
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 29 November 2023
Sec. Pediatric Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1203994
 Federica D’Antonio1
Federica D’Antonio1 Sabrina Rossi2
Sabrina Rossi2 Isabella Giovannoni2
Isabella Giovannoni2 Rita Alaggio2
Rita Alaggio2 Andrea Carai3
Andrea Carai3 Giuseppe M. Milano4
Giuseppe M. Milano4 Antonella Cacchione4Alessandra Cancellieri5Marco Gessi5
Antonella Cacchione4Alessandra Cancellieri5Marco Gessi5 Manila Antonelli6
Manila Antonelli6 Giovanna S. Colafati7
Giovanna S. Colafati7 Giacomina Megaro5
Giacomina Megaro5 Sabina Vennarini8
Sabina Vennarini8 Angela Mastronuzzi4*
Angela Mastronuzzi4*Background: Intracranial mesenchymal tumors are a rare type of neoplasm (0.3% of all soft tissue tumors) characterized by a fusion of a FET family gene (usually EWSR1, rarely FUS) to CREB family genes (CREB1, ATF1, and CREM) with a slow-growing and favorable prognosis. Mesenchymal tumors are most frequently localized in the subcutaneous tissue (typically in the limbs and hands) of young adults and have rarely been diagnosed in the central nervous system. Surgery is the gold standard treatment; adjuvant radiation therapy and chemotherapy with sarcoma-based regimens have been used in rare cases when complete surgical excision was not recommended. In terms of prognosis, these tumors show a tendency for local relapse. The longest patient outcomes reported in the literature are five years.
Case description: This case describes a 27-year-old woman with unconventional extracranial metastatic sites of myxoid intracranial mesenchymal tumor FET::CREB fusion-positive and high expression of PD-1 (40%) and PD-L1 (30%). Based on clinical, molecular, and histological characteristics, she underwent various local and systemic therapies, including surgery, proton beam therapy, the use of immune checkpoint inhibitors, and chemotherapy. These treatments led to a complete remission of the disease after eight years from tumor diagnosis.
Conclusions: Our case sheds light on the importance of precision medicine and tailored therapy to explore new treatment opportunities for rare or unknown tumor entities.
Mesenchymal tumors are rare soft connective tissue neoplasms (1, 2). These tumors may arise in all organs originating from mesodermal precursor cells and also in the central nervous system (CNS), specifically from the meninges and rarely from CNS parenchyma (2, 3).
The FET family (usually EWSR1, less frequently FUS) gene’s rearrangements with CREB family genes (CREB1, ATF1, and CREM) have been identified as characterizing a specific group of mesenchymal tumors called angiomatoid fibrous histiocytomas (AFH) (4). These are uncommon soft tissue tumors (typically found in the limbs, hands feet, or pelvis of children and young adults), with an incidence of < 0.5% and are classified as low-intermediate growing neoplasms with a favorable prognosis (2, 5).
More recently, intracranial AFH has been described as an intracranial mesenchymal tumor (6–8). AFH is usually treated with surgical removal, and only in cases of incomplete resection may adjuvant radiation therapy or chemotherapy be necessary (5). We hereby present a unique case of an intracranial mesenchymal tumor with EWSR1::CREM fusion transcript with extra CNS metastatic spreads, treated with different local and systemic therapies, leading to prolonged complete remission of the disease after two years of treatment discontinuation and after eight years from tumor diagnosis.
A 27-year-old woman was admitted to an outside hospital’s emergency department due to a headache and vomiting. She had no previous medical history or family history of cancer. Computed tomography (CT) and magnetic resonance imaging (MRI) revealed a mass on the right cerebellum that involved the transverse venous sinus (Figure 1). She underwent a partial resection, and the histological diagnosis highlighted a medulloblastoma. Following the diagnosis, the patient was referred to our center. The revised histological findings suggested the possibility of a high-grade glioneuronal tumor (HGG), which was later confirmed after a total secondary resection. Proton beam therapy (PBT) was administered at the surgical site with a total planned dose of 54 Grays (Gy) in 25 sessions, along with concomitant (75 mg/mq/day during PBT) and adjuvant (200 mg/mq/day for five days per month for six months) oral temozolomide. Subsequent MRIs showed complete remission of the disease. After 18 months from the suspension of treatment, a cerebral MRI revealed a local relapse at the surgical site (Figure 2). During the diagnostic workup, two metastatic lung lesions (the larger in the upper lingula segment) were discovered with suspicion of a lesion in the right iliac bone (Figures 3, 4). The iliac bone biopsy confirmed the diagnosis of HGG, with 40% PD-1 expression on lymphocytes and 30% PD-L1 expression on neoplastic cells (Figure 5). Subsequently, immunotherapy with intravenous nivolumab (3 mg/kg/day) was started every two weeks and continued for two years without any reported toxicity. The patient achieved complete remission in all sites (Figure 4D), which was confirmed by a biopsy of the lesion in the right iliac bone, which showed only inflammatory tissue without evidence of neoplastic infiltration. After six months of discontinuing therapy, CT and a positron emission tomography (PET) scan revealed a relapse in the lung lesion and in the right iliac bone lesion (Figures 3A, 4). A biopsy of this bone mass and a review of the previous cerebellar lesion allowed for a reevaluation of the entire case. Morphological features were similar in the primary and metastatic lesions: the tumor consisted of sheets of epithelioid cells with abundant eosinophilic to clear cytoplasm, large nuclei, and prominent nucleoli and showed a marked lymphoplasmacytic infiltrate at the periphery (Figures 5A, B). Mitoses were brisk (three mitoses/mm2), and the Ki67 proliferation index was approximately 15%. Although myxoid stroma and blood-filled cystic spaces were not prominent features, the expression of CD99 (membranous), GLUT-1 with a prominent paranuclear/Golgi pattern, focal EMA, and focal GFAP and synaptophysin raised a suspicion of an unusual mesenchymal tumor (Figure 5). A next-generation sequencing (NGS) panel (Archer Custom Fusion Plex Kit, Integrated DNA Technologies, IA) identified the presence of the EWSR1::CREM fusion transcript on both the primary tumor and the metastasis, confirming the diagnosis of metastatic intracranial mesenchymal tumor FET::CREB fusion-positive. (Figure 6).
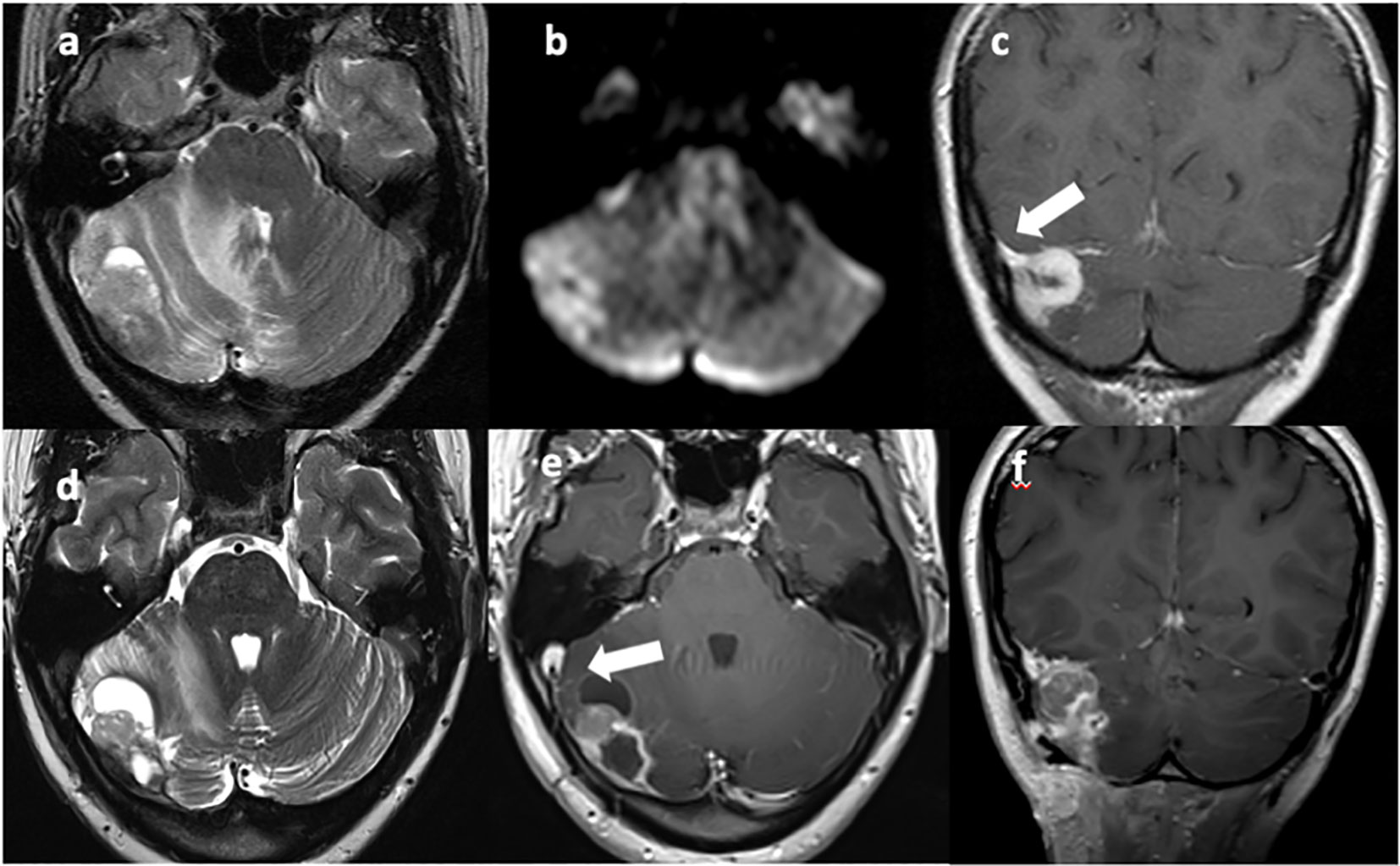
Figure 1 T2w axial (A, D) and T1w axial (E) and coronal (C, F) MRI images show a vascularized solid-cystic edematous lesion in the right cerebellar hemisphere, in continuity with the right transverse sinus [(C), arrow]. Diffusion restriction is present due to the high cellularity of the neoplastic tissue (B). Sinus thrombosis is also present [(E), arrow].
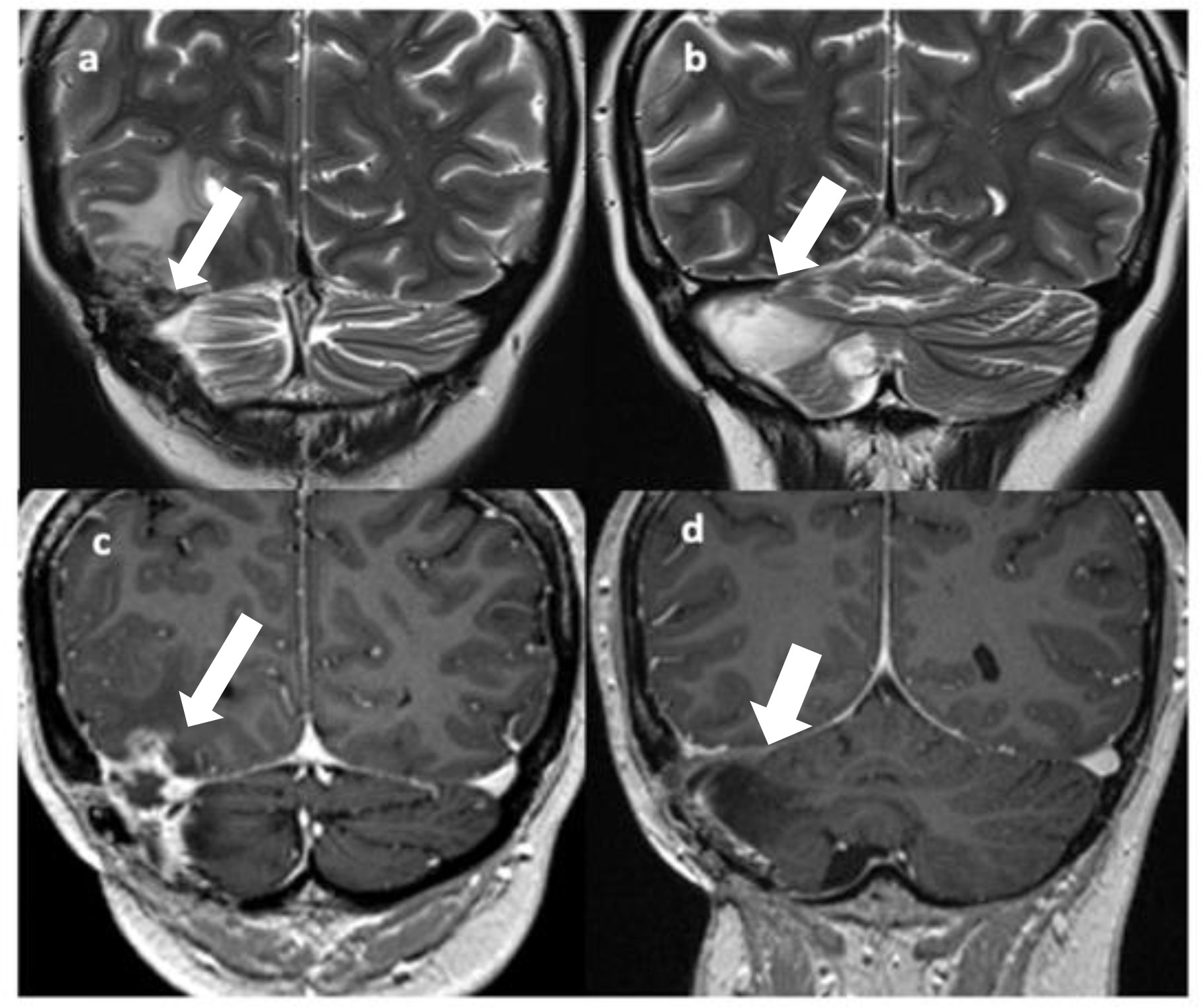
Figure 2 T2 (A, B) and Gd T1 TSE (C, D) coronal images show disease recurrence with a solid-cystic, edematous lesion in the right temporo-occipital and right cerebellar regions with involvement of the right transverse sinus [(A, C), arrows]; gliotic-malacic findings coexist in the right hemicerebellar region [(B, D), arrows].
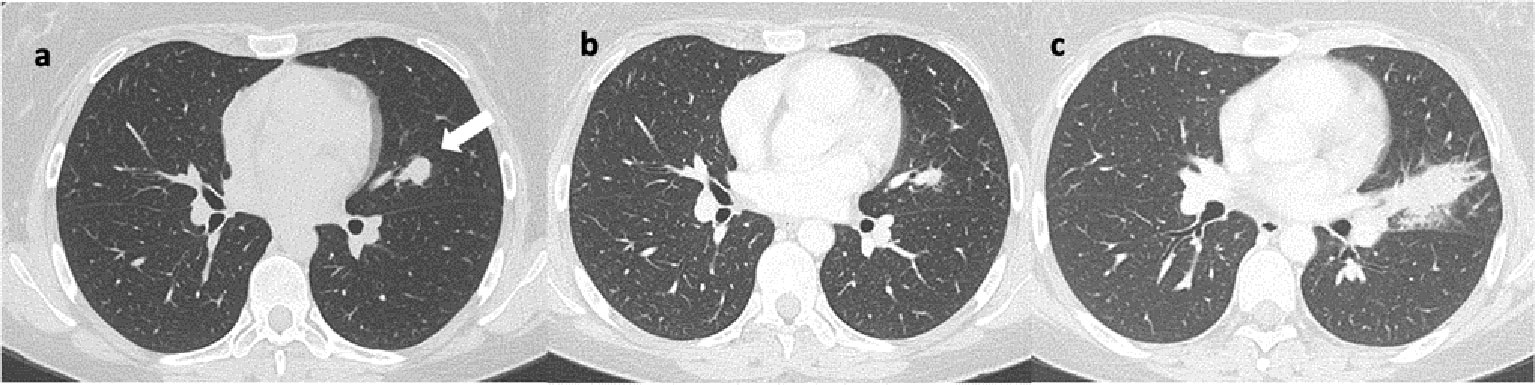
Figure 3 Axial lung CT scans reveal: (A) a nodular lesion in the upper lingular segment, indicated by the arrow. Image (B) shows a reduction in size at the 14-month follow-up after initiation of immunotherapy. Image (C) shows a significant increase in size at the 6-month follow-up after discontinuation of immunotherapy, characterized by spiculated margins.
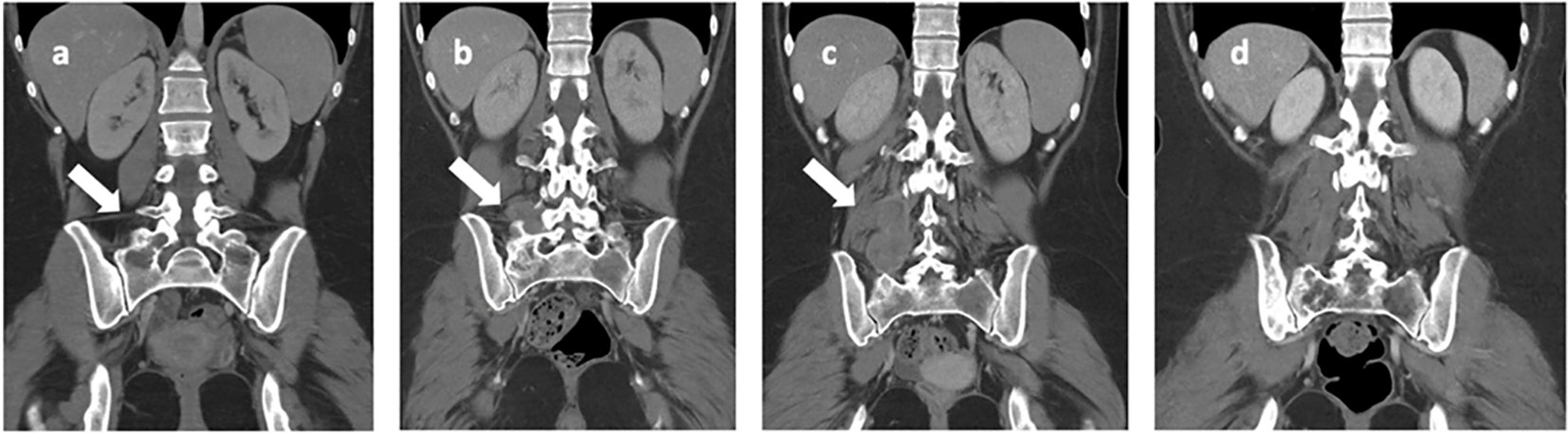
Figure 4 Iliac bone CT scans highlight: (A) a lesion with lytic features located in the right iliac wing and sacral wing (indicated by the arrow) at the onset of bone metastasis. In images (B, C), there is a progressive increase in the extension observed in the right paravertebral soft tissue (indicated by the arrows), as seen at the time of discontinuation of nivolumab. Image (D) depicts the nearly complete resolution observed at the last follow-up, which occurred 8 years after disease onset.
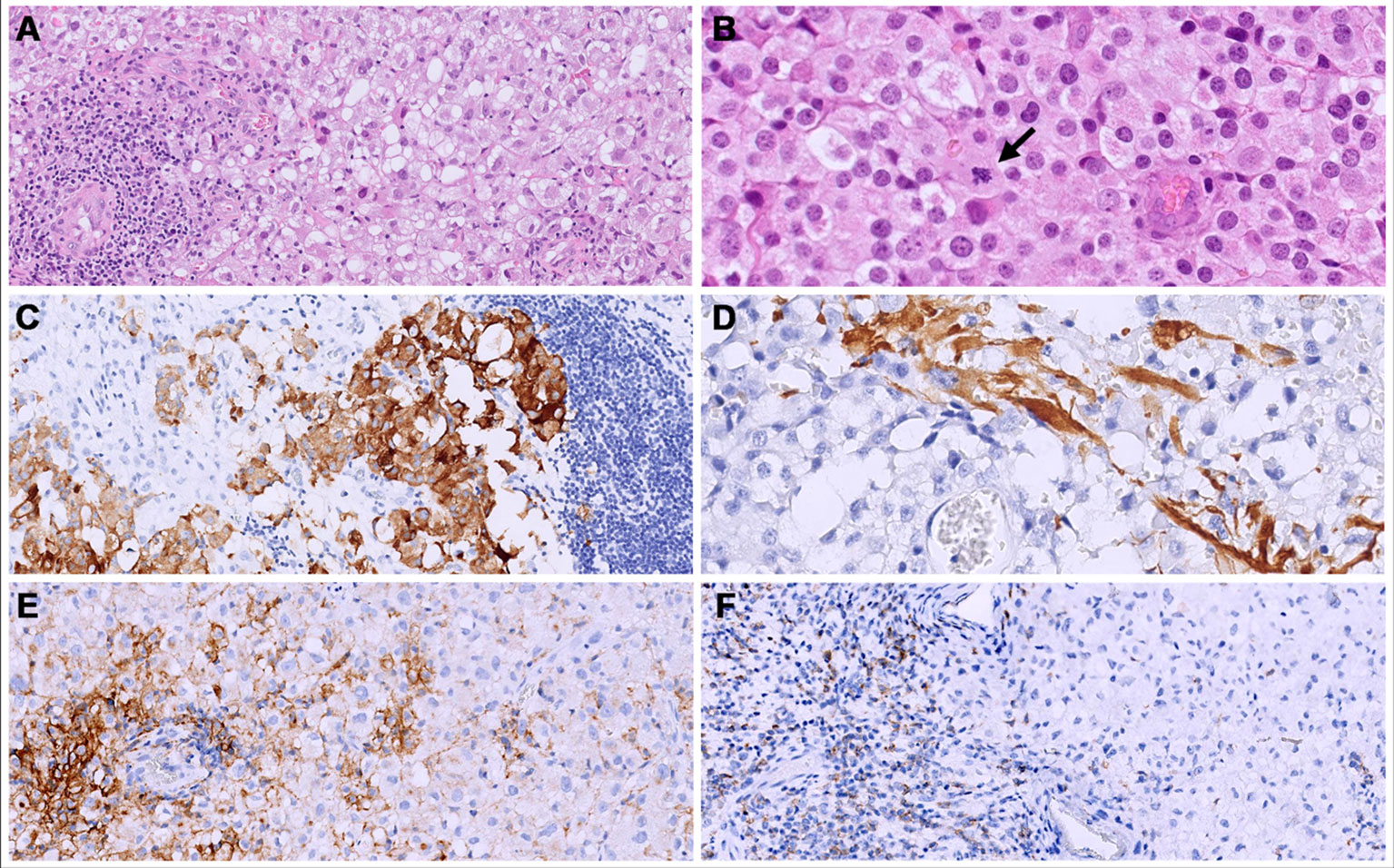
Figure 5 Both primary (cerebral) and metastatic (bone and lung) tumors consisted of solid sheets of epithelioid cells with clear to eosinophilic cytoplasm. (A, B) show the metastatic site, iliac bone); an abundant lymphocytic infiltrate was present mainly at the periphery of the tumors. (B) Mitoses were brisk (arrow). (C, D) The primary tumor (cerebral) showed a focal expression of synaptophysin (C) and GFAP (D). (E) in the bone metastatic neoplasm, a membranous staining for PD-L1 was seen in 30% of the neoplastic cells. (F) PD-1 was expressed in 40% of the lymphocytes.

Figure 6 Analysis of the Archer™ FusionPlex Custom Panel-anchored multiplex PCR result showing an EWSR1 exon 13 and CREM exon 5 gene fusion with reads (#/%) of 1493/22.48.
In accordance with these findings, fluorescent in situ hybridization (FISH) demonstrated the rearrangement of EWSR1.
Additionally, a blood test for cancer predisposition syndromes was conducted using the Twist Custom Panel, which includes assessments for CTNNB1, SMO, PIK3CA, PTEN, ID1, FGFR1, ARID1A, SMARCA4, CHD7, KDM4C, MYC, MYCN, MSH2, TP53, SUFU, PTCH1, PTCH2, ARID1B, and ERBB2 alterations, and no pathogenic/likely pathogenic variants were identified. Based on the previous clinical response to nivolumab, we decided to start a rechallenge treatment. After two cycles, nivolumab was prematurely discontinued due to the development of grade 2 immuno-mediated pneumonia, leading to a decrease in the patient’s performance status (ECOG 2) and iliac bone and lung disease progression. Consequently, PBT was performed on the right iliac bone lesion (with a total planned dose of 59.4 Gy in 48 sessions) with a complete surgical removal of the lung lesion. Thereafter, the patient received consolidation therapy with eight cycles of temozolomide plus irinotecan, achieving a complete remission of the disease.
She is currently still in complete remission with an optimal quality of life, nine years after diagnosis and two years after discontinuation of therapy (Figure 7).
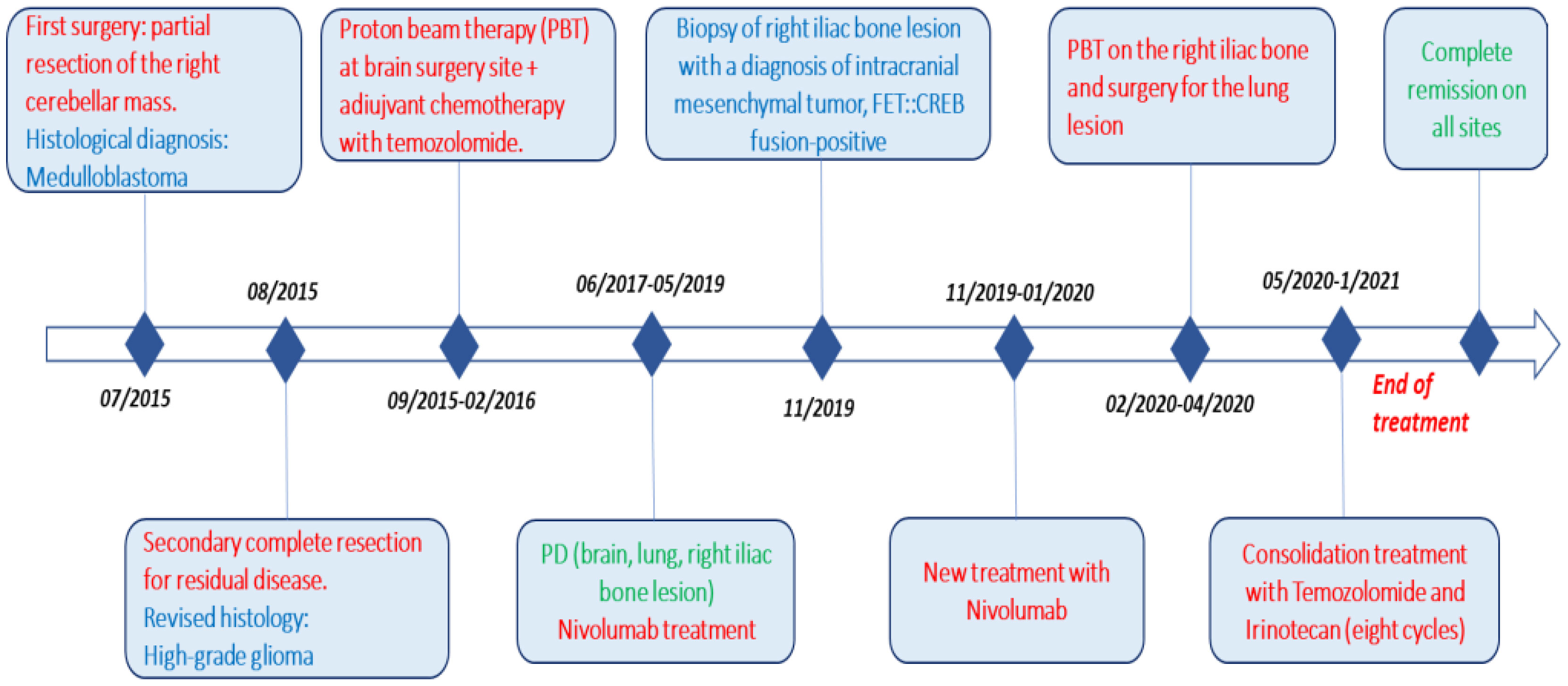
Figure 7 Complete timeline, including pathways to reach the diagnosis (surgery) and treatment (red), different histologies (blue), and response to therapy (green).
AFH is a rare mesenchymal tumor (0.3% of all soft tissue tumors) (3), defined by a fusion of a FET family gene (usually EWSR1, but rarely FUS) with members of the cAMP Response Element-Binding Protein family (ATF1, CREB1, or CREM) (9). These specific gene fusions play a key role in AFH tumorigenesis (9–11). In 1979, Enzinger described “angiomatoid malignant fibrous histiocytoma” as a new histological entity for the first time (12, 13). Although AFH has been known for more than 40 years, only in 2021 was it included in the WHO CNS classification as an intracranial mesenchymal tumor (13). In fact, AFH usually occurs in the extremities of young adults, more frequently in the second decade of life, with a female prevalence, and is rarely diagnosed in the CNS (14, 15). Clinically, patients with AFH can experience, in addition to local symptoms, systemic non-specific signs such as fever, anemia, and weight loss (6, 7, 16). Regional recurrence rates after surgery are relatively low (approximately 15%), but AFH can occasionally metastasize (the most common sites are lymph nodes, lungs, and liver), especially if it is not completely removed (5, 16).
CNS-AFH represents a rare primary site (Tables 1, 2), and the longest patient outcomes reported in the literature are five years with a median progression-free survival of 28 months (8, 9, 11). Patients with subtotal resection showed a local recurrence within 12 months. At microscopic histology examination, AFH is characterized by multinodular proliferation of spindle-shaped, or round cells with syncytial growth, forming bundles surrounded by fibrous pseudocapsules, pericapsular lymphoplasmacytic cuffing, and pseudovascular spaces full of blood. Most AFH cases express desmin but lack positivity for myogenin or MyoD, EMA, and CD68 (4, 29). Regrettably, the definitive histological diagnosis of AFH still presents a challenge (3). In the literature, several unusual clinicopathological presentations and histological variants are described (7, 9). The intracranial mesenchymal tumor is one of these variants, characterized by a prominent collagenous stroma with a dense intracellular matrix resembling the myofibroblastic tumor, poorly differentiated carcinoma, or meningioma. Not all cases contain a myxoid matrix (1, 3, 10).
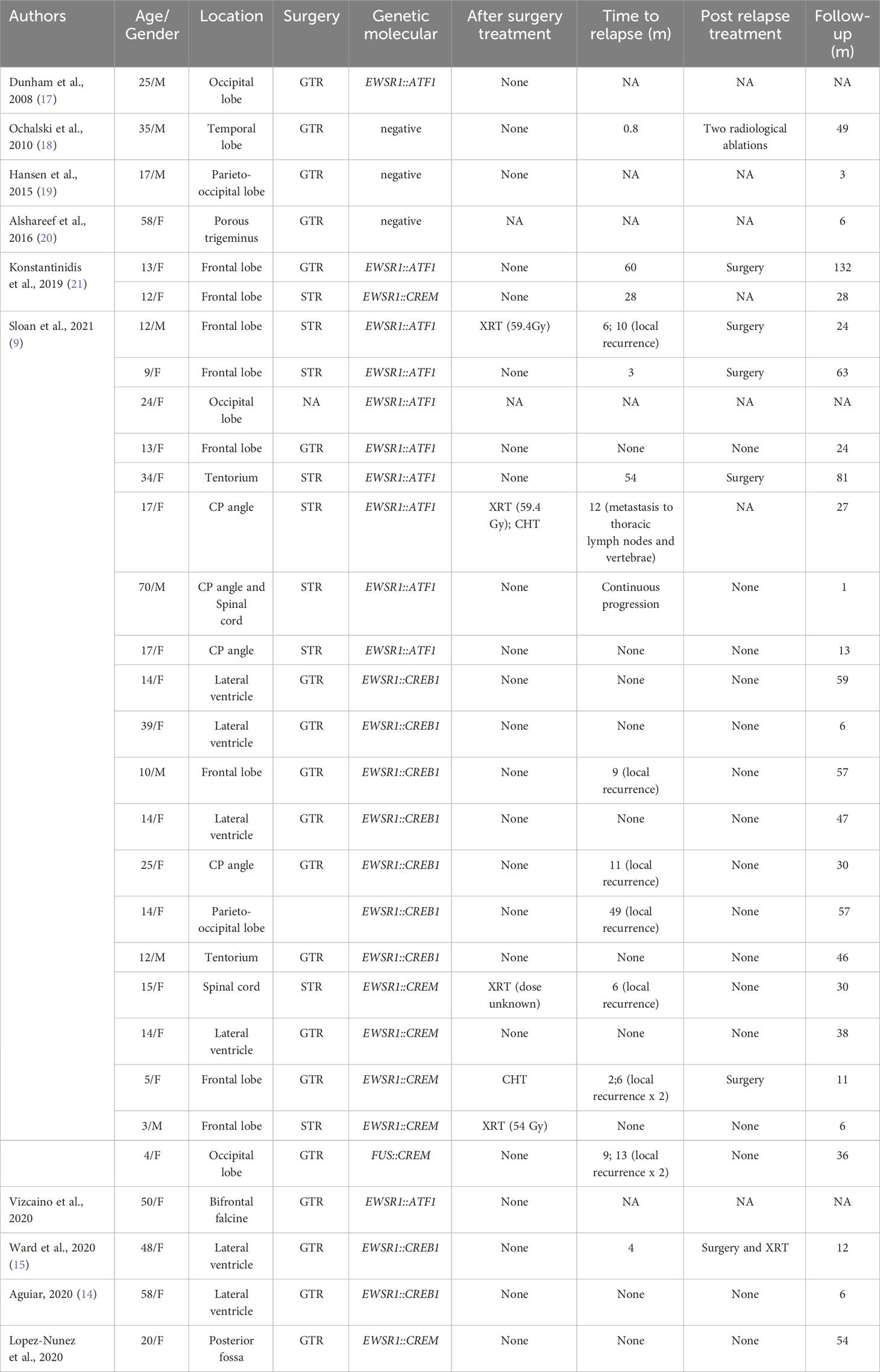
Table 1 Published cases of conventional AFH (NA, not available; GTR, gross total resection; STR, subtotal resection; CHT, chemotherapy; XRT, radiotherapy; CP angle, cerebellopontine angle).
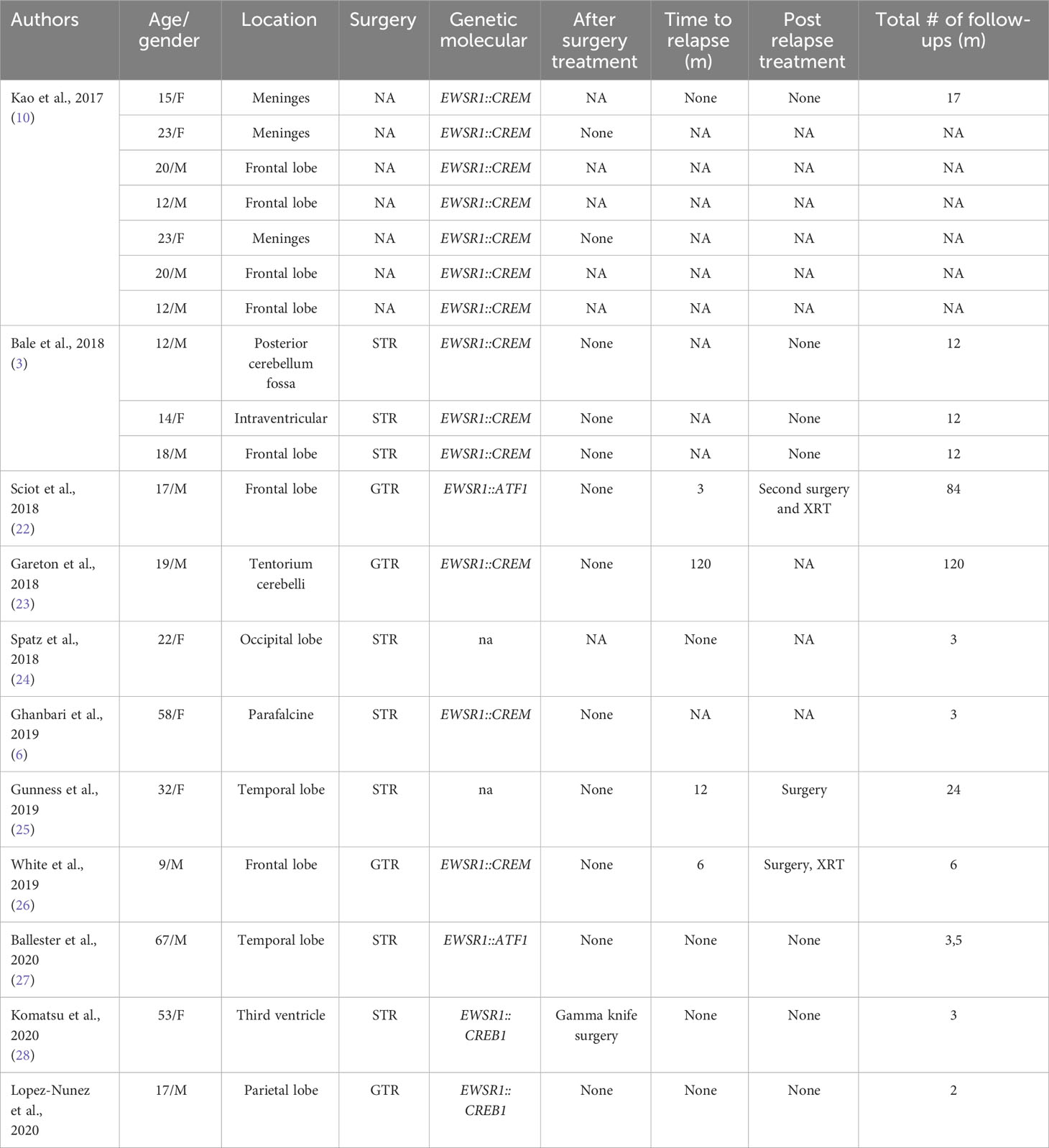
Table 2 Published cases of myxoid mesenchymal AFH (NA, not available; GTR, gross total resection; STR, subtotal resection; CHT, chemotherapy; XRT, radiotherapy; CP angle, cerebellopontine angle).
The intracranial mesenchymal tumor’s radiological aspect shows hypointense T1 signal and hyperintense T2 signal lesions with strong enhancement after gadolinium administration (10). Differential diagnoses include meningioma and schwannoma (Table 3) because intracranial mesenchymal tumors mimic an extra-axial lesion with homogeneous contrast enhancement and a small dural tail in T1 fluid-attenuated inversion recovery (FLAIR) (3). Furthermore, the expression of glial and neural markers is a pitfall for glial and glioneuronal tumors.
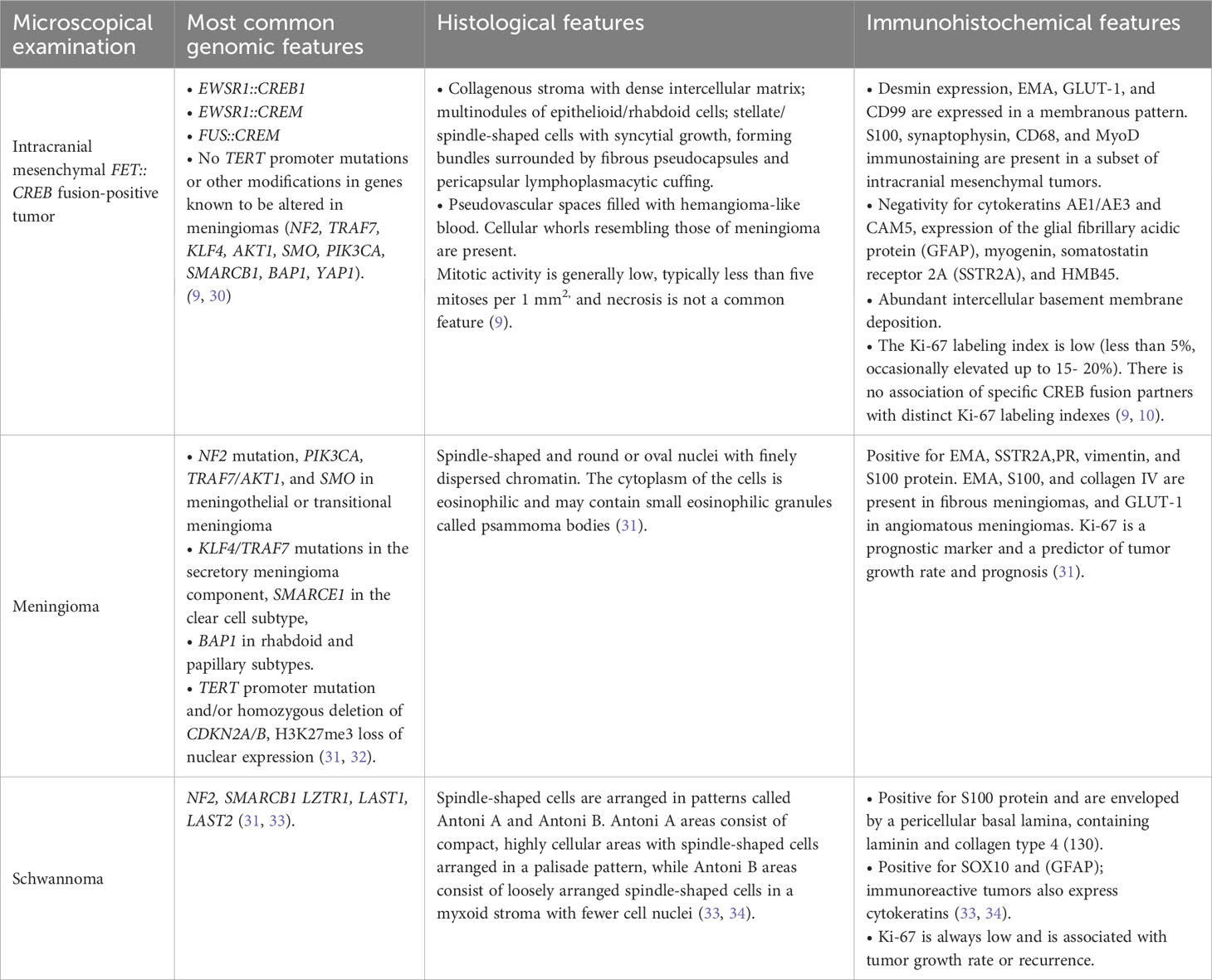
Table 3 Differential diagnosis.
Tauziède-Espariat et al. described 11 cases of CNS mesenchymal tumors with FET::CREB fusion. Six in total were a specific cluster with DNA methylation, and five showed no relation to any of the other classes but were similar to the clusters of extra-CNS angiomatoid fibrous histiocytomas, clear cell sarcomas, or solitary fibrous tumors. Therefore, the authors demonstrated that intracranial FET::CREB-fused tumors did not present a single molecular tumor entity but a primary intracranial mesenchymal tumor, the FET::CREB-fused family (4). Several other authors reported small groups of intracranial mesenchymal cases: Kao et al. described four children and young adults diagnosed with intracranial mesenchymal tumors with myxoid component and EWSR1::CREB1, EWSR1::CREM, or EWSR1::ATF1 fusions (10); Bale et al. described three pediatric cases with similar histology and fusions (3); Sloan et al. reported a series of 20 cases of intracranial mesenchymal tumors with FET::CREB fusion and comprehensively characterized their radiologic, molecular, and clinicopathologic features. Several other intracranial mesenchymal tumor cases without myxoid component and EWSR1::CREB1 fusion have been reported in the literature (Tables 1, 2).
The reported treatment was surgical in all cases; nevertheless, adjuvant radiotherapy and sarcoma-based regimens have also been reported (2). Our case confirms that local treatment (including surgery and proton beam therapy) shows the most favorable outcomes and a more promising prognosis (35).
In the case of the patient described, it was initially difficult to make the correct diagnosis, but by expanding the immunohistochemical panel and integrating it with molecular data, the correct diagnosis of intracranial mesenchymal tumor FET::CREB fusion-positive was made. Due to the rarity of the histopathological and molecular features, this case was previously published as part of a series of EWSR1-rearranged intracranial tumors (29). At the onset, we treated our patient according to the diagnosis of HGG. However, the uncommon occurrence of extracranial metastasis led us to identify a personalized treatment approach. We conducted molecular analysis, which revealed high levels of PD-1 and PD-L1 expression. This information allowed us to initiate treatment with nivolumab, making our case the first to be documented to receive immunotherapy in the medical literature. As a result of this therapy, the patient achieved and maintained a partial response for nearly three years. However, it was only after the most recent disease progression and five years after the initial diagnosis that we were able to identify an intracranial mesenchymal tumor with the EWSR1::CREB1fusion transcript. After this diagnosis, the patient received local treatments (surgery for the lung lesion and proton therapy for the bone lesion) along with consolidation with systemic drug therapy. This consolidation regimen consisted of temozolomide and irinotecan, tailored to sarcoma-specific protocols, with good tolerability and outpatient administration.
In conclusion, our case highlights the necessity and mandatory molecular studies workup for these rare diseases, which are crucial for refining personalized therapies and exploring novel treatment options.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) and/or minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AM: responsible for all published work, FD: responsible for writing paper write, SR, IG, RA, ACan, and MG: critical revision of histological and molecular case diagnosis. ACac and GM: examination, diagnosis and follow-up of the patient, and critical revision of the manuscript. ACar and GMM: critical revision of the manuscript for intellectual content. SC: radiological diagnosis and follow-up of the patient. SV: treatment and follow-up of the patient, and critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP, et al. Morphologic and immunophenotypic diversity in ewing family tumors: A study of 66 genetically confirmed cases. Am J Surg Pathol (2005) 29:1025–33. doi: 10.1097/01.pas.0000167056.13614.62
2. Davis JL, Tihan T, Kilpatrick SE. 14 - mesenchymal tumors of the central nervous system in practical surgical neuropathology: A diagnostic approach. 2nd ed. Perry A, Brat DJ, editors. Elsevier (2018) p. 299–322.
3. Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, Ligon K. Alexandrescu, intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol (2018) 28:183–91. doi: 10.1111/bpa.12504
4. Tauziède-Espariat A, Sievers P, Larousserie F, Benzakoun J, Guillemot D, Pierron G, et al. An integrative histopathological and epigenetic characterization of primary intracranial mesenchymal tumors, FET:CREB-fused broadening the spectrum of tumor entities in comparison with their soft tissue counterparts. Brain Pathol (2022) 32:e13010. doi: 10.1111/bpa.13010
5. Costa MJ, Weiss SW. Angiomatoid Malignant fibrous histiocytoma. A follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol (1990) 14:1126–32. doi: 10.1097/00000478-199012000-00004
6. Ghanbari N, Lam A, Wycoco V, Lee G. Intracranial myxoid variant of angiomatoid fibrous histiocytoma: A case report and literature review. Cureus (2019) 11:e4261. doi: 10.7759/cureus.4261
7. Domingo RA, Vivas-Buitrago T, Jentoft M, Quinones-Hinojosa A. Intracranial myxoid mesenchymal tumor/myxoid subtype angiomatous fibrous histiocytoma: diagnostic and prognostic challenges. Neurosurgery (2020) 88:E114–22. doi: 10.1093/neuros/nyaa357
8. Justin Wong SB, Wee A, Puhaindran ME, Pang B, Lee VKM. Angiomatoid fibrous histiocytoma with prominent myxoid stroma: A case report and review of the literature. Am J Dermatopathol (2015) 37:623–31. doi: 10.1097/DAD.0000000000000263
9. Sloan EA, Chiang J, Villanueva-Meyer JE, Alexandrescu S, Eschbacher JM, Wang W, et al. Intracranial mesenchymal tumor with FET::CREB fusion—A unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol (2021) 31:e12918. doi: 10.1111/bpa.12918
10. Kao Y-C, Sung Y-S, Zhang L, Chen C-L, Vaiyapuri S, Rosenblum MK, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol (2017) 41:482–490. doi: 10.1097/PAS.0000000000000788
11. Garnier L, Fenouil T, Pissaloux D, Ameli R, Ducray F, Meyronet D, et al. Intracranial non-myxoid angiomatoid fibrous histiocytoma with EWSR1-CREB1 transcript fusion treated with doxorubicin: A case report. Mol Clin Oncol (2021) 15:131. doi: 10.3892/mco.2021.2293
12. Enzinger FM. Angiomatoid Malignant fibrous histiocytoma: A distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer (1979) 44:2147–57. doi: 10.1002/1097-0142(197912)44:6<2147::aid-cncr2820440627>3.0.co;2-8
13. Choi JH, Ro JY. The0 WHO classification of tumors of soft tissue: selected changes and new entities. Adv Anat Pathol (2021) 28:44–58. doi: 10.1097/PAP.0000000000000284
14. Valente Aguiar P, Pinheiro J, Lima J, Vaz R, Linhares P. Myxoid mesenchymal intraventricular brain tumour with EWSR1-CREB1 gene fusion in an adult woman. Virchows Arch (2021) 478:1019–24. doi: 10.1007/s00428-020-02885-7
15. Ward B, Wang CP, Macaulay RJB, Liu JKC. Adult intracranial myxoid mesenchymal tumor with EWSR1-ATF1 gene fusion. World Neurosurg (2020) 143:91–6. doi: 10.1016/j.wneu.2020.07.057
16. Fanburg-Smith JC, Miettinen M. Angiomatoid “Malignant” Fibrous histiocytoma: A clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol (1999) 30:1336–43. doi: 10.1016/s0046-8177(99)90065-5
17. Dunham C, Hussong J, Seiff M, Pfeifer J, Perry A. Primary intracerebral angiomatoid fibrous histiocytoma: report of a case with a t(12;22)(Q13;Q12) causing type 1 fusion of the EWS and ATF-1 genes. Am J Surg Pathol (2008) 32:478–84. doi: 10.1097/PAS.0b013e3181453451
18. Ochalski PG, Edinger JT, Horowitz MB, Stetler WR, Murdoch GH, Kassam AB, et al. Intracranial angiomatoid fibrous histiocytoma presenting as recurrent multifocal intraparenchymal hemorrhage. J Neurosurg (2010) 112:978–82. doi: 10.3171/2009.8.JNS081518
19. Hansen JM, Larsen VA, Scheie D, Perry A, Skjøth-Rasmussen J. Primary intracranial angiomatoid fibrous histiocytoma presenting with anaemia and migraine-like headaches and aura as early clinical features. Cephalalgia (2015) 35:1334–6. doi: 10.1177/0333102415583988
20. Alshareef MA, Almadidy Z, Baker T, Perry A, Welsh CT, Vandergrift WA. Intracranial angiomatoid fibrous histiocytoma: case report and literature review. World Neurosurg (2016) 96:403–9. doi: 10.1016/j.wneu.2016.09.059
21. Konstantinidis A, Cheesman E, O’Sullivan J, Pavaine J, Avula S, Pizer B, et al. Intracranial angiomatoid fibrous histiocytoma with EWSR1-CREB family fusions: A report of 2 pediatric cases. World Neurosurg (2019) 126:113–9. doi: 10.1016/j.wneu.2019.02.107
22. Sciot R, Jacobs S, Calenbergh FV, Demaerel P, Wozniak A, Debiec-Rychter M. Primary myxoid mesenchymal tumour with intracranial location: report of a case with an EWSR1-ATan1 fusion. Histopathology (2018) 72:880–3. doi: 10.1111/his.13437
23. Gareton A, Pierron G, Mokhtari K, Tran S, Tauziède-Espariat A, Pallud J, et al. ESWR1-CREM fusion in an intracranial myxoid angiomatoid fibrous histiocytoma-like tumor: A case report and literature review. J Neuropathol Exp Neurol (2018) 77:537–41. doi: 10.1093/jnen/nly039
24. Spatz M, ES N, Lyons L, Greenberg S, Kallmes KM, Nussbaum LA. Primary intracranial angiomatoid fibrous histio- cytoma: A case report and literature review. Br J Neurosurg (2018). doi: 10.1080/02688697.2018.1451823
25. Gunness VR, Munoz I, González-López P, Alshafai N, Mikalkova A, Spears J. Intracranial angiomatoid fibrous histiocytoma with Hodgkin lymphoma. Med J Malaysia (2019) 74:234–6.
26. White MD, McDowell MM, Pearce TM, Bukowinski AJ, Greene S. Intracranial Myxoid mesenchymal tumor with rare EWSR1-CREM translocation. Pediatr Neurosurg (2019) 54:347–53. doi: 10.1159/000501695
27. Ballester LY, Meis JM, Lazar AJ, Prabhu SS, Hoang KB, Leeds NE, et al. Intracranial myxoid mesenchymal tumor with EWSR1-ATF1 Fusion. J Neuropathol Exp Neurol (2020) 79:347–51. doi: 10.1093/jnen/nlz140
28. Komatsu M, Yoshida A, Tanaka K, Matsuo K, Sasayama T, Kojita Y, et al. Intracranial myxoid mesenchymal tumor with EWSR1-CREB1 gene fusion: A case report and literature review. Brain Tumor Pathol (2020) 37:76–80. doi: 10.1007/s10014-020-00359-x
29. Pawel G, Ochalski MD, James T, Edinger MD, Michael B, Horowitz MD, et al. Intracranial angiomatoid fibrous histiocytoma presenting as recurrent multifocal intraparenchymal hemorrhage. J Neurosurg. doi: 10.3171/2009.8.JNS08151
30. Ying L-X, Teng X-D. Myxoid and reticular angiomatoid fibrous histiocytoma: A case confirmed by fluorescence in situ hybridization analysis for EWSR1 rearrangement. Int J Clin Exp Pathol (2018) 11:3186–90.
31. Boulagnon-Rombi C, Caroline Fichel BS, Sophie Lefour BS, Gauchotte G. Immunohistochemical approach to the differential diagnosis of meningiomas and their mimics. J Neuropathol Exp Neurol (2017) 76(4):289–98. doi: 10.1093/jnen/nlx008
32. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol (2016). doi: 10.1007/s10014-016-0271-7
33. Agnihotri S, Jalali S, Wilson MR, Danesh A, Li M, Klironomos G, et al. The genomic landscape of schwannoma. Nat Genet (2016). doi: 10.1038/ng.3688
34. Hilton DA, Hanemann CO. Schwannomas and their pathogenesis. Brain Pathol (2014). doi: 10.1111/bpa.12125
Keywords: intracranial mesenchymal tumor, FET::CREB gene fusion, molecular analysis, rare cancers, challenging diagnosis, immunotherapy, multimodal tailored therapy
Citation: D’Antonio F, Rossi S, Giovannoni I, Alaggio R, Carai A, Milano GM, Cacchione A, Cancellieri A, Gessi M, Antonelli M, Colafati GS, Megaro G, Vennarini S and Mastronuzzi A (2023) Case Report: Remarkable breakthrough: successful treatment of a rare intracranial mesenchymal, FET::CREB fusion-positive tumor treated with patient-tailored multimodal therapy. Front. Oncol. 13:1203994. doi: 10.3389/fonc.2023.1203994
Received: 11 April 2023; Accepted: 26 October 2023;
Published: 29 November 2023.
Edited by:
Atsushi Makimoto, Tokyo Metropolitan Children’s Medical Center, JapanReviewed by:
Kohei Fukuoka, Saitama Children’s Medical Center, JapanCopyright © 2023 D’Antonio, Rossi, Giovannoni, Alaggio, Carai, Milano, Cacchione, Cancellieri, Gessi, Antonelli, Colafati, Megaro, Vennarini and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Mastronuzzi, YW5nZWxhLm1hc3Ryb251enppQG9wYmcubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.