 Richa Sharma1*
Richa Sharma1* Ninad Oak2
Ninad Oak2 Wenan Chen3
Wenan Chen3 Rose Gogal4Martin Kirschner5,6
Rose Gogal4Martin Kirschner5,6 Fabian Beier5,6
Fabian Beier5,6 Michael J. Schnieders4
Michael J. Schnieders4 Maria Spies4
Maria Spies4 Kim E. Nichols2
Kim E. Nichols2 Marcin Wlodarski1
Marcin Wlodarski1- 1Department of Hematology, St. Jude Children´s Research Hospital, Memphis, TN, United States
- 2Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 3Center for Applied Bioinformatics, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 4Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, IA, United States
- 5Department of Hematology, Oncology, Hemostaseology and Stem Cell Transplantation, Medical Faculty, RWTH Aachen University, Aachen, Germany
- 6Center for Integrated Oncology Aachen Bonn Cologne Düsseldorf (CIO ABCD), Bonn, Germany
Replication Protein A (RPA) is single-strand DNA binding protein that plays a key role in the replication and repair of DNA. RPA is a heterotrimer made of 3 subunits – RPA1, RPA2, and RPA3. Germline pathogenic variants affecting RPA1 were recently described in patients with Telomere Biology Disorders (TBD), also known as dyskeratosis congenita or short telomere syndrome. Premature telomere shortening is a hallmark of TBD and results in bone marrow failure and predisposition to hematologic malignancies. Building on the finding that somatic mutations in RPA subunit genes occur in ~1% of cancers, we hypothesized that germline RPA alterations might be enriched in human cancers. Because germline RPA1 mutations are linked to early onset TBD with predisposition to myelodysplastic syndromes, we interrogated pediatric cancer cohorts to define the prevalence and spectrum of rare/novel and putative damaging germline RPA1, RPA2, and RPA3 variants. In this study of 5,993 children with cancer, 75 (1.25%) harbored heterozygous rare (non-cancer population allele frequency (AF) < 0.1%) variants in the RPA heterotrimer genes, of which 51 cases (0.85%) had ultra-rare (AF < 0.005%) or novel variants. Compared with Genome Aggregation Database (gnomAD) non-cancer controls, there was significant enrichment of ultra-rare and novel RPA1, but not RPA2 or RPA3, germline variants in our cohort (adjusted p-value < 0.05). Taken together, these findings suggest that germline putative damaging variants affecting RPA1 are found in excess in children with cancer, warranting further investigation into the functional role of these variants in oncogenesis.
Introduction
Maintenance of genome integrity requires efficient DNA repair. The perturbation of processes engaged in repair of DNA damage by somatic mutations is a well-known mechanism for oncogenesis. Germline biallelic inactivation of genes governing DNA repair leads to classic cancer predisposition syndromes such as Fanconi anemia, ataxia telangiectasia and Bloom syndrome, among others (1–4). Monoallelic mutations impacting some of these genes can also increase the risk for cancer (5–9). We recently discovered that germline heterozygous mutations in the Replication Protein A1 (RPA1) gene cause Telomere Biology Disorder (TBD), a hereditary condition classically associated with pathological shortening of telomeres resulting in bone marrow failure (BMF), pulmonary and liver fibrosis, mucocutaneous fragility, and predisposition to solid tumors, myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) (10).
The RPA1 protein is the largest subunit of Replication Protein A (RPA), a heterotrimeric complex consisting of RPA1 (RPA70), RPA2 (RPA32) and RPA3 (RPA14). As a complex, RPA tightly binds single-strand DNA (ssDNA) to protect it from nucleases while maintaining DNA accessible to essential DNA-DNA and DNA-protein interactions. Consistent with the ubiquitous and ongoing formation of ssDNA, RPA is present and required across almost all cellular processes during replication, recombination, and repair of DNA. In fact, RPA is involved in all ssDNA repair pathways (nucleotide excision, base excision, mismatch) and double strand DNA repair mechanisms (homologous recombination, non-homologous end joining) (11–13). RPA participates in such diverse pathways through its ability to dynamically bind ssDNA while facilitating DNA repair and cell cycle protein interactions (11).
The essential role of RPA in DNA repair might lend RPA to be mutated in cancers. By mining the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (14), we found that somatic mutations in RPA1, RPA2, RPA3 are found in 1.4%, 0.5%, and 0.9% of human cancers, respectively. In our previously published cohort of 4 patients with TBD, one patient who carried a germline RPA1 p.V227A mutation developed advanced MDS requiring hematopoietic stem cell transplantation. All 3 RPA1 germline mutations (p.V227A, p.E240K, p.T270A) identified in the 4 cases were missense and 2 out of 3 exerted a gain-of-function effect, resulting in increased binding to single strand and telomeric DNA (10). Besides these descriptions associating germline RPA1 variants with bone marrow failure or hematologic malignancies, the RPA2 or RPA3 genes have not been linked to any human diseases thus far. Moreover, the landscape of germline variants in RPA heterotrimer in malignancies has not been systematically assessed. To address this knowledge gap, we investigated the occurrence of novel and rare germline variants in RPA1, RPA2 and RPA3 genes, in a cohort of 5,993 children with cancers. We found that ultra-rare and novel germline variants in the RPA1 gene were significantly more common among pediatric cancer patients than non-cancer controls. Furthermore, we examined a separate cohort of 41 young adults with AML and identified potentially deleterious RPA1 germline variants in 3 cases. Our studies indicate that the RPA1 gene may be a novel risk factor for malignancies.
Methods
Data sources
For this study, we used publicly available whole exome sequencing datasets previously collected across studies at St. Jude Children’s Research Hospital or through dbGaP. Specifically, we used the Pediatric Cancer Genome Project (PCGP) (15), real-time clinical genomics (RTCG/G4K) (16), St. Jude Lifetime Cohort (SJLIFE) (17), and TARGET datasets (TARGET URL is https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000218). In sum, we interrogated 5,993 germline samples across 24 cancer types including hematologic, non-central nervous system (CNS) solid tumors, and CNS tumors. Cancers were stratified into hematologic (n = 3,452; 58%), solid (n = 1,974; 33%) or CNS (n = 1,068; 18%) cancers and further subclassified as follows: i) hematologic malignancies: B-cell (B) acute lymphoid leukemia (B-ALL), T-cell ALL (T-ALL), acute myeloid leukemia (AML), Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma; ii) solid tumors: germ cell tumor (GCT), melanoma (MEL), neuroblastoma (NBL), nasopharyngeal carcinoma (NPC), papillary thyroid carcinoma (PTC), sarcomas (Ewing’s (EWS), osteosarcoma (OS), rhabdomyosarcoma (RMS), synovial), Wilms tumor (WT); and iii) CNS tumors: ependymoma (EP), low grade glioma (LGG), medulloblastoma (MB), and high grade glioma (HGG). An external cohort was queried, which consisted of 41 patients with AML from the German Study Alliance Leukemia that met following criteria: age below 35, blast-free remission after chemotherapy, karyotype aberrations (n = 12 with < 3, n = 29 with ≥ 3 aberrations detected in diagnostic karyotype or FISH analysis), and samples of peripheral blood or bone marrow at remission (18). The current study was approved by the Institutional Review Board at St. Jude Children’s Research Hospital.
Variant calling and filtering
Variant calling and genotyping were performed using Genome Analysis Toolkit’s (GATK) best practices workflow with modifications as described previously (19). We retained high quality variants that passed filtering using following criteria: allelic balance > 0.2, genotype quality > 20, variant allelic frequency (VAF) for heterozygous variants between 20-80%, minimum of 10 alternate reads supporting single nucleotide variants (SNVs) and 7 alternate reads supporting InDels, and missingness < 25% of samples. We performed variant annotation using ANNOtate VARiation (ANNOVAR) and variant effect predictor (VEP) tools (20). We also annotated all the variants using InterVar (21) automated clinical interpretation based on the American College of Medical Genetics and Genomics (ACMG) guidelines (22).
We retained coding variants in the RPA heterotrimer (RPA1, RPA2, and RPA3 genes) with genome aggregation database (gnomAD) non-cancer cohort allelic frequency (AF) of < 0.5% (23) of the following classes: missense, frameshift insertions and deletions, stop gain, and splice site. We further filtered to retain missense variants with a computed Combined Annotation-Dependent Depletion (CADD) (24) Phred score > 15.
Computational analysis of RPA mutations
We performed local coordinate minimization followed by global side-chain optimization with the Atomic Multipole Optimized Energetics for Biomolecular Applications (AMOEBA) polarizable force field (25) on 5 high resolution structures of RPA fragments collectively comprising 7 modular domain of RPA heterotrimer. These included X-ray structures of the DNA binding domains A and B, DBD-A and DBD-B (PDB: 1JMC) (26), and the RPA trimerization core composed of DBD-C, D and E (PDB: 1L1O) (27) and NMR structures of the DBD-F (5N8A) (28) and the wing helix domain (PDB: 1DPU) (29). Prior to minimization, the ssDNA was removed from the 1JMC structure and bound peptides were removed from the 2 NMR structures. We then used our optimized structures to predict protein stability differences ΔΔGFold (DDG untrained (DDGun)) (30). DDGun estimates the ΔΔGFold of missense variants from a linear regression of sequence and biochemical features determined from the protein structure. Destabilizing ΔΔGFold values indicate a decrease in the ratio of folded to unfolded protein due to the mutation (we define negative ΔΔGFold values as stabilizing and positive ΔΔGFold values as destabilizing). We established ΔΔGFold cut-offs for mutations highly likely to impact protein folding. Our cut-offs were determined based on a ΔΔGFold that affects the ratio of folded to unfolded protein 12-fold (~1.5 kcal/mol) for both stabilizing and destabilizing mutations.
Statistical analysis
We performed rare-variant burden tests for RPA1, RPA2, RPA3 variants using 5,993 cases from all pediatric cancers in our cohorts (pan-cancer) and within each sub-class of cancers, namely, hematologic (n = 3,452), solid (n = 1,974), and central nervous system CNS (n = 1,068) malignancies. For the control set, we retrieved all variants across RPA1, RPA2, and RPA3 from gnomAD v2 non-cancer subset containing 134,187 individuals with no reported malignancy (23). All variants from control dataset were processed through the same variant annotation and filtering workflow as our cancer cohort (AF < 0.5%). Enrichment tests for cases with and without germline ultra-rare (AF < 0.005%) plus novel (AF 0%) and rare (AF < 0.1%) variants in the 3 genes were performed using both two- and one-sided Fisher exact tests using the statistical package R (v4.3) described in previous studies (31, 32). We used Bonferroni correction to adjust for multiple testing with a significance cutoff of adjusted p-value of < 0.05.
Results
Variants identified among the RPA heterotrimer genes
Within the pan-cancer cohort, we identified 80 cases with 55 germline heterozygous RPA1, RPA2 or RPA3 variants meeting criteria of AF < 0.5% in gnomAD non-cancer cohort and CADD score > 15 for candidate variant selection (Figure 1A). Specifically, 40 RPA1, 7 RPA2 and 8 RPA3 unique heterozygous germline variants were identified in 63, 7 and 10 cases, respectively (Figure 1B). All variants were classified as variant of uncertain significance (VUS) according to the ACMG criteria (Tables 1–3). Majority of the variants (92% of RPA1, 71% of RPA2 and all RPA3 variants) had CADD scores > 20, indicating a higher probability of a deleterious effect (Tables 1–3). In addition, looking at variant burden in population, we found that 98% (54/55) of the identified RPA heterotrimer variants had AF < 0.1% (this includes rare, very-rare, ultra-rare, and novel variants, Figure 1B). All RPA1, RPA2 and RPA3 variants are mutually exclusive and no cases with compound heterozygous or homozygous variants were identified.
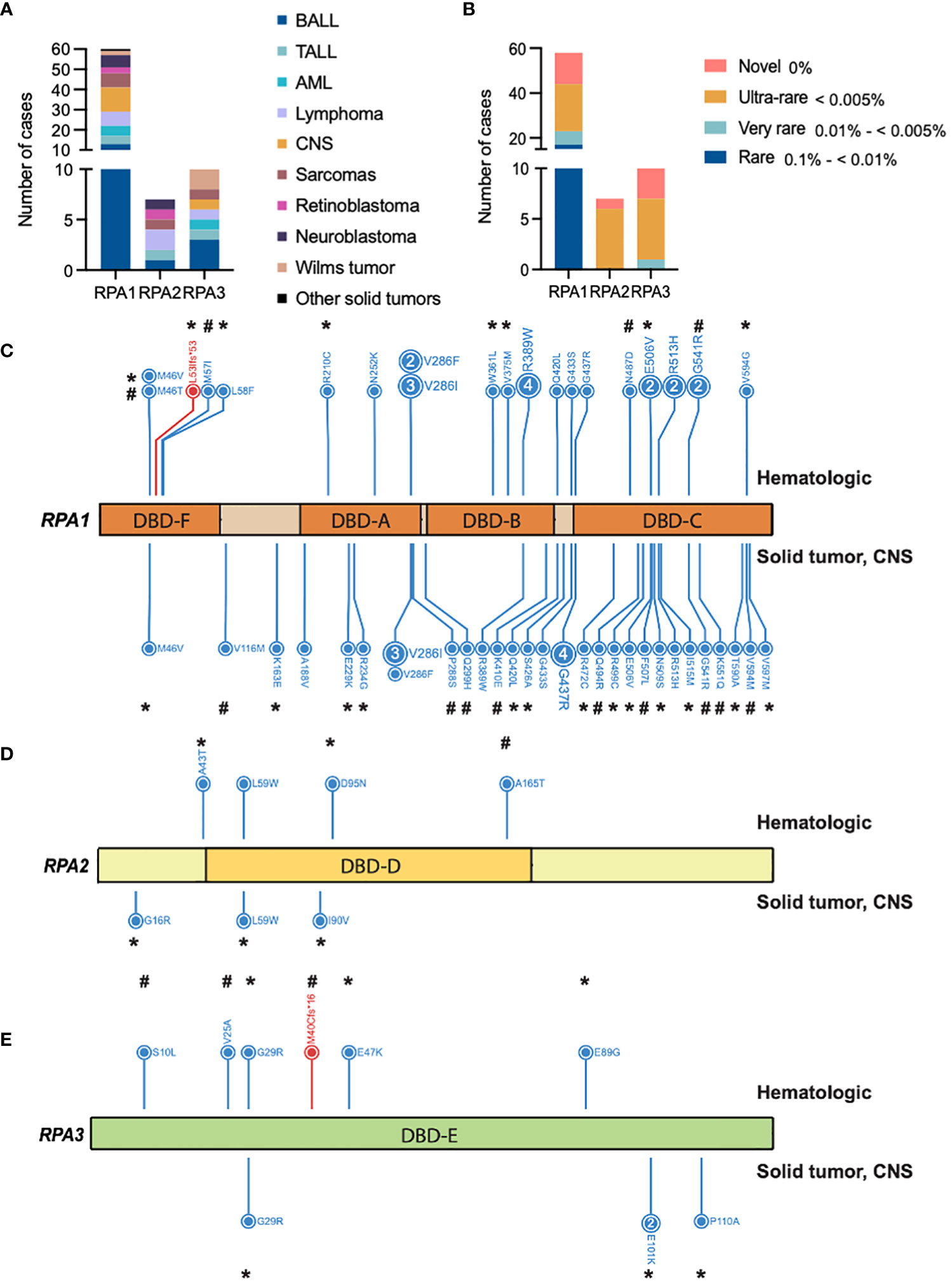
Figure 1 Germline heterozygous variants in the RPA heterotrimer in pediatric cancers. (A) Number of pediatric cancer cases with either RPA1, RPA2 or RPA3 heterozygous germline variants. B-ALL (B cell acute lymphoid leukemia), T-ALL (T cell acute lymphoid leukemia), AML (acute myeloid leukemia). Unique cancers are identified by different colors represented in the legend. (B) Number of cancer cases with either novel (pink), ultra-rare (gold), very rare (light teal), or rare (blue) germline variants in RPA1, RPA2 or RPA3 according to gnomAD allelic frequency. Schematic of human RPA1 (DNA binding domain (DBD- F, A, B, C)) (C), RPA2 (DBD-D) (D) and RPA3 (DBD-E) (E) proteins with germline variants denoted. Blue and red lettering represents missense and frameshift variants, respectively. Numbers within circles represent the number of cases that harbored that variant while lack of numbering denotes one case per variant. Variants found in hematologic cases are represented on top and solid (intra and extra cranial) malignancies are denoted at the bottom of each protein map. * = ultra-rare variant allelic frequency (< 0.005%), # = novel variants.
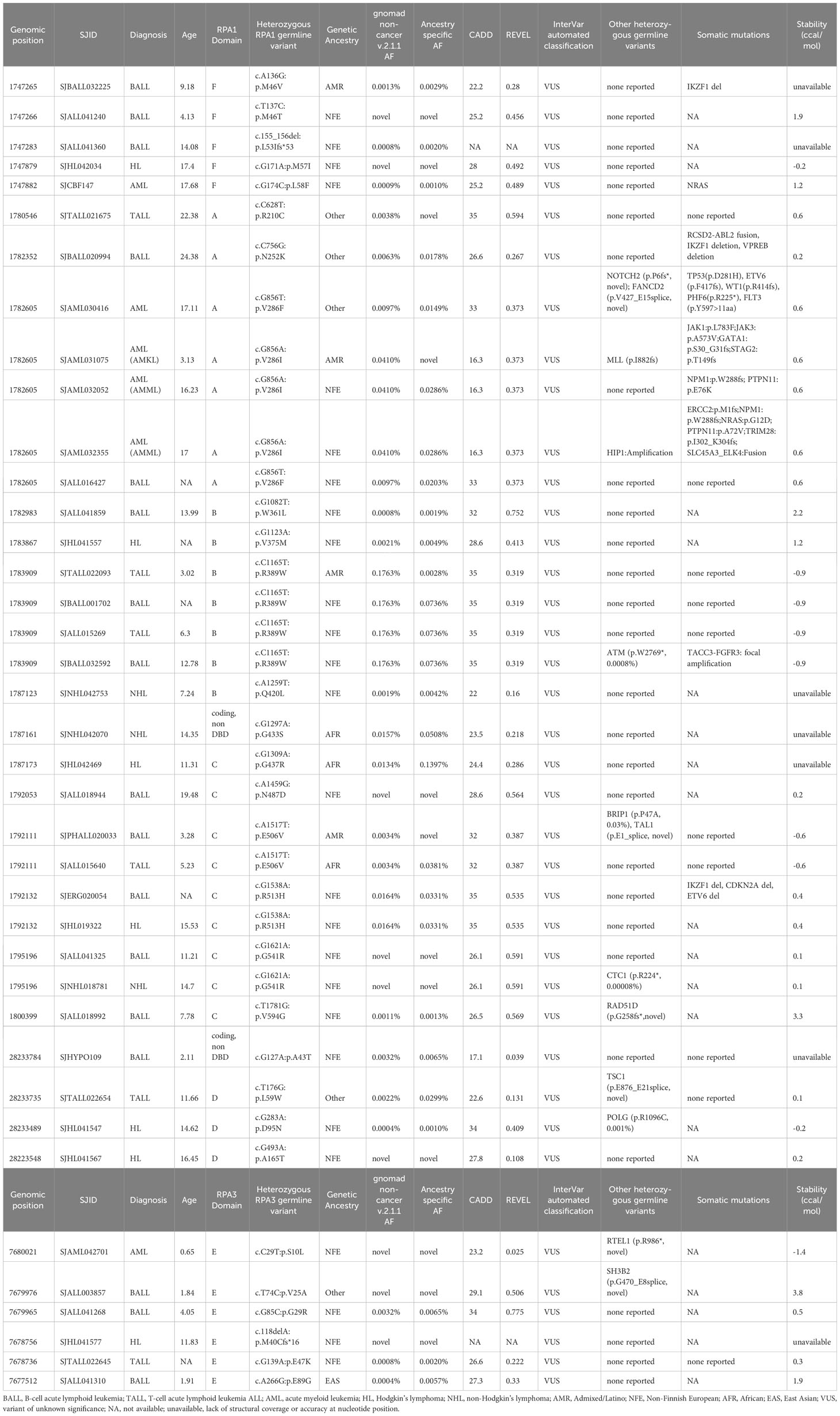
Table 1 Germline heterozygous variants found in RPA1, RPA2 and RPA3 in pediatric hematological malignancies.
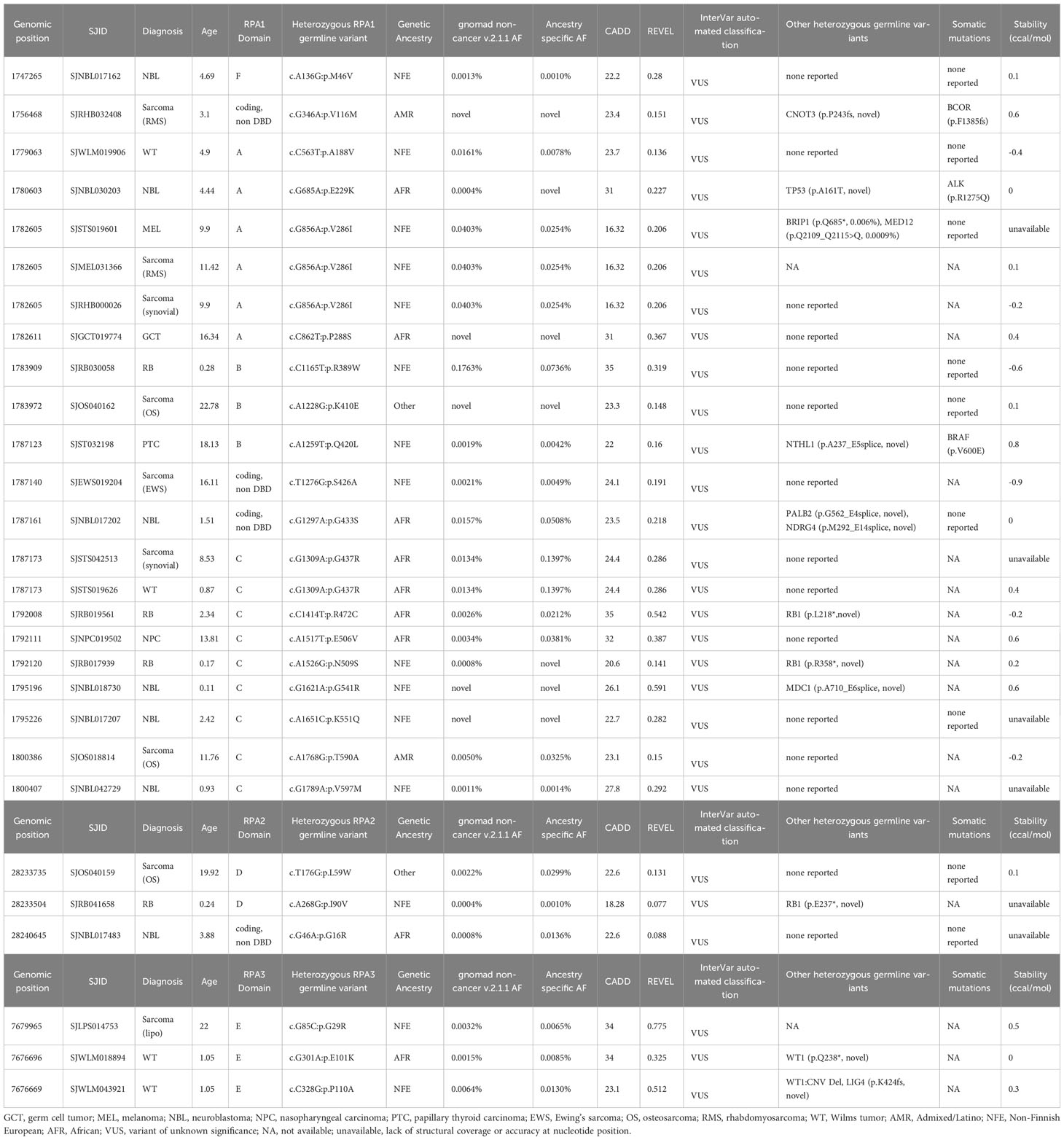
Table 2 Germline heterozygous variants found in RPA1, RPA2 and RPA3 in extra-cranial solid tumors.
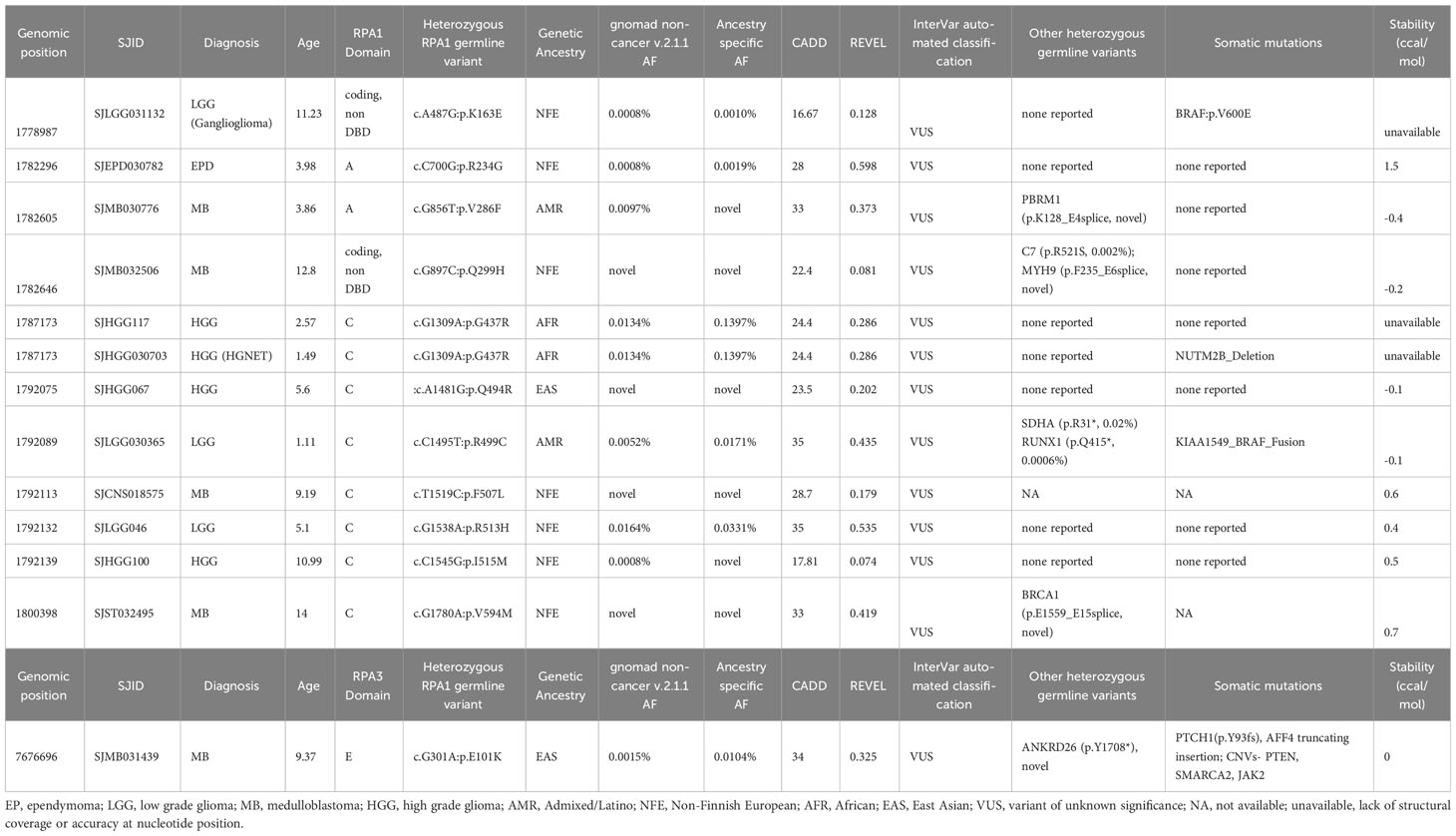
Table 3 Germline heterozygous variants found in RPA1, RPA2 and RPA3 in extra-cranial solid tumors.
RPA1 germline variants and cancers
RPA1 (616 amino acids, 70kDa) is the largest of the 3 subunits of the RPA heterotrimer. We discovered 1.05% (63/5993) of the cohort to harbor heterozygous germline RPA1 variants (Figure 1C; Tables 1–3), which was statistically not significant compared to gnomAD non-cancer controls for all cancers and cancer subtypes (Table 4). RPA1 has 4 modular oligosaccharide binding-fold domains commonly referred to as functional DNA binding domains (DBD): F, A, B and C spanning the N- to C- terminal regions of the protein. RPA1 variants were found across all 4 DBDs as follows: 6 in DBD-F, 15 in DBD-A, 10 in DBD-B, and 26 in DBD-C (Figure 1C). Of note, 6 cases were found to have RPA1 variants in the linker regions between 2 DBDs. All RPA1 variants were missense (Figure 1C) except for p.L53lfs*53 within DBD-F, which was found in 1 case. Three recurrently mutated amino acids were discovered in RPA1 domains DBD-A (p.V286, 9 cases), DBD-B (p.R389, 5 cases), and DBD-C (p.G437, 5 cases). We next focused specifically on novel and ultra-rare RPA1 variants (33), present in 14 and 21 cases, respectively (Figure 1B; Tables 1–3). Notably, we found significant enrichment of RPA1 novel and ultra-rare variants in our cohort (adjusted p-value < 0.05, Table 4).
Prediction of variant structural effect was performed by calculating protein stability change scores (ΔΔGFold) with a ΔΔGFold that affects the ratio of folded to unfolded protein 12-fold (~1.5 kcal/mol) for both stabilizing and destabilizing mutations. Significant scores (>1.5 kcal/mol) were demonstrated for 4 variants (p.M46T in DBD-F, p. R234G in DBD-A, p.W361L in DBD-B, p.V594G in DBD-C) which were novel or ultra-rare (Tables 1–3). RPA1 p.M46T is likely to destabilize folding of DBD-F resulting in the loss of multiple important protein-protein interactions (11). W361 is a key DNA binding residue in DBD-B and human cells with W361A support normal replication but are deficient in DNA repair (12, 34), suggesting that p.W361L may destabilize DBD-B folding resulting in hypomorphic RPA.
We next assessed which types of malignancies were present in patients with RPA1 variants (Figure 1A). We found comparable frequency of cases with RPA1 variants across solid tumors (n = 22, 1.1%), CNS cancers (n = 12, 1.1%) and hematological malignancies (n = 29, 0.8%). Among solid tumor cases with RPA1 variants, 31.8% (7/22) presented with sarcomas and 27.3% (6/22) were diagnosed with neuroblastoma. Notably, the 7 sarcoma cases carried 6 unique RPA1 variants (n = 2 novel and n = 1 ultra-rare) and only one was noted to have a concomitant germline mutation (Table 2). All 6 neuroblastoma cases were found to have unique RPA1 variants (n = 3 novel, n = 2 ultra-rare) of which half were found to have germline variants reported in PALB2/NDRG4, MDC1, or TP53 genes (Table 2). Three cases of retinoblastoma harbored unique RPA1 variants (2 ultra-rare) with 2 cases having concomitant germline RB1 mutation (Table 2). Two cases of Wilms tumor were identified to have germline RPA1 variants. Among the single cases of solid tumors (germ cell tumor, melanoma, nasopharyngeal carcinoma, and papillary thyroid carcinoma), 2 ultra-rare and 1 novel RPA1 variants were found.
Among cases with CNS tumors, 4 patients with medulloblastoma harbored novel (n = 3) or ultra-rare (n = 1) RPA1 variants. Each of these cases also carried other germline mutations (PBRM1, C7 and MYH9, BRCA1, ANKRD26) of which BRCA1 and ANKRD26 are cancer predisposition genes (Table 3). Furthermore, 4 cases with high grade glioma harbored 3 RPA1 variants (n = 1 novel, n = 1 ultra-rare), all clustering within DBD-C domain of RPA1. These patients had no other potentially causative germline variants reported in other predisposition genes. Among the 3 low grade glioma, 3 RPA1 variants (one ultra-rare) were identified, with one harboring other germline mutations in SDHA and RUNX1. Lastly, one ultra-rare RPA1 variant was identified in a case of ependymoma without other germline mutations (Table 3).
From patients with hematologic malignancies, RPA1 variants were most common in B-ALL (n = 13), followed by lymphoma (n = 7), AML (n = 5), and T-ALL (n = 4) (Table 1). Out of 13 B-ALL cases, 3 and 5 were novel and ultra-rare, respectively. Only 2 cases out of the 13 had heterozygous germline variants in cancer predisposition genes (RAD51D, BRIP1). Among lymphomas, we observed 4 Hodgkin’s lymphoma (n = 1 novel, n = 1 ultra-rare) and 3 non-Hodgkin’s lymphoma (n = 1 novel, n = 1 ultra-rare) with RPA1 variants. In the AML sub-cohort, we found 3 unique RPA1 variants in 5 cases, of which 4 were mutated at nucleotide 856 in DBD-A (c.856G>T, c.856G>A coding different amino acids) and 1 ultra-rare variant in DBD-F domain (Table 1). Of the 5 RPA1-mutated AML cases, 3 carried other germline variants (MLL, HIP1, NOTCH2 and FANCD2). Four unique RPA1 variants (n = 2 ultra-rare) were discovered in 4 patients with T-ALL, with one case having additional germline ATM variant (Table 1).
Given the occurrence of MDS/AML in one prior patient with germline RPA1 p.V227A with TBD (10) and 5 AML cases in this study, we queried a cohort of 41 young adults with AML and karyotype aberrations (18) for RPA heterotrimer germline variants. We found 1 ultra-rare (c.460G>A, T154A, AF 0.001%) and 2 rare (c.1397C>G, A466G, AF 0.027%; c.1538G>A, R513H, AF 0.016%) RPA1 heterozygous variants (Supplemental Table 1).
RPA2 and RPA3 germline variants and cancers
RPA2 is the second largest subunit (270 amino acids, 34kDa) of the RPA heterotrimer. We identified 6 heterozygous germline RPA2 variants in 7 cases of pediatric malignancies. Five variants are present in DBD-D (Figure 1D) and did not exhibit dysfunctional protein folding scores (Tables 1, 2). All variants were either ultra-rare (n = 5) or novel (n = 1). Four patients (4/3452, 0.1%) had hematological malignancies (n = 1 B-ALL, n = 1 T-ALL, n = 2 Hodgkin’s lymphoma) and 3 had solid cancers (n = 1 RBL, n = 1 neuroblastoma, n = 1 sarcoma). Other germline mutations were noted in 3 out of 7 cases (Tables 1, 2).
RPA3, although less than half the size of RPA2 (121 amino acids, 14kDa) had 8 unique germline heterozygous variants (n = 4 ultra-rare, n = 3 novel) in 10 cases of pediatric cancers, including 6 hematologic (B-ALL n = 3, T-ALL n = 1, AML n = 1, Hodgkin’s lymphoma n = 1), 3 solid tumors (Wilms tumor n = 2, sarcoma n = 1) and 1 CNS (medulloblastoma) cancers (Figure 1E; Tables 1–3). All were missense except for one frameshift (p.M40Cfs*16). Protein folding scores for 2 out of 7 available RPA3 variants were greater than 1.5 kcal/mol and were either novel or ultra-rare (Tables 1–3). Half of RPA3 mutated cases had other germline variants noted (Tables 1–3). The number of cases with RPA2 or RPA3 germline variants did not reach statistical significance compared to gnomAD non-cancer controls (Table 4).
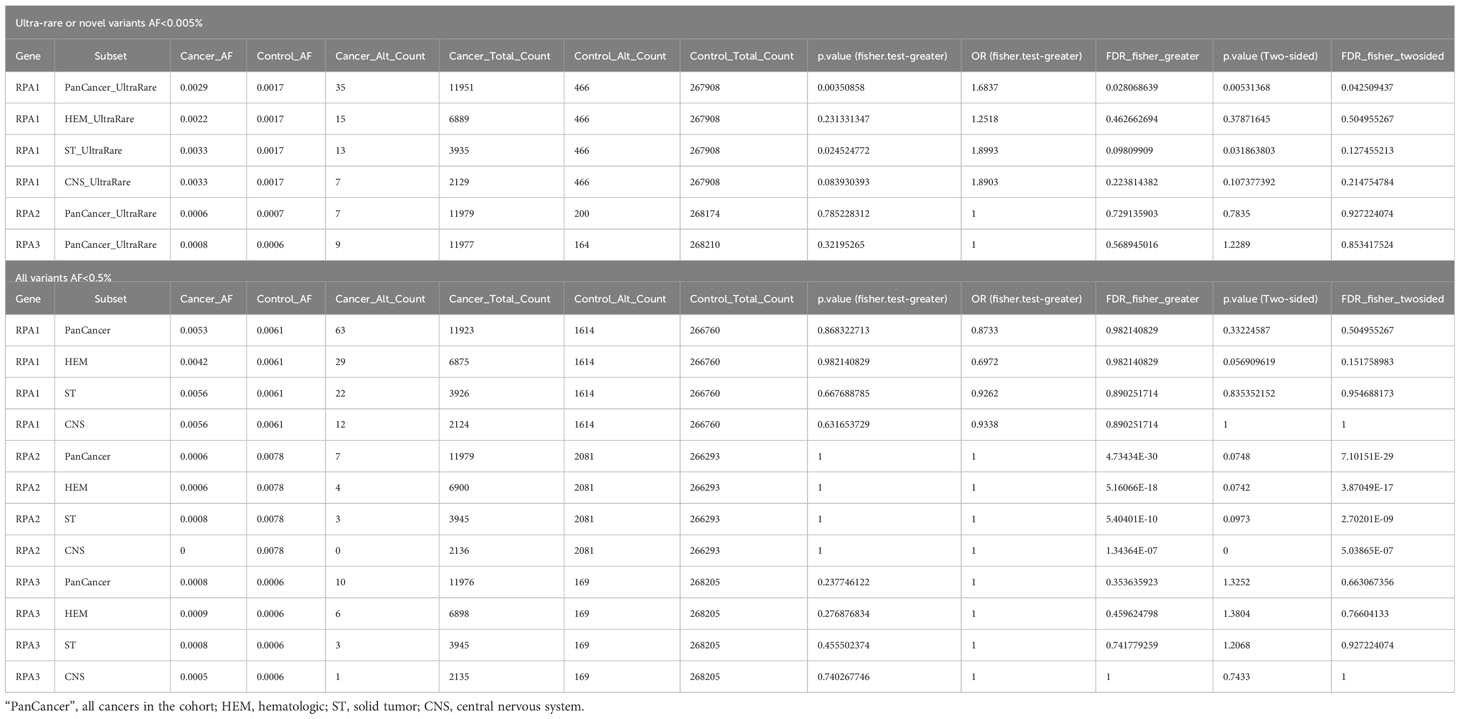
Table 4 Statistical analysis using two- and one-sided Fisher exact tests of ultra-rare plus novel and rare germline heterozygous variants in RPA1, RPA2 and RPA3 across hematologic, extra-cranial solid and CNS tumors.
Discussion
The RPA heterotrimer is an essential protein for binding ssDNA encountered in cellular transactions to facilitate DNA-DNA and DNA-protein interactions during DNA replication, repair, recombination, RNA transcription, and telomere maintenance. As such, mutations in this genome maintenance protein have been linked to cancer formation in mice (35) and are acquired in up to ~1% of human cancers (14). We recently demonstrated that heterozygous germline RPA1 mutations RPA1 c.680T>C p.V227A, c.718G>A p.E240K and c.808A>G p.T270A in DBD-A are associated with TBD, which predisposes to hematologic and solid tumors. In this study, one patient with RPA1-related TBD developed MDS (10). Based on these data, we reasoned that germline defects in RPA1 and possibly also the other 2 components of the RPA heterotrimer (RPA2 and RPA3) might be associated with cancer development. To this end, we investigated comprehensive germline genomic data for the presence of heterozygous variants in RPA1, RPA2 and RPA3 across a large series of pediatric hematologic, solid and CNS malignancies. We discovered significant enrichment of ultra-rare and novel RPA1 germline variants in our pediatric cancer cohort compared to non-cancer controls, positioning RPA1 as a novel candidate predisposition gene. Moreover, in an additional cohort of 41 patients with AML, we identified 3 heterozygous germline RPA1 variants (c460G>A, p.T154A; c.1397C>G, p.A466G; c.1538G>A, p.R513H) with potential pathogenic effect.
RPA1 harbored the most variants likely due to its larger size compared to RPA2 and RPA3. Although we did not observe a statistically significant enrichment of putative damaging variants in RPA2 and RPA3, some of the identified variants were novel or ultra-rare and could possibly have a deleterious effect. Thus, RPA2 and RPA3 could be considered as genes of unknown significance (GUS) yet potentially important in tumor formation. All 3 proteins are required to fold properly to form a functional RPA heterotrimer (13). For this reason, we calculated stabilities of the RPA modular domains harboring mutations to gain insight into the possible effect of identified germline variants on RPA heterotrimer function. Scores greater than 1.5 are highly predictive of protein instability and dysfunction. High protein folding scores were found for 4 unique RPA1 variants in 4 cases, 3 identified in patients with B-ALL and one in a patient with ependymoma. All were either ultra-rare or novel with CADD scores suggesting high likelihood of pathogenicity. Two RPA3 variants also harbored high protein folding scores in patients with B-ALL. This suggests that dysfunctional folding of the RPA heterotrimer may lead to genomic instability in these patients.
In our discovery cohort, we identified 5 AML cases with germline RPA1 variants. One had an ultra-rare RPA1 p.L58F variant in DBD-F and the remaining 4 had variants affecting nucleotide 856 within DBD-A domain (c.G856A, p.V286I in 3 cases and c.G856T, p.V286F in one case). The resulting amino acid changes do not differ in size or charge from wild-type valine and have a neutral protein folding score of 0.6. However, these mutations may disrupt protein-protein, protein-DNA interactions, or post-translational modifications, which are known mechanisms implicated in pathogenicity of RPA1 variants in various experimental models (10–13, 35). Additionally, TBD-associated pathogenic RPA1 variants, p.V227A, p.E240K and p.T270A, have protein folding scores of 1.4, 0.1 and 0.2 (consistent with normal protein folding shown in biochemical assays) yet were shown to exert gain-of-function effect on DNA binding and melting of telomeric G-quadruplexes (10). Three of the 4 AML cases with RPA1 variants in DBD-A domain had additional germline variants in genes (NOTCH2, FANCD2, MLL, HIP1) which, together with RPA1 may have an epistatic effect to cause overall genomic instability. Corroborating data from a small cohort of 41 AML patients in which 3 patients carried RPA1 variants (p.T154A in linker region; p.A466G and p.R513H in DBD-C) deserves further investigation. Beyond RPA1 in the AML cohort, we also found a novel germline missense variant in RPA3 in an infant with AML who also harbored a germline truncating variant in the DNA helicase, RTEL1, which is associated with TBD (36, 37). More functional studies are needed to determine the pathogenicity of RPA1 V286I/F alterations and their role in hematologic malignancy.
Among the 13 CNS tumors with variants in RPA heterotrimer genes, 9 cases were high grade neoplasms, including medulloblastoma and high-grade glioma. Interestingly, 3 of the 5 medulloblastoma cases had novel and one very rare germline RPA1, as well as one ultra-rare RPA3 variant. Notably, even though variants in other unrelated genes were also found in 4 of the 5 medulloblastoma cases, none of these genes have been previously associated with medulloblastomas in the literature. Other studies have identified germline defects in DNA repair genes in medulloblastoma (38, 39). It would stand to reason that germline mutations in the RPA heterotrimer, which functions in almost all DNA repair pathways, could potentiate oncogenic transformation. Further investigation should focus on assessing the function of RPA mutant proteins in DNA repair and their contribution to tumor biology.
Our study has several limitations. Although all cases were assessed using a uniform pipeline, the cohort is skewed towards cases with B-ALL (~4-fold higher number of B-ALL compared to solid and CNS cancers). We included all germline and somatic mutations per case that were reported in previously published studies; however, this information was unavailable for a proportion of cases and therefore we cannot make definitive conclusions about RPA variants being the sole germline driver in these cancers. Although ultra-rare and novel heterozygous germline variants in RPA1 were significantly enriched in pediatric cancers, it is difficult to ascertain pathogenicity and clinical relevance without functional follow-up, which falls beyond the scope of this study. It is plausible that variants with high in-silico protein folding energy, ultra-rare and/or novel allelic frequency and high pathogenicity scores may be clinically relevant and should be among the top variants to explore in future studies.
In summary, evasion of DNA repair mechanisms is a common theme among cancers. RPA is an essential protein for DNA replication and repair. Our study describes novel and rare variants with potentially deleterious effect in the RPA1, RPA2 and RPA3 genes in pediatric malignancies. Moreover, we have identified enrichment of RPA1 variants in cancer cases compared to non-cancer controls, suggesting that this gene potentially acts as a novel cancer driver. We plan to exploit our findings and perform further functional and biochemical characterization of recurrent cancer associated RPA1 variants to assess their potential use as targets for future cancer therapies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
RS, KN, and MW: conceptual design of the study and data interpretation. NO and WC: data analysis and statistics. RG, MJS, and MS: computational analysis of RPA mutations and interpretation. MK and FB: conceptual design and interpretation. All authors contributed to manuscript preparation and editing. All authors approved the submitted version.
Funding
RS was supported by American Society of Hematology Research Training Awards for Fellows and K08 DK134873. MW was supported by Evans MDS Foundation DRG Grant and Dresner Foundation Grant. MS was supported by R35GM131704. FB was supported by “Württembergischer Krebspreis 2019”, START Grant (N° 691743, RWTH Aachen University), “Aachener Krebs-und Leukämiehilfe” and the Deutsche Forschungsgemeinschaft (DFG) through the CRU344. MJS was supported by NIH R01DC012049 and NSF CHE-1751688.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1229507/full#supplementary-material
References
1. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
2. Kennedy RD, D'Andrea AD. DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J Clin Oncol (2006) 24:3799–808. doi: 10.1200/JCO.2005.05.4171
3. Sharma R, Lewis S, Wlodarski MW. DNA repair syndromes and cancer: Insights into genetics and phenotype patterns. Front Pediatr (2020) 8:570084. doi: 10.3389/fped.2020.570084
4. Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, et al. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res (2017) 23:e23–:e31. doi: 10.1158/1078-0432.CCR-17-0465
5. Croitoru ME, Cleary SP, Di Nicola N, Manno M, Selander T, Aronson M, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst (2004) 96:1631–4. doi: 10.1093/jnci/djh288
6. de Voer RM, Hahn MM, Mensenkamp AR, Hoischen A, Gilissen C, Henkes A, et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep (2015) 5:14060. doi: 10.1038/srep14060
7. Hall MJ, Bernhisel R, Hughes E, Larson K, Rosenthal ET, Singh NA, et al. Germline pathogenic variants in the ataxia telangiectasia mutated (ATM) gene are associated with high and moderate risks for multiple cancers. Cancer Prev Res (Phila) (2021) 14:433–40. doi: 10.1158/1940-6207.CAPR-20-0448
8. Kim J, Light N, Subasri V, Young EL, Wegman-Ostrosky T, Barkauskas DA, et al. Pathogenic germline variants in cancer susceptibility genes in children and young adults with rhabdomyosarcoma. JCO Precis Oncol (2021) 5. doi: 10.1200/PO.20.00218
9. Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet (2012) 8:e1002894. doi: 10.1371/journal.pgen.1002894
10. Sharma R, Sahoo SS, Honda M, Granger SL, Goodings C, Sanchez L, et al. Gain-of-function mutations in RPA1 cause a syndrome with short telomeres and somatic genetic rescue. Blood (2022) 139:1039–51. doi: 10.1182/blood.2021011980
11. Caldwell CC, Spies M. Dynamic elements of replication protein A at the crossroads of DNA replication, recombination, and repair. Crit Rev Biochem Mol Biol (2020) 55:482–507. doi: 10.1080/10409238.2020.1813070
12. Haring SJ, Mason AC, Binz SK, Wold MS. Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J Biol Chem (2008) 283:19095–111. doi: 10.1074/jbc.M800881200
13. Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem (1997) 66:61–92. doi: 10.1146/annurev.biochem.66.1.61
14. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res (2019) 47:D941–7. doi: 10.1093/nar/gky1015
15. Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, et al. The pediatric cancer genome project. Nat Genet (2012) 44:619–22. doi: 10.1038/ng.2287
16. Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, et al. Genomes for kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov (2021) 11:3008–27. doi: 10.1158/2159-8290.CD-20-1631
17. Wang Z, Wilson CL, Easton J, Thrasher A, Mulder H, Liu Q, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol (2018) 36:2078–87. doi: 10.1200/JCO.2018.77.8589
18. Kirschner M, Rolles B, Crysandt M, Rollig C, Stolzel F, Kramer M, et al. Impaired overall survival in young patients with acute myeloid leukemia and variants in genes predisposing for myeloid Malignancies. Hemasphere (2022) 6:e787. doi: 10.1097/HS9.0000000000000787
19. Chen W, et al. A rare variant analysis framework using public genotype summary counts to prioritize disease-predisposition genes. Nat Commun (2022) 13:2592. doi: 10.1038/s41467-022-30248-0
20. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
21. Li Q, Wang K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
22. Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med (2008) 10:294–300. doi: 10.1097/GIM.0b013e31816b5cae
23. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
24. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res (2019) 47:D886–94. doi: 10.1093/nar/gky1016
25. Tollefson MR, Litman JM, Qi G, O'Connell CE, Wipfler MJ, Marini RJ, et al. Structural insights into hearing loss genetics from polarizable protein repacking. Biophys J (2019) 117:602–12. doi: 10.1016/j.bpj.2019.06.030
26. Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature (1997) 385:176–81. doi: 10.1038/385176a0
27. Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J (2002) 21:1855–63. doi: 10.1093/emboj/21.7.1855
28. Guilliam TA, Brissett NC, Ehlinger A, Keen BA, Kolesar P, Taylor EM, et al. Molecular basis for PrimPol recruitment to replication forks by RPA. Nat Commun (2017) 8:15222. doi: 10.1038/ncomms15222
29. Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, et al. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell (2000) 103:449–56. doi: 10.1016/S0092-8674(00)00136-7
30. Montanucci L, Capriotti E, Frank Y, Ben-Tal N, Fariselli P. DDGun: an untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinf (2019) 20:335. doi: 10.1186/s12859-019-2923-1
31. Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell (2018) 173:355–370 e14. doi: 10.1016/j.cell.2018.03.039
32. Oak N, Cherniack AD, Mashl RJ, Network TA, Hirsch FR, Ding L, et al. Ancestry-specific predisposing germline variants in cancer. Genome Med (2020) 12:51. doi: 10.1186/s13073-020-00744-3
33. Rasnic R, Linial N, Linial M. Expanding cancer predisposition genes with ultra-rare cancer-exclusive human variations. Sci Rep (2020) 10:13462. doi: 10.1038/s41598-020-70494-0
34. Hass CS, Chen R, Wold MS. Detection of posttranslational modifications of replication protein A. Methods Mol Biol (2012) 922:193–204. doi: 10.1007/978-1-62703-032-8_15
35. Wang Y, Putnam CD, Kane MF, Zhang W, Edelmann L, Russell R, et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet (2005) 37:750–5. doi: 10.1038/ng1587
36. Calado RT, Young NS. Telomere diseases. N Engl J Med (2009) 361:2353–65. doi: 10.1056/NEJMra0903373
37. Marsh JCW, Gutierrez-Rodrigues F, Cooper J, Jiang J, Gandhi S, Kajigaya S, et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv (2018) 2:36–48. doi: 10.1182/bloodadvances.2017008110
38. Trubicka J, Zemojtel T, Hecht J, Falana K, Piekutowska-Abramczuk D, Ploski R, et al. The germline variants in DNA repair genes in pediatric medulloblastoma: a challenge for current therapeutic strategies. BMC Cancer (2017) 17:239. doi: 10.1186/s12885-017-3211-y
Keywords: RPA1, RPA2, RPA3, germline mutation, cancer
Citation: Sharma R, Oak N, Chen W, Gogal R, Kirschner M, Beier F, Schnieders MJ, Spies M, Nichols KE and Wlodarski M (2023) Germline landscape of RPA1, RPA2 and RPA3 variants in pediatric malignancies: identification of RPA1 as a novel cancer predisposition candidate gene. Front. Oncol. 13:1229507. doi: 10.3389/fonc.2023.1229507
Received: 26 May 2023; Accepted: 18 September 2023;
Published: 06 October 2023.
Edited by:
Makiko Mochizuki-Kashio, Tokyo Women’s Medical University, JapanReviewed by:
Lukasz Gondek, Johns Hopkins University, United StatesDragana Vujic, Motehr and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Serbia
Copyright © 2023 Sharma, Oak, Chen, Gogal, Kirschner, Beier, Schnieders, Spies, Nichols and Wlodarski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richa Sharma, cmljaGEuc2hhcm1hQHN0anVkZS5vcmc=