 Selene Cipri
Selene Cipri Giada Del Baldo
Giada Del Baldo Francesco Fabozzi
Francesco Fabozzi Luigi Boccuto
Luigi Boccuto Andrea Carai
Andrea Carai Angela Mastronuzzi
Angela Mastronuzzi- 1Department of Hematology/Oncology, Cell Therapy, Gene Therapies and Hemopoietic Transplant, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 2Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 3Healthcare Genetics Program, School of Nursing, College of Behavioral, Social and Health Sciences, Clemson University, Clemson, SC, United States
- 4Department of Neurosciences, Neurosurgery Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
In the past decade significant advancements have been made in the discovery of targetable lesions in pediatric low-grade gliomas (pLGGs). These tumors account for 30-50% of all pediatric brain tumors with generally a favorable prognosis. The latest 2021 WHO classification of pLGGs places a strong emphasis on molecular characterization for significant implications on prognosis, diagnosis, management, and the potential target treatment. With the technological advances and new applications in molecular diagnostics, the molecular characterization of pLGGs has revealed that tumors that appear similar under a microscope can have different genetic and molecular characteristics. Therefore, the new classification system divides pLGGs into several distinct subtypes based on these characteristics, enabling a more accurate strategy for diagnosis and personalized therapy based on the specific genetic and molecular abnormalities present in each tumor. This approach holds great promise for improving outcomes for patients with pLGGs, highlighting the importance of the recent breakthroughs in the discovery of targetable lesions.
1 Introduction
Pediatric low-grade gliomas (pLGGs) are one of the more frequent pediatric brain tumors accounting for about 30-50% of central nervous system (CNS) tumors of pediatric patients. They carry a favorable prognosis with an overall survival (OS) at 10 years greater than 90%. In a minority of cases an aggressive behaviour is described (1, 2).
To date, complete resection is the most favourable outcome measurement of the patients, but it is not easy to conduct for deep or infiltrative lesions (3), and for progressive residual disease adjuvant chemotherapy or radiation were historically performed (4–12). However, we did not forget that the side effects are far from negligible (5, 13–16). Pediatric LGGs comprise of a heterogeneous group of tumors, and recently molecular studies led to a better clarification and classification of pLGGs, and which paved the way for promising new therapeutic strategies.
Many types of tumors are included under the umbrella of pLGGs. Historically, these types of neoplasms have been classified on the basis of histology, but today we know that the same histologies can underlie different entities and histological classification alone is no longer useful (17). The molecular characterization advancements have revealed that appear similar under a microscope can have different genetic and molecular characteristics, so the new classification system divides pLGGs into several distinct subtypes based on these characteristics, rather than solely on their histological appearance. Better knowledge of the molecular characteristics, technological advances, and new applications in molecular diagnostics of pLGGs have helped overcome these challenges (18).
The updated 2021 World Health Organization (WHO) Classification of Tumors of the CNS has reflected the focus on the integration of histopathological and molecular characteristics to facilitate a more accurate diagnosis (19). In the new classification of pLGGs places a strong emphasis on the molecular characterization of these tumors for significant implications on the prognosis, diagnosis, management, and finally development of personalized treatment (19).
This classification describes three families of tumors that encompass pLGGs and glioneuronal tumors (GNTs) (Table 1), which are now defined by their driver molecular alterations rather than by histopathological features alone: “Glioneuronal and neuronal tumor”, “Circumscribed astrocytic gliomas” and “Pediatric type diffuse low-grade gliomas” (17, 19).
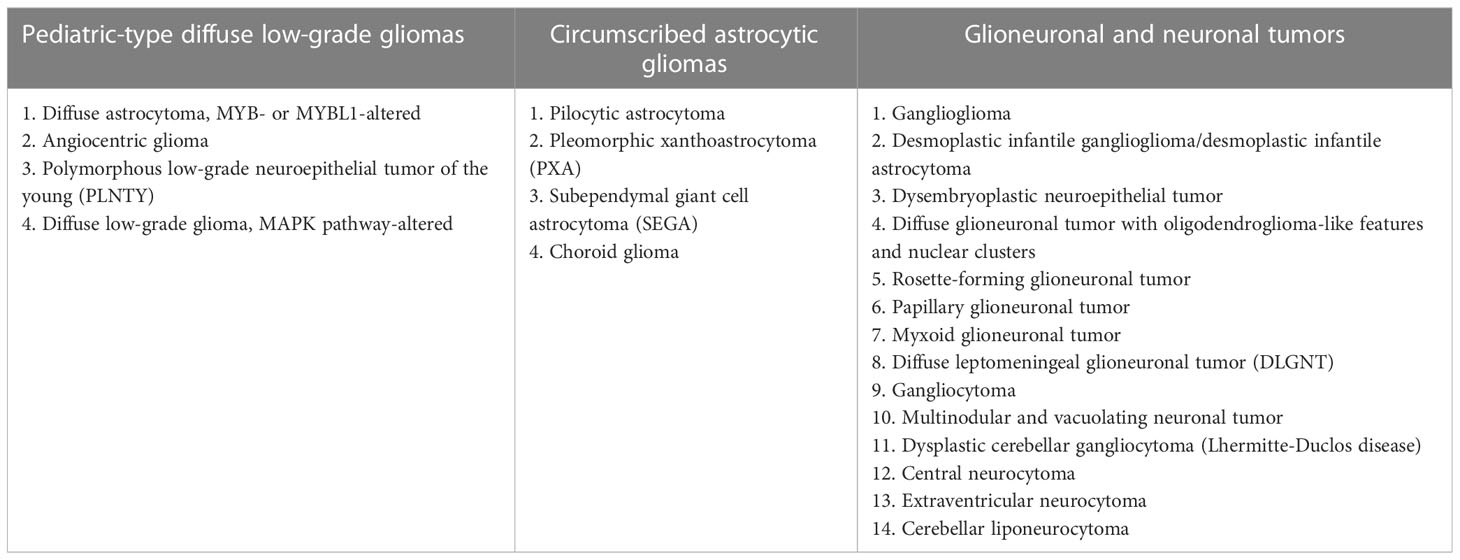
Table 1 WHO 2021 classification for pLGG/low-grade GNTs (19).
In this review, we described the major molecular alterations detected in pLGGs and the molecular target therapy available to date.
2 MAPK/ERK and PI3K/AKT/mTOR signaling pathway alterations in pediatric low-grade gliomas
2.1 MAPK/ERK and PI3K/AKT/mTOR signaling pathway in physiological conditions
In the Mitogen-Activated Protein Kinase/extracellular signal-regulated kinases (MAPK/ERK) signaling pathway (Figure 1), stimulation of receptor tyrosine kinases (RTKs) in physiological conditions causes MAPK activation. The activation of Ras enabled the activity of the serine/threonine-protein kinase B-raf, which homodimerizes or heterodimerizes by phosphorylating and triggering mitogen-activated protein kinase kinase (MEK1 and MEK2), which in turn phosphorylates and trigger ERK 1 and ERK2. Finally, the latter boost dedifferentiation, proliferation and cell survival by scalable transcriptional asset within the nucleus; consequently, downstream activation of ERK causes feedback inhibition of the upstream pathway (20–23).
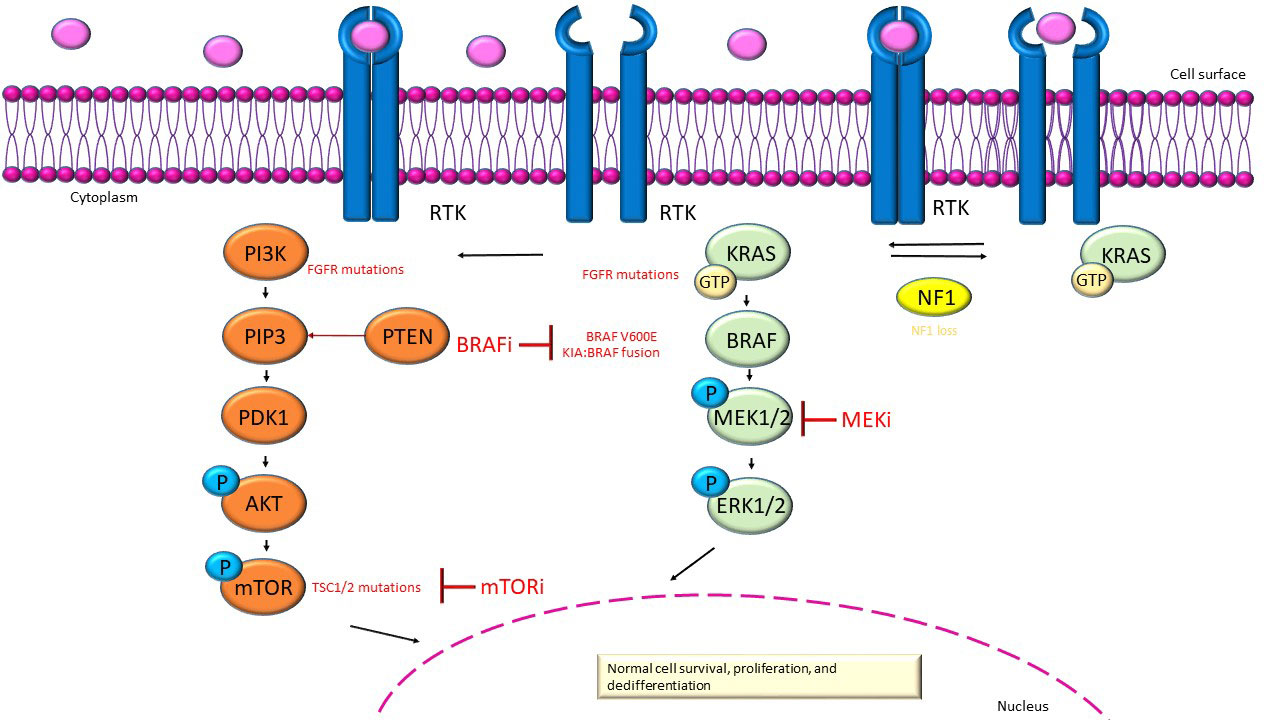
Figure 1 MAPK/ERK (green) and PI3K/AKT/mTOR (orange) signaling pathway. In red show the inhibitors and the alterations in gene that are causative to dysregulation of these pathways in pLGGs.
The activation of the phosphatidylinositol 3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/AKT/mTOR) pathway (Figure 1) is mediated by transmembrane receptor tyrosine kinases of growth factors (24). The Phosphatidylinositol 3-kinase (PI3K) is triggered from the bond of oncogenes or growth factors (24). PI3K transfom phosphatidylinositol-4,5-phosphate (PIP2) to phosphatidylinositol-3,4,5-phosphate (PIP3) (24). The lipid Phosphatase and tensin homolog (PTEN) has the function of countering the build-up of PIP3 and enroll to the membrane protein kinase B (PKB or Akt) and phosphoinositide-dependent kinase 1 (PDK1), which are phosphorylated and triggered (24). The molecular complexes mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2) have both a catalytic subunit mTOR which is negatively regulates by the heterodimer of tuberous sclerosis proteins TSC1 (hamartin) and TSC2 (tuberin) [a GTPase-activating complex (GAP) to Rheb (homolog of Ras enriched in the brain)], in contrast the activation of the PI3K pathway, AKT phosphoryl TSC2 and disable the TSC1/TSC2 complex (25–27). Mechanistic target of rapamycin (mTOR) can even be triggered by the MAPK pathway via RAS/MEK/ERK (28).The phosphorylation of TSC2 by ERK and ribosomal S6 kinase (RSK) can induce mTORC1 activation; instead, RSK can target the mTORC1 complex by directly promoting the kinase activity of the complex (28). Aberrant activation of mTOR may be related to various mutations that activate the mTOR pathway, such as alterations at mTOR negative regulators or mTOR pathway components (28). PI3K activation facilitates the activation of mTORC1 and mTORC2. Activation of mTORC1 downstream of PI3K and protein- kinase B (AKT) promotes cell survival, growth and proliferation. Moreover, mTORC2 increases cell proliferation and survival through regulation of protein kinases, including AKT, which provides significant motivation for further studies on therapeutic targeting of mTOR complexes in cancer, as mTOR plays an important role in tumor progression (29).
2.2 BRAF alterations and targeted therapy
Within pLGG a notorious troublemaker has been identified: the B-Raf proto-oncogene, serine/threonine kinase (BRAF) gene. This gene encodes a protein from the RAF family that is responsible for regulating the MAPK/ERK pathway (30). Pediatric LGGs often harbor alterations in the BRAF gene, such as the p.V600E point mutation and the translocation between BRAF and KIAA1549. These alterations result in a hyperactive protein that wreaks havoc on the MAPK pathway, leading to uncontrolled cell division and tumorigenesis (31–41).
Most of sporadic pLGGs are characterize by BRAF mutations (2, 42). A three-class system was defined based on the result of BRAF mutations on the activity of the encoded protein. RAS-independent as monomers represent the class I mutations RAS-independent as dimers belong to class II mutations, and RAS-dependent with altered kinase activity are class III mutations (24).
Class I mutations, which include the mutation on 600 codon of BRAF, hyperactivate kinases through promotion of MEK/ERK activation regardless of the protein dimerization (for example with Raf has low effect) and activation of RAS (24). In fact, inhibition of upstream ERK feedback has any impact on class I mutations because, although BRAF p.V600E dimerization stays Ras dependent and is blocked by upstream ERK response, but it can yet turn on the pathway like monomer (43, 44). A point mutation c.1799T>A causes the replacement of valine with glutamic acid at codon 600 (p.V600E) within the gene’s activation region. The occurrence of BRAF p.V600E in non-pilocytic pLGGs varies significantly depending on the tumor’s histology and location. Ganglioglioma (25-45%) and pleomorphic xanthoastrocytomas (40-80%) frequently exhibit the variant, while it is less commonly observed in pilocytic astrocytoma (PA) (5-10%) and GNTs (5%) (45–52). Combining histological and molecular data helps to achieve a more precise diagnosis. For example, identifying BRAF p.V600E, along with the detection of a mildly and minimally atypical glial proliferation without eosinophilic granular bodies and Rosenthal fibers (RFs), enables categorizing the tumor as a “low-grade diffuse glioma” (19). In a retrospective study, 17% of children with LGGs carried the BRAF p.V600E variant and presented a 10-year progression-free survival (PFS) rate of about 27% versus a 60% rate for those without the same variant (53). This trend is confirmed by several studies (53–57). However, almost one-third of patients who experienced complete resection relapsed, indicating that BRAF p.V600E is the most interfering phenotype than other mutation known in patients with pLGGs (53). A progression-free survival of 5-year are reported in a study on children with low diencephalic astrocytomas carried BRAF p.V600E (22%) versus the children without BRAF mutations (52%) (58). The BRAF p.V600E variants were more frequently detected in pLGGs that transform into high-grade gliomas (59). Several studies have demonstrated that 25% of patients with pLGGs exhibit BRAF p.V600E in conjunction with deletions of CDKN2A, which probably operates as a second hit, altering the regulation of cell cycle (38, 45, 53, 60–62). Tumors with BRAF p.V600E and a CDKN2A deletion represent a separate subtype of pLGGs which are inclined to change into HGG (59). Reports show that both these mutations are related with oncogene-induced senescence escape and poorer OS and PFS (38, 45, 62). Therefore, pLGGs with CDKN2A deletions, particularly those with p.V600E or possible high-grade histological characteristics, should be considered high-risk tumors requiring close clinical follow-up (63). Finally, some studies have reported rare cases of BRAF missense variants at the p.V600 residue, in which valine is replaced with other amino acids such as lysine (p.V600K), aspartic acid (p.V600D), or arginine (p.V600R). Desmoplastic infant astrocytomas/gliomas exhibit the p.V600K variant, while the BRAF p.V504_R506dup variant was reported in cases with PA. Supratentorial lesions are more frequently associated with BRAF p.V600E, while cerebellar lesions more commonly present KIAA1549-BRAF (47, 51, 64).
Class II mutations involve BRAF-KIAA1549 fusion and other gene fusions. They trigger both intermediate and high kinase activity, requiring dimerization of the protein to activate the MEK/ERK pathway (24). The KIAA1549-BRAF fusion is a great slice of gene fusions involving BRAF in pLGGs, accounting for a whopping 30-40% of cases (65). The KIAA1549 gene belongs to the mysterious UPF0606 family and we are still trying to understand what it does (66). KIAA1549-BRAF fusion is a major player in a variety of CNS tumors. It is particularly prevalent in infratentorial and midline PAs, although it shows up less often in supratentorial tumors (34, 38, 67–72). Interestingly, early studies have proven that fusions that involved these genes are correlated to tandem duplication that creates a brand-new oncogenic fusion. This rearrangement messes with domain at the N-terminal regulatory region of the BRAF protein, which in turn causes RAS/MAPK pathway altered regulation (35, 36, 73). But despite these complicated genetics, one thing is clear: the presence of the KIAA1549-BRAF fusion is associated with better OS and PFS in pLGGs that cannot be fully removed and do not tend to progress too quickly (53, 69, 70, 72). Unfortunately, in cases where the tumor is located in a difficult-to-reach part of the brain, progression is more likely (53).
Other alterations in addition to the BRAF-KIAA1549 fusion, such as CDKN2A deletions, and tumor location may alter the outcome of the patient (45, 74).
Other rearrangements that activate the RAS/MAPK pathway and involving BRAF are the MKRN1 (Makorin Ring Finger Protein 1), SRGAP (SLIT-ROBO Rho GTPase Activating Protein 2), GIT2 (GIT ArfGAP 2), FAM131B (Family with Sequence Similarity 131 Member B), RNF130 (Ring Finger Protein 130), CLCN6 (Chloride Voltage-Gated Channel 6), GNAI1 (G Protein Subunit Alpha I1), and FXR1 (FMR1 Autosomal Homolog 1) mergers involving deletion of BRAF N-regulatory domain (34, 74–76). These non-canonical fusions in particular manifest in older children and adolescents, frequently in brainstem lesions and hemispheres, and are also observed in a series of rare histological profiles (67, 75–77).
Class III mutations are found to be linked to poor or no kinase activity and need both the activation of upstream RAS and dimerization with CRAF to further induce induction of MER/ERK pathway activation (24). In literature are reported a few cases of BRAF p.D594G and p.G466V mutations (78).
Finally, BRAF p.V600E mutations and BRAF fusions enable molecular characterization of nearly 2/3 of pLGGs (2).
BRAF inhibitors (BRAFi), including vemurafenib, dabrafenib, and encorafenib, are drugs that selectively bind to mutated B-Raf proteins and block the activation of MEK by inhibiting the MAPK/ERK cascade signaling (79). Clinical studies have demonstrated that vemurafenib and dabrafenib first-generation BRAFi are highlyeffective in treating children with LGGs, with numerous case reports showing complete responses (52, 80–89). However, these inhibitors have been found to activate the signaling pathway of RAS/MAPK when used in tumors with the fusions that involved KIAA1549 and BRAF or BRAF wild-type (wt) (90, 91). To address this issue, “paradox-breaker” secondo generation agents have been developed that do not activate the RAS/MAPK pathway (92). Ongoing clinical trials are investigating the use of the dual combination of BRAFi and MEK inhibitors (MEKi) to treat BRAF p.V600 mutation-positive gliomas (Table 2) (93–98). There are also emerging new class II BRAF inhibitors, such as TAK-580, that look promising in treating LGGs (116). Overall, BRAF inhibitors offer a remarkable therapeutic option for pLGGs, particularly in pediatric patients where traditional treatment methods may have long-term effects on brain development.
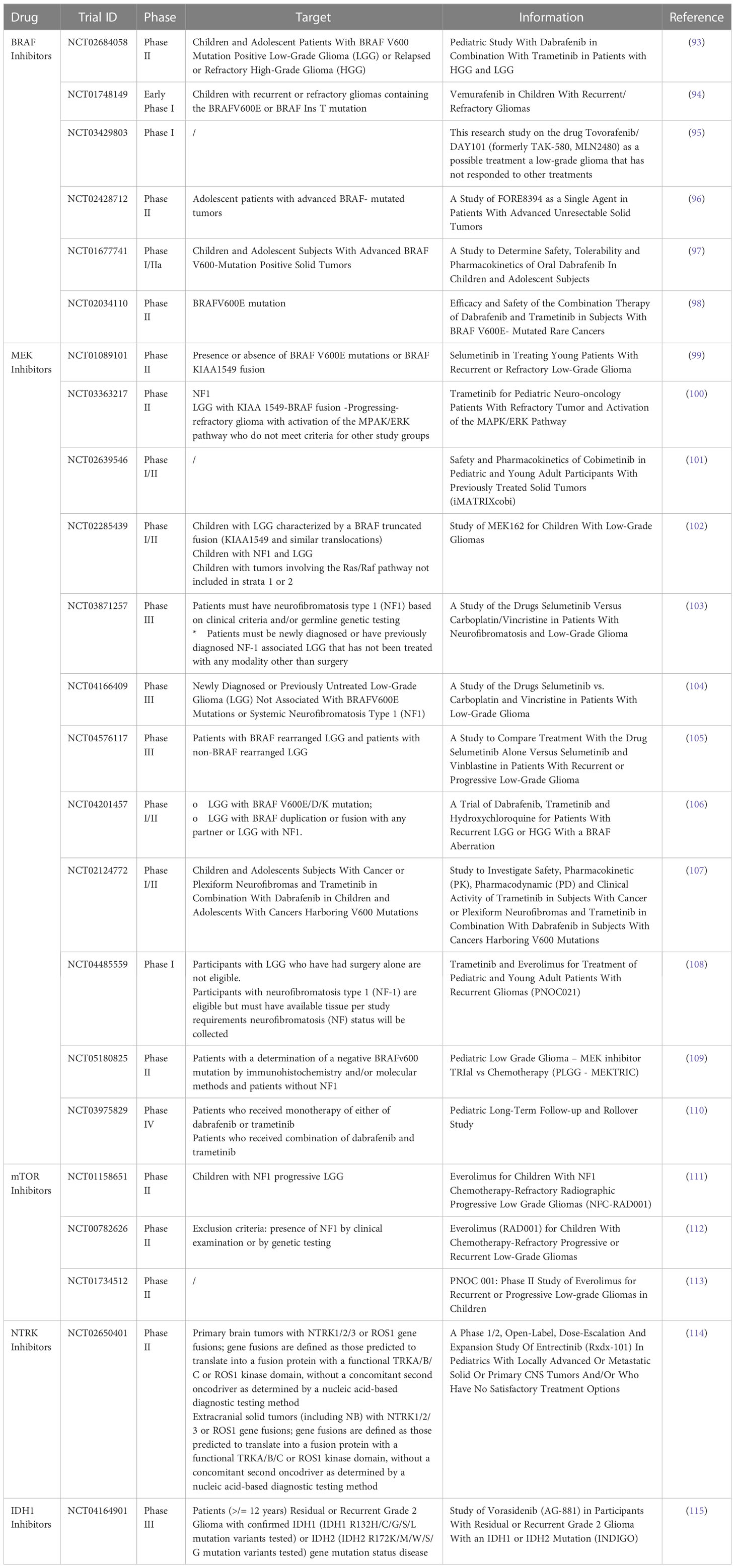
Table 2 List of clinical trials for pLGG using targeted therapy.
In addition, MEKi have emerged as a potential treatment strategy for pLGG patients and ongoing clinical trials are examining the use of several drugs such as selumetinib in treating of young patients with recurrent or refractory LGGs (characterized by the presence or absence of BRAF V600E mutations or BRAF-KIAA1549 fusion); trametinib for pediatric neuro-oncology patients with refractory tumor and activation of the MAPK/ERK pathway causative by a KIAA 1549-BRAF fusion; and a study of MEK162 for children with LGGs characterized by a BRAF truncated fusion (KIAA1549 and similar translocations) (Table 2) (99–102, 105, 108–110).
2.3 FGFR1 alterations
The subunits of the RTKs, which are crucial in transmitting the MAPK signal, are encoded by genes pertaining to the Fibroblast Growth Factor Receptor (FGFR) family (FGFR1-4) (117). Fibroblast Growth Factor Receptor 1 (FGFR1) alterations are common in pLGGs (40, 64, 76), with p.N546K and p.K656E being the most frequent mutations observed in 5-10% of patients, while FGFR1 TKD duplication is detected in 2-23% of tumors. FGFR1 mutations have been identified in various pLGGs, including PA with an unfavorable prognosis, although none of these changes is histologically specific (64, 76, 118, 119). Fusion genes involving FGFR’s N-terminal domain and other genes such as TACC1 (Transforming Acidic Coiled-Coil Containing Protein 1), KIAA1598 (Shootin 1), TACC2 (Transforming Acidic Coiled-Coil Containing Protein 2), TACC3 (Transforming Acidic Coiled-Coil Containing Protein 3) and KIAA1598 (Shootin 1) characterize pLGGs (120).
All these changes lead to FGFR1 self-phosphorylation, are correlated to the up alteration of the RAS/MAPK pathway and PI3K/AKT/mTOR pathway (76). FGFR1 alterations’ clinical manifestations are not yet fully understood and can be the product of more alterations in the genes that are mentioned earlier (64, 76, 119).
2.4 Other alterations in pLGGs and targeted therapy
The neurotrophic receptor of tyrosine kinase (NTRK) family and the ALK gene have significant roles in the development and fuctions of the CNS (81–88). In pLGGs, NTRK gene fusions including NTRK1/2/3, SLMAP-NTRK2, TPM3-NTRK1, RBPMS-NTRK3 and ETV6-NTRK3 are rare (64, 76, 121, 122). ALK alterations are also uncommon in pLGGs, but the fusions that involved CCDC88A and PPP1CB with AKT being the more prevalent and resulting from chromothripsis (123–125). These changes cause tumorigenesis by modifying the RAS/MAPK and PI3K/AKT/mTOR pathways via abnormal NTRK kinase domain dimerization or ectopic expression of the product fusion involved (125–129). Alterations in NTRK gene are rare in pLGGs, while they are common in adult cancers and this has enabled the development and testing of drugs already approved by the FDA. Entrectinib was approved for treatment of solid tumors when patients carrier a NTRK gene fusion and larotrectinib for both population of patients with solid tumors who carrier a fusion that involved TRK without a mutation known as related to acquired resistance, who are metastatic or in whom surgical excision may cause significant morbidity and who have no suitable treatment options or progressed after therapy (130–134).
In pediatric gliomas, in particular, both entrectinib and larotrectinib showed potent antitumor effects (135–137). These findings resulted to a phase I/II study presently ongoing in children to assess entrectinib in primary tumors of CNS (114).
To date there is a lack of data on the role of larotrectinib in primary CNS tumors, as few case reports have been published in particular on pediatric high-grade gliomas (pHGGs) and clinical trials have not yet been completed (138–143).
Finally, another rarely reported alteration in pLGGs, involves IDH1 whose role in these types of pediatric cancers is unclear to date (64, 144). A study of patients with LGGs and mutation in IDH1 found excellent short-term survival, but with a 5-year PFS of less than 43% and mortality after 10 years (145). To date, Vorasidenib (Ag-881), a new inhibitor against IDH1 and IDH2 mutation with high brain penetration, show a good results in clinical trial on adult patients with LGG (above/equal to 18 years of age) and IDH1 mutations (146–150). Consequently, Ag-881 was tested in a phase III clinical trial (INDIGO) in patients up/equal to 12 years of age and with residual or recurrent grade 2 Glioma who carried an IDH1 R132H/C/G/S/L or IDH2 mutation (115).
Another IDH1 inhibitor is FT-2102, used specifically in the treatment of myelodysplastic syndromes and AML, was tested in the adult population with solid tumors and gliomas in which mutation in IDH1 was found (151, 152). Other studies are on going in the adult population (153).
2.5 Cancer predisposition syndrome associated with pLGG: from alterations involving the RAS/MAPK and mTOR signaling pathway to targeted therapy
Alterations involving the RAS/MAPK pathway in pLGG pathogenesis have been studied in patients with Neurofibromatosis type 1 (NF1), of which 10-15% develop low-grade gliomas (154–156).
About 20% of patients with NF1 develop pLGGs (157): they often present with optically induced tumors that are not biopsied, NF1-pLGGs are asymptomatic and indolent, do not require any treatment, and in some cases regress without treatment; however, in case of clinical deterioration (more frequently vision loss), the first line of therapy used is chemotherapy (158–161). In addition, some studies have repositioned NF1-pLGGs patients with other additional genetic alterations of the RAS/MAPK pathway (162). Seventy-five percent of NF1-pLGGs carried a genetic mutation in one or more genes that are involved in biological process (162). Finally, in pLGGs involving NF1-associated alterations, BRAF variants are rare (68, 162).
Target therapies have also been attempted and described in patients with NF1. MEKi have emerged as a potential treatment strategy for pLGG patients who are unresponsive to BRAFi, such as those with KIAA1549-BRAF or NF1-pLGG (163). Ongoing clinical trials are exploring efficacy of treatment with selumetinib, trametinib, cobimetinib, and binimetinib in young patients with refractory pLGGs (Table 2) (99–102). Phase I/II trials on selumetinib have demonstrated its stability or reduction of tumor size in pediatric patients with NF1-associated and sporadic form of pLGGs, with similar results observed in a study of children with progressive/recurrent PA (Table 2) (164, 165). Phase III clinical trial are currently exploring the efficacy of selumetinib as a frontline therapy for both NF1-associated and NF1-non-associated pLGGs (Table 2) (103, 104).Trametinib and binimetinib have also shown promise in small studies, with trametinib appearing effective as a single drug or in compound with dabrafenib (107, 166–171). Broader studies are required to assess the tolerability of MEKi in pLGG patients (159). Overall, MEKi showed a promising therapeutic alternative for pLGGs, particularly for children with NF1-associated tumors without BRAF gene alterations (172).
of the vaste majority of children with tuberous sclerosis have a germline pathogenetic variant in tuberous sclerosis genes (TSC1 or TSC2), that increase the risk of developing subependymal giant cell astrocytomas,subependymal nodules and cortical tubers, as some pathogenetic variants in these genes lead to mTOR pathway activation (173). Subependymal giant cell astrocytomas are led by mTOR activation; mTORi are active drugs that may induce the regression of the tumor in children affected by these tumors (173). Finally, germline mutations in genes (more than 10) involved in the RAS-MAPK pathway are causative of Noonan syndrome (NS), an autosomal dominant congenital condition (174). Noonan Syndrome is correlated to develop a brain tumors (174). Our group described 13-year-old patient with NS who developed a cerebellar PA, an optic pathway glioma (OPG) and a left temporal lobe glioneuronal neoplasm. A pathogenetic variant in the PTPN11 gene was found and the molecular characterization of the GNT revealed elevated levels of phosphorylated mTOR (pMTOR) (175). Tyrosine phosphatase adaptor protein is encoded by PTPN11 gene and it is involved, as reported before, in the RAS/MAPK pathway (78). Additionally, PTPN11 overexpression alone does not significantly activate the RAS/MAPK pathway, and further alterations like mutations in the FGFR1 gene, which activate the PI3K/AKT/mTOR pathway, are required (154).
mTOR was found to be excessively activated in pLGGs associated with syndromic conditions like TS and NF1 and this prompted to the support for the use of mTORi such as everolimus in clinical treatment alternative strategy (161, 176–181). Studies have highlighted that inhibiting the mTOR pathway is a promising therapeutic strategy for pLGG, and experimental evidence is emerging that suggests mTOR pathway activation may be a feature of most pLGGs (111, 173, 182–185). Everolimus has been successful in treating subependymal giant cell astrocytoma, a subtype of pLGG, and has demonstrated seizure control and tumor volume reduction in TS patients with SEGAs (173, 183, 186–188).
Our group suggested using everolimus for patients with RASopathies and brain tumors that have overactive mTOR signaling, and a phase II study is ongoing for recurrent or progressive pLGGs children. Everolimus has also been shown to provide a significant therapeutic alternative to immediate surgery in TSC patients, allowing for the postponement of a neurosurgical resection (Table 2) (112, 113, 175, 189). Moreover, we reported the first use of everolimus in children with pLGGs who were chemo- and radiotherapy-naïve (190). The results showed a lack of progression with a manageable toxicity profile, providing preliminary support for everolimus as a therapy for pLGG (190).
Overall, more studies are needed to develop innovative therapies for pLGG patients based on oncological mechanisms related to tumor development.
3 Discussion
Pediatric LGGs represent 30-50% of CNS childhood tumors; their prognosis greatly differed between tumor clusters and is dictated by a variety of factors, which include age at diagnosis, localization, and extension of resection surgery. pLGG represent a chronic disease and despite the treatments available to date, associated long-term morbidity remain of paramount importance.
Surgery is currently the standard of care, and children who undergo gross tumor resection (GTR) often do not demand additional action other than regularly follow-up. In a cohort of 518 patients, the 5-year PFS rate for children who have undergone GTR was high (94%) with an OS rate of 99%; any degree of residual tumor predicted a worse PFS, with up to 44% of patients with limited residual disease progressing within 5 years (3). However, a cluster of patients who are not susceptible to GTR subset exists, primarily because of tumor location. Although the low-grade biological malignancy of these tumors, products patients with both unresectable and clinically progressive disease masses receive either chemotherapy or radiotherapy, experiencing the toxicities associated with these regimens in the short and long term. The principal advances in treatment of traditional chemotherapy for LGG include carboplatin and vincristine, TPCV (thioguanine, procarbazine, lomustine, and vincristine), and weekly vinblastine monotherapy (191, 192). These conventional chemotherapeutic approaches used in pLGG patients are associated with side effects such as myelosuppression, alopecia, and less frequently ototoxicity (carboplatin) and decreased fertility potential (procarbazine) (193, 194). In addition, bevacizumab is another promising approach as it has shown improvements in the treatment of OPGs (195). Radiotherapy, a historical standard of care and time-tested efficacious therapy, has long been abandoned as a primary treatment for pLGGs. Radiation-induced late effects can be particularly devastating and include vasculopathy, stroke, endocrinopathy, cognitive impairment, and secondary malignancies (196). The decision to avoid radiotherapy in pLGG is that progression of disease may be of little consequence when there are multiple systemic treatment options available, OS remains excellent, and the risks of radiotherapy-associated late effects, particularly secondary malignancy, outweigh any potential benefits of improved progression-free survival (197). Young patients are mostly susceptible, in fact, the actual cut-off age for radiation therapy is moved beyond 12 years (15).
Recently, advancements in how we have gained an insight into pLGG biology have sparked a new promising treatment in the field of pediatric neuro-oncology. Multiple investigations repeatedly affirmed that the great majorities of the pLGG exhibit alterations in their drivers that are commonly found to lead to the dual activation of the MAPK pathway and to downstream mTOR pathway (76). In addition, novel technologies in NGS have allowed the discovery of additional new altered drivers, including FGFR (76). Following these findings, in the last few years, have been developed several drugs targeting the pLGG MAPK and mTOR pathway (79–102, 116, 138–143, 154–156).
Dabrafenib and vemurafenit were demonstrated to have an outstanding efficacy on pLGG mutant BRAF p.V600E patients in early phase clinical trials. Vemurafenib is a small competing drug that selectively recognizes the ATP-binding domain of the BRAF p.V600E mutant. It has been proven efficacious in the management of metastatic melanoma, a malignancy frequently mutated for BRAF. This drug’s function has more recently been shown to be successful in BRAF p.V600E mutated pediatric malignant astrocytomas, while less data is currently available on its use in LGG patients, of which there are more studies on the combination of dabrafenib and trametinib (Table 3) (88, 106, 198–200, 205). The efficacy of the vemurafenib treatment in our small cohort of patients affected by pLGG is promising, with a rate of response of about 60% (88).
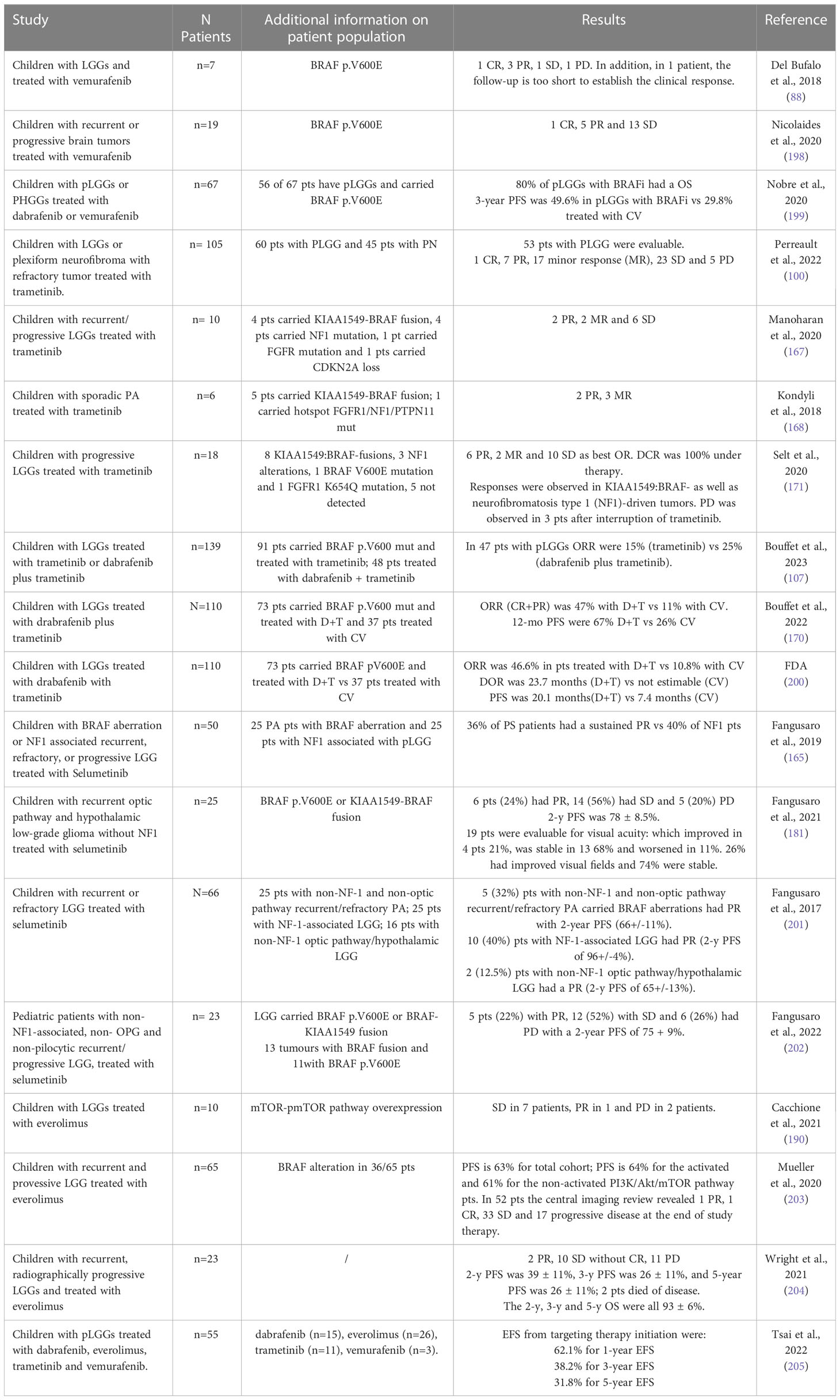
Table 3 List of results of targeted therapy in pLGG.
Furthermore, BRAF-fusions in pLGGs drive resistance/escape mechanisms to targeted inhibitors. For example, KIAA1549-BRAF has innate resistence to first-generation BRAFi vemurafenib as well as paradoxically triggered by PLX4720 treatment resulting in faster growth of tumor (90), while it shows a strong response to clinically available MEKi (e.g., trametinib) (206). Several studies showed that trametinib seems a appropriate choice in refractory as well as in progressive pLGG with KIAA1549-BRAF fusion and suggested that warrants further investigations in case of progression (Table 3) (167, 168, 171). The data of the study on progressive pLGGs lend weight to the class MEKi efficacy in pLGGs and the necessity of a randomized upfront trial of trametinib over current chemotherapy standard regimens (171). A phase 2 trial on patients with refractory/progressing LGG (NF1 patients and patients who carried KIA11549-BRAF fusion) and treated with trametinib, will investigate the molecular biological mechanisms that drive tumor development and progression, and the involvement of these mechanisms in resistance to therapy (100). Bouffet et al. showed the results of a phase II trial in which was compared the ORR in patients with pLGG who carried BRAF p.V600E mutation treated with both dabrafenib and trametinib (47%) or standard chemotherapy (CV) treatment (11%) (170).
In addition, in a cohort of both children and young adults treated for refractory tumors that have mutations or fusions resulting in activation of the MAPK pathway showed restricted selumetinib efficacy, suggesting that the mutation status of the pathway alone is sufficient to provide a predictor of the response to monotherapy with selumetinib for those tumors (207). In contrast, a phase II clinical trial on selumetinib among pediatric patients with relapsed and refractory LGG demonstrated impressive outcomes in sporadic OPG and hypothalamic LGG patients, with 24% partial response rate and 56% of patients showing long-term stability (104). In Table 3 are showed results of selumetinib on pLGG (165, 181, 201, 202).
With the discovery that many relapsed/refractory pLGGs have activation of mTOR pathway more treatment options may be possible for patients, including everolimus, a brain-penetrant drug already approved by the FDA for the treatment of SEGA in children (Table 3) (184, 190, 203, 204, 208–210). In our published experience, everolimus is a feasible treatment for p.V600E wild-type non-TSC pLGG patients (210). Interestingly, everolimus has been shown to synergize with carboplatin in preclinical models in vitro and in vivo by suppressing the conversion of glutamine and glutamate into glutathione (211). The PI3K-AKT-mTOR signaling cascade has been considered the major escape mechanism for BRAF-fusion. Jain and colleagues have shown that combinatorial targeting using MEKi and mTORi for BRAF-fusion-driven tumors is effective in overcoming such emergent resistance to single-agent therapy, highlighting preclinical rationales for using MEKi and mTORi. Very limited experience exists for combination therapies. However, in BRAF WT cells, everolimus and AZD6244 (MEK1/2 inhibitor) have proven to be superior compared to respective monotherapies (212).
To date, due to the results obtained from the various trials, the Children’s Oncology is investigating the possibility of the first-line treatment with MEKi, both as a single agent and in combination with chemotherapy in children with pLGG and relapsed cases.
Currently, the combination therapy development for pLGG patients is under investigation, and in particular, to date, the benefits of personalized therapies based on the administration of a single drug or with multiple combination drugs in non-resectable pLGG patients are unknown. In pLGGs patients, PNOC021 is the first study evaluating the combination of an mTORi (everolimus) and a MEKi (trametinib) to see if there is a possibility of achieving a safe MTD for this combination therapy strategy.
Tergeted drugs have less side systemic effects, however their fairly recent use precludes yet a comprehensive characterization regarding their long-term effects. The majority of short-term adverse events of targeted therapies are temporary and easily manageable, including creatine phosphokinase (CPK) elevation, cutaneous, cardiologic and ocular sequence and toxicities (164, 213). Long-term impact of BRAF, MEK, and mTOR inhibitors on mental and growth consequence in children remain uncertain due to the short follow up described to date. Of note, a limitation of target therapy remains the rebound effect of tumor growth at treatment suspension.
4 Conclusion
This literature review shows that targeted therapy is a feasible approach for pLGGs. Advances in cancer therapies including chemotherapy, radiation therapy, and surgery have significantly improved cancer treatment and outcomes for patients. However, these treatments can lead to a number of toxicities, which are related to a negative impact on their long-term health as well as quality of life. A greater understanding of tumor biology and a germline and somatic genomic approach will play a central role in the therapy strategy of pLGG for the development of increasingly tailored therapies. Limitations still exist regarding the adverse effects of long-term treatment.
Author contributions
AM and AC conceptualized the work. SC, GB and FF wrote the manuscript. AM and AC contributed to the finishing of the work and revised it critically for important intellectual content. All authors finally approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Acknowledgments
The authors thank Megan Eckley for revising the English language for the final version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Raabe E, Kieran MW, Cohen KJ. New strategies in pediatric gliomas: molecular advances in pediatric low-grade gliomas as a model. Clin Cancer Res (2013) 19:4553–8. doi: 10.1158/1078-0432.CCR-13-0662
2. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun (2020) 8:30. doi: 10.1186/s40478-020-00902-z
3. Wisoff JH, Sanford RA, Heier LA, Sposto R, Burger PC, Yates AJ, et al. Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the children’s oncology group. Neurosurgery (2011) 68:1548–55. doi: 10.1227/NEU.0b013e318214a66e
4. Bergthold G, Bandopadhayay P, Bi WL, Ramkissoon L, Stiles C, Segal RA, et al. Pediatric low-grade gliomas: how modern biology reshapes the clinical field. Biochim Biophys Acta BBA - Rev Cancer (2014) 1845:294–307. doi: 10.1016/j.bbcan.2014.02.004
5. Erkal HŞ, Serin M, Çakmak A. Management of optic pathway and chiasmatic-hypothalamic gliomas in children with radiation therapy. Radiother Oncol (1997) 45:11–5. doi: 10.1016/s0167-8140(97)00102-3
6. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: a Canadian pediatric brain tumor consortium study. J Clin Oncol (2016) 34:3537–43. doi: 10.1200/JCO.2016.68.1585
7. Lefkowitz IB, Packer RJ, Sutton LN, Siegel KR, Bruce DA, Evans AE, et al. Results of the treatment of children with recurrent gliomas with lomustine and vincristine. Cancer (1988) 61:896–902. doi: 10.1002/1097-0142(19880301)61:5<896::aid-cncr2820610507>3.0.co;2-c
8. Marcus KJ, Goumnerova L, Billett AL, Lavally B, Scott RM, Bishop K, et al. Stereotactic radiotherapy for localized low-grade gliomas in children: final results of a prospective trial. Int J Radiat Oncol (2005) 61:374–9. doi: 10.1016/j.ijrobp.2004.06.012
9. Merchant TE, Kun LE, Wu S, Xiong X, Sanford RA, Boop FA. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol (2009) 27:3598–604. doi: 10.1200/JCO.2008.20.9494
10. Packer RJ, Lange B, Ater J, Nicholson HS, Allen J, Walker R, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol (1993) 11:850–6. doi: 10.1200/JCO.1993.11.5.850
11. Rosenstock JG, Evans AE, Schut L. Response to vincristine of recurrent brain tumors in children. J Neurosurg (1976) 45:135–40. doi: 10.3171/jns.1976.45.2.0135
12. Saran FH, Baumert BG, Khoo VS, Adams EJ, Garré ML, Warrington AP, et al. Stereotactically guided conformal radiotherapy for progressive low-grade gliomas of childhood. Int J Radiat Oncol (2002) 53:43–51. doi: 10.1016/s0360-3016(02)02734-7
13. Grabenbauer GG, Schuchardt U, Buchfelder M, Rödel CM, Gusek G, Marx M, et al. Radiation therapy of optico–hypothalamic gliomas (OHG) – radiographic response, vision and late toxicity. Radiother Oncol (2000) 54:239–45. doi: 10.1016/s0167-8140(00)00149-3
14. Krishnatry R, Zhukova N, Guerreiro Stucklin AS, Pole JD, Mistry M, Fried I, et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: a population-based study: determinants of long-term survival in PLGG. Cancer (2016) 122:1261–9. doi: 10.1002/cncr.29907
15. Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol (2009) 27:3691–7. doi: 10.1200/JCO.2008.21.2738
16. Nageswara Rao AA, Packer RJ. Advances in the management of low-grade gliomas. Curr Oncol Rep (2014) 16:398. doi: 10.1007/s11912-014-0398-9
17. Bale TA, Rosenblum MK. The 2021 WHO classification of tumors of the central nervous system: an update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol (2022) 32:e13060. doi: 10.1111/bpa.13060
18. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell (2020) 37:569–83. doi: 10.1016/j.ccell.2020.03.011
19. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
20. Cutler RE, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the raf-1 serine/threonine kinase. Proc Natl Acad Sci (1998) 95:9214–9. doi: 10.1073/pnas.95.16.9214
21. Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR. The ins and outs of raf kinases. Trends Biochem Sci (1994) 19:474–80. doi: 10.1016/0968-0004(94)90133-3
22. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417:949–54. doi: 10.1038/nature00766
23. Schreck KC, Grossman SA, Pratilas CA. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers (2019) 11:1262. doi: 10.3390/cancers11091262
24. Talloa D, Triarico S, Agresti P, Mastrangelo S, Attinà G, Romano A, et al. BRAF and MEK targeted therapies in pediatric central nervous system tumors. Cancers (2022) 14:4264. doi: 10.3390/cancers14174264
25. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124:471–84. doi: 10.1016/j.cell.2006.01.016
26. Cantley LC. The phosphoinositide 3-kinase pathway. Science (2002) 296:1655–7. doi: 10.1126/science.296.5573.1655
27. Manning B. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci (2003) 28:573–6. doi: 10.1016/j.tibs.2003.09.003
28. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol OncolJ Hematol Oncol (2019) 12:71. doi: 10.1186/s13045-019-0754-1
29. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi: 10.1126/science.1106148
30. Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell (2001) 93:53–62. doi: 10.1016/s0248-4900(01)01125-x
31. Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of b-RAF. Cell (2004) 116:855–67. doi: 10.1016/s0092-8674(04)00215-6
32. Garnett MJ, Marais R. Guilty as charged. Cancer Cell (2004) 6:313–9. doi: 10.1016/j.ccr.2004.09.022
33. Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol (2008) 67:878–87. doi: 10.1097/NEN.0b013e3181845622
34. Jones DTW, Kocialkowski S, Liu L, Pearson DM, Bäcklund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097
35. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest (2008) 118:1739–49. doi: 10.1172/JCI33656
36. Jones DTW, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549: BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene (2009) 28:2119–23. doi: 10.1038/onc.2009.73
37. Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol (Berl) (2009) 118:401–5. doi: 10.1007/s00401-009-0550-z
38. Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res (2011) 17:3590–9. doi: 10.1158/1078-0432.CCR-10-3349
39. Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. giugno (2018) 37(24):3183–99. doi: 10.1038/s41388-018-0171-x
40. Ahrendsen JT, Sinai C, Meredith DM, Malinowski SW, Cooney TM, Bandopadhayay P, et al. Molecular alterations in pediatric low-grade gliomas that led to death. J Neuropathol Exp Neurol (2021) 80(11):1052–9. doi: 10.1093/jnen/nlab097
41. Del Baldo G, Cacchione A, Dell’Anna VA, Merli P, Colafati GS, Marrazzo A, et al. Rethinking the management of optic pathway gliomas: a single center experience. Front Surg (2022) 9:890875. doi: 10.3389/fsurg.2022.890875
42. Bouchè V, Aldegheri G, Donofrio CA, Fioravanti A, Roberts-Thomson S, Fox SB, et al. BRAF signaling inhibition in glioblastoma: which clinical perspectives? Front Oncol (2021) 11:772052. doi: 10.3389/fonc.2021.772052
43. Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell (2015) 28:370–83. doi: 10.1016/j.ccell.2015.08.001
44. Freeman AK, Ritt DA, Morrison DK. The importance of raf dimerization in cell signaling. Small GTPases (2013) 4:180–5. doi: 10.4161/sgtp.26117
45. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro-Oncol (2012) 14:777–89. doi: 10.1093/neuonc/nos077
46. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro-Oncol (2010) 12:621–30. doi: 10.1093/neuonc/noq007
47. Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. schrijver I, curatore. PloS One (2011) 6:e17948. doi: 10.1371/journal.pone.0017948
48. López GY, Van Ziffle J, Onodera C, Grenert JP, Yeh I, Bastian BC, et al. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol (Berl) (2019) 137:139–50. doi: 10.1007/s00401-018-1906-z
49. Dimitriadis E, Alexiou GA, Tsotsou P, Simeonidi E, Stefanaki K, Patereli A, et al. BRAF alterations in pediatric low grade gliomas and mixed neuronal–glial tumors. J Neurooncol (2013) 113:353–8. doi: 10.1007/s11060-013-1131-5
50. Gierke M, Sperveslage J, Schwab D, Beschorner R, Ebinger M, Schuhmann MU, et al. Analysis of IDH1-R132 mutation, BRAF V600 mutation and KIAA1549–BRAF fusion transcript status in central nervous system tumors supports pediatric tumor classification. J Cancer Res Clin Oncol (2016) 142:89–100. doi: 10.1007/s00432-015-2006-2
51. Dodgshun AJ, SantaCruz N, Hwang J, Ramkissoon SH, Malkin H, Bergthold G, et al. Disseminated glioneuronal tumors occurring in childhood: treatment outcomes and BRAF alterations including V600E mutation. J Neurooncol. giugno (2016) 128(2):293–302. doi: 10.1007/s11060-016-2109-x
52. Lassaletta A, Guerreiro Stucklin A, Ramaswamy V, Zapotocky M, McKeown T, Hawkins C, et al. Profound clinical and radiological response to BRAF inhibition in a 2-month-old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr Blood Cancer (2016) 63:2038–41. doi: 10.1002/pbc.26086
53. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol (2017) 35:2934–41. doi: 10.1200/JCO.2016.71.8726
54. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAFV600E mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol (Berl) (2013) 125:901–10. doi: 10.1007/s00401-013-1120-y
55. Donson AM, Kleinschmidt-DeMasters BK, Aisner DL, Bemis LT, Birks DK, Levy JMM, et al. Pediatric brainstem gangliogliomas show BRAF V600E mutation in a high percentage of cases: BRAF V600E mutation in brainstem gangliogliomas. Brain Pathol (2014) 24:173–83. doi: 10.1111/bpa.12103
56. Chen X, Pan C, Zhang P, Xu C, Sun Y, Yu H, et al. BRAF V600E mutation is a significant prognosticator of the tumour regrowth rate in brainstem gangliogliomas. J Clin Neurosci (2017) 46:50–7. doi: 10.1016/j.jocn.2017.09.014
57. Pagès M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D, et al. Co-Occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma: histone H3 K27M and BRAF V600E mutations in GG. Brain Pathol (2018) 28:103–11. doi: 10.1111/bpa.12473
58. Ho CY, Mobley BC, Gordish-Dressman H, VandenBussche CJ, Mason GE, Bornhorst M, et al. A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol (Berl) (2015) 130:575–85. doi: 10.1007/s00401-015-1467-3
59. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol (2015) 33:1015–22. doi: 10.1200/JCO.2014.58.3922
60. Broniscer A, Baker SJ, West AN, Fraser MM, Proko E, Kocak M, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol (2007) 25:682–9. doi: 10.1200/JCO.2006.06.8213
61. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MYC, Yu M, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res (2010) 70:512–9. doi: 10.1158/0008-5472.CAN-09-1851
62. Jacob K, Quang-Khuong DA, Jones DTW, Witt H, Lambert S, Albrecht S, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res (2011) 17:4650–60. doi: 10.1158/1078-0432.CCR-11-0127
63. Coutant M, Lhermitte B, Guérin E, Chammas A, Reita D, Sebastia C, et al. Retrospective and integrative analyses of molecular characteristics and their specific imaging parameters in pediatric grade 1 gliomas. Pediatr Blood Cancer (2022) 69(8):e29575. doi: 10.1002/pbc.29575
64. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol (Berl) (2016) 131:833–45. doi: 10.1007/s00401-016-1539-z
65. Ryall S, Arnoldo A, Krishnatry R, Mistry M, Khor K, Sheth J, et al. Multiplex detection of pediatric low-grade glioma signature fusion transcripts and duplications using the NanoString nCounter system. J Neuropathol Exp Neurol (2017) 76:562–70. doi: 10.1093/jnen/nlx042
66. Nagase T. Prediction of the coding sequences of unidentified human genes. XVIII. the complete sequences of 100 new cDNA clones from brain which code for Large proteins in vitro. DNA Res (2000) 7:271–81. doi: 10.1093/dnares/7.4.271
67. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol (2009) 218:172–81. doi: 10.1002/path.2558
68. Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, et al. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology (2009) 73:1526–31. doi: 10.1212/WNL.0b013e3181c0664a
69. Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol (Berl) (2010) 119:641–9. doi: 10.1007/s00401-009-0634-9
70. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res (2011) 17:4790–8. doi: 10.1158/1078-0432.CCR-11-0034
71. Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549: BRAF fusion variants. J Neuropathol Exp Neurol (2012) 71:66–72. doi: 10.1097/NEN.0b013e31823f2cb0
72. Becker AP, Scapulatempo-Neto C, Carloni AC, Paulino A, Sheren J, Aisner DL, et al. KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytomas. J Neuropathol Exp Neurol (2015) 74:743–54. doi: 10.1097/NEN.0000000000000213
73. Deshmukh H, Yeh TH, Yu J, Sharma MK, Perry A, Leonard JR, et al. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene (2008) 27:4745–51. doi: 10.1038/onc.2008.110
74. Roth JJ, Fierst TM, Waanders AJ, Yimei L, Biegel JA, Santi M. Whole chromosome 7 gain predicts higher risk of recurrence in pediatric pilocytic astrocytomas independently from KIAA1549-BRAF fusion status. J Neuropathol Exp Neurol (2016) 75:306–15. doi: 10.1093/jnen/nlw001
75. Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DTW, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol (Berl) (2011) 121:763–74. doi: 10.1007/s00401-011-0817-z
76. The St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet (2013) 45:602–12. doi: 10.1038/ng.2611
77. Helgager J, Lidov HG, Mahadevan NR, Kieran MW, Ligon KL, Alexandrescu S. A novel GIT2-BRAF fusion in pilocytic astrocytoma. Diagn Pathol (2017) 12:82. doi: 10.1186/s13000-017-0669-5
78. Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist (2017) 22:1478–90. doi: 10.1634/theoncologist.2017-0242
79. Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs (2012) 72:2207–22. doi: 10.2165/11640870-000000000-00000
80. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380:358–65. doi: 10.1016/S0140-6736(12)60868-X
81. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N Engl J Med (2012) 366:707–14. doi: 10.1056/NEJMoa1112302
82. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation–positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res (2019) 25:7303–11. doi: 10.1158/1078-0432.CCR-19-2177
83. Chamberlain MC. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol. settembre (2013) 114(2):237–40. doi: 10.1007/s11060-013-1176-5
84. Rush S, Foreman N, Liu A. Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol (2013) 31:e159–60. doi: 10.1200/JCO.2012.44.1568
85. del Bufalo F, Carai A, Figà-Talamanca L, Pettorini B, Mallucci C, Giangaspero F, et al. Response of recurrent BRAFV600E mutated ganglioglioma to vemurafenib as single agent. J Transl Med (2014) 12:356. doi: 10.1186/s12967-014-0356-1
86. Skrypek M, Foreman N, Guillaume D, Moertel C. Pilomyxoid astrocytoma treated successfully with vemurafenib: pilomyxoid astrocytoma and vemurafenib. Pediatr Blood Cancer (2014) 61:2099–100. doi: 10.1002/pbc.25084
87. Aguilera D, Janss A, Mazewski C, Castellino RC, Schniederjan M, Hayes L, et al. Successful retreatment of a child with a refractory brainstem ganglioglioma with vemurafenib: vemurafenib response in recurrent ganglioglioma. Pediatr Blood Cancer (2016) 63:541–3. doi: 10.1002/pbc.25787
88. Del Bufalo F, Ceglie G, Cacchione A, Alessi I, Colafati GS, Carai A, et al. BRAF V600E inhibitor (Vemurafenib) for BRAF V600E mutated low grade gliomas. Front Oncol (2018) 8:526. doi: 10.3389/fonc.2018.00526
89. Petruzzellis G, Valentini D, del Bufalo F, Ceglie G, Carai A, Colafati GS, et al. Vemurafenib treatment of pleomorphic xanthoastrocytoma in a child with down syndrome. Front Oncol (2019) 9:277. doi: 10.3389/fonc.2019.00277
90. Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci (2013) 110:5957–62. doi: 10.1073/pnas.1219232110
91. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro-Oncol (2014) 16:1408–16. doi: 10.1093/neuonc/nou059
92. Tutuka CSA, Andrews MC, Mariadason JM, Ioannidis P, Hudson C, Cebon J, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer (2017) 16:112. doi: 10.1186/s12943-017-0684-x
93. Novartis Pharmaceuticals. Phase II open-label global study to evaluate the effect of dabrafenib in combination with trametinib in children and adolescent patients with BRAF V600 mutation positive low grade glioma (LGG) or relapsed or refractory high grade glioma (HGG) (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT02684058.
94. University of California, San Francisco, Pediatric Neuro-Oncology Consortium. (PNOC)-002: safety, phase 0, and pilot efficacy study of vemurafenib, an oral inhibitor of BRAFV600E, in children and young adults with Recurrent/Refractory BRAFV600E- or BRAF ins T mutant brain tumors (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT01748149.
95. Wright KD. A phase I study of Tovorafenib/DAY101 (Formerly TAK-580, MLN2480) for children with low-grade gliomas and other RAS/RAF/MEK/ERK pathway activated tumors (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT03429803.
96. Fore Biotherapeutics. A phase 1/2a study to assess the safety, pharmacokinetics, and pharmacodynamics of FORE8394 in patients with advanced unresectable solid tumors (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT02428712.
97. Novartis Pharmaceuticals. Phase I/IIa, 2-part, multi-center, single-arm, open-label study to determine the safety, tolerability and pharmacokinetics of oral dabrafenib in children and adolescent subjects with advanced BRAF V600-mutation positive solid tumors (2021). Available at: https://clinicaltrials.gov/ct2/show/NCT01677741.
98. Novartis Pharmaceuticals. A phase II, open-label, study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT02034110.
99. National Cancer Institute (NCI). A phase 1 and phase II and re-treatment study of AZD6244 for recurrent or refractory pediatric low grade glioma (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT01089101.
100. Perreault D. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT03363217.
101. Hoffmann-La Roche F. A phase I/II, multicenter, open-label, dose-escalation study of the safety and pharmacokinetics of cobimetinib in pediatric and young adult patients with previously treated solid tumors (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT02639546.
102. Robison N. Phase I study of MEK162 for children with progressive or recurrent cancer and a phase II study for children with low-grade gliomas and other Ras/Raf/MAP pathway activated tumors (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT02285439.
103. National Cancer Institute (NCI). A study of the drugs selumetinib versus carboplatin/Vincristine in patients with neurofibromatosis and low-grade glioma. (USA: clinicaltrials.gov) (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03871257.
104. National Cancer Institute (NCI). A phase 3 randomized non-inferiority study of carboplatin and vincristine versus selumetinib (NSC# 748727) in newly diagnosed or previously untreated low-grade glioma (LGG) not associated with BRAFV600E mutations or systemic neurofibromatosis type 1 (NF1). (USA: clinicaltrials.gov) (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT04166409.
105. National Cancer Institute (NCI). A phase 3 study of selumetinib (NSC# 748727) or selumetinib in combination with vinblastine for non-NF1, non-TSC patients with recurrent or progressive low-grade gliomas (LGGs) lacking BRAFV600E or IDH1 mutations (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT04576117.
106. Pediatric Brain Tumor Consortium. Phase I/II trial of dabrafenib, trametinib, and hydroxychloroquine (HCQ) for BRAF V600E-mutant or trametinib and HCQ for BRAF Fusion/Duplication positive or NF1-associated recurrent or progressive gliomas in children and young adults (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT04201457.
107. Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600–mutant low-grade glioma. J Clin Oncol (2023) 41(3):664–74. doi: 10.1200/JCO.22.01000
108. Mueller S. PNOC021: a phase I trial evaluating the combination of trametinib and everolimus in pediatric and young adult patients with recurrent low-grade gliomas and high grade gliomas (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT04485559.
109. University Hospital, Strasbourg, France. A randomized and controlled phase II national protocol in non NF1 pediatric and AYA (Adolescent and young adults) patients bearing a wild type BRAF gene newly diagnosed comparing a daily oral MEK inhibitor (Trametinib) versus weekly vinblastine during 18 months (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT05180825.
110. Novartis Pharmaceuticals. An open label, multi-center roll-over study to assess long-term effect in pediatric patients treated with tafinlar (Dabrafenib) and/or mekinist (Trametinib) (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03975829.
111. Korf B. A phase II study of RAD001 (Everolimus) for children with NeurF1 and chemotherapy-refractory radiographic progressive low grade gliomas. Birmingham: University of Alabama (2020). Available at: https://clinicaltrials.gov/ct2/show/NCT01158651.
112. Wright KD, Yao X, London WB, Kao P-C, Gore L, Hunger S, et al. A phase II study of everolimus (RAD001) for children with chemotherapy and/or radiation-refractory progressive or recurrent low-grade gliomas. Pediatr Blood Cancer (2021) 68(2):e28787.
113. University of California, San Francisco. PNOC 001: phase II study of everolimus for recurrent or progressive low-grade gliomas in children. (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT01734512.
114. Hoffmann-La Roche F. A phase 1/2, open-label, dose-escalation and expansion study of entrectinib (Rxdx-101) in pediatrics with locally advanced or metastatic solid or primary CNS tumors And/Or who have no satisfactory treatment options (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT02650401.
115. Institut de Recherches Internationales Servier. A phase 3, multicenter, randomized, double-blind, placebo-controlled study of AG-881 in subjects with residual or recurrent grade 2 glioma with an IDH1 or IDH2 mutation (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT04164901.
116. Wright KD, Zimmerman MA, Fine E, Aspri T, Kieran MW, Chi S. LGG-26. type ii braf inhibitor tak-580 shows promise for upcoming clinal trial as evidenced by single patient ind study. Neuro-Oncol (2018) 20(suppl_2):i110. doi: 10.1093/neuonc/noy059.367
117. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer (2010) 10:116–29. doi: 10.1038/nrc2780
118. Gessi M, Moneim YA, Hammes J, Goschzik T, Scholz M, Denkhaus D, et al. FGFR1 mutations in rosette-forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol (2014) 73:580–4. doi: 10.1097/NEN.0000000000000080
119. Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R, et al. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun (2016) 4:93. doi: 10.1186/s40478-016-0353-0
120. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science (2012) 337:1231–5. doi: 10.1126/science.1220834
121. Torre M, Jessop N, Hornick JL, Alexandrescu S. Expanding the spectrum of pediatric NTRK -rearranged fibroblastic tumors to the central nervous system: a case report with RBPMS-NTRK3 fusion. Neuropathology (2018) 38:624–30. doi: 10.1111/neup.12513
122. Deland L, Keane S, Olsson Bontell T, Sjögren H, Fagman H, Øra I, et al. Discovery of a rare GKAP1-NTRK2 fusion in a pediatric low-grade glioma, leading to targeted treatment with TRK-inhibitor larotrectinib. Cancer Biol Ther (2021) 22:184–95. doi: 10.1111/neup.12513
123. Olsen TK, Panagopoulos I, Meling TR, Micci F, Gorunova L, Thorsen J, et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro-Oncol (2015) 17:1365–73. doi: 10.1093/neuonc/nov039
124. Aghajan Y, Levy ML, Malicki DM, Crawford JR. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep (2016) 2016:bcr2016217189. doi: 10.1136/bcr-2016-217189
125. Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun (2019) 10:4343. doi: 10.1038/s41467-019-12187-5
126. Miyake I, Hakomori Y, Shinohara A, Gamou T, Saito M, Iwamatsu A, et al. Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene (2002) 21:5823–34. doi: 10.1038/sj.onc.1205735
127. Armstrong F, Duplantier MM, Trempat P, Hieblot C, Lamant L, Espinos E, et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene (2004) 23:6071–82. doi: 10.1038/sj.onc.1207813
128. Nguyen N, Lee SB, Lee YS, Lee KH, Ahn JY. Neuroprotection by NGF and BDNF against neurotoxin-exerted apoptotic death in neural stem cells are mediated through trk receptors, activating PI3-kinase and MAPK pathways. Neurochem Res (2009) 34:942–51. doi: 10.1007/s11064-008-9848-9
129. Khotskaya YB, Holla VR, Farago AF, Mills Shaw KR, Meric-Bernstam F, Hong DS. Targeting TRK family proteins in cancer. Pharmacol Ther (2017) 173:58–66. doi: 10.1016/j.pharmthera.2017.02.006
130. Liu D, Offin M, Harnicar S, Li BT, Drilon A. Entrectinib: an orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Ther Clin Risk Manag (2018) 14:1247–52. doi: 10.2147/TCRM.S147381
131. Hong DS, Bauer TM, Lee JJ, Dowlati A, Brose MS, Farago AF, et al. Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann Oncol (2019) 30:325–31. doi: 10.1093/annonc/mdy539
132. Scott LJ. Larotrectinib: first global approval. Drugs (2019) 79:201–6. doi: 10.1007/s40265-018-1044-x
133. FDA Approves entrectinib for tumors with NTRK fusions - NCI (2019). Available at: https://www.cancer.gov/news-events/cancer-currents-blog/2019/fda-entrectinib-ntrk-fusion.
134. 210861s006lbl.pdf. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/210861s006lbl.pdf.
135. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
136. Drilon A, Siena S, Ou SHI, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discovery (2017) 7:400–9. doi: 10.1158/2159-8290.CD-16-1237
137. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
138. Ziegler DS, Wong M, Mayoh C, Kumar A, Tsoli M, Mould E, et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br J Cancer (2018) 119:693–6. doi: 10.1038/s41416-018-0251-2
139. Di Ruscio V, Carai A, Del Baldo G, Vinci M, Cacchione A, Miele E, et al. Molecular landscape in infant high-grade gliomas: a single center experience. Diagnostics (2022) 12:372. doi: 10.3390/diagnostics12020372
140. Children’s Oncology Group. Larotrectinib (LOXO-101, NSC# 788607) for previously untreated TRK fusion pediatric solid tumors and TRK fusion relapsed pediatric acute leukemias (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03834961.
141. National Cancer Institute (NCI). NCI-COG pediatric MATCH (Molecular analysis for therapy choice) - phase 2 subprotocol of LOXO-101 (Larotrectinib) in patients with tumors harboring actionable NTRK fusions (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03213704.
142. National Cancer Institute (NCI). NCI-COG pediatric MATCH (Molecular analysis for therapy choice) screening protocol (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03155620.
143. Nationwide Children’s Hospital. A pilot and surgical study of larotrectinib for treatment of children with newly-diagnosed high-grade glioma with NTRK fusion. Available at: https://clinicaltrials.gov/ct2/show/NCT04655404.
144. Ferris SP, Goode B, Joseph NM, Kline CN, Samuel D, Gupta N, et al. IDH1 mutation can be present in diffuse astrocytomas and giant cell glioblastomas of young children under 10 years of age. Acta Neuropathol (Berl) (2016) 132(1):153–5. doi: 10.1007/s00401-016-1579-4
145. Yeo KK, Alexandrescu S, Cotter JA, Vogelzang J, Bhave V, Li MM, et al. Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro-Oncol (2023) 25(1):199–210. doi: 10.1093/neuonc/noac132
146. Institut de Recherches Internationales Servier. A phase 1, multicenter, randomized, controlled, open-label, perioperative study of AG-120 and AG-881 in subjects with recurrent, non-enhancing, IDH1 mutant, low grade glioma (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT03343197.
147. Institut de Recherches Internationales Servier. A phase 1, multicenter, randomized, controlled, open-label, perioperative study of AG-120 and AG-881 in subjects with recurrent, non-enhancing, IDH1 mutant, low grade glioma. (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT03343197.
148. Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I, et al. Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat Med (2023) 29(3):615–22. doi: 10.1038/s41591-022-02141-2
149. Konteatis Z, Artin E, Nicolay B, Straley K, Padyana AK, Jin L, et al. Vorasidenib (AG-881): a first-in-Class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med Chem Lett (2020) 11(2):101–7. doi: 10.1021/acsmedchemlett.9b00509
150. Institut de Recherches Internationales Servier. A phase 1, multicenter, open-label, dose-escalation and expansion, safety, pharmacokinetic, pharmacodynamic, and clinical activity study of orally administered AG-881 in patients with advanced solid tumors, including gliomas, with an IDH1 and/or IDH2 mutation (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT02481154.
151. Megías-Vericat JE, Ballesta-López O, Barragán E, Montesinos P. IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood Lymphat Cancer Targets Ther (2019) 9:19–32. doi: 10.2147/BLCTT.S177913
152. Forma Therapeutics, Inc. A phase 1b/2 study of FT-2102 in patients with advanced solid tumors and gliomas with an IDH1 mutation (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03684811.
153. Daiichi Sankyo Co., Ltd. A phase 1 study of DS-1001b in patients with IDH1 mutated gliomas (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT03030066.
154. Sellmer L, Farschtschi S, Marangoni M, Heran MKS, Birch P, Wenzel R, et al. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J Rare Dis (2017) 12:34. doi: 10.1186/s13023-017-0588-2
155. Blanchard G, Lafforgue MP, Lion-François L, Kemlin I, Rodriguez D, Castelnau P, et al. Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. study and outcome of a French cohort. Eur J Paediatr Neurol (2016) 20:275–81. doi: 10.1016/j.ejpn.2015.12.002
156. Uusitalo E, Rantanen M, Kallionpää RA, Pöyhönen M, Leppävirta J, Ylä-Outinen H, et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol (2016) 34:1978–86. doi: 10.1200/JCO.2015.65.3576
157. Sturm D, Pfister SM, Jones DTW. Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol (2017) 35:2370–7. doi: 10.1200/jco.2017.73.0242
158. Hoyt WF, Baghdassarian SA. Optic glioma of childhood. natural history and rationale for conservative management. Br J Ophthalmol (1969) 53:793–8. doi: 10.1136/bjo.53.12.793
159. Kuenzle CH, Weissert M, Roulet E, Bode H, Schefer S, Th H, et al. Follow-up of optic pathway gliomas in children with neurofibromatosis type 1. Neuropediatrics (1994) 25:295–300. doi: 10.1055/s-2008-1073043
160. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol (2007) 61:189–98. doi: 10.1002/ana.21107
161. Parsa CF. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol (2001) 119:516. doi: 10.1001/archopht.119.4.516
162. D’Angelo F, Ceccarelli M, Tala, Garofano L, Zhang J, Frattini V, et al. The molecular landscape of glioma in patients with neurofibromatosis 1. Nat Med (2019) 25:176–87. doi: 10.1038/s41591-018-0263-8
163. de Blank P, Li N, Fisher MJ, Ullrich NJ, Bhatia S, Yasui Y, et al. Late morbidity and mortality in adult survivors of childhood glioma with neurofibromatosis type 1: report from the childhood cancer survivor study. Genet Med (2020) 22:1794–802. doi: 10.1038/s41436-020-0873-7
164. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro-Oncol (2017) 19:1135–44. doi: 10.1093/neuonc/now282
165. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol (2019) 20:1011–22. doi: 10.1016/S1470-2045(19)30277-3
166. Woodfield SE, Zhang L, Scorsone KA, Liu Y, Zage PE. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer (2016) 16:172. doi: 10.1186/s12885-016-2199-z
167. Manoharan N, Choi J, Chordas C, Zimmerman MA, Scully J, Clymer J, et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J Neurooncol (2020) 149:253–62. doi: 10.1007/s11060-020-03592-8
168. Kondyli M, Larouche V, Saint-Martin C, Ellezam B, Pouliot L, Sinnett D, et al. Trametinib for progressive pediatric low-grade gliomas. J Neurooncol (2018) 140:435–44. doi: 10.1007/s11060-018-2971-9
169. Wen PY, Stein A, van den Bent M, De Greve J, Wick A, de Vos FYFL, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol (2022) 23(1):53–64. doi: 10.1016/S1470-2045(21)00578-7
170. Bouffet E, Hansford J, Garré ML, Hara J, Plant-Fox A, Aerts I, et al. Primary analysis of a phase II trial of dabrafenib plus trametinib (dab + tram) in BRAF V600–mutant pediatric low-grade glioma (pLGG). J Clin Oncol (2022) 40(17_suppl):LBA2002–LBA2002. doi: 10.1200/JCO.2022.40.17_suppl.LBA2002
171. Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, Ecker J, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neurooncol. (2020) 149(3):499–510. doi: 10.1007/s11060-020-03640-3
172. Ronsley R, Hounjet CD, Cheng S, Rassekh SR, Duncan WJ, Dunham C, et al. Trametinib therapy for children with neurofibromatosis type 1 and life-threatening plexiform neurofibroma or treatment-refractory low-grade glioma. Cancer Med (2021) 10(11):3556–64. doi: 10.1002/cam4.3910
173. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2013) 381(9861):125–32. doi: 10.1016/S0140-6736(12)61134-9
174. Smpokou P, Zand DJ, Rosenbaum KN, Summar ML. Malignancy in noonan syndrome and related disorders: malignancy in noonan spectrum disorders. Clin Genet (2015) 88(6):516–22. doi: 10.1111/cge.12568
175. Lodi M, Boccuto L, Carai A, Cacchione A, Miele E, Colafati GS, et al. Low-grade gliomas in patients with noonan syndrome: case-based review of the literature. Diagnostics (2020) 10(8):582. doi: 10.3390/diagnostics10080582
176. Opocher E, Kremer LCM, Da Dalt L, van de Wetering MD, Viscardi E, Caron HN, et al. Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer. (2006) 42(12):1807–16. doi: 10.1016/j.ejca.2006.02.022
177. Avery RA, Mansoor A, Idrees R, Biggs E, Alsharid MA, Packer RJ, et al. Quantitative MRI criteria for optic pathway enlargement in neurofibromatosis type 1. Neurology (2016) 86(24):2264–70. doi: 10.1212/WNL.0000000000002771
178. Robert-Boire V, Rosca L, Samson Y, Ospina LH, Perreault S. Clinical presentation and outcome of patients with optic pathway glioma. Pediatr Neurol (2017) 75:55–60. doi: 10.1016/j.pediatrneurol.2017.06.019
179. Shofty B, Ben-Sira L, Kesler A, Jallo G, Groves ML, Iyer RR, et al. Isolated optic nerve gliomas: a multicenter historical cohort study. J Neurosurg Pediatr (2017) 20(6):549–55. doi: 10.3171/2017.6.PEDS17107
180. Packer RJ, Iavarone A, Jones DTW, Blakeley JO, Bouffet E, Fisher MJ, et al. Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro-Oncol (2020) 22(6):773–84. doi: 10.1093/neuonc/noaa036
181. Fangusaro J, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman N, et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: a pediatric brain tumor consortium study. Neuro-Oncol (2021) 23(10):1777–88. doi: 10.1093/neuonc/noab047
182. Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res (2008) 68(5):1520–8. doi: 10.1158/0008-5472.CAN-07-5916
183. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med (2010) 363(19):1801–11. doi: 10.1056/NEJMoa1001671
184. Hütt-Cabezas M, Karajannis MA, Zagzag D, Shah S, Horkayne-Szakaly I, Rushing EJ, et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro-Oncol (2013) 15(12):1604–14. doi: 10.1093/neuonc/not132
185. Pachow D, Wick W, Gutmann DH, Mawrin C. The mTOR signaling pathway as a treatment target for intracranial neoplasms. Neuro-Oncol (2015) 17(2):189–99. doi: 10.1093/neuonc/nou164
186. Guerreiro Stucklin A, Tabori U, Grotzer M. The changing landscape of pediatric low-grade gliomas: clinical challenges and emerging therapies. Neuropediatrics (2016) 47(02):070–83. doi: 10.1055/s-0035-1570491
187. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet (2016) 388(10056):2153–63. doi: 10.1016/S0140-6736(16)31419-2
188. Filbin M, Sturm D. Gliomas in children. Semin Neurol (2018) 38(01):121–30. doi: 10.1055/s-0038-1635106
189. Moavero R, Carai A, Mastronuzzi A, Marciano S, Graziola F, Vigevano F, et al. Everolimus alleviates obstructive hydrocephalus due to subependymal giant cell astrocytomas. Pediatr Neurol (2017) 68:59–63. doi: 10.1016/j.pediatrneurol.2016.11.003
190. Cacchione A, Lodi M, Carai A, Miele E, Tartaglia M, Megaro G, et al. Upfront treatment with mTOR inhibitor everolimus in pediatric low-grade gliomas: a single-center experience. Int J Cancer. (2021) 148(10):2522–34. doi: 10.1002/ijc.33438
191. Chalil A, Ramaswamy V. Low grade gliomas in children. J Child Neurol (2016) 31(4):517–22. doi: 10.1177/0883073815599259
192. Bouffet E, Jakacki R, Goldman S, Hargrave D, Hawkins C, Shroff M, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol (2012) 30(12):1358–63. doi: 10.1200/JCO.2011.34.5843
193. Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg (1997) 86(5):747–54. doi: 10.3171/jns.1997.86.5.0747
194. Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the children’s oncology group. J Clin Oncol (2012) 30(21):2641–7. doi: 10.1200/JCO.2011.36.6054
195. Avery RA, Hwang EI, Jakacki RI, Packer RJ. Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab. JAMA Ophthalmol (2014) 132(1):111. doi: 10.1001/jamaophthalmol.2013.5819
196. Qaddoumi I, Sultan I, Gajjar A. Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the surveillance, epidemiology, and end results database. Cancer (2009) 115(24):5761–70. doi: 10.1002/cncr.24663
197. Klein M, Heimans J, Aaronson N, van der Ploeg H, Grit J, Muller M, et al. Effect of radiotherapy and other treatment-related factors on mid-term to long-term cognitive sequelae in low-grade gliomas: a comparative study. Lancet (2002) 360(9343):1361–8. doi: 10.1016/s0140-6736(02)11398-5
198. Nicolaides T, Nazemi KJ, Crawford J, Kilburn L, Minturn J, Gajjar A, et al. Phase I study of vemurafenib in children with recurrent or progressive BRAFV600E mutant brain tumors: pacific pediatric neuro-oncology consortium study (PNOC-002). Oncotarget (2020) 11(21):1942–52. doi: 10.18632/oncotarget.27600
199. Nobre L, Zapotocky M, Ramaswamy V, Ryall S, Bennett J, Alderete D, et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis Oncol (2020) 4:561–71. doi: 10.1200/PO.19.00298
200. Center for Drug Evaluation and Research. FDA Approves dabrafenib with trametinib for pediatric patients with low-grade glioma with a BRAF V600E mutation. FDA (2023). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-trametinib-pediatric-patients-low-grade-glioma-braf-v600e-mutation.
201. Fangusaro J, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman N, et al. LGG-08. a phase ii prospective study of selumetinib in children with recurrent or refractory low-grade glioma (lgg): a pediatric brain tumor consortium (pbtc) study. Neuro-Oncol (2017) 19(4):34–5. doi: 10.1093/neuonc/nox083.141
202. Fangusaro J, Onar-Thomas A, Poussaint TY, Lensing S, Wu S, Ligon AH, et al. LGG-06. selumetinib in pediatric patients with non-neurofibromatosis type 1-associated, non-optic pathway (OPG) and non-pilocytic recurrent/progressive low-grade glioma harboring BRAFV600E mutation or BRAF-KIAA1549 fusion: a multicenter prospective pediatric brain tumor consortium (PBTC) phase 2 trial. Neuro-Oncol (2022) 24:i88–8. doi: 10.1093/neuonc/noac079.322
203. Mueller S, Aboian M, Nazemi K, Gauvain K, Yoon J, Minturn J, et al. DDRE-12. PNOC001 (NCT01734512): a phase ii study of everolimus for recurrent or progressive pediatric low-grade gliomas (plgg). Neuro-Oncol (2020) 22:ii63–4. doi: 10.1093/neuonc/noaa215.257
204. Wright KD, Yao X, London WB, Kao P, Gore L, Hunger S, et al. A POETIC phase II study of continuous oral everolimus in recurrent, radiographically progressive pediatric low-grade glioma. Pediatr Blood Cancer (2021) 68(2):e28787. doi: 10.1002/pbc.28787
205. Tsai JW, Choi JJ, Ouaalam H, Murillo EA, Yeo KK, Vogelzang J, et al. Integrated response analysis of pediatric low-grade gliomas during and after targeted therapy treatment. Neuro-Oncol Adv (2023) 5(1):vdac182. doi: 10.1093/noajnl/vdac182
206. Jain P, Silva A, Han HJ, Lang SS, Zhu Y, Boucher K, et al. Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget (2017) 8(49):84697–713. doi: 10.18632/oncotarget.20949
207. Eckstein OS, Allen CE, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey B, et al. Phase II study of selumetinib in children and young adults with tumors harboring activating mitogen-activated protein kinase pathway genetic alterations: arm e of the NCI-COG pediatric MATCH trial. J Clin Oncol (2022) 40(20):2235–45. doi: 10.1200/JCO.21.02840
208. Rodriguez FJ, Raabe EH. mTOR: a new therapeutic target for pediatric low-grade glioma? CNS Oncol (2014) 3(2):89–91. doi: 10.2217/cns.14.4
209. Franz DN, Agricola K, Mays M, Tudor C, Care MM, Holland-Bouley K, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol (2015) 78(6):929–38. doi: 10.1002/ana.24523
210. Cacchione A, Miele E, Lodi MC, Carai A, Colafati GS, Camassei FD, et al. lgg-18. everolimus treatment in pediatric patients affected by low-grade gliomas (plgg) non-tsc, braf v600-wt. Neuro-Oncol (2020) 22(Supplement_3):iii369–9. doi: 10.1093/neuonc/noaa222.400
211. Poore BA. Inhibition of glutamine metabolism sensitizes cancer to further pharmacological disruption and yields clinically viable treatment options. Baltimore: Johns Hopkins University (2018).
212. Olow A, Mueller S, Yang X, Hashizume R, Meyerowitz J, Weiss W, et al. BRAF status in personalizing treatment approaches for pediatric gliomas. Clin Cancer Res (2016) 22(21):5312–21. doi: 10.1158/1078-0432.CCR-15-1101
213. Kieran MW, Geoerger B, Dunkel IJ, Broniscer A, Hargrave D, Hingorani P, et al. A phase I and pharmacokinetic study of oral dabrafenib in children and adolescent patients with recurrent or refractory BRAF V600 mutation-positive solid tumors. Clin Cancer Res (2019) 25:7294–302. doi: 10.1158/1078-0432.ccr-17-3572
Keywords: pediatric low-grade glioma, brain tumors, neuro-oncology, molecular diagnostic, clinical trials, targeted therapies, risk stratification, glioma
Citation: Cipri S, Del Baldo G, Fabozzi F, Boccuto L, Carai A and Mastronuzzi A (2023) Unlocking the power of precision medicine for pediatric low-grade gliomas: molecular characterization for targeted therapies with enhanced safety and efficacy. Front. Oncol. 13:1204829. doi: 10.3389/fonc.2023.1204829
Received: 12 April 2023; Accepted: 01 June 2023;
Published: 15 June 2023.
Edited by:
Jaroslav Sterba, Masaryk University, CzechiaReviewed by:
Pasqualino De Antonellis, University of Toronto, CanadaDomenico Roberti, University of Campania Luigi Vanvitelli, Italy
Copyright © 2023 Cipri, Del Baldo, Fabozzi, Boccuto, Carai and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Mastronuzzi, YW5nZWxhLm1hc3Ryb251enppQG9wYmcubmV0