
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 07 November 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1273305
This article is part of the Research TopicInborn Errors of Immunity (IEI) Breaking Immune Homeostasis and Tolerance: A Key Role for T Regulatory CellsView all 8 articles
 Federico Scarmozzino1†Marco Pizzi1†*Filippo Pelizzaro2Valentina Angerilli1Angelo Paolo Dei Tos1Francesco Piazza3Edoardo Vincenzo Savarino2Fabiana Zingone2‡Matteo Fassan1,4‡
Federico Scarmozzino1†Marco Pizzi1†*Filippo Pelizzaro2Valentina Angerilli1Angelo Paolo Dei Tos1Francesco Piazza3Edoardo Vincenzo Savarino2Fabiana Zingone2‡Matteo Fassan1,4‡Refractory celiac disease (RCD) and enteropathy-associated T-cell lymphoma (EATL) are rare, yet severe complications of celiac disease (CD). Over the last decades, several studies have addressed the biology and clinical-pathological features of such conditions, highlighting unique disease patterns and recurrent genetic events. Current classification proposals identify two forms of RCD, namely: (i) type 1 RCD (RCD-I), characterized by phenotypically normal intra-epithelial lymphocytes (IELs); and (ii) type 2 RCD (RCD-II), featuring phenotypically aberrant IELs. While RCD-I likely represents a gluten-independent dysimmune reaction against small bowel epithelial cells, RCD-II is better considered an in situ aggressive T-cell lymphoma, with high rates of progression to overt EATL. The diagnosis of RCD and EATL is often challenging, due to misleading clinical-pathological features and to significant overlap with several CD-unrelated gastro-intestinal disorders. Similarly, the treatment of RCD and EATL is an unmet clinical need for both gastroenterologists and hematologists. Moving from such premises, this review aims to provide a comprehensive view of RCD and EATL, specifically considering their pathogenesis and the many still open issues concerning their diagnosis and clinical management.
Celiac disease (CD) is a T-cell mediated small intestinal autoimmune-like disease triggered by ingestion of gluten proteins in genetically susceptible individuals. CD is one of the most common autoimmune diseases, affecting approximately 1% of the Western population. Although CD can occur at virtually any age, most cases are diagnosed in children and young adults (1). Almost all CD patients carry one or both of the human leukocyte antigens (HLA) DQ2 and DQ8. Rare HLA-DQ2/DQ8-negative cases are positive for HLA-DQ7.5 (<1% of patients) (2, 3).
The clinical presentation of CD is broad, ranging from fully asymptomatic cases to very morbid conditions. Most symptoms are related to malabsorption, micronutrient deficiency and failure to thrive, as a result of intestinal mucosa damage by gluten-induced dysimmunity. Non-classical presentations can involve extra-gastro-intestinal (GI) sites and include neurological symptoms, endocrinopathies, cutaneous lesions, osteopenia and changes in reproductive function (4, 5). Finally, long-lasting and/or untreated CD can undergo severe complications including small bowel adenocarcinoma and an aggressive form of peripheral T-cell lymphoma, referred to as enteropathy-associated T-cell lymphoma (EATL) (6).
The diagnosis of CD rests on a combination of serologic testing and histological findings (1). Serological diagnosis requires the documentation of CD-specific auto-antibodies (auto-Ab) of either IgA or IgG class (i.e. anti-deamidated gliadin peptide [anti-DGP] auto-Ab; anti-tissue transglutaminase [anti-tTG] auto-Ab; anti-endomysial Ab [EMA]). Testing for IgA auto-Ab is routinely performed in the diagnostic workup of all suspected CD patients, while IgG auto-Ab are mainly tested in cases with selective IgA deficiency (5).
Duodenal biopsy should be performed in all adult patients with suspected CD and positive CD-specific auto-Ab. In cases with negative serology, histological examination is recommended only if clinical data are highly suspicious for CD. In children, duodenal biopsy can be avoided if high titers of IgA anti-tTG auto-Ab and EMA are detected (7). A minimum of 4 biopsy samples are required for histological evaluation (2 biopsies from the duodenal bulb and 2 from the second duodenal portion) (1). Biopsy samples should be correctly orientated (possibly on filter paper) and should contain ≥3–4 consecutive villi-crypt units (7). Histologically, the diagnosis of CD requires the documentation of increased intraepithelial lymphocytes (IELs) at duodenal biopsy (≥25 lymphocytes/100 epithelial cells) with variable degrees of villous atrophy and/or crypt hyperplasia. IELs typically disclose a ‘crescendo pattern’, whereby lymphocytes mostly locate in the upper two thirds of villous epithelium. Based on the severity of mucosal changes, CD is histologically graded according to Corazza-Villanacci and Marsh-Oberhuber schemes (8–11).
Once the diagnosis of CD is established, the only proven treatment consists in strict adhesion to life-long gluten-free diet (GFD) (7). In most cases, GFD leads to complete remission of clinical, serological and histological alterations, although this may take months to years to occur (12). Poor response to GFD is mainly due to poor patient compliance and/or inadvertent food contamination with gluten (13–16). In rare instances, however, GFD failure depends on CD-intrinsic factors, which are responsible of so-called refractory celiac disease (RCD).
According to international consensus reports, RCD is defined as any CD with clinical and histological unresponsiveness to ≥12 months of strict GFD (17, 18). This broad definition encompasses different types of RCD, with variable biological, clinical and prognostic features. As such, the diagnosis and sub-categorization of RCD is often challenging, and its management is still an unmet clinical need. Moreover, the boundaries between RCD and EATL are often blurred, likely as a result of the biological continuum between these entities.
Moving from such premises, this review aims at providing a comprehensive view of the pathogenesis and clinical-pathological features of RCD and EATL, specifically focusing on the most recent biological achievements and on their clinical implications.
According to a systematic review published in 2016, RCD has a prevalence of 0.3-0.4% and a cumulative incidence of 1-4% among CD patients (19). RCD usually affects adult to elderly patients, with most cases being diagnosed between 40 and 60 years of age (20, 21). Compared to GFD-responsive cases, RCD has a longer interval between onset of enteropathy-related symptoms and CD diagnosis, suggesting a direct role for protracted gluten exposure in the pathogenesis of RCD (22).
In the last decades, the incidence of RCD has progressively decreased, possibly as a result of timelier diagnoses of CD and of wider availability of gluten free products (22). Besides gluten exposure, the main risk factors for RCD include male gender and old age at diagnosis, classical symptomatic CD at presentation, negativity for CD-related auto-Ab at the time of diagnosis (23, 24).
RCD can be classified into two subtypes, depending on the immunophenotype of intraepithelial lymphocytes (IELs): (i) RCD type I (RCD-I), characterized by normal (surface CD3 [sCD3]+/CD8+) IELs; and (ii) RCD type II (RCD-II), characterized by phenotypically aberrant (sCD3-/cytoplasmic CD3 [cCD3]+/CD8-) IELs. In most studies, RCD-I occurs one decade earlier than RCD-II (mean age at diagnosis: 40-50 versus 50-60 years) (20–22, 25). . The proportion of RCD subtypes is inconsistent across series and remains largely undefined (26). The biological differences between RCD-I and RCD-II subtend relevant differences in terms of prognosis and treatment options.
Clinically, RCD-I and RCD-II present with symptoms of untreated CD, including long-lasting diarrhea, abdominal pain, weight loss, fatigue and malaise (21, 27). Symptom burden is usually worse in RCD-II, due to extensive bowel involvement (28) and mucosal ulcerations (20). Concurrent autoimmune/dysimmune diseases are frequently reported (e.g. Hashimoto’s thyroiditis; microscopic colitis; autoimmune hepatopathies), being slightly more common in RCD-II than RCD-I (20). Systemic symptoms (i.e. drenching night sweats, fever, and weight loss), small bowel strictures and occlusions are hallmark of EATL progression (20).
Laboratory tests typically disclose anemia, multiple vitamin deficiencies, chronic hyper-transaminasemia (21). The latter correlates with intestinal mucosal damage (20) more frequently reported in RCD-II than RCD-I (70% versus 21% of cases) (29). Although most patients have negative CD-specific antibodies at the time of RCD, positive auto-Ab does not necessarily exclude the diagnosis (20, 27). Compared to uncomplicated CD, RCD-I/RCD-II usually disclose higher Chromogranin A (CgA), β2-microglobulin (B2M) and lactate dehydrogenase (LDH) serum levels (30). B2M and LDH likely parallel lymphoid cell expansion, while CgA correlates with neuroendocrine cell hyperplasia (CgA) (31). As such, serum CgA, B2M and LDH testing may serve as cost-effective strategies for an early diagnosis of RCD.
The diagnosis of RCD is often challenging and, in most cases, one of exclusion (Figure 1). The first step in the diagnostic workup is confirming the original diagnosis of CD. This is usually achieved by re-evaluation of clinical, genetic and histological data, as well as by confirmation of CD-specific auto-Ab (17, 32).
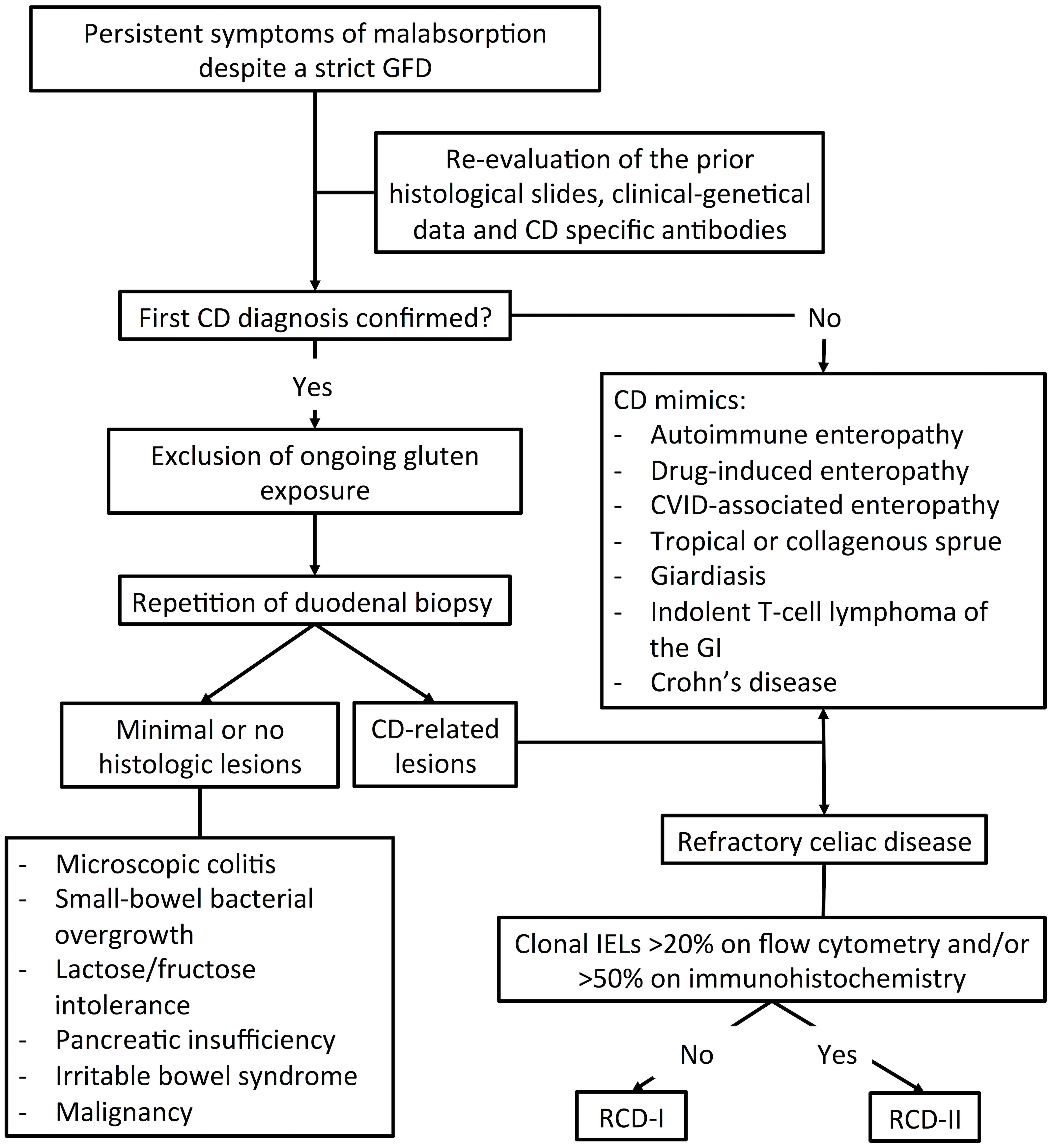
Figure 1 Diagnostic workup for Refracrory Celiac Disease (RCD).
Once the diagnosis of CD is confirmed, adherence to GFD should be carefully assessed. By far, the most common cause of symptom persistence in CD is ongoing gluten exposure with diet. This is documented in roughly 50% of patients with putative RCD (13–15) and should be investigated by dietary interview, testing for CD-specific auto-Ab and/or for gluten peptides in urine/stool samples (32–34). Persistence of anti-tTG auto-Ab and/or EMA should specifically raise concern of ongoing gluten exposure (20, 21).
If adherence to GFD is proven, endoscopic exams and biopsies should be repeated (2). The documentation of CD-related lesions suggests ongoing gluten exposure, RCD or any of its mimickers associated to villous atrophy (see paragraph 4.1) (2). However, if minimal or no microscopic changes are observed, other causes of abdominal discomfort should be considered (30), such as microscopic colitis, small-bowel bacterial overgrowth (SIBO), lactose intolerance, pancreatic insufficiency, or irritable bowel syndrome (32, 35, 36) (Figure 1). According to the latest ESGE guidelines, both a standard esophagogastroduodenoscopy (EGDS) and capsule endoscopy (CE) should be performed in non-responsive CD-patients after excluding gluten ingestion (37). CE allows exploring the small bowel beyond the Treitz ligament, where lesions suggestive of RCD-II and EATL often locate. In particular, the finding of ulcerative jejunitis and/or of large (≥1 cm) ulcerations should specifically raise concern for RCD-II or EATL (20). Further device-assisted enteroscopy (DAE) allows to obtain tissue samples for accurate diagnosis and subsequent treatment (37). Therefore, both standard EGDS and CE/DAE are crucial for patients suspected of having RCD. In all suitable cases, CE should be preceded by small bowel-directed radiological studies (e.g. CT or MR enterography) to detect intestinal strictures or wall thickening that may hamper endoscopic evaluation. Imaging studies may also aid disclosing abdominal masses and mesenteric lymphadenopathies (27, 38), heralding RCD-II and EATL-related complications (Table 1). Splenic atrophy is a further finding, especially in cases of RCD-II and EATL (39).
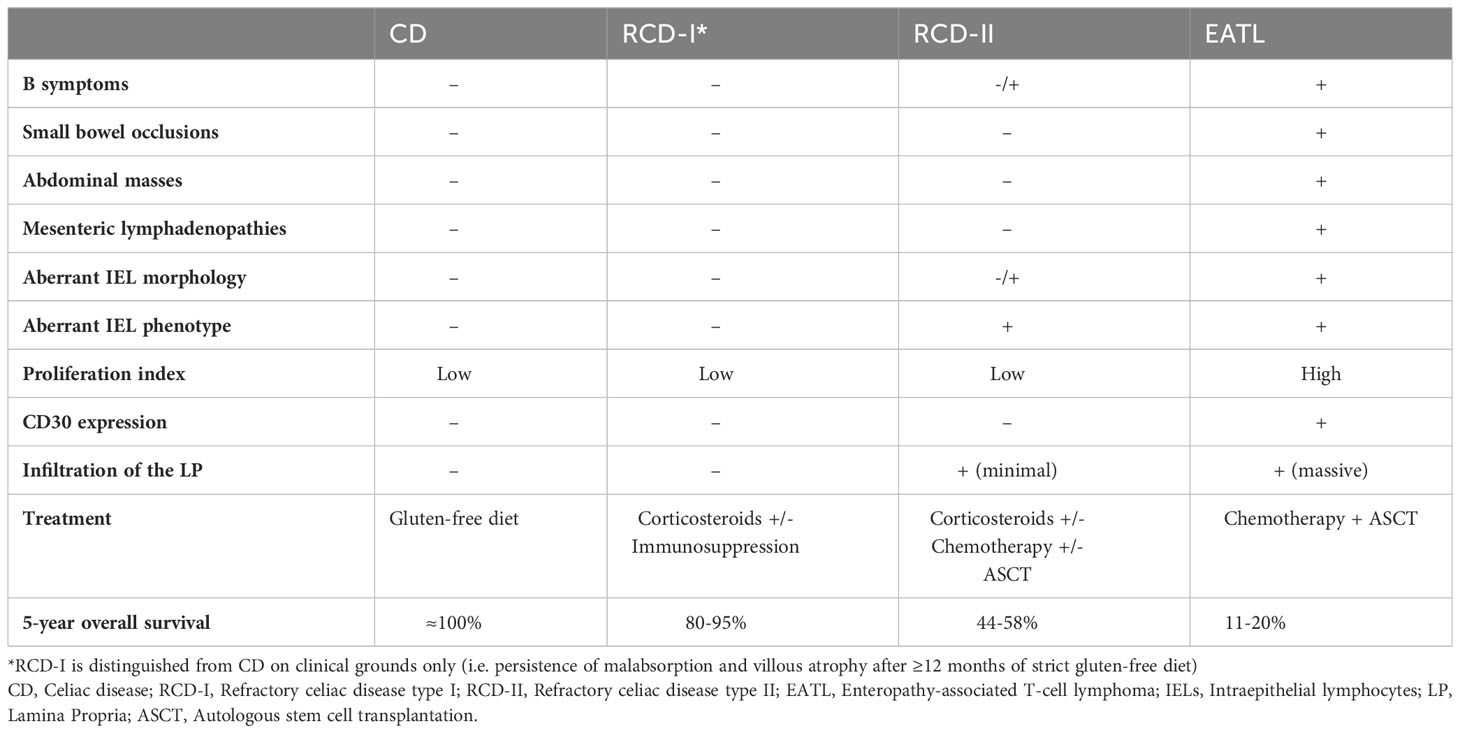
Table 1 Distinguishing features of CD, RCD-I, RCD-II, and EATL.
Endoscopic studies should be integrated with biopsy sampling and histological re-evaluation. A definite diagnosis of RCD can be made only when CD-related lesions are documented and all CD-mimickers are confidently excluded.
The treatment of RCD is challenging and varies depending on disease subtype. Nutritional support and corticosteroids (i.e. open capsule budesonide or prednisone) are the first line therapies for RCD-I, being associated with clinical improvement in most cases (2, 20, 40, 41). The 2019 European Society for the Study of Coeliac Disease (ESsCD) guidelines recommend adding immunosuppressive drugs such as thiopurines (specifically azathioprine or 6-Thioguanine) following a response to steroids, as this may lead to better healing of histological lesions. If the patient responds, annual follow-up with endoscopic exams and biopsies should be performed. If not, dosage of thiopurines should be optimized or the diagnosis of RCD-I should be carefully reconsidered (2).
In RCD-II, steroids are also recommended as first-line therapy (32, 40), being associated with a favourable clinical response (20). Second line therapies generally include multimodality chemotherapy (e.g. cladribine, pentostatine, or fludarabine) to eliminate the aberrant RCD-II IELs. If symptoms worsen, high-dose chemotherapy followed by autologous stem cell transplantation (ASCT) is recommended (2, 42–44). The latter shows high response rates (85% of cases) with 4-year overall survival of 66% (45, 46).
The prognosis of RCD varies depending on disease subtype. In general, RCD-II fares much worse than RCD-I (5-year survival rates: 80-95% in RCD-I; 44–58% in RCD-II), due to the severity of the clinical picture and to the higher risk of EATL evolution (21, 25, 47). In fact, RCD-II can be regarded as an aggressive in situ T-cell lymphoma of the GI tract (i.e. “in situ EATL”), closely related and rapidly progressing to overt EATL (48). Malignancies and starvation represent the main causes of death among RCD-II patients (17).
Over the last decades, several studies have explored the biology and pathophysiology of RCD. It is now clear that RCD-I and RCD-II are very different diseases, sharing a common antigenic trigger and following different pathogenic pathways. This may explain the different epidemiology, clinical features and outcome of RCD-I and RCD-II patients.
The pathophysiology of RCD-I is largely unexplored. Like RCD-II, some environmental factors may be associated with RCD-I, such as poor adherence to a GFD (49, 50) and viral infections. The mechanisms of such association are still hypothetical, yet viral infections may increase the production of type I interferon, thus promoting the proliferation of CD8+ T-cells and natural killer (NK) cells, either directly or via the induction of IL-15. This, in turn, may foster anti-gluten immunological reactions, prompting their evolution to a fully autoimmune (i.e. gluten-independent) disease (26, 51). This scenario may exist also for other environmental and/or host-related factors, but further studies are needed to investigate this possibility.
In the last years, several studies have disclosed genetic and immunological determinants of RCD-II and EATL (Figure 2).
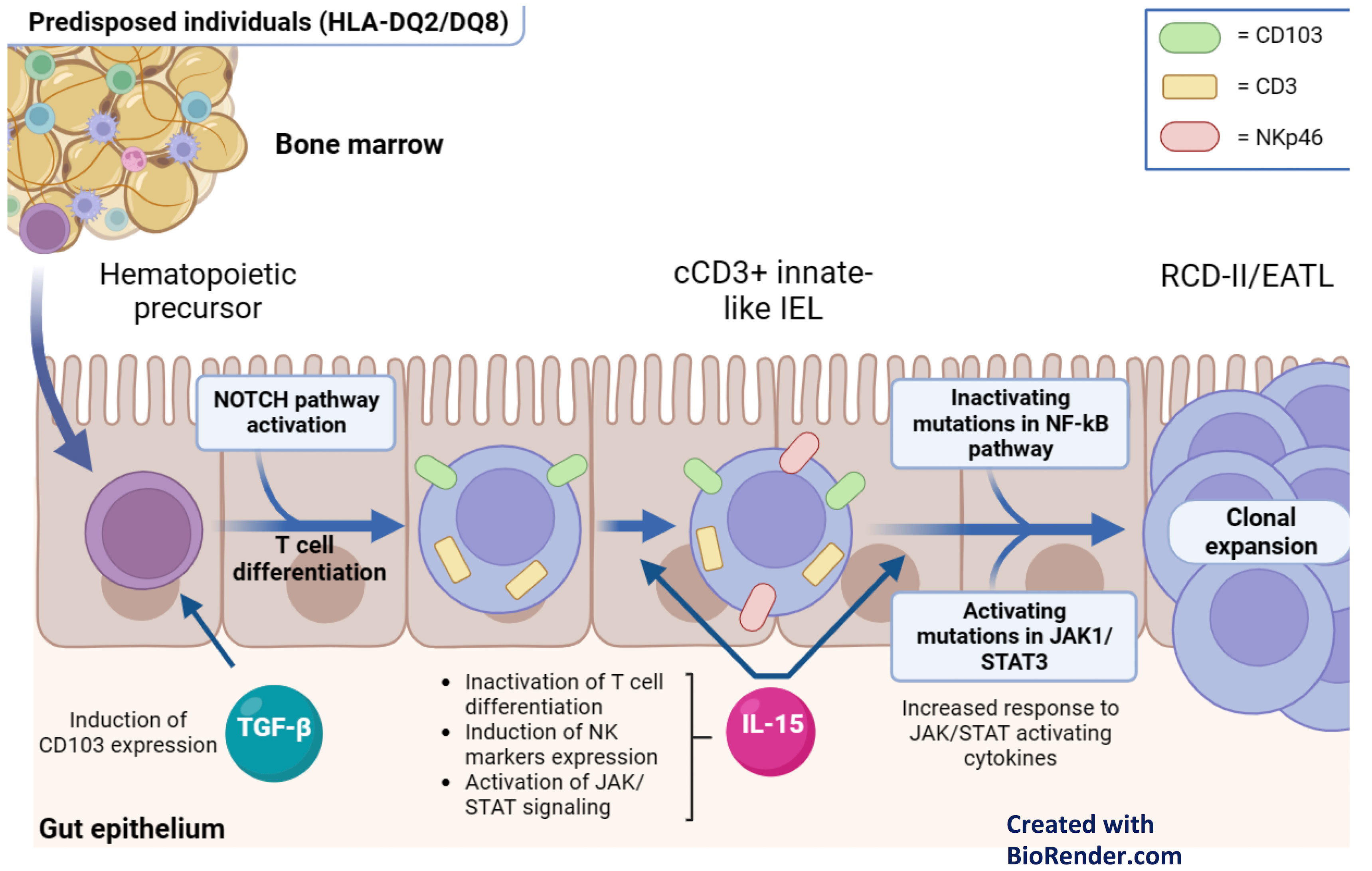
Figure 2 Pathophysiology of RCD-II and EATL. Hematopoietic precursors migrate into the gut epithelium and initiate T cell differentiation in response to NOTCH1 signals. Additionally, they express CD103 in response to TGF-b. In the presence of IL-15, which is overexpressed in the lamina propria and intestinal epithelium of patients with active CD and RCD, these cells inactivate T cell differentiation and express NK cell markers. As a result, these innate-like lymphocytes manifest at the same time T cell (cCD3+) and NK (NKp46+) features. Subsequently, their clonal expansion is driven by gain-of-function somatic mutation in the JAK1/STAT3 pathway, which enhance their responsiveness to IL-15, and/or by loss-of-function mutations in negative regulators of the NF-Kb signaling. .
As for the genetic factors, both RCD-II and EATL are strongly associated with homozygosity of HLA-DQ2. This is reported in 44-65% of RCD-II and in 53.3% of EATL, while it is documented in only 25.5% of RCD-I and in 20.7% of uncomplicated CD (52). Besides HLA haplotypes, RCD-II and EATL are frequently associated with the rs7259292 single nucleotide polymorphism (SNP) of the MYO9B (53). Progression to RCD-II has also been linked to specific SNPs on chromosome 7 (rs2041570) (54). The biological bases of such genetic associations are still under investigation.
As for the immunological determinants, recent studies have shown a link between the neoplastic IELs of RCD-II/EATL and innate-like lymphocytes (ILLs) of normal intestinal mucosa. ILLs are a unique immune cell subset, deriving from immature hematopoietic precursors that migrate into the gut epithelium, start T-cell differentiation in response to NOTCH1 signals, and underwent cell fate reprogramming after IL-15 exposure. Like neoplastic IELs of RCD-II/EATL, ILLs manifest dual T and NK cell traits (55), lack sCD3 and express cCD3, CD103 and various NK receptors (56).
In RCD-II and EATL, the clonal expansion of ILLs is likely driven by somatic gain-of-function mutations of the JAK-STAT pathway (i.e. JAK1 and STAT3 mutations), which enhance response to several cytokines, including IL-15. This is overexpressed in the intestinal mucosa of active CD and RCD and stimulates the proliferation of mutated ILLs (50, 51). Besides the JAK-STAT pathway, RCD-II and EATL bear frequent loss-of-function mutations in negative regulators of the NF-kB pathway (i.e. TNFAIP3 and TNIP3) (57). This supports the expansion of ILL clones, since the NF-kB pathway enhances JAK-STAT-regulated transcriptional programs (58). Finally, the NF-kB and JAK-STAT pathways are sustained by the production of TNFα by IELs (59), by the secretion of several cytokines from gliadin-specific CD4+ T cells (60) and by extra-cellular signals mediated by Smad7 (61).
In RCD, the pathogenic role of IL-15 spans well beyond the pro-survival signals provided to neoplastic IELs. IL-15 is indeed largely responsible of GFD-independent mucosal damage and severe villous atrophy, since it induces a NK-like cytotoxic phenotype in IELs (55). In keeping with this, recent studies have documented the expression of NKp46 (a NK-related marker) in most IELs of RCD-II, in 83% of EATL and 100% of monomorphic epitheliotropic intestinal T-cell lymphomas (MEITL), suggesting a shared biology for these conditions (see paragraph 5.4) (62). Thus, besides its pathogenic relevance, NKp46 may serve as a new marker for the differential diagnosis between RCD-I and RCD-II and might represent a target for future therapies in RCD-II/EATL and MEITL (Figure 2). Similarly, the identification of the pathogenic role of IL-15 may lead to the future use of anti-IL-15 monoclonal antibodies (2).
Histology is the mainstay of RCD diagnosis. Despite this, the microscopic changes of RCD are not entirely specific and overlap with a broad range of conditions, which must be taken into account when facing long-lasting villous atrophy with or without increased IELs. In the following paragraphs the key histological findings of RCD-I and RCD-II will be addressed, specifically considering the differential diagnosis of these entities.
The microscopic changes of RCD-I are virtually indistinguishable from those of uncomplicated CD (48). These include villous atrophy, crypt hypertrophy and increased IELs (>25 IELs/100 epithelial cells) with regular expression of pan-T cell markers and positivity for CD8 (48) (Figure 3). Molecular studies usually show polyclonal TCR gene rearrangements (32, 48, 63).
The differential diagnosis of RCD-I encompasses RCD-II and several CD-unrelated enteropathies, including autoimmune enteropathy (AIE), drug-induced enteropathy (DIE), common variable immunodeficiency (CVID)-associated enteropathy, tropical and collagenous sprue, Giardiasis and Crohn’s disease. While the differential diagnosis with RCD-II relies on IELs phenotype (see below), distinction from other enteropathies requires integration of clinical-epidemiological, histological and laboratory data (Table 2) (64).
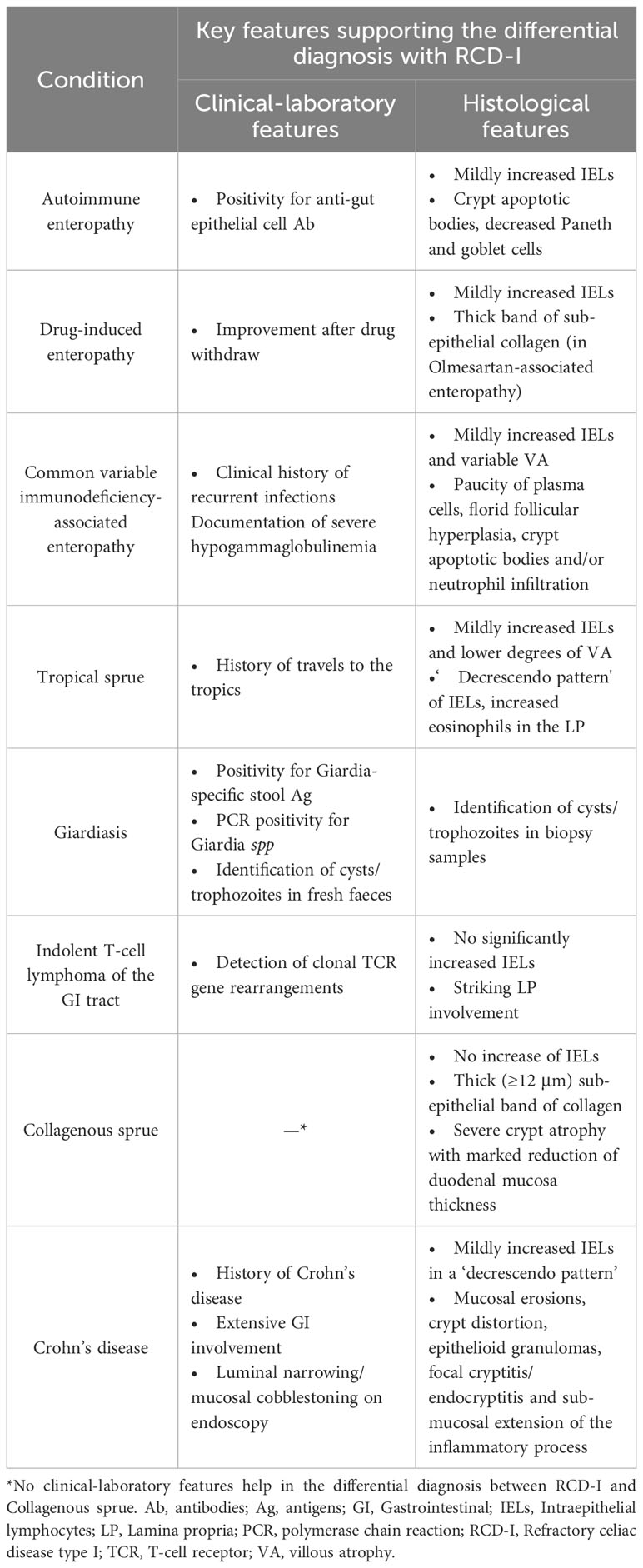
Table 2 Differential diagnosis of RCD-I.
Unlike RCD-I, AIE usually affects young adult patients (mean age at diagnosis: 44 years) with slight male predominance (M:F ratio= 1.5) (65). Histologically, RCD-I and AIE disclose similar degrees of villous atrophy, but IELs are usually lower in the latter. Crypt apoptotic bodies, loss or marked reduction of Paneth and goblet cells support the diagnosis of AIE (65). Finally, AIE is invariably associated with positivity for anti-gut epithelial cell (i.e. anti-enterocyte or anti-goblet cell) auto-Ab, which are never documented in RCD (66).
Among DIE, Olmesartan-associated enteropathy (OAE) and Mycophenolate Mofetil-associated enteropathy (MMAE) closely mimic RCD-I (67). OAE is an extremely rare condition (68, 69), characterized by CD-like symptoms after long-lasting consumption of Olmesartan (69). A similar enteropathy has been associated with Valstartan and Irbesartan use (70, 71). The endoscopic and histological findings of OAE may be indistinguishable from RCD-I, although a thick band of collagen may occasionally be observed in the former (69). MMEA presents with persistent diarrhea and villous atrophy due to inhibition of enterocyte proliferation. Like other DIE (i.e. Methotrexate and Azathioprine-induced enteropathy), MMAE has low numbers of IELs, supporting the differential diagnosis with RCD-I (72–74). In all such cases, the diagnosis of DIE is definitely confirmed by trials of drug withdraw after careful consideration of ongoing and prior treatments (67).
CVID-associated enteropathy may mimic CD/RCD-I both clinically and histologically (75). Small bowel biopsies reveal a moderate increase in IELs (75.6% of cases) with variable villous atrophy (31.2%-87.5% of cases) (75–77). Distinctive morphological features of CVID-associated enteropathy include extreme paucity of plasma cells, florid follicular hyperplasia in the lamina propria, crypt apoptotic bodies and/or neutrophil infiltration (75). The documentation of severe hypogammaglobulinemia and the history of repeated infections further support the diagnosis (78).
Tropical Sprue is a malabsorption syndrome likely caused by long-lasting infections contracted by natives or travelers to the tropics (79). Compared to CD/RCD-I, tropical sprue features lower degrees of villous atrophy, less numerous IELs, a ‘decrescendo pattern’ of IELs (i.e. main location in the villous basal third and in crypt epithelium), and increased eosinophils in the lamina propria. These findings and the history of travels to the tropics support the diagnosis (80).
Giardiasis is another infective enteropathy caused by Giardia lamblia that can mimic CD. Giardiasis may display a wide histological spectrum with variable villous atrophy and IELs, therefore its diagnosis relies on the documentation of Giardia-specific stool antigens, on PCR studies for Giardia-specific nucleic acids or on the microscopic detection of cysts/trophozoites in fresh faeces or biopsy samples (64, 81).
Careful histological evaluation also contributes to the differential diagnosis between RCD-I and collagenous sprue. This is indeed characterized by a thick (≥12 μm wide) sub-epithelial band of collagen, entrapping the blood vessels and stromal cells of the lamina propria. In collagenous sprue, villous distortion is usually accompanied by severe crypt atrophy, resulting in a markedly reduced thickness of duodenal mucosa. These findings and the lack of increased IELs favor the diagnosis of collagenous sprue (82).
Indolent T-cell lymphoproliferative disorders of the GI tract must be included in the differential diagnosis of CD/RCD, as they manifest with variable villous atrophy and crypt hypertrophy. Striking involvement of the lamina propria, lack of increased IELs and detection of clonal TCR gene rearrangements support the diagnosis of these conditions (48, 64).
Finally, duodenal involvement by Crohn’s disease may closely mimic CD/RCD-I. In such cases, thorough clinical-pathological correlations are mandatory to make the correct diagnosis (83). On clinical grounds, Crohn’s duodenitis is usually associated with more conventional ileal and colonic presentations. On histology, IELs are usually fewer and mainly arranged in a ‘decrescendo pattern’ (84). Mucosal erosions, crypt distortion, epithelioid granulomas, focal cryptitis/endocryptitis and sub-mucosal extension of the inflammatory process further support Crohn’s disease (1, 85).
Although RCD-I and RCD-II have overlapping morphology, they are biologically distinct disorders with different malignant potential. RCD-II is indeed a pre-lymphomatous condition characterized by clones of phenotypically aberrant IELs. Phenotypic aberrancies in RCD-II are defined by negativity for sCD3 and CD8, with positivity for cCD3 (48) (Table 1; Figure 3). Of note, clonal TCR rearrangements are documented in most RCD-II, but they are neither specific nor required for the diagnosis. In fact, clonal TCR rearrangement can be detected in a minority of CD and RCD-I patients (63). Clonal testing can also provide false negative results when atypical clones are small (63, 86, 87) and/or have incomplete/non-functional TCR rearrangements (70% of RCD-II) (63, 88, 89).
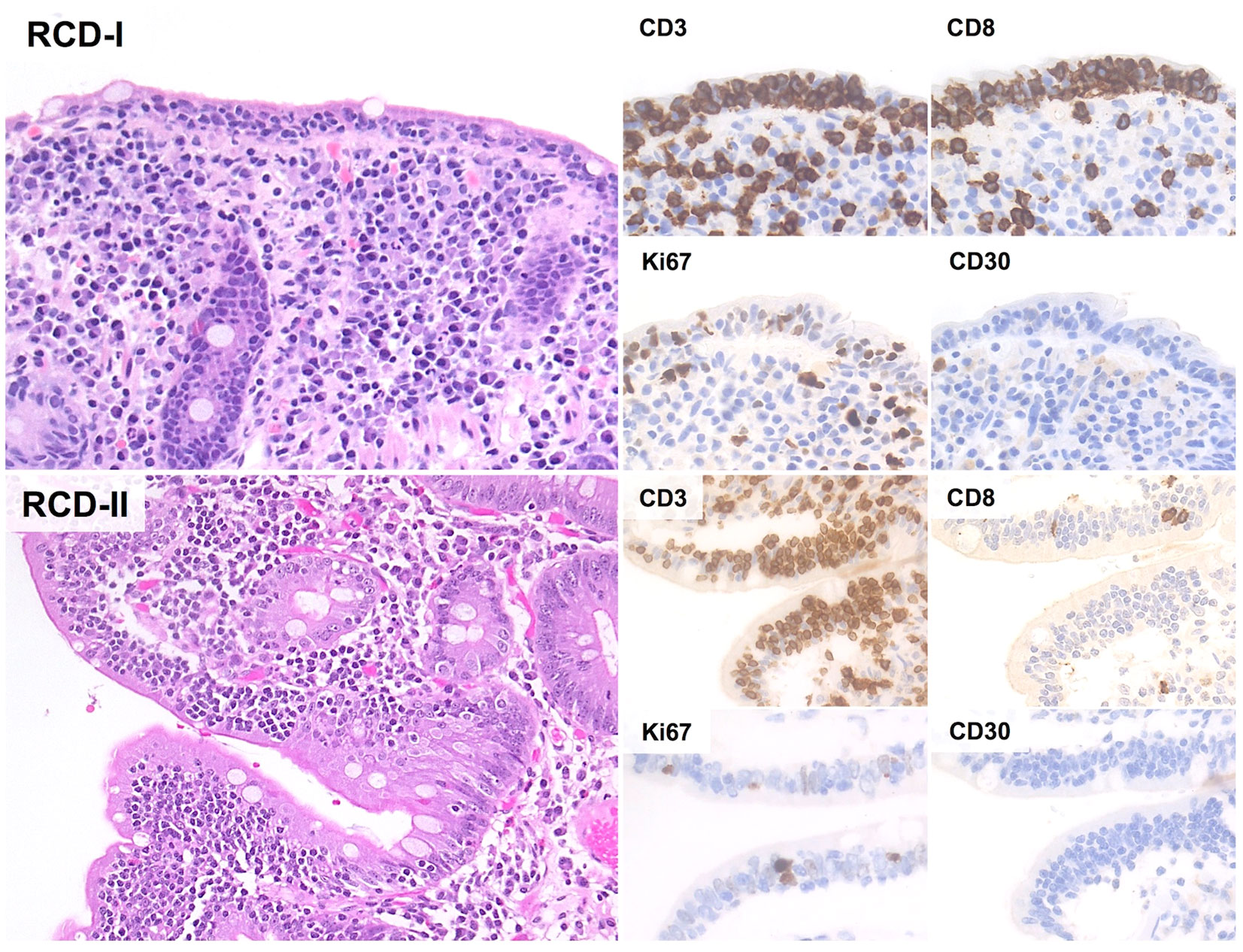
Figure 3 Histological and Immunohistochemical features of RCD-I and RCD-II. In both RCD-I and RCD-II, microscopic examination shows increased IELs without significant cytological atypia. The Ki67 proliferation index is low and CD30 immunostain is negative. However, IELs of RCD-II have an aberrant immunophenotype with negativity for both CD4 and CD8 (H&E and immunoperoxidase stains; original magnification 20x).
Phenotypic aberrancies in RCD-II can be documented by either flow cytometry or immunohistochemistry (IHC). As a general rule, flow cytometry is more sensitive and accurate, although it is not as widely applicable as IHC (48). As such, both techniques can be used to make a diagnosis of RCD-II, but different thresholds for aberrant IELs should be considered (i.e. ≥20% of total IELs for FC; ≥50% of total IELs for IHC) (87, 90).
Morphological assessment of duodenal biopsy in RCD-II shows marked villous atrophy, usually at a greater degree than RCD-I (moderate/severe villous atrophy: 96% of RCD-II and 50% of RCD-I) (20), together with a predominantly intra-epithelial infiltrate of atypical lymphocytes. Minimal sub-epithelial infiltration is frequently observed, constituting up to 20% of lymphocytes in the lamina propria (91). Notably, atypical IELs can be detected all throughout the GI tract, as well as in peripheral blood, mesenteric lymph nodes, lung parenchyma, skin and bone marrow (21, 91–94). In keeping with this, lymphocytic gastritis and lymphocytic colitis with abnormal IELs are reported in roughly 30-50% of RCD-II (21). Such widespread distribution also provides an explanation to the extra-intestinal presentations of EATL, which constitute up to one-third of cases (95).
Phenotypically, IELs of RCD-II lack CD4, CD8, sCD3 and TCRαβ/TCRγδ, while retaining CD7, CD103 and cCD3 expression (93). CD30 is characteristically negative and its expression suggests evolution to EATL (96). Alternative phenotypes are occasionally seen, including positivity for sCD3, CD8, TCRαβ and/or TCRγδ (48).
The differential diagnosis of RCD-II mainly includes RCD-I and EATL. Distinction from RCD-I relies primarily on IEL phenotyping, while the differential diagnosis with EATL is more challenging. In fact, RCD-II and EATL represents two ends of a biological continuum and a diagnosis of overt EATL should only be considered when the neoplastic population massively invades the duodenal/small intestinal wall, with clear-cut evidence of tumor lesions, bowel perforation or strictures (see paragraph 5.4).
EATL is an extremely aggressive peripheral T-cell lymphoma (PTCL), arising from IELs of the small bowel and representing the invasive form of RCD-II. In line with this, EATL and RCD-II share several pathophysiological features, including a common genetic background (i.e. homozygosity for HLA-DQ2; common allelic variants of MYO9B gene) (52, 53) and overlapping mutations in the JAK-STAT and NF-kB pathway (57). Additional events in the pathogenesis of EATL include oncogenic mutations in TET2, POT1, DDX3X, PRDM1/BLIMP1 and KMT2D (57, 97), deletions of 16q12.1 and gains of 1q, 5q and 9q (98). All of this contributes to the acquisition of an aggressive phenotype, whereby intra-mucosal lymphocytes of RCD-II undergo uncontrolled proliferation, invading the intestinal wall, disseminating throughout the GI tract and, ultimately, to extra-intestinal sites.
Despite being the most common intestinal T-cell lymphoma in Western countries, EATL is an exceedingly rare disease with a reported incidence of 0.2-1.0/1.000.000/year. It accounts for 5% of all GI lymphomas (99–101) and for only 3% of PTCLs (102). Virtually all cases arise in the setting of CD and the geographic distribution of the disease likely reflects the higher prevalence of CD in the Western world (48). Depending on the time relationship with CD, two forms of EATL are reported, namely primary EATL (i.e. EATL diagnosed concurrently with CD) and secondary EATL (i.e. EATL arising in patients with prior diagnosis of CD or RCD-II).
EATL affects adult to elderly patients (median age at diagnosis: 61 years) (95) and likely develops several months to years after the onset of pre-malignant IEL clones, which may remain clinically silent for a long time. In keeping with this observation, up to 50% of EATLs arise in the setting of RCD-II (95), thus confirming a tight connection between the two entities.
Clinically, EATL presents with small intestine lesions in about 90% of cases, the jejunum being most frequently involved. Multifocality is observed in 30-55% of cases and advanced-stage disease (Lugano stage II2-IV) is present in about half of the patients (95, 102). As previously reported, a subset of cases presents primarily in extra-intestinal sites, such as the spleen, the lung and the liver (95) (Figure 4). Typical signs and symptoms include abdominal pain, weight loss and diarrhea (102, 103) perforations, obstructions and/or GI bleeding (95). B symptoms (besides weight loss) are reported in one third of the patients (48). Laboratory tests are non-specific with anemia, high LDH and B2M levels and low serum albumin due to starvation (30, 95). Imaging studies often reveal enteric strictures, perforations or mass lesions, as well as mesenteric adenopathies and/or splenomegaly (27, 39). Although these findings are highly suggestive of EATL in the setting of CD, a definite diagnosis is only posed by histological evaluation of endoscopic biopsies or resection specimens.
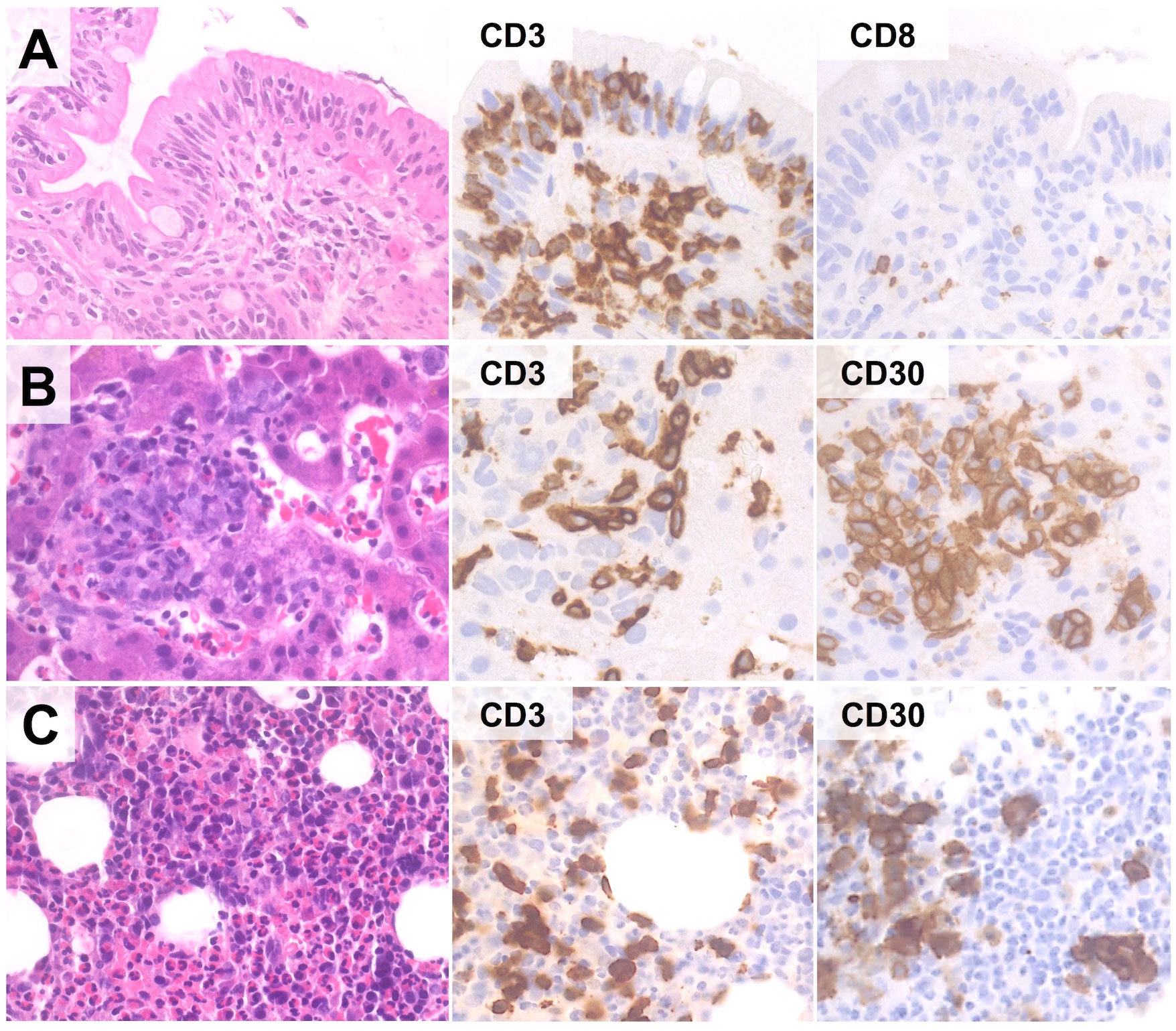
Figure 4 Extra-instestinal presentation of EATL. In this case (67-year old female with history of RCD-II), duodenal biopsy discloses only IELs with aberrant phenotype (CD3+/CD4-/CD8-/CD30-) (A). However, liver (B) and bone marrow (C) biopsies reveal an atypical lymphoid infiltrate comprising numerous CD30+ blasts, suggesting the diagnosis of extra-intestinal progression to EATL. (H&E and immunoperoxidase stains; original magnification 40x).
At present, high-dose chemotherapy followed by ASCT is the mainstay of treatment (104). Unfortunately, only a minority of patients can be treated with such an aggressive approach and the outcome remains poor (5-year OS: 11-20%) (105, 106). To refine the prognostic stratification of patients, a multi-parametric score has been recently proposed by integrating the International Prognostic Index and the presence of B-symptoms (i.e. EATL Prognostic Index [EPI]). The EPI identifies the following risk groups: (i) low-risk EATL (IPI score <2 and no B-symptoms; median OS: 34 months); (ii) intermediate-risk EATL (IPI score ≥2 and no B-symptoms; median OS: 7 months); and (iii) high-risk EATL (presence of B-symptoms irrespective of IPI score; median OS: 2 months) (107). In addition to EPI, the time relationship between CD and EATL likely influences outcome, in that primary EATL seems to fare better than secondary (i.e. post-RCD-II) disease (5-year OS: 60% versus <5%) (95). These prognostic parameters and the recent identification of new therapeutic targets (e.g. CD30 and NKp46 expression on neoplastic cells; IL15 in the tumor microenvironment) will hopefully contribute to improve patient management (108, 109).
Histologically, EATL is characterized by a diffuse infiltrate of atypical T-cells in a rich inflammatory background of histiocytes, plasma cells and granulocytes. Reactive cells may be as many as to obscure the neoplastic population, which consists of medium to large cells with pleomorphic, immunoblastic or anaplastic morphology (95, 102, 110). The infiltrate may be confined to the mucosa/submucosa or may extend throughout the intestinal wall (Figure 5); angioinvasion and angiodestruction can also be observed (48). The adjacent mucosa usually discloses features of CD/RCD-II (102, 110).
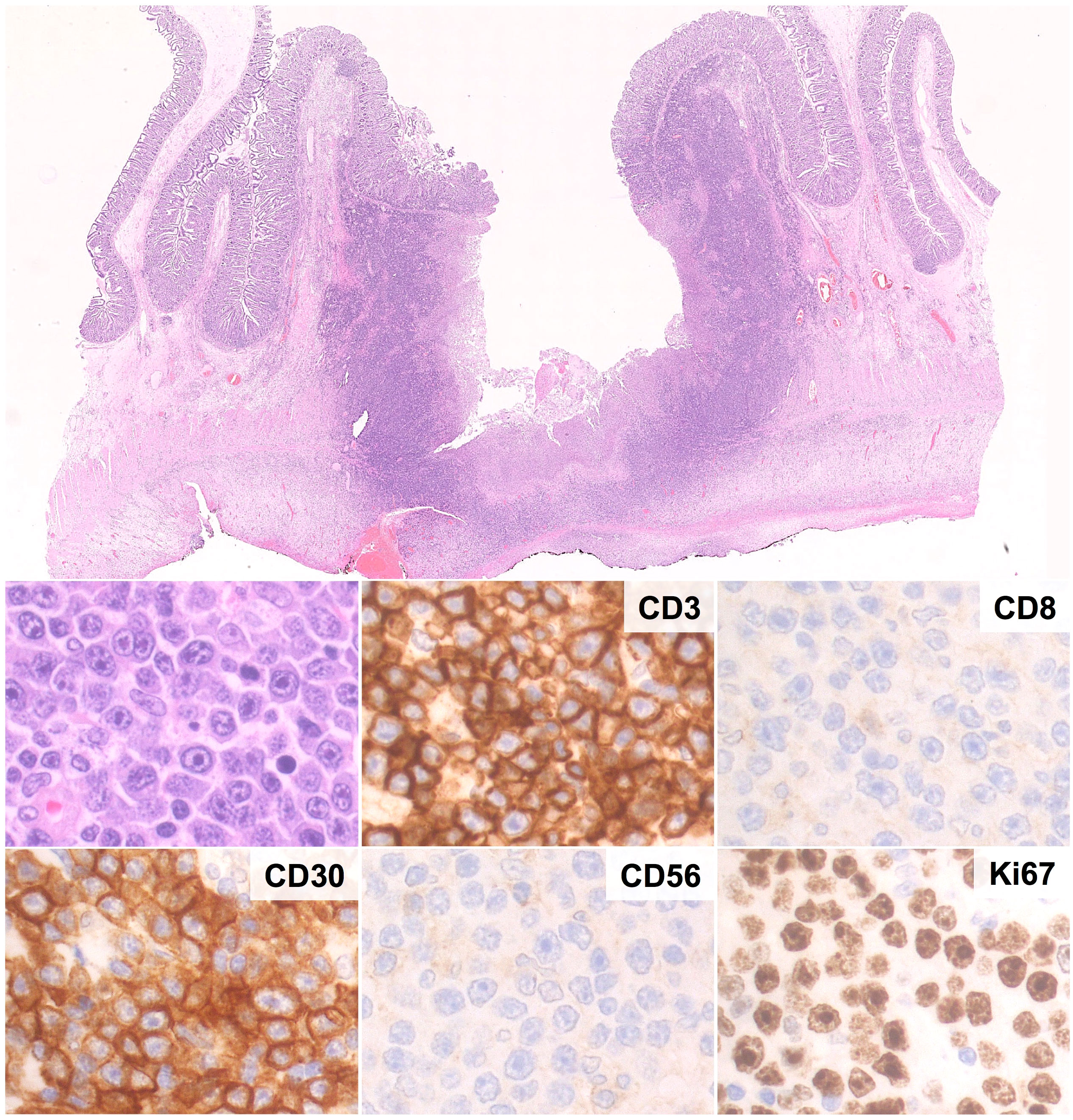
Figure 5 Histological and Immunohistochemical features of EATL. Microscopic examination shows a massive infiltration of the intestinal wall by sheets of CD3+ cells with immunoblastic morphology, high prolifration index, diffuse positivity for CD30 and negativity for CD8 and CD56. (H&E and immunoperoxidase stains; original magnification 1, 25x and 40x).
The phenotype of EATL largely recapitulates that of RCD-II cells, with combined expression of T-cell and NK-cell markers (i.e. positivity for cCD3, CD7, NKp46 and CD103; variable expression of CD2; negativity for CD4, CD5, CD8, CD56, ALK, EBER and TCRαβ/TCRγδ) (62, 102, 111). Neoplastic cells usually show positivity for cytotoxic markers (TIA1, granzyme B, perforin), a high proliferation index (>50%) and CD30, mostly in cases with anaplastic/immunoblastic morphology (95, 102). A minority of cases shows aberrant phenotypes with positivity for CD8 (25% of cases) and/or TCR proteins (48). Molecular analyses disclose clonal TCR rearrangements in most cases (57).
The differential diagnosis of EATL mainly includes RCD-II and MEITL. As previously outlined, distinction from RCD-II relies on the degree of infiltration by neoplastic cells, which is usually massive in EATL and limited to the lamina propria in RCD-II. However, separating early-stage (i.e. mucosa/submucosa-limited) EATL from RCD-II may be matter of subjectivity, especially on small biopsy samples. In such cases, the diagnosis of EATL should be favored in presence of B symptoms, abdominal masses, or specific phenotypic findings (e.g. positivity for CD30; high Ki67 index).
Likewise, distinction from MEITL relies on clinical, morphological, phenotypic and genetic criteria. Unlike EATL, MEITL is rarely associated with CD/RCD-II (98, 112, 113) and consists of a monomorphic population of small-to-medium lymphocytes with little inflammatory background and sharp epitheliotropism (48). The phenotype of MEITL also differs from EATL, in that the neoplastic cells are positive for CD8, CD56, SYC and TCR proteins (usually of γδ type). Despite these differences, EATL and MEITL likely share a common origin from intestinal IELs, as indicated by their clear-cut epitheliotropism, by CD103 and NKp46 expression (62) and by similar activating mutations in the JAK-STAT pathway (114, 115). Unlike EATL, however, MEITL shows frequent SETD2 alterations, which may support the correct diagnosis (116).
Distinction of EATL from other primary GI lymphomas (i.e. aggressive B-cel lymphomas; intestinal T-cell lymphoma NOS; indolent T-cell lymphoproliferative disorders of the GI) and from GI involvement by systemic PTCL is usually straightforward and relies on a combination of morphology, phenotypic studies and clinical correlations.
Over the last decades, a batter characterization of the biology of RCD and EATL has improved our knowledge of these conditions. RCD-I and RCD-II are distinct disorders stemming from a disease initially driven by abnormal T-cell immune responses against gluten-derived peptides in genetically susceptible individuals. In particular, RCD-I represents a gluten-independent dysimmune reaction of the small bowel, while RCD-II can be regarded as an aggressive in situ T-cell lymphoma with high risk of EATL progression. In keeping with this view, several studies have highlighted the complex pathogenesis and kinship of RCD-II and EATL. All of this has been formally acknowledged also by the 2022 WHO and ICC classifications of lymphoid tumors, which include both EATL and RCD-I/RCD-II in the list of intestinal T-cell lymphoproliferative disorders (48).
Despite these achievements, the diagnosis of RCD and EATL remains challenging and the prognosis of RCD-II and EATL is poor. New molecular targets for tailored therapies will hopefully compensate for such dismal outcome. For the time being, the proper recognition and management of RCD and EATL relies on a high degree of suspicion, on careful differential diagnoses, and on the collaboration of gastroenterologists, hematologists and pathologists with specific expertise on GI lymphomas and dysimmune disorders. This teamwork still represents the best strategy for any further development on these conditions and for the appropriate management of patients.
FS: Writing – original draft. MP: Writing – original draft, Supervision, Writing – review & editing. FPe: Writing – review & editing. VA: Writing – review & editing. AD: Supervision, Writing – review & editing. FPi: Writing – review & editing. ES: Writing – review & editing. FZ: Supervision, Writing – original draft. MF: Supervision, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Villanacci V, Vanoli A, Leoncini G, Arpa G, Salviato T, Bonetti LR, et al. Celiac disease: histology-differential diagnosis-complications: A Pract approach. Pathologica. (2020) 112(3):186–96. doi: 10.32074/1591-951X-157
2. Al-Toma A, Volta U, Auricchio R, Castillejo G, Sanders DS, Cellier C, et al. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur Gastroenterol J (2019) 7(5):583–613. doi: 10.1177/2050640619844125
3. Megiorni F, Pizzuti A. HLA-DQA1 and HLA-DQB1 in Celiac disease predisposition: practical implications of the HLA molecular typing. J BioMed Sci (2012) 19(1):88. doi: 10.1186/1423-0127-19-88
4. Caio G, Volta U, Sapone A, Leffler DA, De Giorgio R, Catassi C, et al. Celiac disease: a comprehensive current review. BMC Med (2019) 17(1):142. doi: 10.1186/s12916-019-1380-z
5. Gujral N, Freeman HJ, Thomson ABR. Celiac disease: prevalence, diagnosis, pathogenesis and treatment. World J Gastroenterol (2012) 18(42):6036–59. doi: 10.3748/wjg.v18.i42.6036
6. Pelizzaro F, Marsilio I, Fassan M, Piazza F, Barberio B, D’Odorico A, et al. The risk of Malignancies in celiac disease—A literature review. Cancers (Basel). (2021) 13(21):5288. doi: 10.3390/cancers13215288
7. Zingone F, Maimaris S, Auricchio R, Caio GPI, Carroccio A, Elli L, et al. Guidelines of the Italian societies of gastroenterology on the diagnosis and management of coeliac disease and dermatitis herpetiformis. Digestive Liver Disease. (2022) 54(10):1304–19. doi: 10.1016/j.dld.2022.06.023
8. Marsh MN. Grains of truth: evolutionary changes in small intestinal mucosa in response to environmental antigen challenge. Gut. (1990) 31(1):111–4. doi: 10.1136/gut.31.1.111
9. Oberhuber G, Granditsch G, Vogelsang H. The histopathology of coeliac disease. Eur J Gastroenterol Hepatol (1999) 11(10):1185. doi: 10.1097/00042737-199910000-00019
11. Corazza GR, Villanacci V, Zambelli C, Milione M, Luinetti O, Vindigni C, et al. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastroenterol Hepatology. (2007) 5(7):838–43. doi: 10.1016/j.cgh.2007.03.019
12. Aljada B, Zohni A, El-Matary W. The gluten-free diet for celiac disease and beyond. Nutrients. (2021) 13(11):3993. doi: 10.3390/nu13113993
13. Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systematic approach. Am J Gastroenterol (2002) 97(8):2016–21. doi: 10.1111/j.1572-0241.2002.05917.x
14. Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol (2007) 5(4):445–50. doi: 10.1016/j.cgh.2006.12.006
15. Stasi E, Marafini I, Caruso R, Soderino F, Angelucci E, Del Vecchio Blanco G, et al. Frequency and cause of persistent symptoms in celiac disease patients on a long-term gluten-free diet. J Clin Gastroenterol (2016) 50(3):239–43. doi: 10.1097/MCG.0000000000000392
16. Vahedi K, Mascart F, Mary JY, Laberenne JE, Bouhnik Y, Morin MC, et al. Reliability of antitransglutaminase antibodies as predictors of gluten-free diet compliance in adult celiac disease. Am J Gastroenterology. (2003) 98(5):1079–87. doi: 10.1111/j.1572-0241.2003.07284.x
17. Hujoel IA, Murray JA. Refractory celiac disease. Curr Gastroenterol Rep (2020) 22(4):18. doi: 10.1007/s11894-020-0756-8
18. Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fasano A, Green PHR, et al. The Oslo definitions for coeliac disease and related terms. Gut. (2013) 62(1):43–52. doi: 10.1136/gutjnl-2011-301346
19. Rowinski SA, Christensen E. Epidemiologic and therapeutic aspects of refractory coeliac disease - a systematic review. Dan Med J (2016) 63(12):A5307.
20. Elli L, Soru P, Roncoroni L, Rossi FG, Ferla V, Baldini L, et al. Clinical features of type 1 and 2 refractory celiac disease: Results from a large cohort over a decade. Digestive Liver Disease. (2023) 55(2):235–42. doi: 10.1016/j.dld.2022.08.022
21. Malamut G, Afchain P, Verkarre V, Lecomte T, Amiot A, Damotte D, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology. (2009) 136(1):81–90. doi: 10.1053/j.gastro.2008.09.069
22. Eigner W, Bashir K, Primas C, Kazemi-Shirazi L, Wrba F, Trauner M, et al. Dynamics of occurrence of refractory coeliac disease and associated complications over 25 years. Aliment Pharmacol Ther (2017) 45(2):364–72. doi: 10.1111/apt.13867
23. Ilus T, Kaukinen K, Virta LJ, Huhtala H, Mäki M, Kurppa K, et al. Refractory coeliac disease in a country with a high prevalence of clinically-diagnosed coeliac disease. Aliment Pharmacol Ther (2014) 39(4):418–25. doi: 10.1111/apt.12606
24. Tursi A, Elisei W, Giorgetti GM, Brandimarte G, Aiello F. Complications in celiac disease under gluten-free diet. Dig Dis Sci (2009) 54(10):2175–82. doi: 10.1007/s10620-008-0595-1
25. Rubio–Tapia A, Kelly DG, Lahr BD, Dogan A, Wu T, Murray JA. Clinical staging and survival in refractory celiac disease: a single center experience. Gastroenterology. (2009) 136(1):99–107. doi: 10.1053/j.gastro.2008.10.013
26. Malamut G, Cellier C. Refractory celiac disease. Gastroenterol Clin North Am (2019) 48(1):137–44. doi: 10.1016/j.gtc.2018.09.010
27. Rubio-Tapia A, Murray JA. Classification and management of refractory coeliac disease. Gut. (2010) 59(4):547–57. doi: 10.1136/gut.2009.195131
28. Zammit SC, Sanders DS, Cross SS. Capsule endoscopy in the management of refractory coeliac disease. J Gastrointestinal Liver Diseases. (2019) 28(1):15–22. doi: 10.15403/jgld.2014.1121.281.cel
29. Rubio-Tapia A, Malamut G, Verbeek WHM, van Wanrooij RLJ, Leffler DA, Niveloni SI, et al. Creation of a model to predict survival in patients with refractory coeliac disease using a multinational registry. Aliment Pharmacol Ther (2016) 44(7):704–14. doi: 10.1111/apt.13755
30. Lenti MV, Aronico N, Giuffrida P, Antoci V, Santacroce G, Vanoli A, et al. Serum markers of refractoriness and enteropathy-associated T-cell lymphoma in coeliac disease. Cancers (Basel). (2021) 13(10):2289. doi: 10.3390/cancers13102289
31. Di Sabatino A, Giuffrida P, Vanoli A, Luinetti O, Manca R, Biancheri P, et al. Increase in neuroendocrine cells in the duodenal mucosa of patients with refractory celiac disease. Am J Gastroenterology. (2014) 109(2):258–69. doi: 10.1038/ajg.2013.426
32. Green PHR, Paski S, Ko CW, Rubio-Tapia A. AGA clinical practice update on management of refractory celiac disease: expert review. Gastroenterology. (2022) 163(5):1461–9. doi: 10.1053/j.gastro.2022.07.086
33. Comino I, Fernández-Bañares F, Esteve M, Ortigosa L, Castillejo G, Fambuena B, et al. Fecal gluten peptides reveal limitations of serological tests and food questionnaires for monitoring gluten-free diet in celiac disease patients. Am J Gastroenterology. (2016) 111(10):1456–65. doi: 10.1038/ajg.2016.439
34. Moreno M de L, Cebolla Á, Muñoz-Suano A, Carrillo-Carrion C, Comino I, Pizarro Á, et al. Detection of gluten immunogenic peptides in the urine of patients with coeliac disease reveals transgressions in the gluten-free diet and incomplete mucosal healing. Gut. (2017) 66(2):250–7. doi: 10.1136/gutjnl-2015-310148
35. Tursi A, Brandimarte G, Giorgetti G. High prevalence of small intestinal bacterial overgrowth in celiac patients with persistence of gastrointestinal symptoms after gluten withdrawal. Am J Gastroenterol (2003) 98(4):839–43. doi: 10.1111/j.1572-0241.2003.07379.x
36. Rubio-Tapia A, Barton SH, Rosenblatt JE, Murray JA. Prevalence of small intestine bacterial overgrowth diagnosed by quantitative culture of intestinal aspirate in celiac disease. J Clin Gastroenterol (2009) 43(2):157–61. doi: 10.1097/MCG.0b013e3181557e67
37. Pennazio M, Rondonotti E, Despott EJ, Dray X, Keuchel M, Moreels T, et al. Small-bowel capsule endoscopy and device-assisted enteroscopy for diagnosis and treatment of small-bowel disorders: European Society of Gastrointestinal Endoscopy (ESGE) Guideline – Update 2022. Endoscopy. (2023) 55(01):58–95. doi: 10.1055/a-1973-3796
38. Al-Bawardy B, Barlow JM, Vasconcelos RN, Kim ST, Bruining DH, Hansel SL, et al. Cross-sectional imaging in refractory celiac disease. Abdominal Radiology. (2017) 42(2):389–95. doi: 10.1007/s00261-016-1032-0
39. Gils T, Nijeboer P, Waesberghe JHT, Coupé VM, Janssen K, Zegers JA, et al. Splenic volume differentiates complicated and non-complicated celiac disease. United Eur Gastroenterol J (2017) 5(3):374–9. doi: 10.1177/2050640616663571
40. Mukewar SS, Sharma A, Rubio-Tapia A, Wu TT, Jabri B, Murray JA. Open-capsule budesonide for refractory celiac disease. Am J Gastroenterology. (2017) 112(6):959–67. doi: 10.1038/ajg.2017.71
41. Brar P, Lee S, Lewis S, Egbuna I, Bhagat G, Green PHR. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol (2007) 102(10):2265–9. doi: 10.1111/j.1572-0241.2007.01380.x
42. Al–toma A, Goerres MS, Meijer JWR, von Blomberg BME, Wahab PJ, Kerckhaert JAM, et al. Cladribine therapy in refractory celiac disease with aberrant T cells. Clin Gastroenterol Hepatology. (2006) 4(11):1322–7. doi: 10.1016/j.cgh.2006.07.007
43. Dray X. A severe but reversible refractory sprue. Gut. (2005) 55(8):1210–1. doi: 10.1136/gut.2005.089987
44. Maheshwari PK, Feeley I, Oleary H, Goulding C. A 37-year-old woman with refractory coeliac disease type II disease treated by stem cell transplantation. BMJ Case Rep (2015) 24:bcr2015209363. doi: 10.1136/bcr-2015-209363
45. Tack GJ, Wondergem MJ, Al-Toma A, Verbeek WHM, Schmittel A, MaChado MV, et al. Auto-SCT in refractory celiac disease type II patients unresponsive to cladribine therapy. Bone Marrow Transplant. (2011) 46(6):840–6. doi: 10.1038/bmt.2010.199
46. Al-toma A, Visser OJ, van Roessel HM, von Blomberg BME, Verbeek WHM, Scholten PET, et al. Autologous hematopoietic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood. (2007) 109(5):2243–9. doi: 10.1182/blood-2006-08-042820
47. Al-toma A, Verbeek WHM, Hadithi M, von Blomberg BME, Mulder CJJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut. (2007) 56(10):1373–8. doi: 10.1136/gut.2006.114512
48. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IB de O, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia [Internet]. (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
49. Holmes GK, Prior P, Lane MR, Pope D, Allan RN. Malignancy in coeliac disease–effect of a gluten free diet. Gut. (1989) 30(3):333–8. doi: 10.1136/gut.30.3.333
50. Mention JJ, Ben Ahmed M, Bègue B, Barbe U, Verkarre V, Asnafi V, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. (2003) 125(3):730–45. doi: 10.1016/S0016-5085(03)01047-3
51. Foxman EF, Iwasaki A. Genome–virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol (2011) 9(4):254–64. doi: 10.1038/nrmicro2541
52. Al–Toma A, Goerres MS, Meijer JWR, Peña AS, Crusius JBA, Mulder CJJ. Human leukocyte antigen–DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatology. (2006) 4(3):315–9. doi: 10.1016/j.cgh.2005.12.011
53. Wolters VM, Verbeek WHM, Zhernakova A, Onland–Moret C, Schreurs MWJ, Monsuur AJ, et al. The MYO9B gene is a strong risk factor for developing refractory celiac disease. Clin Gastroenterol Hepatology. (2007) 5(12):1399–405. doi: 10.1016/j.cgh.2007.08.018
54. Hrdlickova B, Mulder CJ, Malamut G, Meresse B, Platteel M, Kamatani Y, et al. A locus at 7p14.3 predisposes to refractory celiac disease progression from celiac disease. Eur J Gastroenterol Hepatol (2018) 30(8):828–37. doi: 10.1097/MEG.0000000000001168
55. Ettersperger J, Montcuquet N, Malamut G, Guegan N, Lopez-Lastra S, Gayraud S, et al. Interleukin-15-dependent T-cell-like innate intraepithelial lymphocytes develop in the intestine and transform into lymphomas in celiac disease. Immunity. (2016) 45(3):610–25. doi: 10.1016/j.immuni.2016.07.018
56. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. (2014) 41(3):354–65. doi: 10.1016/j.immuni.2014.09.005
57. Cording S, Lhermitte L, Malamut G, Berrabah S, Trinquand A, Guegan N, et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. Gut. (2022) 71(3):497–508. doi: 10.1136/gutjnl-2020-322935
58. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev (2010) 21(1):11–9. doi: 10.1016/j.cytogfr.2009.11.005
59. O’Keeffe J, Lynch S, Whelan A, Jackson J, Kennedy NP, Weir DG, et al. Flow cytometric measurement of intracellular migration inhibition factor and tumour necrosis factor alpha in the mucosa of patients with coeliac disease. Clin Exp Immunol (2001) 125(3):376–82. doi: 10.1046/j.1365-2249.2001.01594.x
60. Kooy-Winkelaar YMC, Bouwer D, Janssen GMC, Thompson A, Brugman MH, Schmitz F, et al. CD4 T-cell cytokines synergize to induce proliferation of Malignant and nonmalignant innate intraepithelial lymphocytes. Proc Natl Acad Sci (2017) 114(6):E980–E989. doi: 10.1073/pnas.1620036114
61. Sedda S, De Simone V, Marafini I, Bevivino G, Izzo R, Paoluzi OA, et al. High Smad7 sustains inflammatory cytokine response in refractory coeliac disease. Immunology. (2017) 150(3):356–63. doi: 10.1111/imm.12690
62. Cheminant M, Bruneau J, Malamut G, Sibon D, Guegan N, van Gils T, et al. NKp46 is a diagnostic biomarker and may be a therapeutic target in gastrointestinal T-cell lymphoproliferative diseases: a CELAC study. Gut. (2019) 68(8):1396–405. doi: 10.1136/gutjnl-2018-317371
63. Hussein S, Gindin T, Lagana SM, Arguelles-Grande C, Krishnareddy S, Alobeid B, et al. Clonal T cell receptor gene rearrangements in coeliac disease: implications for diagnosing refractory coeliac disease. J Clin Pathol (2018) 71(9):825–31. doi: 10.1136/jclinpath-2018-205023
64. Schiepatti A, Sanders DS, Baiardi P, Caio G, Ciacci C, Kaukinen K, et al. Nomenclature and diagnosis of seronegative coeliac disease and chronic non-coeliac enteropathies in adults: the Paris consensus. Gut. (2022) 71(11):2218–25. doi: 10.1136/gutjnl-2021-326645
65. Sharma A, Choung RS, Wang XJ, Russo PA, Wu TT, Nehra V, et al. Features of adult autoimmune enteropathy compared with refractory celiac disease. Clin Gastroenterol Hepatology. (2018) 16(6):877–83. doi: 10.1016/j.cgh.2017.12.044
66. Akram S, Murray JA, Pardi DS, Alexander GL, Schaffner JA, Russo PA, et al. Adult autoimmune enteropathy: mayo clinic rochester experience. Clin Gastroenterol Hepatology. (2007) 5(11):1282–90. doi: 10.1016/j.cgh.2007.05.013
67. Marietta EV, Cartee A, Rishi A, Murray JA. Drug-induced enteropathy. Digestive Diseases. (2015) 33(2):215–20. doi: 10.1159/000370205
68. Padwal R, Lin M, Etminan M, Eurich DT. Comparative effectiveness of olmesartan and other angiotensin receptor blockers in diabetes mellitus. Hypertension. (2014) 63(5):977–83. doi: 10.1161/HYPERTENSIONAHA.113.02855
69. Rubio-Tapia A, Herman ML, Ludvigsson JF, Kelly DG, Mangan TF, Wu TT, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc (2012) 87(8):732–8. doi: 10.1016/j.mayocp.2012.06.003
70. Marthey L, Cadiot G, Seksik P, Pouderoux P, Lacroute J, Skinazi F, et al. Olmesartan-associated enteropathy: results of a national survey. Aliment Pharmacol Ther (2014) 40(9):1103–9. doi: 10.1111/apt.12937
71. Herman M, Rubio-Tapia A, Marietta E, Wu TT, Murray J. Severe enteropathy in a patient on valsartan. Am J Gastroenterology. (2013) 108:S302. doi: 10.14309/00000434-201310001-01011
72. Kamar N, Faure P, Dupuis E, Cointault O, Joseph-Hein K, Durand D, et al. Villous atrophy induced by mycophenolate mofetil in renal-transplant patients. Transplant Int (2004) 17(8):463–7. doi: 10.1111/j.1432-2277.2004.tb00471.x
73. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol (2012) 30(21):2691–7. doi: 10.1200/JCO.2012.41.6750
74. DeGaetani M, Tennyson CA, Lebwohl B, Lewis SK, Abu Daya H, Arguelles-Grande C, et al. Villous atrophy and negative celiac serology: a diagnostic and therapeutic dilemma. Am J Gastroenterology. (2013) 108(5):647–53. doi: 10.1038/ajg.2013.45
75. Malamut G, Verkarre V, Suarez F, Viallard JF, Lascaux AS, Cosnes J, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterology. (2010) 105(10):2262–75. doi: 10.1038/ajg.2010.214
76. Mannon PJ, Fuss IJ, Dill S, Friend J, Groden C, Hornung R, et al. Excess IL-12 but not IL-23 Accompanies the Inflammatory Bowel Disease Associated With Common Variable Immunodeficiency. Gastroenterology. (2006) 131(3):748–56. doi: 10.1053/j.gastro.2006.06.022
77. Luzi G, Zullo A, Iebba F, Rinaldi V, Mete LS, Muscaritoli M, et al. Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. Am J Gastroenterol (2003) 98(1):118–21. doi: 10.1111/j.1572-0241.2003.07159.x
78. Rosen FS, Wedgwood RJP, Eibl M, Griscelli C, Seligmann M, Aiuti F, et al. Primary immunodeficiency diseases. Report of a WHO scientific group. Clin Exp Immunol (1997) 109 Suppl 1:1–28.
79. Ghoshal UC, Srivastava D, Verma A, Ghoshal U. Tropical sprue in 2014: the new face of an old disease. Curr Gastroenterol Rep (2014) 16(6):391. doi: 10.1007/s11894-014-0391-3
80. Sharma P, Baloda V, Gahlot GP, Singh A, Mehta R, Vishnubathla S, et al. Clinical, endoscopic, and histological differentiation between celiac disease and tropical sprue: a systematic review. J Gastroenterol Hepatol (2019) 34(1):74–83. doi: 10.1111/jgh.14403
81. Shen M, Voltaggio L, Robertson S. Giardia is often overlooked on histopathologic examination: a high-volume, single-institution experience. Int J Surg Pathol (2021) 29(3):257–62. doi: 10.1177/1066896920947795
82. Jain A, Sebastian K, Quigley B. Collagenous sprue, an enigma in the spectrum of celiac disease. Clin Gastroenterol Hepatol (2014) 12(1):e2–3; quiz e4-6. doi: 10.1016/j.cgh.2013.05.031
83. Kővári B, Pai RK. Upper gastrointestinal tract involvement in inflammatory bowel diseases: histologic clues and pitfalls. Adv Anat Pathol (2022) 29(1):2–14. doi: 10.1097/PAP.0000000000000311
84. Patterson ER, Shmidt E, Oxentenko AS, Enders FT, Smyrk TC. Normal villous architecture with increased intraepithelial lymphocytes. Am J Clin Pathol (2015) 143(3):445–50. doi: 10.1309/AJCPBKQND4SHVX9Q
85. AbdullGaffar B, Quraishi H. Histopathologic manifestations of crohn disease in duodenal endoscopy biopsy: the value of different patterns of involvement of brunner glands. Int J Surg Pathol (2021) 29(7):710–5. doi: 10.1177/1066896921998438
86. Derrieux C, Trinquand A, Bruneau J, Verkarre V, Lhermitte L, Alcantara M, et al. A single-tube, euroClonality-inspired, TRG clonality multiplex PCR aids management of patients with enteropathic diseases, including from formaldehyde-fixed, paraffin-embedded tissues. J Mol Diagnostics. (2019) 21(1):111–22. doi: 10.1016/j.jmoldx.2018.08.006
87. van Wanrooij RLJ, Müller DMJ, Neefjes-Borst EA, Meijer J, Koudstaal LG, Heideman DAM, et al. Optimal strategies to identify aberrant intra-epithelial lymphocytes in refractory coeliac disease. J Clin Immunol (2014) 34(7):828–35. doi: 10.1007/s10875-014-0075-7
88. Soderquist CR, Hsiao S, Mansukhani MM, Alobeid B, Green PH, Bhagat G. Refractory celiac disease type II: An atypical case highlighting limitations of the current classification system. Hematol Oncol (2020) 38(3):399–405. doi: 10.1002/hon.2720
89. Tack GJ, van Wanrooij RLJ, Langerak AW, Tjon JML, von Blomberg BME, Heideman DAM, et al. Origin and immunophenotype of aberrant IEL in RCDII patients. Mol Immunol (2012) 50(4):262–70. doi: 10.1016/j.molimm.2012.01.014
90. Verbeek WHM, Goerres MS, von Blomberg BME, Oudejans JJ, Scholten PET, Hadithi M, et al. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T-cell clonality analysis in Refractory Celiac Disease. Clin Immunol (2008) 126(1):48–56. doi: 10.1016/j.clim.2007.09.002
91. Verbeek WHM, von Blomberg BME, Coupe VMH, Daum S, Mulder CJJ, Schreurs MWJ. Aberrant T-lymphocytes in refractory coeliac disease are not strictly confined to a small intestinal intraepithelial localization. Cytometry B Clin Cytom (2009) 76B(6):367–74. doi: 10.1002/cyto.b.20481
92. Verkarre V. Refractory coeliac sprue is a diffuse gastrointestinal disease. Gut. (2003) 52(2):205–11. doi: 10.1136/gut.52.2.205
93. Cellier C, Patey N, Mauvieux L, Jabri B, Delabesse E, Cervoni J, et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology. (1998) 114(3):471–81. doi: 10.1016/S0016-5085(98)70530-X
94. Cellier C, Delabesse E, Helmer C, Patey N, Matuchansky C, Jabri B, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. Lancet (2000) 356(9225):203–8. doi: 10.1016/S0140-6736(00)02481-8
95. Malamut G, Chandesris O, Verkarre V, Meresse B, Callens C, Macintyre E, et al. Enteropathy associated T cell lymphoma in celiac disease: A large retrospective study. Digestive Liver Disease. (2013) 45(5):377–84. doi: 10.1016/j.dld.2012.12.001
96. Farstad IN. Heterogeneity of intraepithelial lymphocytes in refractory sprue: potential implications of CD30 expression. Gut. (2002) 51(3):372–8. doi: 10.1136/gut.51.3.372
97. Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med (2017) 214(5):1371–86. doi: 10.1084/jem.20160894
98. deLeeuw RJ, Zettl A, Klinker E, Haralambieva E, Trottier M, Chari R, et al. Whole-genome analysis and HLA genotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma subtypes. Gastroenterology. (2007) 132(5):1902–11. doi: 10.1053/j.gastro.2007.03.036
99. Sharaiha RZ, Lebwohl B, Reimers L, Bhagat G, Green PH, Neugut AI. Increasing incidence of enteropathy-associated T-cell lymphoma in the United States, 1973-2008. Cancer. (2012) 118(15):3786–92. doi: 10.1002/cncr.26700
100. Verbeek WHM, Van De Water JMW, Al-Toma A, Oudejans JJ, Mulder CJJ, Coupé VMH. Incidence of enteropathy - associated T-cell lymphoma: A nation-wide study of a population-based registry in The Netherlands. Scand J Gastroenterol (2008) 43(11):1322–8. doi: 10.1080/00365520802240222
101. Catassi C, Bearzi I, Holmes GKT. Association of celiac disease and intestinal lymphomas and other cancers. Gastroenterology. (2005) 128(4):S79–86. doi: 10.1053/j.gastro.2005.02.027
102. Delabie J, Holte H, Vose JM, Ullrich F, Jaffe ES, Savage KJ, et al. Enteropathy-associated T-cell lymphoma: clinical and histological findings from the International Peripheral T-Cell Lymphoma Project. Blood. (2011) 118(1):148–55. doi: 10.1182/blood-2011-02-335216
103. Wierdsma NJ, Nijeboer P, de van der Schueren MAE, Berkenpas M, van Bodegraven AA, Mulder CJJ. Refractory celiac disease and EATL patients show severe malnutrition and malabsorption at diagnosis. Clin Nutr (2016) 35(3):685–91. doi: 10.1016/j.clnu.2015.04.014
104. Sieniawski M, Angamuthu N, Boyd K, Chasty R, Davies J, Forsyth P, et al. Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood. (2010) 115(18):3664–70. doi: 10.1182/blood-2009-07-231324
105. Gale J, Simmonds PD, Mead GM, Sweetenham JW, Wright DH. Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol (2000) 18(4):795–5. doi: 10.1200/JCO.2000.18.4.795
106. Egan LJ, Walsh SV, Stevens FM, Connolly CE, Egan EL, McCarthy CF. Celiac-associated lymphoma. A single institution experience of 30 cases in the combination chemotherapy era. J Clin Gastroenterol (1995) 21(2):123–9. doi: 10.1097/00004836-199509000-00012
107. de Baaij LR, Berkhof J, van de Water JMW, Sieniawski MK, Radersma M, Verbeek WHM, et al. A new and validated clinical prognostic model (EPI) for enteropathy-associated T-cell lymphoma. Clin Cancer Res (2015) 21(13):3013–9. doi: 10.1158/1078-0432.CCR-14-2195
108. Khalaf WF, Caldwell ME, Reddy N. Brentuximab in the treatment of CD30-positive enteropathy-associated T-cell lymphoma. J Natl Compr Cancer Network. (2013) 11(2):137–40. doi: 10.6004/jnccn.2013.0021
109. Voorhees TJ, Ghosh N, Grover N, Block J, Cheng C, Morrison K, et al. Long-term remission in multiply relapsed enteropathy-associated T-cell lymphoma following CD30 CAR T-cell therapy. Blood Adv (2020) 4(23):5925–8. doi: 10.1182/bloodadvances.2020003218
110. Isaacson PG, Du MQ. Gastrointestinal lymphoma: where morphology meets molecular biology. J Pathol (2005) 205(2):255–74. doi: 10.1002/path.1703
111. Tjon JML, Verbeek WHM, Kooy-Winkelaar YMC, Nguyen BH, van der Slik AR, Thompson A, et al. Defective synthesis or association of T-cell receptor chains underlies loss of surface T-cell receptor–CD3 expression in enteropathy-associated T-cell lymphoma. Blood. (2008) 112(13):5103–10. doi: 10.1182/blood-2008-04-150748
112. Chott A, Haedicke W, Mosberger I, Födinger M, Winkler K, Mannhalter C, et al. Most CD56+ Intestinal lymphomas are CD8+CD5– T-cell lymphomas of monomorphic small to medium size histology. Am J Pathol (1998) 153(5):1483–90. doi: 10.1016/S0002-9440(10)65736-7
113. Lenti MV, Biagi F, Lucioni M, Di Sabatino A, Paulli M, Corazza GR. Two cases of monomorphic epitheliotropic intestinal T-cell lymphoma associated with coeliac disease. Scand J Gastroenterol (2019) 54(8):965–8. doi: 10.1080/00365521.2019.1647455
114. Nairismägi ML, Tan J, Lim JQ, Nagarajan S, Ng CCY, Rajasegaran V, et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia. (2016) 30(6):1311–9. doi: 10.1038/leu.2016.13
115. Nicolae A, Xi L, Pham TH, Pham TA, Navarro W, Meeker HG, et al. Mutations in the JAK/STAT and RAS signaling pathways are common in intestinal T-cell lymphomas. Leukemia. (2016) 30(11):2245–7. doi: 10.1038/leu.2016.178
Keywords: coeliac disease, refractory coeliac disease, enteropathy-associated T-cell lymphoma, gastrointestinal lymphomas, differential diagnosis
Citation: Scarmozzino F, Pizzi M, Pelizzaro F, Angerilli V, Dei Tos AP, Piazza F, Savarino EV, Zingone F and Fassan M (2023) Refractory celiac disease and its mimickers: a review on pathogenesis, clinical-pathological features and therapeutic challenges. Front. Oncol. 13:1273305. doi: 10.3389/fonc.2023.1273305
Received: 05 August 2023; Accepted: 20 October 2023;
Published: 07 November 2023.
Edited by:
Rosa Bacchetta, Stanford University, United StatesReviewed by:
Maria Raffaella Ambrosio, University of Siena, ItalyCopyright © 2023 Scarmozzino, Pizzi, Pelizzaro, Angerilli, Dei Tos, Piazza, Savarino, Zingone and Fassan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Pizzi, marco.pizzi.1@unipd.it
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.