 Baiqi Liu
Baiqi Liu Zefang Sun1,2†
Zefang Sun1,2† Shuai Zhu
Shuai Zhu Gengwen Huang
Gengwen Huang- 1Department of Hernia and Abdominal Wall Surgery, General Surgery, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
Desmoid tumor (DT) is a rare neoplasm characterized by the proliferation of myofibroblastic cells that infiltrates and invades adjacent tissues. Due to its locally aggressive and recurrent nature, DT often causes local symptoms and can be challenging to manage clinically. Therefore, identifying biomarkers that can predict the progression of DT and guide treatment decisions is critical. This review summarizes several biomarkers that have been implicated in active surveillance (AS) and the prediction of postoperative recurrence and attempts to elucidate their underlying mechanisms. Some of these novel markers could provide prognostic value for clinicians, and ultimately help facilitate optimal and accurate therapeutic decisions for DT.
1 Introduction
Desmoid tumor (DT), also known as aggressive fibromatosis (AF), is a rare and locally invasive soft tissue tumor, which occurs in approximately 3-5 individuals per million per year (1). It is estimated that 85% of DT cases are sporadic, while 3.5-32% of cases are related to familial adenomatous polyposis (FAP) or Gardner’s variant (2).
DT is characterized with monoclonal myofibroblast proliferation, which originates from musculoaponeurotic structures and may occur in the abdominal, chest walls, mesenteric root and extremities (3). Previous trauma history, genetic factors and pregnancy are all closely related to the etiology of DT (3).
Despite 20-30% of spontaneous regression or resolution in DT, the high recurrence rate(25-77%) of DT poses a long-term treatment dilemma (3, 4). DT patients face substantial challenges due to the unpredictable course and the uncertainty of treatment effect. While surgical resection was once the preferred treatment, it is now considered invasive, complex, and prone to recurrence. Even with negative resection margins, the recurrence rate was as high as 39.3% (5). Surgical resection with a safety margin is recommended only when tumor is large and causing significant symptoms. Currently, active surveillance (AS) is recommended based on results from numerous clinical trials and observational studies (3). AS and surgical treatments did not differ significantly in RFS over three years (6). If patients with DT have minimal or no symptoms, 2-3 monthly clinical and radiographic observation is recommended. Nevertheless, the unpredictable natural history of DT and lack of monitoring biomarkers make clinical decisions difficult during AS. Therefore, it is essential to discover stable biomarkers that can be used to predict progression and guide treatment direction. In the current review, we summarized a series of novel biomarkers with the aim of providing options for monitoring the clinical progression and recurrence of DT.
2 Pathway
2.1 Wnt pathway
Wnt pathway is an evolutionarily conserved signaling pathway that plays a key role in organ development and the function of various tissues (7). When the Wnt is inactivated, the complex consisting of GSK-3β, APC, CK1α and Axin performs sequential phosphorylation reactions at β-catenin. The phosphorylation of the β-catenin results in the ubiquitination and subsequent degradation by β-TrCP and its proteasome, which in turn increases the β-catenin steady-state level (8, 9). While binding with activated Wnt, Dvl and LRP5/6 phosphorylate and inactivate GSK-3β, preventing β-catenin from being phosphorylated and degraded. These processes cause β-catenin to accumulate in the nucleus, which binds with TCF/LEF to activate downstream target genes including MYC, COX, Cyclin D, PDGF, VEGF, etc. (8, 9) (Figure 1).
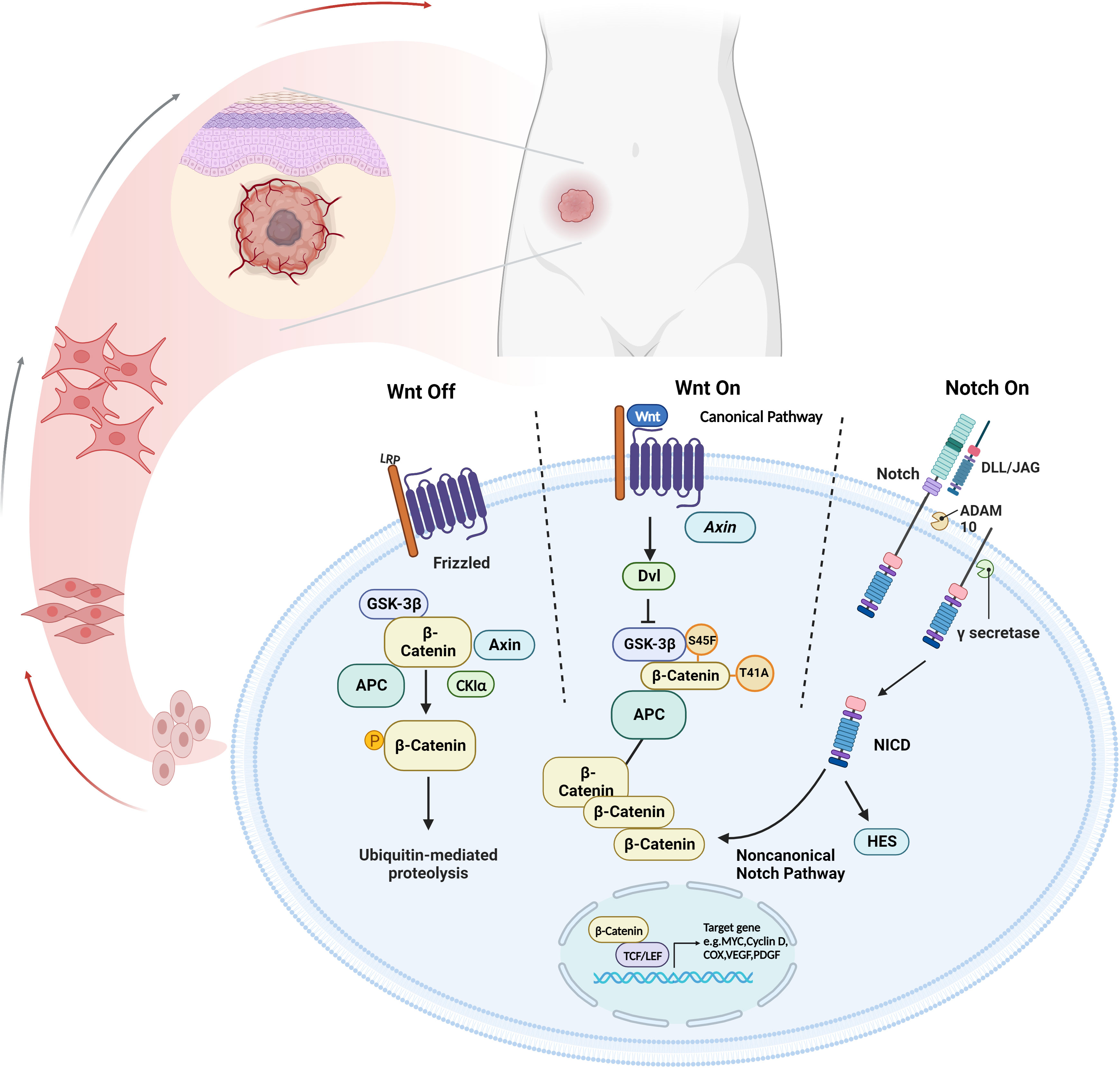
Figure 1 The cross-talk between Wnt/β-catenin and Notch signaling pathway. The canonical Wnt signaling pathway suppresses β-catenin ubiquitination to stabilize β-catenin expression, allowing it to contact TCF/LEF to upregulate pro-tumor factors. Notch signaling results in the progression disease progression through NICD in a non-canonical Notch pathway. Created with Biorender.com.
The activated Wnt pathway is closely associated with tumorigenesis (10–12). Numerous studies have shown that the Wnt signaling components, including β-catenin and APC, are essential in DT. Approximately 85-90% of sporadic DT accompanies with the mutation of CTNNB1, and most of the remaining cases are related to APC mutations (3). COX, VEGF and Cyclins are shown as representative downstream signaling molecules, contributing to the progression of DT. They are implicated in several pathophysiological mechanisms, including cell proliferation, invasion, angiogenesis and apoptosis (13). Thus, the molecules involved in the Wnt pathway may provide valuable prognostic targets for DT.
2.2 Notch pathway
A series of studies have shown that the Notch signaling pathway participated in cancer development by regulating cell proliferation, apoptosis, and differentiation (14–16). A canonical Notch signaling consists of Notch 1–4, DLL-1/3/4, JAG1/2 and CBF-1. Initiated by either ADAM10 or ADAM17, Ligand-activated Notch receptors undergo multiple proteolytic cleavages, forming the transmembrane fragment Notch. The proteolytic product transforms into the NICD after a second proteolytic cleavage, which is controlled by γ-secretase. NICDs enter the nucleus and interact directly with the CSL complex, regulating the expression of downstream genes, such as HEY and HES (14, 17).
The Notch-related molecules, including HES and ADAM, can be used as clinical markers for the diagnosis of DT. These molecules have been used to further distinguish DT from hypertrophic scars (18, 19). As for the treatment of advanced and progressive DT, preliminary data from the clinical trial showed 71% of advanced DT patients partially responded to oral γ-secretase inhibitors (GSI) PF-03084014 (20). Additionally, several clinical trials(NCT01981551, NCT00878189) observed positive results from advanced and recurrent DT treated with GSI (21–23). These results indicated that some key elements in the Notch pathway might contribute to the progression and recurrence of DT (24).
Previous studies have shown that the crosstalk between the Notch signaling pathway and the Wnt signaling pathway facilitated tumor progression (24, 25) (Figure 1). Peignon G et al. elucidated that Notch activation was an early event in Wnt-induced intestinal tumorigenesis, and maintained throughout downstream from the Wnt/β-catenin cascade (26). In patients with FAP, Notch signaling was activated by β-catenin-mediated upregulation of JAG1 (27). Among the complex Wnt and Notch crosstalk, many abnormally expressed specific molecules may be potential significant biomarkers.
2.3 Other potential pathways
In addition to the Wnt pathway and the Notch signaling pathway, other pathways have been shown to be involved in DT initiation and progression. The PI3K/Akt pathway has been considered as a therapeutic target for DT, as the tyrosine-kinase receptors (e.g., VEGFR and PDGFR) are detected in DT (28). Some tyrosine kinase inhibitors, including imatinib, sunitinib and sorafenib, are currently being tested in different phases of clinical trials for DT (29, 30). Expression of estrogen receptor-β (ERβ) in DT samples suggests the specific functions of estrogen signaling, dictating distinct therapeutic options for DT (31–33). However, the individual response to anti-estrogen agents varies and evidence from prospective studies is limited. Besides the above, recent studies have indicated that several signaling pathways, including TGF-β signaling pathway, JAK/STAT signaling pathway and Hedgehog signaling pathway, might also be involved in the transformation and progression of DT (34–36).
3 Biomarkers in AS
3.1 Tumor tissue
3.1.1 CTNNB1 S45F mutation
CTNNB1 mutations are common in DT patients. It is estimated that two types of CTNNB1 mutations, T41 and S45F, account for 35% and 55% of DT patients respectively. Other types of mutations are relatively infrequent, including S45P, D32G, T41A, S45C, T42_K49delinsQ and H36del (4, 37, 38). Accumulating evidence indicates that the S45F mutation is associated with poor prognosis in DT patients (39–42) (Table 1). Several studies reveal that S45F mutation is more likely to occur in the extremities, and the prognosis of these cases is worse than other sites (37, 43). A recent prospective study has shown that S45F mutation is associated with tumor progression (HR = 6.24 [95% CI 1.92–20.30]) and suggests the onset of active treatment (AT) during the AS (44). Compared with tumor diameter, gender, recurrence cases and other clinical factors, S45F mutation is significantly related to 3-year RFS (45). It has been reported that CTNNB1 mutation types was associated with tumor progression and aggressive treatment with adjuvant radiotherapy was administered accordingly. Follow-up showed no recurrence over 38 months, highlighting the value of CTNNB1 mutation type for guiding treatment strategies in DT (46). These findings suggested that S45F might be the most significant prognostic factor during the monitoring period. Further prospective studies with large sample sizes will give more solid evidence to guide clinical usage, especially for predicting recurrence and active surveillance.
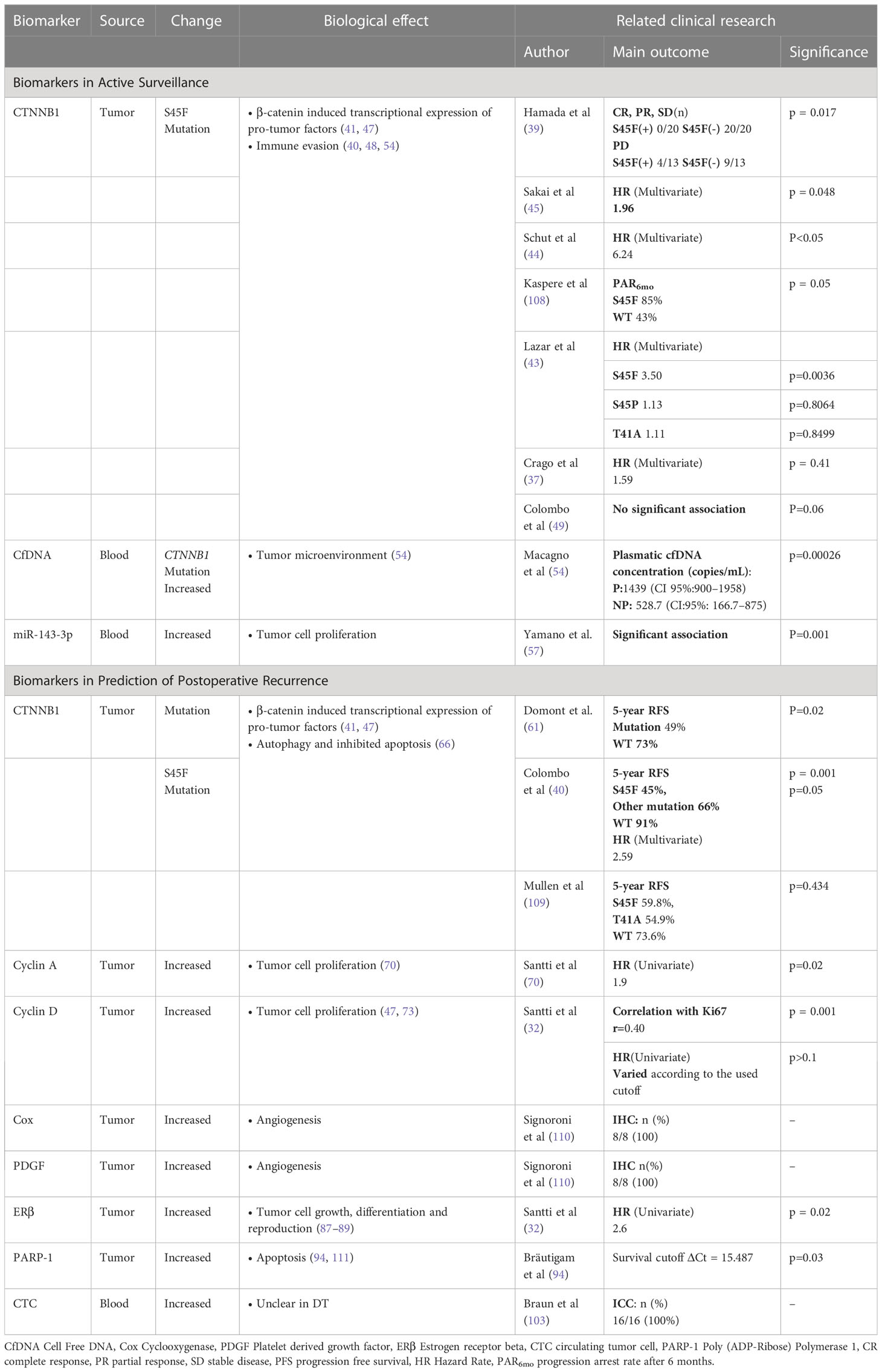
Table 1 Biomarkers of progression in desmoid tumor.
To account for the strong tendency of S45F mutation toward the progression, the function of CK1α in the Wnt/β-catenin pathway may be relevant. β-catenin ubiquitination begins with the phosphorylation in position 45 amino acid residue by CK1α. With the mutation of this specific residue, the ubiquitination process can not completely begin, leading to the imbalance of β-catenin (40). A study showed that the CBNNT1 S45F mutant allelic replication promoted the overexpression of β-catenin in DT. Correspondingly, the downstream factors, including COX, MYC, VEGF, and PDGF, show increased expression patterns, which are involved in tumor progression (41, 47).
In addition to affecting its own expression, the S45F mutation also works by influencing the immune response in the tumor microenvironment (TME). Colombo et al. observed that the two genetic lines (S45F/T41) exhibited different enrichment in immune-related genes (40). Inflammatory-defense-humoral immune response and antigen-binding related genes were enriched in T41F mutation cases, which corresponded with a high proportion of T cells at the tumor margin by immunohistochemical analysis. Instead, the mutation of S45F was found to be capable of increasing Treg cells and diminishing effector T-cell numbers, thereby promoting tumor progression by immune evasion (48). The discrepancy between S45F and other mutations may be due to the differences in the TME. However, the effect of TME on DT has not been clearly elaborated and deserves more exploration (Figure 2).
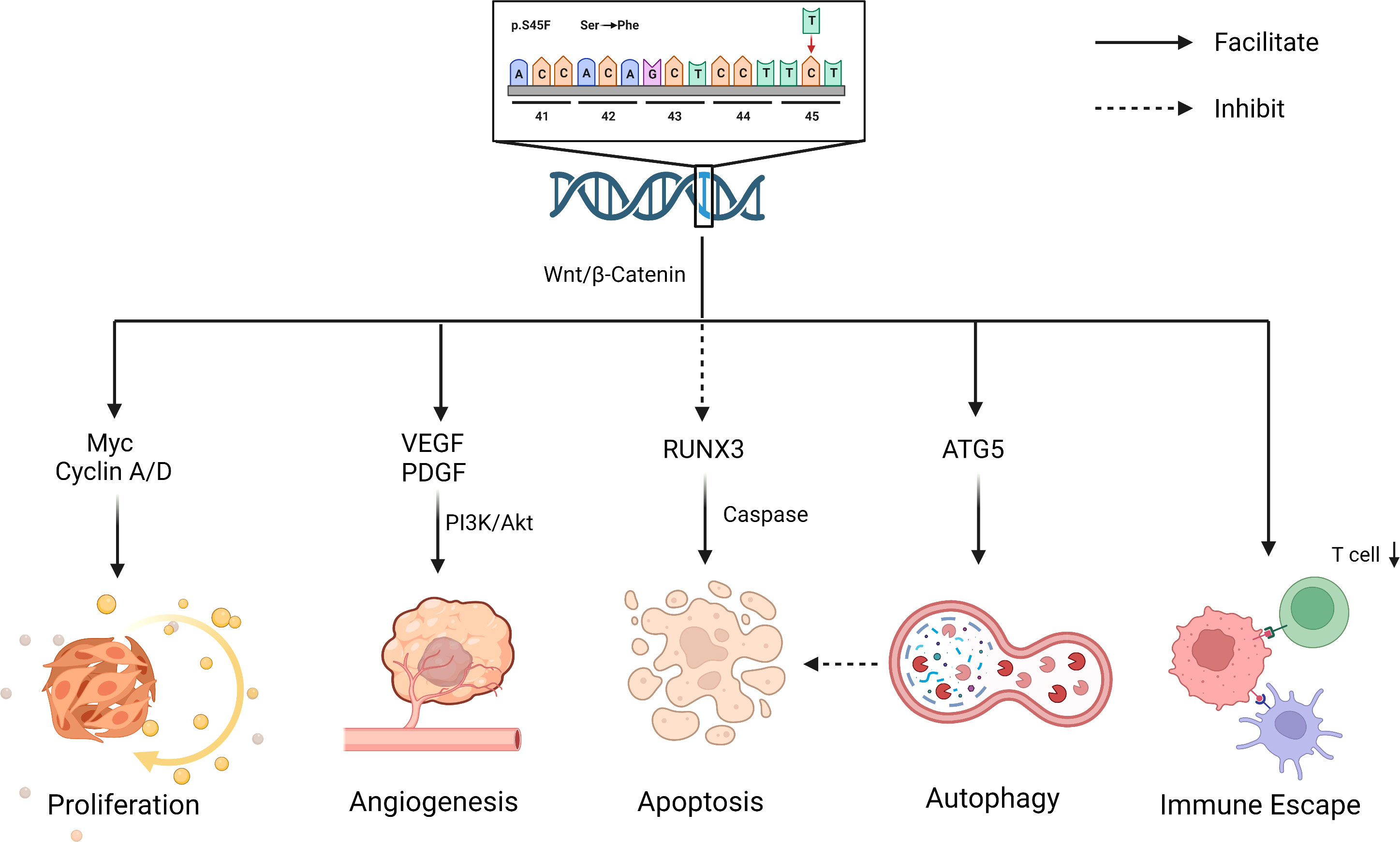
Figure 2 Biological effects for S45F mutation of CTNNB1. S45F mutation is involved in the progression and recurrence of DT, including autophagy, immune escape, proliferation, angiogenesis an inhibited apoptosis, etc. Created with Biorender.com.
Although many studies support the association between CTNNB1 and the poor prognosis of DT, the relationship between CTNNB1 mutation status and the clinicopathological characteristics of DT remains controversial. A follow-up prospective study detected that despite a trend toward the switch to AT, there is no significant association between RFS and the S45F mutation (49). Crago et al. found that “Wild types” defined by Sanger sequencing actually had CTNNB1, APC and other rare mutations(chromosome 6 loss and BMI1 mutation) using next generation sequencing (37). Colombo et al. first detected two different large deletions of about 190 bp involving exon 3 of CTNNB1 in two DT cases through separate analysis of unmapped reads and subsequent validation using PCR, which are difficult to be detected by conventional whole-exome sequencing (WES) analysis (50). These findings emphasize the challenges in detecting these deletions and a high level of tumor heterogeneity not previously described in DT. Because of these properties, larger samples and more precise sequencing methods are needed to confirm the clinical value of the CTNNB1 mutation.
3.2 Peripheral blood
3.2.1 Cell free DNA
CfDNA is a fragment of DNA released from tumor cells during necrosis or apoptosis. The cfDNA levels in the peripheral blood of most tumor patients are significantly higher than normal individuals (51). It has been demonstrated that cfDNA can be used to diagnose pancreatic cancer, colorectal cancer and other cancers (52, 53). Macagno et al. determined plasmatic cfDNA concentration and mutation from DT patients’ blood using a targeted Digital-droplet PCR (54). A significant correlation was found between the concentration of cfDNA and tumor progression (54). CfDNA level greater than 900 DNA copies/m provided 100% sensitivity and 76.5% specificity as the poor prognostic factor. When the cfDNA level was greater than 1375 DNA copies/m, the sensitivity was 57.14% and the specificity was 100%. Using these two thresholds(900 DNA copies/m and 1375 DNA copies/m), cfDNA could accurately predict the prognosis of DT in 65% of cases (54). However, it is noteworthy that there were inconsistent correlations between CTNNB1 status in cfDNA and prognosis. This could be attributed to the fact that cfDNA is derived from the TME and the adjacent tissues rather than tumor cells. Based on the local invasive characterization, surrounding cells and inflammatory cells in the TME released wild-type cfDNA into the blood. CfDNA is more indicative of the invasion intensity of DT rather than tumor size (54). The finding indicates that cfDNA analysis may be clinically useful for DT patients, especially those who are under AS management.
3.2.1 Circulating microRNA
MiRNAs are small non-coding RNA molecules that participate in RNA silencing and gene regulation post-transcriptionally, which are widely present in multiple diseases (55). There is growing evidence that circulating miRNAs can be used as a stable and reliable serological biomarker (56). Given the rarity of DT, the assessments of miRNAs on DT are limited. A recent study has investigated that the levels of circulating miR-143-3p were screened out as a candidate biomarker for FAP, compared with healthy controls (57). Notably, among these FAP patients, the miR-143-3p expression was strongly upregulated in DT tissues while reduced in colorectal cancer (CRC) tissues. Moreover, the miR-143-3p expression in DT tissues is consistent with plasma levels in FAP patients. In previous studies, miR-143-3p has been identified as highly expressed in mesenchymal cells (58). Bulk levels are based on the aggregation of sources, so the plasma miR-143-3p concentration might be influenced by the production or uptake of DT tissue. Furthermore, it has been reported that the expression of miR-143-3p is related to cell proliferation (59, 60). Thus, circulating miR-143-3p might be a potential diagnostic and prognostic biomarker for DT, which requires more experiments to confirm, especially in sporadic DT.
4 Biomarkers in prediction of postoperative recurrence
4.1 Tumor tissue
4.1.1 CTNNB1 mutation
In addition to its specific role in AS, CTNNB1 also has a predictive role in the prediction of postoperative recurrence. Domont et al. performed genetic testing on 155 frozen specimens of DT tissue to analyze whether the recurrence of DT was related to the mutation of CTNNB1. The results showed that regardless of the specific genotype, CTNNB1 mutated tumors had a worse prognosis than those with wild-type CTNNB1 (61). Furthermore, S45F mutation was identified as a significant risk factor for recurrence. A multicenter study found that the 5-year RFS were 45%, 91% and 66% for patients with the S45F mutation, WT and other mutations respectively (40). S45F mutation was an independent prognostic factor for patients with DT, rather than marginal status, tumor size, or disease site (40). Another retrospective report also described an association between S45F and the risk factors of relapse in pediatric patients. All of these evidences indicated that CTNNB1 mutation might be a predictive biomarker in postoperative recurrence (42). Although many studies have shown the relationship between the S45F mutation and relapse, the retrospective nature of the current studies constitutes an inherent limitation, which requires prospective studies for validation.
The contribution of the specific mutation on DT local recurrence is uncertain. S45F mutation completely blocks ubiquitination, leading to a massive increase in β-catenin, which partly explains the higher recurrence. In addition, there exists indirect evidence that the recurrence of DT is related to autophagy and inhibited apoptosis. Apoptosis and autophagy are two forms of programmed cell death, promoting or inhibiting tumorigenesis in response to a tumor’s type and stage (62, 63). Braggio et al. observed that autophagy gene overexpression promoted resistance to sorafenib in CTNNB1 S45F mutation in vitro and ex vivo (64). Previous research elucidated that the overexpression of antiapoptotic genes inhibited apoptosis induction, leading to resistance to therapeutics (65). The drug resistance of S45F mutation patients may be associated with their poor prognosis, in which apoptosis and autophagy may play a role. In addition, studies have shown that RUNX3, a transcription factor within the Wnt pathway, might be involved in caspase-3-dependent apoptosis (66) (Figure 2). All these suggest that apoptosis and autophagy may work in the process of CTNNB1 mutation affecting the outcome of DT patients, which deserves more attention.
4.1.2 Cyclins
Cyclin A is essential for the passage of cells through the S and G2M phases, which is usually accompanied with abnormal proliferation or tumor growth (67–69). As a downstream product, Cyclin A contributes to the regulation of cell cycle progression by the Wnt signaling pathway. Studies have shown that Cyclin A can influence the prognosis of DT patients. Santti K et al. observed that Cyclin A expression was significantly associated with decreased RFS (HR =1.9, P = 0.02) in a study enrolling 76 DT patients (70). However, there is fewer data about the association between Cyclin A and the recurrence of DT, and further investigations are needed to confirm these findings.
The cyclin D regulates pRB in the G1 phase of the cell cycle. During G1 phase, the pRB binds to transcription factors like E2F to regulate cell growth (71, 72). Researches show that cyclin D1 overexpression and CTNNB1 mutation are correlated in DT (p = 0.029; p = 0.034, respectively) (73, 74). However, recent studies revealed that excessive Cyclin D could not predict a high risk of recurrence and local progression (32, 70). A possible explanation for this contradiction is that the progression is caused by inhibited apoptosis rather than cell proliferation driven by Cyclin D. Upon recurrence of the tumor, increased expression of the anti-apoptotic protein Bcl-2, Bcl-XL, Survivin and transcription factor NF-κB was observed, but no cell proliferation occurred (75, 76). Therefore, the function of Cyclin D in DT remains to be determined. More comprehensive studies are required to evaluate and confirm the prognostic value of Cyclin D and investigate apoptosis in DT.
4.1.3 COX2 and PDGFβ
As a downstream target of the Wnt pathway, COX2 is a key enzyme responsible for prostaglandin synthesis. It plays a significant role in CRC progression with angiogenesis and invasion by modulating the PDGF (77, 78). In DT, COX2 immunoreactivity is significantly higher than in hypertrophic scars and normal fibrous tissue (18). Mignemi et al. found that the COX2 expression correlated with PDGFβ expression and increased its activity (18). PDGFβ expression was observed in all DT samples (27/27) with a significant immunoreactivity compared to normal tissues (28). Matano et al. investigated that the recurrent DT had higher microvessel density compared with normal samples, indicating that angiogenesis was an essential component in tumor recurrence (79). These two molecules might be underlying biomarkers for the prediction of recurrence by participating in angiogenesis.
4.1.4 Estrogen signaling related molecules
The estrogen-driven pathway participates in various physiological functions by regulating gene expression, which serves as the basis for many therapeutic interventions (80). Many studies have demonstrated an involvement of the estrogen receptor in the progression of the tumor, making it a common prognostic factor and an attractive therapeutic target (81–83). Most estrogen-related studies on DT are based on clinical observations. Females are more likely to develop DT, particularly during the fertile period. Epidemiological and clinical studies indicated that ERβ is an effective biomarker for predicting outcomes. Several studies demonstrated that DT overexpressed mainly ERβ instead of ERα, with an estimated expression rate of 54.5–90% (84–86). Santti et al. analyzed 83 consecutive DT samples immunohistochemically for ERβ, Cyclin D, and Ki67. A significant correlation was found between ERβ expression and the high risk of recurrence (HR=2.6) (32). Furthermore, several studies elucidated that targeted therapy on ERβ prolonged the RFS and reached complete response (CR) in 6-14 months (87, 88). In a meta-analysis of 168 DT patients, the complete and partial response rate was 51%, with the anti-ERβ therapy or combination with the NSAID therapy (31). Another clinical trial observed only one patient (134 patients who completed treatment at least 1 year) experienced a relapse after 10 years (89). These results indicate that ERβ has a close relationship with the recurrence of DT, but the underlying mechanism remains unclear. Potential downstream targets may provide clinical value.
4.1.5 Poly ADP-ribose polymerase 1
PARP-1 is an enzyme belonging to the PARP family, and it accounts for more than 90% of the enzyme activity in its family. PARP-1 is essential for repairing DNA damage, including single-strand breaks and double-strand breaks. PARP-1 binds broken DNA to its N-terminal zinc finger structure, thereby producing poly ADP-ribose chains involved in DNA repair (90, 91). PARP-1 function in DT may be promising, as PARP-1 inhibitors have been used as chemo/radiosensitizers in Ewing sarcoma (92, 93). Bräutigam et al. investigated the mRNA levels of PARP-1, ERβ, progesterone receptor (PR) and androgen receptor (AR) in DT samples, and found only PARP-1 was related to early relapse (94). Although PARP-1 may promote DT recurrence, the detailed mechanism is unknown. Previous research elucidated that cells disassembled and underwent apoptosis, as PARP-1 was cleaved (95). PARP-1 requires NAD+ as a substrate for DNA repair, which means overexpressing PARP-1 might consume available NAD+ and create a metabolic vulnerability that can be targeted (96). The related metabolomics profiles for DT showed that 1-methylnicotinamide, involved in NAD metabolism, was highly expressed in the S45F tumor cell line (97), which indicated PARP-1 might have an impact on DT progression through oxidative metabolism. More experiments are needed to confirm these assumptions.
4.1.6 Potential biomarkers through sequencing
Recently, with the continuous development of sequencing technology, researchers can efficiently distinguish differentially expressed genes, which allows screening of potential markers to assess the recurrent risk in DT. Using WES, Kohsaka et al. identified three genes for prognosis, namely IFI6, CKLF and LGMN (98). IFI6 was the only statistically significant gene. Salas et al. screened out FECH, STOML2 and TRIP6 which were able to predict RFS (99). In addition to coding genes, Cavallini et al. found the dysregulation of miR-21-3pg and miR-197-3p also associated with CTNNB1 mutation might affect the progression of DT (100). However, these molecules need to be further validated in animal models and tumor samples.
4.2 Peripheral blood
4.2.1 Circulating tumor cells
As the precursors of tumor dissemination and metastasis, CTCs are associated with cancer metastasis and poor prognosis (101). CTCs and circulating tumor microemboli in peripheral blood have been reported as early indicators for tumor invasion (102). In contrast, the role of CTCs in mesenchymal neoplasms is poorly investigated and remains unclear. In a recent study, CTCs were identified in the peripheral blood of patients with DT after AT, especially the recurrent cases (103). This outcome looks contradictory since DT is thought to lack metastasis potential. This may be inferred from two perspectives. On the one hand, as this study focused on patients following surgery, surgical manipulation may affect the CTCs release, called intraoperative tumor metastasis. Moreover, it is also possible for CTCs to colonize their tumors of origin, a process called “tumor self-seeding” (104). CTCs could reinfiltrate and promote angiogenesis in the primary tumor, which means they can easily survive in the tumor environment from their primary organs with fewer adaptations (105). This process could have consequences for tumor growth and progression, which might be a potential biomarker for DT recurrence. Several studies have associated CTC count with survival outcomes after metastatic cancer and high-CTC counts have been reported in conjunction with poor prognosis (106, 107). In DT, in addition to clinical application assessment, baseline CTC counts which have prognostic value remain to be determined experimentally.
5 Perspectives and concluding remarks
High clinical variability and unclear mechanism of DT make it a clinical dilemma. Thus, it is important to find molecules as reliable predictors of recurrence and progression. The current review summarizes potential biomarkers in DT progression and prognosis, focusing on the underlying mechanisms. Among these biomarkers, CTNNB1 mutations have been demonstrated to have promising clinical value in assessing relapse and prognosis, particularly in AS. However, the biological roles of these molecules will require more comprehensive research in the future. Increasing sample sizes and standardization are necessary for future research to determine causality and long-term effects.
Author contributions
ZS and BL designed this study. BL drafted the manuscript. RZ, ZS, DS and SZ revised the manuscript. GH supervised this study and contributed to manuscript revision with contributions from all of the other authors. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that this review was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Zhong X, Hu X, Zhao P, Wang Y, Fang XF, Shen J, et al. The efficacy of low-power cumulative high-intensity focused ultrasound treatment for recurrent desmoid tumor. Cancer Med (2022) 11:2079–84. doi: 10.1002/cam4.4573
2. Libertini M, Mitra I, van der Graaf WTA, Miah AB, Judson I, Jones RL, et al. Aggressive fibromatosis response to tamoxifen: lack of correlation between MRI and symptomatic response. Clin Sarcoma Res (2018) 8:13. doi: 10.1186/s13569-018-0100-3
3. Kasper B, Baumgarten C, Garcia J, Bonvalot S, Haas R, Haller F, et al. An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol (2017) 28:2399–408. doi: 10.1093/annonc/mdx323
4. Huss S, Nehles J, Binot E, Wardelmann E, Mittler J, Kleine MA, et al. β-catenin (CTNNB1) mutations and clinicopathological features of mesenteric desmoid-type fibromatosis. Histopathology (2013) 62:294–304. doi: 10.1111/j.1365-2559.2012.04355.x
5. Nuyttens JJ, Rust PF, Thomas CR, Turrisi AT. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors: A comparative review of 22 articles. Cancer (2000) 88:1517–23. doi: 10.1002/(SICI)1097-0142(20000401)88:7<1517::AID-CNCR3>3.0.CO;2-9
6. Bonvalot S, Eldweny H, Haddad V, Rimareix F, Missenard G, Oberlin O, et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol (2008) 34:462–8. doi: 10.1016/j.ejso.2007.06.006
7. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
8. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol (2012) 4:a008052. doi: 10.1101/cshperspect.a008052
9. Huang P, Yan R, Zhang X, Wang L, Ke X, Qu Y. Activating Wnt/β-catenin signaling pathway for disease therapy: Challenges and opportunities. Pharmacol Ther (2019) 196:79–90. doi: 10.1016/j.pharmthera.2018.11.008
10. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene (2017) 36:1461–73. doi: 10.1038/onc.2016.304
11. Zhao H, Ming T, Tang S, Ren S, Yang H, Liu M, et al. Wnt signaling in colorectal cancer: pathogenic role and therapeutic target. Mol Cancer (2022) 21:144. doi: 10.1186/s12943-022-01616-7
12. Singla A, Wang J, Yang R, Geller DS, Loeb DM, Hoang BH. Wnt signaling in osteosarcoma. Adv Exp Med Biol (2020) 1258:125–39. doi: 10.1007/978-3-030-43085-6_8
13. Salas S, Chibon F. Biology and signaling pathways involved in the oncogenesis of desmoid tumors. Bull Cancer (2020) 107:346–51. doi: 10.1016/j.bulcan.2019.12.003
14. South AP, Cho RJ, Aster JC. The double-edged sword of notch signaling in cancer. Semin Cell Dev Biol (2012) 23:458–64. doi: 10.1016/j.semcdb.2012.01.017
15. Wu WKK, Wang XJ, Cheng ASL, Luo MXM, Ng SSM, To KF, et al. Dysregulation and crosstalk of cellular signaling pathways in colon carcinogenesis. Crit Rev Oncol Hematol (2013) 86:251–77. doi: 10.1016/j.critrevonc.2012.11.009
16. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther (2012) 11:1565–75. doi: 10.1158/1535-7163.MCT-11-0938
17. Bertrand FE, Angus CW, Partis WJ, Sigounas G. Developmental pathways in colon cancer: crosstalk between WNT, BMP, Hedgehog and Notch. Cell Cycle (2012) 11:4344–51. doi: 10.4161/cc.22134
18. Mignemi NA, Itani DM, Fasig JH, Keedy VL, Hande KR, Whited BW, et al. Signal transduction pathway analysis in desmoid-type fibromatosis: transforming growth factor-β, COX2 and sex steroid receptors. Cancer Sci (2012) 103:2173–80. doi: 10.1111/cas.12037
19. Misemer BS, Skubitz APN, Carlos Manivel J, Schmechel SC, Cheng EY, Henriksen JC, et al. Expression of FAP, ADAM12, WISP1, and SOX11 is heterogeneous in aggressive fibromatosis and spatially relates to the histologic features of tumor activity. Cancer Med (2014) 3:81–90. doi: 10.1002/cam4.160
20. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res (2015) 21:60–7. doi: 10.1158/1078-0432.CCR-14-0607
21. Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J (2015) 5:e350. doi: 10.1038/bcj.2015.80
22. Pfizer. A phase I trial of PF-03084014 in patients with advanced solid tumor malignancy and t-cell acute lymphoblastic leukemia/lymphoblastic lymphoma. (2019). Available at: https://clinicaltrials.gov; NCT00878189.
23. M. D AC. Phase II trial of the gamma-secretase inhibitor PF-03084014 in adults with desmoid tumors/aggressive fibromatosis. (2022). Available at: https://clinicaltrials.gov; NCT01981551.
24. Andersen P, Uosaki H, Shenje L, Kwon C. Non-canonical notch signaling: emerging role and mechanism. Trends Cell Biol (2012) 22:257–65. doi: 10.1016/j.tcb.2012.02.003
25. Carothers AM, Rizvi H, Hasson RM, Heit YI, Davids JS, Bertagnolli MM, et al. Mesenchymal stromal cell mutations and wound healing contribute to the etiology of desmoid tumors. Cancer Res (2012) 72:346–55. doi: 10.1158/0008-5472.CAN-11-2819
26. Peignon G, Durand A, Cacheux W, Ayrault O, Terris B, Laurent-Puig P, et al. Complex interplay between -catenin signalling and Notch effectors in intestinal tumorigenesis. Gut (2011) 60:166–76. doi: 10.1136/gut.2009.204719
27. Federman N. Molecular pathogenesis of desmoid tumor and the role of γ-secretase inhibition. NPJ Precis Onc (2022) 6:62. doi: 10.1038/s41698-022-00308-1
28. Cates JMM, Black JO, Itani DM, Fasig JH, Keedy VL, Hande KR, et al. Signal transduction pathway analysis in fibromatosis: receptor and nonreceptor tyrosine kinases. Hum Pathol (2012) 43:1711–8. doi: 10.1016/j.humpath.2011.12.021
29. Weiss A. A pilot study evaluating the use of mTor inhibitor sirolimus in children and young adults with desmoid-type fibromatosis. (2022). Available at: https://clinicaltrials.gov; NCT01265030.
30. Kim HS. A phase 1b/2a, open-label, multicentre study to assess the safety, tolerability, pharmacokinetics, and antitumor activity of vactosertib in combination with imatinib in patients with advanced desmoid tumor (Aggressive Fibromatosis). (2022). Available at: https://clinicaltrials.gov; NCT03802084.
31. Bocale D, Rotelli MT, Cavallini A, Altomare DF. Anti-oestrogen therapy in the treatment of desmoid tumours: a systematic review. Colorectal Dis (2011) 13:e388–395. doi: 10.1111/j.1463-1318.2011.02758.x
32. Santti K, Ihalainen H, Rönty M, Karlsson C, Haglund C, Sampo M, et al. Estrogen receptor beta expression correlates with proliferation in desmoid tumors. J Surg Oncol (2019) 119:873–9. doi: 10.1002/jso.25407
33. Quast DR, Schneider R, Burdzik E, Hoppe S, Möslein G. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: a single-center long-term observational study in 134 patients. Fam Cancer (2016) 15:31–40. doi: 10.1007/s10689-015-9830-z
34. Yun K-H, Park C, Ryu HJ, Ock C-Y, Lee YH, Baek W, et al. Therapeutic implications of TGF-β Pathway in desmoid tumor based on comprehensive molecular profiling and clinicopathological properties. Cancers (Basel) (2022) 14:5975. doi: 10.3390/cancers14235975
35. Tjandra SS, Hsu C, Goh YI, Gurung A, Poon R, Nadesan P, et al. IFN-{beta} signaling positively regulates tumorigenesis in aggressive fibromatosis, potentially by modulating mesenchymal progenitors. Cancer Res (2007) 67:7124–31. doi: 10.1158/0008-5472.CAN-07-0686
36. Timbergen MJM, Smits R, Grünhagen DJ, Verhoef C, Sleijfer S, Wiemer EAC. Activated signaling pathways and targeted therapies in desmoid-type fibromatosis: A literature review. Front Oncol (2019) 9:397. doi: 10.3389/fonc.2019.00397
37. Crago AM, Chmielecki J, Rosenberg M, O’Connor R, Byrne C, Wilder FG, et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer (2015) 54:606–15. doi: 10.1002/gcc.22272
38. Aitken SJ, Presneau N, Kalimuthu S, Dileo P, Berisha F, Tirabosco R, et al. Next-generation sequencing is highly sensitive for the detection of beta-catenin mutations in desmoid-type fibromatoses. Virchows Arch (2015) 467:203–10. doi: 10.1007/s00428-015-1765-0
39. Hamada S, Futamura N, Ikuta K, Urakawa H, Kozawa E, Ishiguro N, et al. CTNNB1 S45F mutation predicts poor efficacy of meloxicam treatment for desmoid tumors: A pilot study. PloS One (2014) 9:e96391. doi: 10.1371/journal.pone.0096391
40. Colombo C, Miceli R, Lazar AJ, Perrone F, Pollock RE, Le Cesne A, et al. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence. Cancer (2013) 119:3696–702. doi: 10.1002/cncr.28271
41. Kim G, Kurnit KC, Djordjevic B, Singh C, Munsell MF, Wang W-L, et al. Nuclear β-catenin localization and mutation of the CTNNB1 gene: a context-dependent association. Mod Pathol (2018) 31:1553–9. doi: 10.1038/s41379-018-0080-0
42. Bo N, Wang D, Wu B, Chen L, Ma R. Analysis of β-catenin expression and exon 3 mutations in pediatric sporadic aggressive fibromatosis. Pediatr Dev Pathol (2012) 15:173–8. doi: 10.2350/10-07-0866-OA.1
43. Lazar AJF, Tuvin D, Hajibashi S, Habeeb S, Bolshakov S, Mayordomo-Aranda E, et al. Specific mutations in the β-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol (2008) 173:1518–27. doi: 10.2353/ajpath.2008.080475
44. Schut A-RW, Timbergen MJM, van Broekhoven DLM, van Dalen T, van Houdt WJ, Bonenkamp JJ, et al. A nationwide prospective clinical trial on active surveillance in patients with non-intra-abdominal desmoid-type fibromatosis: the GRAFITI trial. Ann Surg (2022) 277(4):689–696. doi: 10.1097/SLA.0000000000005415
45. Sakai T, Nishida Y, Ito K, Ikuta K, Urakawa H, Koike H, et al. Clinical results of active surveillance for extra-abdominal desmoid-type fibromatosis. Cancer Med 12(5):5245–5254. doi: 10.1002/cam4.5329
46. Lee JM, Kim HG, Shin SY, Lee SH. Clinical application of next-generation sequencing for the management of desmoid tumors: A case report and literature review. Med (Baltimore) (2021) 100:e24238. doi: 10.1097/MD.0000000000024238
47. Hamada S, Urakawa H, Kozawa E, Arai E, Ikuta K, Sakai T, et al. Characteristics of cultured desmoid cells with different CTNNB1 mutation status. Cancer Med (2016) 5:352–60. doi: 10.1002/cam4.582
48. Siozopoulou V, Marcq E, Jacobs J, Zwaenepoel K, Hermans C, Brauns J, et al. Desmoid tumors display a strong immune infiltration at the tumor margins and no PD-L1-driven immune suppression. Cancer Immunol Immunother (2019) 68:1573–83. doi: 10.1007/s00262-019-02390-0
49. Colombo C, Fiore M, Grignani G, Tolomeo F, Merlini A, Palassini E, et al. A prospective observational study of active surveillance in primary desmoid fibromatosis. Clin Cancer Res (2022) 28:4027–32. doi: 10.1158/1078-0432.CCR-21-4205
50. Colombo C, Urbini M, Astolfi A, Collini P, Indio V, Belfiore A, et al. Novel intra-genic large deletions of CTNNB1 gene identified in WT desmoid-type fibromatosis. Genes Chromosomes Cancer (2018) 57:495–503. doi: 10.1002/gcc.22644
51. Jung K, Fleischhacker M, Rabien A. Cell-free DNA in the blood as a solid tumor biomarker–a critical appraisal of the literature. Clin Chim Acta (2010) 411:1611–24. doi: 10.1016/j.cca.2010.07.032
52. Jones RP, Pugh SA, Graham J, Primrose JN, Barriuso J. Circulating tumour DNA as a biomarker in resectable and irresectable stage IV colorectal cancer; a systematic review and meta-analysis. Eur J Cancer (2021) 144:368–81. doi: 10.1016/j.ejca.2020.11.025
53. Ma X, Zhu L, Wu X, Bao H, Wang X, Chang Z, et al. Cell-free DNA provides a good representation of the tumor genome despite its biased fragmentation patterns. PloS One (2017) 12:e0169231. doi: 10.1371/journal.pone.0169231
54. Macagno N, Fina F, Penel N, Bouvier C, Nanni I, Duffaud F, et al. Proof of concept: prognostic value of the plasmatic concentration of circulating cell free DNA in desmoid tumors using ddPCR. Oncotarget (2018) 9:18296–308. doi: 10.18632/oncotarget.24817
55. Felekkis K, Touvana E, Stefanou C, Deltas C. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia (2010) 14:236–40. doi: 10.1111/j.1749-4486.2009.01952.x
56. Terrinoni A, Calabrese C, Basso D, Aita A, Caporali S, Plebani M, et al. The circulating miRNAs as diagnostic and prognostic markers. Clin Chem Lab Med (2019) 57:932–53. doi: 10.1515/cclm-2018-0838
57. Yamano T, Kubo S, Sonoda E, Kominato T, Kimura K, Yasuhara M, et al. Assessment of circulating microRNA specific for patients with familial adenomatous polyposis. PloS One (2021) 16:e0250072. doi: 10.1371/journal.pone.0250072
58. Kent OA, McCall MN, Cornish TC, Halushka MK. Lessons from miR-143/145: the importance of cell-type localization of miRNAs. Nucleic Acids Res (2014) 42:7528–38. doi: 10.1093/nar/gku461
59. Zhao L, Li Y, Song A. Inhibition of lncRNA TMPO-AS1 suppresses proliferation, migration and invasion of colorectal cancer cells by targeting miR-143-3p. Mol Med Rep (2020) 22:3245–54. doi: 10.3892/mmr.2020.11427
60. Liu M, Jia J, Wang X, Liu Y, Wang C, Fan R. Long non-coding RNA HOTAIR promotes cervical cancer progression through regulating BCL2 via targeting miR-143-3p. Cancer Biol Ther (2018) 19:391–9. doi: 10.1080/15384047.2018.1423921
61. Dômont J, Salas S, Lacroix L, Brouste V, Saulnier P, Terrier P, et al. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: a potential molecular tool for disease management. Br J Cancer (2010) 102:1032–6. doi: 10.1038/sj.bjc.6605557
62. Liu M, Yue Z, Zhang B, Li F, Liu L, Li F. mTORC1 mediates the processes of lysine regulating satellite cells proliferation, apoptosis, and autophagy. Metabolites (2022) 12:788. doi: 10.3390/metabo12090788
63. Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer (2012) 131:548–57. doi: 10.1002/ijc.26374
64. Braggio D, Koller D, Jin F, Siva N, Zewdu A, Lopez G, et al. Autophagy inhibition overcomes sorafenib resistance in S45F-mutated desmoid tumors. Cancer (2019) 125(15):2693–2703, cncr.32120. doi: 10.1002/cncr.32120
65. McCarty MF. Targeting multiple signaling pathways as a strategy for managing prostate cancer: multifocal signal modulation therapy. Integr Cancer Ther (2004) 3:349–80. doi: 10.1177/1534735404270757
66. Braggio D, Zewdu A, Londhe P, Yu P, Lopez G, Batte K, et al. β-catenin S45F mutation results in apoptotic resistance. Oncogene (2020) 39:5589–600. doi: 10.1038/s41388-020-1382-5
67. Miao C, Wang Z, Yang J, Li J, Gao X. Expression and mutation analysis of Cyclin A and Ki-67 in glioma and their correlation with tumor progression. Oncol Lett (2015) 10:1716–20. doi: 10.3892/ol.2015.3474
68. Huuhtanen RL, Blomqvist CP, Böhling TO, Wiklund TA, Tukiainen EJ, Virolainen M, et al. Expression of cyclin A in soft tissue sarcomas correlates with tumor aggressiveness. Cancer Res (1999) 59:2885–90.
69. Tökés A-M, Szász AM, Geszti F, Lukács LV, Kenessey I, Turányi E, et al. Expression of proliferation markers Ki67, cyclin A, geminin and aurora-kinase A in primary breast carcinomas and corresponding distant metastases. J Clin Pathol (2015) 68:274–82. doi: 10.1136/jclinpath-2014-202607
70. Santti K, Ihalainen H, Rönty M, Böhling T, Karlsson C, Haglund C, et al. High cyclin A expression, but not Ki67, is associated with early recurrence in desmoid tumors: SANTTI et al. J Surg Oncol (2018) 118:192–8. doi: 10.1002/jso.25121
71. Han SH, Park KM, Bae BN, Ryu SY, Kim KH, Kim HJ, et al. Interrelation of cyclin D1, cyclin E, and p27Kip1 expression on tissue arrays of breast cancer. Cancer Res Treat (2002) 34:388–93. doi: 10.4143/crt.2002.34.5.388
72. Irawan C, Cahyanur R, Lisnawati L, Abdullah M, Yunus RE. The difference in the Cyclin D1 expression in advanced stage nasopharyngeal cancer based on treatment response: A retrospective cohort study. Acta Med Indones (2020) 52:147–54. doi: 10.1111/j.1749-4486.2009.01952.x
73. Saito T, Oda Y, Tanaka K, Matsuda S, Tamiya S, Iwamoto Y, et al. beta-catenin nuclear expression correlates with cyclin D1 overexpression in sporadic desmoid tumours. J Pathol (2001) 195:222–8. doi: 10.1002/path.942
74. Matono H, Oda Y, Nakamori M, Tamiya S, Yamamoto H, Yokoyama R, et al. Correlation between beta-catenin widespread nuclear expression and matrix metalloproteinase-7 overexpression in sporadic desmoid tumors. Hum Pathol (2008) 39:1802–8. doi: 10.1016/j.humpath.2008.05.005
75. Jilong Y, Jian W, Xiaoyan Z, Xiaoqiu L, Xiongzeng Z. Analysis of APC/beta-catenin genes mutations and Wnt signalling pathway in desmoid-type fibromatosis. Pathology (2007) 39:319–25. doi: 10.1080/00313020701329823
76. Sharma H, Sen S, Sheriff AK, Bal S, Mathur M, Singh N. Characterization of apoptosis-related molecular changes in a desmoid tumor of the chest wall: report of a case. Surg Today (2003) 33:358–62. doi: 10.1007/s005950300081
77. Zhang Y, Pu W, Bousquenaud M, Cattin S, Zaric J, Sun L, et al. Emodin inhibits inflammation, carcinogenesis, and cancer progression in the AOM/DSS model of colitis-associated intestinal tumorigenesis. Front Oncol (2021) 10:564674. doi: 10.3389/fonc.2020.564674
78. Dempke W, Rie C, Grothey A, Schmoll HJ. Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer Res Clin Oncol (2001) 127:411–7. doi: 10.1007/s004320000225
79. Matono H, Tamiya S, Yokoyama R, Saito T, Iwamoto Y, Tsuneyoshi M, et al. Abnormalities of the Wnt/β-catenin signalling pathway induce tumour progression in sporadic desmoid tumours: correlation between β-catenin widespread nuclear expression and VEGF overexpression. Histopathology (2011) 59:368–75. doi: 10.1111/j.1365-2559.2011.03945.x
80. Wang T, Jin J, Qian C, Lou J, Lin J, Xu A, et al. Estrogen/ER in anti-tumor immunity regulation to tumor cell and tumor microenvironment. Cancer Cell Int (2021) 21:295. doi: 10.1186/s12935-021-02003-w
81. Pannier D, Cordoba A, Ryckewaert T, Robin Y-M, Penel N. Hormonal therapies in uterine sarcomas, aggressive angiomyxoma, and desmoid-type fibromatosis. Crit Rev Oncol Hematol (2019) 143:62–6. doi: 10.1016/j.critrevonc.2019.08.007
82. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol (2013) 75:225–40. doi: 10.1146/annurev-physiol-030212-183708
83. Bartels S, Christgen M, Luft A, Persing S, Jödecke K, Lehmann U, et al. Estrogen receptor (ESR1) mutation in bone metastases from breast cancer. Mod Pathol (2018) 31:56–61. doi: 10.1038/modpathol.2017.95
84. McLean TD, Duchi S, Di Bella C. Molecular pathogenesis of sporadic desmoid tumours and its implications for novel therapies: A systematised narrative review. Targ Oncol (2022) 17:223–52. doi: 10.1007/s11523-022-00876-z
85. Santos GAC, Cunha IW, Rocha RM, Mello CAL, Guimarães GC, Fregnani JH, et al. Evaluation of estrogen receptor alpha, estrogen receptor beta, progesterone receptor, and cKIT expression in desmoids tumors and their role in determining treatment options. Biosci Trends (2010) 4:25–30. doi: 10.1016/s0065-1281(04)00036-4
86. Zhang H-Y, Ke Q, Zhang Z, Zhang R, Fu J, Chen H-J, et al. Expression of beta-catenin and estrogen receptor in desmoid-type fibromatosis. Sichuan Da Xue Xue Bao Yi Xue Ban (2010) 41:101–5.
87. Maseelall P, Robins JC, Williams DB, Thomas MA. Stabilization and regression of a recurrent desmoid tumor with the antiestrogen toremifene. Fertil Steril (2005) 84:509. doi: 10.1016/j.fertnstert.2005.03.030
88. Ohashi T, Shigematsu N, Kameyama K, Kubo A. Tamoxifen for recurrent desmoid tumor of the chest wall. Int J Clin Oncol (2006) 11:150–2. doi: 10.1007/s10147-005-0543-4
89. Mir O, Honoré C, Chamseddine AN, Dômont J, Dumont SN, Cavalcanti A, et al. Long-term outcomes of oral vinorelbine in advanced, progressive desmoid fibromatosis and influence of CTNNB1 mutational status. Clin Cancer Res (2020) 26:6277–83. doi: 10.1158/1078-0432.CCR-20-1847
90. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer (2010) 10:293–301. doi: 10.1038/nrc2812
91. Davar D, Beumer JH, Hamieh L, Tawbi H. Role of PARP inhibitors in cancer biology and therapy. Curr Med Chem (2012) 19:3907–21. doi: 10.4161/cbt.26160
92. Lee H-J, Yoon C, Schmidt B, Park DJ, Zhang AY, Erkizan HV, et al. Combining PARP-1 inhibition and radiation in Ewing sarcoma results in lethal DNA damage. Mol Cancer Ther (2013) 12:2591–600. doi: 10.1158/1535-7163.MCT-13-0338
93. Lee S-G, Kim N, Kim S-M, Park IB, Kim H, Kim S, et al. Ewing sarcoma protein promotes dissociation of poly(ADP-ribose) polymerase 1 from chromatin. EMBO Rep (2020) 21:e48676. doi: 10.15252/embr.201948676
94. Bräutigam K, Lindner J, Budczies J, Pahl S, Kunitz A, Melcher I, et al. PARP-1 expression as a prognostic factor in Desmoid-type fibromatosis. Ann Diagn Pathol (2020) 44:151442. doi: 10.1016/j.anndiagpath.2019.151442
95. Banerjee J, Lodhi N, Nguyen B-N. The role of Poly(ADP-Ribose) Polymerase-1 in cutaneous wound healing. Adv Wound Care (New Rochelle) (2019) 8:634–43. doi: 10.1089/wound.2018.0821
96. Nagashima H, Lee CK, Tateishi K, Higuchi F, Subramanian M, Rafferty S, et al. Poly(ADP-ribose) glycohydrolase inhibition sequesters NAD+ to potentiate the metabolic lethality of alkylating chemotherapy in IDH mutant tumor cells. Cancer Discovery (2020) 10:1672–89. doi: 10.1158/2159-8290.CD-20-0226
97. Mercier KA, Al-Jazrawe M, Poon R, Acuff Z, Alman B. A metabolomics pilot study on desmoid tumors and novel drug candidates. Sci Rep (2018) 8:584. doi: 10.1038/s41598-017-18921-7
98. Kohsaka S, Hirata M, Ikegami M, Ueno T, Kojima S, Sakai T, et al. Comprehensive molecular and clinicopathological profiling of desmoid tumours. Eur J Cancer (2021) 145:109–20. doi: 10.1016/j.ejca.2020.12.001
99. Salas S, Brulard C, Terrier P, Ranchere-Vince D, Neuville A, Guillou L, et al. Gene expression profiling of desmoid tumors by cDNA microarrays and correlation with progression-free survival. Clin Cancer Res (2015) 21:4194–200. doi: 10.1158/1078-0432.CCR-14-2910
100. Cavallini A, Rotelli MT, Lippolis C, Piscitelli D, Digennaro R, Covelli C, et al. Human microRNA expression in sporadic and FAP-associated desmoid tumors and correlation with beta-catenin mutations. Oncotarget (2017) 8:41866–75. doi: 10.18632/oncotarget.16383
101. Wang X, Jaimes M, Gu H, Shults K, Putta S, Sharma V, et al. Cell by cell immuno- and cancer marker profiling of non-small cell lung cancer tissue: Checkpoint marker expression on CD103+, CD4+ T-cells predicts circulating tumor cells. Transl Oncol (2020) 14:100953. doi: 10.1016/j.tranon.2020.100953
102. Paterlini-Brechot P, Benali NL. Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer Lett (2007) 253:180–204. doi: 10.1016/j.canlet.2006.12.014
103. Ac B, Fab C, Ea A, Apc R, Tds M, Ms T, et al. Circulating tumor cells in desmoid tumors: new perspectives. Front Oncol (2021) 11:622626. doi: 10.3389/fonc.2021.622626
104. Kim M-Y, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH-F, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell (2009) 139:1315–26. doi: 10.1016/j.cell.2009.11.025
105. Leung CT, Brugge JS. Tumor self-seeding: bidirectional flow of tumor cells. Cell (2009) 139:1226–8. doi: 10.1016/j.cell.2009.12.013
106. SY C, Lim B, Kyung YS, Kim Y, Kim BM, Jeon BH, et al. Circulating tumor cell counts in patients with localized prostate cancer including those under active surveillance. Vivo (2019) 33:1615–20. doi: 10.21873/invivo.11645
107. Yoneda K, Kuwata T, Chikaishi Y, Mori M, Kanayama M, Takenaka M, et al. Detection of circulating tumor cells with a novel microfluidic system in malignant pleural mesothelioma. Cancer Sci (2019) 110:726–33. doi: 10.1111/cas.13895
108. Kasper B, Gruenwald V, Reichardt P, Bauer S, Hohenberger P, Haller F. Correlation of CTNNB1 mutation status with progression arrest rate in RECIST progressive desmoid-type fibromatosis treated with imatinib: translational research results from a phase 2 study of the german interdisciplinary Sarcoma Group (GISG-01). Ann Surg Oncol (2016) 23:1924–7. doi: 10.1245/s10434-016-5132-4
109. Mullen JT, DeLaney TF, Rosenberg AE, Le L, Iafrate AJ, Kobayashi W, et al. β-catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist (2013) 18:1043–9. doi: 10.1634/theoncologist.2012-0449
110. Signoroni S, Frattini M, Negri T, Pastore E, Tamborini E, Casieri P, et al. Cyclooxygenase-2 and platelet-derived growth factor receptors as potential targets in treating aggressive fibromatosis. Clin Cancer Res (2007) 13:5034–40. doi: 10.1158/1078-0432.CCR-07-0336
Keywords: Desmoid tumor, predictive markers, progression, recurrence, active surveillance
Citation: Liu B, Sun Z, Zhou R, Shen D, Zhu S, Chen L and Huang G (2023) Novel insights into biomarkers of progression in Desmoid tumor. Front. Oncol. 13:1206800. doi: 10.3389/fonc.2023.1206800
Received: 16 April 2023; Accepted: 19 July 2023;
Published: 03 August 2023.
Edited by:
Manpreet (Meena) Bedi, Medical College of Wisconsin, United StatesReviewed by:
Milena Urbini, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyFrancesco Fiore, G. Pascale National Cancer Institute Foundation (IRCCS), Italy
Copyright © 2023 Liu, Sun, Zhou, Shen, Zhu, Chen and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gengwen Huang, aHVhbmdnZW5nd2VuQGNzdS5lZHUuY24=
†These authors have contributed equally to this work