
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 10 July 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1199105
This article is part of the Research Topic Pharmaceutically Targeting Hypoxia in the Breast Cancer Microenvironment: Mechanistic and Translational Approaches View all 10 articles
 Nityanand Srivastava1*†
Nityanand Srivastava1*† Salman Sadullah Usmani2*†
Salman Sadullah Usmani2*† Rajasekaran Subbarayan3,4
Rajasekaran Subbarayan3,4 Rashmi Saini5
Rashmi Saini5 Pranav Kumar Pandey6
Pranav Kumar Pandey6Triple-negative breast cancer (TNBC) is one of the deadliest subtypes of breast cancer (BC) for its high aggressiveness, heterogeneity, and hypoxic nature. Based on biological and clinical observations the TNBC related mortality is very high worldwide. Emerging studies have clearly demonstrated that hypoxia regulates the critical metabolic, developmental, and survival pathways in TNBC, which include glycolysis and angiogenesis. Alterations to these pathways accelerate the cancer stem cells (CSCs) enrichment and immune escape, which further lead to tumor invasion, migration, and metastasis. Beside this, hypoxia also manipulates the epigenetic plasticity and DNA damage response (DDR) to syndicate TNBC survival and its progression. Hypoxia fundamentally creates the low oxygen condition responsible for the alteration in Hypoxia-Inducible Factor-1alpha (HIF-1α) signaling within the tumor microenvironment, allowing tumors to survive and making them resistant to various therapies. Therefore, there is an urgent need for society to establish target-based therapies that overcome the resistance and limitations of the current treatment plan for TNBC. In this review article, we have thoroughly discussed the plausible significance of HIF-1α as a target in various therapeutic regimens such as chemotherapy, radiotherapy, immunotherapy, anti-angiogenic therapy, adjuvant therapy photodynamic therapy, adoptive cell therapy, combination therapies, antibody drug conjugates and cancer vaccines. Further, we also reviewed here the intrinsic mechanism and existing issues in targeting HIF-1α while improvising the current therapeutic strategies. This review highlights and discusses the future perspectives and the major alternatives to overcome TNBC resistance by targeting hypoxia-induced signaling.
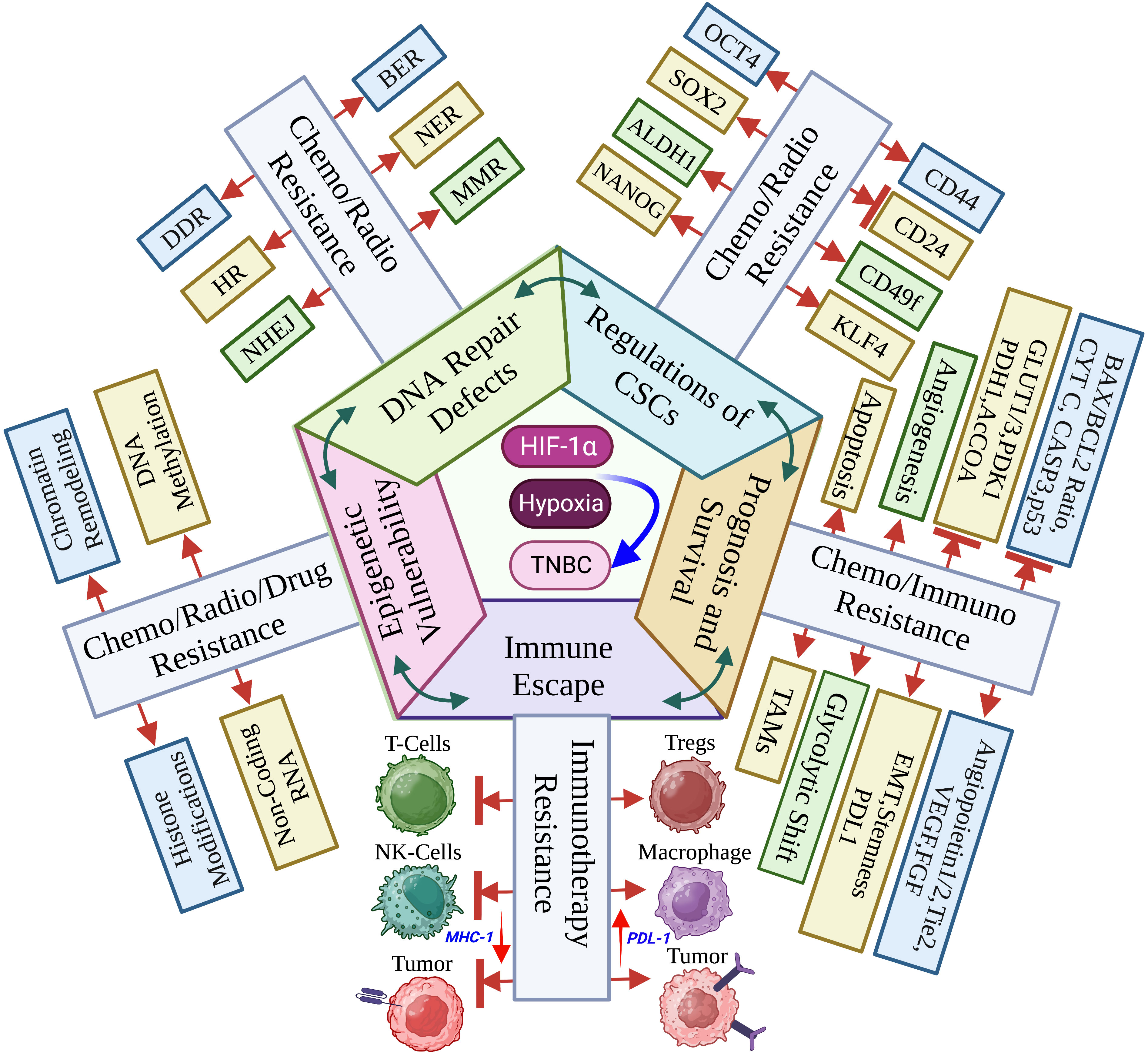
Graphical Abstract Hypoxia within the TNBC tumor microenvironment stabilizes the HIF-1α to decide the cancer cell fate, genetics, metabolism, immune response and clinicopathology. (Created via BioRender.com).
Breast cancer (BC) is the 2nd most common leading cause of cancer-related deaths, mostly diagnosed in young women. It accounts for over 43,000 estimated deaths annually among women in the US alone (1, 2). The stratification of breast carcinoma involves histological features, including the expression of markers such as estrogen receptor (ER) (3), progesterone receptor (PR) (4), and human epidermal growth factor receptor 2 (hEGFR2) (5). Furthermore, six intrinsic subtypes of TNBC, such as basal like, HER2 enriched, luminal A, luminal B, normal like and claudin low have been identified by high-throughput transcriptomic and genomic sequencing (6). These subtypes display distinct features in molecular portraits as well as clinical outcomes (5, 7, 8). TNBC is the small claudin-low subset of BC. It has a high histological grade, high epithelial-mesenchymal transition (EMT) marker enrichment, and high metastasis rates, including aggressive cancer stem cell-like features. In addition, they also have low luminal differentiation power and low expression of cell-cell adhesion molecules but are highly hypoxic in nature, making TNBC the most aggressive and deadliest subtype of BC (9–12). The term “negative” in TNBC refers to a very uncommon BC subtype that does not express ER, PR, and hEGFR2 (11, 13–16).
Epidemiological data analysis reveals that premenopausal women under the age of 40 are the primary suspects of TNBC occurrence, and approximately 20% of all BC patients are under the young age (13, 14). The TNBC patient’s survival time is comparably shorter than that of patients with another subtype of BC. The mortality rate of TNBC patients is also significantly high, and around 40% of deaths occur in TNBC patients within five years after the first diagnosis (13, 15). TNBC is a highly heterogenous subtype, and because of its aggressiveness and invasiveness, approximately 46% of TNBC patients have distant metastasis. Patients diagnosed with TNBC are more likely to develop distant metastasis within three years of diagnosis. Besides, the overall survival rate of metastatic patients is low, and based on the available data, the average median survival is only 13.3 months. The studies also demonstrate that the chances of tumor recurrence after surgery are as high as 25%. Brain and visceral organ metastasis also have been reported in metastatic TNBC patients. Most distant metastasis happen in the third year following diagnosis (16–18). TNBC patients have a shorter average time to relapse (19–40 months) than non-TNBC patients (35–67 months). According to published statistics, the death rate of TNBC patients after tumor recurrence is as high as 75% compared to non-TNBCs (16, 19).
Since heterogeneity, aggressiveness, and hypoxia create a favourable microenvironment for TNBC to grow and spread faster than other types of invasive BC, therefore, planning an effective treatment strategy for TNBC patients’ remains a herculean task. Although, in recent years, much research has been focused on identifying the specific targets of TNBC, the need for well-defined molecular targets in TNBC has resulted in limited therapeutic options. Currently, TNBC patients treatment mainly relies on standard therapies for TNBC, such as surgery, chemotherapy (CT), radiotherapy (RT), and photodynamic therapy (PDT) (20, 21). Therefore, identifying new therapeutic targets for TNBC is an urgent need and a high priority for society. Studies have investigated and identified several therapeutic molecules targeting oncogenic signaling pathways, including the PI3K/AKT/mTOR pathway and Src/Wnt signaling, to check their effectiveness in treating TNBC (22, 23). In addition, the alterations of BC genes 1 and 2 (BRCA1/2) and DNA damage-responsive (DDR) genes, including dysfunction of epigenetic and immune regulators, have also been used as an inhibitory index to predict treatment response in TNBCs (13, 24). Moreover, several studies also demonstrated a promising result by following a combination drug therapy strategy where they use targeted cancer drugs combined with chemotherapy or radiotherapy, and a few are in clinical trials (25, 26). Although the current treatment strategies are significantly effective, unfortunately, the overall treatment outcomes are highly variable, and it could be because of the highly heterogeneous nature of TNBC. Therefore, there is an urgent demand for alternative and accurate therapeutic strategies with improved efficiency, either alone or in combination with other therapies. HIF-1 is a key heterodimeric transcription factor of hypoxia. It consists of an oxygen-sensitive α subunit and a constitutively expressed β subunit. It is the master regulator to induce oncogenes and inactivate tumor suppressor genes functionality. It is widely regulated by inflammatory mediators released by tumor stromal cells TNBC that allow cellular adaptation against hypoxia (27, 28). Several studies established and proved that an intra-tumoral hypoxic environment creates a negative impact on the survival of BC patients and is associated with tumor aggressiveness and heterogenic phenotypes, which further induce a high risk of metastasis and provide a shielding barrier against various therapies such as chemotherapy, radiotherapy, and immunotherapy which suggest that hypoxia makes TNBC resistant to different treatments (28–31). Available evidence also supports the hypothesis that the elevation of HIF-1α expression in TNBC may provide a suitable environment for TNBC to grow in hypoxic conditions (32). Therefore, targeting hypoxic cancer cells seems to be a plausible idea for treating TNBC. Studies have also revealed that in TNBC, HIF-1α regulates the various complex biological processes and activates the transcription of several target genes involved in regulating angiogenesis, cellular metabolism, stem cell differentiation, and immune cell migration. The activation of these pathways further induces the expression of downstream gene products associated with stemness and EMT that have been further proven to be hyperactivated in TNBC by various research groups (33–35). Therefore, targeting HIF-1α could be a significant potential therapeutic option.
The pattern of HIF-1 expression in TNBC as well as the mechanism by which HIF-1 accelerates the disease are reviewed. This review also examines how breast cancer stem cell (BCSC) enrichment and immune evasion are affected by HIF-1 in the regulation of angiogenesis, invasion, and metastasis. It has also been investigated how HIF-1 affects TNBC through chemotherapy, immunotherapy, anti-angiogenic therapy, adjuvant therapy, PDT, adoptive cell therapy, antibody drug conjugates, cancer vaccines and also in combination therapies. The internal mechanisms as well as prospective therapeutic medicines that target HIF-1 are also reviewed.
The hypoxia-related mechanism is one of the distinguishing features of the cancer signaling system (34, 36). Each stage of the metastatic process is constrained by the hypoxic tumor microenvironment, which also regulates different cancer phenotypes (31, 37). Intratumorally, hypoxia is the major critical microenvironmental factor that is associated with TNBC and its invasiveness, metastasis and mortality (38). Additionally, disorganization of the tumor vasculature during tumor growth, is also associated with fluctuation of oxygen and glucose levels, leading to a heterogenous state of hypoxia, aerobic and anaerobic glycolysis (39, 40). Therefore, it is fundamental to correctly define the term ‘hypoxia’, presumed as an unusual system accompanying advanced malignancy by absolutely different mechanisms like chronic permanent inflammation or cell death pathways. Yet, the local hypoxia is deliberately produced by tumor cells to induce angiogenesis, hence directing the growth factors (30, 41). In chronic hypoxia, cells remain in a state above the diffusion limit of oxygen due to increased distance caused by tumor expansion (42, 43). This oxygen fluctuation within the tumor stipulates cancer cells for both aerobic and anaerobic glycolysis (44). Amplified glycolysis with or without oxygen is an important indicator for cancer and serves as a connecting link between TNBC and hypoxia (45, 46). Hypoxic microenvironment response in TNBC is tightly regulated by HIFs, which contain either HIF-1α or HIF-2α with a constituent expression of the HIF-1β subunit. Their elevation is associated with an increased risk of metastasis and mortality (34, 47). HIF-1α, HIF-2α, or both cause the activation of hypoxia-inducible genes, and their translational product is involved in several steps of the TNBC invasion and metastasis (48, 49). Under normoxic conditions, HIF-1α subunits are finally degraded by the proteosome, whereas hypoxia inhibits prolyl hydroxylases (PHD) and factor-inhibiting HIF-1 (FIH-1), key components required in the steps involved in the proteasomal degradation of HIF-1α, leading to HIF-1α stabilization and translocation to the nucleus, where they dimerize with HIF-1β and finally bind with hypoxia response elements within the promoters of target genes (50–52). In TNBCs, HIF target genes are highly expressed, whereas the expression of progesterone, estrogen and human epidermal growth factor receptors are deficient. Thus, TNBCs respond poorly to several current therapeutic regimens (53, 54). Giatromanolaki et al. claimed that the overexpression of HIF-1α is closely related to the immune response and adverse prognosis of BC and also inhibits the proliferation and survival of cytotoxic T cells and the expression of IL-2 and IFN-γ cytokines (55).
In TNBC, hypoxia plays a critical role in the prognosis and survival of cancer cells via the mediation of angiogenesis, glycolytic shift, apoptosis, and the recruitment of tumor-associated macrophages (TAMs). Hypoxic conditions upregulate the angiogenic growth factors and their receptors, leading to increased vascular permeability due to endothelial cell migration, and thus mediating TNBC angiogenesis. This phenomenon of angiogenesis is achieved via HIF pathways by regulating several pro-angiogenic genes such as angiopoietin-1 and 2, tunica intima endothelial kinase 2 (Tie2), vascular endothelial growth factor (VEGF) and their receptor, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), etc. (56). Besides, hypoxia causes the accumulation of TAMs, exhibiting the cancerous phenotype. TAMs also secrete angiogenic growth factors, leading to angiogenesis and prognosis (57, 58).
Hypoxia induces apoptosis by regulating several pro- and anti-apoptotic pathways either by HIF-dependent or independent mechanisms. Hypoxia reduces the bax/bcl-2 ratio as well as cytochrome c release and caspase-3 activity, thus inhibiting the pro-apoptosis pathways. In addition, hypoxic conditions favor the selection of p53 mutant cells having elevated levels of bcl-2, which is a well-known inhibitor of apoptosis, thus causing the decline in the p53 and bcl-2 ratio (p53/bcl-2), which further increases the mutation rates in clone populations (59, 60). This endless cycle promotes the prognosis of TNBCs (61).
Hypoxia modulates the expression of glucose transporters like GLUT-1 and GLUT-3 as well as glycolytic enzymes such as hexokinase 1, hexokinase 2, and phosphoglycerate kinase 1 (PDK1), causing a glycolytic shift from oxidative phosphorylation to glycolysis. HIF-1α plays a fundamental role in this metabolic adaptation (62). HIF-1α induces the expression of PDK1, which after phosphorylation inhibits pyruvate dehydrogenase (PDH), a key enzyme converting pyruvate to acetyl-CoA. In anaerobic glycolysis, this pyruvate is forced to be metabolized into lactate. Thus, hypoxic conditions reduce the amount of acetyl-CoA available to enter the Krebs cycle or TCA cycle, leading to a reduced amount of substrate availability for mitochondrial respiration as well as oxygen consumption (63–65). It is a well-known fact that cancer cells immediately use glycolysis, even when sufficient oxygen is available. This dependency on inefficient aerobic glycolysis is known as the Warburg effect, which promotes tumor prognosis and survival (Figure 1).
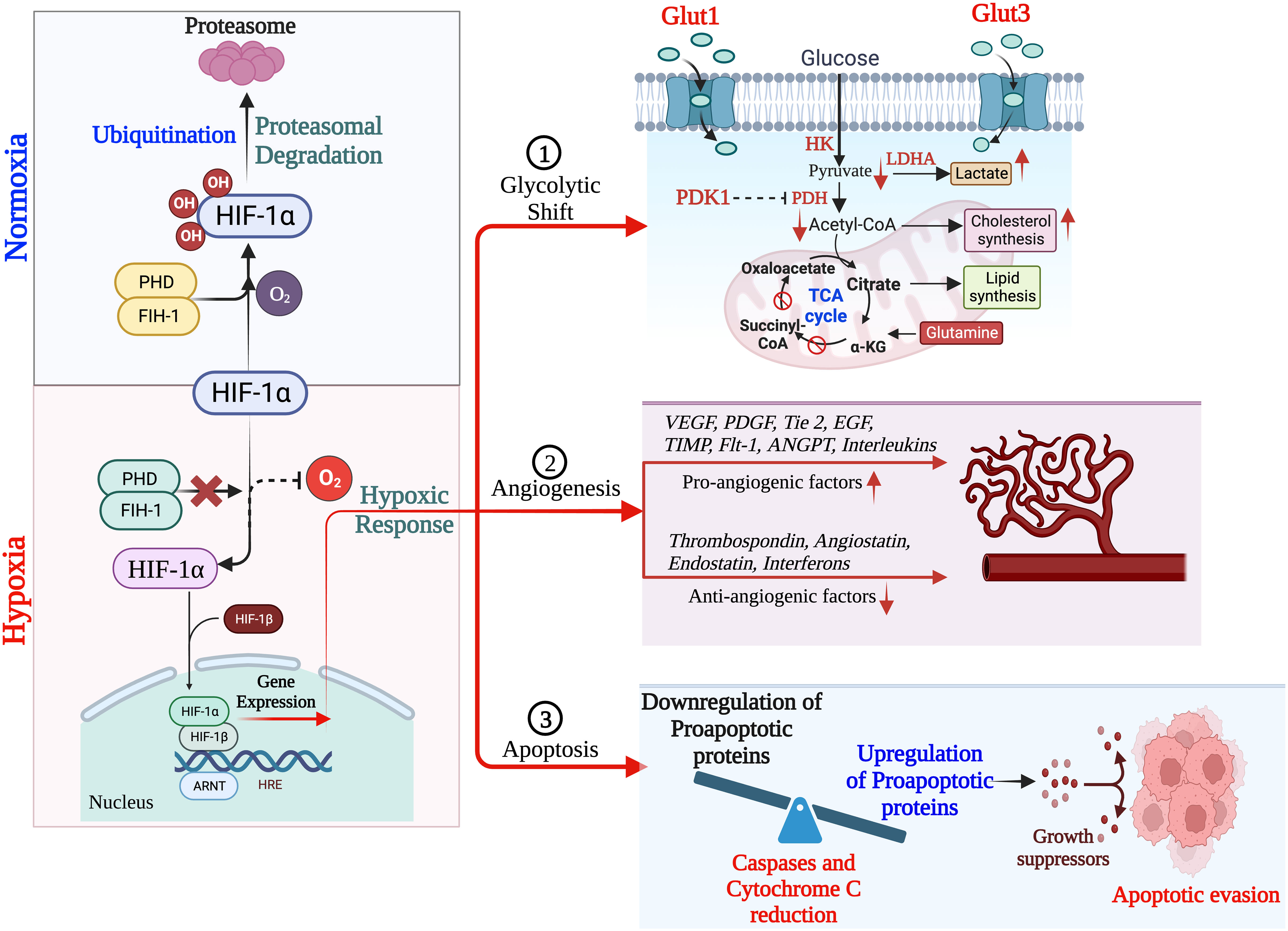
Figure 1 Schematic overview of HIF-1α regulation leading to hypoxia, resulting into adaptation of metabolic pathways, inducing angiogenesis and apoptosis. (Created via BioRender.com).
CSCs are a small heterogenous subset with self-renewal characteristics. These cells have a tremendous power to differentiate into all other specific cell types within the tumor tissues and can survive after therapy (66, 67). Specific biomarkers such as CD44high/CD24low, CD49f, and aldehyde dehydrogenase 1 (ALDH1) define the BCSCs in TNBC and are predominantly associated with a poor survival rate in TNBC patients (68, 69). Several analytical reports on human breast carcinomas have proved that CD44highCD24lowALDH1high CSCs predominantly associated with TNBCs and are significantly associated with tumor recurrence (69, 70). The hypoxia environment in TNBCs induces several stress-responsive genes which modulate the CSCs to activate their self-renewal and anti-apoptotic phenotype properties. These properties play a crucial role in tumor growth, immune evasion, metabolic reprogramming, drug resistance, and constraining clinical outcomes by modulating the transcription of several target genes (71). Hypoxia-activated pathways in the tumor microenvironment, such as HIF, CD133, CD24, CD47, DLK1, and mixed lineage leukemia 1 (MLL1), are the most essential contributing vital factors to CSC generation and maintenance (72–74). Increasing published evidences has supported and proved that HIF-1α is the central regulator of induction and maintenance of self-renewal and anti-apoptotic phenotypic properties of various CSCs such as octamer-binding transcription factor 4 (OCT4), SRY (sex determining region Y)-box 2 (SOX2), NANOG (encodes an NK2-family homebox transcripton factor), and Krüppel-like factor 4 (KLF4) (35, 75, 76). Published reports strongly suggests that HIF-1α directly binds to the promoter region of CD24 and induces CD24 overexpression which further accelerates tumor formation and metastasis (77, 78). Additionally, the direct binding of HIF-1α to CD47 and CD133 activate several gene transcription factors that inhibit the phagocytic activity of macrophages and promote the production of CD133+, respectively, which maintain the OCT4 and SOX2-mediated CSC pool of TNBC (79). However, there is controversial evidence in gastrointestinal cancer cells where hypoxia-induced HIF-1α expression decreases CD133 expression. Still, during normoxic states, inhibition of mTOR signaling in gastrointestinal cancer cells reduces the HIF-1α expression that overexpresses CD133 (79).
Moreover, increasing evidence also suggests that HIF-1 transactivates the RNA demethylase ALKBH5 to encode N6-methyladenosine demethylase and increases the stability of NANOG mRNA in BC (80, 81). Additionally, HIF-1 also induced A2BR and activates protein kinase C to transcribe IL-6, IL8, and NANOG, which further promotes stemness, as Lan et al. reported (82). HIF-1α also regulates the 4-trimethylaminobutyraldehyde dehydrogenase, which is associated with cancer cells metastasis, self-renewal, and resistance in BC (80, 83). In turn, HIF-1α expression is also regulated by aldehyde dehydrogenase 1A1(ALDH1A1) via retinoic acid signaling in TNBC (83). Studies also suggested that HIF-1α induces the JAK/STAT3 signaling pathway, which can upregulate IL-6 and NANOG while promoting the production of VEGF, responsible for the self-renewal ability and maintenance of the CSC phenotype (81, 84). Another study reported by Lee et al., showed that the production of reactive oxygen species (ROS) in TNBC via amplification of MYC and MCL1 overexpress the HIF-1α expression and promotes stemness and chemoresistance in TNBC (85). Crowder et al. discussed the correlation between the antioxidative enzyme superoxide dismutase 2 (SOD2) expression and the expansion of BCSCs in hypoxic conditions. They suggested that TNBCSCs might resist radiation via a SOD2-mediated mechanism (86). The tumor microenvironment pH is also associated with CSC’s survival in various cancer types, including TNBC. Interestingly, the pH of TME is tightly regulated by the hypoxia-inducible protein carbonic anhydrase IX (CAIX) by improving the acids transport within the tumor, further increasing the BCSCs survival, expansion and tumor invasiveness (87, 88). Published reports suggest that hypoxia upregulates the CAIX, further enhancing their downstream mTORC1 signaling pathway responsible for regulating triple negative breast cancer stem cells (TNBCSCs) stemness and EMT genes such as Snail and NOTCH (89).
HIF-1α also regulates the expression of ERs, which is a critical indicator of the hypoxic response of BCSCs. Harrison et al. have shown that higher expression of estrogen receptors (ER) is also regulated by HIF-1α and activates the hypoxic responsive factor for the maintenance and proliferation of BCSCs and stimulates the upregulation of Notch genes (90). An interesting study was conducted by Xing et al. group to analyze the expression pattern of Notch ligands in BC patients. They revealed that the expression of Notch and Jagged2 is significantly upregulated in the hypoxic breast tumors, which suggests that they might also regulate the TNBCs maintenance and proliferation and provide critical evidence that Notch and Jagged2 should act as a potential prognostic marker for future clinical applications (91).
Recent advances in BC research have also shown that hypoxia induces the involvement of microRNAs (miRNAs) in regulating the response of BCSCs (35, 92, 93). Hwang-Verslues et al. reported for the first time that miRNA-495 increases the colony-formation ability, invasive capacity and tumor formation capacity, which further regulates the tumor aggressiveness and hypoxic response of BCSCs (94). Several studies have shown that HIF-1α upregulation in TNBC controls cancer metastasis, CSC self-renewal, and invasion. As a result, the data imply that HIF-1α acts as a direct or indirect upstream regulator of BCSCs in TNBC under low oxygen conditions. This suggests that HIF-1α could be a new target for removing CSCs, which would enhance therapeutic approaches (Figure 2).
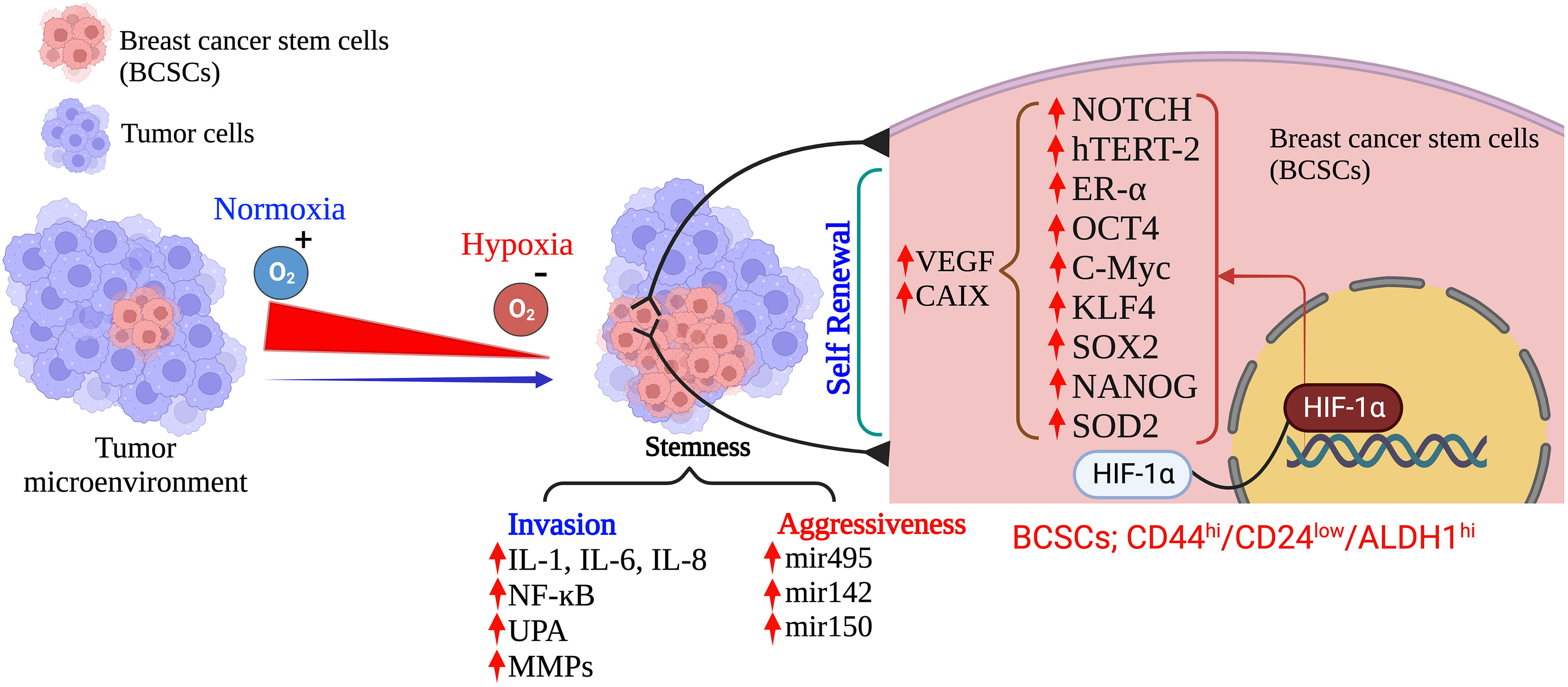
Figure 2 Hypoxia: Determinant of the fate of BCSCs in TNBC. (Created via BioRender.com).
Tumor immune escape allows tumor cells to survive and grow after evading the host immune system by several mechanisms. It has been reported that hypoxia may induce immunogenic cell death (ICD) within a tumor (95). Earlier investigations have shown that immune escape caused by hypoxia has poor prognostic results and have highlighted the hypoxia pathways as prospective therapeutic targets. Hypoxia signaling has intricate and contradictory functions in triggering immunological escape, encouraging tumor growth and the potential for metastasis, as well as boosting some immunogenic aspects of the tumor microenvironment. Under hypoxic conditions, tumor immune escape involves HIF-1α overexpression (96). HIF-1α inhibits immune cells tumor killing function, which is mediated by regulatory cytokines, granulocyte macrophage colony-stimulating factor (GM-CSF), colony stimulating factor-1 (CSF-1), transforming growth factor-β (TGF-β), CC-chemokine ligand 5 (CCL5), and VEGF in TNBC. In addition, the transcription factor Foxp3 and immune checkpoint molecules like PD-1 also participate in the tumor immune escape mechanism through activation and infiltration of immunosuppressive cells (97).
Myeloid-derived suppressor cells (MDSCs) in TNBC can control tumor-killing cells and immunosuppressive cells by secreting cytokines and suppressing anti-tumor immunity. HIF-1 drives immune evasion and encourages MDSC recruitment. There isn’t much research in TNBC on HIF-1’s modulation of myeloid suppressor cells. HIF-1 controls the communication between TNBC cells and MSCs, leading to the control of MDSCs recruitment. MSCs create CCL5 and bind to CCR5 on TNBC cells in a mouse model (98). The CSF1 receptor on MSCs is simultaneously bound by the cytokine CSF1 produced by basal cells (49). To encourage the recruitment of MDSCs, HIF-1 increases CSF1 and CCL5 signaling.
TNBC has higher levels of TAMs, which are strongly linked to a poor prognosis. By producing immunosuppressive molecules, including IL-10 and TGF-β, M2 macrophages have an immunosuppressive effect (99). HIF-1 drives the development of an immunosuppressive milieu and stimulates the polarization of TAMs towards the M2 phenotype (100). Granulocyte-monocyte stimulating factor plays a variety of activities in TAMs, according to prior research. A high concentration of GM-CSF has an immunosuppressive impact by enriching M2 macrophages, whereas a low concentration has an anti-tumor effect by stimulating dendritic cells (DCs) (101). High levels of GM-CSF are generated in TNBC cells under the control of HIF-1 and NF-κB, attracting additional macrophages, and polarizing them into M2-type macrophages (102). Macrophage CSF1, which is secreted by MDA-MB-231 TNBC cells and binds to its receptor on mesenchymal stem cells (MSCs), aids in the attraction of TAMs and MDSCs. Through preserving CCL5/chemokine receptor type 5 (CCR5) communication between MSCs and MDA-MB-231 TNBC cells, HIF-1 controls the expression of CSF-1 (52). As a result, HIF-1 can promote the polarisation of TAMs to the M2 type via regulating GM-CSF and CSF1 in TNBC. The T-regulatory (Tregs) cell transcription factor Foxp3 is essential. HIF-1 can regulate the aggregation of immunosuppressive Tregs in TNBC via regulating forkhead box P3 (FoxP3) and the C-X-C motif chemokine receptor 4 (CXCR4). Via co-regulatory proteins like co-stimulators, transcription factors, co-repressors, and chromatin remodelers, Foxp3 controls the restrictive activity of Tregs (103). CXCR4 is widely expressed on the Treg cell surface and regulates the recruitment of Tregs (104). In TNBC, HIF-1 directs downstream Foxp3 expression by binding to HREs while indirectly enhancing CXCR4 expression by acting on regulatory regions upstream of the CXCR4 transcription start site (105). In patients with TNBC, enrichment of CD8+T is directly linked with improved clinical prognosis and a higher immunological response because CD8+T cells are essential anti-tumor immune cells (106). The tumor-killing ability of HIF-1 in CD8+ T cells is controversial because HIF-1 overexpression in CD8+ T cells increases the level of infiltration and tumor-killing ability of CD8+ T cells (101). The dysfunction of CD8+ T cells was caused by HIF-1’s suppression of immunological effector gene expression under hypoxic settings through histone deacetylase (HDAC-1) and polycomb repressive complex 2 (PCR2)-mediated histone alterations (102). Moreover, under the control of HIF-1, tumor cells produce more adenosine in a hypoxic microenvironment. Adenosine inhibits T cell proliferation and toxicity, promotes T cell death, and inhibits anti-tumor immunological function via interacting with adenosine A2A receptors (103). In summary, HIF-1 inhibits CD8+ T cell activation and immune infiltration in TNBC while largely promoting immunological escape. By generating the overexpression of VEGF and PD-1 and encouraging the release of adenosine from tumor cells, HIF-1 regulates the epigenetic mechanism of immune effector genes. This reduces infiltration and impairs CD8+ T lymphocytes capacity to attack tumors (Figure 3).
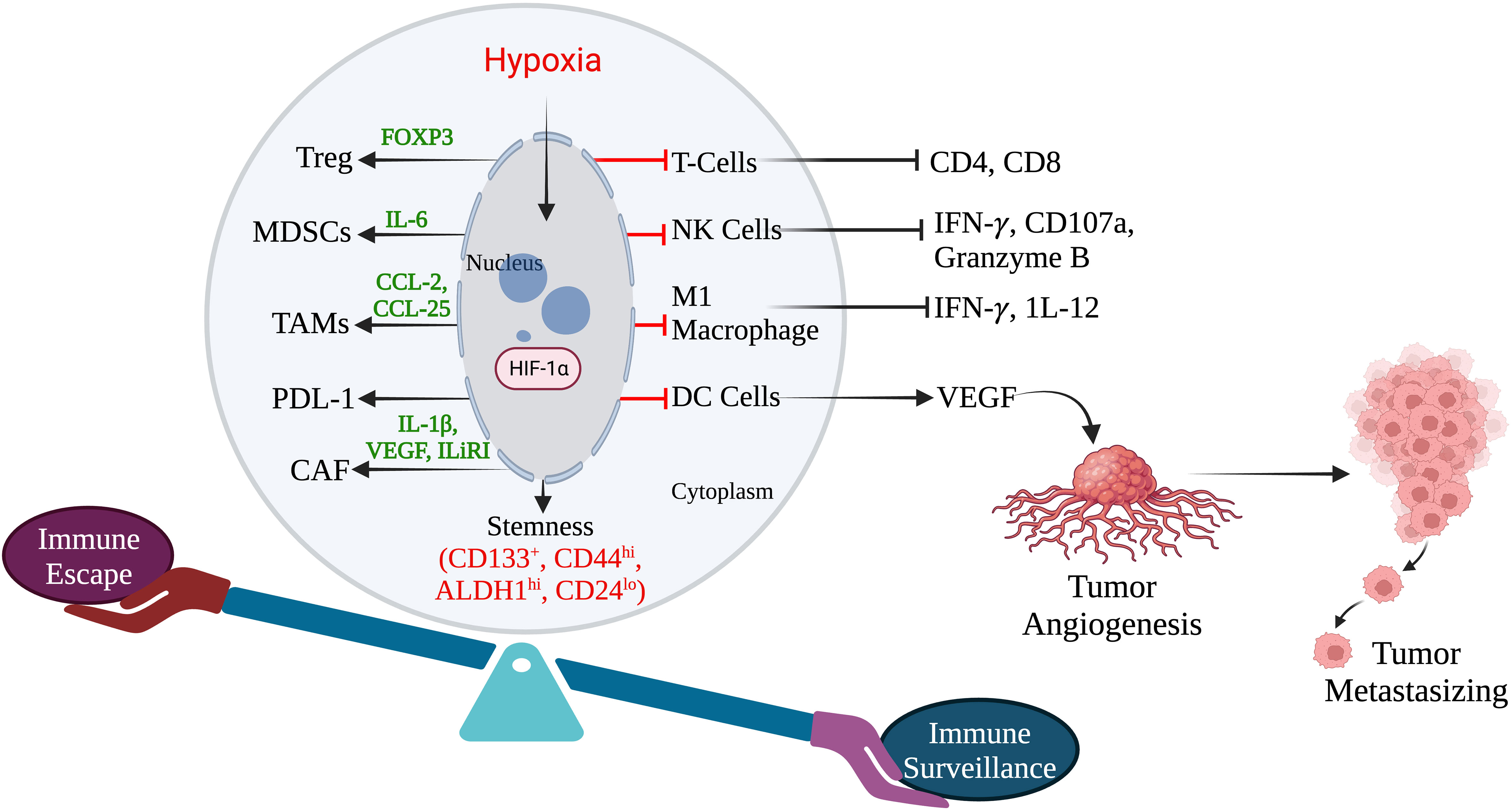
Figure 3 Hypoxia induces immune escape in TNBC. (Created via BioRender.com).
Accumulating evidence has also demonstrated that hypoxia-mediated increased expression of HIF is closely linked with manipulating epigenetic plasticity in the tumor cellular system and subsequently induces immune dysfunction (104, 105). Recently, transcriptional and epigenomic analyses by Cong et al. group have shown that Bromodomain-containing protein 4 (BRD4) is the epigenetic regulator. Its dysfunction is critical in mediating several transcription factors by HIF-1α in hypoxic conditions. The study also proved a close association of BRD4 dysfunction with the malignant progression of various tumors; thus, it could be a promising target for different cancer treatments (107). They have also shown that selective degradation of BRD4 subsequently down-regulates the expression of CAIX, a crucial hypoxia-mediated pH gene regulator. Further, it has been proved that overexpression of CAIX induces the acidic environment in various tumors, including TNBC, to adapt the hypoxic environment. Hypoxia-mediated CAIX overexpression also causes a marked reduction in VEGF levels, a master regulator of angiogenesis (107). Another study by Ma et al. demonstrated that hypoxia induces chromatin remodeling in TNBC by the interaction between HIF-1α with HDAC1 and the concurrent PRC2. This interaction epigenetically suppresses the effector gene’s function, which subsequently impacts immune homeostasis and disturbs immune tolerance (102). Chromatin immunoprecipitation (ChIP) assay data demonstrated that hypoxia induces the enrichment of HDAC1 and PRC2. However, it does not change in HDAC 2 and HDAC3 at the effector gene IFN- γ and TNF promoters in the T cells and NK cells (102). Some data suggest that enrichment of HDAC1 and PRC2 by HIF1α-mediated pathways suppresses the functionality of various effector immune cells. Therefore, targeting these pathways through either genetic depletion or therapeutic intervention may be a revolutionary strategy to overcome alteration in immune cell homeostasis. In addition, it might inhibit the disturbance of immune tolerance in TNBC and enhance checkpoint immunotherapy responses. Further, several studies have shown that bromodomain and extra-terminal domain (BET) protein reads histone-acetylation and recruits various transcription factors in TNBC to adapt to hypoxic conditions (108, 109). Ongoing investigations have also shown that hypoxia increases H4ac and H3K27ac, which are associated with transcription and BET protein binding in the HIF targets such as CA9, VEGFA, and LOX promoters (107, 110, 111). Luo et al. discussed and demonstrated selective and specific interactions of HIF-1α with histone demethylase jumonji domain-containing protein 2C but not with HIF-2α (112). By this selective interaction, HIF-1α recruits JMJD2C to the hypoxia response elements of HIF-1 target genes, further reducing the histone H3 trimethylation at lysine 9, and enhancing HIF-1 binding to hypoxia response elements. The enhancement of HIF-1 binding to hypoxia response elements activates the transcription of PDK1, L1CAM, GLUT1, LOX, LOXL2, and LDHA mRNA in human BC biopsies (112, 113). Lambert et al. suggested that lysine demethylases 4C (KDM4C) encoded by JMJD2C interact with HIF1α and are involved in metabolic remodeling and metastasis (114). Overall, based on emerging research, epigenetic plasticity plays a critical role in stimulating HIF-1, which further mediates the transactivation of genes that code for proteins implicated in immunological and metabolic tolerance reprogramming in TNBC.
Hypoxic tumor microenvironment down-regulates or deregulates the DNA repair pathways by inducing modifications in several transcriptional, translational, post-translational and epigenetic mechanisms (115). This down-regulation or deregulation causes defects in DNA repair pathways linked to extensive genomic rearrangements with a high rate of mutation burden, DNA hyper-replication stress, fragile site induction, and microsatellite instability (MSI). These DNA repair anomalies further lead to the tumor becoming more aggressive and are associated with a significantly worse prognosis/survival in TNBC (116). Therefore to protect the cell against hypoxia-induced replication stress and DNA damage, three primary DNA damage response (DDR) kinases, which include DNA-dependent protein kinase, ataxia telangiectasia and Rad3-related (ATR) protein, and ataxia-telangiectasia-mutated (ATM) kinases are responsible and becomes activated through post-translational modifications (117). Studies have demonstrated that genetic or chemical depletion of ATM or Chk2 in tumor cells has reduced clonogenic survival and increased apoptosis after exposure to hypoxia (118–120). Additionally, hypoxia also activates the ATR/Chk1 response, which subsequently causes pan-nuclear induction of phosphorylation of H2AX (γH2AX) and p53 (121, 122). Further, an emerging study has also demonstrated that depletion of ATR/Chk1 followed by exposure to hypoxia or re-oxygenation significantly reduces cell survival by increasing apoptosis. The study has also unraveled that DNA-PK is activated in hypoxic cells and phosphorylates at Ser2056 of the catalytic subunit that regulates HIF-1 expression (123). The study has demonstrated that ER, PR, and HER2 deficiency in TNBC leads to ATM response hyperactivates (124). Hyperactivation of ATM is predominantly associated with high invasiveness and metastasis of TNBCs by inducing the expression of EMT markers such as Snail and vimentin and reducing the expression of E-cadherin and cytokeratin, which exclusively characterized the epithelial cells (124). Another interesting study demonstrated that hypoxia induces the activation of oxidized ATM, which is independent of DNA damage-mediated ATM activation in TNBCs. Hypoxia-dependent activation of oxidized ATM accumulates the citrate in the cytoplasm. This extracellular accumulation of citrate stimulates the signaling pathway to activate the AKT/ERK/MMP2/9 crucial signaling axis for cell growth, survival, motility and metabolism in TNBC. These findings unravel that oxidized ATM is significantly responsible for TNBC hyperproliferation, invasion and metastasis (125). In addition, available reports have also shown a significant overexpression of ATR and CHK1 in TNBC tissues and promoted tumor progression (126). Meyer et al. demonstrated that an ATR/CHK1 mediated-DDR response prevents the replication stress and induces the resistance of homologous recombination-deficient (HRD) TNBC to mitomycin C (127). This study also suggests that ATR/Chk1 DDR might be a primary mechanism that induces chemoresistance in HR-deficient TNBC (127). Emerging studies have also observed aberrant genetic alteration in other DDR pathways, such as a high prevalence of p53 functional insufficiency and BRCA1/2 mutations (126, 128, 129).
Moreover, HRD is a crucial clinicopathological feature of the BRCA1/2 mutated TNBC, and several studies have proven that specific ATR inhibitors are highly efficient in sensitizing HR-proficient as well as HR-deficient TNBC cells against radiotherapy and inhibit the TNBC proliferation (130, 131). Rad51 is an essential protein for HR that triggers the initiation of the HR process at the sites of DNA damage to repair the damaged DNA. Studies have demonstrated that the knockdown of Rad51 or treatment with a Rad51 inhibitor can enhance the sensitivity to proton therapy and induce proton-mediated clonogenic cell death in TNBC cells (132). Several investigations also revealed that complement 1q binding protein (C1QBP) is highly expressed in hypoxic TNBC and promotes the progression of TNBC. In addition, hypoxia mediated high expression of C1QBP, stabilizes the MRE11 protein (DDR protein) in MRN complex (MRE11/RAD50/NBS1) and inhibiting MRE11 exonuclease activity which makes TNBC resistance to chemotherapy (133). Study has also demonstrated that blocking RAD50 within the MRN complex sensitizes CSCs and chemo-resistant BT-549 and MDA-MB-231 TNBC cell lines to chemotherapeutic drugs (3, 134). Recent studies have also shown that BRCA1 is critical in resolving DNA double-strand breaks (DSB) by HR repair, particularly DSB associated with cross-links at the end of DNA replication forks. Moreover, available reports suggest that mutation or deficiency of BRCA1 genes altered the HR-related gene, which further leads to HR deficiency in TNBC, makes TNBC more sensitive to the specific therapies that generate cross-links or DSBs fragments, including platinum drugs, alkylating agents, anthracyclines and PARP inhibitors (134, 135).
Non-homologous end joining (NHEJ) repair pathway is another crucial pathway that recognizes DSBs via Ku70/80 heterodimer in association with DNA-PKcs to repair damaged DNA in various cancer cell normal cells (136). Although it’s still debated, the exact effects of hypoxia on NHEJ still need to be explored. It is most likely that several hypoxia-mediated molecular mechanisms altered the crucial NHEJ protein expression and deregulated or downregulated functionality of NHEJ-associated proteins. Studies have demonstrated that long noncoding RNA (lncRNA) overexpressed in TNBC and hyperactivated the NHEJ pathway by supporting Ku80 and DNA-PKcs to repair of DSBs and promote tumorigenesis (137). Interestingly, the hypoxia-mediated amplification of EGFR activity and P53 mutation in TNBC is also responsible for the high expression of lncRNA in non-homologous end-joining pathway 1 (LINP1) in TNBC (137). Zhang et al. demonstrated by RNA-immunoprecipitation assays (RNA-IP) that the association between LINP1 and Ku80 or DNA-PKcs induced by ionizing radiation (IR) exposure to tumor cells. However, blocking LINP1 radio sensitizes the TNBC tumor for radiotherapy (137).
Hypoxia also alters the mismatch repair (MMR) pathway by mutating the major MMR proteins MLH1 and MSH2, including MSH6 (138, 139). Severe hypoxic conditions downregulate MLH1 and MSH2 in a HIF-independent manner. However, in moderate hypoxic conditions, MSH2 and MSH6 downregulate in HIF and P53-dependent manner (138, 140). Although it’s unclear, available reports suggest that hypoxia-mediated MMR downregulation also requires HDAC activity (141). These mutations or deregulation further led to microsatellite instability and are characterized by decreased or increased repeated nucleotide sequences (138–140).
Moreover, a high incidence of MSI observes in various tumor models, such as colorectal, ovarian, stomach, urothelial, central nervous system, and adrenal gland. The high incidence of MSI is further responsible for developing several malignant mutations and tumor evasion. However, based in published reports so far, only limited data are available on disease prevalence, and the prognostic significance of MMR-d/MSI-H in BC. Additionally, in TNBC, a low incidence rate of MMR-D/MSI-H is observed. Recent clinicopathological studies conducted in 440 patients with TNBC demonstrated no correlation between MMR-d/MSI-H and clinicopathological parameters such as PD1/PDL-l immune checkpoint expression and survival. Another study also revealed that low expression MMR-D/MSI-H characteristics in TNBC may not be a practical predictive marker for immunotherapy by using immune checkpoint inhibitors of PD1/PDL-l (139).
Emerging studies also demonstrated the hypoxia mediated reduction of several base excision repair (BER) factors expression such as OGG1, MYH, POLB, APE1, RPA, PCNA and ASCIZ/ATMIN in TNBC which leads the TNBC BER-deficient (BER-d). Moreover, induction of oxidative or alkylating glycosylation via low protein production in TNBC is significantly associated with oxidative or alkylating glycosylases through low protein production (142, 143). The available report suggests that a combination of PARP inhibitors that directly impact BER signaling and other relevant therapies that generate ROS and induce defective glycosylation would be more precise and targeted therapies for TNBC treatment (142, 144).
The effects of hypoxia on nucleotide excision repair (NER) remain debatable. Several NER gene expressions, such as XPA, XPB, XPD and XPG, do not change after hypoxia exposure (115). However, some evidence indicates that severe hypoxia suppresses the NER capacity and demonstrates hypermutability to UV irradiation (145). Another study conducted in HCC1806 and MDA-468 TNBC cells showed the inactivation of major NER proteins such as ERCC1, XPA, and XPF (146). These proteins play an essential role in recognizing and excision bulky base lesions. The inactivation of scaffold protein, XPA, induces severe sensitivity to UV radiation and a high risk of carcinogenesis (147). These studies suggest that the induction of deregulation of NER in TNBC could offer revolutionary new targets for the treatment of TNBC either alone or in combination with other therapies. In summary, understanding of DNA repair defects in TNBC can potentially be used to overcome resistance to treatment.
TNBC harbors a highly heterogenous and aggressive behavior with a distinct metastatic pattern. It also contains a high mutational burden and activates various tumor initiation signaling pathways. TNBC is the highest malignant BC subtype with a poor clinical outcome. Therefore, the current treatment options are limited to surgery, chemotherapy, and radiotherapy. The limited clinical ramifications of TNBC have lagged other types of BC. Due to its hyper-progression and aggressiveness, it remains challenging BC to treat and minimal options are available for treating this form of BC. Many efficacious treatments for most BC are limited to inhibiting the growth-stimulating effects of PR, ER, and HER2. Finding novel and potent therapies for TNBC remains a crucial clinical need because it lacks these growth-stimulating receptors. To develop specific efficacious drugs, new experimental approaches need to be adequately investigated in pre-clinical and followed by clinical trials platform on patients diagnosed with TNBC. Figure 4 summarizes the tumor hypoxia mechanism and current treatment approaches for TNBC.
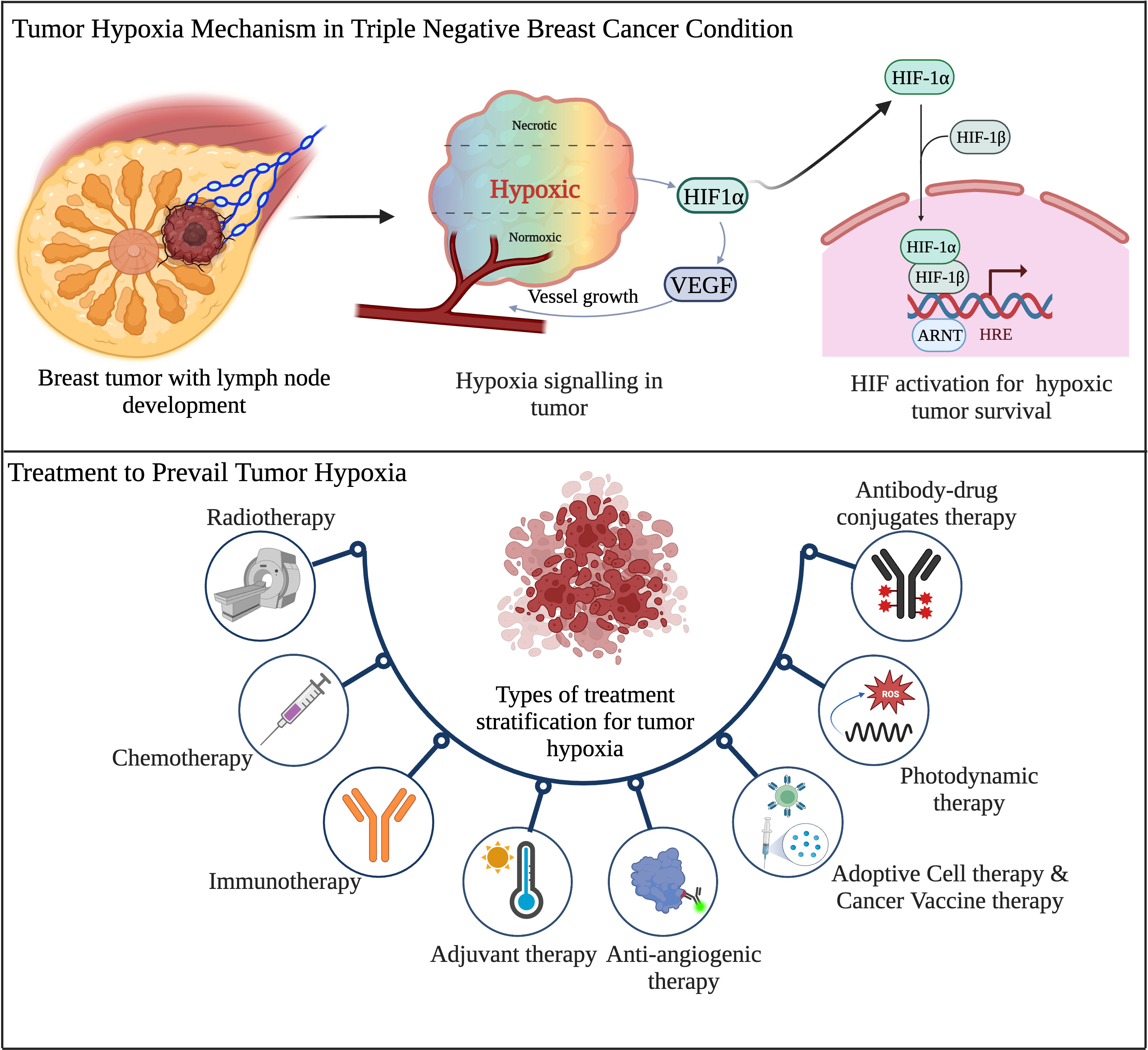
Figure 4 Schematic overview of tumor hypoxia mechanism and existing therapeutic regimens in TNBC. (Created via BioRender.com).
In 1920, Hall et al. showed that the tissue inadequately supplied with oxygen was highly resistant to ionizing radiation (IR) (148, 149). In 1953, Gray’s seminal paper demonstrated that the preclinical study reported that decreasing hypoxia enhanced oxygen distribution and increased radio sensitivity. However, these findings facilitate the way for several studies summarizing the mechanism of radio resistance induced by hypoxia. On the whole, HIF-1 and radiotherapy have a complicated, two-sided relationship.
Following radiotherapy, which induces hypoxia-dependent HIF-1 expression in the tumor, causes vasculature damage and a consequent oxygen level deficit. Earlier, Moller et al. demonstrated that following IR, the murine tumor model showed increased HIF-1 expression. As a result, the irradiated mouse tumors gave the impression of radiation-induced reoxygenation and displayed relatively high oxygenation levels in the tissue (150). Authors found two unexpected results were found in the same study: (a) ROS activation led to HIF-1 stabilization, and (b) the increased translation of HIF-1 transcripts owing to reoxygenation caused the breakdown of monomers referred to as “stress granules". Previous studies endorsed ROS-directed HIF-1 stabilization, most likely by decreasing PHD enzyme activity (151, 152). However, another hypothesis emerged as P13/AKT/mTOR pathways, which result in increased HIF-1α expression, and Ras/Raf/ERK/MEK pathways, which always attribute ROS-mediated regulation of HIF-1 expression.
Additionally, the stabilization of the HIF-1 protein during IR is linked to heat shock protein 90 (Hsp90) (153, 154). In response to IR, endothelial cells underwent a stress reaction that resulted in either recovery or function loss and cell death. The critical destiny is determined by a number of variables, such as intrinsic TME characteristics, fractionation schedule, and total dose (155). VEGF is at the centre of this interaction as an HIF-1 target gene and a crucial regulator of tumor vascularization (156). Radiation-induced fibrosis, or RIF, is a dose-limiting postradiotherapy consequence. In reaction to radiation exposure, the body undergoes an aberrant wound-healing process (RIF), which leads to a self-replicating fibroproliferative condition (157). IR-induced DNA damage triggers an initial inflammatory response, which is followed by endothelial cell failure and hypoxia. This causes aberrant collagen and other extracellular matrix protein buildup as well as an atypical activation of fibroblasts (sometimes referred to as the activated state of myofibroblasts). TGF- is an important participant in this process, and one of the critical RIF-initiating events is ROS-mediated post-translational activation of TGF- β (158).
One of the most notable instances of how HIF-1-mediated metabolic reprogramming might directly counteract radio-resistance in relation to radiation is glycolysis-induced activation of the pentose phosphate pathway (PPP). PPP activation decreases oxidized glutathione, restores nicotinamide adenine dinucleotide phosphate (NADPH), and shields cancer cells from ROS (159). In a recent clinical trial looking at first-time tumor hypoxia in SBRT, Song et al. first showed a clinical study investigating tumor hypoxia in SBRT with high doses of single fraction radiation delivered to patients with lung cancer (160). Overall, there is still much to learn about the importance of tumor hypoxia in the era of hypofractionation therapy. Stereotactic body radiation (SBRT) replaced traditional fractionated radiotherapy techniques, and this change sparked a new quest for hypoxia-modifying radiotherapy drugs. Image-guided radiation, intensity-modulated radiotherapy, volumetric modulated arc treatment, targeted combinatorial drug therapies, and immunotherapy are examples of contemporary innovations in radiotherapy delivery and imaging approaches that have improved radiotherapy’s therapeutic index (161, 162).
The conventional radiotherapy involves the usage of either external beam, like that of a regular x-ray or more feasible internal radiation known as brachytherapy, in which a sealed radiation source is inserted to target the cancerous area. However, Smith et al. demonstrated that a high increased risk of subsequent mastectomy after brachytherapy treatment compared with external beam therapy (163). Proton beam therapy (PBT) has a lower administrative dose than conventional radiotherapy and allows the majority of the radiation dose to be explicitly focused on the tumor, is also more frequently used. This can reduce the needless irradiation of nearby normal tissues, which will lessen the likelihood of side effects. Emerging studies demonstrated a dosimetric comparison between brachytherapy and PBT and intensity modulated proton radiotherapy technique (IMPT) and suggested a comparability similar dose effect in BC patients and also suggested to use of PBT for significant outcome (164) (Table 1). Modern developments in clinical radiotherapy technology are aimed at enhancing the capabilities of the radiotherapy machines and altering the local mode of radiotherapy to maximize the accuracy of irradiating tumor tissue while minimizing damage to healthy tissue.
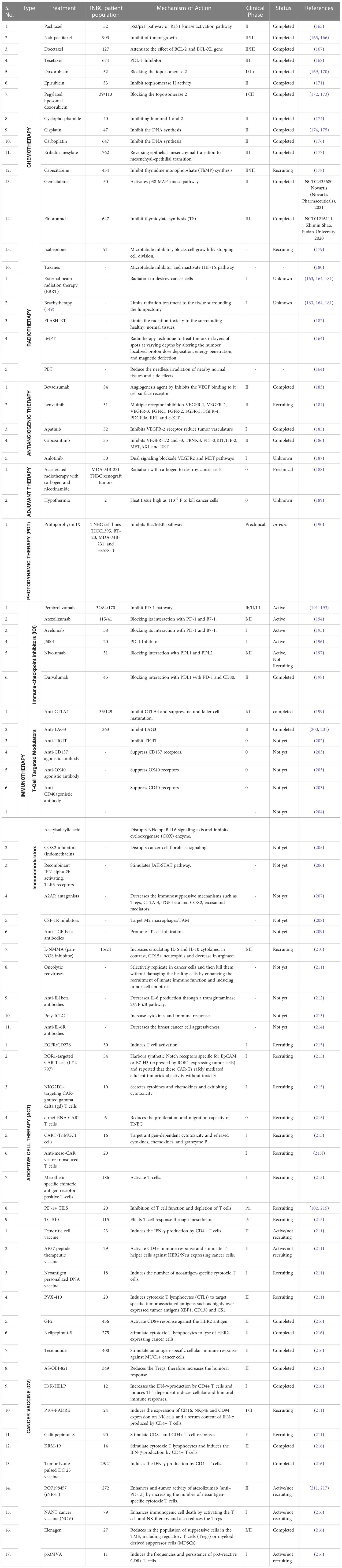
Table 1 Clinical trials evaluating different therapeutic approaches in patients with TNBC.
Ultra-high dose rate (UHDR) radiotherapy called FLASH radiotherapy (FLASH-RT) has been expected as a new method in recent years (Table 1). In multiple trials, radiation toxicity to the surrounding healthy, normal tissues was markedly decreased, and tumor growth was suppressed, with tumor control on par with conventional dose rate irradiation. It is generally acknowledged that FLASH irradiation has great future potential and is perhaps the most significant discovery in the history of radiation treatment, despite some researchers’ skepticism over FLASH-effectiveness RT’s in treating cancer patients (182).
Chemotherapy aims to weaken the cancer cell defenses against apoptosis, mitotic catastrophe, autophagy, and necrosis in order to promote cancer cell death. Apoptosis and autophagy, to start, are genetically programmed. Second, passive reactions to extreme cellular mistreatment include necrosis and mitotic catastrophe (218). The majority of chemotherapy medicines cause DNA damage. Apoptosis is the main method of cell death in reaction to DNA damage, even if medications that cause DNA damage can also cause necrosis as an alkylating agent or autophagy as an etoposide (219). It is well known that hypoxia lowers the effectiveness of chemotherapy treatments since these drugs need oxygen to act as an electron acceptor in order to kill cells.
Hypoxic tumor cells are discussed in this review as a way to avoid chemotherapy (220). Hypoxia drastically altered the transcription of cells, principally by activating HIF-1. HIF-1 is made up of two subunits: HIF-1/ARNT, which is constitutively stable and HIF-1, which is oxygen-sensitive. Low oxygen levels enable the development of active HIF-1 by preventing post-translational changes of the HIF-1 subunit. Through controlling expression, angiogenesis, autocrine growth factor signaling, invasion, and treatment failure often due to the presence of ABC transporters, HIF-1 contributes to metabolic reprogramming (221, 222). In addition, the HIF-1 subunit helps control p53. Its main function is to control the expression of numerous genes that code for proteins, which helps to control apoptosis. The preservation of genomic integrity depends heavily on the transcription factor p53. An et al. were the first to discover that HIF-1 is essential to the p53 pathway. The stability of the p53 protein under extreme hypoxia conditions was determined by the authors in previous study (223). In addition, they noted that the phosphorylation of HIF-1 has a dual role function in controlling apoptosis. According to Suzuki et al., the dephosphorylated form of HIF-1, which predominates in severe hypoxia and has a greater affinity for ARNT than the phosphorylated version of HIF-1, is necessary for p53 stabilization through HIF-1. On the other hand, research by Pan et al. and others have demonstrated that even intense hypoxia is insufficient to stabilize the p53 gene without the secondary holding provided by rigorous hypoxia, such as food shortage and pH collapse (224, 225). For many years, the only way to completely eradicate tumor cells and prevent their development and proliferation by chemical agents used in cancer therapy was through chemotherapy. Chemotherapy’s main strength and most compelling flaw is its inability to distinguish between cancer cells and healthy cells, which results in severe toxicity and side effects. Cancer treatment has changed significantly during the past 20 years from broad-spectrum cytotoxic medications to tailored treatments (226). Targeted medications now have a higher potency and lower toxicity as compared to traditional chemotherapeutic drugs since they can directly target cancer cells while protecting healthy cells. Targeted medications can be broadly categorised into two groups: (a) small compounds like imatinib, which the US Food and Drug Administration (FDA) licenced for clinical use in 2001 (227); and (b) macromolecules such monoclonal antibodies, polypeptides, nucleic acids, and antibody-drug conjugates (228, 229). It is a well-known medication that will be easily developed and has entered a golden stage of development, which has been supported by the approval of targeted drugs.
Over the last 20 years, there has been a significant increase in targeted FDA-approved medicines for cancer treatment. Small-molecule targeted drugs, on the other hand, have several advantages over macromolecule targeted drugs in terms of cost, pharmacokinetic properties (PK), patient compliance, drug storage, and market availability. In the United States and China, 89 small anticancer molecules have been approved. Small molecules for anticancer drugs face numerous challenges, including drug resistance and a low response rate. Many strategies for administering chemotherapeutic medications can now be used to extend life. Chemotherapeutic drugs are commonly administered in a combinational approach. For various types of cancer, various combinations are available. In this review, we will focus on a few drugs that are commonly used to treat various cancer types, such as doxorubicin, cyclophosphamide, cisplatin, 5-fluorouracil, and others (Table 1). In general, chemotherapeutic agents are administered to patients who may be able to withstand the treatment. Because less cell death is observed in tumor masses, current clinical setup chemotherapeutic regimens are used to treat cyclic tumors. As a result, dose reputation is required to reduce tumor size. There are a few drawbacks to the duration and frequency of chemotherapies, which are limited by patient toxicity (230).
Hypoxia refers to solid tumors and attributes the selection of intrusive and destructive malignant clones displaying resistance to RT, traditional chemotherapy, or small molecule targeted therapy. The recent clinically applicable immunotherapy-based checkpoint inhibitors (ICPIs) and chimeric antigen receptor (CAR) T- cells, has evidently altered the prognosis for certain tumors (231). Notably, hypoxia triggers the angiogenesis and causes immunosuppression, which is termed another dilemma of hypoxia-induced immune resistance. While these treatment strategies reveal both a promise and a despair in terms of efficacy and safety in phases of clinical trials, they correspond to the future solution to appreciate the efficacy of immunotherapy in contrast to hypoxic and therapy-resistant solid tumors.
However, based on the prediction, tumor hypoxia has shown poor outcomes across all types of cancer. Despite the success of T-cell immune checkpoint blockade in treating melanoma, abrasive adenocarcinomas of the prostate and pancreas are mostly resistant to CTLA-4 and PD-1 antibody treatment in the mice and humans. Previously, Midan et al. reported that hypoxic zones of the tumors endure infiltration by T cells, even in the context of vigorous infiltration of T cells in normoxic regions of the same tumor (4). Beyond the dearth of admissibility to tumor-specific T-cells, hypoxia energizes the foundation of an extremely interdependent network of immunosuppressive stromal cells. Based on the Midan et al. finding it was noted that the critical population of myeloid-derived suppressor cells (MDSCs) and myofibroblasts which act together to suppress T-cell responses and intervene in immunotherapy resistance (4).
Tumor hypoxia primarily affects antitumoral immune activity by inhibiting the native immune system and immune killing mechanisms. Many studies have concentrated on immunosuppressive elements in the tumor microenvironment, including MDSCs, Treg cells, and TAMs in the hypoxic zone of solid tumors (232). Inside the hypoxic TME, HIF-1, a key hypoxia transcriptional factor, controls MDSC activity and differentiation. According to research by Norman et al. on this subject, enhanced HIF-1-dependent arginase activity and nitric oxide production in tumor-dependent MDSCs make them more immunosuppressive than splenic-derived MDSCs (233). Another study discovered evidence that HIF-1 controls PD-L1 expression by directly attaching to components that have hypoxia-responsive properties in the proximal promoter of PD-L1 (234). Atezolizumab is the first FDA approved ICI monoclonal antibody for the treatment of mTNBC which targets PDL-1 and later pembrolizumab is also approved for mTNBC treatment in combination with chemotherapy based on the positive clinical trials result with atezolizumab and pembrolizumab monotherapy in TNBC (215). There are several clinical trial studies are registered on clinicaltrial.gov which implies that using ICI either alone or in combination with other therapy could be a promising strategy in TNBC treatment which is summarizes in Table 1. In addition to ICI several other combinations with immunotherapies such as immunomodulators (acetylsalicylic acid, indomethacin, IFN-α2b etc.), T-cell targeted modulators (CART-TnMUC1, TC-510 etc.) are still under investigation and these may contribute to the development of precision immunotherapy for TNBC (215) (Table 1).
The main challenge in overcoming tumor hypoxia in a clinical setting is to increase oxygen delivery. Horsman et al. previously showed that hyperbaric oxygen (HBO) treatment involves breathing 100% oxygen 2-4 times daily at normal atmospheric pressure. The results showed increased saturation of hemoglobin and oxygen levels in the circulation (235–237). In general, HBO treatment is administered during or shortly before radiation therapy. Previously, in the 1970s, Chaplin et al. reported that, when compared to normal air, patients with head and neck squamous cell carcinoma responded better to HBO treatment in terms of local control in a multi-center randomized trial (238).
On the other hand, carbogen breathing produced disparate results, which might be explained by variations in the number of patients who underwent carbogen breathing (239). Although the treatment for high-risk brain stem glioma in pediatric patients was well tolerated, there was no evidence of any benefit when radiation therapy was added. Siemann et al. previously reported that the combination of nicotinamide, a vitamin B3-derived molecule, and radiation appears to target both acute and chronic hypoxia (240). Additional research suggests that nicotinamide reduces ACT hypoxia by sporadically preventing vascular shut-down. Van Laarhoven et al. conducted ARCON trials, and other groups demonstrated improved patient survival, particularly in bladder and laryngeal cancer (241–243). However, the ARCON trials have demonstrated the efficacy of carbogen breathing as an adjuvant therapeutic regimen (244).
Interestingly, one of the adjuvant treatments called hyperthermia (HT) involves heating tissue over physiological temperatures (40–450°C). Although HT can be given directly to tumor masses, it is typically utilized as an adjuvant therapy alongside chemotherapy or radiation therapy due to technical challenges in obtaining cytotoxic temperatures (153, 154). However, in patients, ultrasonography is used to administer HT superficially or intravenously. Microwaves, radio frequencies, and electromagnetic radiation are all examples of electromagnetic radiation. In the 1970s and 80s, HT’s positive impacts were seen in primary and secondary cells in the culture system, as well as in the preclinical evaluation of animal models and patients. Previously, five randomized trials combining radiation and HT exhibited benefits in recurrent melanoma, cervical and BC patients (155–157). HT has also been shown in studies to be beneficial in adolescent and pediatrics patients with various types of tumors, including soft tissue sarcoma, malignant germ cell tumors, and chondrosarcomas.
The impact of HT in DDR is another alternative to improve the current radiotherapy strategy, and it can significantly radiosensitize the tumor cells. According to earlier studies, HT stimulates the ATM and γ-H2AX pathways and increases the expression of p53 (158, 159). As a result, HT induction is an important pathway in DDR and plays an early role in DDR responses. Another intriguing study found that HT has a direct beneficial effect in combination with DDR-targeted therapy by inhibiting the homologous recombinant repair pathway. In addition, it also deactivates the NHEJ pathway by suppressing the interaction between Ku80 and BRCA2 at DSB damage sites (160). A clinical trial is underway with HT and Olaparib combination therapy for BC patients (NCT03955640). The hypothesis is that HT could modify the immune system via systemic treatments, promoting the expansion of the MHC class I. Published reports demonstrated that HT induces the significant infiltration of cytotoxic T, B, and NK cells (161, 162, 245). The preceding studies demonstrated that combining HT with immuno- and radiation therapy may improve treatment efficacy. Mild HT is one of the potential adjuvant treatments to overcome tumor hypoxia. To gain a more precise understanding, we must investigate the molecular-level relationship between HT and tumor oxygenation in depth. Furthermore, to identify therapeutic targets and understand the underlying mechanisms (HIF pathways, ROS, heat shock proteins, and EMT) of heat resistance pathways that could be used as therapeutic targets in cancer patients (218–220).
As already discussed in Section 2.1, the role of angiogenesis in cancer survival and progression, we can estimate that targeting the angiogenesis could be a possible approach to combat TNBC. While reviewing the literature, we found that several angiogenesis inhibitors are clinically available against different types of advanced solid cancers. These inhibitors are generally either monoclonal antibodies or small molecule-based tyrosine kinase inhibitor, which target the VEGF and receptors. Angiogenesis inhibitors act by blocking the activity and expression of pro-angiogenic factors, secreted by tumor cells by targeting their receptors. Consequently, these inhibitors reduce the amount of nutrients available for tumor growth, and promote tumor vasculature normalization, and increase the delivery of cytotoxic chemotherapy (246–248). Unfortunately, these angiogenesis inhibitors failed to respond against BC when comparing the patient’s survival outcome to that of other solid tumors. Although research is ongoing, several clinical trials are underway to explore the angiogenesis inhibitors clinical outcomes in BC and TNBC patients.
In clinical trials, in the subgroup analysis, TNBC patients had shown a significant improvement in overall response rate in the E2100 and Avado trials; however, no statistical differences were observed in the Ribbon 1 trial, in which bevacizumab, humanized anti-VEGF monoclonal antibody, was given in combination with either second-line treatment by using chemotherapy or bevacizumab plus paclitaxel for first-line treatment or bevacizumab is added to neoadjuvant chemotherapy (249–251). In these three trials, 684 patients with TNBC were enrolled, and a meta-analysis was performed. In these three trials, 684 patients with TNBC were enrolled, and a meta-analysis was performed. This exciting study has clearly shown a marginal increase in progression-free survival. The overall objective response rate was also statistically increased, and there was a trend towards improved overall survival (226). Similarly, several other monoclonal antibodies, such as ramucirumab, have also been studied in several clinical trials, but no improvement in the overall survival of patients has been observed (227).
Besides, few small molecule-based tyrosine kinase inhibitors were studied in several clinical trials. There are several inhibitors such as bevacizumab, lenvatinib, apatinib, cabozantinib have been shown a positive clinical response in BC patients including TNBCs which are summarized in Table 1. In addition, a few other examples include, sorafenib, vandetanib, sunitinib, axitinib, pazopanib and cediranib, which are approved in several other cancers, like advanced renal cell carcinoma, hepatocellular carcinoma, soft-tissue sarcoma, gastrointestinal stromal tumors, advanced pancreatic neuroendocrine tumors, medullary thyroid carcinoma etc. Till now, these inhibitors were studied either as alone or in combination of first- and second-line treatment in various studies, but no significant improvement in overall survival in BC patients has been observed. All these inhibitors generally target the classical angiogenic pathway by targeting VEGF and VEGFR, and gave suboptimal results (228, 229). Thus, in our view, there is a need to explore novel anti-angiogenic approaches, such as targeting pericytes for vascular normalization, miRNA utilization and usage of immunotherapeutic drugs.
Another emerging and constantly developing method to treat cancer is photodynamic therapy (PDT), which involves using low to medium-energy monochromatic light to photo-excite subsequently applied photosensitizers (PS) interacting with the oxygen and producing ROS. The interaction between light and tissue is via absorption, scattering, reflection and refraction. Tissue’s optical properties determine the distribution of treatment light, as most of the light is transmitted at near-infrared wavelengths. PDT uses light with a wavelength of 600-800 nm, and it is a well-known fact that light with longer wavelengths has been absorbed to a greater extent; therefore, one of the limitations of PDT is its therapeutic depth, which is less than a centimeter (252–254).
At its early stage, PDT is well established and accepted in dermatology such as non-melanoma skin cancers, pre-malignant conditions like actinic keratosis and Bowen’s disease (255). Besides, it is also accepted in non-dermatologic condition like head and neck cancer (256), low grade prostate cancer (257) and pancreatic cancer (258, 259). But now, there have been reports describing PDT as suitable options for treating cutaneous metastases from BC as well as primary BC (239, 260). PDT combined with traditional antitumor therapies show much promising effect in improving patient outcome and reducing the unwanted side effects. The combination of light with rhodamine 123 and its platinum complex, indocyanine green (261), meso-tetra hydroxyphenyl chlorine and zinc phthalocyanine has been proven very effective in in -vitro studies (262–265). Recently, Chou et al. study the effect of combination of PDT and bio reductive therapy in targeting TNBC with an aptamer functionalized nano formulation (23). This new therapeutic strategy, which utilized the combination of protoporphyrin IX and tirapazamine, performed well in both hypoxia and normoxia, and hence could be a promising medical procedure for effective treatment of TNBC (Table 1). In summary, the synergistic effect of PDT and traditional therapies could enhance the therapeutic effect and even can prove to be a better way to tackle TNBCs.
Recently, adoptive cell therapy (ACT) and cancer vaccines have been proposed as future therapy approaches, which can cure various cancer stages, including TNBC. Adoptive cell therapy in TNBC mainly covers three types of ACT which include three types of therapy: tumor-infiltrating lymphocyte (TILs), engineered T cell receptor, and chimeric antigen receptor therapy (CAR-T) which is strongly correlated with the infiltration of T-cells in TNBC (266). These all ACT based on similar principles where patients’ natural t-cells have been modified genetically in ex-vivo condition and injected back into the patient’s body to make them tumor antigen-specific and accelerate their ability to kill cancer cells by triggering the cytotoxic immune response (266, 267). CART-T cells improve the effective tumor transport of engineered activated T-cells and overcome antigenic heterogeneity and the broad repertoire of immune escape mechanisms occurring in advanced TNBC. However, certain issues need to be addressed, such as identifying tumor-specific antigens (TSAs) rather than tumor-associated antigens (TAs) and optimizing the adverse effects of cell lysis for immune hyper-activation (215). Currently, CAR-T cell therapies have been FDA-approved for the treatment of various cancer-type patients, including TNBC, and a considerable number of clinical trials are testing CAR constructs against multiple tumor antigens in TNBC, which are summarized in Table 1.
Cancer vaccines also target TAs to accelerate tumor-specific immune responses through active immunization by generating cytotoxic CD8+ T-cell (CTLs) and other effector immune responses such as NK and dendritic cell responses (266). These vaccines consist of either peptides, carbohydrates, recombinant DNA or RNA, whole cells, or dendritic cells (DC), which summarizes in Table 1. In addition, neoantigen vaccines use peptides that are specific to mutations in the tumor and not present in normal cells, therefore have been shown to elicit robust immunogenic responses because of high tumor mutational burden (TMB) and further activates tumor antigen specific CD8+ and CD4+ T cells (266). Emerging evidence suggests that these cancer vaccines, in combination with ICI and chemotherapeutic agents, may boost the anti-tumor immune response. The current clinical trials using cancer vaccines in combination with ICI and chemotherapeutic agents are summarized in Table 1.
ADC are immunoconjugative drugs which are specifically engineered by using three pre-defined immune components a) cytotoxic drugs, b) a chemical linker moiety and c) a humanized monoclonal antibody specifically recognizing neoplastic epitopes on tumor cells and overexpressed definite antigens {trophoblast cell surface antigen 2 (trop-2), receptor tyrosine kinase-like orphan receptor (180), human epidermal growth factor receptor (HER) etc.} (215, 268). These ADC drugs are degraded once it recognizes and conjugates with specific antigens in the highly acidic metabolic TME (268). ADC’s high target specificity and potency feature defines its novelty in personalized therapeutic approaches. Emerging evidence also suggests that a high therapeutic index compared to traditional chemotherapies and their specificity against selective tumor populations make the ADCs a promising partner for targeted agents in combination therapies (268, 269). However, several preclinical and clinical data have shown and suggested high pharmacological properties and improved survival benefits, several limitations still need to be improved, such as recognition of specific binding antigens, optimization of the drug-to-antibody ratio (DAR) and release of the chemical linker in tumor cells and their toxicities etc. Song Hua et al., 2010 have shown that novel anti-HIF-1α ADC nano micelles filled with paclitaxel precisely target and selectively kill the stomach cancer cells having high expression of HIF-1α and suggesting that HIF-1 ADC could be great potential in various clinical settings (270). Several clinical studies have so far been ongoing based on preclinical antitumor activity in both neoadjuvant and metastatic settings in the TNBC cohort, and trop-2 targeted sacituzumab govitecan is the first FDA-approved ADC for the mTNBC treatment (215, 269). Table 2. summarizes the ongoing clinical trials of ADCs and their analogues in locally advanced or metastatic TNBC.
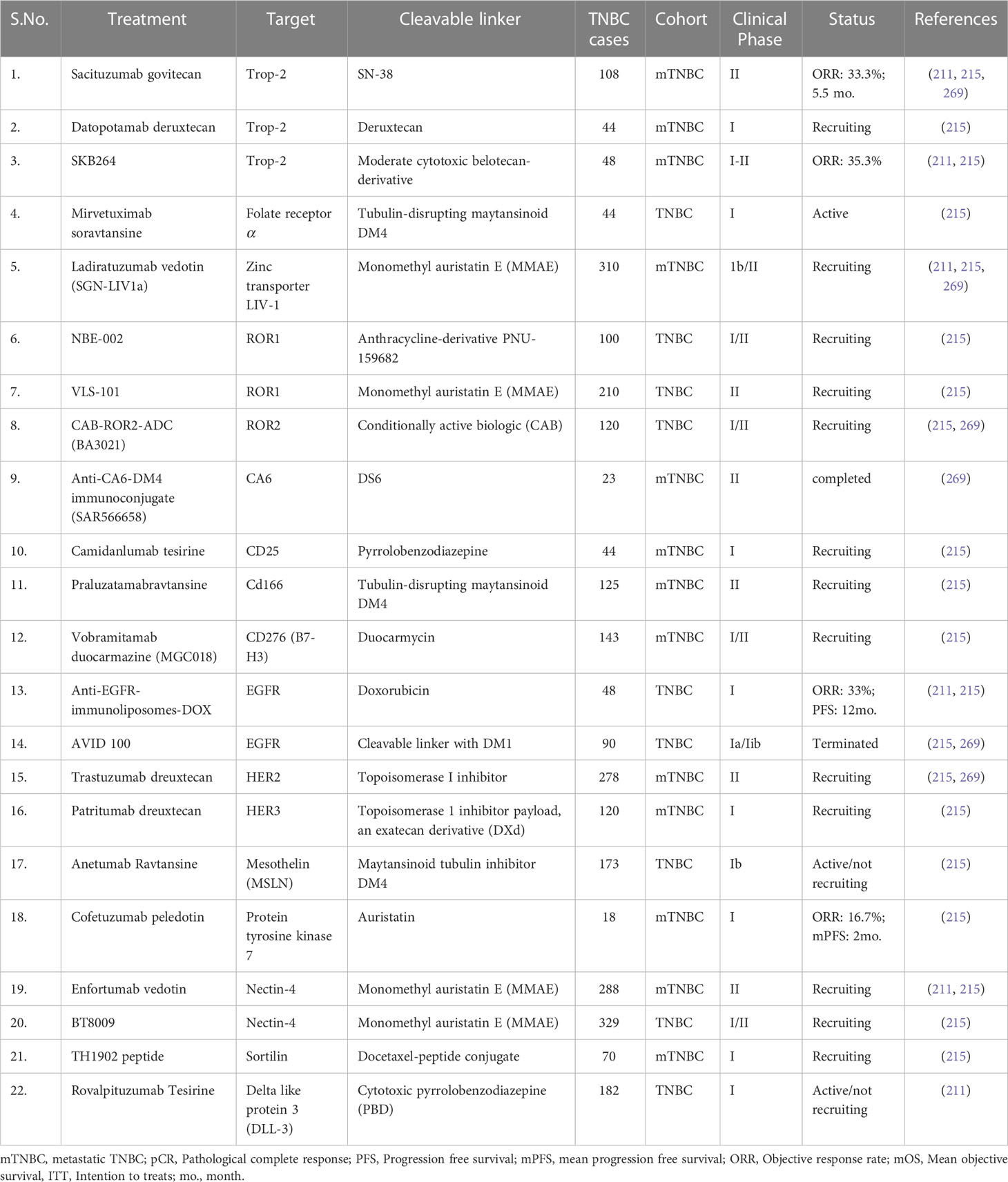
Table 2 Development of antibody-drug conjugates (ADC) and ongoing clinical trials for TNBC treatment.
TNBC lacks expression of some generalized targeted receptors such as estrogen, progesterone, and HER2 receptors, making it difficult to target with conventional therapies. However, combination therapy involves the simultaneous use of multiple treatment modalities, such as chemotherapy, targeted radiotherapy, and immunotherapies, to enhance efficacy and overcome resistance mechanisms by targeting multiple signaling pathways and tumor vulnerabilities. The combination treatment approaches mostly involved tailored strategies based on individual patient characteristics and the tumor’s molecular profile, leading to precise therapy to improve patient outcomes. Paclitaxel and nab-paclitaxel are among the frequently used chemotherapy options, but their resistance is one of the major reasons for the failure and relapse of TNBC (271). Therefore, currently, several clinical trials are undergoing where the combination of paclitaxel or nab-paclitaxel with immune checkpoint inhibitors such as atezolizumab, cobimetinib, or PARP, AKT, PI3K, or VEGF inhibitors has been administered, leading to a significant increase in mean objective survival and response rate. Besides, the combination of chemotherapy and immune checkpoint inhibitors followed by adjuvant therapy are also under clinical trials, exhibiting significant positive responses. Similarly, the combination of anti-angiogenic therapy like lenvatinib, apatinib etc., with several inhibitors also exhibits positive responses in undergoing clinical trials. Ongoing research and clinical trials continue to explore innovative combination regimens, offering hope for improved survival rates and a brighter future for TNBC patients, and we have summarized such clinical trials revolving around combination therapy in Table 3.
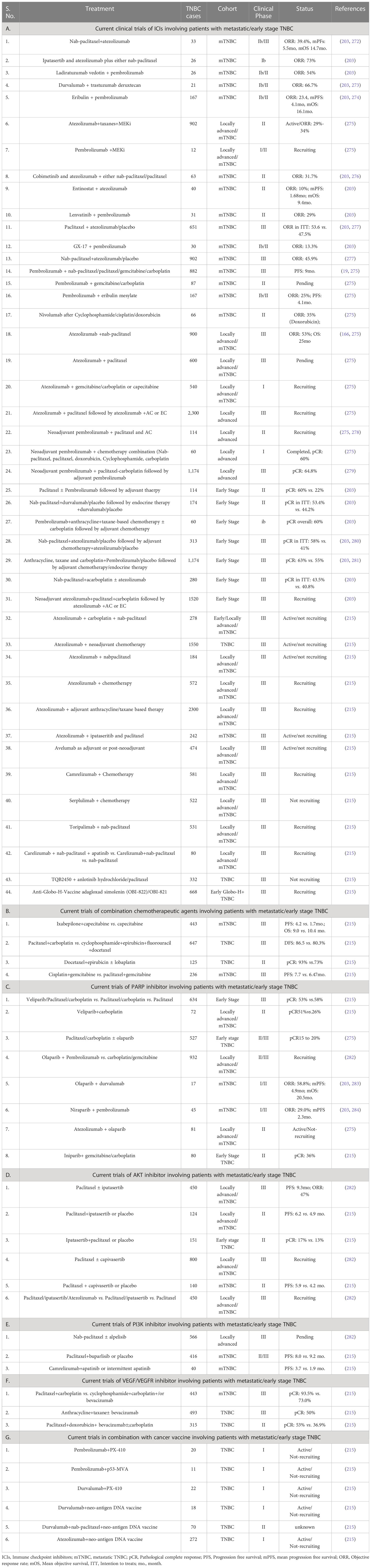
Table 3 Current combination treatments in TNBCs.
Normal tissues generally require a steady supply of oxygen and nutrients to stay alive and remove waste through metabolism. The solid tumor, unlike normal tissue, has dysfunctional vasculature (285, 286). The rate of tumor progression, stroma composition, and pathological vasculature all contribute to a hypoxic environment in the tumor microenvironment, which impairs immune cell function. Furthermore, hypoxia creates selection pressure by promoting cell growth alongside genetic machinery having malignant potential (287). As a result, hypoxia causes EMT, which leads to cell mobility and metastasis (288, 289). Moreover, metabolism of tumor cell reforms after hypoxia, leading to cell quiescence (30, 64). This condition alters transport or distribution and is resistant to radiotherapy, chemotherapy, immunotherapy, and adjuvant therapy (31, 290). Chemotherapy and radiotherapy affect proliferating tumor cells, especially in normoxic states, but hypoxic cells survive these antineoplastic therapies. Thomlinson and Gray et al. previously proposed that hypoxia is a “diffusion-limited chronic hypoxia” (291).
The top preclinical evaluation studies demonstrated increasing tumor oxygenation by modifying oxygen delivery by allowing tumor-bearing rodent models to inhale either (95% O2 + 5% CO2) carbogen or 100% oxygen. According to the data, the tumor grew significantly after radiation (292). Many preclinical studies previously reported that cancer cells were more malignant in hypoxic conditions. Earlier, Young et al. in-vitro studies demonstrated that cells were kept for 18–24h in hypoxic conditions and injected into the mice (293). In such cases, injected cells reach the lungs and form lung nodules; additionally, they reported that the level of hypoxia in the primary tumor directly increases the number of metastases in tumor-injected mice, regardless of whether the hypoxia was natural or induced (294, 295). According to the previous report, two separate clinical trial studies on how hypoxia influences the malignant progression of cancer cells were conducted. The study’s findings revealed that the oxygenation status of the patients was assessed using the Eppendorf electrode before the regimen. Previously, only one study on a cervix cancer patient who underwent surgery was reported (296). In the other group, most soft-tissue sarcoma patients underwent surgery (297). In such cases, injected cells reach the lungs and develop lung nodules. In addition, they also reported that the level of hypoxia in the primary tumor directly aggravated the number of metastases in tumor-injected mice regardless of whether that hypoxia was natural or induced (294, 295). Based on the previous report, two separate clinical trial studies were conducted on how hypoxia influences cancer cells’ malignant progression. The study outcome revealed that the oxygenation status of the patient’s assessed by applying the Eppendorf electrode before the regimen. Earlier, only one study was reported on a cervix cancer patient who underwent surgery (296). In the other group, most patients with soft-tissue sarcoma underwent surgery (297). Both studies found that patients who had previously received oxygenation treatment had an overall higher survival rate. Patients with higher levels of hypoxia had significantly worse survival outcomes based on their pretreatment oxygenation status (296, 297). Later clinical studies revealed that using an eppendorf electrode causes significant hypoxia in leiomyomas, myometrium, and leiomyosarcomas, all originating in premenopausal women (298). Solid tumors directly contribute to cancer’s malignant properties and are a hallmark of hypoxia (299, 300). It is well understood that HIFs instantly activate tumors through HIF transcription factors, which promote changes in the expression of VEGF and CAIX levels, both of which are required for unstable and anaerobic energy production (301). In general, HIFs promote the expression of multiple genes involved in metabolic management, pH balance, angiogenesis, and cell apoptosis, all of which contribute to tumor survival.
As a result, Figure 5 clearly explains the mechanism, as mentioned earlier. HIFs play a role in tumor blood recovery via vascular protection and promote nutrient supply to solid tumors, which become one of the most difficult to treat, leading to resistance to chemotherapy, radiotherapy, immunotherapy, and adjuvant therapy.
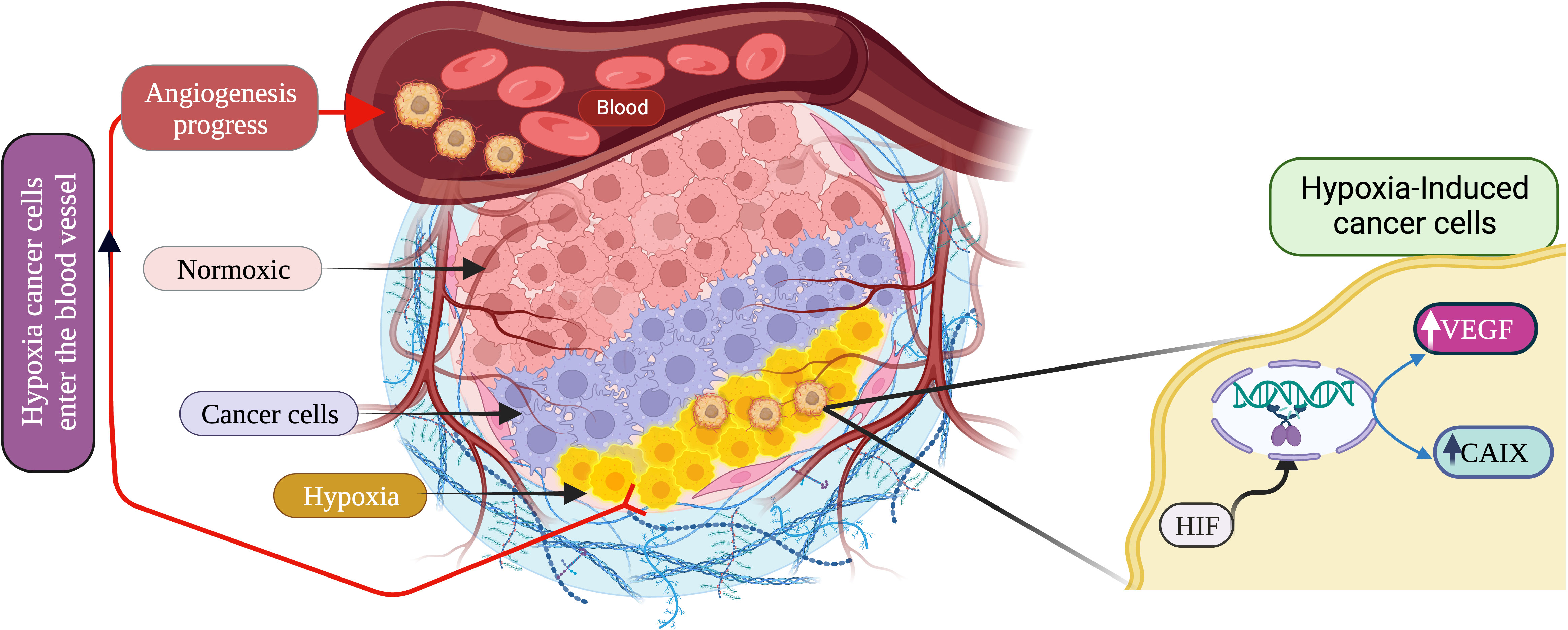
Figure 5 Schematic description of hypoxic cancer microenvironment and angiogenesis progression. (Created via BioRender.com).
A key goal of TNBC therapy has been to target hypoxia, which inhibits several tumor characteristics, including metastasis, radio-resistance, and chemoresistance (302, 303). A study that has been published suggests that many hypoxia-related genes (HRGs) and their mediators, HIFs, may be used as therapeutic targets and prognostic indicators in BC (34, 303).
HIF1α is a well-established key target regulating the TNBC, and its expression is regulated by various signaling pathways like NF-κB, PI3K/Akt/mTOR, RAS-RAF-ME-ERK and JAK-STAT. Studies have found that under hypoxic conditions, HIF1α induced STAT3 via JAK or adenylate receptor 2B pathway, which upregulates the IL-6 and NANOG to maintain the CSC phenotype and also enhances the production of VEGF, required for the self-renewal ability of CSCs (79). Studies have also demonstrated that in hypoxic conditions, HIF-1α activates the Sonic Hedgehog signaling pathway to induce the production of CSC markers in cholangiocarcinoma cells which can be blocked by HIF-1α inhibition (304). Emerging studies also demonstrated that HIF1α suppress the ERK activity and induces the P38 activity, which further upregulates NANOG and KLF4 to promote the development of breast CSCs. Several studies have been conducted to identify targetable molecules from these signaling pathways that characterize various inhibitors or drug molecules (304, 305). The astonishing fact is that some of these signaling pathways can be targeted by already approved therapeutics or inhibitors under clinical trials alpelisib is an approved inhibitor while buparlisib is under clinical trial, and both inhibit class I PI3K. Similarly, several inhibitors target VEGF, EGFR, PARP, and cell cycle and have shown significant outcomes in TNBC patients. Table 4 summarizes potential inhibitors and drug molecules against molecular targets and signaling pathways involved in the progression of hypoxia-induced TNBC.
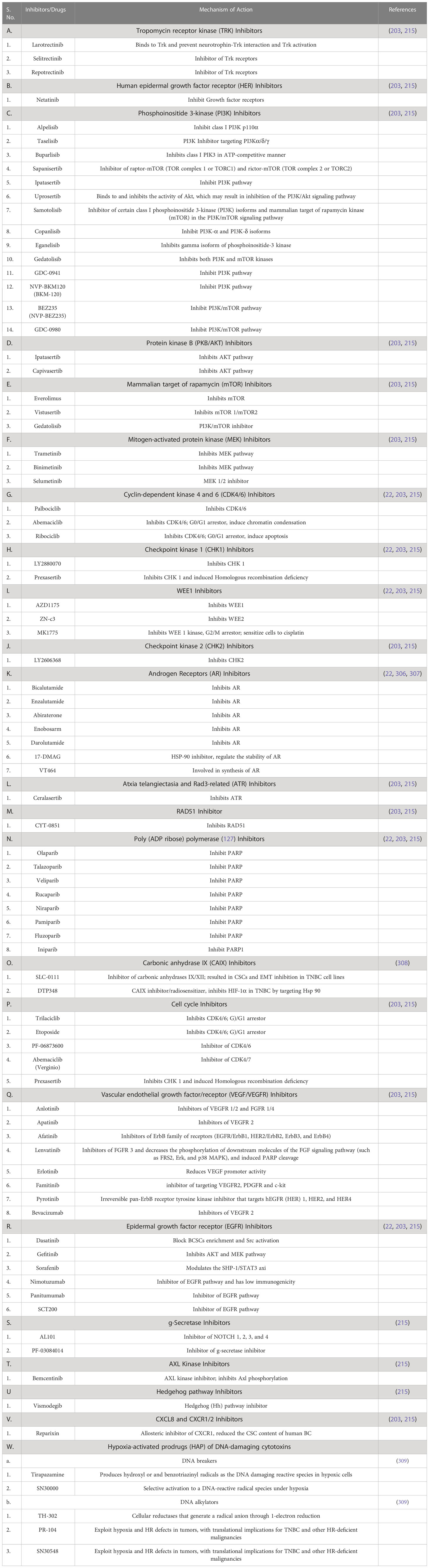
Table 4 Potential inhibitors and drugs for hypoxia induced TNBC.
TNBC patients exhibit higher mortality rates, and it has already been studied that overexpression of HIF-1α is associated with poor prognosis in various cancer (302). Here, we have explored a publicly available gene expression dataset (GSE103091, subseries GSE58812) to study the effect of hypoxia-related gene expression on mortality (303, 304). This dataset contains gene expression molecular subtyping of TNBC samples from 107 patients (78 alive and 29 dead). We have explored the expression of HIF and VEGFs and genes for glucose transporters. As shown in Figure 6, the overall presentation of HIF-1α is significantly higher in the patients who died due to TNBC than in the alive patients. However, there is no significant relationship between HIF-3α with the mortality.
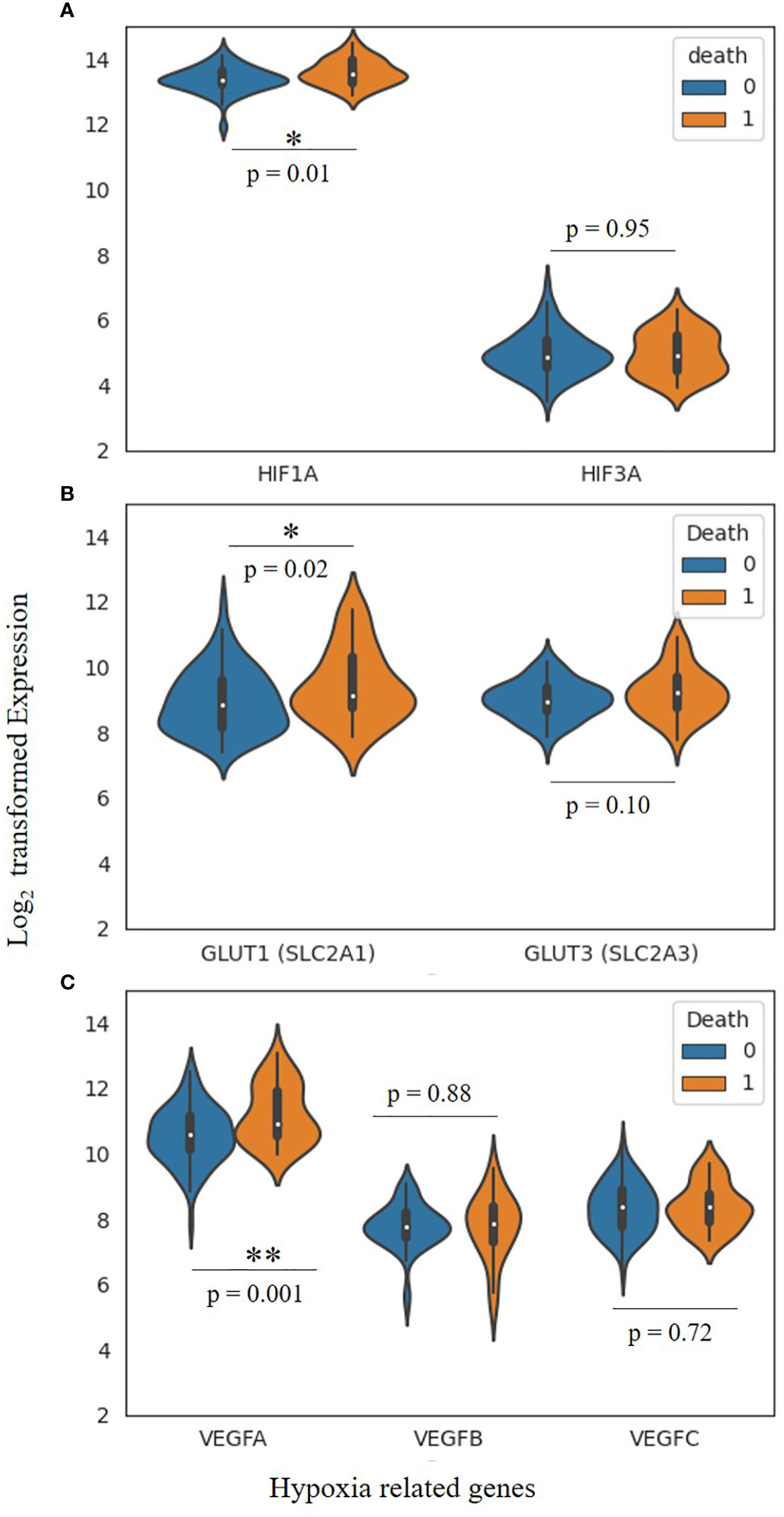
Figure 6 Hypoxia-related gene expression and morality in TNBC patients. (A) Violin plot exhibiting the expression of HIF-1α and HIF-3 α, (B) GLUT-1 and GLUT-3 Glucose transporters (genes SLC2A1 and SLC2A3) and (C) Vascular endothelial growth factors, VEGFA, VEGFB as well as VEGFC. Two-tailed T-test has been aplied for the significance (*, P < 0.05, **, P < 0.005). Cohort contains n=107 TNBC pateint samples (alive 79, dead 28).
Similarly, the expression of VEGF-A and GLUT-1 significantly (p= 0.001 and 0.02, respectively) differs in both cohorts. This study suggests a possible association between hypoxia-related gene expression and mortality. However, other factors like age, cancer grade, metastatic etc., haven’t been considered and may impact the conclusion. But hypothetically, there is a strong correlation between hypoxia-related gene expression and mortality, and it needs to be validated in larger cohorts.
The association between HIF-1α and TNBC strongly suggests the possibility of novel targeted therapy in combination with chemotherapy, anti-angiogenic therapy, and immunotherapy for TNBC treatment (97, 102). HIF-1α reflects its potential to improve the current therapeutic outcome because of its extensive biological activities, particularly its function in angiogenesis, activation, and enhancement of tumor stem cells among other processes. The enrichment of BCSCs in tumors generated by various chemotherapeutic treatments is highly correlated with the increase of HIF-1, which is the major hurdle against chemotherapy. Clinical evidences support that some molecules like selenium, docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) in combination with low-dose chemotherapeutic agents significantly induced the degradation of HIF-1α and limits the BCSCs enrichment which may increase TNBC chemotherapy resistance (97, 310).
It is well documented that HIF-1 is stabilized in hypoxic conditions, and transcriptionally controls the lactate dehydrogenase A (LDHA) gene, which is associated to glycolysis and supports an acidic milieu. In patients with TNBC, this acidic milieu changes increases the CD8+ T cell counts and the generation of IFN, which is linked to a better clinical result and a stronger immunological response (311). Therefore, to improve the acidic microenvironment through HIF-1/LDHA targeting may restore the cytotoxic effect of CD8 cells to enhance the impact of immunotherapy in TNBC (97, 312). HIF-1α interaction with HDAC1 and concurrent PRC2 dependency epigenetically suppress the effector genes and induces the immune dysfunction in TNBC which results resistance to immunotherapy. A recent study in syngeneic and humanized TNBC mouse model has shown the efficacy of PD-1 blockade combined with HIF-1α and HDAC1 inhibition by PX478 and ENT respectively to reverse the anti-PD-1-resistant TNBC and significantly reduces tumor metabolic activity and metastasis (102, 313). Some clinical studies have showed that anti-angiogenic therapy alone is not recommended as the first-line treatment for metastatic TNBC since it increases the likelihood of TNBC invasion and metastasis. Consequently, inhibiting HIF-1 can enhance clinical efficacy by preventing invasion and metastasis-induced anti-angiogenic treatment. When used with the anti-angiogenic drug avastin, Guo et al. discovered that selenium with omega-3 polyunsaturated fatty acids decrease angiogenesis and metastasis via preventing COX-2 overexpression induced by HIF-1 (168). Furthermore, the anti-angiogenic drug bevacizumab hyperactivates the Wnt/β-catenin signaling in response to HIF-1α’s high expression in TNBC because of aberrant expression of frizzled 7 (Fzd7), a key receptor for Wnt/β-catenin signaling’s key receptor that induces cell invasiveness and metastasis (314, 315). Consequently, using an anti-Fzd7 antibody (SHH002-hu1) to target hypoxia adaptation-related proteins VEGFA and Glut1 expression as well as HIF-1 transcriptional activity will decrease TNBC cells’ acclimation to hypoxia and counteract the negative effects of anti-angiogenic medicines (314).
Under hypoxic conditions, irradiation can increase HIF-1 expression, which would lead to radio resistance (316). There are some confirmed reports suggesting that after radiation therapy, the availability of oxygen and glucose is increased in solid tumors, which activate HIF-1 and promote EMT that is HIF-1 dependent. Because of the translocating cells’ proximity to the blood arteries, which allows them to absorb nutrition and oxygen, tumor recurrence may be made easier (317, 318). The HIF-1 dependent translocation and migration of the surviving cells towards radio-protected blood vessels may indicate a specific role for HIF-1 in both local tumor recurrence and distant tumor metastasis after radiation therapy. Recent findings also suggest that HIF-1 inhibition utilizing HIF-1 inhibitors, may enhance radiosensitivity, chemosensitivity, immunosenstivity that could potentially provide advantages to methods of therapeutic treatment for hypoxic malignancies (316, 319). Table 5 summarizes several potential synthetic compounds and natural products that have clinically proven to inhibit the HIF-1α activity at the transcriptional and translational level in TNBC, such as inhibiting the mRNA level of HIF-1α and their dimerization with HIF-1β as well as accelerating the degradation of the HIF-1α protein.
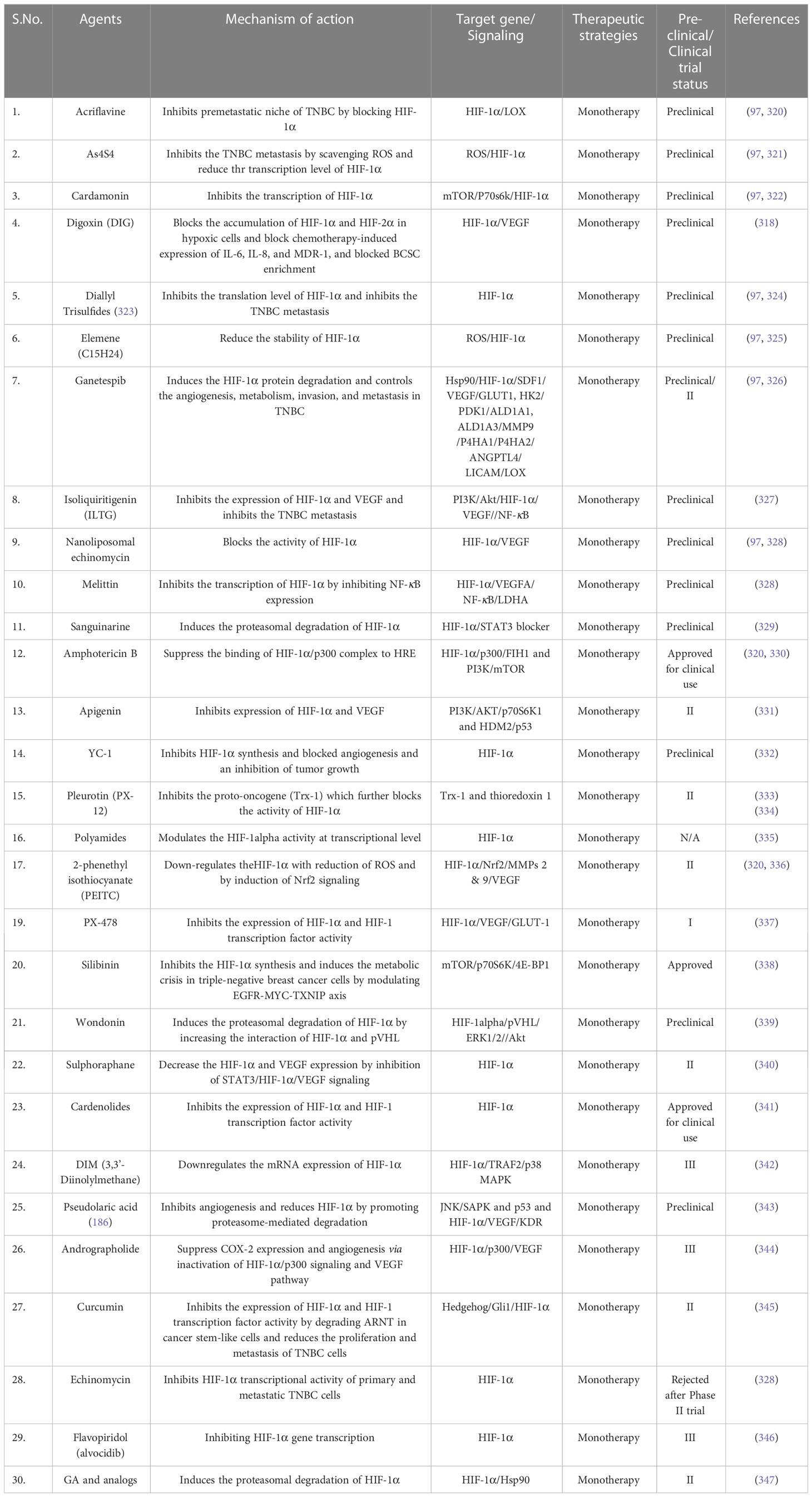
Table 5 Summary of related drugs/inhibitors targeting HIF-1α.
A preponderance of evidence supports the notion that different histological and molecular subtypes of TNBC signify its heterogeneity and aggressiveness. Several genetic and transcriptomic alterations define each subtype of TNBC, and they can be potentially targeted for a unique therapy. Recent advancements in targeting hypoxic-tumor microenvironments by suppressing HIF-1α transcription and oxidative phosphorylation have yielded promising results. Besides, anti-angiogenesis inhibitors and hypoxia-activated pro-drugs gained a lot of attention. Moreover, recent studies confirmed that TNBC also causes hypoxia-dependent genetic changes in DDR pathways, which suggests the possibility of predictive biomarkers. Combining DDR inhibitors with other therapy, including radiotherapy, chemotherapy, immunotherapy, PDT, and adjuvant therapies, can optimize their efficacy in TNBC treatment. TNBC also characterize by complex immunological landscape vulnerability through defects in the DDR pathway, which induces high TMB, anti-tumor immune suppressive features, as well as adaptive immune resistance via the expression of corresponding inhibitory ligands against immune checkpoints such as the PD1-PDL1 interaction. Therefore, DDR deficiencies offer potential therapeutic leverage for TNBC treatment by combining DNA/DDR-targeted therapies with cytotoxic anti-tumor immune cells, leading to favorable immune effects. Combining immune-checkpoint inhibitors, chemotherapy, and radiotherapy with HIF-1α inhibitors or its downstream target inhibitors like Trk, PI3K, PARP, CAIX etc., maybe a significant potential to match the high standard of clinical benefit in TNBC. In addition, combining these inhibitors with emerging antibody-drug conjugates, cancer vaccines, or adoptive cell therapy followed with existing treatments may be a significant step towards precision therapy and extend overall clinical benefits. Therefore, to determine the solid TNBC combination therapy regimens, it is pertinent to access the immune-molecular expression of HIF-1α and its associated mutational analysis in hypoxic TNBC. Although, HIFs have already been largely explored, but their downstream effector signaling, as well as other pathways like MYC, TP53, and KRAS, should be further explored in the surge of potential therapeutic targets. A more in-depth understanding of the TNBC hypoxic microenvironment, its molecular nature and its effect on tumor prognosis and survival will surely help in early detection and accurate treatment. 10,000 human genome projects will definitely aid in designing precise medicine based on the individual genome as well as tumor specificity.
Conceptualization: NS and SU; writing and original draft preparation: NS, SU, RSS, and PKP; writing—review and editing: NS, SU, RSS, and RS; visualization: NS, SU, RSS, and RS; supervision and critically reviewed: NS, SU, RSS, and RS. All authors contributed to the article and approved the submitted version.
There is no funding involved in this project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated projection of US cancer incidence and death to 2040. JAMA Network Open (2021) 4(4):e214708–e. doi: 10.1001/jamanetworkopen.2021.4708
2. Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Internal Med (2013) 274(2):113–26. doi: 10.1111/joim.12084
3. Abad E, Civit L, Potesil D, Zdrahal Z, Lyakhovich A. Enhanced DNA damage response through RAD50 in triple negative breast cancer resistant and cancer stem-like cells contributes to chemoresistance. FEBS J (2021) 288(7):2184–202. doi: 10.1111/febs.15588
4. Ai M, Budhani P, Sheng J, Balasubramanyam S, Bartkowiak T, Jaiswal AR, et al. Tumor hypoxia drives immune suppression and immunotherapy resistance. J ImmunoTherapy Cancer (2015) 3:1–. doi: 10.1186/2051-1426-3-S2-P392
5. Allred DC. Issues and updates: evaluating estrogen receptor-α, progesterone receptor, and HER2 in breast cancer. Modern Pathology (2010) 23(2):S52–S9. doi: 10.1038/modpathol.2010.55
6. Pommier RM, Sanlaville A, Tonon L, Kielbassa J, Thomas E, Ferrari A, et al. Comprehensive characterization of claudin-low breast tumors reflects the impact of the cell-of-origin on cancer evolution. Nat Commun (2020) 11(1):3431. doi: 10.1038/s41467-020-17249-7
7. Polyak K. Breast cancer: origins and evolution. J Clin Invest (2007) 117(11):3155–63. doi: 10.1172/JCI33295
8. Colleoni M, Rotmensz N, Maisonneuve P, Mastropasqua M, Luini A, Veronesi P, et al. Outcome of special types of luminal breast cancer. Ann Oncol (2012) 23(6):1428–36. doi: 10.1093/annonc/mdr461
9. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. New Engl J Med (2010) 363(20):1938–48. doi: 10.1056/NEJMra1001389
10. Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. New Engl J Med (2021) 384(16):1529–41. doi: 10.1056/NEJMoa2028485
11. Bernardi R, Gianni L. Hallmarks of triple negative breast cancer emerging at last? Cell Res (2014) 24(8):904–5. doi: 10.1038/cr.2014.61
12. Zheng S, Zou Y, Liang Jy, Xiao W, Yang A, Meng T, et al. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol Oncol (2020) 14(11):2814–33. doi: 10.1002/1878-0261.12747
13. Yin L, Duan J-J, Bian X-W, Yu S-c. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res (2020) 22:1–13. doi: 10.1186/s13058-020-01296-5
14. Almansour NM. Triple-negative breast cancer: a brief review about epidemiology, risk factors, signaling pathways, treatment and role of artificial intelligence. Front Mol Biosciences (2022) 9:32. doi: 10.3389/fmolb.2022.836417
15. Wu Q, Siddharth S, Sharma D. Triple negative breast cancer: a mountain yet to be scaled despite the triumphs. Cancers (2021) 13(15):3697. doi: 10.3390/cancers13153697
16. Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer (2008) 113(10):2638–45. doi: 10.1002/cncr.23930
17. Yao Y, Chu Y, Xu B, Hu Q, Song Q. Risk factors for distant metastasis of patients with primary triple-negative breast cancer. Bioscience Rep (2019) 39(6). doi: 10.1042/BSR20190288
18. Riggio AI, Varley KE, Welm AL. The lingering mysteries of metastatic recurrence in breast cancer. Br J cancer (2021) 124(1):13–26. doi: 10.1038/s41416-020-01161-4
19. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet (2020) 396(10265):1817–28. doi: 10.1016/S0140-6736(20)32531-9
20. Al-Mahmood S, Sapiezynski J, Garbuzenko OB, Minko T. Metastatic and triple-negative breast cancer: challenges and treatment options. Drug delivery Trans Res (2018) 8:1483–507. doi: 10.1007/s13346-018-0551-3
21. Yagata H, Kajiura Y, Yamauchi H. Current strategy for triple-negative breast cancer: appropriate combination of surgery, radiation, and chemotherapy. Breast Cancer (2011) 18:165–73. doi: 10.1007/s12282-011-0254-9
22. Jhan J-R, Andrechek ER. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics (2017) 18(17):1595–609. doi: 10.2217/pgs-2017-0117
23. Chou Y-T, Lin C-Y, Wen J-W, Hung L-C, Chang Y-F, Yang C-M, et al. Targeting triple-negative breast cancer with an aptamer-functionalized nanoformulation: a synergistic treatment that combines photodynamic and bioreductive therapies. J Nanobiotechnology (2021) 19(1):1–15. doi: 10.1186/s12951-021-00786-8
24. Pohl-Rescigno E, Hauke J, Loibl S, Moebus V, Denkert C, Fasching PA, et al. Association of germline variant status with therapy response in high-risk early-stage breast cancer: a secondary analysis of the GeparOcto randomized clinical trial. JAMA Oncol (2020) 6(5):744–8. doi: 10.1001/jamaoncol.2020.0007
25. Lee A, Djamgoz MB. Triple negative breast cancer: emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev (2018) 62:110–22. doi: 10.1016/j.ctrv.2017.11.003
26. Mir MA, Qayoom H, Mehraj U, Nisar S, Bhat B, Wani NA. Targeting different pathways using novel combination therapy in triple negative breast cancer. Curr Cancer Drug Targets (2020) 20(8):586–602. doi: 10.2174/1570163817666200518081955
27. Liu Z-j, Semenza GL, Zhang H-f. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang University-SCIENCE B (2015) 16(1):32–43. doi: 10.1631/jzus.B1400221
28. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest (2022) 132(11):e159839. doi: 10.1172/JCI159839
29. Shi R, Liao C, Zhang Q. Hypoxia-driven effects in cancer: characterization, mechanisms, and therapeutic implications. Cells (2021) 10(3):678. doi: 10.3390/cells10030678
30. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83. doi: 10.2147/HP.S93413
31. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol cancer (2019) 18:1–15. doi: 10.1186/s12943-019-1089-9
32. Mori N, Mironchik Y, Wildes F, Wu SY, Mori K, Krishnamachary B, et al. HIF and COX-2 expression in triple negative breast cancer cells with hypoxia and 5-fluorouracil. Curr Cancer Rep (2020) 2(1):54. doi: 10.25082/CCR.2020.01.005
33. Mimeault M, Batra SK. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer-and metastasis-initiating cells. J Cell Mol Med (2013) 17(1):30–54. doi: 10.1111/jcmm.12004
34. Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-inducible factors and cancer. Curr sleep Med Rep (2017) 3:1–10. doi: 10.1007/s40675-017-0062-7
35. Fultang N, Chakraborty M, Peethambaran B. Regulation of cancer stem cells in triple negative breast cancer. Cancer Drug Resistance (2021) 4(2):321. doi: 10.20517/cdr.2020.106
36. Qiu G-Z, Jin M-Z, Dai J-X, Sun W, Feng J-H, Jin W-L. Reprogramming of the tumor in the hypoxic niche: the emerging concept and associated therapeutic strategies. Trends Pharmacol Sci (2017) 38(8):669–86. doi: 10.1016/j.tips.2017.05.002
37. Zheng H, Siddharth S, Parida S, Wu X, Sharma D. Tumor microenvironment: key players in triple negative breast cancer immunomodulation. Cancers (2021) 13(13):3357. doi: 10.3390/cancers13133357
38. Zarrilli G, Businello G, Dieci MV, Paccagnella S, Carraro V, Cappellesso R, et al. The tumor microenvironment of primitive and metastatic breast cancer: implications for novel therapeutic strategies. Int J Mol Sci (2020) 21(21):8102. doi: 10.3390/ijms21218102
39. Ezdakova M, Andreeva E, Gurieva T, Dadasheva O, Orlova V, Buravkova L. Effects of hypoxia and growth factors on the angiogenic activity of multipotent mesenchymal stromal cells. Aviakosmicheskaia i Ekologicheskaia Meditsina= Aerospace Environ Med (2015) 49(5):29–35.
40. Fu Y, Mackowiak B, Feng D, Lu H, Guan Y, Lehner T, et al. MicroRNA-223 attenuates hepatocarcinogenesis by blocking hypoxia-driven angiogenesis and immunosuppression. Gut (2023). doi: 10.1136/gutjnl-2022-327924
41. Zhang Y, Zhang H, Wang M, Schmid T, Xin Z, Kozhuharova L, et al. Hypoxia in breast cancer–scientific translation to therapeutic and diagnostic clinical applications. Front Oncol (2021) 11:652266. doi: 10.3389/fonc.2021.652266
42. Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int (2021) 21(1):1–26. doi: 10.1186/s12935-020-01719-5
43. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Institute (2001) 93(4):266–76. doi: 10.1093/jnci/93.4.266
44. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget (2016) 7(25):38908. doi: 10.18632/oncotarget.7676
45. Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging roles of aerobic glycolysis in breast cancer. Clin Trans Oncol (2020) 22:631–46. doi: 10.1007/s12094-019-02187-8
46. Doigneaux C, Pedley AM, Mistry IN, Papayova M, Benkovic SJ, Tavassoli A. Hypoxia drives the assembly of the multienzyme purinosome complex. J Biol Chem (2020) 295(28):9551–66. doi: 10.1074/jbc.RA119.012175
47. Bharti SK, Mironchik Y, Wildes F, Penet M-F, Goggins E, Krishnamachary B, et al. Metabolic consequences of HIF silencing in a triple negative human breast cancer xenograft. Oncotarget (2018) 9(20):15326. doi: 10.18632/oncotarget.24569
48. Gilkes DM, Semenza GL. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol (2013) 9(11):1623–36. doi: 10.2217/fon.13.92
49. Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci (2014) 111(20):E2120–E9. doi: 10.1073/pnas.1406655111
50. Pezzuto A, Carico E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr Mol Med (2018) 18(6):343–51. doi: 10.2174/1566524018666181109121849
51. Lu H, Zhu Y, Liu W, Yan Y, Jiang X, Wang Q, et al. A comprehensive description of hypoxia-inducible factor 2α inhibitors as anticancer agents: a mini-review. Curr Medicinal Chem (2023) 30(25):2835–49. doi: 10.2174/0929867329666220829095334
52. Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M, et al. Up-down regulation of HIF-1α in cancer progression. Gene (2021) 798:145796. doi: 10.1016/j.gene.2021.145796
53. Chen J-Q, Russo J. ERα-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta (BBA)-Reviews Cancer (2009) 1796(2):162–75. doi: 10.1016/j.bbcan.2009.06.003
54. Yao H, He G, Yan S, Chen C, Song L, Rosol TJ, et al. Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget (2017) 8(1):1913. doi: 10.18632/oncotarget.12284
55. Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter K, et al. Hypoxia inducible factor 1α and 2α overexpression in inflammatory bowel disease. J Clin pathology (2003) 56(3):209–13. doi: 10.1136/jcp.56.3.209
56. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes cancer (2011) 2(12):1117–33. doi: 10.1177/1947601911423654
57. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17(8):457–74. doi: 10.1038/nrc.2017.51
58. Russo M, Nastasi C. Targeting the tumor microenvironment: a close up of tumor-associated macrophages and neutrophils. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.871513
59. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. nature (1996) 379(6560):88–91. doi: 10.1038/379088a0
60. Das S, Shukla N, Singh SS, Kushwaha S, Shrivastava R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis (2021), 1–22. doi: 10.1007/s10495-021-01687-9
61. Ozretic P, Alvir I, Sarcevic B, Vujaskovic Z, Rendic-Miocevic Z, Roguljic A, et al. Apoptosis regulator bcl-2 is an independent prognostic marker for worse overall survival in triple-negative breast cancer patients. Int J Biol markers (2018) 33(1):109–15. doi: 10.5301/ijbm.5000291
62. Lee S-H, Golinska M, Griffiths JR. HIF-1-independent mechanisms regulating metabolic adaptation in hypoxic cancer cells. Cells (2021) 10(9):2371. doi: 10.3390/cells10092371
63. Paredes F, Williams HC, San Martin A. Metabolic adaptation in hypoxia and cancer. Cancer letters (2021) 502:133–42. doi: 10.1016/j.canlet.2020.12.020
64. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer (2017) 8(17):3430. doi: 10.7150/jca.21125
65. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
66. Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol (2013) 61(1):14.25. 1–14.25. 14. doi: 10.1002/0471141755.ph1425s61
67. Liang Y, Zhong Z, Huang Y, Deng W, Cao J, Tsao G, et al. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem (2010) 285(7):4931–40. doi: 10.1074/jbc.M109.048397
68. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res (2012) 22(3):457–72. doi: 10.1038/cr.2012.13
69. Dionísio MR, Vieira AF, Carvalho R, Conde I, Oliveira M, Gomes M, et al. BR-BCSC signature: the cancer stem cell profile enriched in brain metastases that predicts a worse prognosis in lymph node-positive breast cancer. Cells (2020) 9(11):2442. doi: 10.3390/cells9112442
70. Jaggupilli A, Elkord E. Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol (2012) 2012:708036. doi: 10.1155/2012/708036
71. Miranda-Galvis M, Teng Y. Targeting hypoxia-driven metabolic reprogramming to constrain tumor progression and metastasis. Int J Mol Sci (2020) 21(15):5487. doi: 10.3390/ijms21155487
72. Collina F, Di Bonito M, Li Bergolis V, De Laurentiis M, Vitagliano C, Cerrone M, et al. Prognostic value of cancer stem cells markers in triple-negative breast cancer. BioMed Res Int (2015) 2015. doi: 10.1155/2015/158682
73. Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet (2021) 53(9):1334–47. doi: 10.1038/s41588-021-00911-1
74. Zhang Y-L, Deng L, Liao L, Yang S-Y, Hu S-Y, Ning Y, et al. Chromatin complexes subunit BAP18 promotes triple-negative breast cancer progression through transcriptional activation of oncogene S100A9. Cell Death Disease (2022) 13(4):408. doi: 10.1038/s41419-021-04490-1
75. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal transduction targeted Ther (2020) 5(1):8. doi: 10.1038/s41392-020-0110-5
76. Fath MK, Garousi S, Mottahedi M, Ghasemzadeh N, Salmani K, Olfati F, et al. The role of hypoxia-inducible factors in breast cancer stem cell specification. Pathology-Research Pract (2023) 154349.
77. Thomas S, Harding MA, Smith SC, Overdevest JB, Nitz MD, Frierson HF, et al. CD24 is an effector of HIF-1–driven primary tumor growth and metastasis. Cancer Res (2012) 72(21):5600–12. doi: 10.1158/0008-5472.CAN-11-3666
78. Qian J, Rankin EB. Hypoxia-induced phenotypes that mediate tumor heterogeneity. Hypoxia Cancer metastasis (2019), 43–55. doi: 10.1007/978-3-030-12734-3_3
79. Zhang Q, Han Z, Zhu Y, Chen J, Li W. Role of hypoxia inducible factor-1 in cancer stem cells. Mol Med Rep (2021) 23(1):1–. doi: 10.3892/mmr.2020.11655
80. Shiraishi A, Tachi K, Essid N, Tsuboi I, Nagano M, Kato T, et al. Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer science (2017) 108(3):362–72. doi: 10.1111/cas.13147
81. Jin W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial–mesenchymal transition. Cells (2020) 9(1):217. doi: 10.3390/cells9010217
82. Lan J, Lu H, Samanta D, Salman S, Lu Y, Semenza GL. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci (2018) 115(41):E9640–E8. doi: 10.1073/pnas.1809695115
83. Ciccone V, Terzuoli E, Donnini S, Giachetti A, Morbidelli L, Ziche M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J Exp Clin Cancer Res (2018) 37:1–16. doi: 10.1186/s13046-018-0975-0
84. Wei X, Chen Y, Jiang X, Peng M, Liu Y, Mo Y, et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol cancer (2021) 20(1):1–18. doi: 10.1186/s12943-020-01288-1
85. Lee K-m, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab (2017) 26(4):633–47.e7. doi: 10.1016/j.cmet.2017.09.009
86. Crowder SW, Balikov DA, Hwang Y-S, Sung H-J. Cancer stem cells under hypoxia as a chemoresistance factor in the breast and brain. Curr pathobiology Rep (2014) 2:33–40. doi: 10.1007/s40139-013-0035-6
87. Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev (2019) 38:65–77. doi: 10.1007/s10555-019-09799-0
88. Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol (2014) 4:400. doi: 10.3389/fphys.2013.00400
89. Lock F, McDonald P, Lou Y, Serrano I, Chafe S, Ostlund C, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene (2013) 32(44):5210–9. doi: 10.1038/onc.2012.550
90. Harrison H, Simões BM, Rogerson L, Howell SJ, Landberg G, Clarke RB. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and notch signalling. Breast Cancer Res (2013) 15(2):1–12. doi: 10.1186/bcr3396
91. Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, Liu W, et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene (2011) 30(39):4075–86. doi: 10.1038/onc.2011.122
92. Humphries B, Wang Z, Yang C. MicroRNA regulation of breast cancer stemness. Int J Mol Sci (2021) 22(7):3756. doi: 10.3390/ijms22073756
93. Khan AQ, Ahmed EI, Elareer NR, Junejo K, Steinhoff M, Uddin S. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells (2019) 8(8):840. doi: 10.3390/cells8080840
94. Hwang-Verslues W, Chang P, Wei P, Yang C, Huang C, Kuo W, et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of e-cadherin and REDD1. Oncogene (2011) 30(21):2463–74. doi: 10.1038/onc.2010.618
95. Vito A, El-Sayes N, Mossman K. Hypoxia-driven immune escape in the tumor microenvironment. Cells (2020) 9(4):992. doi: 10.3390/cells9040992
96. Wu Q, You L, Nepovimova E, Heger Z, Wu W, Kuca K, et al. Hypoxia-inducible factors: master regulators of hypoxic tumor immune escape. J Hematol Oncol (2022) 15(1):1–18. doi: 10.1186/s13045-022-01292-6
97. Liu Q, Guan C, Liu C, Li H, Wu J, Sun C. Targeting hypoxia-inducible factor-1alpha: a new strategy for triple-negative breast cancer therapy. Biomedicine Pharmacotherapy (2022) 156:113861. doi: 10.1016/j.biopha.2022.113861
98. Naik A, Monjazeb AM, Decock J. The obesity paradox in cancer, tumor immunology, and immunotherapy: potential therapeutic implications in triple negative breast cancer. Front Immunol (2019) 10:1940. doi: 10.3389/fimmu.2019.01940
99. Zhang P, Liu G, Hu J, Chen S, Wang B, Peng P, et al. Tenascin-c can serve as an indicator for the immunosuppressive microenvironment of diffuse low-grade gliomas. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.824586
100. Ryan D, Bogan D, Davies J, Koziol J, ElShamy WM. A niche that triggers aggressiveness within BRCA1-IRIS overexpressing triple negative tumors is supported by reciprocal interactions with the microenvironment. Oncotarget (2017) 8(61):103182–103206. doi: 10.18632/oncotarget.20892
101. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell (2017) 32(5):669–83.e5. doi: 10.1016/j.ccell.2017.10.003
102. Ma S, Zhao Y, Lee WC, Ong L-T, Lee PL, Jiang Z, et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat Commun (2022) 13(1):4118. doi: 10.1038/s41467-022-31764-9
103. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
104. Terry S, Buart S, Chouaib S. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front Immunol (2017) 8:1625. doi: 10.3389/fimmu.2017.01625
105. Li T, Mao C, Wang X, Shi Y, Tao Y. Epigenetic crosstalk between hypoxia and tumor driven by HIF regulation. J Exp Clin Cancer Res (2020) 39(1):1–25. doi: 10.1186/s13046-020-01733-5
106. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell (2019) 179(5):1191–206. e21. doi: 10.1016/j.cell.2019.10.028
107. Do TC, Lau JW, Sun C, Liu S, Kha KT, Lim ST, et al. Hypoxia deactivates epigenetic feedbacks via enzyme-derived clicking proteolysis-targeting chimeras. Sci Advances (2022) 8(50):eabq2216. doi: 10.1126/sciadv.abq2216
108. da Motta LL, Ledaki I, Haider S, De Bastiani M, Baban D, Stefan K, et al. PO-276 histone acetylation readers BET in hypoxia adaptation in triple negative breast cancer (TNBC). ESMO Open (2018) 3:A335.
109. Khandekar D, Tiriveedhi V. Role of BET inhibitors in triple negative breast cancers. Cancers (2020) 12(4):784. doi: 10.3390/cancers12040784
110. Andrikopoulou A, Liontos M, Koutsoukos K, Dimopoulos M-A, Zagouri F. The emerging role of BET inhibitors in breast cancer. Breast (2020) 53:152–63. doi: 10.1016/j.breast.2020.08.005
111. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell (2022) 13(12):877–919. doi: 10.1007/s13238-021-00846-7
112. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci (2012) 109(49):E3367–E76. doi: 10.1073/pnas.1217394109
113. Nicolini A, Ferrari P, Diodati L, Carpi A. Recent advances in comprehending the signaling pathways involved in the progression of breast cancer. Int J Mol Sci (2017) 18(11):2321. doi: 10.3390/ijms18112321
114. Lambert M, Jambon S, Depauw S, David-Cordonnier M-H. Targeting transcription factors for cancer treatment. Molecules (2018) 23(6):1479. doi: 10.3390/molecules23061479
115. Kaplan AR, Glazer PM. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis (2020) 35(1):61–8. doi: 10.1093/mutage/gez019
116. Bolland H, Ma TS, Ramlee S, Ramadan K, Hammond EM. Links between the unfolded protein response and the DNA damage response in hypoxia: a systematic review. Biochem Soc Trans (2021) 49(3):1251–63. doi: 10.1042/BST20200861
117. Mirza-Aghazadeh-Attari M, Recio MJ, Darband SG, Kaviani M, Safa A, Mihanfar A, et al. DNA Damage response and breast cancer development: possible therapeutic applications of ATR, ATM, PARP, BRCA1 inhibition. DNA repair (2021) 98:103032. doi: 10.1016/j.dnarep.2020.103032
118. Olcina MM, Grand RJ, Hammond EM. ATM Activation in hypoxia-causes and consequences. Mol Cell Oncol (2014) 1(1):e29903. doi: 10.4161/mco.29903
119. Gibson SL, Bindra RS, Glazer PM. Hypoxia-induced phosphorylation of Chk2 in an ataxia telangiectasia mutated–dependent manner. Cancer Res (2005) 65(23):10734–41. doi: 10.1158/0008-5472.CAN-05-1160
120. Freiberg RA, Hammond EM, Dorie MJ, Welford SM, Giaccia AJ. DNA Damage during reoxygenation elicits a Chk2-dependent checkpoint response. Mol Cell Biol (2006) 26(5):1598–609. doi: 10.1128/MCB.26.5.1598-1609.2006
121. Hammond EM, Denko NC, Dorie MJ, Abraham RT, Giaccia AJ. Hypoxia links ATR and p53 through replication arrest. Mol Cell Biol (2002) 22(6):1834–43. doi: 10.1128/MCB.22.6.1834-1843.2002
122. Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem (2003) 278(14):12207–13. doi: 10.1074/jbc.M212360200
123. Bouquet F, Ousset M, Biard D, Fallone F, Dauvillier S, Frit P, et al. A DNA-dependent stress response involving DNA-PK occurs in hypoxic cells and contributes to cellular adaptation to hypoxia. J Cell science (2011) 124(11):1943–51. doi: 10.1242/jcs.078030
124. Guo X, Lei T, Sun M, Yang C, Fu L, Xu B. ATM Is hyperactivated in triple-negative breast cancer tissues. Cancer Res (2011) 71(8_Supplement):2270–. doi: 10.1158/1538-7445.AM2011-2270
125. Peng M, Yang D, Hou Y, Liu S, Zhao M, Qin Y, et al. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death disease (2019) 10(3):228. doi: 10.1038/s41419-019-1475-7
126. Jin J, Tao Z, Cao J, Li T, Hu X. DNA Damage response inhibitors: an avenue for TNBC treatment. Biochim Biophys Acta (BBA)-Reviews Cancer (2021) 1875(2):188521. doi: 10.1016/j.bbcan.2021.188521
127. Meyer F, Becker S, Classen S, Parplys AC, Mansour WY, Riepen B, et al. Prevention of DNA replication stress by CHK1 leads to chemoresistance despite a DNA repair defect in homologous recombination in breast cancer. Cells (2020) 9(1):238. doi: 10.3390/cells9010238
128. O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MHZ, Weiner-Gorzel K, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA clinical (2015) 3:257–75. doi: 10.1016/j.bbacli.2015.03.003
129. Derakhshan F, Reis-Filho JS. Pathogenesis of triple-negative breast cancer. Annu Rev Pathology: Mech Disease (2022) 17:181–204. doi: 10.1146/annurev-pathol-042420-093238
130. Tan AR, Swain SM. Therapeutic strategies for triple-negative breast cancer. Cancer J (2008) 14(6):343–51. doi: 10.1097/PPO.0b013e31818d839b
131. Clark CA, Yang ES. Harnessing DNA repair defects to augment immune-based therapies in triple-negative breast cancer. Front Oncol (2021) 11:703802. doi: 10.3389/fonc.2021.703802
132. Choi C, Cho WK, Park S, Shin S-W, Park W, Kim H, et al. Checkpoint kinase 1 (CHK1) inhibition enhances the sensitivity of triple-negative breast cancer cells to proton irradiation via Rad51 downregulation. Int J Mol Sci (2020) 21(8):2691. doi: 10.3390/ijms21082691
133. Wu H, Chu Y, Sun S, Li G, Xu S, Zhang X, et al. Hypoxia-mediated complement 1q binding protein regulates metastasis and chemoresistance in triple-negative breast cancer and modulates the PKC-NF-κB-VCAM-1 signaling pathway. Front Cell Dev Biol (2021) 9:607142. doi: 10.3389/fcell.2021.607142
134. Mo W, Liu Q, Lin CC-J, Dai H, Peng Y, Liang Y, et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin Cancer Res (2016) 22(7):1699–712. doi: 10.1158/1078-0432.CCR-15-1772
135. Huang R-X, Zhou P-K. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal transduction targeted Ther (2020) 5(1):60. doi: 10.1038/s41392-020-0150-x
136. Chang HH, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol (2017) 18(8):495–506. doi: 10.1038/nrm.2017.48
137. Zhang Y, He Q, Hu Z, Feng Y, Fan L, Tang Z, et al. Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer. Nat Struct Mol Biol (2016) 23(6):522–30. doi: 10.1038/nsmb.3211
138. Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol (2003) 23(9):3265–73. doi: 10.1128/MCB.23.9.3265-3273.2003
139. Wu S, Shi X, Wang J, Wang X, Liu Y, Luo Y, et al. Triple-negative breast cancer: intact mismatch repair and partial co-expression of PD-L1 and LAG-3. Front Immunol (2021) 12:561793. doi: 10.3389/fimmu.2021.561793
140. Koshiji M, To KK-W, Hammer S, Kumamoto K, Harris AL, Modrich P, et al. HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol Cell (2005) 17(6):793–803. doi: 10.1016/j.molcel.2005.02.015
141. Scanlon SE, Glazer PM. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA repair (2015) 32:180–9. doi: 10.1016/j.dnarep.2015.04.030
142. Lee KJ, Piett CG, Andrews JF, Mann E, Nagel ZD, Gassman NR. Defective base excision repair in the response to DNA damaging agents in triple negative breast cancer. PloS One (2019) 14(10):e0223725. doi: 10.1371/journal.pone.0223725
143. Alli E, Sharma VB, Sunderesakumar P, Ford JM. Defective repair of oxidative dna damage in triple-negative breast cancer confers sensitivity to inhibition of poly (ADP-ribose) polymerase. Cancer Res (2009) 69(8):3589–96. doi: 10.1158/0008-5472.CAN-08-4016
144. Chan N, Ali M, McCallum GP, Kumareswaran R, Koritzinsky M, Wouters BG, et al. Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol Cancer Res (2014) 12(10):1407–15. doi: 10.1158/1541-7786.MCR-14-0246
145. Yuan J, Narayanan L, Rockwell S, Glazer PM. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res (2000) 60(16):4372–6.
146. Lee KJ, Mann E, Wright G, Piett CG, Nagel ZD, Gassman NR. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Ther Adv Med Oncol (2020) 12:1758835920958354. doi: 10.1177/1758835920958354
147. Van Steeg H, De Vries A, Van Oostrom CTM, Van Benthem J, Beems RB, Van Kreijl CF, et al. And Xpa/p53+/-Knock-Out mice: nature of the models. Toxicologic Pathol (2001) 29(1_suppl):109–16. doi: 10.1080/019262301753178519
148. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. 6th Edition, Lippincott Williams and Wilkins, Philadelphia. (2006).
149. Crabtree H, Cramer W. The action of radium on cancer cells. II.–some factors determining the susceptibility of cancer cells to radium. Proc R Soc London Ser B Containing Papers Biol Character (1933) 113(782):238–50.
150. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell (2004) 5(5):429–41. doi: 10.1016/S1535-6108(04)00115-1
151. Klimova T, Chandel N. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differentiation (2008) 15(4):660–6. doi: 10.1038/sj.cdd.4402307
152. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem (2000) 275(33):25130–8. doi: 10.1074/jbc.M001914200
153. Brizel DM, Hage WD, Dodge RK, Munley MT, Piantadosi CA, Dewhirst MW. Hyperbaric oxygen improves tumor radiation response significantly more than carbogen/nicotinamide. Radiat Res (1997) 147(6):715–20. doi: 10.2307/3579485
154. Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia (2005) 21(8):779–90. doi: 10.1080/02656730500271668
155. Overgaard J, Gonzalez DG, Hulshof M, Arcangeli G, Dahl O, Mella O, et al. Hyperthermia as an adjuvant to radiation therapy of recurrent or metastatic malignant melanoma. a multicentre randomized trial by the European society for hyperthermic oncology. Int J hyperthermia (1996) 12(1):3–20. doi: 10.3109/02656739609023685
156. van der Zee J, González D, van Rhoon GC, van Dijk JD, van Putten WL, Hart AA. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Lancet (2000) 355(9210):1119–25. doi: 10.1016/S0140-6736(00)02059-6
157. Jones EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, et al. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol (2005) 23(13):3079–85. doi: 10.1200/JCO.2005.05.520
158. Dewhirst MW, Viglianti B, Lora-Michiels M, Hanson M, Hoopes P. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int J hyperthermia (2003) 19(3):267–94. doi: 10.1080/0265673031000119006
159. Moon EJ, Sonveaux P, Porporato PE, Danhier P, Gallez B, Batinic-Haberle I, et al. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc Natl Acad Sci (2010) 107(47):20477–82. doi: 10.1073/pnas.1006646107
160. van Leeuwen CM, Oei AL, Chin KW, Crezee J, Bel A, Westermann AM, et al. A short time interval between radiotherapy and hyperthermia reduces in-field recurrence and mortality in women with advanced cervical cancer. Radiat Oncol (2017) 12:1–8. doi: 10.1186/s13014-017-0813-0
161. Hua W, Ten Dijke P, Kostidis S, Giera M, Hornsveld M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci (2020) 77(11):2103–23. doi: 10.1007/s00018-019-03398-6
162. Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res (2007) 67(7):3010–7. doi: 10.1158/0008-5472.CAN-06-4328
163. Smith GL, Jiang J, Buchholz TA, Xu Y, Hoffman KE, Giordano SH, et al. Benefit of adjuvant brachytherapy versus external beam radiation for early breast cancer: impact of patient stratification on breast preservation. Int J Radiat Oncol Biol Physics (2014) 88(2):274–84. doi: 10.1016/j.ijrobp.2013.07.011
164. Musielak M, Suchorska WM, Fundowicz M, Milecki P, Malicki J. Future perspectives of proton therapy in minimizing the toxicity of breast cancer radiotherapy. J Personalized Med (2021) 11(5):410. doi: 10.3390/jpm11050410
165. Arpino G, De Placido S, De Angelis C. Nab-paclitaxel for the management of triple-negative metastatic breast cancer: a case study. Anti-Cancer Drugs (2015) 26(1):117. doi: 10.1097/CAD.0000000000000159
166. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2020) 21(1):44–59. doi: 10.1016/S1470-2045(19)30689-8
167. Ademuyiwa FO, Chen I, Luo J, Rimawi MF, Hagemann IS, Fisk B, et al. Immunogenomic profiling and pathological response results from a clinical trial of docetaxel and carboplatin in triple-negative breast cancer. Breast Cancer Res Treat (2021) 189(1):187–202. doi: 10.1007/s10549-021-06307-3
168. Tolaney SM, Bondarenko I, Chan A, Dacosta NA, Izarzugaza Y, Kim GM, et al. CONTESSA TRIO: a multinational, multicenter, phase (P) II study of tesetaxel (T) plus three different PD-(L) 1 inhibitors in patients (Pts) with metastatic triple-negative breast cancer (TNBC) and tesetaxel monotherapy in elderly pts with HER2-metastatic breast cancer (MBC). J Clin Oncol (2020) 38 (15_suppl). doi: 10.1200/JCO.2020.38.15_suppl.TPS1111
169. Basho RK, Gilcrease M, Murthy RK, Helgason T, Karp DD, Meric-Bernstam F, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol (2017) 3(4):509–15. doi: 10.1001/jamaoncol.2016.5281
170. Basho RK, Yam C, Gilcrease M, Murthy RK, Helgason T, Karp DD, et al. Comparative effectiveness of an mTOR-based systemic therapy regimen in advanced, metaplastic and nonmetaplastic triple-negative breast cancer. Oncologist (2018) 23(11):1300–9. doi: 10.1634/theoncologist.2017-0498
171. Zheng F, Du F, Wang W, Wang Y, Li M, Zhao J, et al. Updated efficacy of adjuvant epirubicin plus cyclophosphamide followed by taxanes versus carboplatin plus taxanes in early triple-negative breast cancer in phase 2 trial: 8.1-year median follow-up. Breast Cancer Res Treat (2022), 1–9. doi: 10.1007/s10549-021-06401-6
172. Khallaf SM, Roshdy J, Ibrahim A. Pegylated liposomal doxorubicin in patients with metastatic triple-negative breast cancer: 8-year experience of a single center. J Egyptian Natl Cancer Institute (2020) 32:1–10. doi: 10.1186/s43046-020-00034-4
173. Lien M-Y, Liu L-C, Wang H-C, Yeh M-H, Chen C-J, Yeh S-P, et al. Safety and efficacy of pegylated liposomal doxorubicin-based adjuvant chemotherapy in patients with stage I-III triple-negative breast cancer. Anticancer Res (2014) 34(12):7319–26.
174. Telli ML, Tolaney SM, Shapiro GI, Middleton M, Lord SR, Arkenau HT, et al. Phase 1b study of berzosertib and cisplatin in patients with advanced triple-negative breast cancer. NPJ Breast Cancer (2022) 8(1):45. doi: 10.1038/s41523-022-00406-0
175. Baek DW, Park J-Y, Lee SJ, Chae YS. Impressive effect of cisplatin monotherapy on a patient with heavily pretreated triple-negative breast cancer with poor performance. Yeungnam Univ J Med (2020) 37(3):230–5. doi: 10.12701/yujm.2019.00423
176. Yu K-D, Ye F-G, He M, Fan L, Ma D, Mo M, et al. Effect of adjuvant paclitaxel and carboplatin on survival in women with triple-negative breast cancer: a phase 3 randomized clinical trial. JAMA Oncol (2020) 6(9):1390–6. doi: 10.1001/jamaoncol.2020.2965
177. Twelves C, Cortes J, Vahdat LT, Wanders J, Akerele C, Kaufman PA. Phase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic breast cancer. Clin Breast cancer (2010) 10(2):160–3. doi: 10.3816/CBC.2010.n.023
178. Wang X, Wang S-S, Huang H, Cai L, Zhao L, Peng R-J, et al. Effect of capecitabine maintenance therapy using lower dosage and higher frequency vs observation on disease-free survival among patients with early-stage triple-negative breast cancer who had received standard treatment: the SYSUCC-001 randomized clinical trial. Jama (2021) 325(1):50–8. doi: 10.1001/jama.2020.23370
179. Hyder T, Rosenzweig MQ, Brufsky A. Ixabepilone efficacy and tolerability in metastatic breast cancer (MBC) patients in a real-world setting. J Clin Oncol (2020) 38(15_suppl):e13067–e13067. doi: 10.1200/JCO.2020.38.15_suppl.e13067
180. Carbonaro M, Escuin D, O’Brate A, Thadani-Mulero M, Giannakakou P. Microtubules regulate hypoxia-inducible factor-1α protein trafficking and activity: implications for taxane therapy. J Biol Chem (2012) 287(15):11859–69. doi: 10.1074/jbc.M112.345587
181. Azoury F, Misra S, Barry A, Helou J. Role of radiation therapy in triple negative breast cancer: current state and future directions–a narrative review. Precis Cancer Med (2022) 5(3):21–29. doi: 10.21037/pcm-21-9
182. Vozenin M-C, De Fornel P, Petersson K, Favaudon V, Jaccard M, Germond J-F, et al. The advantage of FLASH radiotherapy confirmed in mini-pig and cat-cancer PatientsThe advantage of flash radiotherapy. Clin Cancer Res (2019) 25(1):35–42. doi: 10.1158/1078-0432.CCR-17-3375
183. Mohamed RF, Bakri HM, Eid S. Does bevacizumab carry a hope for metastatic triple-negative breast cancer in the era of immunotherapy? Anti-Cancer Drugs (2022) 33(1):e604–e9. doi: 10.1097/CAD.0000000000001192
184. Chung HC, Saada-Bouzid E, Munoz F, Yanez E, Im S-A, Castanon E, et al. Lenvatinib plus pembrolizumab for previously treated, advanced triple-negative breast cancer: early results from the multicohort phase 2 LEAP-005 study. Cancer Res (2021) 81(4_Supplement):PS12–07. doi: 10.1158/1538-7445.SABCS20-PS12-07
185. Zhang Q, Shao B, Tong Z, Ouyang Q, Wang Y, Xu G, et al. A phase ib study of camrelizumab in combination with apatinib and fuzuloparib in patients with recurrent or metastatic triple-negative breast cancer. BMC Med (2022) 20(1):321. doi: 10.1186/s12916-022-02527-6
186. Leone JP, Duda DG, Hu J, Barry WT, Trippa L, Gerstner ER, et al. A phase II study of cabozantinib alone or in combination with trastuzumab in breast cancer patients with brain metastases. Breast Cancer Res Treat (2020) 179:113–23. doi: 10.1007/s10549-019-05445-z
187. Wang J, Sun T, Ouyang Q, Han Y, Xu B. A phase Ib study of TQB2450 plus anlotinib in patients with advanced triple-negative breast cancer. iScience (2023) 26(6):106876. doi: 10.1016/j.isci.2023.106876
188. Mast JM, Kuppusamy P. Hyperoxygenation as a therapeutic supplement for treatment of triple negative breast cancer. Front Oncol (2018) 8:527. doi: 10.3389/fonc.2018.00527
189. Sawai H, Kurimoto M, Suzuki Y, Yamaguchi Y, Murata A, Suganuma E, et al. Efficacy of hyperthermia in treatment of recurrent metastatic breast cancer after long-term chemotherapy: a report of 2 cases. Am J Case Rep (2020) 21:e926647–1. doi: 10.12659/AJCR.926647
190. Palasuberniam P, Kraus D, Mansi M, Howley R, Braun A, Myers KA, et al. Small molecule kinase inhibitors enhance aminolevulinic acid-mediated protoporphyrin IX fluorescence and PDT response in triple negative breast cancer cell lines. J Biomed Optics (2021) 26(9):098002–. doi: 10.1117/1.JBO.26.9.098002
191. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase ib KEYNOTE-012 study. J Clin Oncol (2016) 34(21):2460. doi: 10.1200/JCO.2015.64.8931
192. Adams S, Loi S, Toppmeyer D, Cescon D, De Laurentiis M, Nanda R, et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort b of the phase II KEYNOTE-086 study. Ann Oncol (2019) 30(3):405–11. doi: 10.1093/annonc/mdy518
193. Adams S, Schmid P, Rugo H, Winer E, Loirat D, Awada A, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort a of the phase II KEYNOTE-086 study. Ann Oncol (2019) 30(3):397–404. doi: 10.1093/annonc/mdy517
194. Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: a phase 1 study. JAMA Oncol (2019) 5(1):74–82. doi: 10.1001/jamaoncol.2018.4224
195. Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau H-T, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN solid tumor study. Breast Cancer Res Treat (2018) 167:671–86. doi: 10.1007/s10549-017-4537-5
196. Bian L, Zhang H, Wang T, Zhang S, Song H, Xu M, et al. JS001, an anti-PD-1 mAb for advanced triple negative breast cancer patients after multi-line systemic therapy in a phase I trial. Ann Trans Med (2019) 7(18). doi: 10.21037/atm.2019.09.08
197. Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med (2019) 25(6):920–8. doi: 10.1038/s41591-019-0432-4
198. Howard FM, Pearson AT, Nanda R. Clinical trials of immunotherapy in triple-negative breast cancer. Breast Cancer Res Treat (2022) 195(1):1–15. doi: 10.1007/s10549-022-06665-6
199. Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor MicroenvironmentAdenosine impairs proliferation of terminal NK cells. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
200. Bottai G, Raschioni C, Losurdo A, Di Tommaso L, Tinterri C, Torrisi R, et al. An immune stratification reveals a subset of PD-1/LAG-3 double-positive triple-negative breast cancers. Breast Cancer Res (2016) 18(1):1–10. doi: 10.1186/s13058-016-0783-4
201. Kok M. LAG-3: another brake to release in breast cancer? Ann Oncol (2017) 28(12):2907–8. doi: 10.1093/annonc/mdx708
202. Boissière-Michot F, Chateau M-C, Thezenas S, Guiu S, Bobrie A, Jacot W. Correlation of the TIGIT-PVR immune checkpoint axis with clinicopathological features in triple-negative breast cancer. Front Immunol (2022) 13:1058424. doi: 10.3389/fimmu.2022.1058424
203. Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer–expanded options, evolving needs. Nat Rev Clin Oncol (2022) 19(2):91–113. doi: 10.1038/s41571-021-00565-2
204. Sambi M, Samuel V, Qorri B, Haq S, Burov SV, Markvicheva E, et al. A triple combination of metformin, acetylsalicylic acid, and oseltamivir phosphate impacts tumour spheroid viability and upends chemoresistance in triple-negative breast cancer. Drug Design Dev Ther (2020), 1995–2019. doi: 10.2147/DDDT.S242514
205. Somasundaram V, Ridnour LA, Cheng RY, Walke AJ, Kedei N, Bhattacharyya DD, et al. Systemic Nos2 depletion and cox inhibition limits TNBC disease progression and alters lymphoid cell spatial orientation and density. Redox Biol (2022) 58:102529. doi: 10.1016/j.redox.2022.102529
206. Xiong F, Wang Q, Wu G-h, Liu W-z, Wang B, Chen Y-j. Direct and indirect effects of IFN-α2b in malignancy treatment: not only an archer but also an arrow. Biomark Res (2022) 10(1):69. doi: 10.1186/s40364-022-00415-y
207. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia–A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res (2014) 2(7):598–605. doi: 10.1158/2326-6066.CIR-14-0075
208. Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J immunotherapy cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
209. Yi M, Li T, Niu M, Wu Y, Zhao Z, Wu K. TGF-β: a novel predictor and target for anti-PD-1/PD-L1 therapy. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1061394
210. Chung AW, Anand K, Anselme AC, Chan AA, Gupta N, Venta LA, et al. A phase 1/2 clinical trial of the nitric oxide synthase inhibitor l-NMMA and taxane for treating chemoresistant triple-negative breast cancer. Sci Trans Med (2021) 13(624):eabj5070. doi: 10.1126/scitranslmed.abj5070
211. Bai X, Ni J, Beretov J, Graham P, Li Y. Immunotherapy for triple-negative breast cancer: a molecular insight into the microenvironment, treatment, and resistance. J Natl Cancer Center (2021) 1(3):75–87. doi: 10.1016/j.jncc.2021.06.001
212. Wilson BE, Shen Q, Cescon DW, Reedijk M. Exploring immune interactions in triple negative breast cancer: IL-1β inhibition and its therapeutic potential. Front Genet (2023) 14. doi: 10.3389/fgene.2023.1086163
213. Criscitiello C, Viale G, Curigliano G. Peptide vaccines in early breast cancer. Breast (2019) 44:128–34. doi: 10.1016/j.breast.2019.02.003
214. Vecchi L, Mota STS, Zóia MAP, Martins IC, de Souza JB, Santos TG, et al. Interleukin-6 signaling in triple negative breast cancer cells elicits the annexin A1/Formyl peptide receptor 1 axis and affects the tumor microenvironment. Cells (2022) 11(10):1705. doi: 10.3390/cells11101705
215. Li Y, Zhang H, Merkher Y, Chen L, Liu N, Leonov S, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol (2022) 15(1):121. doi: 10.1186/s13045-022-01341-0
216. Hosseini M, Seyedpour S, Khodaei B, Loghman A-H, Seyedpour N, Yazdi M-H, et al. Cancer vaccines for triple-negative breast cancer: a systematic review. Vaccines (2023) 11(1):146. doi: 10.3390/vaccines11010146
217. Lopez JS, Camidge R, Iafolla M, Rottey S, Schuler M, Hellmann M, et al. Abstract CT301: a phase ib study to evaluate RO7198457, an individualized neoantigen specific immunoTherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. Cancer Res (2020) 80(16_Supplement):CT301–CT. doi: 10.1158/1538-7445.AM2020-CT301
218. Willis W, Jackman M, Bizeau M, Pagliassotti M, Hazel J. Hyperthermia impairs liver mitochondrial function in vitro. Am J Physiology-Regulatory Integr Comp Physiol (2000) 278(5):R1240–R6. doi: 10.1152/ajpregu.2000.278.5.R1240
219. Ito A, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol Immunother (2001) 50(10):515–22. doi: 10.1007/s00262-001-0233-7
220. Mace TA, Zhong L, Kokolus KM, Repasky EA. Effector CD8+ T cell IFN-γ production and cytotoxicity are enhanced by mild hyperthermia. Int J Hyperthermia (2012) 28(1):9–18. doi: 10.3109/02656736.2011.616182
221. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev cancer (2003) 3(10):721–32. doi: 10.1038/nrc1187
222. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol (2006) 70(5):1469–80. doi: 10.1124/mol.106.027029
223. An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature (1998) 392(6674):405–8. doi: 10.1038/32925
224. Pan Y, Oprysko PR, Asham AM, Koch CJ, Simon MC. p53 cannot be induced by hypoxia alone but responds to the hypoxic microenvironment. Oncogene (2004) 23(29):4975–83. doi: 10.1038/sj.onc.1207657
225. Fels DR, Koumenis C. HIF-1α and p53: the ODD couple? Trends Biochem Sci (2005) 30(8):426–9. doi: 10.1016/j.tibs.2005.06.009
226. Brufsky A, Valero V, Tiangco B, Dakhil S, Brize A, Rugo HS, et al. Second-line bevacizumab-containing therapy in patients with triple-negative breast cancer: subgroup analysis of the RIBBON-2 trial. Breast Cancer Res Treat (2012) 133:1067–75. doi: 10.1007/s10549-012-2008-6
227. Vahdat LT, Layman R, Yardley DA, Gradishar W, Salkeni MA, Joy AA, et al. Randomized phase II study of ramucirumab or icrucumab in combination with capecitabine in patients with previously treated locally advanced or metastatic breast cancer. oncologist (2017) 22(3):245–54. doi: 10.1634/theoncologist.2016-0265
228. Ning W-J, Liu X, Zeng H-Y, Z-q An, Luo W-X, Xia N-S. Recent progress in antibody-based therapeutics for triple-negative breast cancer. Expert Opin Drug Delivery (2022) 19(7):815–32. doi: 10.1080/17425247.2022.2093853
229. Ayoub NM, Jaradat SK, Al-Shami KM, Alkhalifa AE. Targeting angiogenesis in breast cancer: current evidence and future perspectives of novel anti-angiogenic approaches. Front Pharmacol (2022) 13. doi: 10.3389/fphar.2022.838133
230. Green M, Manikhas G, Orlov S, Afanasyev B, Makhson A, Bhar P, et al. Abraxane®, a novel cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann Oncol (2006) 17(8):1263–8. doi: 10.1093/annonc/mdl104
231. Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resistance Updates (2021) 100787. doi: 10.1016/j.drup.2021.100787
232. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
233. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
234. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
235. Horsman M, Sørensen B, Busk M, Siemann D. Therapeutic modification of hypoxia. Clin Oncol (2021) 33(11):e492–509. doi: 10.1016/j.clon.2021.08.014
236. Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med (2009) 9(4):442–58. doi: 10.2174/156652409788167087
237. Horsman MR, Overgaard J. The impact of hypoxia and its modification of the outcome of radiotherapy. J Radiat Res (2016) 57(S1):i90–i8. doi: 10.1093/jrr/rrw007
238. Chaplin D, Horsman M, Siemann D. Further evaluation of nicotinamide and carbogen as a strategy to reoxygenate hypoxic cells in vivo: importance of nicotinamide dose and pre-irradiation breathing time. Br J cancer (1993) 68(2):269–73. doi: 10.1038/bjc.1993.326
239. Banerjee SM, El-Sheikh S, Malhotra A, Mosse CA, Parker S, Williams NR, et al. Photodynamic therapy in primary breast cancer. J Clin Med (2020) 9(2):483. doi: 10.3390/jcm9020483
240. Siemann DW, Hill RP, Bush RS. The importance of the pre-irradiation breathing times of oxygen and carbogen (5% CO2: 95% O2) on the in vivo radiation response of a murine sarcoma. Int J Radiat Oncol Biol Phys (1977) 2(9-10):903–11. doi: 10.1016/0360-3016(77)90188-2
241. van Laarhoven HW, Bussink J, Lok J, Punt CJ, Heerschap A, van der Kogel AJ. Effects of nicotinamide and carbogen in different murine colon carcinomas: immunohistochemical analysis of vascular architecture and microenvironmental parameters. Int J Radiat Oncol Biol Physics (2004) 60(1):310–21. doi: 10.1016/j.ijrobp.2004.05.014
242. Kaanders JH, Bussink J, van der Kogel AJ. ARCON: a novel biology-based approach in radiotherapy. Lancet Oncol (2002) 3(12):728–37. doi: 10.1016/S1470-2045(02)00929-4
243. Hoskin P, Rojas A, Saunders M. Accelerated radiotherapy, carbogen, and nicotinamide (ARCON) in the treatment of advanced bladder cancer: mature results of a phase II nonrandomized study. Int J Radiat Oncol Biol Physics (2009) 73(5):1425–31. doi: 10.1016/j.ijrobp.2008.06.1950
244. Janssens GO, Rademakers SE, Terhaard CH, Doornaert PA, Bijl HP, van den Ende P, et al. Improved recurrence-free survival with ARCON for anemic patients with laryngeal CancerARCON and cancer-related anemia. Clin Cancer Res (2014) 20(5):1345–54. doi: 10.1158/1078-0432.CCR-13-1730
245. Oei AL, van Leeuwen CM, ten Cate R, Rodermond HM, Buist MR, Stalpers LJ, et al. Hyperthermia selectively targets human papillomavirus in cervical tumors via p53-dependent apoptosis. Cancer Res (2015) 75(23):5120–9. doi: 10.1158/0008-5472.CAN-15-0816
246. Petrazzuolo A, Maiuri MC, Zitvogel L, Kroemer G, Kepp O. Trial watch: combination of tyrosine kinase inhibitors (TKIs) and immunotherapy. Oncoimmunology (2022) 11(1):2077898. doi: 10.1080/2162402X.2022.2077898
247. Ruan L, Zhang S, Chen X, Liang W, Xie Q. Role of anti-angiogenic factors in the pathogenesis of breast cancer: a review of therapeutic potential. Pathology-Research Practice (2022) 153956. doi: 10.1016/j.prp.2022.153956
248. Ribatti D, Nico B, Ruggieri S, Tamma R, Simone G, Mangia A. Angiogenesis and antiangiogenesis in triple-negative breast cancer. Trans Oncol (2016) 9(5):453–7. doi: 10.1016/j.tranon.2016.07.002
249. Brufsky AM, Hurvitz S, Perez E, Swamy R, Valero V, O’Neill V, et al. RIBBON-2: a randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of bevacizumab in combination with chemotherapy for second-line treatment of human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol (2011) 29(32):4286–93. doi: 10.1200/JCO.2010.34.1255
250. Robert NJ, Saleh MN, Paul D, Generali D, Gressot L, Copur MS, et al. Sunitinib plus paclitaxel versus bevacizumab plus paclitaxel for first-line treatment of patients with advanced breast cancer: a phase III, randomized, open-label trial. Clin Breast cancer (2011) 11(2):82–92. doi: 10.1016/j.clbc.2011.03.005
251. Bear HD, Tang G, Rastogi P, Geyer CE Jr., Robidoux A, Atkins JN, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. New Engl J Med (2012) 366(4):310–20. doi: 10.1056/NEJMoa1111097
252. Czarnecka-Czapczyńska M, Aebisher D, Oleś P, Sosna B, Krupka-Olek M, Dynarowicz K, et al. The role of photodynamic therapy in breast cancer–a review of in vitro research. Biomedicine Pharmacotherapy (2021) 144:112342. doi: 10.1016/j.biopha.2021.112342
253. Sun J, Zhao H, Fu L, Cui J, Yang Y. Global trends and research progress of photodynamic therapy in skin cancer: a bibliometric analysis and literature review. Clinical Cosmetic Investigational Dermatol (2023), 479–98. doi: 10.2147/CCID.S401206
254. Huis In 't Veld RV, Heuts J, Ma S, Cruz LJ, Ossendorp FA, Jager MJ. Current challenges and opportunities of photodynamic therapy against cancer. Pharmaceutics (2023) 15(2):330. doi: 10.3390/pharmaceutics15020330
255. Tampa M, Sarbu M-I, Matei C, Mitran C-I, Mitran M-I, Caruntu C, et al. Photodynamic therapy: a hot topic in dermato-oncology. Oncol letters (2019) 17(5):4085–93. doi: 10.3892/ol.2019.9939
256. Civantos FJ, Karakullukcu B, Biel M, Silver CE, Rinaldo A, Saba NF, et al. A review of photodynamic therapy for neoplasms of the head and neck. Adv Ther (2018) 35:324–40. doi: 10.1007/s12325-018-0659-3
257. Xue Q, Zhang J, Jiao J, Qin W, Yang X. Photodynamic therapy for prostate cancer: recent advances, challenges and opportunities. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.980239
258. Karimnia V, Slack FJ, Celli JP. Photodynamic therapy for pancreatic ductal adenocarcinoma. Cancers (2021) 13(17):4354. doi: 10.3390/cancers13174354
259. Yang H, Liu R, Xu Y, Qian L, Dai Z. Photosensitizer nanoparticles boost photodynamic therapy for pancreatic cancer treatment. Nano-Micro Letters (2021) 13:1–16. doi: 10.1007/s40820-020-00561-8
260. Morrison SA, Hill SL, Rogers GS, Graham RA. Efficacy and safety of continuous low-irradiance photodynamic therapy in the treatment of chest wall progression of breast cancer. J Surg Res (2014) 192(2):235–41. doi: 10.1016/j.jss.2014.06.030
261. Montazerabadi AR, Sazgarnia A, Bahreyni-Toosi MH, Ahmadi A, Aledavood A. The effects of combined treatment with ionizing radiation and indocyanine green-mediated photodynamic therapy on breast cancer cells. J Photochem Photobiol B: Biol (2012) 109:42–9. doi: 10.1016/j.jphotobiol.2012.01.004
262. Lamberti MJ, Vittar NBR, Rivarola VA. Breast cancer as photodynamic therapy target: enhanced therapeutic efficiency by overview of tumor complexity. World J Clin Oncol (2014) 5(5):901. doi: 10.5306/wjco.v5.i5.901
263. Zhang B, Xue R, Lyu J, Gao A, Sun C. Tumor acidity/redox hierarchical-activable nanoparticles for precise combination of X-ray-induced photodynamic therapy and hypoxia-activated chemotherapy. J Materials Chem B (2022) 10(20):3849–60. doi: 10.1039/D2TB00303A
264. Mayahi S, Neshasteh-Riz A, Pornour M, Eynali S, Montazerabadi A. Investigation of combined photodynamic and radiotherapy effects of gallium phthalocyanine chloride on MCF-7 breast cancer cells. JBIC J Biol Inorganic Chem (2020) 25:39–48. doi: 10.1007/s00775-019-01730-w
265. Sazgarnia A, Montazerabadi AR, Bahreyni-Toosi MH, Ahmadi A, Aledavood A. In vitro survival of MCF-7 breast cancer cells following combined treatment with ionizing radiation and mitoxantrone-mediated photodynamic therapy. Photodiagnosis Photodyn Ther (2013) 10(1):72–8. doi: 10.1016/j.pdpdt.2012.06.001
266. Abdou Y, Goudarzi A, Yu JX, Upadhaya S, Vincent B, Carey LA. Immunotherapy in triple negative breast cancer: beyond checkpoint inhibitors. NPJ Breast Cancer (2022) 8(1):121. doi: 10.1038/s41523-022-00486-y
267. Fuentes-Antrás J, Guevara-Hoyer K, Baliu-Piqué M, García-Sáenz J, Pérez-Segura P, Pandiella A, et al. Adoptive cell therapy in breast cancer: a current perspective of next-generation medicine. Front Oncol (2020) 10:605633. doi: 10.3389/fonc.2020.605633
268. Fuentes-Antrás J, Genta S, Vijenthira A, Siu LL. Antibody–drug conjugates: in search of partners of choice. Trends Cancer (2023) 9(4):339–54. doi: 10.1016/j.trecan.2023.01.003
269. Nagayama A, Vidula N, Ellisen L, Bardia A. Novel antibody–drug conjugates for triple negative breast cancer. Ther Adv Med Oncol (2020) 12:1758835920915980. doi: 10.1177/1758835920915980
270. Song H, He R, Wang K, Ruan J, Bao C, Li N, et al. Anti-HIF-1α antibody-conjugated pluronic triblock copolymers encapsulated with paclitaxel for tumor targeting therapy. Biomaterials (2010) 31(8):2302–12. doi: 10.1016/j.biomaterials.2009.11.067
271. Han J, Yun J, Quan M, Kang W, Jung J-G, Heo W, et al. JAK2 regulates paclitaxel resistance in triple negative breast cancers. J Mol Med (2021) 99:1783–95. doi: 10.1007/s00109-021-02138-3
272. Adams S, Diamond JR, Hamilton E, Pohlmann PR, Tolaney SM, Chang C-W, et al. Atezolizumab plus nab-paclitaxel in the treatment of metastatic triple-negative breast cancer with 2-year survival follow-up: a phase 1b clinical trial. JAMA Oncol (2019) 5(3):334–42. doi: 10.1001/jamaoncol.2018.5152
273. Schmid P, Im S-A, Armstrong A, Park YH, Chung W-P, Nowecki Z, et al. BEGONIA: phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)–initial results from arm 1, d+ paclitaxel (P), and arm 6, d+ trastuzumab deruxtecan (T-DXd). Wolters Kluwer Health (2021) 39:1023. doi: 10.1200/JCO.2021.39.15_suppl.1023
274. Tolaney SM, Kalinsky K, Kaklamani VG, D’Adamo DR, Aktan G, Tsai ML, et al. Eribulin Plus Pembrolizumab in Patients with Metastatic Triple-Negative Breast Cancer (ENHANCE 1): A Phase Ib/II Study. Clin Cancer Res (2021) 27(11):3061–8. doi: 10.1158/1078-0432.CCR-20-4726
275. Thomas R, Al-Khadairi G, Decock J. Immune checkpoint inhibitors in triple negative breast cancer treatment: promising future prospects. Front Oncol (2021) 10:600573. doi: 10.3389/fonc.2020.600573
276. Brufsky A, Kim S, Zvirbule Ž, Eniu A, Mebis J, Sohn J, et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): primary analysis. Ann Oncol (2021) 32(5):652–60. doi: 10.1016/j.annonc.2021.01.065
277. Miles D, Gligorov J, André F, Cameron D, Schneeweiss A, Barrios C, et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol (2021) 32(8):994–1004. doi: 10.1016/j.annonc.2021.05.801
278. Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol (2020) 6(5):676–84. doi: 10.1001/jamaoncol.2019.6650
279. Schmid P, Salgado R, Park Y, Muñoz-Couselo E, Kim S, Sohn J, et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann Oncol (2020) 31(5):569–81. doi: 10.1016/j.annonc.2020.01.072
280. Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet (2020) 396(10257):1090–100. doi: 10.1016/S0140-6736(20)31953-X
281. Schmid P, Cortes J, Dent R, Pusztai L, McArthur H, Kümmel S, et al. VP7-2021: KEYNOTE-522: phase III study of neoadjuvant pembrolizumab+ chemotherapy vs. placebo+ chemotherapy, followed by adjuvant pembrolizumab vs. placebo for early-stage TNBC. Ann Oncol (2021) 32(9):1198–200. doi: 10.1016/j.annonc.2021.06.014
282. Won KA, Spruck C. Triple−negative breast cancer therapy: current and future perspectives. Int J Oncol (2020) 57(6):1245–61. doi: 10.3892/ijo.2020.5135
283. Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol (2020) 21(9):1155–64. doi: 10.1016/S1470-2045(20)30324-7
284. Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol (2019) 5(8):1132–40. doi: 10.1001/jamaoncol.2019.1029
285. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat Rev (2011) 37(1):63–74. doi: 10.1016/j.ctrv.2010.05.001
286. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death disease (2018) 9(2):115. doi: 10.1038/s41419-017-0061-0
287. Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC. The clinical importance of assessing tumor hypoxia: relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxidants Redox Signaling (2014) 21(10):1516–54. doi: 10.1089/ars.2013.5378
288. Hapke RY, Haake SM. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer letters (2020) 487:10–20. doi: 10.1016/j.canlet.2020.05.012
289. Yeo CD, Kang N, Choi SY, Kim BN, Park CK, Kim JW, et al. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: a possible link to epigenetic regulation. Korean J Internal Med (2017) 32(4):589–99. doi: 10.3904/kjim.2016.302
290. Graham K, Unger E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J nanomedicine (2018) 13:6049. doi: 10.2147/IJN.S140462
291. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J cancer (1955) 9(4):539. doi: 10.1038/bjc.1955.55
292. Du Sault LA. The effect of oxygen on the response of spontaneous tumours in mice to radiotherapy. Br J radiology (1963) 36(430):749–54. doi: 10.1259/0007-1285-36-430-749
293. Young S, Marshall R, Hill R. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci (1988) 85(24):9533–7. doi: 10.1073/pnas.85.24.9533
294. Rofstad EK, Galappathi K, Mathiesen B, Ruud E-BM. Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin Cancer Res (2007) 13(7):1971–8. doi: 10.1158/1078-0432.CCR-06-1967
295. Hill R, De Jaeger K, Jang A, Cairns R. (2001). “pH, hypoxia and metastasis.” In The Tumour Microenvironment: Causes and Consequences of Hypoxia and Acidity: Novartis Foundation Symposium 240:pp. 154–68. Chichester, UK: John Wiley & Sons, Ltd.
296. Höckel M, Schlenger K, Aral B, Mitze M, Schäffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res (1996) 56(19):4509–15.
297. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. Int multi-center study Radiotherapy Oncol (2005) 77(1):18–24. doi: 10.1016/j.radonc.2005.06.038
298. Mayer A, Höckel M, Wree A, Leo C, Horn L-C, Vaupel P. Lack of hypoxic response in uterine leiomyomas despite severe tissue hypoxia. Cancer Res (2008) 68(12):4719–26. doi: 10.1158/0008-5472.CAN-07-6339
299. Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal (2007) 9(8):1221–35. doi: 10.1089/ars.2007.1628
300. Moulder JE, Rockwell S. Hypoxic fractions of solid tumors: experimental techniques, methods of analysis, and a survey of existing data. Int J Radiat Oncol Biol Physics (1984) 10(5):695–712. doi: 10.1016/0360-3016(84)90301-8
301. Zeng W, Liu P, Pan W, Singh SR, Wei Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer letters (2015) 356(2):263–7. doi: 10.1016/j.canlet.2014.01.032
302. Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduction Targeted Ther (2023) 8(1):70. doi: 10.1038/s41392-023-01332-8
303. Sun X, Luo H, Han C, Zhang Y, Yan C. Identification of a hypoxia-related molecular classification and hypoxic tumor microenvironment signature for predicting the prognosis of patients with triple-negative breast cancer. Front Oncol (2021) 11:700062. doi: 10.3389/fonc.2021.700062
304. Bhuria V, Xing J, Scholta T, Bui KC, Nguyen MLT, Malek NP, et al. Hypoxia induced sonic hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp Cell Res (2019) 385(2):111671. doi: 10.1016/j.yexcr.2019.111671
305. Lu H, Tran L, Park Y, Chen I, Lan J, Xie Y, et al. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy-induced breast cancer stem cell EnrichmentHIF1 regulates DUSP9 and DUSP16 to induce BCSC enrichment. Cancer Res (2018) 78(15):4191–202. doi: 10.1158/0008-5472.CAN-18-0270
306. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest (2011) 121(7):2750–67. doi: 10.1172/JCI45014
307. Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, et al. Phase II trial of bicalutamide in patients with androgen receptor–positive, estrogen receptor–negative metastatic breast cancer. Clin Cancer Res (2013) 19(19):5505–12. doi: 10.1158/1078-0432.CCR-12-3327
308. Liu J, Hu X, Feng L, Lin Y, Liang S, Zhu Z, et al. Carbonic anhydrase IX-targeted h-APBC nanosystem combined with phototherapy facilitates the efficacy of PI3K/mTOR inhibitor and resists HIF-1α-dependent tumor hypoxia adaptation. J Nanobiotechnology (2022) 20(1):187. doi: 10.1186/s12951-022-01394-w
309. Hunter FW, Hsu H-L, Su J, Pullen SM, Wilson WR, Wang J. Dual targeting of hypoxia and homologous recombination repair dysfunction in triple-negative breast CancerTargeting hypoxia and HR repair defects in TNBC. Mol Cancer Ther (2014) 13(11):2501–14. doi: 10.1158/1535-7163.MCT-14-0476
310. Guo C-H, Hsia S, Chung C-H, Lin Y-C, Shih M-Y, Chen P-C, et al. Nutritional supplements in combination with chemotherapy or targeted therapy reduces tumor progression in mice bearing triple-negative breast cancer. J Nutr Biochem (2021) 87:108504. doi: 10.1016/j.jnutbio.2020.108504
311. Abou Khouzam R, Zaarour RF, Brodaczewska K, Azakir B, Venkatesh GH, Thiery J, et al. The effect of hypoxia and hypoxia-associated pathways in the regulation of antitumor response: friends or foes? Front Immunol (2022) 13:337. doi: 10.3389/fimmu.2022.828875
312. Verma NK, Wong BHS, Poh ZS, Udayakumar A, Verma R, Goh RKJ, et al. Obstacles for T-lymphocytes in the tumour microenvironment: therapeutic challenges, advances and opportunities beyond immune checkpoint. EBioMedicine (2022) 83:104216. doi: 10.1016/j.ebiom.2022.104216
313. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature (2020) 579(7798):284–90. doi: 10.1038/s41586-020-2054-x
314. Xie W, Zhao H, Wang F, Wang Y, He Y, Wang T, et al. A novel humanized frizzled-7-targeting antibody enhances antitumor effects of bevacizumab against triple-negative breast cancer via blocking wnt/β-catenin signaling pathway. J Exp Clin Cancer Res (2021) 40:1–18. doi: 10.1186/s13046-020-01800-x
315. Yehia L, Boulos F, Jabbour M, Mahfoud Z, Fakhruddin N, El-Sabban M. Expression of HIF-1α and markers of angiogenesis are not significantly different in triple negative breast cancer compared to other breast cancer molecular subtypes: implications for future therapy. PloS One (2015) 10(6):e0129356. doi: 10.1371/journal.pone.0129356
316. Zhang J, Zhang Y, Mo F, Patel G, Butterworth K, Shao C, et al. The roles of HIF-1α in radiosensitivity and radiation-induced bystander effects under hypoxia. Front Cell Dev Biol (2021) 9:637454. doi: 10.3389/fcell.2021.637454
317. Harada H, Inoue M, Itasaka S, Hirota K, Morinibu A, Shinomiya K, et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat Commun (2012) 3(1):783. doi: 10.1038/ncomms1786
318. Sharma A, Sinha S, Shrivastava N. Therapeutic targeting hypoxia-inducible factor (HIF-1) in cancer: cutting Gordian knot of cancer cell metabolism. Front Genet (2022) 13. doi: 10.3389/fgene.2022.849040
319. Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B (2015) 5(5):378–89. doi: 10.1016/j.apsb.2015.05.007
320. Ghosh R, Samanta P, Sarkar R, Biswas S, Saha P, Hajra S, et al. Targeting HIF-1α by natural and synthetic compounds: a promising approach for anti-cancer therapeutics development. Molecules (2022) 27(16):5192.
321. Wang T, Meng J, Wang C, Wen T, Jia M, Ge Y, et al. Inhibition of murine breast cancer metastases by hydrophilic As4S4 nanoparticles is associated with decreased ROS and HIF-1α downregulation. Front Oncol (2019) 9:333. doi: 10.3389/fonc.2019.00333
322. Jin J, Qiu S, Wang P, Liang X, Huang F, Wu H, et al. Cardamonin inhibits breast cancer growth by repressing HIF-1α-dependent metabolic reprogramming. J Exp Clin Cancer Res (2019) 38(1):377. doi: 10.1186/s13046-019-1351-4
323. Minami T, Matsueda S, Takedatsu H, Tanaka M, Noguchi M, Uemura H, et al. Identification of SART3-derived peptides having the potential to induce cancer-reactive cytotoxic T lymphocytes from prostate cancer patients with HLA-A3 supertype alleles. Cancer immunology Immunother (2007) 56(5):689–98. doi: 10.1007/s00262-006-0216-9
324. Li X, Yue Y, Zhou Y, Fan Y, Fan C, Huang Y, et al. An oil-free microemulsion for intravenous delivery of diallyl trisulfide: formulation and evaluation. Int J pharmaceutics (2011) 407(1-2):158–66. doi: 10.1016/j.ijpharm.2011.01.008
325. Han B, Wang T, Xue Z, Wen T, Lu L, Meng J, et al. Elemene nanoemulsion inhibits metastasis of breast cancer by ROS scavenging. Int J Nanomedicine (2021) 16:6035. doi: 10.2147/IJN.S327094
326. Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N, et al. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (2014) 92:151–64. doi: 10.1007/s00109-013-1102-5
327. Wang K-L, Hsia S-M, Chan C-J, Chang F-Y, Huang C-Y, Bau D-T, et al. Inhibitory effects of isoliquiritigenin on the migration and invasion of human breast cancer cells. Expert Opin Ther targets (2013) 17(4):337–49. doi: 10.1517/14728222.2013.756869
328. Bailey CM, Liu Y, Peng G, Zhang H, He M, Sun D, et al. Liposomal formulation of HIF-1α inhibitor echinomycin eliminates established metastases of triple-negative breast cancer. Nanomedicine: Nanotechnology Biol Med (2020) 29:102278. doi: 10.1016/j.nano.2020.102278
329. Su Q, Wang J, Wu Q, Ullah A, Ghauri MA, Sarwar A, et al. Sanguinarine combats hypoxia-induced activation of EphB4 and HIF-1α pathways in breast cancer. Phytomedicine (2021) 84:153503. doi: 10.1016/j.phymed.2021.153503
330. Yeo E-J, Ryu J-H, Cho Y-S, Chun Y-S, Huang LE, Kim M-S, et al. Amphotericin b blunts erythropoietin response to hypoxia by reinforcing FIH-mediated repression of HIF-1. Blood (2006) 107(3):916–23. doi: 10.1182/blood-2005-06-2564
331. Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang B-H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J (2005) 19(3):342–53. doi: 10.1096/fj.04-2175com
332. Yu K-H, Hung H-Y. Synthetic strategy and structure–activity relationship (SAR) studies of 3-(5′-Hydroxymethyl-2′-Furyl)-1-benzyl indazole (YC-1, lificiguat): a review. RSC advances (2022) 12(1):251–64. doi: 10.1039/D1RA08120A
333. Ramanathan RK, Abbruzzese J, Dragovich T, Kirkpatrick L, Guillen JM, Baker AF, et al. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer chemotherapy Pharmacol (2011) 67:503–9. doi: 10.1007/s00280-010-1343-8
334. Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol Cancer Ther (2003) 2(3):235–43.
335. Olenyuk BZ, Zhang G-J, Klco JM, Nickols NG, Kaelin WG Jr., Dervan PB. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc Natl Acad Sci (2004) 101(48):16768–73. doi: 10.1073/pnas.0407617101
336. Cavell BE, Syed Alwi SS, Donlevy AM, Proud CG, Packham G. Natural product-derived antitumor compound phenethyl isothiocyanate inhibits mTORC1 activity via TSC2. J Natural Products (2012) 75(6):1051–7. doi: 10.1021/np300049b
337. Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol Cancer Ther (2004) 3(3):233–44. doi: 10.1158/1535-7163.233.3.3
338. Garcia-Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1α and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene (2009) 28(3):313–24. doi: 10.1038/onc.2008.398
339. Jun H-O, Kim Y, Kwon Y-W, Hong S-S, Kim K-W, Shin J, et al. Wondonin, a novel compound, inhibits hypoxia-induced angiogenesis through hypoxia-inducible factor 1 alpha. FEBS letters (2007) 581(25):4977–82. doi: 10.1016/j.febslet.2007.09.034
340. Kim DH, Sung B, Kang YJ, Hwang SY, Kim MJ, Yoon J-H, et al. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int J Oncol (2015) 47(6):2226–32. doi: 10.3892/ijo.2015.3200
341. Zheng Z, Zhou Z, Zhang Q, Zhou X, Yang J, Yang M-R, et al. Non-classical cardenolides from calotropis gigantea exhibit anticancer effect as HIF-1 inhibitors. Bioorganic Chem (2021) 109:104740. doi: 10.1016/j.bioorg.2021.104740
342. Bhowmik A, Chakravarti S, Ghosh A, Shaw R, Bhandary S, Bhattacharyya S, et al. Anti-SSTR2 peptide based targeted delivery of potent PLGA encapsulated 3, 3’-diindolylmethane nanoparticles through blood brain barrier prevents glioma progression. Oncotarget (2017) 8(39):65339–65358. doi: 10.18632/oncotarget.18689
343. Li M-H, Miao Z-H, Tan W-F, Yue J-M, Zhang C, Lin L-P, et al. Pseudolaric acid b inhibits angiogenesis and reduces hypoxia-inducible factor 1α by promoting proteasome-mediated degradation. Clin Cancer Res (2004) 10(24):8266–74. doi: 10.1158/1078-0432.CCR-04-0951
344. Cui K, Liu J, Huang L, Qin B, Yang X, Li L, et al. Andrographolide attenuates choroidal neovascularization by inhibiting the HIF-1α/VEGF signaling pathway. Biochem Biophys Res Commun (2020) 530(1):60–6. doi: 10.1016/j.bbrc.2020.06.130
345. Sarighieh MA, Montazeri V, Shadboorestan A, Ghahremani MH, Ostad SN. The inhibitory effect of curcumin on hypoxia inducer factors (Hifs) as a regulatory factor in the growth of tumor cells in breast cancer stem-like cells. Drug Res (2020) 70(11):512–8. doi: 10.1055/a-1201-2602
346. Awan FT, Jones JA, Maddocks K, Poi M, Grever MR, Johnson A, et al. A phase 1 clinical trial of flavopiridol consolidation in chronic lymphocytic leukemia patients following chemoimmunotherapy. Ann hematology (2016) 95:1137–43. doi: 10.1007/s00277-016-2683-1
Keywords: Hypoxia, HIF-1, TNBC, immune escape, DNA damage response, chemotherapy, immunotherapy, cancer vaccines
Citation: Srivastava N, Usmani SS, Subbarayan R, Saini R and Pandey PK (2023) Hypoxia: syndicating triple negative breast cancer against various therapeutic regimens. Front. Oncol. 13:1199105. doi: 10.3389/fonc.2023.1199105
Received: 02 April 2023; Accepted: 05 June 2023;
Published: 10 July 2023.
Edited by:
Subhadeep Roy, National Institute of Pharmaceutical Education and Research, IndiaReviewed by:
Abhishek Basu, National Institutes of Health (NIH), United StatesCopyright © 2023 Srivastava, Usmani, Subbarayan, Saini and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nityanand Srivastava, bml0eWFuYW5kLnNyaXZhc3RhdmFAZWluc3RlaW5tZWQuZWR1; Salman Sadullah Usmani, c2FsbWFuLnVzbWFuaUBlaW5zdGVpbm1lZC5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.