 Teng Tu
Teng Tu Hongying Zhang2†
Hongying Zhang2† Huanji Xu
Huanji Xu- 1Department of Medical Oncology, Cancer Center and Laboratory of Molecular Targeted Therapy in Oncology, West China Hospital, Sichuan University, Chengdu, China
- 2Laboratory of Oncogene, West China Hospital, Sichuan University, Chengdu, China
Cholesterol esterification is often dysregulated in cancer. Sterol O-acyl-transferase 1 (SOAT1) plays an important role in maintaining cellular cholesterol homeostasis by catalyzing the formation of cholesterol esters from cholesterol and long-chain fatty acids in cells. Many studies have implicated that SOAT1 plays a vital role in cancer initiation and progression and is an attractive target for novel anticancer therapy. In this review, we provide an overview of the mechanism and regulation of SOAT1 in cancer and summarize the updates of anticancer therapy targeting SOAT1.
Introduction
Sterol O-acyl-transferase 1 (SOAT1), also known as acyl-CoA:cholesterol acyltransferase 1 (ACAT1), is an exclusively intracellular enzyme that catalyzes the formation of cholesterol esters from cholesterol and long-chain fatty acids in cells. It plays an important role in maintaining cellular cholesterol homeostasis (1–3). In the early time, SOAT1 has been studied extensively as a potential drug target in atherosclerosis and Alzheimer’s disease (4, 5). In recent years, many studies have found that tumors such as glioblastoma and pancreatic cancer exhibit high expression of SOAT1 accompanied by high cholesteryl esters, indicating that SOAT1 is also crucial for cancer cells to regulate cholesterol metabolic homeostasis (6). Compelling evidence have implicated that targeting SOAT1 is a promising therapeutic strategy for cancer management. In this review, we mainly discuss the cholesterol metabolism centered on SOAT1 and mechanisms that regulate SOAT1 expression in cancers. Finally, the latest developments that target SOAT1 for cancer treatment are summarized.
Cholesterol metabolism centered on SOAT1
Cholesterol can maintain membrane fluidity and integrity and form membrane microstructures as an essential lipid component of the mammalian cell membrane (7). Intracellular cholesterol metabolic homeostasis is determined by a complex network that regulates cholesterol biosynthesis, uptake, export, and esterification (Figure 1) (8). Almost all mammalian cells can de novo synthesize cholesterol from acetyl-CoA to cholesterol through more than 20 enzymatic reactions. Besides that way, most cells absorb cholesterol from low-density lipoprotein (LDL) via LDLR-mediated endocytosis (9), where the complex is internalized into the cell endosome by endocytosis with the binding of LDL to LDLR. In the endosome, LDL is dissociated from LDLR and further transferred to the lysosome, where the free cholesterol is released from LDL.
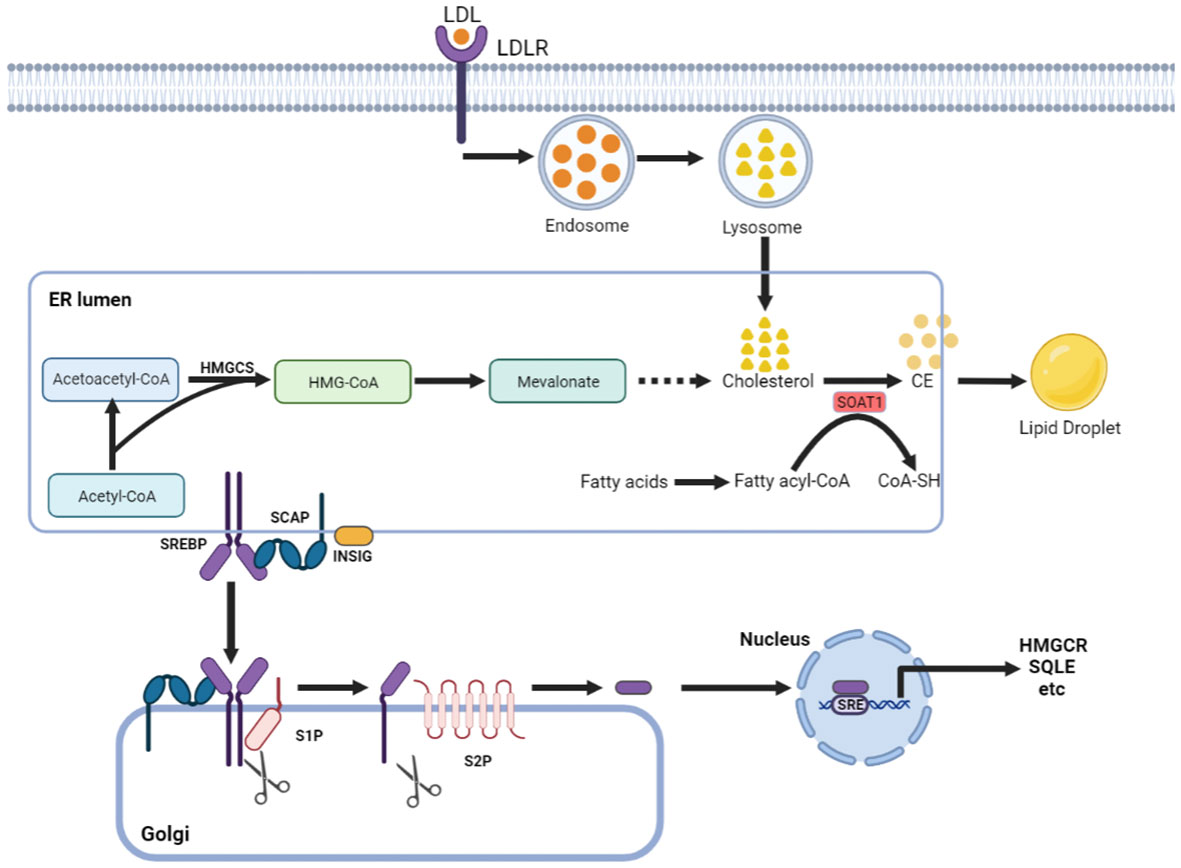
Figure 1 Cholesterol metabolism centered on SOAT1. Cholesterol acquisition by cells occurs through de novo synthesis in the endoplasmic reticulum (ER) and via LDLR-mediated endocytosis of LDL. SOAT1 is an ER-associated enzyme responsible for cholesterol storage. SOAT1-mediated esterification of increased cholesterol and fatty acids results in the formation of CE, which is stored in lipid droplets. SREBP cleavage-activating protein (SCAP) is a cholesterol sensing protein and forms a complex with SREBP2 and an ER membrane anchor protein insulin-induced gene (INSIG). A decrease in cholesterol levels within the endoplasmic reticulum (ER) results in the dissociation of INSIG from SCAP, thereby releasing the SCAP/SREBP2 complex. Subsequently, SREBP2 is transported to the Golgi complex and undergoes proteolytic activation by the site-1 and site-2 proteases (S1P and S2P). The N-terminal domain of SREBP2 then translocates to the nucleus to initiate gene transcription necessary for cholesterol synthesis and uptake.
Increased cholesterol can be converted to cholesteryl ester (CE) by sterol O-acyltransferase (SOAT) and stored in lipid droplets (10). There are two genes encoding the two SOAT enzymes, SOAT1 and SOAT2 (11). SOAT1 is widely expressed in most cell types and highly expressed in some tumors (12). SOAT2 is mainly distributed in hepatocytes and intestinal epithelial cells (13). Despite being essential for cell viability, cholesterol is not always beneficial; in contrast, when in excess, it is highly toxic, and its levels must therefore be tightly controlled. This is, in part, achieved by SOAT1-mediated cholesterol esterification (14). Sterol regulatory element-binding proteins (SREBPs) are core transcription factors that can sense cholesterol content in endoplasmic reticulum (ER) membrane and control the expression of important genes in cholesterol synthesis and uptake (15, 16). Elevated cholesterol content can inhibit the activity of SREBPs. SOAT1 can alleviate the inhibitory effect of SREBPs activity by converting cholesterol into CE (17), thus promoting the synthesis and absorption of cholesterol.
The precise control of free cholesterol content makes SOAT1 as a key intrinsic driver of cholesterol metabolism. Previous studies showed that SOAT1 plays a crucial role in the accumulation of foam cells from macrophages, the key pathological process in atherosclerosis. Besides, SOAT1 can promote the formation of very low-density lipoprotein (VLDL) in the liver, and the absorption of dietary cholesterol from the intestines (18). Targeting SOAT can directly lower the level of plasma cholesterol and inhibit the formation of arterial plaque, which may significantly reduce the risk of atherosclerosis and other cardiovascular diseases, although relevant phase III clinical trials were failed (19). Cholesterol metabolism is also closely associated with Alzheimer’s disease at several stages (5). Cholesterol levels affect the processing of amyloid protein and its precursors (20). Genetic knockout or pharmacological inhibition of SOAT1 have also been proved to provide several beneficial effects on Alzheimer’s disease.
Clinical relevance of SOAT1 in cancer
In recent years, several studies have indicated that cholesterol esterification is deregulated in cancers, and SOAT1 has received increasing attention for its association with cancer (17). SOAT1 is widely expressed across various cancers, such as breast cancer (21), renal cancer (22), liver cancer (12), glioma cancer (23), pancreatic cancer (24) and adrenocortical cancer (25). SOAT1 can play an essential role in cancer cell proliferation, migration, invasion and metastasis (26). As reported in previous studies, deregulated SOAT1 is associated with tumor aggressiveness and therapy resistance, indicating poor prognosis in different kinds of cancers (17).
Typically, high expression of SOAT1 is a specific signature of hepatocellular carcinoma (HCC), which can regulate the distribution of cellular cholesterol and promote HCC cell proliferation (27). Meanwhile, SOAT1 knockdown suppresses cell proliferation and migration in HCC (12, 28). In gastric cancer (GC), overexpression of SOAT1 could promote cholesterol ester synthesis and GC cell proliferation. Meanwhile, SOAT1 can promote gastric cancer lymph node metastasis via regulating lipid synthesis (29). Mechanistically, SOAT1 can upregulate the activities of SREBPs, which induces lymphangiogenesis by increasing the expression of VEGF-C.
In addition, high SOAT1 expression is also associated with lymph node metastasis, which indicates poor patient disease-free survival and overall survival in colorectal cancer (CRC) (30, 31). In prostate cancer, high expression of SOAT1 upregulates the expression levels of SREBPs and LDLR and eventually promotes cancer proliferation (32). A recent study found a significantly shorter median biochemical recurrence (BCR)-free survival of 93 months in patients with high SOAT1 compared to 134 months with low SOAT1 (33). In HCC, the protein expression of SOAT1 was significantly increased in the tumor compared with adjacent tissue (34). A proteomics study found that HCC patients with disrupted cholesterol metabolism and high expression of SOAT1 tend to have a poorer prognosis (12). Collectively, these results show that high expression of SOAT1 is associated with poor prognosis and promotes the growth and migration of different kinds of tumors.
Mechanisms that regulate SOAT expression in cancer
SOAT1, as the main enzyme that converts cholesterol into CE, is regulated exquisitely by a complex network, including the transcription program and the posttranscriptional program. Recent studies have reported some novel mechanisms that result in the high expression of SOAT in cancer.
Transcriptional regulation
Previous studies have found that the SOAT1 promoter can be activated by IFNγ, TNF, insulin and glucocorticoids (14, 35, 36). However, no binding site for the classic cholesterol metabolism transcription factors SREBPs or LXRs has been identified in the SOAT1 promoter (14). Recent studies have shown that the mRNA levels of SOAT1 are also directly regulated by P53, β-catenin and piRNA-6426.
P53, the tumor suppressor, plays a central role in cancer development. Over 60% of human cancers carry P53 gene missense mutations or deletions (37). P53 has the ability to repress cholesterol biosynthesis and restrict tumor growth by blocking cholesterol efflux (38). The recent study also indicated that P53 can directly bind to the promoters of SOAT1 genes and lead to its transcriptional attenuation (39). The oncogene β-catenin can also regulate cholesterol esterification by directly activating the transcriptional expression of SOAT1 (30). piRNAs are small RNAs with a length of approximately 30 nt that can guide PIWI proteins to silence transposable factors by binding to DNA methyltransferases (40). The overexpression of piRNA-6426 could increase the methylation level of the SOAT1 promoter, which can reduce the expression level of SOAT1 mRNA and protein, while the interference of piRNA-6426 can enhance the expression levels of SOAT1 mRNA and protein (41).
Posttranscriptional regulation
SOAT1 is regulated not only by the transcription program but also by the posttranscriptional program. Studies have shown that P53 can not only directly regulate the mRNA levels of SOAT1 involved in cholesterol metabolism but can also regulate the protein levels of SOAT1 by regulating the mRNA levels of USP19 (39). Ubiquitin-specific peptidase 19 (USP19) is a deubiquitinating enzyme (DUB) that stabilizes SOAT1 by decreasing its ubiquitination (42). Potential ubiquitination of SOAT1 includes the ϵ-amino groups of the protein’s seven lysine residues (K6, K11, K27, K29, K33, K48, and K63). USP19 can stabilize SOAT1 by decreasing K33/K48-linked ubiquitination (38). In addition, a previous study have also reported that SOAT2, the homologous protein of SOAT1 can be regulated by cholesterol and fatty acids (FAs). High levels of cholesterol and FA can prevent SOAT2 from ubiquitination on C277 cite and degradation via inducing ROS (43). However, whether cholesterol and FA have the same effect on SOAT1 is still unknown.
Tumor promotion by high SOAT1 expression
Cholesterol metabolism has been considered to play essential roles in tumor growth and metastasis (17). As the key enzyme in the steps of cholesteryl ester synthesis, the activity of SOAT determines the abundance of cholesterol in cancer. The excess free cholesterol, together with the fatty acyl CoA substrate can be converted to CE by SOAT1 and stored in LDs. Some studies also emphasized that it is the role of cholesteryl ester rather than cholesterol that promotes tumor growth (44–47). Given that cholesterol and fatty acids are important components of the cell membrane, cancer cells may use these stored CEs for new cell membranes and signaling molecules, which can promote the growth of tumors. Studies have showed that high SOAT1 expression elevated lipid availability, which can promote tumor aggressiveness (12, 32).
There are three SREBP isoforms. SREBP-1a and SREBP-1c mainly regulate fatty acid synthesis, and SREBP-2 controls cholesterol synthesis (48, 49). They all have the function of controlling lipid and cholesterol homeostasis (50). Normally, SREBP activity is tightly regulated by negative feedback, which is controlled by endoplasmic reticulum (ER) membrane cholesterol (14). However, studies found that tumor cells could evade high levels of cholesterol-induced negative feedback inhibition as well as maintain SREBP activity (51, 52). In PTEN-deficient tumors, the PI3K/AKT/mTOR pathway is activated, which in turn upregulates SREBPs-mediated cholesterol synthesis and absorption. However, elevated cholesterol content can inhibit the activity of SREBPs. SOAT1 could alleviate the inhibitory effects of SREBPs via converting cholesterol into CE, thus promoting the growth of PTEN-deficient tumors. The deficient of SOAT1 or SOAT1 inhibition would disturb cholesterol homeostasis, and then inhibits the activities of SREBPs and eventually suppresses tumor development (32).
In addition, SOAT1 can suppress the function of CD8+ T cells. CD8+ T cells have a central role in antitumor immunity. However, numerous studies have shown that their activity is suppressed in the tumor microenvironment (TME) (53–56). Inhibiting cholesterol esterification in T cells by SOAT1 inhibitors can enhance the proliferation of CD8+ T cells and their effector function. This is because when the cholesterol level of CD8+ T cells decreases, T-cell receptor clustering and signaling as well as efficient formation of the immunological synapse were suppressed (57). SOAT1 expression in tumors might also affect immune infiltration in the TME. A recent study noted that SOAT1 expression was positively correlated with various tumor infiltrating immune cells in glioma. More importantly, SOAT1 expression was found to be positively correlated with multiple chemokine/chemokine receptor gene and various checkpoint genes, including PD-L1. These results indicated that SOAT1 overexpression may affect immune infiltration and tumor immune escape via increasing the secretion of chemokines and the levels of checkpoint genes (58). Nevertheless, both the mechanism by which SOAT1 promotes tumor growth and immune escape need to be further studied.
SOAT1-targeted therapeutic strategies for cancer treatment
The process by which SOAT1 converts cholesterol into CE occurs in many types of aggressive cancer. High SOAT1 expression in tumors has been considered to be associated with higher grades of breast and renal cancer, respectively (21, 22). In addition, high expression of SOAT1 has also been considered to be associated with poor prognosis for patients with liver (12), glioma (23), pancreatic (24) and adrenocortical cancers (25). Thus, SOAT1 inhibition could be pursued as a therapeutic strategy for cancers (Table 1).

Table 1 SOAT1-targeted therapies in cancer.
Avasimibe, first discovered in 1996, is the first and most frequently used SOAT1 inhibitor (62, 63). It has been clinically verified to be safe and effective for cancer treatment in preclinical studies (64). Therefore, targeting SOAT1 with avasimibe may be a safe and effective method to disrupt cholesterol metabolic homeostasis in cancer treatment. In prostate cancer and glioblastoma, avasimibe could increase intracellular cholesterol levels, thus inhibiting the activities of SREBPs and eventually suppressing cancer proliferation (32, 44). In pancreatic cancer, avasimibe can promote tumor cell apoptosis by increasing intracellular cholesterol-induced ER stress (59). In HCC, SOAT1 inhibition by avasimibe dramatically repressed high-cholesterol high-fat diet (HCHFD)-induced HCC tumorigenesis and decreased cholesterol esterification, which markedly reduced tumor growth in HCC with high SOAT1 expression (12, 39). In gastric cancer, the inhibition of SOAT1 by avasimibe could suppress GC cell proliferation, cholesterol ester synthesis, and lymphangiogenesis (29). In addition, SOAT deficiency in cytotoxic T lymphocyte (CTL) leads to enhanced T-cell receptor clustering and increased cholesterol content in the plasma membrane, which can enhance CTL cytotoxicity, and synergize with immunotherapy in controlling cancer development and growth (57, 65, 66).
In addition to avasimibe, there are also some other compounds that can suppress SOAT1 activity. ATR-101 (nevanimibe) is a SOAT1-specific inhibitor that has been tested against adrenocortical carcinoma in clinical trial. This study demonstrated the safety of nevanimibe, but with limited efficacy in patients with advanced ACC (60). A recent study also screened three compounds, nilotinib, ABT-737, and evacetrapib, that exhibited optimal binding with SOAT1 and inhibitive activities. In particular, nilotinib, a second-generation tyrosine kinase inhibitor used in the clinic, displayed a high affinity for SOAT1 protein and significantly inhibited tumor activity both in vitro and in vivo by reprogramming the intracellular cholesterol metabolism of tumor cells and enhancing the effect of CD8+ T cells and neutrophils (61).
In addition, the latest research has also explored combination strategies based on avasimibe for cancer treatment. A previous study showed that simultaneously targeting SOAT1 and CPT1A by avasimibe and etomoxir had cooperative anticancer efficacy in HCC in vitro and in vivo via increasing the intracellular levels of cholesterol and fatty acids (28). Nystatin, a polyene antifungal drug for managing cutaneous or mucosal candidiasis, can synergize with avasimibe in suppressing the viability of colon cancer cells in vitro and in vivo via cholesterol sequestration and decreasing cholesterol-promoted oncogenic signals (67).
Conclusion
SOAT1 plays an important role in maintaining cellular cholesterol homeostasis by meeting the requirement of cancer cells for cholesterol. The high abundance of SOAT1 in tumors indicates worse prognosis. Therefore, SOAT1 seems to be an attractive target for novel anticancer therapy. It is glad that an increasing number of preclinical studies have revealed the antitumor effects of SOAT1 inhibition and relative mechanisms. Several SOAT1 inhibitors, such as avasimibe and nevanimibe, have shown a good human safety profile in clinical trials, making them promising approach to fight against tumors. However, these clinical trials have proven to be ineffective in retarding atherosclerosis (68, 69), probably due to the nonspecific binding and low absorptivity. To date, clinical trials have not extensively evaluated the efficacy of SOAT1 inhibitors for cancer treatment, with the exception of nevanimibe in a phase I study (60). To facilitate their clinical implementation, it is imperative to conduct large-scale clinical trials and explore combination therapy. The elucidation of the SOAT1 protein crystal structure provides an opportunity to develop a more potent and safer SOAT1 inhibitor with specific binding and high absorptivity, potentially paving the way for the next generation of SOAT1-targeted therapy in cancer (70).
Author contributions
HX made contributions to the conception and design. TT and HX wrote the manuscript. HZ helped to revise the manuscript and collect the data. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (82103364); the Sichuan Science and Technology Program (2022YFS0204); the Post-doctor Research Project, West China Hospital (2023HXBH127); the Postdoctoral Innovative Talents Support Program of China (BX2021203); and the China Postdoctoral Science Foundation (2021M702353).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang TY, Chang CC, Cheng D. Acyl-coenzyme a:cholesterol acyltransferase. Annu Rev Biochem (1997) 66:613–38. doi: 10.1146/annurev.biochem.66.1.613
2. Yu C, Chen J, Lin S, Liu J, Chang CC, Chang TY. Human acyl-CoA:cholesterol acyltransferase-1 is a homotetrameric enzyme in intact cells and in vitro. J Biol Chem (1999) 274(51):36139–45. doi: 10.1074/jbc.274.51.36139
3. Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme a:cholesterol acyltransferases. Am J Physiol Endocrinol Metab (2009) 297(1):E1–9. doi: 10.1152/ajpendo.90926.2008
4. Hai Q, Smith JD. Acyl-coenzyme a: cholesterol acyltransferase (ACAT) in cholesterol metabolism: from its discovery to clinical trials and the genomics era. Metabolites (2021) 11(8):543. doi: 10.3390/metabo11080543
5. Shibuya Y, Chang CC, Chang TY. ACAT1/SOAT1 as a therapeutic target for alzheimer’s disease. Future Med Chem (2015) 7(18):2451–67. doi: 10.4155/fmc.15.161
6. Geng F, Guo D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern Med Rev (2017) 3(5):10.18103/imr.v3i5.443. doi: 10.18103/imr.v3i5.443
7. Espinosa G, López-Montero I, Monroy F, Langevin D. Shear rheology of lipid monolayers and insights on membrane fluidity. Proc Natl Acad Sci (2011) 108(15):6008–13. doi: 10.1073/pnas.1018572108
8. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol (2006) 22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656
9. Wijers M, Kuivenhoven JA, van de Sluis B. The life cycle of the low-density lipoprotein receptor: insights from cellular and in-vivo studies. Curr Opin Lipidol (2015) 26(2):82–7. doi: 10.1097/MOL.0000000000000157
10. Kou Y, Geng F, Guo D. Lipid metabolism in glioblastoma: from De novo synthesis to storage. Biomedicines (2022) 10(8):1943. doi: 10.3390/biomedicines10081943
11. Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, et al. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med (2000) 6(12):1341–7. doi: 10.1038/82153
12. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature (2019) 567(7747):257–61. doi: 10.1038/s41586-019-0987-8
13. Chang CC, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem (2000) 275(36):28083–92. doi: 10.1074/jbc.M003927200
14. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol (2020) 21(4):225–45. doi: 10.1038/s41580-019-0190-7
15. Li Y, Wu S, Zhao X, Hao S, Li F, Wang Y, et al. Key events in cancer: dysregulation of SREBPs. Front Pharmacol (2023) 14:1130747. doi: 10.3389/fphar.2023.1130747
16. Zou Y, Zhang H, Bi F, Tang Q, Xu H. Targeting the key cholesterol biosynthesis enzyme squalene monooxygenasefor cancer therapy. Front Oncol (2022) 12:938502. doi: 10.3389/fonc.2022.938502
17. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim Biophys Acta Rev Cancer (2020) 1874(1):188394. doi: 10.1016/j.bbcan.2020.188394
18. Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme a:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol (2001) 12(3):289–96. doi: 10.1097/00041433-200106000-00008
19. Bhattacharjee P, Rutland N, Iyer MR. Targeting sterol O-Acyltransferase/Acyl-CoA : cholesterol acyltransferase (ACAT): a perspective on small-molecule inhibitors and their therapeutic potential. J Med Chem (2022) 65(24):16062–98. doi: 10.1021/acs.jmedchem.2c01265
20. Shibuya Y, Chang CC, Huang LH, Bryleva EY, Chang TY. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance. J Neurosci (2014) 34(43):14484–501. doi: 10.1523/JNEUROSCI.2567-14.2014
21. Kuldeep S, Soni S, Srivastava A, Mishra A, Sharma LK, Mandal CC. Dysregulated cholesterol regulatory genes as a diagnostic biomarker for cancer. J Gene Med (2023) 25(4):e3475. doi: 10.1002/jgm.3475
22. Matsumoto K, Fujiwara Y, Nagai R, Yoshida M, Ueda S. Expression of two isozymes of acyl-coenzyme a: cholesterol acyltransferase-1 and -2 in clear cell type renal cell carcinoma. Int J Urol (2008) 15(2):166–70. doi: 10.1111/j.1442-2042.2007.01947.x
23. Löhr M, Härtig W, Schulze A, Kroiß M, Sbiera S, Lapa C, et al. SOAT1: a suitable target for therapy in high-grade astrocytic glioma? Int J Mol Sci (2022) 23(7):3726. doi: 10.3390/ijms23073726
24. Oni TE, Biffi G, Baker LA, Hao Y, Tonelli C, Somerville TDD, et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med (2020) 217(9):e20192389. doi: 10.1084/jem.20192389
25. Lacombe AMF, Soares IC, Mariani BMP, Nishi MY, Bezerra-Neto JE, Charchar HDS, et al. Sterol O-acyl transferase 1 as a prognostic marker of adrenocortical carcinoma. Cancers (2020) 12(1):247. doi: 10.3390/cancers12010247
26. Websdale A, Kiew Y, Chalmers P, Chen X, Cioccoloni G, Hughes TA, et al. Pharmacologic and genetic inhibition of cholesterol esterification enzymes reduces tumour burden: a systematic review and meta-analysis of preclinical models. Biochem Pharmacol (2022) 196:114731. doi: 10.1016/j.bcp.2021.114731
27. Khatib SA, Wang XW. Proteomic heterogeneity reveals SOAT1 as a potential biomarker for hepatocellular carcinoma. Transl Gastroenterol Hepatol (2019) 4:37. doi: 10.21037/tgh.2019.05.09
28. Ren M, Xu H, Xia H, Tang Q, Bi F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discovery (2021) 7(1):125. doi: 10.1038/s41420-021-00504-1
29. Zhu T, Wang Z, Zou T, Xu L, Zhang S, Chen Y, et al. SOAT1 promotes gastric cancer lymph node metastasis through lipid synthesis. Front Pharmacol (2021) 12:769647. doi: 10.3389/fphar.2021.769647
30. Zhu Y, Gu L, Lin X, Zhang J, Tang Y, Zhou X, et al. Ceramide-mediated gut dysbiosis enhances cholesterol esterification and promotes colorectal tumorigenesis in mice. JCI Insight (2022) 7(3):e150607. doi: 10.1172/jci.insight.150607
31. Wang XC, Luo LM, Huang TS, Feng LF. SOAT1 is a new prognostic factor of colorectal cancer. Ir J Med Sci (2022) 191(4):1549–54. doi: 10.1007/s11845-021-02746-5
32. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab (2014) 19(3):393–406. doi: 10.1016/j.cmet.2014.01.019
33. Eckhardt C, Sbiera I, Krebs M, Sbiera S, Spahn M, Kneitz B, et al. High expression of sterol-O-Acyl transferase 1 (SOAT1), an enzyme involved in cholesterol metabolism, is associated with earlier biochemical recurrence in high risk prostate cancer. Prostate Cancer Prostatic Dis (2022) 25(3):484–90. doi: 10.1038/s41391-021-00431-3
34. Chen Y, Yang X, Chen Y, Chen G, Winkler CA, An P, et al. Impacts of the SOAT1 genetic variants and protein expression on HBV-related hepatocellular carcinoma. BMC Cancer (2021) 21(1):615. doi: 10.1186/s12885-021-08245-1
35. Ge J, Zhai W, Cheng B, He P, Qi B, Lu H, et al. Insulin induces human acyl-coenzyme a: cholesterol acyltransferase1 gene expression via MAP kinases and CCAAT/enhancer-binding protein α. J Cell Biochem (2013) 114(9):2188–98. doi: 10.1002/jcb.24568
36. Lei L, Xiong Y, Chen J, Yang JB, Wang Y, Yang XY, et al. TNF-alpha stimulates the ACAT1 expression in differentiating monocytes to promote the CE-laden cell formation. J Lipid Res (2009) 50(6):1057–67. doi: 10.1194/jlr.M800484-JLR200
37. Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discovery (2023) 22(2):127–44. doi: 10.1038/s41573-022-00571-8
38. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JP 4th, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell (2019) 176(3):564–80. doi: 10.1016/j.cell.2018.11.011
39. Zhu Y, Gu L, Lin X, Zhou X, Lu B, Liu C, et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology (2023) 77(5):1499–511. doi: 10.1002/hep.32518
40. Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell (2008) 31(6):785–99. doi: 10.1016/j.molcel.2008.09.003
41. Zhong N, Nong X, Diao J, Yang G. piRNA-6426 increases DNMT3B-mediated SOAT1 methylation and improves heart failure. Aging (2022) 14(6):2678–94. doi: 10.18632/aging.203965
42. Rossi FA, Rossi M. Emerging role of ubiquitin-specific protease 19 in oncogenesis and cancer development. Front Cell Dev Biol (2022) 10:889166. doi: 10.3389/fcell.2022.889166
43. Wang YJ, Bian Y, Luo J, Lu M, Xiong Y, Guo SY, et al. Cholesterol and fatty acids regulate cysteine ubiquitylation of ACAT2 through competitive oxidation. Nat Cell Biol (2017) 19(7):808–19. doi: 10.1038/ncb3551
44. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-Mediated lipogenesis. Clin Cancer Res (2016) 22(21):5337–48. doi: 10.1158/1078-0432.CCR-15-2973
45. Wei L, Lu X, Weng S, Zhu S, Chen Y. Cholesteryl ester promotes mammary tumor growth in MMTV-PyMT mice and activates akt-mTOR pathway in tumor cells. Biomolecules (2021) 11(6):853. doi: 10.3390/biom11060853
46. Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci Transl Med (2018) 10(437):eaap9840. doi: 10.1126/scitranslmed.aap9840
47. Benitez-Amaro A, Martínez-Bosch N, Manero-Rupérez N, Claudi L, La Chica Lhoëst MT, Soler M, et al. Peptides against low density lipoprotein (LDL) aggregation inhibit intracellular cholesteryl ester loading and proliferation of pancreatic tumor cells. Cancers (2022) 14(4):890. doi: 10.3390/cancers14040890
48. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109:1125–31. doi: 10.1172/JCI0215593
49. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell (2006) 124(1):35–46. doi: 10.1016/j.cell.2005.12.022
50. Jiang T, Zhang G, Lou Z. Role of the sterol regulatory element binding protein pathway in tumorigenesis. Front Oncol (2020) 10:1788. doi: 10.3389/fonc.2020.01788
51. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discovery (2011) 1(5):442–56. doi: 10.1158/2159-8290.CD-11-0102
52. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal (2009) 2(101):ra82. doi: 10.1126/scisignal.2000446
53. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12(4):298–306. doi: 10.1038/nrc3245
54. Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med (2021) 27(9):856–67. doi: 10.1016/j.molmed.2021.06.014
55. van Weverwijk A, de Visser KE. Mechanisms driving the immunoregulatory function of cancer cells. Nat Rev Cancer (2023) 23(4):193–215. doi: 10.1038/s41568-022-00544-4
56. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
57. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
58. Guo X, Zhou S, Yang Z, Li ZA, Hu W, Dai L, et al. Comprehensive analysis of sterol O-acyltransferase 1 as a prognostic biomarker and its association with immune infiltration in glioma. Front Oncol (2022) 12:896433. doi: 10.3389/fonc.2022.896433
59. Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene (2016) 35(50):6378–88. doi: 10.1038/onc.2016.168
60. Smith DC, Kroiss M, Kebebew E, Habra MA, Chugh R, Schneider BJ, et al. A phase I study of nevanimibe HCl, a novel adrenal-specific sterol O-acyltransferase 1 (SOAT1) inhibitor, in adrenocortical carcinoma. Invest New Drugs (2020) 38(5):1421–9. doi: 10.1007/s10637-020-00899-1
61. Wang Z, Wang M, Zhang M, Xu K, Zhang X, Xie Y, et al. High-affinity SOAT1 ligands remodeled cholesterol metabolism program to inhibit tumor growth. BMC Med (2022) 20(1):292. doi: 10.1186/s12916-022-02436-8
62. Lee HT, Sliskovic DR, Picard JA, Roth BD, Wierenga W, Hicks JL, et al. Inhibitors of acyl-CoA: cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. CI-1011: an acyl sulfamate with unique cholesterol-lowering activity in animals fed noncholesterol-supplemented diets. J Med Chem (1996) 39(26):5031–4. doi: 10.1021/jm960674d
63. Insull W Jr, Koren M, Davignon J, Sprecher D, Schrott H, Keilson LM, et al. Efficacy and short-term safety of a new ACAT inhibitor, avasimibe, on lipids, lipoproteins, and apolipoproteins, in patients with combined hyperlipidemia. Atherosclerosis (2001) 157(1):137–44. doi: 10.1016/S0021-9150(00)00615-8
64. Tardif JC, Grégoire J, L’Allier PL, Anderson TJ, Bertrand O, Reeves F, et al. Effects of the acyl coenzyme a: cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation (2004) 110(21):3372–7. doi: 10.1161/01.CIR.0000147777.12010.EF
65. Hao M, Hou S, Li W, Li K, Xue L, Hu Q, et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci Transl Med (2020) 12(571):eaaz6667. doi: 10.1126/scitranslmed.aaz6667
66. Zhao L, Li J, Liu Y, Kang L, Chen H, Jin Y, et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J Immunother (2018) 41(2):45–52. doi: 10.1097/CJI.0000000000000207
67. Xu H, Xia H, Zhou S, Tang Q, Bi F. Cholesterol activates the Wnt/PCP-YAP signaling in SOAT1-targeted treatment of colon cancer. Cell Death Discov (2021) 7(1):38. doi: 10.1038/s41420-021-00421-3
68. Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, et al. ACAT intravascular atherosclerosis treatment evaluation (ACTIVATE) investigators. effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med (2006) 354(12):1253–63. doi: 10.1056/NEJMoa054699
69. Fazio S, Linton M. Failure of ACAT inhibition to retard atherosclerosis. N Engl J Med (2006) 354(12):1307–9. doi: 10.1056/NEJMe068012
Keywords: sterol-O-acyltransferase 1, cholesterol metabolism, cancer therapy, avasimibe, cholesterol esterification
Citation: Tu T, Zhang H and Xu H (2023) Targeting sterol-O-acyltransferase 1 to disrupt cholesterol metabolism for cancer therapy. Front. Oncol. 13:1197502. doi: 10.3389/fonc.2023.1197502
Received: 31 March 2023; Accepted: 05 June 2023;
Published: 20 June 2023.
Edited by:
Olivia Crociani, University of Florence, ItalyReviewed by:
Antonella Mannini, Department of Experimental and Clinical Medicine University of Florence, ItalyCopyright © 2023 Tu, Zhang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huanji Xu, eHVodWFuamkxOTkwQHNjdS5lZHUuY24=
†These authors have contributed equally to this work