
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 January 2024
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1289222
This article is part of the Research Topic Integrating Transcriptional Modulation in Systemic Tumor Therapy View all 7 articles
 Dennis Christoph Harrer1
Dennis Christoph Harrer1 Florian Lüke1,2
Florian Lüke1,2 Tobias Pukrop1,3
Tobias Pukrop1,3 Lina Ghibelli4
Lina Ghibelli4 Christopher Gerner5
Christopher Gerner5 Albrecht Reichle1*†
Albrecht Reichle1*† Daniel Heudobler1,3
Daniel Heudobler1,3A series of seven clinical trials on relapsed or refractory (r/r) metastatic neoplasias followed the question: Are networks of ligand-receptor cross-talks that support tumor-specific cancer hallmarks, druggable with tumor tissue editing approaches therapeutically exploiting tumor plasticity? Differential recombinations of pioglitazone, a dual peroxisome-proliferator activated receptorα/γ (PPARα/γ) agonist, with transcriptional modulators, i.e., all-trans retinoic acid, interferon-α, or dexamethasone plus metronomic low-dose chemotherapy (MCT) or epigenetic modeling with azacitidine plus/minus cyclooxygenase-2 inhibition initiated tumor-specific reprogramming of cancer hallmarks, as exemplified by inflammation control in r/r melanoma, renal clear cell carcinoma (RCCC), Hodgkin’s lymphoma (HL) and multisystem Langerhans cell histiocytosis (mLCH) or differentiation induction in non-promyelocytic acute myeloid leukemia (non-PML AML). Pioglitazone, integrated in differentially designed editing schedules, facilitated induction of tumor cell death as indicated by complete remission (CR) in r/r non-PML AML, continuous CR in r/r RCCC, mLCH, and in HL by addition of everolimus, or long-term disease control in melanoma by efficaciously controlling metastasis, post-therapy cancer repopulation and acquired cell-resistance and genetic/molecular-genetic tumor cell heterogeneity (M-CRAC). PPARα/γ agonists provided tumor-type agnostic biomodulatory efficacy across different histologic neoplasias. Tissue editing techniques disclose that wide-ranging functions of PPARα/γ agonists may be on-topic focused for differentially unlocking tumor phenotypes. Low-dose MCT facilitates targeted reprogramming of cancer hallmarks with transcriptional modulators, induction of tumor cell death, M-CRAC control and editing of non-oncogene addiction. Thus, pioglitazone, integrated in tumor tissue editing protocols, is an important biomodulatory drug for addressing urgent therapeutic problems, such as M-CRAC in relapsed or refractory tumor disease.
Pioglitazone is a thiazolidine-2, 4-dione compound and an approved dual peroxisome proliferator-activated receptor (PPAR)α/γ agonist for the treatment of insulin resistance (1, 2). Besides this limited indication, more and more pre-clinical data reveal a broad, multileveled activity profile in cancer tissue, modulating cancer-associated inflammation, immune response, sustained proliferative signaling, cancer metabolism, angiogenesis, i.e., tissue functions that are described by the hallmarks of cancer (3, 4).
Promising experimental findings on the anti-tumor activity of pioglitazone face the difficulty of missing monoactivity in metastatic tumor disease, despite of the very low mutation rate of PPARα/γ in human tumor cells (5–7). Additionally, the use of glitazones in clinical settings is pejorative because dual PPARα/γ agonists may induce histologically different tumors in rodents (8). However, the discussion about pioglitazone and bladder cancer induction in patients with type II diabetes is still not based on sufficient evidence (9). Research for novel, more active PPARα/PPARγ/PPARα/γ agonists did not bring more agonists to clinical approval (10).
Two rationales worked hand in hand to successfully integrate pioglitazone in novel therapy designs for relapsed/refractory (r/r) metastatic neoplasias. Firstly, a general therapeutic problem is pending for solution. Maximized apoptosis induction with maximum tolerable doses of pulsed therapies for rescuing r/r tumor states may inevitably re-establish in most neoplasias cancer promoting hallmarks, metastasis, post-therapy cancer repopulation and acquired cell-resistance and genetic/molecular-genetic tumor cell heterogeneity (M-CRAC) and limits initially induced tumor responses (11, 12). Thus, approved pulsed therapy approaches- irrespectively of their composition – are extremely challenged to induce continuous complete remission (cCR) and, therefore, demand novel therapy techniques to overcome M-CRAC (13).
Secondly, novel therapy techniques resolving disease traits of M-CRAC must unlock the tumor phenotype by reprogramming hallmarks of cancer, just those included in M-CRAC, that means, multiple tumor cell compartments and their communication profiles must be targeted, as hypothesized, by the concerted use of transcriptional modulators, including pioglitazone (14–19).
As clinically shown, unlocking the tumor’s plasticity with therapy approaches editing the tumor’s growth-promoting phenotype in a therapeutically relevant way, i.e., tumor tissue editing, facilitates the successful clinical integration of combined transcriptional modulation with pioglitazone (20–24). Tumor tissue editing techniques concertedly reprogram hallmarks of cancer for establishing biologic hallmarks that may control tumor growth, and even induce tumor cell death (11).
Tissue editing techniques include synergistic combinations of bioactive drugs, such as metronomic low-dose chemotherapies, transcriptional modulators, i.e., pioglitazone, interferon-α, dexamethasone or all-trans retinoic acid (ATRA) and cyclooxygenase-2 (COX-2) inhibitors, and may induce long-term tumor control, objective response, even continuous complete remission (cCR) in r/r neoplasias (21–23, 25–28). On the top, edited stress reponse pathways, referred to as non-oncogene addiction in tumor tissues, provide a novel, repurposed activity profile for approved targeted therapies (20, 29).
Inflammation control, reestablishing immunosurveillance, metabolic reprogramming, enhancing tumor growth suppression, differentiation induction and consecutively M-CRAC control are now the contributions of pioglitazone within editing approaches, as shown in a large series of clinical trials (21–23, 30–32).
The present review categorizes the reprogramming activity of pioglitazone in recombination with additional transcriptional modulators, also that of metronomic low-dose chemotherapy, by analyzing trials on tumor tissue editing, designed for promoting inflammation control or differentiation induction in r/r metastatic cancer or r/r non-promyelocytic acute myelocytic leukemia (non-PML AML).
The novel treatment concept ‘tumor tissue editing’ adopts to the use of tissue editing techniques for correcting epigenetic or genetic aberrations in tumor tissues (20). Tumor tissue editing methods aim at therapeutically exploiting tumor phenotypes by reprogramming hallmarks of cancer (28).
Tumor tissue editing is defined by therapy-guided targeted evolution of tumor tissues for establishing biologic functions in tumor tissues facilitating tumor control or initiating complete remission in relapsed or refractory tumor disease.
Tumor tissue editing techniques are supported by bioactive drugs with no or poor monoactivity that are therapeutically involving tumor cells but as well, the whole repertoire of stroma cells, i.e., hematopoietic cells, such as myeloid-derived suppressive cells, tumor-associated macrophages (TAMs) and T-cells, mesenchymal cells, cancer-associated fibroblasts (CAFs), lipocytes, and endothelial cells (26, 33). The target of metronomic low dose chemotherapy plus transcriptional modulators is the tumor tissue’s phenotypic plasticity to be therapeutically exploited (Figure 1).
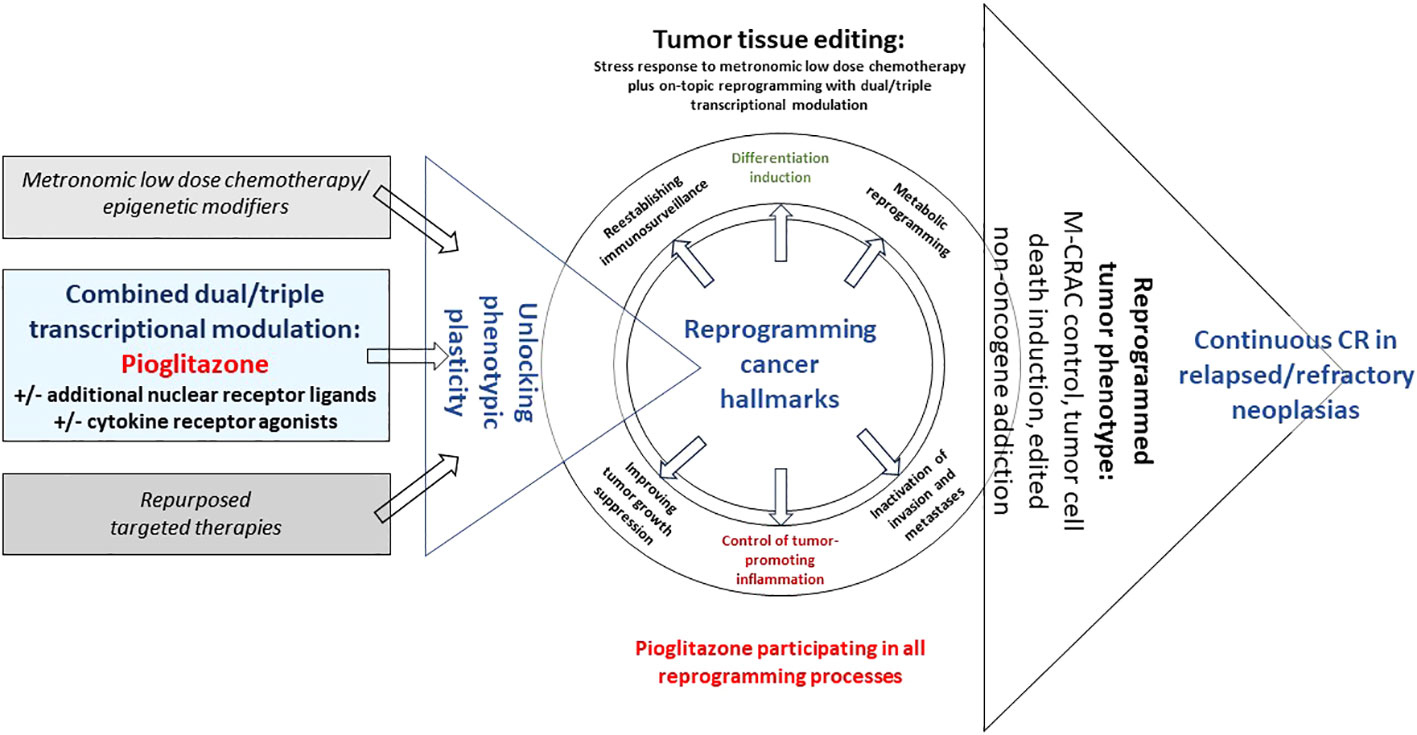
Figure 1 Unlocking tumor phenotype with editing schedules including pioglitazone. Editing with pioglitazone may reprogram hallmarks of cancer via tumor-specific ligand-receptor networks, thereby, inducing tumor cell death, M-CRAC control and edited non-oncogene addiction in relapsed/refractory metastatic tumor disease or hematologic neoplasias.
Backbone of the editing concept is pioglitazone and metronomic low dose chemotherapy. The addition of selected transcriptional modulators, such as ATRA, interferon-a or dexamethasone contributes to differentiation induction in r/r non-PML AML, to inflammation control in r/r Hodgkin’s lymphoma, Langerhans cell histiocytosis and RCCC, and clinically documented impressive immune response in CRPC (22, 23, 34, 35). All these biologic surrogates, inflammation control, differentiation induction and immune response may be associated with objective tumor response, CR or cCR in r/r neoplasias (Table 1).
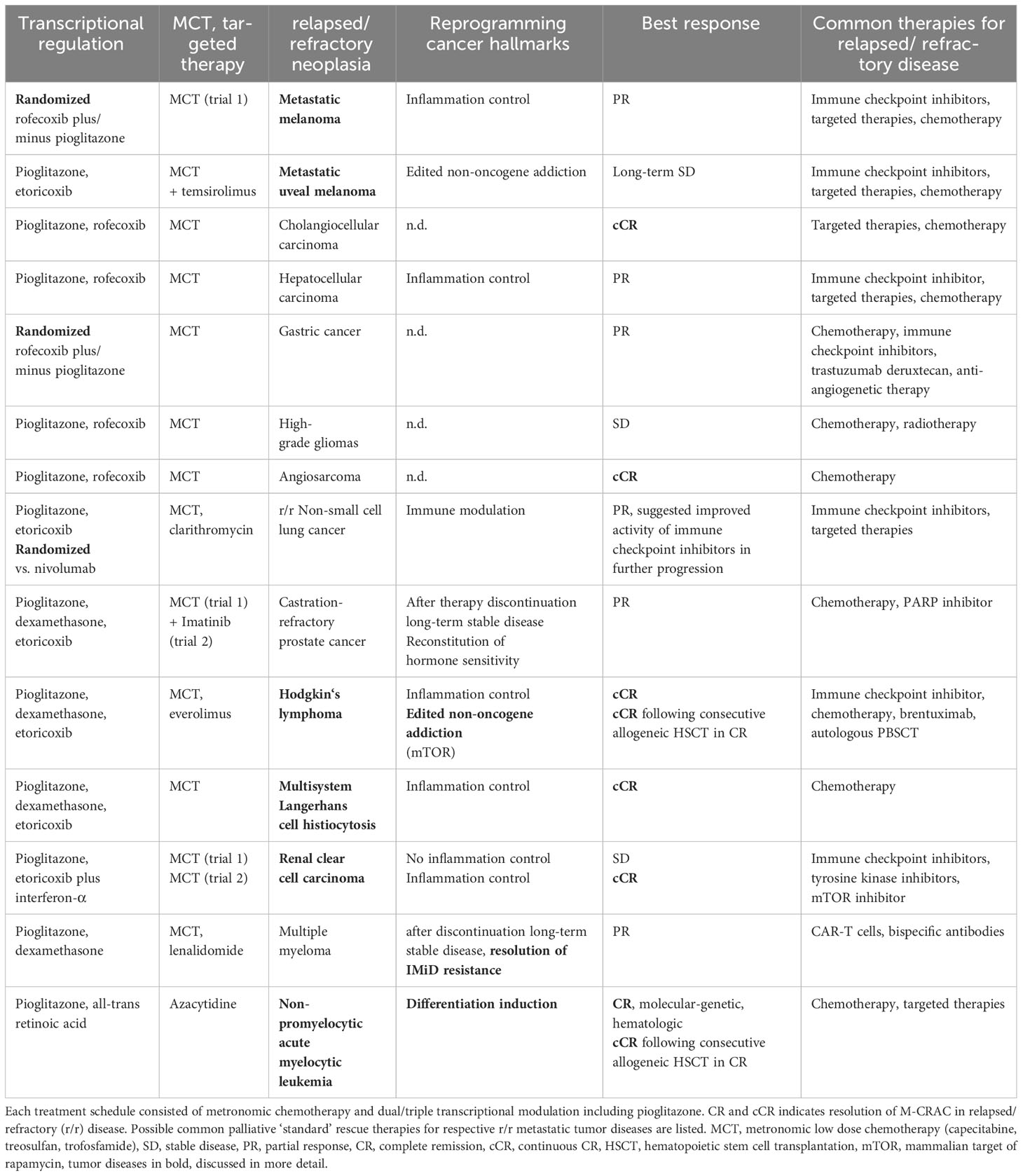
Table 1 Pioglitazone contributing to M-CRAC control or resolution in carcinomas, sarcomas and hematologic diseases with tissue editing approaches.
Currently, the concerted activity profile of the biomodulatory drug combinations cannot be pinned down to single pathways or to the contribution of single cell types of the tumor tissue. The necessity to use drug combinations including pioglitazone has been exemplarily pre-clinically and clinically tested in case of non-PML AML or clinically with consecutively performed trials with identical inclusion criteria (r/r RCCC) or randomized phase II trials (34, 36–39).
Thus, the clinical results rely on a concerted activity profile of drugs without monoactivity in the respective histologic tumor type. The suggested mechanism of action is a communicative reprogramming of tumor phenotypes. That is a novel approach for controlling r/r neoplasias and M-CRAC development.
Although, the activity of the single drugs used in editing approaches is basically pharmacologically well-defined, their interaction with additional transcriptional modulators is mostly poorly understood, especially on the background that metronomic low dose chemotherapy is essential to establish the striking activity of the combinations of transcriptional modulators. The drug combinations only, lead to clinical effects, such as CR or cCR in r/r neoplasias (Table 2).

Table 2 Editing schedules for differentiation induction in relapsed/refractory (r/r) acute myelocytic leukemia and inflammation control in r/r renal clear cell carcinoma, r/r Hodgkin’s lymphoma and r/r metastatic melanoma are exemplarily shown.
‘Normalizing’ tumor-promoting pro-inflammatory signals from quite different sources or redirecting sustained proliferation by differentiation induction are activity profiles of pioglitazone in vitro and in animal models (40–43). Unsurprisingly, in heterogeneous and dynamically evolving tumor systems, the activities of drugs that are not directly targeting oncogenic events, tumor cell specific targets or non-oncogene addiction, but predominantly participate in the realignment of dysbalanced homeostatic processes among cancer hallmarks, show poor or clinically irrelevant monoactivity, like pioglitazone and other agonistically active transcriptional modulators (26).
Pioglitazone plus/minus recombined transcriptional modulators contributes to phenotype plasticity of metastatic r/r neoplasias by facilitating phenotype switches, associated with inflammation control or differentiation, under tumor tissue conditions established by metronomic low-dose chemotherapy (Table 2). Immediate therapeutically induced phenotype switches may be functionally or morphologically comprehensible, but are not based on genetic changes as those frequently discussed in terms of ‘phenotype changes’ (36, 44).
Tumor tissue editing as systems-biologic therapy approach targets tumor-specific networks of ligand-receptor crosstalks, i.e., pro-anakoinotic activity, and reprograms complex homeostatic dysbalances of cancer hallmarks in a therapeutically meaningful way (Figure 1) (17, 22, 28, 45).
Therapeutically reprogramming sustained tumor cell proliferation by control of tumor-linked inflammation, by improving immunosurveillance, correcting tumor tissue’s metabolic processes, reestablishing tumor suppression, inducing differentiation, proved to be systematically druggable approaches by the introduction of tumor tissue editing techniques that may achieve tumor cell death, attenuation, resolution or bypassing of M-CRAC and edited non-oncogene addiction in r/r neoplasias of quite different histologic origin (Table 3; Figure 1) (3, 22, 28).
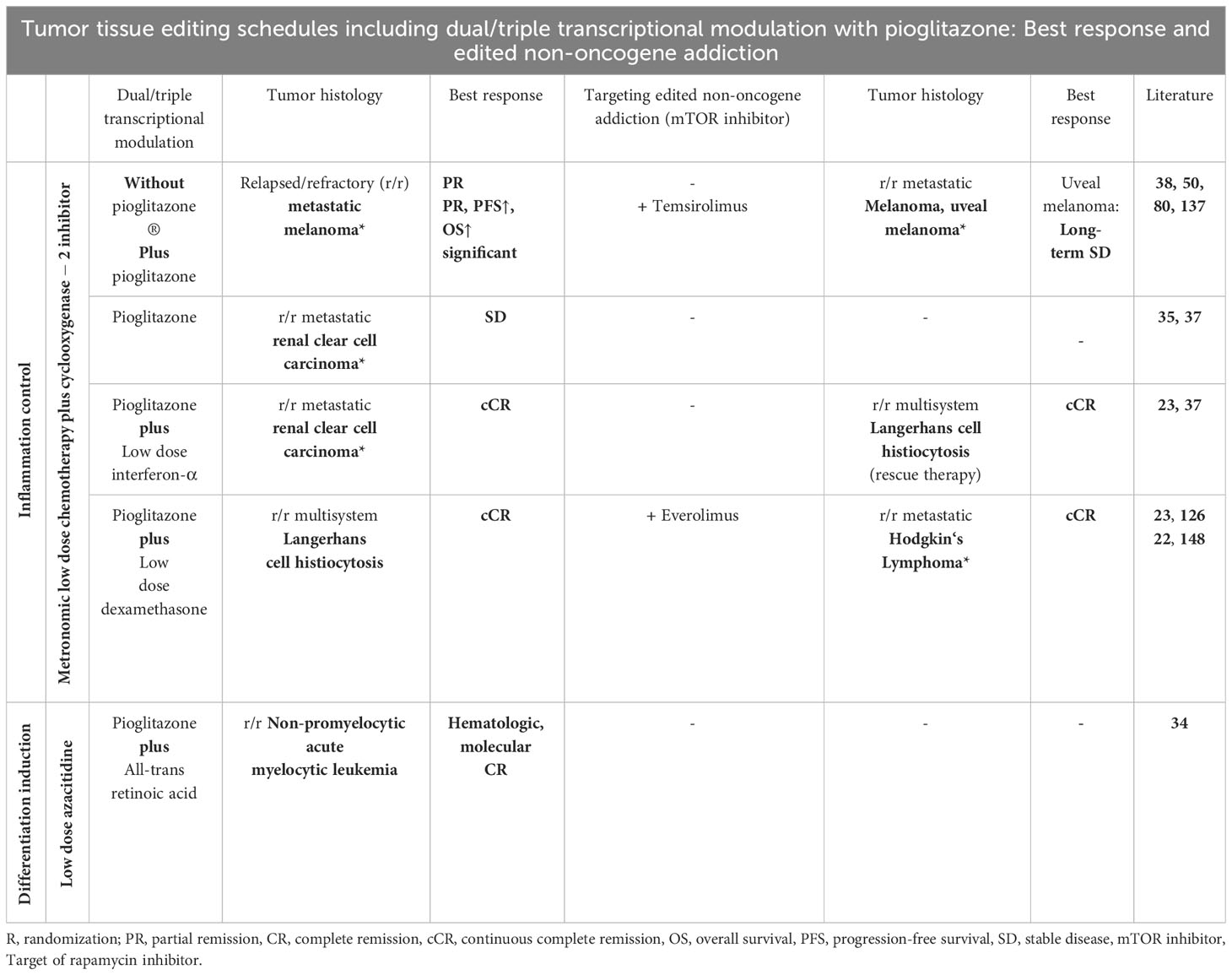
Table 3 Tumor tissue editing trials for different relapsed/refractory tumors and hematologic neoplasias are categorized according to metronomic low-dose chemotherapy/azacitidine, transcriptional modulation with pioglitazone and respective recombinations with further transcriptional modulators aiming at inflammation control, differentiation induction, and at editing non-oncogene addiction, here, *mTOR addiction in r/r Hodgkin’s lymphoma and uveal melanoma.
Tumor tissue editing provides considerable clinical advantages, establishes tumor cell death by targeting the tumor’s systems biology, and the homeostatic dysbalances constituted between hallmarks of cancer. Editing saves toxicity, as only regulatorily active doses must be used. The single drugs in editing schedules must not show any clinical monoactivity, such as pioglitazone (20, 21).
The dual nuclear transcription factor agonist pioglitazone regulates homeostatic balances maintained by all cell compartments of the tumor tissue, the tumor cells, the tumor microenvironment with immune cells, cancer-associated fibroblasts, endothelial cells, and tumor-related adipocytes (10, 46–48). The importance of PPARγ expression in tumor cells for tumor pathophysiology is determined by the expression of the PPARγ receptor which varies dependent on tumor stage and histology n (49, 50). Receptor-independent activities of pioglitazone must be considered, additionally (26).
In contrast to most drugs used for tumor therapy, pioglitazone has an activating activity and modulates intra- and intercellular communication lines, including Wnt signaling (43). Contradictory results on pioglitazone concerning its activity derived from experimental studies are not astonishing as the cellular context and context-dependent interpretation of signals decisively guides the activity profile of pioglitazone, also in non-oncologic disease (26, 51). The dual activity of pioglitazone decisively extends the activity profile also in non-oncologic disease, as shown by the withdrawal of rosiglitazone, a specific PPARγ agonist (3, 52, 53). PPARα activation adds a strong anti-inflammatory effect (54, 55).
The role of PPARα/γ expression in tumor cells for response to tumor tissue editing approaches remains open, especially as metronomic chemotherapy may enhance PPARγ expression in stress response to metronomic chemotherapy (56). For example, in r/r cholangiocarcinoma, r/r Hodgkin’s lymphoma (HL) with a principally weak PPARγ expression, cCR may be achieved with editing schedules including pioglitazone, in contrast, in r/r non-small cell lung cancer with commonly relative intensive expression, only partial remission (21, 22, 39, 49). In r/r metastatic melanoma significant improved PFS was observed for patients with high PPARγ expression in respective tumor probes (49, 50).
The interaction of pioglitazone with interferon-α, dexamethasone or all-trans retinoic acid has been recently described (26). Type I interferons show synergistic antiproliferative activity if combined with glitazones in pancreatic cancer cell lines (57). In the two indicated phase II trials on RCCC, starting dose of interferon-α was only 4.5 million U thrice weekly with scheduled de-escalation dependent on tolerability as compared to the approved dose of up to 18 million U thrice weekly in monotherapy for RCCC (58). ATRA and pioglitazone show synergistic activity in AML in vitro and in vivo (30, 34, 36). The glucocorticoid receptor functions in a combinatorial manner with PPARα/γ by reprogramming and integrating local and systemic responses to inflammation (59). How the immune response of glucocorticoids and pioglitazone is modified by metronomic low dose chemotherapy cannot be pinned down, yet. Metronomic chemotherapy may additionally improve immunesurveillance, as shown by the combination of immune checkpoint inhibitors in a randomized phase III trial (60).
Tables 2, 3 outline the major therapy elements of tissue editing approaches including pioglitazone that have been introduced for unlocking tumor phenotypes in r/r neoplasias. How do single drugs without significant monoactivity contribute to tumor tissue editing approaches?
Drug interactions may be considered in a traditional way. Steel et al. introduced the term ‘coalism’ for drugs that are not active alone, or active in ‘cooperation’ if the combined effect is directed on a range of biologic systems (61, 62), (63). This applies for pioglitazone. In the next step, the targeted biologic systems, and their target profiles available for reprogramming hallmarks of cancer, are of pivotal interest.
Anakoinosis outlines a novel systems-therapeutic anticancer treatment paradigm, the therapeutic unlocking and exploitation of tumor specific phenotypes for controlling r/r metastatic disease by reprogramming cancer hallmarks and ‘normalizing’ dysbalanced tumor tissue homeostasis. The selected editing techniques, and on tumor site the specific patterns of pro-anakoinotically druggable communicative tissue networks, and homeostatically balancing hallmarks of cancer, are determining the qualitative outcome of pro-anakoinotic reprogramming techniques (Table 3).
Still insufficiently evaluated are the tumor-specific network characteristics coordinating hallmarks of cancer or the key parameters determining the specific relevance of distinct hallmarks in the systems context, and the systems-biologic prerequisites how to specifically unlock the tumor-promoting phenotypes. Therefore, it is only possible to draw on an effect-based description of anakoinosis, that records quantitative and qualitative changes in tumor phenotypes, here inflammation control, differentiation induction, and clinical outcome parameters, such as long-term disease control, CR and cCR in metastatic r/r neoplasias (28).
For the assessment of drug interactions in tumor tissue editing schedules, it is decisive that the single drugs have no or limited monoactivity, that CR or cCR induction with single components of the editing schedules may be excluded in r/r neoplasias, and that the tissue targets are communicatively linked tumor networks whose network characteristics must be readjusted at multiple heterogeneous localizations in case of metastatic r/r neoplasias. Thus, each therapeutic element, metronomic low-dose chemotherapy/azacitidine, and dual/triple transcriptional modulation pro-anakoinotically contributes to reprogram hallmarks of cancer for induction of tumor cell death, and for achieving M-CRAC control by re-integration of on-topic edited hallmarks in the tumor systems context (Table 3) (11, 40, 64–67).
General side effects of pioglitazone are weight gain and fluid retention (2). Therefore, creatinine at inclusion had to be <132.6 µmol/L and serum albumin >25g/L (39). Only in rare cases pioglitazone had to be discontinued due to fluid retention (68). Hypoglycemia did not occur in normoglycemic patients. In case of patients with diabetes mellitus, the additional anti-diabetic medication was successfully adapted. Elevation of serum creatinine occurred if pioglitazone was combined with a cyclooxygenease-2 inhibitor (2, 69).
Scheduled dose reductions were intended for each drug within the respective editing schedules. If clinically necessary, the COX-2 inhibitor was discontinued after preceding scheduled dose reduction. Also, patients with reduction of pioglitazone to 15 mg daily during treatment achieved CR in r/r neoplasia. Therefore, the initial dose of 60mg pioglitazone in early editing trials was reduced to 45mg as starting dose (39, 70). The minimal active dose of pioglitazone could not be evaluated. Fluid retention during additional dexamethasone treatment caused scheduled dose reduction of dexamethasone in the first step. Discontinuation of pioglitazone treatment due to pioglitazone related side effects led to study termination in rare cases (68). Urothelial carcinoma was not observed during pioglitazone treatment and follow-up (Table 1).
All discussed contributions on PPARα/γ activation with pioglitazone are to be considered conditionally, even if interactions with other cytokines or nuclear transcription factors have been studied experimentally (59). All these dual or triple transcriptional modulation therapies alone, would not induce CR or cCR in r/r neoplasias. However, induction of tumor stress response with low dose chemotherapy in addition to tumor specific transcriptional modulation facilitates control of r/r neoplasias (Figure 1).
M-CRAC re-establishes tumor promoting hallmarks of cancer, particularly, following maximized apoptosis induction with pulsed tumor therapies. Tumor cell death inducing therapies promote inflammation, hypoxia, ROS production and may additionally activate the Phoenix rising - caspase-3- cytosolic phospholipase A (2) alpha (cPLA-2)-COX-2-PGE-2-STAT3 pathway to reestablish compensatory tumor regrowth by establishing all disease traits described with M-CRAC (12, 71–73).
As countermeasure, therapeutic PPARα/γ activation reprograms in context with the concerted activity profile of tumor tissue editing approaches hallmarks of cancer, represses important transcription factors, like STAT3, NF-kB, AP-1, PI3K/Akt, HIF1α and NFAT and decreases the expression of TNF-α, TGF-β, IL-6, IL-8, VEGF, iNOS, as indicated by preclinical data (40, 74–76).
PPAR α and γ are positioned at crossing points between lipid metabolism and transcription, balancing and reciprocally cross-linking developmental homeostatic processes, that are established between classic cancer hallmarks, e.g., between tumor cell differentiation and immune surveillance, or immune response and the inflammatory status (26, 77–79). The expression profiles of PPARs in tumor tissues under changing phenotypic conditions are not well studied, yet (56).
M-CRAC control with pioglitazone was feasible independently of the chosen editing procedure, that was either directed at inflammation control or differentiation induction. Responses to edited inflammation control comprised cCR in metastatic r/r RCCC, mLCH and HL and long-term tumor control, even without objective response, e.g., in uveal melanoma (21–23, 30, 80). Therefore, M-CRAC control with pioglitazone provides a unique treatment quality of tumor tissue editing schedules.
Pioglitazone, both the PPARα and PPARγ agonistic component, inhibit carcinogenesis, tumor progression, proliferative capacity of metastasis-initiating stem cells, migration, invasion and remodel the extracellular matrix and angiogenesis (3, 77, 81). Attenuation of the Wnt/β-Catenin signaling, and reduction of the non-canonical NF-κB activity contribute to the M-CRAC inhibiting profile (43, 82–86) (Figure 1).
Clinically, pioglitazone inhibits colony-formation of stem cells in chronic myelocytic leukemia and may induce differentiation via CD44, an epitope, frequently expressed on cancer stem cells (63). Metabolic dependencies between tumor cells and the adjacent microenvironment promote heterogeneous metabolic phenotypes during development of therapy resistance and metastases. Metabolic reprogramming with glitazones might lower in experimental models the efficacy of the metastatic process (87–90).
The addition of pioglitazone in tumor tissue editing schedules may control metastatic spread in histologically different r/r tumor treated. In >60% of patients with tumor progression following tumor tissue editing, progression took place at the original tumor sites (21).
Restoring the expression of tumor suppressors, such as PTEN with PPARγ agonists attenuates tumor repopulation, prevents residual tumor cells and aggressivity of tumor cells (86, 91) (Figure 1). Simultaneously, PPARγ agonists reduce the activity of PI3K/Akt pathway and down-regulate Bcl-2 (92).
Less recognized is the potent immunoregulatory role of PPARγ regarding all immune cells that contributes to improved immune surveillance and growth attenuation (93–96) (Figure 1). PPARγ agonists are decisively shaping the molecular phenotype of the whole T-cell repertoire, of macrophages, and dendritic cells inclusively their communicative behavior (97, 98) and favor the M2 phenotype of macrophages which is associated with the expression of TGF-β, that is involved in M-CRAC promotion (99, 100). In all subtypes of T cells and macrophages, PPARγ agonists regulate the expression of genes involved in lipid metabolism and transport, e.g., the class B scavenger receptor CD36, besides FABP4, LXRA, and PGAR (101–103). Differentiation induction in non-PML AML by including pioglitazone could alter the antigenicity of leukemia cells (30, 34).
Enhanced PPARγ-mediated lipid antigen presentation facilitates the activation of iNKT cells (76, 104–107). Promoted by PPARγ agonists, fatty acid up-take and oxygenation may derepress effector T-cells and favor immunologic tumor response (108, 108).
PPARα and PPARγ belong to ferroptosis related genes (109). Clinical trials on tumor editing, including PPARalpha/gamma agonists have shown that apoptosis resistance may be overcome by reprogramming cancer hallmarks. Apoptosis may be bypassed via differentiation induction (30). Pro-apoptotic effects of PPARγ ligands have been proven by multiple pre-clinical studies (110). PPARγ activation decreases the expression of cyclin D1, thereby stopping the cytosolic β-catenin accumulation and may induce G2/M cell cycle arrest (111).
Pioglitazone alone may not induce clinically relevant differentiation (84). Nevertheless, pioglitazone contributes to balance proliferation and differentiation. Highly differential pathways may be involved in differentiation induction dependent on tumor histology, favoring the use of combined biomodulatory therapies including pioglitazone (Figure 1) (30, 112, 113).
Tumor cell death, and hypoxia following apoptosis inducing therapies contribute to a phenotype favoring drug resistance (114–116). In the first step phenotypic alterations of tumor and stroma cells arise on epigenetic basis (117, 118). PPARγ activation may normalize epigenetic and transcriptional regulation related to altered lipid metabolism (119, 120). The development of genetic resistance is based on genetic instability of tumor cells.
By attenuating the detrimental effects of pro-inflammatory cytokines, pioglitazone contributes to avoid the development of resistance in a concerted approach with other biomodulators (121). Further, therapeutic attenuation of Wnt signaling may be an important approach for resolving resistance (122).
In a clinical trial on r/r multiple myeloma the addition of pioglitazone in IMiD resistant disease resolved IMiD resistance while continuing IMiD therapy combined with pioglitazone (123). Experimentally and clinically studied is the method to overcome imatinib resistance in CML, defined as no achievement of MRD negativity. By targeting CML stem cells and the STAT system, pioglitazone even allows the discontinuation of the combination therapy pioglitazone, imatinib (124, 125).
The unimpeded passage of pioglitazone through the blood-brain barrier must be suggested, as cCR may be achieved in multisystem Langerhans cell histiocytosis (mLCH) and cerebral involvement (23, 126).
Huge timely and spatially diversified activity profiles of pioglitazone in heterogeneously constituted tumor cell niches might be expected when considering the context-dependent activity profiles of pioglitazone, as indicated by pre-clinical data (26, 127, 128). In contrast, concertedly targeting cancer-associated hallmarks by reprogramming techniques including pioglitazone may induce cCR, even in r/r HL, although all HL patients received prior local irradiation (21–23, 80). Control of obstacles given by molecular-genetic/genetic tumor cell and stroma heterogeneity in metastatic tumor disease, is facilitated by the concerted action of metronomic low-dose chemotherapy that provides important prerequisites for the activity profile of dual/triple transcriptional modulation with pioglitazone (21–23, 44).
Metronomic low dose chemotherapy plus pioglitazone alone or in combination with additional transcriptional modulators, i.e., dexamethasone, ATRA, or interferon-α contributes to therapeutically unlocking the tumor tissues’ phenotype via anakoinosis, causing CR and cCR and long-term tumor control in r/r neoplasias by attenuating tumor-promoting inflammation, differentiation induction, M-CRAC control, and non-oncogene addiction in edited tumor tissue (Tables 1–3; Figure 1) (21–23, 25–28). Trials on tumor tissue editing were performed as indicated in Table 1 (21–23, 30, 35, 38, 39, 68, 70, 80, 123, 129–134).
Control of tumor-promoting inflammation could be achieved with histology adapted and individually designed editing protocols in four histologically quite different r/r metastatic neoplasias, melanoma, RCCC, hepatocellular carcinoma, mLCH and HL (Table 3) (22, 23, 37, 38).
In a randomized comparison for r/r metastatic melanoma, the addition of a COX-2 inhibitor for inflammation control to metronomic low-dose chemotherapy emerged significantly inferior compared to intensified anti-inflammatory therapy with COX-2 inhibitor plus pioglitazone concerning PFS (Table 3) (38, 135). OS in r/r metastatic melanoma was significantly correlated with CRP serum response (38, 136). In a preceding trial including pioglitazone, one cCR was reported in r/r melanoma (137). COX2/PPARγ tissue immunoreactivity significantly increases stage-dependently from primary melanoma to metastases. Strong PPARγ immunoreactivity in melanoma cells was associated with improved PFS in retrospective analysis (50). Further, it could be shown that improvement of ECOG status and cachexia control in melanoma patients may be mediated by pioglitazone/COX-2-related disruption of platelet derived aberrant serum protein and lipid crosstalk between lipolysis of fat tissue and muscle wasting associated oxidative stress, that are both mediating cachexia (138, 139).
Intensification of anti-inflammatory therapy with low-dose interferon-α in addition to pioglitazone in a trial on r/r metastatic RCCC led to early CRP serum response in objective responders, also in patients with delayed CR (Tables 2, 3) (21, 35, 37, 140–144). In contrast to stable disease (SD) as best response in the preceding trial without low-dose interferon-α, cCRs were achieved with intensified inflammation control (35, 37, 145). Interferon-α, prednisone and 5-FU are only moderately active in RCCC (142).
A strong inflammation-triggered tumor promotion via activation of the nuclear factor kappa B (NFκB) pathway is well known in mLCH (146, 147). Pioglitazone plus dexamethasone induced early CRP response in some patients followed by cCR, even in case of cerebral involvement (21, 23).
The editing schedule for Hodgkin’s lymphomas was identical to the schedule used in r/r mLCH but supplemented by an mTOR inhibitor (Table 2). Response to editing therapy was closely related to serum CRP response (148–150). In contrast to mLCH, only the addition of everolimus to the mLCH schedule induced cCR in r/r HL (22, 23, 151, 152). Importantly, mTOR inhibition has been reported to be inefficacious in addition to pulsed chemotherapy (Table 2) (22, 151, 153). Editing non-oncogene addiction for successful clinical access of mTOR inhibition might also enhance immunosurveillance in r/r HL (154).
By adding temsirolimus to the pioglitazone arm of the melanoma editing schedule, long-term melanoma control was achieved in patients with extensive liver metastases of uveal melanoma by efficacious M-CRAC control (79, 80, 155).
Differentiation induction in leukemias with driver mutation is suggested to be locked for therapeutic reprogramming. The prototype for successful differentiation induction is the PML. PML may be controlled by the classic drug, ATRA. In case of ATRA therapy for PML, only the combined use with an additional drug, e.g., arsenic trioxide, may induce cCR (156). In another disease with a typical driver mutation, chronic myelocytic leukemia (CML), pioglitazone combined with imatinib may overcome minimal residual disease in patients not achieving molecular CR (124).
Differentiation induction in non-PML AML without typical driver mutation Also non-PML AMLs without actionable mutation are accessible for differentiation induction. R/r non-PML AML patients may achieve molecular-genetic or hematologic remission with azacitidine, ATRA and pioglitazone (Tables 2, 3) (30, 34). In the experimental setting, only the addition of pioglitazone to the combined editing approach facilitates differentiation and regain of phagocytic activity of differentiated, neutrophil-like blasts in vitro (36).
Moreover, blasts differentiated to neutrophil-like cells regain in vivo phagocytic activity and may resolve during study treatment prior to study medication acquired pneumonia. The clinical observation impressively demonstrates the generally low-toxicity range of tissue editing approaches and the regain of functionality by differentiation induction in AML blasts (30, 34).
Differentiation induction with pioglitazone in tumors In an animal model on breast cancer, it could be shown that the combination of pioglitazone with a MEK inhibitor may induce tumor differentiation, transdifferentiation of tumor cells to adipocytes by epithelial-mesenchymal transission (42, 157).
Thus, editing techniques including pioglitazone may induce specific types of differentiation in quite different neoplasias. However, editing schedules must be specifically adapted to tumor histology. Although, pioglitazone is discussed as apoptosis or ferroptosis inducer, differentiation inducing editing schedules may bypass common cell death pathways via differentiation (30) Figure 1.
Due to treatment failure to an inflammation suppressing editing schedule including pioglitazone, an individual therapeutic adaption was successfully performed in refractory mLCH by the addition of low-dose interferon-α to pioglitazone and discontinuation of dexamethasone (Tables 2, 3) (22). The adaption was considered due to the observed strong inflammation control of interferon-α and pioglitazone, administered in a RCCC editing schedule. In the mLCH schedule, only the cytotoxic drug of metronomic chemotherapy was substituted by a continuously administered alkylating agent, trofosfamide, instead of capecitabine, as used in the RCCC trial. Adaption of the editing schedule led to cCR, here, in a little child with severe multisystem involvement of refractory LCH, including LCH mediated severe bone marrow and liver failure, disease traits, which were primarily intended for combined liver and bone marrow transplantation (22, 37).
Histology-related accessibility of editing approaches controlling inflammation or inducing differentiation and obviously shared phenotype-maintaining ligand-receptor cross-talks among histologically quite different tumors show that therapeutic editing including pioglitazone for reprogramming tumor hallmarks is tumor-type agnostic, independently of the editing strategy, inflammation control or differentiation induction (Table 2).
CR or cCR induction with dual/triple transcriptional modulation in histologically quite different r/r neoplasias without driver mutation was only possible in combination with metronomic low-dose chemotherapy (11, 63, 156).
The possibility for unlocking tumor phenotypes via the tumor-type agnostic activity of pioglitazone with differential recombinations of additional transcriptional modulators underlines the novel unique activity profile of low-dose metronomic chemotherapy, namely, providing pleiotropic cancer hallmark-related tumor tissue responses, i.e., altered cytokine and transcriptional repertoires in tumor tissues, including altered PPARγ expression, as prerequisite for specified tumor tissue editing with ligands of nuclear transcription factors or cytokine receptors (21–23, 25, 37, 56, 158–160) (Table 3). The fact that induction of cCR was possible also in cases with scheduled dose reductions up to > 66% of the metronomic starting dose, irrespectively of the used cytotoxic drug, trofosfamide, treosulfan or capecitabine, underlines that apoptosis induction could not be the primary purpose of low-dose metronomic chemotherapy (Table 2) (21–23, 37). Importantly, at the end of a 3 or 4-week cycle the trials did not achieve the cumulative dose of corresponding pulsed chemotherapy schedules, and scheduled dose reductions were frequently performed, in some patients primarily, due to multiple preceding therapies (Tables 2, 3) (21–23, 30, 38, 44, 56, 80, 161, 162).
Drawing on observations in biology, the clinical data on low-dose metronomic chemotherapy suggest that pleiotropic stress responses to metronomic chemotherapy limit tumor tissue plasticity, probably decrease functional heterogeneity of tumor cell niches, as tissue stress generally induces a tighter phenotype. Metronomic chemotherapy could promote via stress response phenotypic integration of inflammation or differentiation within editing schedules, and consecutively may serve as an enhancer of pro-anakoinotic effects induced by combined treatment with nuclear receptor agonists or cytokines (Figure 1) (26, 162–164).
In diseases with driver mutation, such as chronic myelocytic leukemia (CML) the addition of pioglitazone to imatinib is sufficient to eliminate minimal residual disease (63, 124). Metronomic chemotherapy is no prerequisite for the biomodulatory activity of pioglitazone in CML.
Whether a MEK inhibitor plus pioglitazone, probably in combination with metronomic chemotherapy, is sufficient to control metastatic breast cancer in humans via differentiation induction must be further evaluated (42).
Metronomic low-dose chemotherapy or epigenetic modeling with azacitidine combined with differential recombinations of pioglitazone with transcriptional modulators initiate tumor-specific reprogramming of cancer hallmarks, i.e., anakoinosis, here exemplified by tumor-associated inflammation control or differentiation induction. Thus, tissue editing techniques disclose that the wide-ranging functions of PPARα/γ agonists in tumor tissues may be selectively focused on differential reprogramming patterns of cancer hallmarks, induction of tumor cell death, and on facilitating edited non-oncogene addiction. Now, pioglitazone reaches clinical relevance in oncology if combined with appropriate additional biomodulators, even in relapsed or refractory neoplasias (Figure 1).
Therapeutic control of M-CRAC in r/r neoplasias affords tumor tissue editing with metronomic low dose chemotherapy and histology-adapted dual or triple transcriptional modulation, particularly in case of r/r tumors without driver mutation and complex genetic aberrations. By selecting histology-specific editing protocols tumor phenotypes may be specifically unlocked with respective dual/triple transcriptional modulation. Clinically, main therapeutic emphasis may be, as shown, either differentiation induction, enhancement of immunosurveillance, metabolic reprogramming or inflammation control.
Thus, editing procedures repurpose the function of metronomic low-dose chemotherapy and add an important, yet less considered activity profile of metronomic chemotherapy, induction of stress response in tumor tissues as prerequisite for combined transcriptional editing (Figure 1) (56).
Editing techniques reveal that tumor-specific networks of ligand-receptor cross-talks for maintaining tumor phenotypes provide unique tumor systems characteristics. Tumor phenotype-maintaining cross-talks facilitate concerted ‘targeted’ pro-anakoinotic tumor systems access. Anakoinosis may be initiated by induction of tumor stress response combined with dual/triple transcriptional modulation including pioglitazone.
Drugability of tumor phenotypes for therapeutically exploiting tumor plasticity is in line with experimental data derived from Zebrafish models showing that e.g., the re-establishment of embryonic microenvironment may determine tumor cell fate (165).
Editing of cancer hallmarks with pioglitazone may directly induce tumor cell death or alternatively, provides novel access for tumor cell death induction via edited non-oncogene addiction (21–23, 38, 80). However, editing may also facilitate control of metastasis, post-therapy cancer repopulation and acquired cell-resistance and genetic/molecular-genetic tumor cell heterogeneity (M-CRAC) (11). That means tumor promoting hallmarks remain long-term silenced, as shown in uveal melanoma (80). These differential response patterns reveal that on-topic edited hallmarks of cancer are both, differentially constituted and interconnected within the pattern of cancer hallmarks, therefore, specifically accessible with recombinations of transcriptional modulators. The differential activity profiles of pioglitazone in on-topic editing approaches highlights a further individual tumor characteristic, namely, the specific communicative integration of distinct cancer hallmarks within the whole pattern of hallmarks (22, 23, 30, 38, 163).
The unlocking technique of tumor phenotypes with pioglitazone plus/minus additional transcriptional modulators must be currently selected according to tumor histology. However, ligand-receptor cross-talks, maintaining cancer hallmarks, may be shared among quite different tumor histologies revealing a tumor-type agnostic therapeutic access of pioglitazone across different histologic tumor types, e.g., melanoma, renal clear cell carcinoma, Langerhans cell histiocytosis, Hodgkin’s lymphoma and acute myelocytic leukemia (166).
Tissue editing approaches, and therapies including maximum tolerable doses to induce maximized apoptosis induction, seem to be mutually exclusive, as exemplified from the literature for mTOR inhibitors in r/rHL, interferon-α in RCCC or epigenetic modifiers (21, 22, 30, 37, 118, 153, 167–169).
Metronomic low-dose chemotherapy provides prerequisites for the therapeutic dual/triple transcriptional modulation to unfold combined transcriptional systems activity in tumors. The configuration of cancer hallmarks, their ‘integration quality’, i.e., how hallmarks of cancer are communicatively cross-linked, and their particular importance within the tumor systems context specify outcome of tumor tissue editing and the context-dependent therapeutic use of pioglitazone. Monitoring these tumor-promoting hallmark qualities may specify tumor tissue editing and also the context-dependent therapeutic use of pioglitazone (22, 23, 30, 38, 163).
Basic research is now challenged to provide data on network-based ligand-receptor cross-talks in tumor tissue compartments for specifying editing schedules according to the individual configuration and integration of hallmarks, to evaluate the most suitable pro-anakoinotically druggable hallmarks and to exploit the most powerful activity profiles for pioglitazone to finally efficaciously reprogram the tumor tissue’s network-based transcriptional ‘Achilles heel’ (44, 163, 170).
Experimental data derived from tumor tissues show that hallmarks of cancer are in homeostatic balance by spatially organized, multicellular inflammatory and immune modulatory hubs and that cellular heterogeneity in tumor tissues is not at random (171, 172). These hubs might serve as organized access for tumor tissue editing schedules, also for pioglitazone as immune modulator, inflammation inhibitor, metabolic regulator and differentiation inducer (172). Particularly, proteomic platforms may enable to test differentiation induction or inflammation control and to uncover ways for edited non-oncogene addiction in a highly personalized manner (20, 29, 173).
Integration of pioglitazone in tissue editing approaches marks the starting point for concertedly targeting the communicative context of multiple tumor cell compartments and their tumor-typical networks of ligand-receptor cross-talks for systematically controlling metastatic r/r neoplasias up to cCR. The pro-anakoinotic active magic bullets including pioglitazone unlock the tumor phenotype, thereby addressing urgent therapeutic problems, such as functional, topographic, and genetic heterogeneity of tumor and stroma cells, tumor cell dormancy, dynamically changing phenotypic competences of tumor cell niches, and M-CRAC (24, 36, 44, 127, 174–176). Alongside, tumor tissue editing with pioglitazone may induce tumor cell differentiation. As differentiated blasts gain phagocytic competence, a broad variety of cell death pathways might be induced in those cells, similar to the repertoire of cell death pathways in granulocytes, including necroptosis and pyroptosis with disintegration of the cellular membrane and non-lytic apoptosis or NETosis (24, 36, 44, 127, 174–177).
The therapeutic paradigm change, namely, evolving tumor phenotypes with tumor-type agonistic tumor tissue editing approaches including pioglitazone in therapeutically stressed tumor tissue, provides a novel expedient technique for controlling r/r metastatic tumors of quite different histologic origin (178).
DH: Conceptualization, Writing – original draft, Writing – review & editing. FL: Conceptualization, Writing – original draft, Writing – review & editing. TP: Conceptualization, Writing – review & editing. LG: Conceptualization, Writing – review & editing. CG: Conceptualization, Writing – review & editing. AR: Conceptualization, Writing – original draft, Writing – review & editing. DCH: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Liao H-W, Saver JL, Wu Y-L, Chen T-H, Lee M, Ovbiagele B. Pioglitazone and cardiovascular outcomes in patients with insulin resistance, pre-diabetes and type 2 diabetes: a systematic review and meta-analysis. BMJ Open (2017) 7:e013927. doi: 10.1136/bmjopen-2016-013927
2. Bansal G, Thanikachalam PV, Maurya RK, Chawla P, Ramamurthy S. An overview on medicinal perspective of thiazolidine-2,4-dione: A remarkable scaffold in the treatment of type 2 diabetes. J Adv Res (2020) 23:163–205. doi: 10.1016/j.jare.2020.01.008
3. Wagner N, Wagner K-D. Peroxisome proliferator-activated receptors and the hallmarks of cancer. Cells (2022) 11. doi: 10.3390/cells11152432
4. Walczak K, Gerkowicz A, Krasowska D. PPARs and the kynurenine pathway in melanoma—Potential biological interactions. IJMS (2023) 24:3114. doi: 10.3390/ijms24043114
5. Raman P, Koenig RJ. Pax-8-PPAR-γ fusion protein in thyroid carcinoma. Nat Rev Endocrinol (2014) 10:616–23. doi: 10.1038/nrendo.2014.115
6. Ondrey F. Peroxisome proliferator-activated receptor gamma pathway targeting in carcinogenesis: implications for chemoprevention. Clin Cancer Res (2009) 15:2–8. doi: 10.1158/1078-0432.CCR-08-0326
7. Fenner MH, Elstner E. Peroxisome proliferator-activated receptor-gamma ligands for the treatment of breast cancer. Expert Opin Investig Drugs (2005) 14:557–68. doi: 10.1517/13543784.14.6.557
8. Vitale SG, Laganà AS, Nigro A, La Rosa VL, Rossetti P, Rapisarda AM, et al. Peroxisome proliferator-activated receptor modulation during metabolic diseases and cancers: master and minions. PPAR Res (2016) 2016:6517313. doi: 10.1155/2016/6517313
9. Papaetis GS. Pioglitazone, bladder cancer, and the presumption of innocence. Curr Drug Saf (2022) 17:294–318. doi: 10.2174/1574886317666220304124756
10. Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JY, Tan NS. Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. IJMS (2019) 20. doi: 10.3390/ijms20205055
11. Heudobler D, Ghibelli L, Reichle A. Editorial: Anakoinosis for promoting tumor tissue editing: Novel therapeutic opportunities for establishing clinically relevant tumor control by targeting tumor plasticity. Front Oncol (2022) 12:1005381. doi: 10.3389/fonc.2022.1005381
12. Corsi F, Capradossi F, Pelliccia A, Briganti S, Bruni E, Traversa E, et al. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of phoenix rising in prostate cancer. IJMS (2022) 23. doi: 10.3390/ijms23031152
13. Gökbuget N, Zugmaier G, Dombret H, Stein A, Bonifacio M, Graux C, et al. Curative outcomes following blinatumomab in adults with minimal residual disease B-cell precursor acute lymphoblastic leukemia. Leuk Lymphoma (2020) 61:2665–73. doi: 10.1080/10428194.2020.1780583
14. Saha S, Pradhan N BN, Mahadevappa R, Minocha S, Kumar S. Cancer plasticity: Investigating the causes for this agility. Semin Cancer Biol (2023) 88:138–56. doi: 10.1016/j.semcancer.2022.12.005
15. McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol (2014) 16:717–27. doi: 10.1038/ncb3015
16. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
17. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell (2009) 136:823–37. doi: 10.1016/j.cell.2009.02.024
18. Tao Z, Wu X. Targeting transcription factors in cancer: from "Undruggable" to "Druggable". Methods Mol Biol (2023) 2594:107–31. doi: 10.1007/978-1-0716-2815-7_9
19. Dzobo K, Senthebane DA, Dandara C. The tumor microenvironment in tumorigenesis and therapy resistance revisited. Cancers (Basel) (2023) 15. doi: 10.3390/cancers15020376
20. Lüke F, Harrer DC, Pantziarka P, Pukrop T, Ghibelli L, Gerner C, et al. Drug repurposing by tumor tissue editing. Front Oncol (2022) 12:900985. doi: 10.3389/fonc.2022.900985
21. Reichle A, Vogt T. Systems biology: a therapeutic target for tumor therapy. Cancer Microenviron (2008) 1:159–70. doi: 10.1007/s12307-008-0012-5
22. Lüke F, Harrer DC, Menhart K, Wolff D, Holler E, Hellwig D, et al. Biomodulatory treatment regimen, MEPED, rescues relapsed and refractory classic hodgkin's disease. Front Pharmacol (2021) 12:599561. doi: 10.3389/fphar.2021.599561
23. Harrer DC, Jakob M, Vogelhuber M, Lüke F, Utpatel K, Corbacioglu S, et al. Biomodulatory therapy induces durable remissions in multi-system Langerhans cell histiocytosis. Leuk Lymphoma (2022) 63:2858–68. doi: 10.1080/10428194.2022.2095627
24. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
25. Cazzaniga ME, Cordani N, Capici S, Cogliati V, Riva F, Cerrito MG. Metronomic chemotherapy. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13092236
26. Heudobler D, Rechenmacher M, Lüke F, Vogelhuber M, Pukrop T, Herr W, et al. Peroxisome proliferator-activated receptors (PPAR)γ Agonists as master modulators of tumor tissue. IJMS (2018) 19. doi: 10.3390/ijms19113540
27. Heudobler D, Rechenmacher M, Lüke F, Vogelhuber M, Klobuch S, Thomas S, et al. Clinical efficacy of a novel therapeutic principle, anakoinosis. Front Pharmacol (2018) 9:1357. doi: 10.3389/fphar.2018.01357
28. Heudobler D, Lüke F, Vogelhuber M, Klobuch S, Pukrop T, Herr W, et al. Anakoinosis: correcting aberrant homeostasis of cancer tissue-going beyond apoptosis induction. Front Oncol (2019) 9:1408. doi: 10.3389/fonc.2019.01408
29. Kleszcz R. Advantages of the combinatorial molecular targeted therapy of head and neck cancer—A step before anakoinosis-based personalized treatment. Cancers (Basel) (2023) 15:4247. doi: 10.3390/cancers15174247
30. Heudobler D, Klobuch S, Lüke F, Hahn J, Grube M, Kremers S, et al. Low-dose azacitidine, pioglitazone and all-trans retinoic acid versus standard-dose azacitidine in patients ≥ 60 years with acute myeloid leukemia refractory to standard induction chemotherapy (AMLSG 26-16/AML-viVA): results of the safety run-in phase I. Blood (2019) 134:1382. doi: 10.1182/blood-2019-129977
31. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
32. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science (2013) 339:286–91. doi: 10.1126/science.1232227
33. Chen Y-L, Chang M-C, Cheng W-F. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett (2017) 400:282–92. doi: 10.1016/j.canlet.2017.01.040
34. Heudobler D, Luke F, Hahn J, Grube M, Schlosser P, Kremers S, et al. Low-dose azacitidine, pioglitazone and all-trans retinoic acid is safe in patients aged ≥ 60 years with acute myeloid leukemia refractory to standard induction chemotherapy (AMLSG 26-16/AML-ViVA): results of the safety run-in phase. Haematologica (2023). doi: 10.3324/haematol.2023.283864
35. Reichle A, Grassinger J, Bross K, Wilke J, Suedhoff T, Walter B, et al. C-reactive protein in patients with metastatic clear cell renal carcinoma: an important biomarker for tumor-associated inflammation. biomark Insights (2007) 1:87–98.
36. Klobuch S, Steinberg T, Bruni E, Mirbeth C, Heilmeier B, Ghibelli L, et al. Biomodulatory treatment with azacitidine, all-trans retinoic acid and pioglitazone induces differentiation of primary AML blasts into neutrophil like cells capable of ROS production and phagocytosis. Front Pharmacol (2018) 9:1380. doi: 10.3389/fphar.2018.01380
37. Walter B, Schrettenbrunner I, Vogelhuber M, Grassinger J, Bross K, Wilke J, et al. Pioglitazone, etoricoxib, interferon-α, and metronomic capecitabine for metastatic renal cell carcinoma: final results of a prospective phase II trial. Med Oncol (2012) 29:799–805. doi: 10.1007/s12032-011-9982-0
38. Reichle A, Vogt T, Coras B, Terheyden P, Neuber K, Trefzer U, et al. Targeted combined anti-inflammatory and angiostatic therapy in advanced melanoma: a randomized phase II trial. Melanoma Res (2007) 17:360–4. doi: 10.1097/CMR.0b013e3282f1d2c8
39. Heudobler D, Schulz C, Fischer JR, Staib P, Wehler T, Südhoff T, et al. A randomized phase II trial comparing the efficacy and safety of pioglitazone, clarithromycin and metronomic low-dose chemotherapy with single-agent nivolumab therapy in patients with advanced non-small cell lung cancer treated in second or further line (ModuLung). Front Pharmacol (2021) 12:599598. doi: 10.3389/fphar.2021.599598
40. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
41. Hou J, Karin M, Sun B. Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age? Nat Rev Clin Oncol (2021) 18:261–79. doi: 10.1038/s41571-020-00459-9
42. Ishay-Ronen D, Diepenbruck M, Kalathur RK, Sugiyama N, Tiede S, Ivanek R, et al. Gain fat-lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell (2019) 35:17–32.e6. doi: 10.1016/j.ccell.2018.12.002
43. Vallée A, Lecarpentier Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol (2018) 9:745. doi: 10.3389/fimmu.2018.00745
44. Pagliuca C, Di Leo L, Zio D. New insights into the phenotype switching of melanoma. Cancers (Basel) (2022) 14. doi: 10.3390/cancers14246118
45. Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM, et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature (2022) 603:166–73. doi: 10.1038/s41586-022-04437-2
46. Cheng HS, Yip YS, Lim EK, Wahli W, Tan NS. PPARs and tumor microenvironment: the emerging roles of the metabolic master regulators in tumor stromal-epithelial crosstalk and carcinogenesis. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13092153
47. Gionfriddo G, Plastina P, Augimeri G, Catalano S, Giordano C, Barone I, et al. Modulating tumor-associated macrophage polarization by synthetic and natural PPARγ Ligands as a potential target in breast cancer. Cells (2020) 9. doi: 10.3390/cells9010174
48. Kennel KB, Bozlar M, de Valk AF, Greten FR. Cancer-associated fibroblasts in inflammation and anti-tumor immunity. Clin Cancer Res (2022). doi: 10.1158/1078-0432.CCR-22-1031
49. Meyer S, Vogt T, Landthaler M, Berand A, Reichle A, Bataille F, et al. Cyclooxygenase 2 (COX2) and peroxisome proliferator-activated receptor gamma (PPARG) are stage-dependent prognostic markers of Malignant melanoma. In: Reichle A, editor. From Molecular to Modular Tumor Therapy. Dordrecht: Springer Netherlands (2010). p. 433–65.
50. Meyer S, Vogt T, Landthaler M, Berand A, Reichle A, Bataille F, et al. Cyclooxygenase 2 (COX2) and peroxisome proliferator-activated receptor gamma (PPARG) are stage-dependent prognostic markers of Malignant melanoma. PPAR Res (2009) 2009:848645. doi: 10.1155/2010/848645
51. de Bosscher K, Desmet SJ, Clarisse D, Estébanez-Perpiña E, Brunsveld L. Nuclear receptor crosstalk - defining the mechanisms for therapeutic innovation. Nat Rev Endocrinol (2020) 16:363–77. doi: 10.1038/s41574-020-0349-5
52. Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet (2009) 373:2125–35. doi: 10.1016/S0140-6736(09)60953-3
53. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med (2016) 374:1321–31. doi: 10.1056/NEJMoa1506930
54. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev (2018) 39:760–802. doi: 10.1210/er.2018-00064
55. Tan Y, Wang M, Yang K, Chi T, Liao Z, Wei P. PPAR-α Modulators as current and potential cancer treatments. Front Oncol (2021) 11:599995. doi: 10.3389/fonc.2021.599995
56. Doloff JC, Waxman DJ. Transcriptional profiling provides insights into metronomic cyclophosphamide-activated, innate immune-dependent regression of brain tumor xenografts. BMC Cancer (2015) 15:375. doi: 10.1186/s12885-015-1358-y
57. Dicitore A, Caraglia M, Gaudenzi G, Manfredi G, Amato B, Mari D, et al. Type I interferon-mediated pathway interacts with peroxisome proliferator activated receptor-γ (PPAR-γ): at the cross-road of pancreatic cancer cell proliferation. Biochim Biophys Acta (2014) 1845:42–52. doi: 10.1016/j.bbcan.2013.11.003
58. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med (2007) 356:2271–81. doi: 10.1056/NEJMoa066838
59. Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell (2005) 122:707–21. doi: 10.1016/j.cell.2005.06.029
60. Patil VM, Noronha V, Menon N, Rai R, Bhattacharjee A, Singh A, et al. Low-dose immunotherapy in head and neck cancer: A randomized study. JCO (2023) 41:222–32. doi: 10.1200/JCO.22.01015
61. Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys (1979) 5:85–91. doi: 10.1016/0360-3016(79)90044-0
62. Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect (2015) 3:e00149. doi: 10.1002/prp2.149
63. Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature (2015) 525:380–3. doi: 10.1038/nature15248
64. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell (2011) 147:992–1009. doi: 10.1016/j.cell.2011.11.016
65. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer (2018) 18:533–48. doi: 10.1038/s41568-018-0038-z
66. Welch DR, Hurst DR. Defining the hallmarks of metastasis. Cancer Res (2019) 79:3011–27. doi: 10.1158/0008-5472.CAN-19-0458
67. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
68. Walter I, Schulz U, Vogelhuber M, Wiedmann K, Endlicher E, Klebl F, et al. Communicative reprogramming non-curative hepatocellular carcinoma with low-dose metronomic chemotherapy, COX-2 inhibitor and PPAR-gamma agonist: a phase II trial. Med Oncol (2017) 34:192. doi: 10.1007/s12032-017-1040-0
69. Reinhold SW, Reichle A, Leiminger S, Bergler T, Hoffmann U, Krüger B, et al. Renal function during rofecoxib therapy in patients with metastatic cancer: retrospective analysis of a prospective phase II trial. BMC Res Notes (2011) 4:2. doi: 10.1186/1756-0500-4-2
70. Vogt T, Hafner C, Bross K, Bataille F, Jauch K-W, Berand A, et al. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced Malignant vascular tumors. Cancer (2003) 98:2251–6. doi: 10.1002/cncr.11775
71. Labrie M, Brugge JS, Mills GB, Zervantonakis IK. Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat Rev Cancer (2022) 22:323–39. doi: 10.1038/s41568-022-00454-5
72. Morana O, Wood W, Gregory CD. The apoptosis paradox in cancer. IJMS (2022) 23. doi: 10.3390/ijms23031328
73. Khalaf K, Hana D, Chou JT-T, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the tumor microenvironment involved in immune resistance and drug resistance. Front Immunol (2021) 12:656364. doi: 10.3389/fimmu.2021.656364
74. Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol (2013) 33 Suppl 1:S79–84. doi: 10.1007/s10875-012-9847-0
75. Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature (2015) 520:368–72. doi: 10.1038/nature14336
76. Hernandez-Quiles M, Broekema MF, Kalkhoven E. PPARgamma in metabolism, immunity, and cancer: unified and diverse mechanisms of action. Front Endocrinol (Lausanne) (2021) 12:624112. doi: 10.3389/fendo.2021.624112
77. Deyell M, Garris CS, Laughney AM. Cancer metastasis as a non-healing wound. Br J Cancer (2021) 124:1491–502. doi: 10.1038/s41416-021-01309-w
78. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
79. Falletta P, Goding CR, Vivas-García Y. Connecting metabolic rewiring with phenotype switching in melanoma. Front Cell Dev Biol (2022) 10:930250. doi: 10.3389/fcell.2022.930250
80. Hart C, Vogelhuber M, Hafner C, Landthaler M, Berneburg M, Haferkamp S, et al. Biomodulatory metronomic therapy in stage IV melanoma is well-tolerated and may induce prolonged progression-free survival, a phase I trial. J Eur Acad Dermatol Venereol (2016) 30:e119–21. doi: 10.1111/jdv.13391
81. Liu H, Zang C, Fenner MH, Possinger K, Elstner E. PPARgamma ligands and ATRA inhibit the invasion of human breast cancer cells. vitro. Breast Cancer Res Treat (2003) 79:63–74. doi: 10.1023/a:1023366117157
82. Ninomiya I, Yamazaki K, Oyama K, Hayashi H, Tajima H, Kitagawa H, et al. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol Lett (2014) 8:2709–14. doi: 10.3892/ol.2014.2553
83. Takano S, Kubota T, Nishibori H, Hasegawa H, Ishii Y, Nitori N, et al. Pioglitazone, a ligand for peroxisome proliferator-activated receptor-gamma acts as an inhibitor of colon cancer liver metastasis. Anticancer Res (2008) 28:3593–9.
84. Chi T, Wang M, Wang X, Yang K, Xie F, Liao Z, et al. PPAR-γ Modulators as current and potential cancer treatments. Front Oncol (2021) 11:737776. doi: 10.3389/fonc.2021.737776
85. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature (2017) 550:402–6. doi: 10.1038/nature24050
86. Teresi RE, Shaiu C-W, Chen C-S, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by Lovastatin and Rosiglitazone. Int J Cancer (2006) 118:2390–8. doi: 10.1002/ijc.21799
87. Gottfried E, Rogenhofer S, Waibel H, Kunz-Schughart LA, Reichle A, Wehrstein M, et al. Pioglitazone modulates tumor cell metabolism and proliferation in multicellular tumor spheroids. Cancer Chemother Pharmacol (2011) 67:117–26. doi: 10.1007/s00280-010-1294-0
88. Nascentes Melo LM, Lesner NP, Sabatier M, Ubellacker JM, Tasdogan A. Emerging metabolomic tools to study cancer metastasis. Trends Cancer (2022) 8:988–1001. doi: 10.1016/j.trecan.2022.07.003
89. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368. doi: 10.1126/science.aaw5473
90. Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer (2022) 22:157–73. doi: 10.1038/s41568-021-00427-0
91. Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature (2011) 476:163–9. doi: 10.1038/nature10275
92. Shiau C-W, Yang C-C, Kulp SK, Chen K-F, Chen C-S, Huang J-W, et al. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Res (2005) 65:1561–9. doi: 10.1158/0008-5472.CAN-04-1677
93. Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell (1998) 93:229–40. doi: 10.1016/S0092-8674(00)81574-3
94. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med (2001) 7:48–52. doi: 10.1038/83336
95. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med (2001) 7:53–8. doi: 10.1038/83348
96. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi: 10.1038/nature05894
97. Vogt T, Coras B, Hafner C, Landthaler M, Reichle A. Antiangiogenic therapy in metastatic prostate carcinoma complicated by cutaneous lupus erythematodes. Lancet Oncol (2006) 7:695–7. doi: 10.1016/S1470-2045(06)70798-7
98. Gehrmann M, Brunner M, Pfister K, Reichle A, Kremmer E, Multhoff G. Differential up-regulation of cytosolic and membrane-bound heat shock protein 70 in tumor cells by anti-inflammatory drugs. Clin Cancer Res (2004) 10:3354–64. doi: 10.1158/1078-0432.CCR-03-0382
99. Goswami KK, Bose A, Baral R. Macrophages in tumor: An inflammatory perspective. Clin Immunol (2021) 232:108875. doi: 10.1016/j.clim.2021.108875
100. van Ginderachter JA, Meerschaut S, Liu Y, Brys L, de Groeve K, Hassanzadeh Ghassabeh G, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood (2006) 108:525–35. doi: 10.1182/blood-2005-09-3777
101. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell (1998) 93:241–52. doi: 10.1016/s0092-8674(00)81575-5
102. Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med (1998) 188:1359–68. doi: 10.1084/jem.188.7.1359
103. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond) (2018) 38:27. doi: 10.1186/s40880-018-0301-4
104. Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity (2004) 21:95–106. doi: 10.1016/j.immuni.2004.06.003
105. Szatmari I, Pap A, Rühl R, Ma J-X, Illarionov PA, Besra GS, et al. PPARγ controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Cell Biol (2006) 175:i1–1. doi: 10.1083/JCB1751OIA1
106. Szatmari I, Töröcsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood (2007) 110:3271–80. doi: 10.1182/blood-2007-06-096222
107. Nencioni A, Grünebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. Dendritic cell immunogenicity is regulated by peroxisome proliferator-activated receptor gamma. J Immunol (2002) 169:1228–35. doi: 10.4049/jimmunol.169.3.1228
108. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res (2018) 6:1375–87. doi: 10.1158/2326-6066.CIR-18-0095
109. Venkatesh D, O'Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev (2020) 34:526–43. doi: 10.1101/gad.334219.119
110. Trindade-da-Silva CA, Reis CF, Vecchi L, Napimoga MH, Sperandio M, Matias Colombo BF, et al. 15-deoxy-Δ(12,14)-prostaglandin J2 induces apoptosis and upregulates SOCS3 in human thyroid cancer cells. PPAR Res (2016) 2016:4106297. doi: 10.1155/2016/4106297
111. Kitamura S, Miyazaki Y, Hiraoka S, Nagasawa Y, Toyota M, Takakura R, et al. PPARgamma agonists inhibit cell growth and suppress the expression of cyclin D1 and EGF-like growth factors in ras-transformed rat intestinal epithelial cells. Int J Cancer (2001) 94:335–42. doi: 10.1002/ijc.1470
112. Zhang X, Zhang S, Wang T. How the mechanical microenvironment of stem cell growth affects their differentiation: a review. Stem Cell Res Ther (2022) 13:415. doi: 10.1186/s13287-022-03070-0
113. Bar-Hai N, Ishay-Ronen D. Engaging plasticity: Differentiation therapy in solid tumors. Front Pharmacol (2022) 13:944773. doi: 10.3389/fphar.2022.944773
114. Zhao Y-Z, Liu X-L, Shen G-M, Ma Y-N, Zhang F-L, Chen M-T, et al. Hypoxia induces peroxisome proliferator-activated receptor γ expression via HIF-1-dependent mechanisms in HepG2 cell line. Arch Biochem Biophys (2014) 543:40–7. doi: 10.1016/j.abb.2013.12.010
115. Wang M, Li G, Yang Z, Wang L, Zhang L, Wang T, et al. Uncoupling protein 2 downregulation by hypoxia through repression of peroxisome proliferator-activated receptor γ promotes chemoresistance of non-small cell lung cancer. Oncotarget (2017) 8:8083–94. doi: 10.18632/oncotarget.14097
116. Xu R, Luo X, Ye X, Li H, Liu H, Du Q, et al. SIRT1/PGC-1α/PPAR-γ Correlate with hypoxia-induced chemoresistance in non-small cell lung cancer. Front Oncol (2021) 11:682762. doi: 10.3389/fonc.2021.682762
117. Chang L, Guo R, Huang Q, Yen Y. Chromosomal instability triggered by Rrm2b loss leads to IL-6 secretion and plasmacytic neoplasms. Cell Rep (2013) 3:1389–97. doi: 10.1016/j.celrep.2013.03.040
118. Kumar VE, Nambiar R, de Souza C, Nguyen A, Chien J, Lam KS. Targeting epigenetic modifiers of tumor plasticity and cancer stem cell behavior. Cells (2022) 11. doi: 10.3390/cells11091403
119. Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med (2018) 10. doi: 10.1126/scitranslmed.aao0303
120. Aouali N, Broukou A, Bosseler M, Keunen O, Schlesser V, Janji B, et al. Epigenetic activity of peroxisome proliferator-activated receptor gamma agonists increases the anticancer effect of histone deacetylase inhibitors on multiple myeloma cells. PloS One (2015) 10:e0130339. doi: 10.1371/journal.pone.0130339
121. Zhang Y, Guo H, Zhang Z, Lu W, Zhu J, Shi J. IL-6 promotes chemoresistance via upregulating CD36 mediated fatty acids uptake in acute myeloid leukemia. Exp Cell Res (2022) 415:113112. doi: 10.1016/j.yexcr.2022.113112
122. Barzegar Behrooz A, Talaie Z, Jusheghani F, Łos MJ, Klonisch T, Ghavami S. Wnt and PI3K/akt/mTOR survival pathways as therapeutic targets in glioblastoma. IJMS (2022) 23. doi: 10.3390/ijms23031353
123. Heudobler D, Elger T, Mayer S, Hart C, Vogelhuber M, Grube M, et al. Biomodulatory therapy approach with lenalidomide in combination with pioglitazone, dexamethasone, and metronomic low-dose chemotherapy with treosulfan in patients with relapsed/refractory multiple myeloma > second-line. JCO (2019) 37:8037. doi: 10.1200/JCO.2019.37.15_suppl.8037
124. Rousselot P, Prost S, Guilhot J, Roy L, Etienne G, Legros L, et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer (2017) 123:1791–9. doi: 10.1002/cncr.30490
125. Li Y-J, Zhang C, Martincuks A, Herrmann A, Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer (2023). doi: 10.1038/s41568-022-00537-3
126. Reichle A, Vogt T, Kunz-Schughart L, Bretschneider T, Bachthaler M, Bross K, et al. Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans' cell histiocytosis of adults. Br J Haematol (2005) 128:730–2. doi: 10.1111/j.1365-2141.2004.05359.x
127. Karras P, Bordeu I, Pozniak J, Nowosad A, Pazzi C, van Raemdonck N, et al. A cellular hierarchy in melanoma uncouples growth and metastasis. Nature (2022) 610:190–8. doi: 10.1038/s41586-022-05242-7
128. Hossain SM, Eccles MR. Phenotype switching and the melanoma microenvironment; impact on immunotherapy and drug resistance. IJMS (2023) 24. doi: 10.3390/ijms24021601
129. Reichle A, Lugner A, Ott C, Klebl F, Vogelhuber M, Berand A, et al. Control of cancer-associated inflammation and survival: Results from a prospective randomized phase II trial in gastric cancer. JCO (2009) 27:e15584–4. doi: 10.1200/jco.2009.27.15_suppl.e15584
131. Hau P, Kunz-Schughart L, Bogdahn U, Baumgart U, Hirschmann B, Weimann E, et al. Low-dose chemotherapy in combination with COX-2 inhibitors and PPAR-gamma agonists in recurrent high-grade gliomas - a phase II study. Oncology (2007) 73:21–5. doi: 10.1159/000120028
132. Walter B, Rogenhofer S, Vogelhuber M, Berand A, Wieland WF, Andreesen R, et al. Modular therapy approach in metastatic castration-refractory prostate cancer. World J Urol (2010) 28:745–50. doi: 10.1007/s00345-010-0567-x
133. Vogelhuber M, Feyerabend S, Stenzl A, Suedhoff T, Schulze M, Huebner J, et al. Biomodulatory treatment of patients with castration-resistant prostate cancer: A phase II study of imatinib with pioglitazone, etoricoxib, dexamethasone and low-dose treosulfan. Cancer Microenviron (2015) 8:33–41. doi: 10.1007/s12307-014-0161-7
134. Hart C, Vogelhuber M, Wolff D, Klobuch S, Ghibelli L, Foell J, et al. Anakoinosis: communicative reprogramming of tumor systems - for rescuing from chemorefractory neoplasia. Cancer Microenviron (2015) 8:75–92. doi: 10.1007/s12307-015-0170-1
135. Sun W-H, Chen G-S, Ou X-L, Yang Y, Luo C, Zhang Y, et al. Inhibition of COX-2 and activation of peroxisome proliferator-activated receptor gamma synergistically inhibits proliferation and induces apoptosis of human pancreatic carcinoma cells. Cancer Lett (2009) 275:247–55. doi: 10.1016/j.canlet.2008.10.023
136. Fang S, Sui D, Wang Y, Liu H, Chiang Y-J, Ross MI, et al. Association of vitamin D levels with outcome in patients with melanoma after adjustment for C-reactive protein. JCO (2016) 34:1741–7. doi: 10.1200/JCO.2015.64.1357
137. Reichle A, Bross K, Vogt T, Bataille F, Wild P, Berand A, et al. Pioglitazone and rofecoxib combined with angiostatically scheduled trofosfamide in the treatment of far-advanced melanoma and soft tissue sarcoma. Cancer (2004) 101:2247–56. doi: 10.1002/cncr.20574
138. Muqaku B, Eisinger M, Meier SM, Tahir A, Pukrop T, Haferkamp S, et al. Multi-omics analysis of serum samples demonstrates reprogramming of organ functions via systemic calcium mobilization and platelet activation in metastatic melanoma. Mol Cell Proteomics (2017) 16:86–99. doi: 10.1074/mcp.M116.063313
139. Aguilar-Cazares D, Chavez-Dominguez R, Marroquin-Muciño M, Perez-Medina M, Benito-Lopez JJ, Camarena A, et al. The systemic-level repercussions of cancer-associated inflammation mediators produced in the tumor microenvironment. Front Endocrinol (Lausanne) (2022) 13:929572. doi: 10.3389/fendo.2022.929572
140. Jabs WJ, Busse M, Krüger S, Jocham D, Steinhoff J, Doehn C. Expression of C-reactive protein by renal cell carcinomas and unaffected surrounding renal tissue. Kidney Int (2005) 68:2103–10. doi: 10.1111/j.1523-1755.2005.00666.x
141. Angelo LS, Talpaz M, Kurzrock R. Autocrine interleukin-6 production in renal cell carcinoma: evidence for the involvement of p53. Cancer Res (2002) 62:932–40.
142. Haarstad H, Jacobsen AB, Schjølseth SA, Risberg T, Fosså SD. Interferon-alpha, 5-FU and prednisone in metastatic renal cell carcinoma: a phase II study. Ann Oncol (1994) 5:245–8. doi: 10.1093/oxfordjournals.annonc.a058801
143. Kerr C. Inflammatory response predicts survival in renal cancer. Lancet Oncol (2006) 7:284. doi: 10.1016/s1470-2045(06)70632-5
144. Enocsson H, Sjöwall C, Skogh T, Eloranta M-L, Rönnblom L, Wetterö J. Interferon-alpha mediates suppression of C-reactive protein: explanation for muted C-reactive protein response in lupus flares? Arthritis Rheum (2009) 60:3755–60. doi: 10.1002/art.25042
145. Abuhelwa AY, Bellmunt J, Kichenadasse G, McKinnon RA, Rowland A, Sorich MJ, et al. C-reactive protein provides superior prognostic accuracy than the IMDC risk model in renal cell carcinoma treated with Atezolizumab/Bevacizumab. Front Oncol (2022) 12:918993. doi: 10.3389/fonc.2022.918993
146. Brown RE. The NF-kappaB pathway and the successful application of anti-inflammatory and angiostatic therapy in Langerhans' cell histiocytosis. Br J Haematol (2005) 130:147–8. doi: 10.1111/j.1365-2141.2005.05575.x
147. Haroche J, Cohen-Aubart F, Rollins BJ, Donadieu J, Charlotte F, Idbaih A, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol (2017) 18:e113–25. doi: 10.1016/S1470-2045(17)30031-1
148. Ugocsai P, Wolff D, Menhart K, Hellwig D, Holler E, Herr W, et al. Biomodulatory metronomic therapy induces PET-negative remission in chemo- and brentuximab-refractory Hodgkin lymphoma. Br J Haematol (2016) 172:290–3. doi: 10.1111/bjh.13480
149. Herishanu Y, Perry C, Braunstein R, Metser U, Goor O, Rogowski O, et al. Early-mid treatment C-reactive protein level is a prognostic factor in aggressive non-Hodgkin's lymphoma. Eur J Haematol (2007) 79:150–4. doi: 10.1111/j.1600-0609.2007.00894.x
150. Calabretta E, d'Amore F, Carlo-Stella C. Immune and inflammatory cells of the tumor microenvironment represent novel therapeutic targets in classical hodgkin lymphoma. IJMS (2019) 20. doi: 10.3390/ijms20215503
151. Johnston PB, Inwards DJ, Colgan JP, Laplant BR, Kabat BF, Habermann TM, et al. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol (2010) 85:320–4. doi: 10.1002/ajh.21664
152. Schmitt M, Ceteci F, Gupta J, Pesic M, Böttger TW, Nicolas AM, et al. Colon tumour cell death causes mTOR dependence by paracrine P2X4 stimulation. Nature (2022) 612:347–53. doi: 10.1038/s41586-022-05426-1
153. Gillessen S, Hüttmann A, Vucinic V, Müller H, Plütschow A, Viardot A, et al. Reinduction therapy with everolimus in combination with dexamethasone, high-dose cytarabin and cisplatinum in patients with relapsed or refractory classical Hodgkin lymphoma: an experimental phase I/II multicentre trial of the German Hodgkin Study Group (GHSG HD-R3i). Br J Haematol (2022) 196:606–16. doi: 10.1111/bjh.17878
154. Petroni G, Buqué A, Coussens LM, Galluzzi L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat Rev Drug Discovery (2022) 21:440–62. doi: 10.1038/s41573-022-00415-5
155. Carvajal RD, Sacco JJ, Jager MJ, Eschelman DJ, Olofsson Bagge R, Harbour JW, et al. Advances in the clinical management of uveal melanoma. Nat Rev Clin Oncol (2023) 20:99–115. doi: 10.1038/s41571-022-00714-1
156. Cicconi L, Divona M, Ciardi C, Ottone T, Ferrantini A, Lavorgna S, et al. PML-RARα kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia (2016) 30:1987–92. doi: 10.1038/leu.2016.122
157. Zhang Y, Donaher JL, Das S, Li X, Reinhardt F, Krall JA, et al. Genome-wide CRISPR screen identifies PRC2 and KMT2D-COMPASS as regulators of distinct EMT trajectories that contribute differentially to metastasis. Nat Cell Biol (2022) 24:554–64. doi: 10.1038/s41556-022-00877-0
158. Biziota E, Mavroeidis L, Hatzimichael E, Pappas P. Metronomic chemotherapy: A potent macerator of cancer by inducing angiogenesis suppression and antitumor immune activation. Cancer Lett (2017) 400:243–51. doi: 10.1016/j.canlet.2016.12.018
159. Bocci G, Nicolaou KC, Kerbel RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res (2002) 62:6938–43.
160. Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother (2012) 61:353–62. doi: 10.1007/s00262-011-1106-3
161. Du B, Waxman DJ. Medium dose intermittent cyclophosphamide induces immunogenic cell death and cancer cell autonomous type I interferon production in glioma models. Cancer Lett (2020) 470:170–80. doi: 10.1016/j.canlet.2019.11.025
162. Meng F, Henson R, Patel T. Chemotherapeutic stress selectively activates NF-kappa B-dependent AKT and VEGF expression in liver cancer-derived endothelial cells. Am J Physiol Cell Physiol (2007) 293:C749–60. doi: 10.1152/ajpcell.00537.2006
163. Matesanz S, Blanco-Sánchez M, Ramos-Muñoz M, Cruz M, Benavides R, Escudero A. Phenotypic integration does not constrain phenotypic plasticity: differential plasticity of traits is associated to their integration across environments. New Phytol (2021) 231:2359–70. doi: 10.1111/nph.17536
164. Tian X, Zhang S, Zhou L, Seyhan AA, Hernandez Borrero L, Zhang Y, et al. Targeting the integrated stress response in cancer therapy. Front Pharmacol (2021) 12:747837. doi: 10.3389/fphar.2021.747837
165. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit L-M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer (2007) 7:246–55. doi: 10.1038/nrc2108
166. Pestana RC, Serrano C. Analysis of histology-agnostic targets among soft tissue and bone sarcomas in the AACR GENIE database. Front Oncol (2022) 12:1079909. doi: 10.3389/fonc.2022.1079909
167. Bachy E, Camus V, Thieblemont C, Sibon D, Casasnovas R-O, Ysebaert L, et al. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the ro-CHOP phase III study (Conducted by LYSA). JCO (2022) 40:242–51. doi: 10.1200/JCO.21.01815
168. Canil C, Hotte S, Mayhew LA, Waldron TS, Winquist E. Interferon-alfa in the treatment of patients with inoperable locally advanced or metastatic renal cell carcinoma: a systematic review. Can Urol Assoc J (2010) 4:201–8. doi: 10.5489/cuaj.853
169. Kornblum N, Zhao F, Manola J, Klein P, Ramaswamy B, Brufsky A, et al. Randomized phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer resistant to aromatase inhibitor therapy: results of prE0102. JCO (2018) 36:1556–63. doi: 10.1200/JCO.2017.76.9331
170. Ghoshdastider U, Rohatgi N, Mojtabavi Naeini M, Baruah P, Revkov E, Guo YA, et al. Pan-cancer analysis of ligand-receptor cross-talk in the tumor microenvironment. Cancer Res (2021) 81:1802–12. doi: 10.1158/0008-5472.CAN-20-2352
171. Grünwald BT, Devisme A, Andrieux G, Vyas F, Aliar K, McCloskey CW, et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell (2021) 184:5577–5592.e18. doi: 10.1016/j.cell.2021.09.022
172. Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell (2021) 184:4734–4752.e20. doi: 10.1016/j.cell.2021.08.003
173. Meier-Menches SM, Neuditschko B, Janker L, Gerner MC, Schmetterer KG, Reichle A, et al. A proteomic platform enables to test for AML normalization. In Vitro. Front Chem (2022) 10:826346. doi: 10.3389/fchem.2022.826346
174. Francescangeli F, de Angelis ML, Rossi R, Cuccu A, Giuliani A, de Maria R, et al. Dormancy, stemness, and therapy resistance: interconnected players in cancer evolution. Cancer Metastasis Rev (2023). doi: 10.1007/s10555-023-10092-4
175. Househam J, Heide T, Cresswell GD, Spiteri I, Kimberley C, Zapata L, et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature (2022) 611:744–53. doi: 10.1038/s41586-022-05311-x
176. Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet (2019) 20:404–16. doi: 10.1038/s41576-019-0114-6
177. Singhal A, Kumar S. Neutrophil and remnant clearance in immunity and inflammation. Immunology (2022) 165:22–43. doi: 10.1111/imm.13423
Keywords: pioglitazone, interferon-α, dexamethasone, all-trans retinoic acid, tumor tissue editing, anakoinosis, transcriptional modulation, phenotypic plasticity
Citation: Harrer DC, Lüke F, Pukrop T, Ghibelli L, Gerner C, Reichle A and Heudobler D (2024) Peroxisome proliferator-activated receptorα/γ agonist pioglitazone for rescuing relapsed or refractory neoplasias by unlocking phenotypic plasticity. Front. Oncol. 13:1289222. doi: 10.3389/fonc.2023.1289222
Received: 05 September 2023; Accepted: 15 December 2023;
Published: 11 January 2024.
Edited by:
Daekyu Sun, University of Arizona, United StatesReviewed by:
Gabriele Multhoff, Technical University of Munich, GermanyCopyright © 2024 Harrer, Lüke, Pukrop, Ghibelli, Gerner, Reichle and Heudobler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albrecht Reichle, YWxicmVjaHQucmVpY2hsZUB1a3IuZGU=
†ORCID: Albrecht Reichle, orcid.org/0000-0002-1821-7887
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.