 Hashnu Dutta1,2*
Hashnu Dutta1,2* Nishant Jain1,2*
Nishant Jain1,2*- 1Department of Applied Biology, CSIR-Indian Institute of Chemical Technology, Hyderabad, India
- 2Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, India
Post-translational modifications (PTMs) are crucial regulatory mechanisms that alter the properties of a protein by covalently attaching a modified chemical group to some of its amino acid residues. PTMs modulate essential physiological processes such as signal transduction, metabolism, protein localization, and turnover and have clinical relevance in cancer and age-related pathologies. Majority of proteins undergo post-translational modifications, irrespective of their occurrence in or after protein biosynthesis. Post-translational modifications link to amino acid termini or side chains, causing the protein backbone to get cleaved, spliced, or cyclized, to name a few. These chemical modifications expand the diversity of the proteome and regulate protein activity, structure, locations, functions, and protein-protein interactions (PPIs). This ability to modify the physical and chemical properties and functions of proteins render PTMs vital. To date, over 200 different protein modifications have been reported, owing to advanced detection technologies. Some of these modifications include phosphorylation, glycosylation, methylation, acetylation, and ubiquitination. Here, we discuss about the existing as well as some novel post-translational protein modifications, with their implications in aberrant states, which will help us better understand the modified sites in different proteins and the effect of PTMs on protein functions in core biological processes and progression in cancer.
Introduction
Post-translational modifications are typical biochemical reactions that covalently bind (poly)peptide chains, chemical moieties, lipids, or carbohydrates to amino acids of the target molecule during or after its translation (1). PTMs occur in majority of known proteins, and nearly all amino acids can be changed by one or more of these reactions. The modified proteins acquire uncommon amino acids that can have a significant impact on their structure and function (2). Akin to noncovalent binding, PTM processes may occur at the functional site (orthosteric PTMs) namely sumoylation, or at a distance (allosteric PTMs) namely, ubiquitination and phosphorylation (1).
Post-translational modifications diversify the proteome by altering protein structure, location, interactions, and function and their regulation, thus affecting all facets of cell biology (Figure 1) (3). Currently, the UniProt database contains over 400 distinct PTMs that are covalently bonded to amino acid side chains. PTMs range in size from moderate alterations such as methylation to high-molecular-weight polyprotein chains such as polyubiquitination (4, 5).
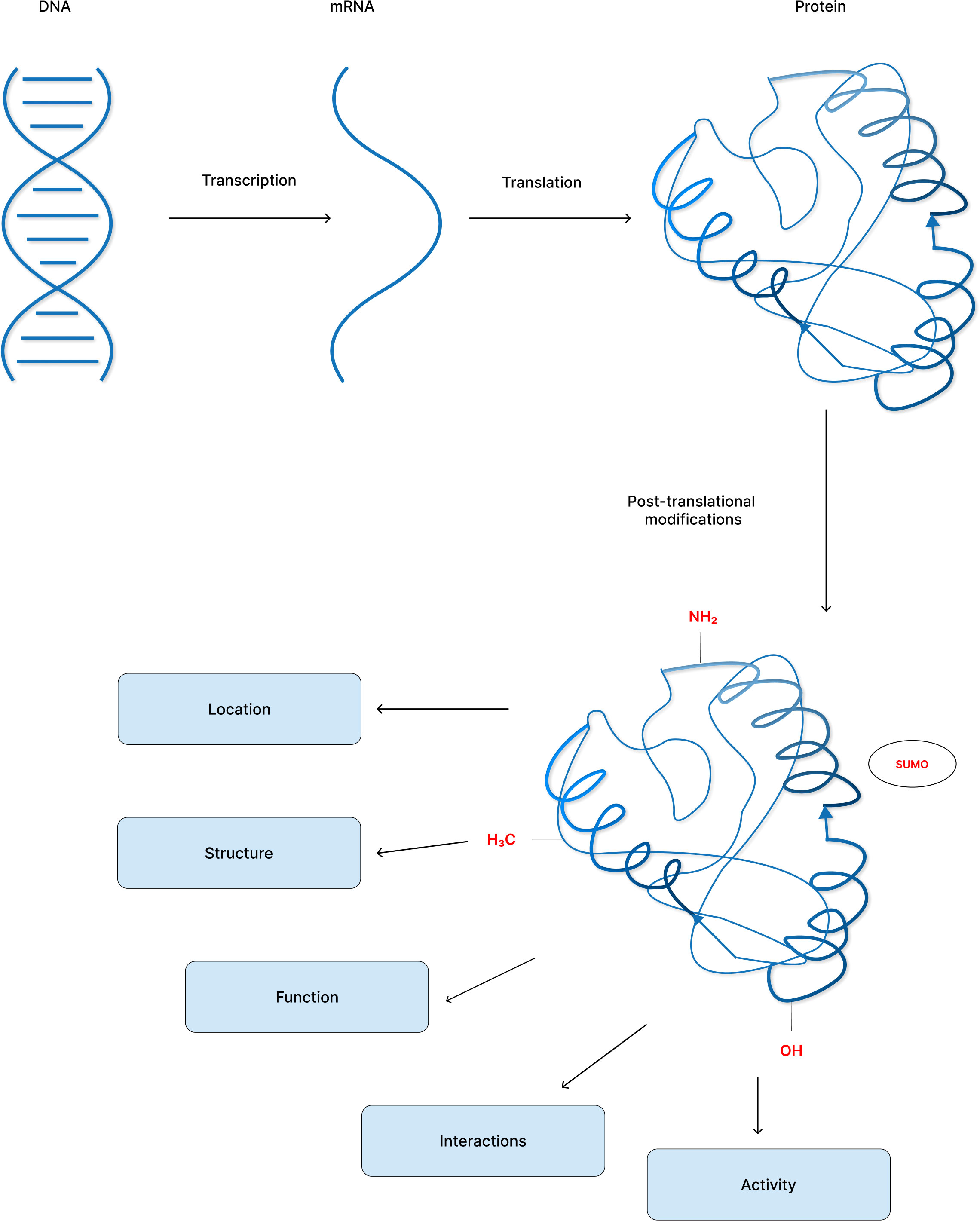
Figure 1 Post-translational modification of proteins increase proteome diversity to perform the core physiological processes.
Protein modifications are commonly catalyzed by enzymes, although a number of reports have stated that some PTMs may happen spontaneous. Non-enzymatic PTMs (that occur spontaneously) can be divided into two subclasses: irreversible and reversible non-enzymatic PTMs, based on their stability and physiological shelf-life. Non-enzymatic irreversible PTMs include stress-induced modifications, such as oxidative and metabolic stresses, that cannot be eliminated by spontaneous dissociation or enzyme-catalyzed mechanisms. These irreversible PTMs occur in a wide spectrum of amino acids, especially when basal levels of reactive oxygen species (ROS) rise and/or when metabolite concentrations approach a physiological imbalance, during aging or in diseases such as cancer, diabetes, and Alzheimer’s disease (6–9). On the contrary, non-enzymatic reversible PTMs are physiologically reversible. The dissociation of these reversible PTMs can be enzyme catalyzed, or from an innate chemical instability, which is involved in various metabolic and signaling pathways.
Eukaryotes exploit post-translational modifications to modulate protein stability, function, subcellular localization, and different protein–protein interactions (10–13). PTMs control protein function of diverse physiological and pathological cellular events. Furthermore, PTMs have been reported to be linked to neurodegenerative disorders, brain tumors, breast neoplasias, and hematological disorders (14, 15). Numerous other forms of PTMs can occur due to metabolic stress. For instance, disruption of Krebs cycle enzymes might increase the fumarate levels followed by succination of cysteine residues by Michael addition reaction (16, 17). Higher succination rates can also be caused by cancer-related mutations influencing fumarate metabolizing proteins, and hence can function as a cancer biomarker (18, 19).
Despite conventional PTMs such as phosphorylation, acetylation, ubiquitination, and methylation that have been extensively studied, the emergence of numerous new modifications, such as hydroxybutyrylation, lactylation, and succinylation, add another dimension to protein regulation, shedding light to many yet-to-be-discovered modifications and their application possibilities per se (20). In this mini-review, we outline a brief overview of both classical (which have been identified within the past few decades) and the novel (newly discovered) post-translational modifications, as well as the important roles these PTMs offer in regulating human cancer hallmarks using relevant examples. Also, the different PTMs that are discussed here have been summarized highlighting the reactions involved and the enzymes catalyzing it (Table 1).
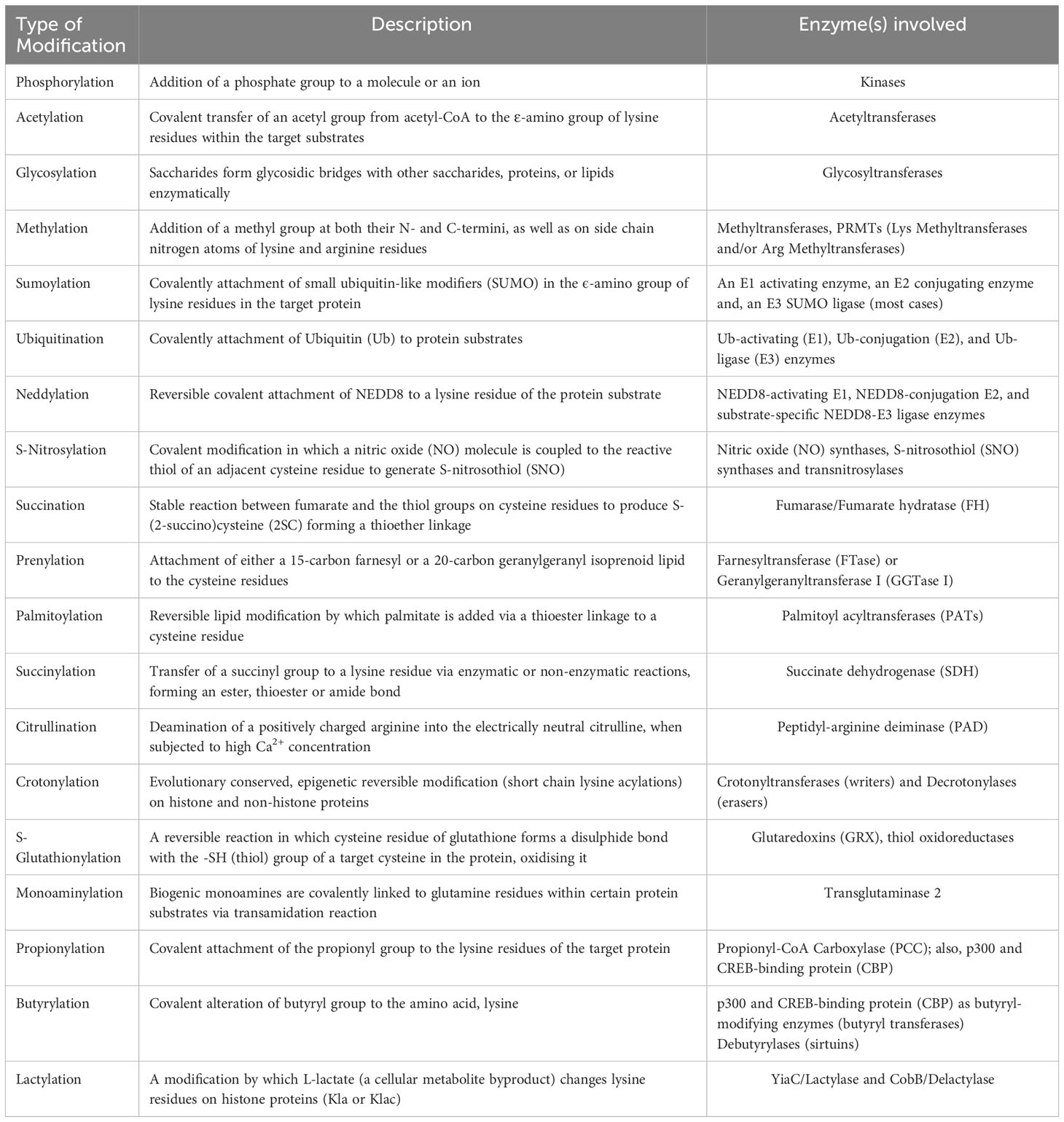
Table 1 An overview of different post-translational modifications and the enzymes catalyzing these reactions.
Classical post-translational modifications
Phosphorylation
Phosphorylation is the most researched and one of the essential types of PTM, which frequently occurs either in nucleus or in cytosol on target proteins in all organisms (21, 22). The kinase enzyme catalyzing the phosphorylation reaction transfers a phosphate group from adenosine triphosphate (ATP) to the receptor residues, thereby altering the protein function in either of the two processes: via allostery or by interacting with the adjoining domains (22). Phosphorylation predominantly occurs on target sites comprising Ser, Thr, Tyr, and His residues (23). In contrast, dephosphorylation or elimination of a phosphate group is a phosphatase-mediated enzymatic reaction (24).
Cancer is one of the most abhorrent disorders in which numerous regulatory gene products are transformed (25). Recent studies have highlighted the involvement of aberrant protein phosphorylation in cancer manifestation (26–29). The primary mechanisms underlying impaired signal transduction are due to uncontrolled phosphorylation (gain- or loss-of -function mutation) (30). Yet, mutations in kinases or phosphatases which induce aberrant phosphorylation and upset the delicate equilibrium of activation or inactivation of cancer-associated proteins are regarded as both the cause and effect of cancer (31).
Acetylation
Acetylation modification is a reversible acylation process catalyzed by acetyltransferase in which an acetyl group from acetyl-CoA is covalently transferred to the ε-amino group of lysine residues (typically) within the target substrates (32, 33). Deacetylase enzymes can also reverse this biochemical process. Acetylation is critical in chromatin remodeling, gene expression, and protein function regulation (34). Many nonhistone proteins, like histones, are sensitive to acetylation and other PTMs (35). Because lysine acetylation was first discovered in histones, many KATs and KDACs are also known as histone acetyltransferases (HATs) and deacetylases (HDACs), respectively (36). Aberrant acetylation of nonhistone proteins is common in several human disorders, particularly cancer (37). Acetylation is also closely associated to active glycolysis in tumors. Phosphoglycerate kinases that include PGK1 and PGK2 are ATP producing glycolytic enzymes and few reports have showed acetylation at K323 in PGK1 increases its enzyme activity and glucose absorption, causing an increase in aberrant hepatic cell growth and carcinogenesis (38). Enolase 2 (ENO2) is another important glycolytic enzyme that is overexpressed in prostate cancer, small-cell lung cancer, metastatic neuroblastoma, and leukemia (39, 40).
Glycosylation
Glycosylation is the most common and diversified type of protein post-translational modifications that is vital for many biological processes (41). Glycosylation is an enzymatic process by which saccharides form glycosidic bridges with other saccharides, proteins, or lipids (42, 43). Protein glycosylation are of two types: N-linked glycosylation (N-glycosylation) and O-linked glycosylation (O-glycosylation), with O-linked being a more difficult modification due to its structural complexity and diversity (41). Different glycoconjugates disrupt essential cancer cell functions as well as the tumor microenvironment, resulting in carcinogenesis. Mostly, cancer cells diverge from the classical glycosylation pathway, resulting in aberrant glycan expression (44). O-GlcNAc modifications have been linked to tumor cell proliferation employing FoxM1, a transcription factor and cyclin D1, both of which are involved in cell cycle progression, cancer cell survival, and angiogenesis (45).
Methylation
Methylation is a classic post-translational modification utilized to transfer information in signal transduction pathways in the cell. Protein methylation can occur at both their N- and C-termini, as well as on side chain nitrogen atoms of lysine and arginine residues (46, 47), mediated by protein methyltransferases (PRMTs) (48). PRMTs, which are a family of enzymes that methylate both histone and nonhistone proteins (49) are of two types, namely, lysine methyltransferases (KMTs) and protein arginine methyltransferases (RMTs). Lysine residues can be mono-, di-, or trimethylated, but arginine residues can only be mono- or can remain demethylated (50). PRMT family members were identified to be overexpressed in a variety of cancers (51, 52). Elevated PRMT3 expression has been linked to poor clinical outcomes in pancreatic cancer (53), colorectal cancer (54), and hepatocellular carcinoma (55). In lung adenocarcinoma, the tumor suppressor DAL-1/4.1B interacts with PRMT3 and suppresses its methyltransferase activity, signifying a putative involvement of PRMT3 regulation in tumor progression (56).
Sumoylation
SUMOylation is an important post-translational protein modification that covalently attaches small ubiquitin-like modifiers (SUMO) in the ϵ-amino group of lysine residues in the target protein, via a multi-enzymatic reaction. SUMO is an 11 kDa protein which has a structural similarity with ubiquitin (57). SUMO is covalently linked to a lysine residue in the substrate protein via three enzymes: activating (SUMO E1), conjugating (SUMO E2), and ligase (SUMO E3) (58). Furthermore, SUMO is dissociated from the target protein by SUMO proteases (sentrin-specific protease 1, SENP1 in humans) (59). The enzymes involved in SUMO pathway are commonly increased in numerous malignancies and have been associated to carcinogenesis and poor patient prognosis. SUMOylation has been linked to chemoresistance and hormone therapy resistance in many studies. Also, the regulation of the oncogene Myc and the SUMOylation machinery has been discovered in pancreatic cancer (60).
Ubiquitination
Ubiquitin (Ub), a 76-amino acid regulatory protein, is covalently attached to protein substrates using a series of enzymatic cascade that involves Ub-activating (E1), Ub-conjugating (E2), and Ub-ligating (E3) enzymes in a highly conserved post-translational modification called as Ubiquitination (or, Ubiquitylation). Ub attaches to substrates in multiple ways, such as a single Ub attached to an amino acid (monoubiquitination) or to many amino acids (multiple monoubiquitination), or as polymeric chains (polyubiquitination). Different Ub chains are produced via isopeptide linkage with the N-terminal methionine (M1) and seven internal lysine (K) residues (61). As a result, mono- or polyubiquitination affects the function of several proteins under healthy and/or pathological events (62, 63). Deubiquitinases (DUBs) inhibit ubiquitination by eliminating Ub modifications from their target substrates, which are crucial for cell cycle, apoptosis, and gene transcription (64, 65). Abnormal ubiquitination may contribute to tumor growth and tumorigenesis (66). Mutations in E3 ligases can rapidly degrade tumor suppressors or, conversely, loss of ubiquitination in growth-promoting oncoproteins (67). Furthermore, E3 ligases and counterbalancing Deubiquitinases are interesting prospective targets for cancer therapies because of their substrate specificity.
Neddylation
Neddylation is a reversible covalent attachment of a ubiquitin-like molecule NEDD8 (neuronal precursor cell-expressed developmentally down-regulated protein 8) to a lysine residue of the protein substrate (68, 69). The NEDD8-activating enzyme E1, the NEDD8-conjugating enzyme E2, and substrate-specific NEDD8-E3 ligases all work together to promote neddylation, similar to ubiquitination (70–72). The key family of multiunit E3 ubiquitin ligases, Cullin-RING ligases (CRLs), regulate 20% of proteasome-mediated protein degradation (73–76). Targeting cullin neddylation looks to be an appealing cancer therapeutic strategy, provided hyperactivation of CRLs support carcinogenesis (77, 78). Aberrant activation of the neddylation pathway results in increased global neddylation of substrates such as cullins, leading to subsequent degradation of tumor suppressors (e.g., p21 and p27) and facilitating carcinogenesis and progression (71, 79). Thus, utilizing the neddylation route to inactivate CRLs can be a feasible anticancer approach.
S-Nitrosylation
S-nitrosylation is a covalent post-translational modification in which a nitric oxide (NO) molecule is coupled to the reactive thiol of an adjacent cysteine residue to generate S-nitrosothiol (SNO) (80). Endogenous NO is synthesized from L-arginine in mammalian cells by three distinct gene-encoded NO synthases (NOSs): neuronal NOS (nNOS/NOS-1), inducible NOS (iNOS/NOS-2), and endothelial NOS (eNOS/NOS-3) (81). S-nitrosylation, like other post-translational modifications, regulates biological activity of a wide range of proteins in vivo, as well as controls transcription (82), DNA repair (83), and apoptosis (84–86). Aberrant S-nitrosylation has been associated with significant pathophysiological events, including reported studies that influence the initiation of cancer and cancer cell proliferation both directly and indirectly (83, 87). Bentz et al. demonstrated in 2000 that nitrotyrosine levels are elevated in reactive and dysplastic types of head and neck squamous cell carcinoma (HNSCC) lesions than in normal squamous mucosa (88).
Succination
Succination is a stable post-translational modification that occurs when the Krebs cycle intermediate fumarate combines with the thiol groups on cysteine residues to produce S-(2-succino)cysteine (2SC) (16). Some studies have found that 2SC is a biomarker for not only mitochondrial stress in obesity and diabetes, but also fumarate hydratase (FH) deficiency in individuals with hereditary leiomyomatosis and renal cell carcinoma (HLRCC) (16, 19, 89, 90). Interestingly, fumarate can enter other cellular and extracellular compartments, most likely because fumarate can be transported from mitochondria via specialized carriers, causing 2SC production and extra-mitochondrial protein impairment. The pool of impaired proteins is referred to as the succinated proteome, which then loses its ability to perform functional and structural activities, possibly leading to cell death (91). There is substantial evidence that shows elevated levels of protein succination in diabetes (17, 92), obesity (89), and cancer (93, 94). As a result, 2SC is identified as a mitochondrial stress biomarker, which may result in cellular dysfunction and even apoptosis in cancer and diabetes (91).
Prenylation
Prenylation is a common covalent post-translational modification that occurs in all eukaryotic cells and is mediated by the enzymes protein farnesyltransferase (FTase) or protein geranylgeranyltransferase I (GGTase I), respectively. Prenylation involves the attachment of either a 15-carbon farnesyl or a 20-carbon geranylgeranyl isoprenoid lipid to the cysteine residues (95). When the farnesyl isoprenoid is involved, this enzyme-catalyzed process is known as farnesylation and when the geranylgeranyl isoprenoid is involved, the reaction is known as geranylgeranylation (96). Prenylation is the first crucial step in membrane targeting and binding, as well as mediating protein-protein interactions for many of these proteins; heterotrimeric G-proteins also depend on prenylation for its function (97). The discovery that protein prenylation was required to maintain the malignant activity of oncogenic Ras GTPases evoked immense interest in this field, as Ras proteins have been known to drive more than 30% of human malignancies (98, 99). Preclinical trials of farnesyltransferase inhibitors (FTIs) in cancer cell lines and animal models were highly effective, which resulted in the entry of four FTIs (as monotherapy or in combination with other anti-cancer drugs) into clinical trials in 2000 (100, 101). This initial victory did not translate into significant anticancer activity in patients with advanced solid tumors or acute myeloid leukaemia (AML), despite promising phase 1 and 2 trials (102). Responses to FTIs, irrespective of cellular or tissue origin, do not appear to rely on RAS mutations, because inhibiting KRAS farnesylation directs to its geranylgeranylation (101). Thus, understanding the molecular mechanism of farnesylated proteins and their inhibition strategies, the therapy regimen for cancer patients responsive to FTI treatment can be augmented.
Palmitoylation
Palmitoylation is an enzyme-mediated reversible lipid modification by which palmitate, a 16-carbon palmitic acid, is added via a thioester linkage to a cysteine (Cys) residue (103). The enzymes regulating both palmitoylation and depalmitoylation are the DHHC protein family named as palmitoyl acyltransferases (PATs) (104) and enzymes such as the acyl protein thioesterase (APT) and alpha beta hydrolase-domain containing protein 17 (ABHD17) protein family showing depalmitoylase activity (105, 106). Palmitoylation occurs on a number of critical cancer-related proteins. The RAS family of small GTPases is one such classic example (107, 108). A report demonstrated that palmitoylation at Cys181 site is required for oncogenic NRAS connection with the plasma membrane, and palmitoylation removal inactivated numerous signaling pathways downstream of NRAS, thereby inhibiting leukemogenesis (109).
Succinylation
Succinylation is a post-translational modification that involves the transfer of a succinyl group (-CO-CH2-CH2-CO2H) to a lysine residue via enzymatic or non-enzymatic reactions (110). Succinylation PTM is predominantly mitochondrial of both prokaryotic and eukaryotic organisms (111, 112) and is involved in the production of mitochondrial energy (113, 114). Aberrant succinylation can influence cancer progression, cardiometabolic disorders, hepatometabolic disorders, and neurological disorders (115). Succinylation of a key glycolytic enzyme, pyruvate kinase muscle isotype M2 (PKM2) has been described to increase its enzymatic activity in the context of cancer, but also a different succinylated lysine residue (K498) has been reported, stating that succinylation may have contrasting effects on the same protein target depending on modified site of lysine residues (116).
Citrullination
Citrullination modification is deamination of a positively charged arginine into the electrically neutral citrulline (a non-coding amino acid), when subjected to an elevated Ca2+ concentration (117) catalyzed by the peptidyl-arginine deiminase (PAD). The peptidyl-arginine deiminase (PAD) enzyme family has five isoenzymes (PAD1-4 and PAD6) that target different tissues (118). Vimentin, actin, collagen, fibronectin, keratin, tubulin, and different histone proteins are amongst the proteins found in the human citrullinome (119, 120). A study found that benign tumors and non-tumor inflamed tissues were devoid of PAD4 expression (121), whereas malignant tumors had significantly higher PAD4 levels than similar primary tumors (122), implying that citrullination plays a role in the transformation from benign neoplasm to invasive malignancy.
S-Glutathionylation
S-glutathionylation is a distinct redox-driven post-translational modification in which the cysteine residue of glutathione forms a disulphide bond with the -SH (thiol) group of a target cysteine in the protein, oxidizing it. The reversible nature of this unique modification can lead to transitory alterations in the target protein’s structure and/or function (123). Few proteins related to cellular metabolism and detoxification are identified to be affected by glutathionylation. One such key metabolic enzyme is pyruvate kinase M2 (PKM2), which is critical for cancer cells due to their reliance on the glycolytic pathway and its potential role in modifying antioxidant responses (124, 125). Anastasiou et al. established that S-glutathionylation of PKM2 Cys358 is important for responding to oxidative stress by permitting PKM2 deactivation (125).
Novel post-translational modifications
Monoaminylation
Monoaminylation is a biochemical process in which biogenic monoamines (serotonin, dopamine, histamine, etc.) are covalently linked to glutamine residues within certain protein substrates, by Transglutaminase 2, an enzyme that catalyzes the transamidation reaction of primary amines to glutamine-carboxamides (126). Monoaminylation is important in several aspects of cellular plasticity (in the brain and maybe elsewhere) (127). Because monoaminylation is one of numerous newly found histone PTMs, further studies are ongoing that may have biological significance in terms of carcinogenesis and cancer therapy.
Crotonylation
Protein crotonylation is one of the newly discovered post-translational modifications that occur primarily on the lysine residue, known as lysine crotonylation (Kcr). Kcr is a conserved modification that is controlled by a number of enzymes and co-enzymes, such as lysine crotonyltransferase (writer), lysine decrotonylase (eraser), specific YEATS proteins (reader), and crotonyl-coenzyme A (donor) (128). Crotonylation was first discovered as a PTM on histone lysines (129). Histone Kcr has been linked to a variety of illnesses, including depression, acute renal damage, HIV latency, and carcinogenesis (130–133). Immunohistochemical staining of tumor specimens with adjacent normal tissues reveals that lysine crotonylation occurs in both the cytoplasm and the nucleus, and that lysine crotonylation level is downregulated in liver cancer, gastric cancer, and renal carcinoma, but upregulated in thyroid, esophageal, colon, pancreatic, and lung cancers (134).
Propionylation
Propionylation (propionyl; C-3 fatty acid chain) is a novel histone acylation modification where in the propionyl group covalent attaches to the lysine residues of the target protein (Kpr). After its discovery in histone proteins, Kpr was also found to be present in nonhistones (135). Propionyl-CoA, the co-substrate for Kpr modification arises from the breakdown product of cholesterol and odd-chain fatty acid oxidation and branched-chain amino acid catabolism, which thereby provides us an insight that Kpr may have a potential role in cellular metabolism that involves chromatin architecture during fasting and catabolism (136, 137). Not much has been observed about the function of propionylation in cancer development yet but it is well-known that most of the acylation modifications act synergistically in tumor pathogenesis (138).
Butyrylation
Butyrylation is one of the newly discovered biochemical acylation modifications, observed in both plants and animals, in which the butyryl group covalently alters the amino acid, lysine (Kbu). Kbu occurs in both histone and nonhistone proteins (135). The butyryl group or butyrate (C-4 fatty acid chain) plays an important role in several human diseases, such as vascular dementia, diabetes, psychiatric disorders, and cancer whereas various plant-based substances have also been potentially associated to Kbu of histones (139–141). p300 and CBP were discovered as butyryl-modifying enzymes, that allows histone H4 to get propionylated and butyrylated. Soon after Kbu was identified, SIRT enzymes (Sirt7) with debutyrylase activities were identified that act as erasers (142). A recent study has made a comparative analysis of histone markers in two types of esophageal squamous cell carcinoma using mass spectrometry, and found that various modifications, such as lysine methylation, acetylation, as well as butyrylation on histones H3 and H4 work together in the development of cancer (143). Since these are novel acylation modifications, further studies are required to explore their roles in different disease pathologies.
Lactylation
Lactylation is another novel post-translational modification that occurs when lactate (L-lactate rather than D-lactate) changes lysine residues on histone proteins (Kla or Klac) (144, 145). Cancer cells rely on aerobic glycolysis to produce energy and metabolites, forming lactate, which supports lysine lactylation (Kla) (145). The role of histone Kla has been investigated in mouse embryo fibroblasts (MEFs), ocular melanoma cells (146), and non-small cell lung cancer (NSCLC) cells (147), suggesting that Kla is important in controlling pluripotency and oncogenesis. Recently it was found that Kla levels were considerably greater in 51 tumor tissues as compared to its corresponding para-cancerous tissues, when pan-Kla levels in gastric tumors and adjacent tissues were evaluated (148). Furthermore, higher Kla levels were associated with poorly differentiated tumors, lymph node metastases, and poorer prognostic rates of patients, thus implying that Kla has prognostic significance in gastric tumors (148).
Discussion
Post-translational modifications (PTMs) affect many essential biological processes by reversibly altering the conformation, function, and/or interaction regions of a protein. Aberrant changes are signs of cellular stress or malfunction, and they have been linked to cancer and a variety of other diseases. Understanding which proteins has changed, at which locations, and the associated biological consequences are therefore critical (Figure 2). Therefore, identification of a multitude of post-translationally modified protein sites has become an important and promising endeavor with the advent of mass spectrometry (MS)-based proteomics, chemical biology tools, fluorescence-based, and bioinformatic techniques (149, 150). However, these cutting-edge approaches present a number of complex challenges. Few of the difficulties may be to determine which proteins are altered, at what locations, and what biological relevance it has. Also, whether cells are biased on non-enzymatic over enzymatic pathway of modification and what is the reason behind that preference can be some of the intriguing questions to consider.
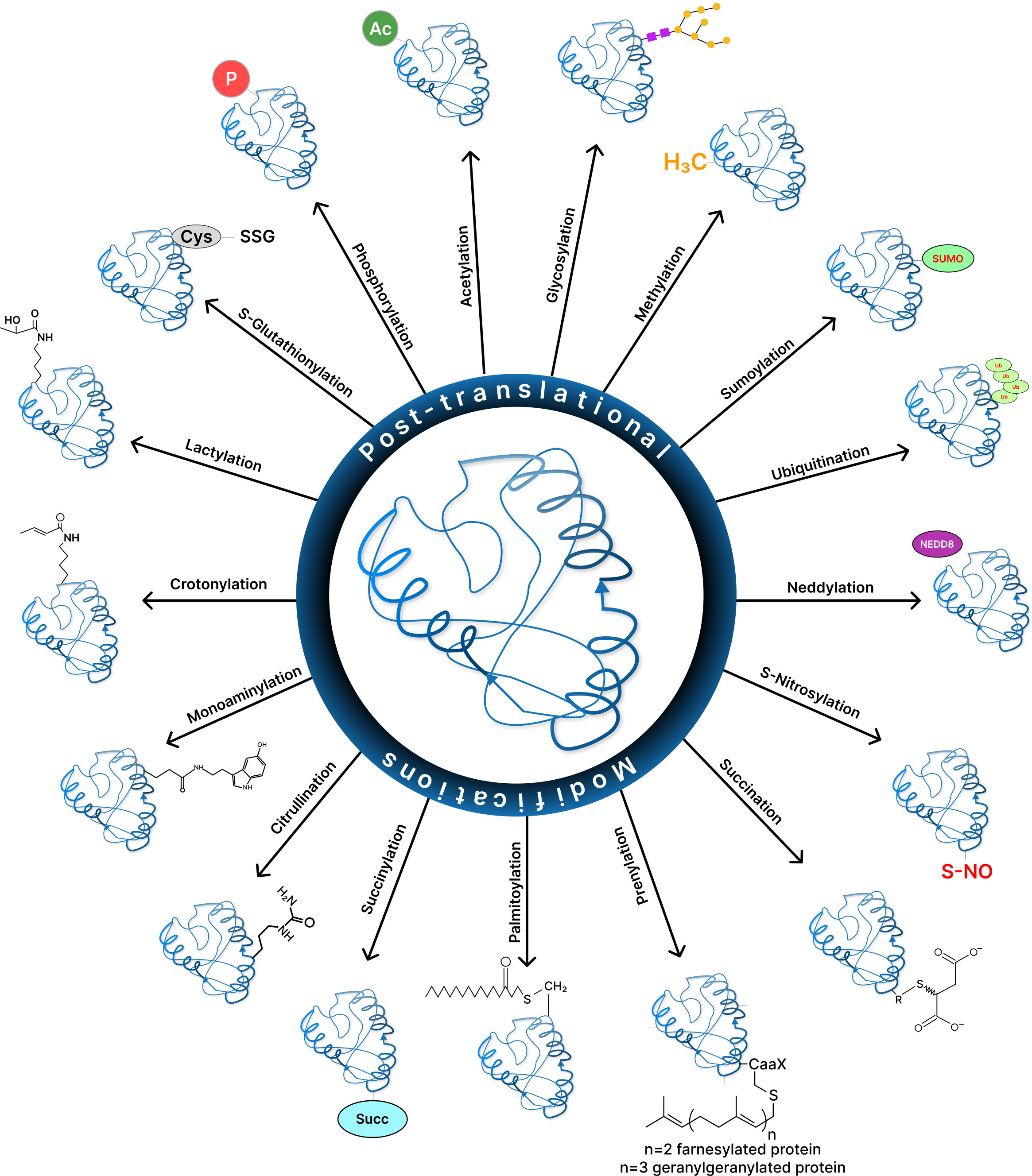
Figure 2 Various post-translational modificalions in cancer.
Exciting breakthroughs in identifying PTMs on a proteome-wide and cellular levels show a growing impact in understanding the proteome’s diversity. Once PTM analysis at the proteome level becomes regular, the role of PTMs in disease can be explored considerably more systematically than before and will promote in the discovery of novel drugs and cancer therapeutic strategies. Few key proteins whose post-translational modifications are clinical targets for cancer therapeutics as well as associated with the hallmarks of cancer or its pathophysiology have been enlisted (Tables 2, 3).

Table 2 Examples of clinically targeted oncoproteins and the associated post-translational modifications responsible for cancer therapeutics.
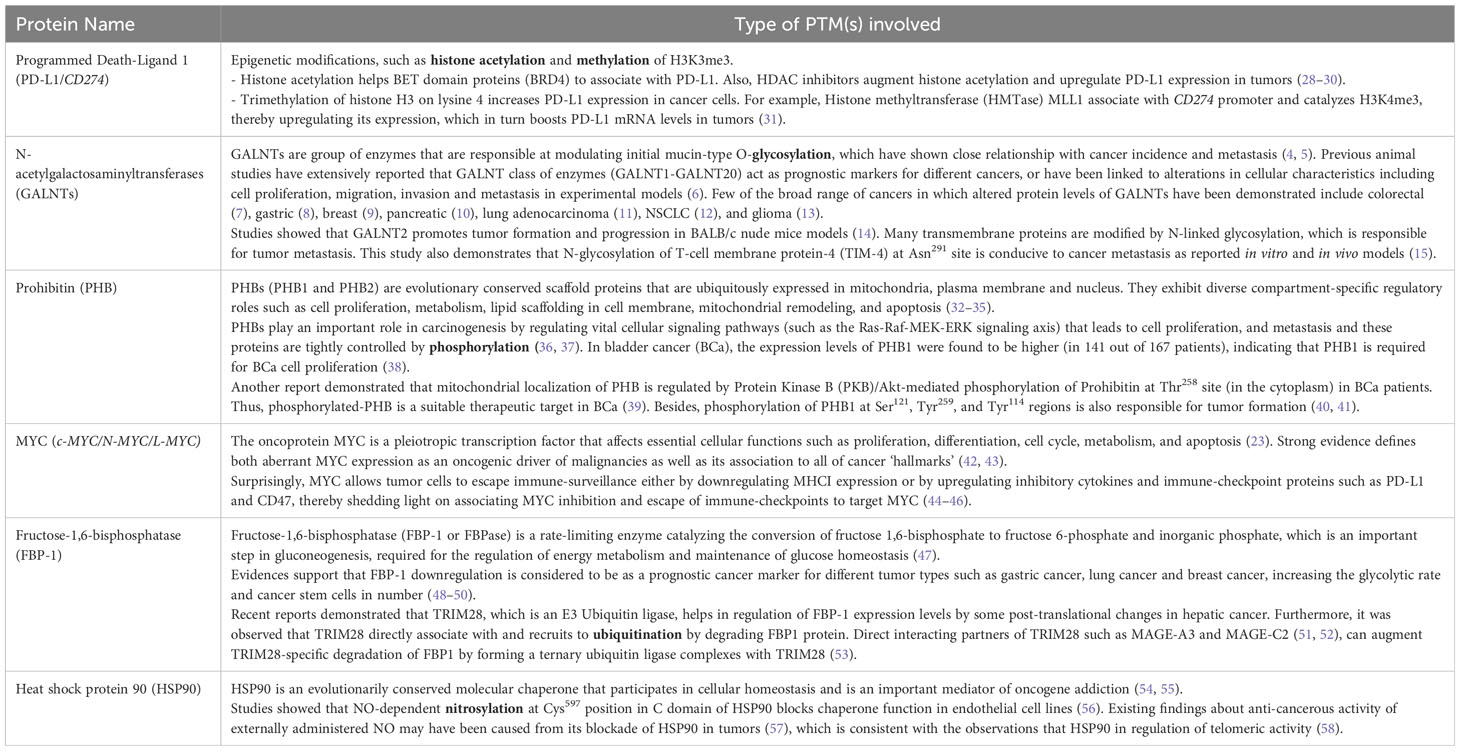
Table 3 Examples of key proteins with its corresponding post-translational modification(s) (in bold) related to cancer hallmarks/responsible for cancer pathophysiology.
A number of small molecule drugs that target PTM regulators, such as kinase, deacetylase, and methyltransferase inhibitors, have been approved for cancer treatment. However, in addition to the therapeutic target, each upstream regulator often has several substrates. Therefore, unbiased inhibition of PTM regulators may result in off-target effects, limiting their therapeutic usage. As a result, ‘small’ molecules with profound selectivity that can modulate a particular PTM of a protein are in huge demand. Many chemical proximity-induced PTM methods, such as proteolysis-targeting chimera (PROTAC), have arrived in the last two decades as having immense potential for targeting ‘undruggable’ proteins and for clinical translation in the treatment of cancer (151, 152).
Author contributions
HD and NJ wrote and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Department of Biotechnology (6242-P69/RGCB/PMD/DBT/NDJN/2015), and the Council of Scientific and Industrial Research (CSIR-IICT/MLP0068), India to NJ. HD is supported by a senior research fellowship from Department of Science and Technology-INSPIRE, India.
Acknowledgments
We thank the Director, CSIR-IICT (Ms. No. IICT/Pubs./2023/215) for providing all the required facilities to carry out the work. To whom correspondence may be addressed: Department of Applied Biology, CSIR-Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad 500007, Telangana State, India. Tel.: 91-40-27191857; Fax: 91-40-27191812; E-mail: H.D,aGFzaG51ZHV0dGFAZ21haWwuY29tand N.J,bmlzaGFudC5paWN0QGdvdi5pbg==.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. T.U. Consortium. The universal protein resource (UniProt) 2009. Nucleic Acids Res (2008) 37:D169–74.
2. Fearon WR. The carbamido diacetyl reaction: a test for citrulline. Biochem J (1939) 33:902–7. doi: 10.1042/bj0330902
3. Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr. Protein posttranslational modifications: the chemistry of proteome diversifications Angewandte Chemie Int Edition (2005) 44:7342–72 doi: 10.1002/anie.200501023
4. Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
5. Meierhofer D, Wang X, Huang L, Kaiser P. Quantitative analysis of global ubiquitination in heLa cells by mass spectrometry. J Proteome Res (2008) 7:4566–76. doi: 10.1021/pr800468j
6. Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia (2001) 44:129–46. doi: 10.1007/s001250051591
7. Verbrugge FH, Tang WHW, Hazen SL. Protein carbamylation and cardiovascular disease. Kidney Int (2015) 88:474–8. doi: 10.1038/ki.2015.166
8. Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med (2003) 9:169–76. doi: 10.1016/S1471-4914(03)00031-5
9. Kalim S, Karumanchi SA, Thadhani RI, Berg AH. Protein carbamylation in kidney disease: pathogenesis and clinical implications. Am J Kidney Dis (2014) 64:793–803. doi: 10.1053/j.ajkd.2014.04.034
10. Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, et al. Systematic functional prioritization of protein posttranslational modifications. Cell (2012) 150:413–25. doi: 10.1016/j.cell.2012.05.036
11. Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol (2006) 24:1241–52. doi: 10.1038/nbt1252
12. Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol (2010) 17:666–72. doi: 10.1038/nsmb.1842
13. Peng Y, Liu H, Liu J, Long J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radical Biol Med (2022) 179:11–23. doi: 10.1016/j.freeradbiomed.2021.12.264
14. Karve TM, Cheema AK. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids (2011) 2011:207691. doi: 10.4061/2011/207691
15. Xu H, Wang Y, Lin S, Deng W, Peng D, Cui Q, et al. PTMD: A database of human disease-associated post-translational modifications. Genomics Proteomics Bioinf (2018) 16:244–51. doi: 10.1016/j.gpb.2018.06.004
16. Alderson NL, Wang Y, Blatnik M, Frizzell N, Walla MD, Lyons TJ, et al. S-(2-Succinyl)cysteine: A novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophysics (2006) 450:1–8. doi: 10.1016/j.abb.2006.03.005
17. Blatnik M, Thorpe SR, Baynes JW. Succination of proteins by fumarate. Ann New York Acad Sci (2008) 1126:272–5. doi: 10.1196/annals.1433.047
18. Adam J, Yang M, Soga T, Pollard PJ. Rare insights into cancer biology. Oncogene (2014) 33:2547–56. doi: 10.1038/onc.2013.222
19. Bardella C, El-Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol (2011) 225:4–11. doi: 10.1002/path.2932
20. Wang H, Yang L, Liu M, Luo J. Protein post-translational modifications in the regulation of cancer hallmarks. Cancer Gene Ther (2023) 30:529–47. doi: 10.1038/s41417-022-00464-3
21. Edwards AS, Scott JD. A-kinase anchoring proteins: protein kinase A and beyond. Curr Opin Cell Biol (2000) 12:217–21. doi: 10.1016/S0955-0674(99)00085-X
22. Duan G, Walther D. The roles of post-translational modifications in the context of protein interaction networks. PloS Comput Biol (2015) 11:e1004049. doi: 10.1371/journal.pcbi.1004049
23. Panni S. Phospho-peptide binding domains in S. cerevisiae model organism. Biochimie (2019) 163:117–27. doi: 10.1016/j.biochi.2019.06.005
24. Skamnaki VT, Owen DJ, Noble MEM, Lowe ED, Lowe G, Oikonomakos NG, et al. Catalytic mechanism of phosphorylase kinase probed by mutational studies. Biochemistry (1999) 38:14718–30. doi: 10.1021/bi991454f
25. Jin J, Pawson T. Modular evolution of phosphorylation-based signalling systems. Philos Trans R Soc B: Biol Sci (2012) 367:2540–55. doi: 10.1098/rstb.2012.0106
26. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
27. Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem (2001) 268:2764–72. doi: 10.1046/j.1432-1327.2001.02225.x
28. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin Ther Targets (2012) 16:103–19. doi: 10.1517/14728222.2011.645805
29. Jaggi M, Rao PS, Smith DJ, Hemstreet GP, Balaji KC. Protein kinase C μ is down-regulated in androgen-independent prostate cancer. Biochem Biophys Res Commun (2003) 307:254–60. doi: 10.1016/S0006-291X(03)01161-6
30. Radivojac P, Baenziger PH, Kann MG, Mort ME, Hahn MW, Mooney SD. Gain and loss of phosphorylation sites in human cancer. Bioinformatics (2008) 24:i241–7. doi: 10.1093/bioinformatics/btn267
31. Lim YP. Mining the tumor phosphoproteome for cancer markers. Clin Cancer Res (2005) 11:3163–9. doi: 10.1158/1078-0432.CCR-04-2243
32. Hu M, He F, Thompson EW, Ostrikov K, Dai X. Lysine acetylation, cancer hallmarks and emerging onco-therapeutic opportunities. Cancers (2022) 14:346. doi: 10.3390/cancers14020346
33. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324:1076–80. doi: 10.1126/science.1164097
34. Harachi M, Masui K, Cavenee WK, Mischel PS, Shibata N. Protein acetylation at the interface of genetics. Epigenet Environ Cancer Metabolites (2021) 11:216. doi: 10.3390/metabo11040216
35. Latham JA, Dent SYR. Cross-regulation of histone modifications. Nat Struct Mol Biol (2007) 14:1017–24. doi: 10.1038/nsmb1307
36. Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell (2007) 131:633–6. doi: 10.1016/j.cell.2007.10.039
37. Shang S, Liu J, Hua F. Protein acylation: mechanisms, biological functions and therapeutic targets. Signal Transduction Targeted Ther (2022) 7:396. doi: 10.1038/s41392-022-01245-y
38. Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology (2017) 65:515–28. doi: 10.1002/hep.28887
39. Gu L, Zhu Y, Lin X, Li Y, Cui K, Prochownik EV, et al. Amplification of glyceronephosphate O-acyltransferase and recruitment of USP30 stabilize DRP1 to promote hepatocarcinogenesis. Cancer Res (2018) 78:5808–19. doi: 10.1158/0008-5472.CAN-18-0340
40. Carney D, Ihde D, Cohen M, Marangos P, Bunn P, Minna J, et al. SERUM NEURON-SPECIFIC ENOLASE: A MARKER FOR DISEASE EXTENT AND RESPONSE TO THERAPY OF SMALL-CELL LUNG CANCER. Lancet (1982) 319:583–5. doi: 10.1016/S0140-6736(82)91748-2
41. Behren S, Schorlemer M, Schmidt G, Aktories K, Westerlind U. Antibodies directed against galNAc- and glcNAc-O-tyrosine posttranslational modifications – a new tool for glycoproteomic detection. Chem – A Eur J (2023) 29:e202300392. doi: 10.1002/chem.202300392
42. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer (2005) 5:526–42. doi: 10.1038/nrc1649
43. Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol (2012) 13:448–62. doi: 10.1038/nrm3383
44. Kakugawa Y, Wada T, Yamaguchi K, Yamanami H, Ouchi K, Sato I, et al. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc Natl Acad Sci (2002) 99:10718–23. doi: 10.1073/pnas.152597199
45. Ma Z, Vosseller K. Cancer metabolism and elevated O-glcNAc in oncogenic signaling*. J Biol Chem (2014) 289:34457–65. doi: 10.1074/jbc.R114.577718
46. Anderson JL, Henriksen BS, Gibbs RA, Hrycyna CA. The isoprenoid substrate specificity of isoprenylcysteine carboxylmethyltransferase: DEVELOPMENT OF NOVEL INHIBITORS*. J Biol Chem (2005) 280:29454–61. doi: 10.1074/jbc.M504982200
47. Aletta JM, Cimato TR, Ettinger MJ. Protein methylation: a signal event in post-translational modification. Trends Biochem Sci (1998) 23:89–91. doi: 10.1016/S0968-0004(98)01185-2
48. Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, et al. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol Cell (2011) 41:331–42. doi: 10.1016/j.molcel.2010.12.030
49. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell (2009) 33:1–13. doi: 10.1016/j.molcel.2008.12.013
50. Wesche J, Kühn S, Kessler BM, Salton M, Wolf A. Protein arginine methylation: a prominent modification and its demethylation. Cell Mol Life Sci (2017) 74:3305–15. doi: 10.1007/s00018-017-2515-z
51. Hwang JW, Cho Y, Bae G-U, Kim S-N, Kim YK. Protein arginine methyltransferases: promising targets for cancer therapy. Exp Mol Med (2021) 53:788–808. doi: 10.1038/s12276-021-00613-y
52. Baldwin RM, Morettin A, Côté J. Role of PRMTs in cancer: Could minor isoforms be leaving a mark? World J Biol Chem (2014) 5:115–29. doi: 10.4331/wjbc.v5.i2.115
53. Hsu M-C, Tsai Y-L, Lin C-H, Pan M-R, Shan Y-S, Cheng T-Y, et al. Protein arginine methyltransferase 3-induced metabolic reprogramming is a vulnerable target of pancreatic cancer. J Hematol Oncol (2019) 12:79. doi: 10.1186/s13045-019-0769-7
54. Zhang X, Wang K, Feng X, Wang J, Chu Y, Jia C, et al. PRMT3 promotes tumorigenesis by methylating and stabilizing HIF1α in colorectal cancer. Cell Death Dis (2021) 12:1066. doi: 10.1038/s41419-021-04352-w
55. Lei Y, Han P, Chen Y, Wang H, Wang S, Wang M, et al. Protein arginine methyltransferase 3 promotes glycolysis and hepatocellular carcinoma growth by enhancing arginine methylation of lactate dehydrogenase A. Clin Trans Med (2022) 12:e686. doi: 10.1002/ctm2.686
56. Singh V, MIranda TB, Jiang W, Frankel A, Roemer ME, Robb VA, et al. DAL-1/4.1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene (2004) 23:7761–71. doi: 10.1038/sj.onc.1208057
57. Mustfa SA, Singh M, Suhail A, Mohapatra G, Verma S, Chakravorty D, et al. SUMOylation pathway alteration coupled with downregulation of SUMO E2 enzyme at mucosal epithelium modulates inflammation in inflammatory bowel disease. Open Biol (2017) 7:170024. doi: 10.1098/rsob.170024
58. Desterro JMP, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1*. J Biol Chem (1999) 274:10618–24. doi: 10.1074/jbc.274.15.10618
59. Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol (2012) 13:755–66. doi: 10.1038/nrm3478
60. Du L, Liu W, Rosen ST. Targeting SUMOylation in cancer. Curr Opin Oncol (2021) 33:520–5. doi: 10.1097/CCO.0000000000000765
61. Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem (2012) 81:291–322. doi: 10.1146/annurev-biochem-051810-094654
62. Shmueli A, Oren M. Life, death, and ubiquitin: taming the mule. Cell (2005) 121:963–5. doi: 10.1016/j.cell.2005.06.018
63. López-Otín C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer (2010) 10:278–92. doi: 10.1038/nrc2823
64. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. EMBO Rep (2008) 9:536–42. doi: 10.1038/embor.2008.93
65. Rajalingam K, Dikic I. Expanding the ubiquitin code. Cell (2016) 164:1074–1074.e1071. doi: 10.1016/j.cell.2016.02.019
66. Morrow JK, Lin H-K, Sun S-C, Zhang S. Targeting ubiquitination for cancer therapies. Future Medicinal Chem (2015) 7:2333–50. doi: 10.4155/fmc.15.148
67. Mansour MA. Ubiquitination: Friend and foe in cancer. Int J Biochem Cell Biol (2018) 101:80–93. doi: 10.1016/j.biocel.2018.06.001
68. Kamitani T, Kito K, Nguyen HP, Yeh ETH. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein*. J Biol Chem (1997) 272:28557–62. doi: 10.1074/jbc.272.45.28557
69. Dimitris P. Xirodimas, Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans (2008) 36:802–6. doi: 10.1042/BST0360802
70. Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin–RING ligases. Nat Rev Mol Cell Biol (2015) 16:30–44. doi: 10.1038/nrm3919
71. Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell signalling (2018) 44:92–102. doi: 10.1016/j.cellsig.2018.01.009
72. Zhao Y, Morgan MA, Sun Y. Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal (2014) 21:2383–400. doi: 10.1089/ars.2013.5795
73. Petroski MD, Deshaies RJ. Function and regulation of cullin–RING ubiquitin ligases. Nat Rev Mol Cell Biol (2005) 6:9–20. doi: 10.1038/nrm1547
74. Deshaies RJ, Joazeiro CAP. RING domain E3 ubiquitin ligases. Annu Rev Biochem (2009) 78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809
75. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer (2006) 6:369–81. doi: 10.1038/nrc1881
76. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature (2009) 458:732–6. doi: 10.1038/nature07884
77. Zhao Y, Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Design (2013) 19:3215–25. doi: 10.2174/13816128113199990300
78. Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets (2011) 11:347–56. doi: 10.2174/156800911794519734
79. Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxidants Redox Signaling (2014) 21:2383–400. doi: 10.1089/ars.2013.5795
80. Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell (2001) 106:675–83. doi: 10.1016/S0092-8674(01)00495-0
81. Nathan C, Xie Q-w. Nitric oxide synthases: Roles, tolls, and controls. Cell (1994) 78:915–8. doi: 10.1016/0092-8674(94)90266-6
82. Marshall HE, Merchant K, Stamler JS. Nitrosation and oxidation in the regulation of gene expression. FASEB J (2000) 14:1889–900. doi: 10.1096/fj.00.011rev
83. Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med (2009) 15:391–404. doi: 10.1016/j.molmed.2009.06.007
84. Mannick JB. Regulation of apoptosis by protein S-nitrosylation. Amino Acids (2007) 32:523–6. doi: 10.1007/s00726-006-0427-6
85. Benhar M, Stamler JS. A central role for S-nitrosylation in apoptosis. Nat Cell Biol (2005) 7:645–6. doi: 10.1038/ncb0705-645
86. Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistic G, Finazzi-Agr A. S-nitrosylation regulates apoptosis. Nature (1997) 388:432–3. doi: 10.1038/41237
87. Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol (2005) 6:150–66. doi: 10.1038/nrm1569
88. Bentz BG, Haines GK III, Radosevich JA. Increased protein nitrosylation in head and neck squamous cell carcinogenesis. Head Neck (2000) 22:64–70. doi: 10.1002/(SICI)1097-0347(200001)22:1<64::AID-HED10>3.0.CO;2-J
89. Thomas SA, Storey KB, Baynes JW, Frizzell N. Tissue distribution of S-(2-succino)cysteine (2SC), a biomarker of mitochondrial stress in obesity and diabetes. Obesity (2012) 20:263–9. doi: 10.1038/oby.2011.340
90. Frizzell N, Lima M, Baynes JW. Succination of proteins in diabetes. Free Radical Res (2011) 45:101–9. doi: 10.3109/10715762.2010.524643
91. Merkley ED, Metz TO, Smith RD, Baynes JW, Frizzell N. The succinated proteome. Mass Spectrometry Rev (2014) 33:98–109. doi: 10.1002/mas.21382
92. Frizzell N, Thomas SA, Carson JA, Baynes JW. Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem J (2012) 445:247–54. doi: 10.1042/BJ20112142
93. Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet (2005) 14:2231–9. doi: 10.1093/hmg/ddi227
94. Yang M, Soga T, Pollard P, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol (2012) 2. doi: 10.3389/fonc.2012.00085
95. Zhang FL, Casey PJ. PROTEIN PRENYLATION: molecular mechanisms and functional consequences. Annu Rev Biochem (1996) 65:241–69. doi: 10.1146/annurev.bi.65.070196.001325
96. Ashby MN. CaaX converting enzymes. Curr Opin Lipidology (1998) 9:99–102. doi: 10.1097/00041433-199804000-00004
97. Placzek AT, Krzysiak AJ, Gibbs RA. Chemical probes of protein prenylation. In: The enzymes, vol. 30. Cambridge, MA: Academic Press Inc (2011). p. 91–127.
98. Jackson JH, Cochrane CG, Bourne JR, Solski PA, Buss JE, Der CJ. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci United States America (1990) 87:3042–6. doi: 10.1073/pnas.87.8.3042
99. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discovery (2014) 13:828–51. doi: 10.1038/nrd4389
100. Cutsem EV, v.d. Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol (2004) 22:1430–8. doi: 10.1200/JCO.2004.10.112
101. Rao S, Cunningham D, d. Gramont A, Scheithauer W, Smakal M, Humblet Y, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol (2004) 22:3950–7. doi: 10.1200/JCO.2004.10.037
102. Harousseau J-L, Martinelli G, Jedrzejczak WW, Brandwein JM, Bordessoule D, Masszi T, et al. f.t.F.-A.-. Investigators, A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood (2009) 114:1166–73. doi: 10.1182/blood-2009-01-198093
103. Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol (2007) 8:74–84. doi: 10.1038/nrm2084
104. Korycka J, Łach A, Heger E, Bogusławska DM, Wolny M, Toporkiewicz M, et al. Human DHHC proteins: A spotlight on the hidden player of palmitoylation. Eur J Cell Biol (2012) 91:107–17. doi: 10.1016/j.ejcb.2011.09.013
105. Duncan JA, Gilman AG. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein α Subunits and p21RAS*. J Biol Chem (1998) 273:15830–7. doi: 10.1074/jbc.273.25.15830
106. Lin DTS, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife (2015) 4:e11306. doi: 10.7554/eLife.11306.017
107. Schmick M, Kraemer A, Bastiaens PIH. Ras moves to stay in place. Trends Cell Biol (2015) 25:190–7. doi: 10.1016/j.tcb.2015.02.004
108. Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol (2010) 6:449–56. doi: 10.1038/nchembio.362
109. Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood (2010) 115:3598–605. doi: 10.1182/blood-2009-03-213876
110. Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol (2011) 7:58–63. doi: 10.1038/nchembio.495
111. Chen H, Xu H, Potash S, Starkov A, Belousov VV, Bilan DS, et al. Mild metabolic perturbations alter succinylation of mitochondrial proteins. J Neurosci Res (2017) 95:2244–52. doi: 10.1002/jnr.24103
112. Gibson GE, Xu H, Chen H-L, Chen W, Denton TT, Zhang S. Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J Neurochemistry (2015) 134:86–96. doi: 10.1111/jnc.13096
113. Smestad J, Erber L, Chen Y, Maher LJ. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience (2018) 2:63–75. doi: 10.1016/j.isci.2018.03.012
114. Wang G, Meyer JG, Cai W, Softic S, Li ME, Verdin E, et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol Cell (2019) 74:844–857.e847. doi: 10.1016/j.molcel.2019.03.021
115. Alleyn M, Breitzig M, Lockey R, Kolliputi N. The dawn of succinylation: a posttranslational modification. Am J Physiology-Cell Physiol (2018) 314:C228–32. doi: 10.1152/ajpcell.00148.2017
116. Xiangyun Y, Xiaomin N, linping G, Yunhua X, Ziming L, Yongfeng Y, et al. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget (2016) 8(4):6984–93. doi: 10.18632/oncotarget.14346
117. Fujisaki M, Sugawara K. Properties of peptidylarginine deiminase from the epidermis of newborn rats1. J Biochem (1981) 89:257–63. doi: 10.1093/oxfordjournals.jbchem.a133189
118. Rogers GE, Simmonds DH. Content of citrulline and other amino-acids in a protein of hair follicles. Nature (1958) 182:186–7. doi: 10.1038/182186a0
119. Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, et al. The rheumatoid arthritis-associated citrullinome. Cell Chem Biol (2018) 25:691–704.e696. doi: 10.1016/j.chembiol.2018.03.002
120. Lee CY, Wang D, Wilhelm M. Mining the human tissue proteome for protein citrullination. Mol Cell Proteomics (2018) 17:1378–91. doi: 10.1074/mcp.RA118.000696
121. Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with Malignant tumors. BMC Cancer (2009) 9:40. doi: 10.1186/1471-2407-9-40
122. Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun (2018) 9:4783. doi: 10.1038/s41467-018-07306-7
123. Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci (2009) 34:85–96. doi: 10.1016/j.tibs.2008.11.002
124. Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, et al. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol Lett (2016) 11:1980–6. doi: 10.3892/ol.2016.4168
125. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (2011) 334:1278–83. doi: 10.1126/science.1211485
126. Bader M. Serotonylation: serotonin signaling and epigenetics. Front Mol Neurosci (2019) 12. doi: 10.3389/fnmol.2019.00288
127. Al-Kachak A, Maze I. Post-translational modifications of histone proteins by monoamine neurotransmitters. Curr Opin Chem Biol (2023) 74:102302. doi: 10.1016/j.cbpa.2023.102302
128. Wang ZA, Kurra Y, Wang X, Zeng Y, Lee Y-J, Sharma V, et al. A versatile approach for site-specific lysine acylation in proteins. Angewandte Chemie Int Edition (2017) 56:1643–7. doi: 10.1002/anie.201611415
129. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell (2011) 146:1016–28. doi: 10.1016/j.cell.2011.08.008
130. Ruiz-Andres O, Sanchez-Niño MD, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, et al. Histone lysine crotonylation during acute kidney injury in mice. Dis Models Mech (2016) 9:633–45. doi: 10.1242/dmm.024455
131. Liu Y, Li M, Fan M, Song Y, Yu H, Zhi X, et al. Chromodomain Y-like protein–mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry (2019) 85:635–49. doi: 10.1016/j.biopsych.2018.11.025
132. Jiang G, Nguyen D, Archin NM, Yukl SA, Méndez-Lagares G, Tang Y, et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest (2018) 128:1190–8. doi: 10.1172/JCI98071
133. Berger K, Moeller MJ. Mechanisms of epithelial repair and regeneration after acute kidney injury. Semin Nephrol (2014) 34:394–403. doi: 10.1016/j.semnephrol.2014.06.006
134. Wan J, Liu H, Ming L. Lysine crotonylation is involved in hepatocellular carcinoma progression. Biomedicine Pharmacotherapy (2019) 111:976–82. doi: 10.1016/j.biopha.2018.12.148
135. Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones*. Mol Cell Proteomics (2007) 6:812–9. doi: 10.1074/mcp.M700021-MCP200
136. William G, Kaelin S, McKnight L. Influence of metabolism on epigenetics and disease. Cell (2013) 153:56–69. doi: 10.1016/j.cell.2013.03.004
137. Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-coA synthetase*. J Biol Chem (2007) 282:30239–45. doi: 10.1074/jbc.M704409200
138. Rousseaux S, Khochbin S. Histone acylation beyond acetylation: terra incognita in chromatin biology. Cell J (Yakhteh) (2015) 17:1–6. doi: 10.22074/cellj.2015.506
139. Chen X-F, Chen X, Tang X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin Sci (2020) 134:657–76. doi: 10.1042/CS20200128
140. Xu H, Wu M, Ma X, Huang W, Xu Y. Function and mechanism of novel histone posttranslational modifications in health and disease. BioMed Res Int (2021) 2021:6635225. doi: 10.1155/2021/6635225
141. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduction Targeted Ther (2022) 7:305. doi: 10.1038/s41392-022-01151-3
142. Aksnes H, Drazic A, Marie M, Arnesen T. First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem Sci (2016) 41:746–60. doi: 10.1016/j.tibs.2016.07.005
143. Zhang K, Li L, Zhu M, Wang G, Xie J, Zhao Y, et al. Comparative analysis of histone H3 and H4 post-translational modifications of esophageal squamous cell carcinoma with different invasive capabilities. J Proteomics (2015) 112:180–9. doi: 10.1016/j.jprot.2014.09.004
144. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
145. Moreno-Yruela C, Zhang D, Wei W, Bæk M, Liu W, Gao J, et al. Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Sci Adv (2022) 8:eabi6696. doi: 10.1126/sciadv.abi6696
146. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol (2021) 22:85. doi: 10.1186/s13059-021-02308-z
147. Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.647559
148. Yang D, Yin J, Shan L, Yi X, Zhang W, Ding Y. Identification of lysine-lactylated substrates in gastric cancer cells. iScience (2022) 25:104630. doi: 10.1016/j.isci.2022.104630
149. Farley AR, Link AJ. Chapter 40 Identification and Quantification of Protein Posttranslational Modifications. In: Burgess RR, Deutscher MP, editors. Methods in Enzymology, vol. 463. Cambridge, MA: Academic Press Inc (2009). p. 725–63.
150. Chuh KN, Pratt MR. Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr Opin Chem Biol (2015) 24:27–37. doi: 10.1016/j.cbpa.2014.10.020
151. Stanton BZ, Chory EJ, Crabtree GR. Chemically induced proximity in biology and medicine. Science (2018) 359:eaao5902. doi: 10.1126/science.aao5902
Keywords: post-translational modifications, protein structure, cancer, proteome diversity, protein function, covalent bond
Citation: Dutta H and Jain N (2023) Post-translational modifications and their implications in cancer. Front. Oncol. 13:1240115. doi: 10.3389/fonc.2023.1240115
Received: 14 June 2023; Accepted: 21 August 2023;
Published: 19 September 2023.
Edited by:
Arun Kumar Trivedi, Central Drug Research Institute (CSIR), IndiaReviewed by:
Linhui Zhai, Chinese Academy of Sciences (CAS), ChinaCopyright © 2023 Dutta and Jain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hashnu Dutta, aGFzaG51ZHV0dGFAZ21haWwuY29t; Nishant Jain, bmlzaGFudC5paWN0QGdvdi5pbg==