
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 31 July 2023
Sec. Breast Cancer
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1240865
 Brando Poggiali1
Brando Poggiali1 Agostino Ponzetti2
Agostino Ponzetti2 Marica Malerba1Fabio Landuzzi1
Marica Malerba1Fabio Landuzzi1 Federica Furia1
Federica Furia1 Debora Charrance1Sara Trova3
Debora Charrance1Sara Trova3 Vittoria Perseghin1Patrizia A. Falcone4Valentina Alliod2
Vittoria Perseghin1Patrizia A. Falcone4Valentina Alliod2 Alessandra Malossi2Pierpaolo Carassai5Ubaldo Familiari5
Alessandra Malossi2Pierpaolo Carassai5Ubaldo Familiari5 Manuela Vecchi3
Manuela Vecchi3 Stefano Gustincich3Marina Schena2Andrea Cavalli1,6*†
Stefano Gustincich3Marina Schena2Andrea Cavalli1,6*† Alessandro Coppe1*†
Alessandro Coppe1*†Breast carcinoma is the most prevalent cancer among women globally. It has variable clinical courses depending on the stage and clinical-biological features. This case report describes a 56-year-old female with invasive breast cancer without estrogen or progesterone receptor expression, with apocrine differentiation, and with no germline variants in the BRCA1 and BRCA2 genes. Throughout the clinical course, the patient exhibited discordant results for HER2 in immunohistochemistry and in situ hybridization. During the second relapse, the disease displayed apocrine microscopic features. The tumor underwent analysis for the androgen receptor, GCDFP-15, RNA-seq, and whole-genome sequencing (WGS) to identify the breast cancer subtype and to characterize the cancer genome. Our bioinformatic analysis revealed 20,323 somatic SNV/Indels, including five mutations in cancer-related genes that are believed to be responsible for the tumor’s development. Two of these mutations were found in the PIK3CA and TP53 genes. Furthermore, the tumor tissue exhibited large copy number alterations to the chromosomes, which could impact gene expression through complex mechanisms and contribute to the tumor phenotype. Clustering algorithms applied on RNA-sequencing data categorized this cancer as a HER2+ subtype. The second-line capecitabine chemotherapy treatment is ongoing, and the patient is responding well. Bioinformatic results support the current treatment decision and open the way to further treatments.
Breast cancer is the most common cancer in women, with over 2.2 million new cases and 684,996 deaths reported in 2020 (1). While early-stage (non-metastatic) breast cancer is considered curable, advanced or metastatic breast cancer remains incurable, with a 5-year survival rate of only 38% (2). However, therapeutic strategies are available. Their main goal is prolonging survival and maintaining quality of life, and even better results can be obtained in the oligometastatic setting (3, 4).
The tumor’s histological and molecular characteristics influence breast cancer treatment decisions. Indeed, breast cancer is a molecularly heterogeneous disease, and several classifications have been developed to group tumors based on their molecular features. Sørlie et al. classified breast cancers based on their gene expression profile (5) into five intrinsic subtypes: Luminal A, Luminal B, HER2-enriched, Basal-like, and Normal breast-like. Each category has well-defined classical immunochemistry markers, such as estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), and Ki-67 (6). Invasive apocrine carcinoma is a rare subtype of non-luminal breast cancer. Invasive apocrine carcinoma is HER2-positive in ~30% of cases and displays significant biological aggressiveness, potentially related to the activation of the androgen receptor (AR) pathway (7, 8). Identifying the correct molecular subtype is crucial for treatment decisions because each subtype has specific therapeutic targets and different prognoses (9). Furthermore, germline mutation in BRCA1/2 and somatic mutations, such as copy number alterations (CNAs) and single nucleotide variants (SNV) in driver genes (e.g. TP53, PIK3CA), have prognostic relevance for the therapy outcome and survival (10–13), including in subtypes like apocrine carcinoma (14). The complexity of breast cancer and its variability in responding to different treatments underscores the need for personalized medicine.
In this paper, we present a case report of a patient with an ER/PR-negative invasive breast cancer, which, from a clinical-molecular perspective, exhibited a discordant HER2 status and expression of the AR. At the second relapse, the tumor displayed partial apocrine features and a pathogenetic PIK3CA mutation. The clinical course could be defined as oligometastatic, and the response was obtained with both first-line paclitaxel and second-line capecitabine combined with radiotherapy.
At the second relapse, we performed both whole-genome and transcriptome sequencing analyses to fully characterize the tumor’s genomic variations and gene expression. Our findings provide insight into the molecular characteristics of this unique breast cancer subtype and may contribute to the development of more effective personalized treatment strategies.
We present the case of a 56-year-old female with no significant medical history or family history of cancer. During routine screening in November 2015, she was diagnosed with localized left breast cancer. The patient underwent a breast-wide excision with sentinel lymph node biopsy, revealing a breast carcinoma not otherwise specified (NOS), grade 2 according to Elston-Ellis classification, without expression of ER or PR, and with a HER2 score of 2+ (Figure 1) but without amplification at fluorescence in situ hybridization (FISH). The Ki-67 labeling index was 40%, and the disease was classified as pT1c-pN1(sn). Post-operative computed tomography scan and bone scintigraphy showed no signs of distant metastasis. From February to August 2016, the patient received adjuvant chemo-radiotherapy with epirubicin, cyclophosphamide, and paclitaxel. This was followed by 45 Gy in 20 fractions on the left breast plus 5 Gy boost on the surgical bed. BRCA1 and BRCA2 analysis showed no mutations.
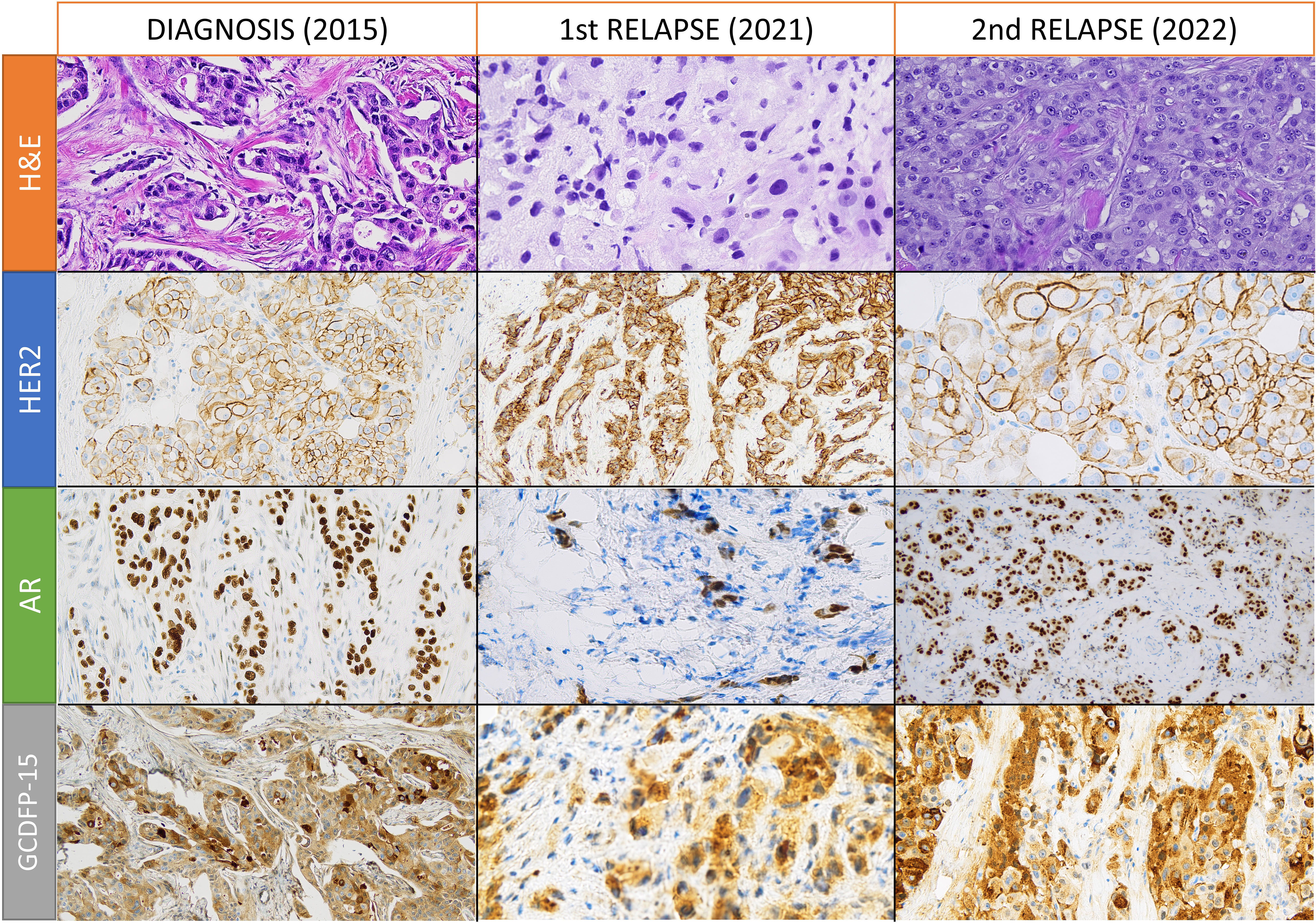
Figure 1 Hematoxylin and eosin (H&E) and immunohistochemical stainings for human epidermal growth factor receptor 2 (HER2), androgen receptor (AR), and gross cystic disease fluid protein 15 (GCDFP-15) in samples from diagnosis, first relapse, and second relapse. All images are obtained with 200X magnification, except for HER2 and AR stainings of the second relapse (obtained with 400X and 100X magnification, respectively).
During follow-up, in December 2020, a left supraclavicular lymph node appeared and an 18-fluorodeoxyglucose (18-FDG) positron emission tomography-computed tomography (PET-CT) scan confirmed the disease relapse in five non-bulky supraclavicular and retropectoral lymph nodes. A lymph-nodal ultrasound-guided core biopsy revealed malignant cells from breast carcinoma that were ER-negative and PR-negative with a HER2-score of 3+ (Figure 1) but without gene amplification at chromogenic in situ hybridization (CISH). PD-L1 staining with Ventana SP142 clone was negative on tumor-infiltrating lymphocytes with an IC score of less than 1%.
After discussing the risks and benefits of a single-agent chemotherapy with the patient and her relatives, she received first-line chemotherapy with Paclitaxel from February 2021 to January 2022. This produced a partial response to the disease; however, the chemotherapy caused persistent maximum grade (maxG) 2 peripheral neuropathy.
In February 2022, ultrasonography and PET-CT confirmed the clinical suspicion of oligoprogression in a new supraclavicular lymph node at a site anterior to the previous site, and in two internal mammary nodes.
After a multidisciplinary discussion, supraclavicular lymph node surgical excision was performed, and histopathological analysis showed a gross cystic disease fluid protein 15 (GCDFP-15) positive breast carcinoma with partial apocrine differentiation and expression of the AR in 95% of cells. The carcinoma was ER-negative, PR-negative, and HER2 3+, but without amplification at CISH and FISH. A revision was performed in a referral center in Turin, Italy, and the HER2 IIC was downstaged to 2+ disease. The histopathological samples from breast disease (2015) and the first lymph nodal relapse were re-assessed and showed positivity for the AR and GCDFP-15.
Next-generation sequencing (NGS) analysis was performed on the metastatic supraclavicular lymph node using Myriapod NGS Cancer Panel DNA. This showed a PIK3CA p.His1047Arg mutation with an allelic frequency of 26.39%. After excluding a dihydropyrimidine dehydrogenase (DPYD) polymorphism, the patient began receiving second-line capecitabine in April 2022. From June to July 2022, consolidation radiotherapy was performed on the left infra-supraclavicular region and a local boost of 9 Gy with maxG2 fatigue, maxG1 hand-foot syndrome grade, and maxG2 radiodermatitis.
In November 2022, after the sixth cycle of capecitabine, a PET-CT scan with 18-FDG showed a metabolic response at all disease sites, without any pathological FDG-capitation.
The second-line chemotherapy is ongoing.
Genomic DNA was extracted from peripheral blood (control) and the metastatic supraclavicular lymph node, obtained from the second relapse. Whole-genome sequencing (WGS) was performed on the two samples using a PCR-free library approach and the Novaseq 6000 System with target coverages of 60X and 120X for the blood and tumor samples, respectively. The NVIDIA Clara Parabricks pipelines were used to identify germline and somatic variants, which were annotated and filtered using an in-house pipeline (see Supplementary Materials).
No pathogenic or likely pathogenic germline variants were found in breast cancer predisposition genes including BRCA1 and BRCA2 (15). After subtracting the germline variants from the metastatic tumor sample, we detected 20,323 somatic mutations (16,544 SNVs and 3,779 Indels). In particular, we identified five mutations in five cancer-related genes (see Table 1) that could be responsible for the tumor’s development. Two of these mutations were classified as pathogenic according to CLINVAR (16): a PIK3CA mutation with a variant allele frequency of 34.17%, and a TP53 mutation with a variant allele frequency of 55.71%. CLINVAR did not classify the mutations in NFB2, ATM, or BTK as clinically significant. CANCERVAR software classified the mutation in NFKB2 as having uncertain significance (tier III) based on three types of evidence. It classified the mutation in ATM as potentially clinically significant (tier II) based on 8 types of evidence. Lastly, the mutation in BTK was classified as having uncertain significance based on seven types of evidence.

Table 1 Somatic variants obtained from the WGS bioinformatic analysis.
To investigate the type of somatic mutations and the processes that generated them, we performed a mutational signatures analysis using R (www.r-project.org) and the MutationalPatterns (17) R/Bioconductor package. We analyzed 16,544 SNVs for mutational changes and sequence context, and generated a plot that represents the abundance of somatic SNVs in trinucleotide contexts, which is also defined as a 96-mutational profile (see Figure 2A). The most abundant nucleotide change was T>G, and the most enriched mutational trinucleotide contexts were ATT and TTT sequences. We decomposed the 96-mutational profile into different mutational signatures stored in COSMIC (18). Their relative contributions are shown in Figure 2B. We detected seven substitution mutational signatures (SBSs) in the tumor sample: SBS1, SBS9, SBS17b, SBS28, SBS37, SBS40, and SBS89. SBS40 contributed the most to the 96-mutational profile. The number of mutations attributed to SBS40 correlates with patient age in different types of human cancer (18), although the etiology is unknown. SBS1 is attributed to the deamination of 5-methylcytosine and is a clock-like signature, with the number of mutations correlating with age in normal cells and cancer cells (19). SBS9 seems to be due to the activity of polymerase η (18). The etiology of SBS17b, SBS28, SBS89, and SBS37 is unknown. However, SBS17b is associated with fluorouracil (5FU) chemotherapy treatment and damage inflicted by reactive oxygen species (ROS) (20).
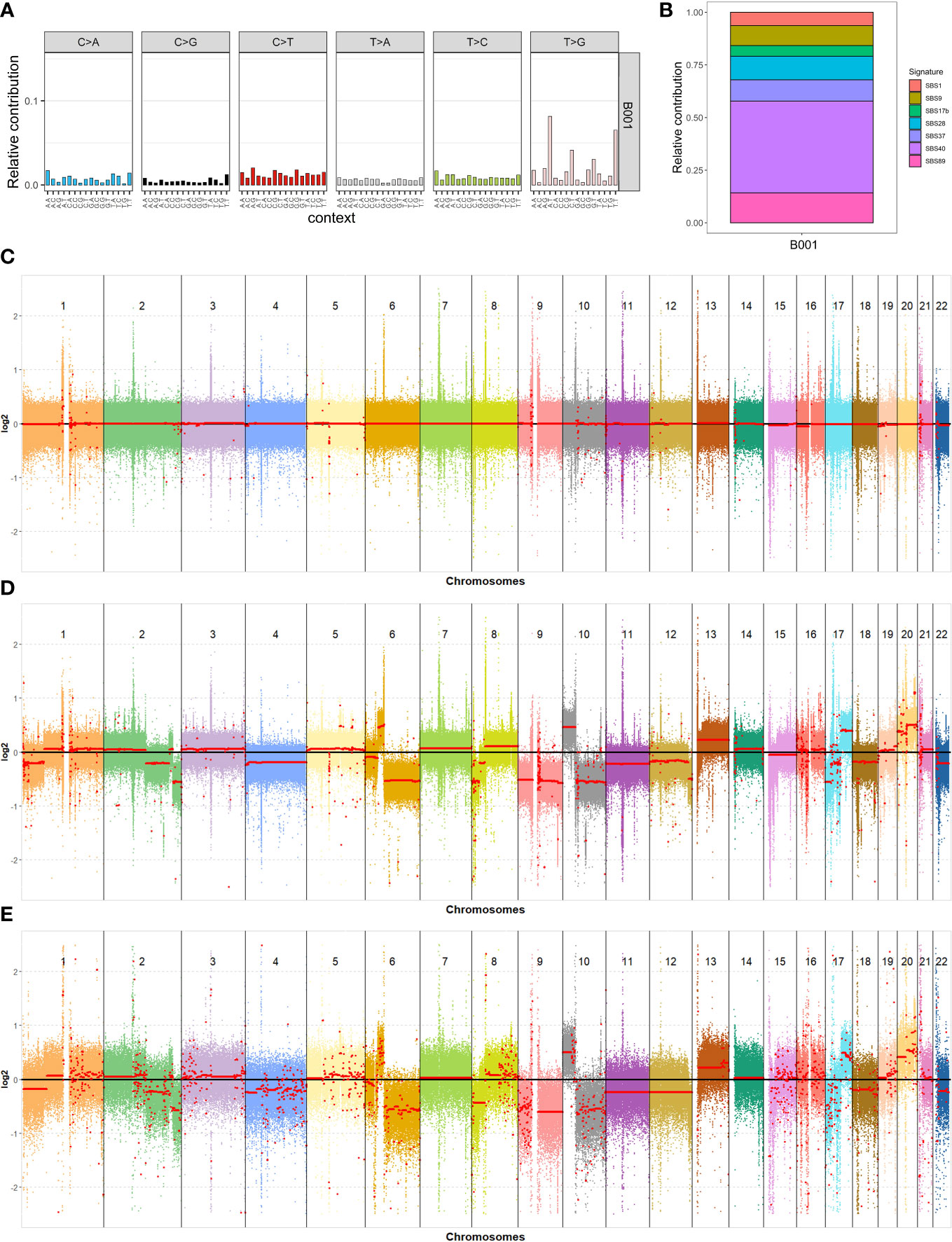
Figure 2 Genomic analysis of the blood and tumor samples. (A) 96-mutational profile; (B) Relative contribution of the different mutational signatures identified after the decomposition of the 96-mutational profile; (C–E) Results of the copy number variations analysis for all autosomal chromosomes: blood and tumor samples using short reads data (C, D) and tumor sample using long reads data (E), respectively.
We used CNVkit software (21) to analyze copy number variations (CNVs) in the genome. The copy number profiles for all autosomal chromosomes in the blood and tumor samples are shown in Figures 2C, D, respectively. Each colored dot represents the log2 ratio in a sequence range of 750 bp, while the red line is the average log2 ratio on a broader region. The red line of blood chromosomes lies exactly on the log2 ratio value of zero, which means that they do not have any CNAs. In contrast, the tumor shows large CNVs in the chromosomes. Our focus was on cancer-associated genes located in chromosomal regions detected with CNV analysis. We identified 2,414 genes in duplicated regions, with 1294 of these coding for proteins, and 1528 genes in deleted regions, with 768 of these coding for proteins (Table S1). To identify potential tumor-specific genes, we filtered these genes using a list of 723 cancer-associated genes from the Cancer Gene Census website (https://cancer.sanger.ac.uk/census), resulting in 45 duplicated and 90 deleted genes (Table S2). We decided to perform CNV analysis in the tumor sample using data from third-generation sequencing technology. This was to reduce problems related to the short-read sequencing methodology in the CNV analyses, such as secondary alignments due to highly repetitive regions and technical duplicates. The tumor sample was sequenced, and we obtained a genome coverage of 27.54X, an N50 length of 45,147 bp, and a mean length of 17,518.9 bp. The CNV analysis was performed by setting a bin of 10,000 bp to reduce the high variance of the log2 ratio in each bin probability due to the lack of a reference and the lower coverage relative to the Illumina experiment. Nevertheless, the CNV analysis obtained with PromethION 24 (Oxford Nanopore Technologies) showed large chromosome alterations to the tumor genome (Figure 2E), which strongly correspond to the alteration detected using Illumina sequencing data (Figure 2D).
Finally, tumor mutational burden (4.65 muts/Mb) and microsatellite instability (0.04% of mutated microsatellites) analyses did not show any relevant results (see Supplementary Materials).
To gain insight into the gene expression profile of the metastatic tumor sample, we conducted a comprehensive analysis using RNA sequencing (RNA-Seq) with the NovaSeq 6000 System (Illumina) and an in-house pipeline (Supplementary Materials). To supplement our analysis, we incorporated publicly available datasets, including a total of 28 healthy breast tissue samples (including 15 adjacent noncancerous tissues) downloaded from NCBI SRA (project accession numbers: PRJNA292118, PRJNA855324, PRJNA839244). We also used RNA-sequencing data from 1085 breast cancer patients obtained from the TCGA datasets (see Supplementary Materials).
Next, we compared the expression levels of the tumor-specific genes located in CNVs in our tumor sample to those of 10 healthy breast tissue samples from the PRJNA855324 and PRJNA839244 projects. In the set of 47 duplicated genes, we detected 24 genes in our tumor sample with higher expression levels than healthy breast tissues (Table S3), while 2 genes had lower expression levels. For the deleted genes, 25 had a lower expression, and 4 had a higher expression (Table S4).
We also examined the expression levels in the tumor sample of critical genes involved in breast cancer, including Progesterone Receptor (PGR), Estrogen Receptor 1 (ESR1), Human Epidermal growth factor Receptor 2 (HER2, also known as ERBB2), Marker Of Proliferation Ki-67 (MKI67), and AR. Subsequently, we compared these levels with those observed in 28 samples of healthy breast tissue. The resulting heatmap (Figure 3A) showed that the tumor sample had low expression levels for PGR and ESR1, medium expression levels for MKI67, and high expression levels for HER2 and AR.
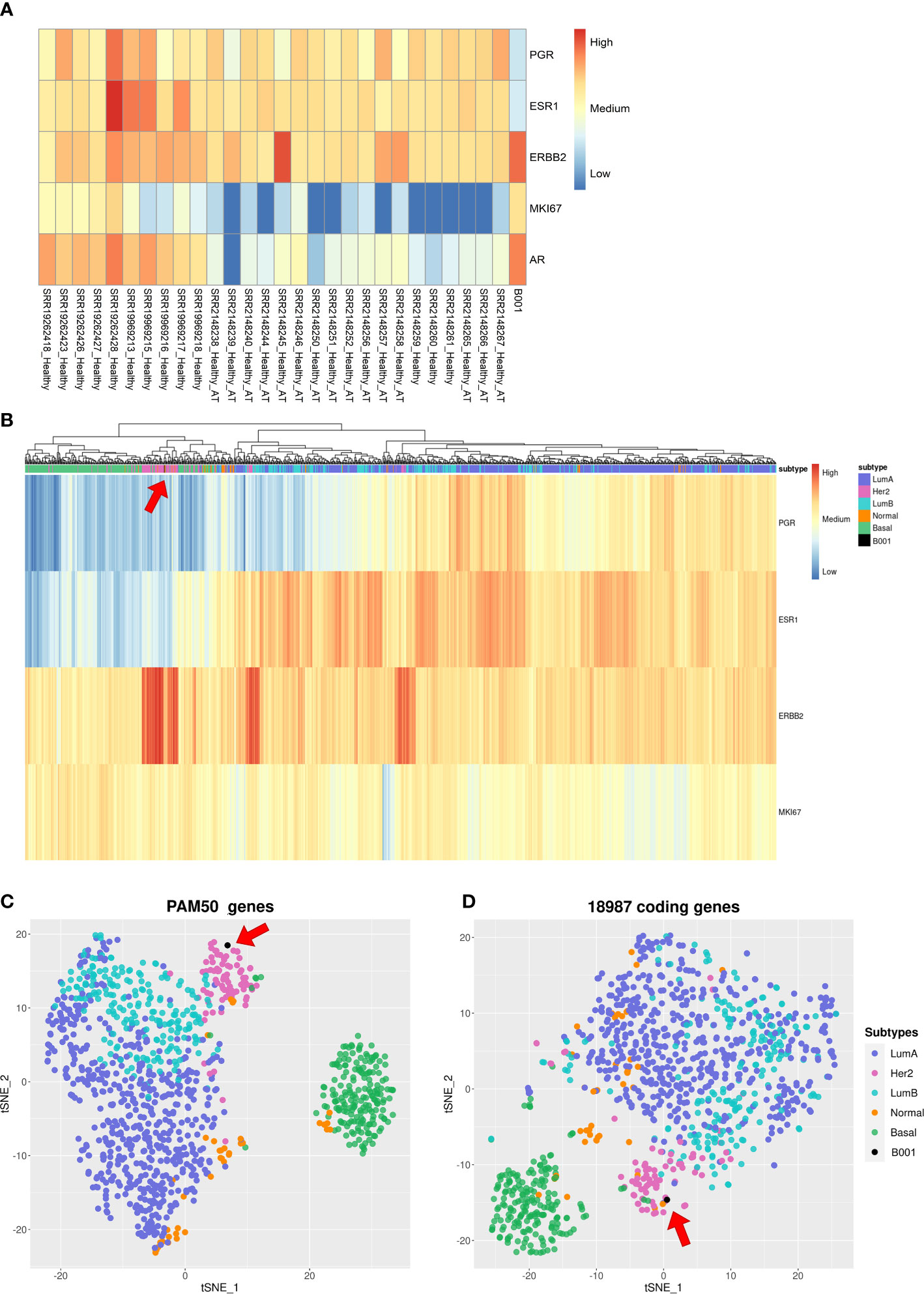
Figure 3 Gene expression analysis of the tumor sample. (A) Hierarchical clustering heatmap with gene expression levels of PGR, ESR1, HER2, MKI67, and AR in breast cancer tissue from 28 healthy patients and from our tumor sample; (B) Hierarchical clustering heatmap showing gene expression levels of PGR, ESR1, HER2, and MKI67 of 1085 breast cancer patients in the TCGA dataset and our tumor sample; (C, D) T-SNE algorithm with 1085 breast cancer patients in the TCGA dataset and our tumor sample using PAM50 genes and 18987 coding genes, respectively.
To further understand the molecular subtype of our tumor sample, we performed cluster analysis of 1085 breast cancer patients from TCGA datasets using different sets of genes. Figure 3B reports a heatmap built with the four genes used to classify the breast cancer subtypes: PGR, ESR1, HER2, and MKI67. These four genes capture a structure in the data: there is a light green cluster containing Basal-like patients, a pink cluster with HER2+ patients, and on the right branch of the dendrogram there are Luminal A and Luminal B samples, which are not separated. Our sample is in the pink cluster close to the HER2+ patients. Figure 3C shows a t-distributed stochastic neighbor embedding (t-SNE) plot built using PAM50 genes. In this case, we see: i) a well-defined cluster related to Basal-like and HER2+ patients; ii) Luminal A and Luminal B clusters that partially overlap; and iii) Normal-like patients with no well-defined location in the plot. Our sample is in the HER2-enriched cluster. To gain a complete picture of the tumor’s gene expression profile, we analyzed 18,987 coding genes in the human genome using the t-SNE statistical method. While the resulting clusters were not well-defined for all subtypes, we were still able to identify a cluster of patients with Basal-like subtypes and a blurry cluster of HER2+ individuals, which contained our sample (Figure 3D).
Adapting the clinical and molecular classification of breast cancer to a single patient’s disease is one of the most difficult tasks for medical and molecular oncologists.
In this study, we investigated the deep molecular issues of a patient with a metastatic ER-negative and PR-negative breast cancer with partial apocrine differentiation, in agreement with GCDFP-15 expression, and an oligometastatic clinical course. Since the diagnosis, the disease exhibited a moderate (2+) or strong (3+) expression of the HER2 protein, but CISH or FISH analyses were negative, thus indicating a HER2-low disease (22). After more than 20 years of clinical use of anti-HER2, emerging data and improved analytical methods mean that the dichotomous classification of a positive or negative HER2 clinical category is evolving towards a continuum (23, 24). Here, our patient did not receive any anti-HER2 antibodies associated with conventional chemotherapy. At diagnosis, this choice was based on the HER2 2+/FISH-negative result. At relapse, despite the HER2 3+ result, CISH was performed (FISH was not available at our center at that time) because of the slow and oligometastatic behavior. Single-agent paclitaxel was then administered with a progression-free interval of nearly one year. At the second oligometastatic relapse, the case was revised in a referral center. Both CISH and FISH for HER2 were performed, again with negative results. Thus, systemic treatment with single-agent capecitabine was chosen because of the possibility of concurrent radiotherapy, obtaining a complete metabolic response.
The tumor genome revealed a complex picture, with 20,323 somatic mutations (16,544 SNVs and 3,779 Indels). We found an enrichment of T>G nucleotide change in the SNVs, and mutational signature analysis associated some of the somatic mutations with the patient’s age. Additionally, we detected SBS17b, associated with fluorouracil chemotherapy treatment and damage inflicted by ROS, which could be due to the patient’s drug treatments. However, most SBSs do not yet have a known etiology. Further studies are needed to identify the etiology of these SBSs and increase the value of mutational signature analysis for personalized medicine. After filtering the variants, we identified five SNVs potentially associated with the development of cancer in our patient: PIK3CA, TP53, NFKB2, ATM, and BTK. Pathogenic mutations in PIK3CA and TP53 were hypothesized to be drivers of the cancer. Because the sample’s tumor purity was higher than 80%, the mutation in TP53 seemed to be present in all the cancer cells (allele frequency 55.71%), while the mutation in PIK3CA was present in a large percentage of them (allele frequency 34.17%). We also observed large chromosomal duplications and deletions in several chromosomes, highlighting the genomic instability of the tumor sample. These alterations presented a log2 between -1 and 1, except in rare cases, which indicated their presence in one or more tumor cell subpopulations but not in the entire population of tumor cells. Given the large size of these alterations, we hypothesize that several genes and regulatory elements are involved and contribute to the cancer phenotype. To the best of our knowledge, this report is the first scientific paper showing chromosome alterations in apocrine breast cancer using short-read and long-read sequencing approaches.
Our RNA-seq analysis characterized the tumor sample’s gene expression profile. The results supported the immunohistochemical analysis in classifying the breast cancer subtype. Three different clustering approaches were used to achieve the goal: 1) Hierarchical clustering using Euclidean distance and complete-linkage method with PGR, ESR1, HER2, and MKI67 genes, 2) t-SNE with PAM50 genes, 3) t-SNE with 18987 coding genes. All three methods clustered our sample with the HER2+ samples. This result, combined with the immunohistochemical analysis, led us to categorize this breast cancer sample as a HER2-positive subtype, although no HER2 amplification at FISH was found. These promising results with clustering approaches highlight the need for a machine learning model running on gene expression data to improve the classification of breast cancer subtypes.
The results from the NGS and genomic/transcriptomic analysis confirmed our previous treatment choice but also opened the way for further treatments. Given the evidence of a strong intracellular driver (i.e. PIK3CA) combined with an inactivating TP53 mutation, conventional chemotherapy is a more suitable candidate than anti-androgen or anti-HER2 therapy to stop neoplastic progression. However, if there is further progression, then the positivity of RNA for HER2 and AR pathways will support the use of anti-HER2 drug conjugates (25) or anti-androgenic treatments, possibly combined with off-label use of anti-PIK3CA treatment (26).
Although WGS and RNA-seq demand sophisticated infrastructure and expertise, they provide a comprehensive molecular characterization of the tumor. Our report and bioinformatics analysis offer an innovative personalized omics approach that could complement standard clinical practices and serve as a foundation for further fundamental research.
This case study presents a complex picture of a 56-year-old woman patient with oligometastatic breast cancer with an intermediate phenotype between HER2-positive and triple-negative. The slow clinical course allowed the use of a sequential rather than all-in approach.
The genomic-bioinformatic analysis revealed five SNVs potentially associated with the development of cancer in the patient, as well as large chromosomal duplications and deletions. Somatic SNV mutations were associated with the patient’s age and chemotherapy treatment. RNA-seq analysis supported immunohistochemical analysis in classifying the breast cancer subtype as HER2-positive, although the disease was clinically defined as HER2-low. Overall, the study provides valuable insights into the complex genomic and molecular landscape of breast cancer with partial apocrine differentiation, and emphasizes the need for personalized and comprehensive approaches in cancer research and treatment.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The research was conducted in accordance with the principles embodied in the Declaration of Helsinki, in accordance with the Guidelines ICH-GCP and the applicable regulations. The study was approved by the Ethical Committee of the Regional Hospital “U. Parini”. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
ACa, BP, ACo, MS, AP, MV, SG conceptualized and designed the research. MV, MM, ST, VP, UF performed sample collection and sequencing. ACo, BP, FL, DC, FF carried out bioinformatic data analysis and tables/figures creation. ACo, BP, AP conducted manuscript drafting. ACa, MS, AP, MV, ST, UF, ACo, BP, PF, VA, AM, PC reviewed the written manuscript and participated in the modification. All authors contributed to the article and approved the submitted version.
The research center CMP3VdA and the Project 5000genomi@VdA are co-founded by “Fondo Europeo di Sviluppo Regionale (FESR) Programma Investimenti per la crescita e l’occupazione 2014/20” (European Social Fund, ESF, and European Regional Development Fund, ERDF), the Autonomous Region of the Aosta Valley, and the Italian Ministry of Labour and Social Policy (CUPB68H19005520007). BP, MM, FF, VP, DC have carried out this work supported by a grant of the EU-ESF, the Autonomous Region of the Aosta Valley, and the Italian Ministry of Labour and Social Policy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1240865/full#supplementary-material
Supplementary Figure 1 | Case report timeline. Presented according to CARE guidelines. The dotted line is for ongoing treatments.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Gennari A, André F, Barrios CH, Cortés J, de Azambuja E, DeMichele A, et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann Oncol (2021) 32(12):1475–95. doi: 10.1016/j.annonc.2021.09.019
3. Dorn PL, Corbin KS, Al-Hallaq H, Hasan Y, Chmura SJ. Feasibility and acute toxicity of hypofractionated radiation in large-breasted patients. Int J Radiat Oncol Biol Phys (2012) 83(1):79–83. doi: 10.1016/j.ijrobp.2011.05.074
4. Kent CL, McDuff SGR, Salama JK. Oligometastatic breast cancer: where are we now and where are we headed?-a narrative review. Ann Palliat Med (2021) 10(5):5954–68. doi: 10.21037/apm-20-1128
5. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA (2001) 98(19):10869–74. doi: 10.1073/pnas.191367098
6. Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res (2015) 5(10):2929–43.
7. Dellapasqua S, Maisonneuve P, Viale G, Pruneri G, Mazzarol G, Ghisini R, et al. Immunohistochemically defined subtypes and outcome of apocrine breast cancer. Clin Breast Cancer. (2013) 13(2):95–102. doi: 10.1016/j.clbc.2012.11.004
8. Vranic S, Feldman R, Gatalica Z. Apocrine carcinoma of the breast: A brief update on the molecular features and targetable biomarkers. Bosn J Basic Med Sci (2017) 17(1):9–11. doi: 10.17305/bjbms.2016.1811
9. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers (2019) 5(1):67. doi: 10.1038/s41572-019-0122-z
10. Hurvitz SA, Gonçalves A, Rugo HS, Lee KH, Fehrenbacher L, Mina LA, et al. Talazoparib in patients with a germline BRCA-mutated advanced breast cancer: detailed safety analyses from the phase III EMBRACA trial. Oncologist (2020) 25(3):e439–50. doi: 10.1634/theoncologist.2019-0493
11. Varna M, Bousquet G, Plassa LF, Bertheau P, Janin A. TP53 status and response to treatment in breast cancers. J Biomedicine Biotechnol (2011) 2011:1–9. doi: 10.1155/2011/284584
12. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for -mutated, hormone receptor-positive advanced breast cancer. N Engl J Med (2019) 380(20):1929–40. doi: 10.1056/NEJMoa1813904
13. Hu W, Li M, Zhang Q, Liu C, Wang X, Li J, et al. Establishment of a novel CNV-related prognostic signature predicting prognosis in patients with breast cancer. J Ovarian Res (2021) 14(1):103. doi: 10.1186/s13048-021-00823-y
14. Sun X, Zuo K, Yao Q, Zhou S, Shui R, Xu X, et al. Invasive apocrine carcinoma of the breast: clinicopathologic features and comprehensive genomic profiling of 18 pure triple-negative apocrine carcinomas. Mod Pathol (2020) 33(12):2473–82. doi: 10.1038/s41379-020-0589-x
15. Lázaro C, Lerner-Ellis J, Spurdle A. Clinical DNA Variant Interpretation: Theory and Practice. Academic Press (2021). p. 436.
16. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
17. Manders F, Brandsma AM, de Kanter J, Verheul M, Oka R, van Roosmalen MJ, et al. MutationalPatterns: the one stop shop for the analysis of mutational processes. BMC Genomics (2022) 23(1):134. doi: 10.1186/s12864-022-08357-3
18. Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer (2004) 91(2):355–8. doi: 10.1038/sj.bjc.6601894
19. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer (2020) doi: 10.1101/322859.
20. Koh G, Degasperi A, Zou X, Momen S, Nik-Zainal S. Mutational signatures: emerging concepts, caveats and clinical applications. Nat Rev Cancer (2021) 21(10):619–37. doi: 10.1038/s41568-021-00377-7
21. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PloS Comput Biol (2016) 12(4):e1004873. doi: 10.1371/journal.pcbi.1004873
22. Tarantino P, Hamilton E, Tolaney SM, Cortes J, Morganti S, Ferraro E, et al. HER2-low breast cancer: pathological and clinical landscape. J Clin Oncol (2020) 38(17):1951–62. doi: 10.1200/JCO.19.02488
23. Marchiò C, Annaratone L, Marques A, Casorzo L, Berrino E, Sapino A. Evolving concepts in HER2 evaluation in breast cancer: Heterogeneity, HER2-low carcinomas and beyond. Semin Cancer Biol (2021) 72:123–35. doi: 10.1016/j.semcancer.2020.02.016
24. Sajjadi E, Venetis K, Ivanova M, Fusco N. Improving HER2 testing reproducibility in HER2-low breast cancer. Cancer Drug Resist (2022) 5(4):882–8. doi: 10.20517/cdr.2022.29
25. Lang Y, Wu B, Liu X. Economic evaluation of trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer in the United States. Breast Cancer. (2022) 14:453–63. doi: 10.2147/BCTT.S389696
Keywords: breast cancer, personalized medicine, NGS, bioinformatic analysis, apocrine carcinoma, case report, WGS, nanopore
Citation: Poggiali B, Ponzetti A, Malerba M, Landuzzi F, Furia F, Charrance D, Trova S, Perseghin V, Falcone PA, Alliod V, Malossi A, Carassai P, Familiari U, Vecchi M, Gustincich S, Schena M, Cavalli A and Coppe A (2023) Multiomic analysis of HER2-enriched and AR-positive breast carcinoma with apocrine differentiation and an oligometastatic course: a case report. Front. Oncol. 13:1240865. doi: 10.3389/fonc.2023.1240865
Received: 15 June 2023; Accepted: 05 July 2023;
Published: 31 July 2023.
Edited by:
Nicola Fusco, University of Milan, ItalyReviewed by:
Carlo Pescia, University of Milan, ItalyCopyright © 2023 Poggiali, Ponzetti, Malerba, Landuzzi, Furia, Charrance, Trova, Perseghin, Falcone, Alliod, Malossi, Carassai, Familiari, Vecchi, Gustincich, Schena, Cavalli and Coppe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Cavalli, YW5kcmVhLmNhdmFsbGlAaWl0Lml0; Alessandro Coppe, YWxlc3NhbmRyby5jb3BwZUBpaXQuaXQ=
†These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.