.1007/s00401-019-02056-2
31. Ellison D WHO classification of tumors editorial board central nervous system tumours. In: Ependymal tumors: introduction. International Agency for Research on Cancer (2021).
32. Pietsch T, Aldape K, Korshunov A, Pajtler K, Taylor M. WHO classification of tumours editorial board central nervous system tumours. In: Spinal ependymoma. International Agency for Research on Cancer (2021).
33. Shatara M, Schieffer KM, Klawinski D, Thomas DL, Pierson CR, Sribnick EA, et al. Clinically aggressive pediatric spinal ependymoma with novel MYC amplification demonstrates molecular and histopathologic similarity to newly described MYCN-amplified spinal ependymomas. Acta Neuropathol Commun (2021) 9(1):192. doi: 10.1186/s40478-021-01296-2
34. Giannini C, Aldape K, Korshunov A, Pajtler KW, Pietsch T, Ramaswamy V, et al. Spinal ependymoma, MYC-N amplified. WHO Classification of Tumours Editorial Board Central Nervous system Tumours. (Lyon: International Agency for Research on Cancer) (2021), pp. 180–2.
35. Scheil S, Brüderlein S, Eicker M, Herms J, Herold-Mende C, Steiner HH, et al. Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol Zurich Switz (2001) 11(2):133–43. doi: 10.1111/j.1750-3639.2001.tb00386.x
36. Bertero L, Ricci AA, Tampieri C, Cassoni P, Modena P. Ependymomas. Pathologica (2022) 114(6):436–46. doi: 10.32074/1591-951X-817
37. Cervoni L, Celli P, Caruso R, Gagliardi FM, Cantore GP. Neurinomas and ependymomas of the cauda equina. a review of the clinical characteristics. Minerva Chir (1997) 52(5):629–33. doi: PMID: 9297152
38. Rosenblum M, Korshunov A, Pajtler K, Pietsch T, Taylor M, Venneti S. WHO classification of tumours editorial board central nervous system tumours. In: Myxopapillary ependymoma (2021).
39. Kraetzig T, McLaughlin L, Bilsky MH, Laufer I. Metastases of spinal myxopapillary ependymoma: unique characteristics and clinical management. J Neurosurg Spine (2018) 28(2):201–8. doi: 10.3171/2017.5.SPINE161164
40. Weber DC, Wang Y, Miller R, Villà S, Zaucha R, Pica A, et al. Long-term outcome of patients with spinal myxopapillary ependymoma: treatment results from the MD anderson cancer center and institutions from the rare cancer network. Neuro-Oncol (2015) 17(4):588–95. doi: 10.1093/neuonc/nou293
41. Bates JE, Choi G, Milano MT. Myxopapillary ependymoma: a SEER analysis of epidemiology and outcomes. J Neurooncol (2016) 129(2):251–8. doi: 10.1007/s11060-016-2167-0
42. Katz SY, Cachia D, Kamiya-Matsuoka C, Olar A, Theeler B, Penas Prado M, et al. Ependymomas arising outside of the central nervous system: A case series and literature review. J Clin Neurosci Off J Neurosurg Soc Australas (2018) 47:202–7. doi: 10.1016/j.jocn.2017.10.026
43. Bockmayr M, Harnisch K, Pohl LC, Schweizer L, Mohme T, Körner M, et al. Comprehensive profiling of myxopapillary ependymomas identifies a distinct molecular subtype with relapsing disease. Neuro-Oncol (2022) 24(10):1689–99. doi: 10.1093/neuonc/noac088
44. Bockmayr M, Körner M, Schweizer L, Schüller U. Cauda equina paragangliomas express HOXB13. Neuropathol Appl Neurobiol (2021) 47(6):889–90. doi: 10.1111/nan.12713
45. Shimada S, Ishizawa K, Horiguchi H, Shimada T, Hirose T. Subependymoma of the spinal cord and review of the literature. Pathol Int (2003) 53(3):169–73. doi: 10.1046/j.1440-1827.2003.01450.x
46. Rosenblum M, Korshunov A, Pajtler K, Pietsch T, Taylor M, Venneti S. WHO classification of tumours editorial board central nervous system tumours. In: Subependymoma. International Agency for Research On Cancer (2021).
47. Spinal ependymoma | radiology reference article | radiopaedia.org. radiopaedia (online website) (2023). Available at: https://radiopaedia.org/articles/spinal-ependymoma?lang=us (Accessed June 7, 2023).
48. Marrazzo A, Cacchione A, Rossi S, Carboni A, Gandolfo C, Carai A, et al. Intradural pediatric spinal tumors: An overview from imaging to novel molecular findings. Diagn Basel Switz (2021) 11(9):1710. doi: 10.3390/diagnostics11091710
49. Koeller KK, Rosenblum RS, Morrison AL. Neoplasms of the spinal cord and filum terminale: radiologic-pathologic correlation. Radiogr Rev Publ Radiol Soc N Am Inc (2000) 20(6):1721–49. doi: 10.1148/radiographics.20.6.g00nv151721
50. Pikis S, Cohen JE, Vargas AA, Gomori JM, Harnof S, Itshayek E. Superficial siderosis of the central nervous system secondary to spinal ependymoma. J Clin Neurosci Off J Neurosurg Soc Australas (2014) 21(11):2017–9. doi: 10.1016/j.jocn.2014.05.020
51. Choi KE, Na SH, Jeong HS, Im JJ, Kim YD. Superficial siderosis of the central nervous system due to spinal ependymoma. Ann Geriatr Med Res (2018) 22(1):43–5. doi: 10.4235/agmr.2018.22.1.43
52. Wein S. Myxopapillary ependymoma | radiology reference article | radiopaedia.org. Radiopaedia. doi: 10.53347/rID-19263
53. Kahan H, Sklar EM, Post MJ, Bruce JH. MR characteristics of histopathologic subtypes of spinal ependymoma. AJNR Am J Neuroradiol (1996) 17(1):143–50. doi: PMID: 8770266
54. Rudrabhatla P, Sabarish S, Nair SS, George T, Divakar G, Sylaja PN. Superficial siderosis due to spinal myxopapillary ependymoma mimicking idiopathic intracranial hypertension. Ann Indian Acad Neurol (2022) 25(1):156–7. doi: 10.4103/aian.AIAN_186_21
55. Toi H, Ogawa Y, Kinoshita K, Hirai S, Takai H, Hara K, et al. Bamboo leaf sign as a sensitive magnetic resonance imaging finding in spinal subependymoma: Case report and literature review. Case Rep Neurol Med (2016) 2016:9108641. doi: 10.1155/2016/9108641
56. Chang UK, Choe WJ, Chung SK, Chung CK, Kim HJ. Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol (2002) 57(2):133–9. doi: 10.1023/a:1015789009058
57. Celano E, Salehani A, Malcolm JG, Reinertsen E, Hadjipanayis CG. Spinal cord ependymoma: a review of the literature and case series of ten patients. J Neurooncol (2016) 128(3):377–86. doi: 10.1007/s11060-016-2135-8
58. Mirza AB, Gebreyohanes A, Knight J, Bartram KJ, Vastani A, Kalaitzoglou D, et al. Prognostic factors for surgically managed intramedullary spinal cord tumours: a single-centre case series. Acta Neurochir (Wien) (2022) 164(10):2605–22. doi: 10.1007/s00701-022-05304-9
59. Volpp PB, Han K, Kagan AR, Tome M. Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys (2007) 69(4):1199–204. doi: 10.1016/j.ijrobp.2007.04.058
60. Oh MC, Kim JM, Kaur G, Safaee M, Sun MZ, Singh A, et al. Prognosis by tumor location in adults with spinal ependymomas. J Neurosurg Spine (2013) 18(3):226–35. doi: 10.3171/2012.12.SPINE12591
61. Klekamp J. Spinal ependymomas. part 1: Intramedullary ependymomas. Neurosurg Focus (2015) 39(2):E6. doi: 10.3171/2015.5.FOCUS15161
62. Skrap B, Tramontano V, Faccioli F, Meglio M, Pinna G, Sala F. Surgery for intramedullary spinal cord ependymomas in the neuromonitoring era: results from a consecutive series of 100 patients. J Neurosurg Spine (2021), 1–11. doi: 10.3171/2021.7.SPINE21148
63. Capo G, Vandenbulcke A, Yves Barrey C. Central nervous system tumors. In: Intramedullary spinal tumors. Feyzi Birol Sarica (2022). doi: 10.5772/intechopen.102249
64. Lee SH, Chung CK, Kim CH, Yoon SH, Hyun SJ, Kim KJ, et al. Long-term outcomes of surgical resection with or without adjuvant radiation therapy for treatment of spinal ependymoma: a retrospective multicenter study by the korea spinal oncology research group. Neuro-Oncol (2013) 15(7):921–9. doi: 10.1093/neuonc/not038
65. Nakamura M, Ishii K, Watanabe K, Tsuji T, Takaishi H, Matsumoto M, et al. Surgical treatment of intramedullary spinal cord tumors: prognosis and complications. Spinal Cord (2008) 46(4):282–6. doi: 10.1038/sj.sc.3102130
66. Oh MC, Ivan ME, Sun MZ, Kaur G, Safaee M, Kim JM, et al. Adjuvant radiotherapy delays recurrence following subtotal resection of spinal cord ependymomas. Neuro-Oncol (2013) 15(2):208–15. doi: 10.1093/neuonc/nos286
67. Nagasawa DT, Smith ZA, Cremer N, Fong C, Lu DC, Yang I. Complications associated with the treatment for spinal ependymomas. Neurosurg Focus (2011) 31(4):E13. doi: 10.3171/2011.7.FOCUS11158
68. Wahab SH, Simpson JR, Michalski JM, Mansur DB. Long term outcome with post-operative radiation therapy for spinal canal ependymoma. J Neurooncol (2007) 83(1):85–9. doi: 10.1007/s11060-006-9310-2
69. Gomez DR, Missett BT, Wara WM, Lamborn KR, Prados MD, Chang S, et al. High failure rate in spinal ependymomas with long-term follow-up. Neuro-Oncol (2005) 7(3):254–9. doi: 10.1215/S1152851704001231
70. Whitaker SJ, Bessell EM, Ashley SE, Bloom HJ, Bell BA, Brada M. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg (1991) 74(5):720–8. doi: 10.3171/jns.1991.74.5.0720
71. Byun HK, Yi S, Yoon HI, Kim SH, Cho J, Suh CO. Clinical outcomes of radiotherapy for spinal cord ependymoma with adverse prognostic features: a single-center study. J Neurooncol (2018) 140(3):649–57. doi: 10.1007/s11060-018-2995-1
72. Shaw EG, Evans RG, Scheithauer BW, Ilstrup DM, Earle JD. Radiotherapeutic management of adult intraspinal ependymomas. Int J Radiat Oncol Biol Phys (1986) 12(3):323–7. doi: 10.1016/0360-3016(86)90345-7
73. Lin YH, Huang CI, Wong TT, Chen MH, Shiau CY, Wang LW, et al. Treatment of spinal cord ependymomas by surgery with or without postoperative radiotherapy. J Neurooncol (2005) 71(2):205–10. doi: 10.1007/s11060-004-1386-y
74. Akyurek S, Chang EL, Yu TK, Little D, Allen PK, McCutcheon I, et al. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. anderson cancer center. J Neurooncol (2006) 80(2):177–83. doi: 10.1007/s11060-006-9169-2
75. Ryu SM, Lee SH, Kim ES, Eoh W. Predicting survival of patients with spinal ependymoma using machine learning algorithms with the SEER database. World Neurosurg (2018), S1878–8750(18)32914-0. doi: 10.1016/j.wneu.2018.12.091
76. Peschel RE, Kapp DS, Cardinale F, Manuelidis EE. Ependymomas of the spinal cord. Int J Radiat Oncol Biol Phys (1983) 9(7):1093–6. doi: 10.1016/0360-3016(83)90402-9
77. Montero AS, Tran S, Amelot A, Berriat F, Lot G, Gaillard S, et al. Clinical characteristics and long-term surgical outcome of spinal myxopapillary ependymoma: a french cohort of 101 patients. J Neurooncol (2021) 152(3):491–9. doi: 10.1007/s11060-021-03717-7
78. Liu T, Yang C, Deng X, Li A, Xin Y, Yang J, et al. Clinical characteristics and surgical outcomes of spinal myxopapillary ependymomas. Neurosurg Rev (2020) 43(5):1351–6. doi: 10.1007/s10143-019-01150-z
79. Schild SE, Wong W, Nisi K. In regard to the radiotherapy of myxopapillary ependymomas. Int J Radiat Oncol Biol Phys (2002) 53(3):787. doi: 10.1016/s0360-3016(02)02776-1
80. Rudà R, Gilbert M, Soffietti R. Ependymomas of the adult: molecular biology and treatment. Curr Opin Neurol (2008) 21(6):754–61. doi: 10.1097/WCO.0b013e328317efe8
81. Tobin MK, Geraghty JR, Engelhard HH, Linninger AA, Mehta AI. Intramedullary spinal cord tumors: a review of current and future treatment strategies. Neurosurg Focus (2015) 39(2):E14. doi: 10.3171/2015.5.FOCUS15158
82. Franceschi E, Frappaz D, Rudà R, Hau P, Preusser M, Houillier C, et al. Rare primary central nervous system tumors in adults: An overview. Front Oncol (2020) 10:996. doi: 10.3389/fonc.2020.00996
83. Gilbert MR, Yuan Y, Wu J, Mendoza T, Vera E, Omuro A, et al. A phase II study of dose-dense temozolomide and lapatinib for recurrent low-grade and anaplastic supratentorial, infratentorial, and spinal cord ependymoma. Neuro-Oncol (2021) 23(3):468–77. doi: 10.1093/neuonc/noaa240
84. Chamberlain MC. Etoposide for recurrent spinal cord ependymoma. Neurology (2002) 58(8):1310–1. doi: 10.1212/wnl.58.8.1310
85. Kim WH, Yoon SH, Kim C-Y, Kim K-J, Lee MM, Choe G, et al. Temozolomide for malignant primary spinal cord glioma: an experience of six cases and a literature review. J Neurooncol (2011) 101(2):247–54. doi: 10.1007/s11060-010-0249-y
86. Fujiwara Y, Manabe H, Izumi B, Shima T, Adachi N. Remarkable efficacy of temozolomide for relapsed spinal myxopapillary ependymoma with multiple recurrence and cerebrospinal dissemination: a case report and literature review. Eur Spine J Off Publ Eur Spine Soc Eur Spinal Deform Soc Eur Sect Cerv Spine Res Soc (2018) 27(Suppl 3). doi: 10.1007/s00586-017-5413-z
87. Tapia Rico G, Townsend A, Price T, Patterson K. Metastatic myxopapillary ependymoma treated with immunotherapy achieving durable response. BMJ Case Rep (2020) 13(12). doi: 10.1136/bcr-2020-236242
88. Kim WH, Yoon SH, Kim CY, Kim KJ, Lee MM, Choe G, et al. Temozolomide for malignant primary spinal cord glioma: an experience of six cases and a literature review. J Neurooncol (2011) 101(2). doi: 10.1007/s11060-010-0249-y
89. Gramatzki D, Felsberg J, Hentschel B, Bähr O, Westphal M, Schackert G, et al. Chemotherapy for adult patients with spinal cord gliomas. Neuro-Oncol Pract (2021) 8(4):475–84. doi: 10.1093/nop/npab017
90. Essayed WI, Bernard A, Kalamarides M. Clinical response associated with radiographic regression of a cervicomedullary ependymoma in a NF2 patient treated by bevacizumab. J Neurooncol (2015) 125(2):445–6. doi: 10.1007/s11060-015-1925-8
91. Saleh AH, Samuel N, Juraschka K, Saleh MH, Taylor MD, Fehlings MG. The biology of ependymomas and emerging novel therapies. Nat Rev Cancer (2022) 22(4):208–22. doi: 10.1038/s41568-021-00433-2
92. Witt DA, Donson AM, Amani V, Moreira DC, Sanford B, Hoffman LM, et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: Implications for PD-1-targeted therapy. Pediatr Blood Cancer (2018) 65(5):e26960. doi: 10.1002/pbc.26960
93. Gatto L, Franceschi E, Di Nunno V, Maggio I, Lodi R, Brandes AA. Engineered CAR-t and novel CAR-based therapies to fight the immune evasion of glioblastoma: gutta cavat lapidem. Expert Rev Anticancer Ther (2021) 21(12):1333–53. doi: 10.1080/14737140.2021.1997599
94. Gatto L, Nunno VD, Franceschi E, Brandes AA. Chimeric antigen receptor macrophage for glioblastoma immunotherapy: the way forward. Immunotherapy (2021) 13(11):879–83. doi: 10.2217/imt-2021-0054
Keywords: ependymoma, spinal ependymoma, radiotherapy, chemotherapy, temozolomide, lapatinib
Citation: Cerretti G, Pessina F, Franceschi E, Barresi V, Salvalaggio A, Padovan M, Manara R, Di Nunno V, Bono BC, Librizzi G, Caccese M, Scorsetti M, Maccari M, Minniti G, Navarria P and Lombardi G (2023) Spinal ependymoma in adults: from molecular advances to new treatment perspectives. Front. Oncol. 13:1301179. doi: 10.3389/fonc.2023.1301179
Received: 24 September 2023; Accepted: 24 October 2023;
Published: 24 November 2023.
Edited by:
Maria Caffo, University of Messina, ItalyReviewed by:
Lorena Gurrieri, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyCopyright © 2023 Cerretti, Pessina, Franceschi, Barresi, Salvalaggio, Padovan, Manara, Di Nunno, Bono, Librizzi, Caccese, Scorsetti, Maccari, Minniti, Navarria and Lombardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Cerretti, Z2l1bGlhLmNlcnJldHRpQGlvdi52ZW5ldG8uaXQ=
†ORCID: Giulia Cerretti, orcid.org/0000-0002-2849-0537
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.