
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 02 November 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1197340
 Laura Rosiñol1*†
Laura Rosiñol1*† Benjamin Hebraud2
Benjamin Hebraud2 Albert Oriol3
Albert Oriol3 Anne-Laurène Colin4
Anne-Laurène Colin4 Rafael Ríos Tamayo5Cyrille Hulin6María Jesús Blanchard7
Rafael Ríos Tamayo5Cyrille Hulin6María Jesús Blanchard7 Denis Caillot8
Denis Caillot8 Anna Sureda9Miguel Teodoro Hernández10
Anna Sureda9Miguel Teodoro Hernández10 Bertrand Arnulf11
Bertrand Arnulf11 Maria-Victoria Mateos12Margaret Macro13Jesús San-Miguel14
Maria-Victoria Mateos12Margaret Macro13Jesús San-Miguel14 Karim Belhadj15
Karim Belhadj15 Juan José Lahuerta14M. Brigid Garelik16Joan Bladé1Philippe Moreau17
Juan José Lahuerta14M. Brigid Garelik16Joan Bladé1Philippe Moreau17Objective: Providing the most efficacious frontline treatment for newly diagnosed multiple myeloma (NDMM) is critical for patient outcomes. No direct comparisons have been made between bortezomib + lenalidomide + dexamethasone (VRD) and bortezomib + thalidomide + dexamethasone (VTD) induction regimens in transplant-eligible NDMM.
Methods: An integrated analysis was performed using patient data from four trials meeting prespecified eligibility criteria: two using VRD (PETHEMA GEM2012 and IFM 2009) and two using VTD (PETHEMA GEM2005 and IFM 2013-04).
Results: The primary endpoint was met, with VRD demonstrating a noninferior rate of at least very good partial response (≥ VGPR) after induction vs VTD. GEM comparison demonstrated improvement in the ≥ VGPR rate after induction for VRD vs VTD (66.3% vs 51.2%; P = .00281) that increased after transplant (74.4% vs 53.5%). Undetectable minimal residual disease rates post induction (46.7% vs 34.9%) and post transplant (62.4% vs 47.3%) support the benefit of VRD vs VTD. Treatment-emergent adverse events leading to study and/or treatment discontinuation were less frequent with VRD (3%, GEM2012; 6%, IFM 2009) vs VTD (11%, IFM 2013-04).
Conclusion: These results supported the benefit of VRD over VTD for induction in transplant-eligible patients with NDMM. The trials included are registered with ClinicalTrials.gov (NCT01916252, NCT01191060, NCT00461747, and NCT01971658).
Multiple myeloma remains an incurable disease with relatively high mortality rates despite availability of multiple treatment options (1–3). Several treatment regimens are recommended for induction therapy in patients with transplant-eligible (TE) newly diagnosed multiple myeloma (NDMM) (4–6). Selection of the optimal frontline therapy is important, as 60% to 70% of patients receive fewer than three lines of therapy (7–10). Therefore, providing the most efficacious frontline therapy is critical to minimizing disease burden and optimizing survival outcomes (7–9, 11). Studies show that achieving at least very good partial response (≥ VGPR) during induction is associated with improved long-term outcomes (12–15). Other goals of induction therapy include achievement of rapid disease control and undetectable minimal residual disease (MRD) without negatively impacting stem cell harvest. Furthermore, low rates of toxicity would enable patients to complete induction, which helps optimize treatment responses.
National and international TE NDMM treatment guidelines, including European Society for Medical Oncology (ESMO), European Myeloma Network (EMN), and American Society of Clinical Oncology/Cancer Care Ontario (ASCO/CCO) guidelines, recommend triplet regimens such as bortezomib + lenalidomide + dexamethasone (VRD) and bortezomib + thalidomide + dexamethasone (VTD) (4–6). In addition, VRD and VTD are both currently being used as backbone therapy in modern quadruplet induction regimens with anti-CD38 monoclonal antibodies. However, in contrast to lenalidomide, the use of thalidomide is limited by peripheral neuropathy (16, 17). Bortezomib has also been associated with peripheral neuropathy, and the combination with thalidomide in VTD led to higher rates of this adverse event vs thalidomide + dexamethasone in phase 3 studies (18–20). The tolerability of bortezomib has improved with subcutaneous administration, which demonstrated noninferior efficacy and a lower incidence of peripheral neuropathy vs intravenous administration (21–23). Thus, VRD with subcutaneous bortezomib offers a treatment option with reduced rates of peripheral neuropathy. In phase 3 studies, VRD provided deep and durable responses during frontline therapy without limiting a patient’s ability to receive further therapy (24–28). Given these results, VRD has been integrated into clinical practice and is a preferred regimen for primary therapy for transplant candidates (4–6).
While both VRD and VTD are included as options for frontline therapy in international guidelines, no direct comparison of the safety and efficacy of VRD vs VTD has been done to date. In the absence of a randomized controlled trial (RCT), an integrated analysis can be performed using propensity score (PS)–based statistical methods (29, 30). This strategy minimizes the effects of observed baseline factors that could confound analysis to improve the comparison between different treatment cohorts. This method was previously used in a cross-trial comparison and regulatory submission to evaluate bortezomib ± dexamethasone in relapsed MM (31).
The goal of this integrated analysis was to compare VRD and VTD induction therapy in patients with TE NDMM. A literature search for phase 3 RCTs that met prespecified eligibility criteria identified two trials using VRD (PETHEMA GEM2012 and IFM 2009) and two trials using VTD (PETHEMA GEM2005 and IFM 2013-04). These four RCTs were included in the integrated analysis.
A comprehensive review of published literature and ongoing clinical studies was performed to identify studies that met the following eligibility criteria: (1) study was a phase 3 RCT evaluating a full-dose VRD or VTD induction regimen (every 3 or 4 weeks) in patients with TE NDMM before autologous stem cell transplant (ASCT), (2) study had reached the primary endpoint for the purpose of the integrated analysis before data transfer, and (3) an agreement was in place for access to patient-level data adequate to conduct an integrated analysis by 31 December 2016. Search details are provided in the Supplement.
The primary endpoint of the integrated analysis was noninferiority of the post induction ≥ VGPR rate based on International Myeloma Working Group criteria. Secondary endpoints were safety, post ASCT ≥ VGPR rate, and ≥ VGPR rate over time during induction. Exploratory endpoints were progression-free survival (PFS), overall survival (OS), and undetectable MRD.
Statistical methods were based on the PS, which is a conditional probability of being treated given observed covariates that could be used to balance the covariates in two treatment cohorts and reduce bias (30). A logistic regression model in which treatment group was regressed based on 11 baseline variables (age, sex, height, weight, performance status, International Staging System disease stage, hemoglobin level, creatinine clearance, albumin level, β2-microglobulin level, and lactate dehydrogenase level) was used to estimate PS (32). Patients with missing baseline values for any of these variables were excluded. Patients were stratified into five equal-sized groups using the quintiles of the estimated PS. Additional details on the PS model and noninferiority hypothesis are provided in the Supplement.
To demonstrate noninferiority of VRD vs VTD, a margin of noninferiority was prespecified using a two-sided 95% confidence interval. For the primary endpoint, post-initial treatment response rate of ≥ VGPR, the noninferiority margin (11.3%) was selected using historical data; a margin of 10% did not represent a substantial difference in treatment effect and was within normal variance between two treatment regimens in similar patient populations.
For PFS and OS, Kaplan-Meier methodology was used for the descriptive statistics. The stratified log-rank test, with the stratum based on the quintiles of the PS, was used to assess statistical significance of treatment effects. The stratified Cox proportional hazards model was used to estimate the hazard ratio and 95% CI for VRD vs VTD.
In GEM2005, MRD was assessed using four-color multiparameter flow cytometry with undetectable MRD defined as < 20 clonal plasma cells after measuring ≥ 200,000 nucleated cells at a sensitivity level of 10−4. GEM2012 assessed MRD at sensitivity levels of 10−4 and 10−6 using next-generation flow following EuroFlow protocols per International Myeloma Working Group, using an optimized eight-color, two-tube antibody panel (33).
The safety analysis was primarily based on the induction phase of treatment, and the safety population included all randomized patients who received at least one dose of study drug. Adverse events were coded using the Medical Dictionary for Regulatory Activities version 15.1 and summarized by system organ class and preferred terms. Adverse events were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events versions 4.03 (PETHEMA GEM2012) and 4.0 (IFM studies). Due to limited access to some data from the PETHEMA GEM2005 clinical trial database, safety data from that study are not included here. Treatment-emergent peripheral neuropathy was summarized using the grouped term (see Supplement).
Sixteen studies of prospective phase 3 RCTs evaluating VRD or VTD induction (every 3 or 4 weeks) in patients with TE NDMM before ASCT were identified. A brief overview of the study details and the criteria not met for inclusion for 12 trials are provided in Table S1. Four studies met the eligibility criteria and were included: PETHEMA GEM2012 and IFM 2009 for VRD and PETHEMA GEM2005 and IFM 2013-04 for VTD (Figure 1A) (16, 18, 24, 25).
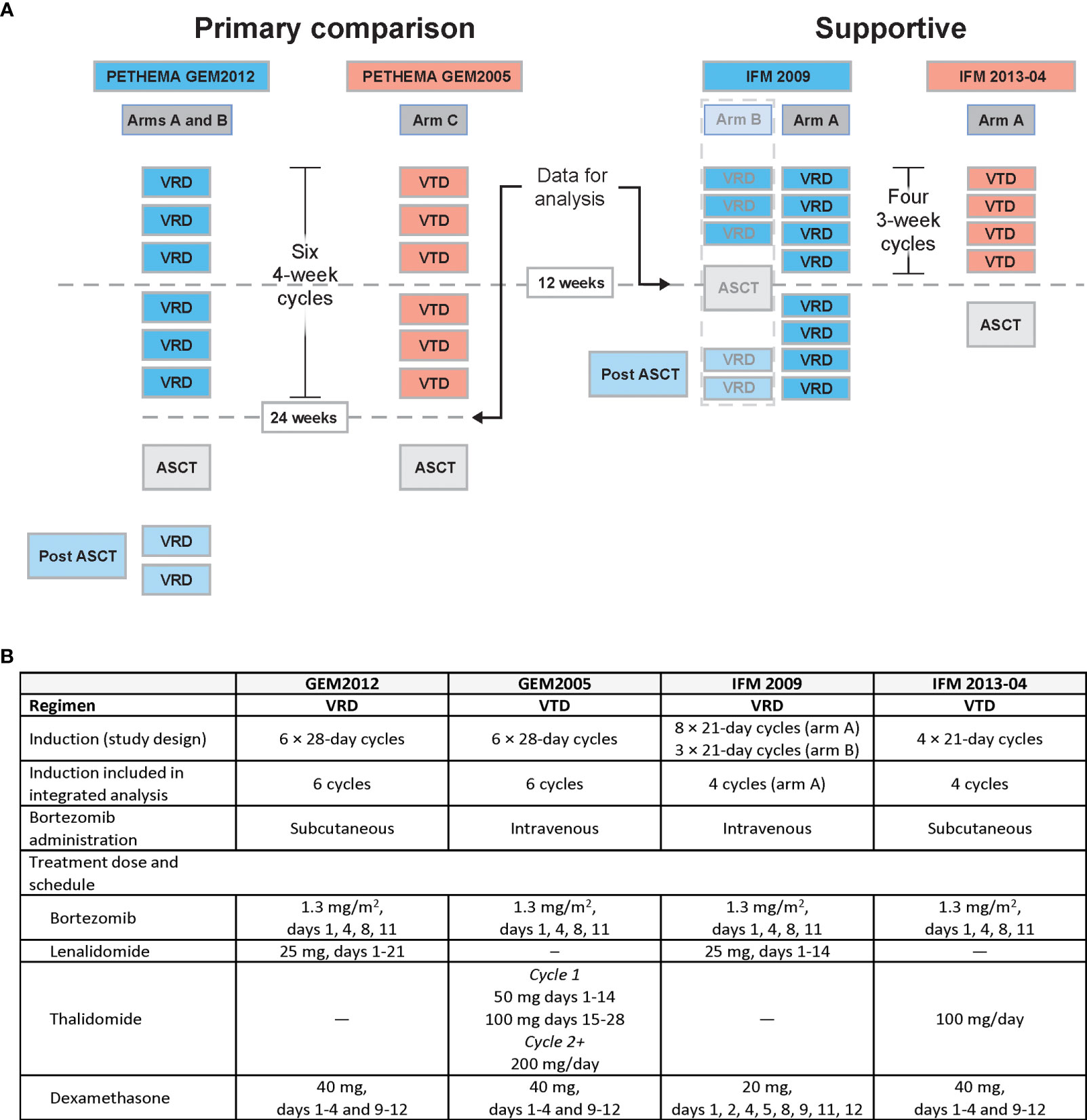
Figure 1 Design of studies meeting eligibility criteria. (A) Graphical overview and (B) tabular summary of GEM and IFM study designs. ASCT, autologous stem cell transplant; MRD, minimal residual disease; VRD, bortezomib, lenalidomide, and dexamethasone; VTD, bortezomib, thalidomide, and dexamethasone.
Similarities and differences in the induction phase (length, number, dose, schedule, and route of bortezomib administration) are summarized for the included studies in Figure 1B. For example, GEM studies used 28-day cycles, whereas IFM studies used 21-day cycles. Additionally, although the bortezomib dose was the same for all four studies, bortezomib was administered subcutaneously in GEM2012 and IFM2013-04 vs intravenously in GEM2005 and IFM 2009. Lenalidomide was given on days 1 to 21 of the 28-day cycle in GEM2012 vs days 1 to 14 of the 21-day cycle in IFM 2009. In GEM2005, the thalidomide dose was escalated from 50 mg/day to 100 mg/day in cycle 1 and 200 mg/day thereafter; whereas, in IFM 2013-04, it was 100 mg/day throughout induction. Furthermore, differences between the induction regimens affected how data were included in the integrated analysis. For example, GEM2012 (VRD) and GEM2005 (VTD) were considered the main studies for the efficacy analysis due to their symmetrical induction regimens (six 4-week cycles of induction followed by ASCT). IFM 2009 (VRD) and IFM 2013-04 (VTD) were considered supportive studies due to differences in design, meaning that these studies were included in the efficacy analysis to support the main efficacy comparisons made for the GEM studies. While both used 3-week cycles, IFM 2009 arm A used eight cycles, arm B used three cycles followed by ASCT and two cycles of consolidation, and IFM 2013-04 used four cycles followed by ASCT. Based on these differences in the IFM studies, only IFM 2009 arm A was used to compare with IFM 2013-04. Only data through four cycles of induction were included. This comparison of studies was possible because of the research agreement granting patient-level data access for each study.
Eligibility criteria were similar between the included studies. The four studies were all conducted in patients < 65 years of age with newly diagnosed, untreated MM with measurable M-protein concentrations (16, 18, 24, 25). Patients in these studies all had an Eastern Cooperative Oncology Group (ECOG) performance status of ≤ 3. GEM2012, GEM2005, and IFM2009 studies included patients with platelet counts of ≥ 50 to 100 × 109/L and neutrophil counts of ≥ 1 × 109/L. Additionally, GEM2012, IFM2009, and IFM2013-04 all excluded patients with grade ≥ 2 peripheral neuropathy.
In the GEM studies, the intent-to-treat populations included 458 patients who received VRD (GEM2012) and 130 patients who received VTD (GEM2005). Due to missing data for at least one baseline variable, 51 and 1 patients were excluded from the respective PS-stratified cohorts, leaving 407 (GEM2012) and 129 (GEM2005) patients in the integrated analysis. Similarly, intent-to-treat populations of the IFM studies had 19 and 15 patients, respectively, excluded from the respective PS-stratified cohorts due to missing data for at least one baseline variable. Thus, as 350 patients received VRD in IFM 2009 arm A and 169 received VTD in IFM 2013-14, 331 (IFM 2009) and 154 (IFM 2013-04) patients remained in the integrated analysis. The distributions of the overall PS of the VRD vs VTD cohorts for both GEM and IFM studies were similar (Table S2). Baseline characteristics were similar between the VRD and VTD PS-stratified cohorts (Table 1) and the respective overall intent-to-treat populations (Table S3).
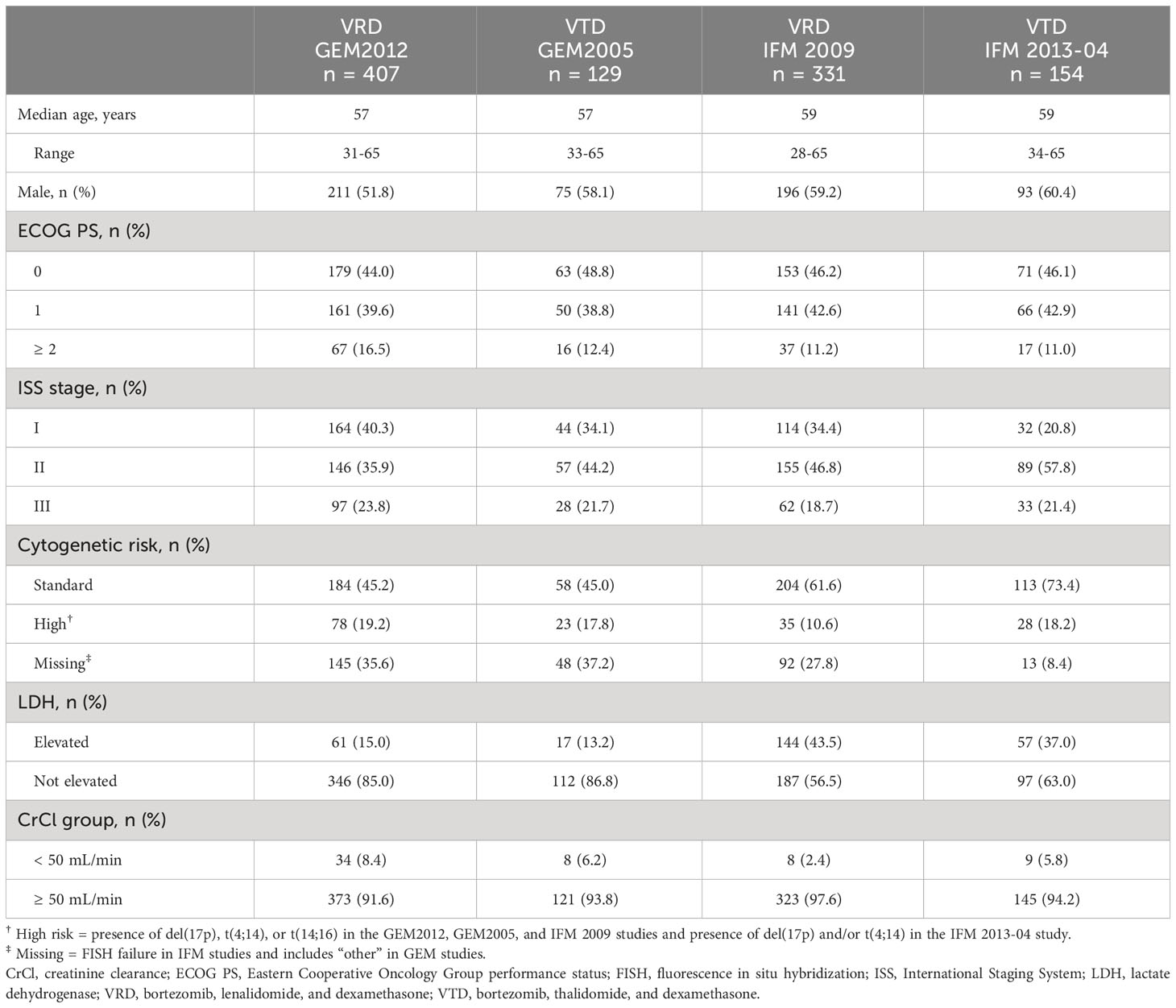
Table 1 Baseline patient and disease characteristics.
The primary endpoint of noninferiority for VRD vs VTD was met in both the GEM and IFM analyses. In the primary comparison (GEM2012 vs GEM2005), the ≥ VGPR rate after six cycles of induction with VRD vs VTD was 66.3% vs 51.2%, respectively (odds ratio, 1.87 [95% CI, 1.23-2.83]; P = .00281) (Table 2), which was similar in most subgroups (Figure S1). In the GEM comparison, the ≥ VGPR difference was 15.0% (95% CI, 5.0%-25.0%) (Figure 2). Since the 95% CI was entirely above zero, superiority of VRD over VTD was concluded. In the IFM studies, the ≥ VGPR rate by four cycles (12 weeks) was noninferior with VRD (57.1%) vs VTD (56.5%) in the PS-stratified population, with similar results in patient subgroups (Figure S2).
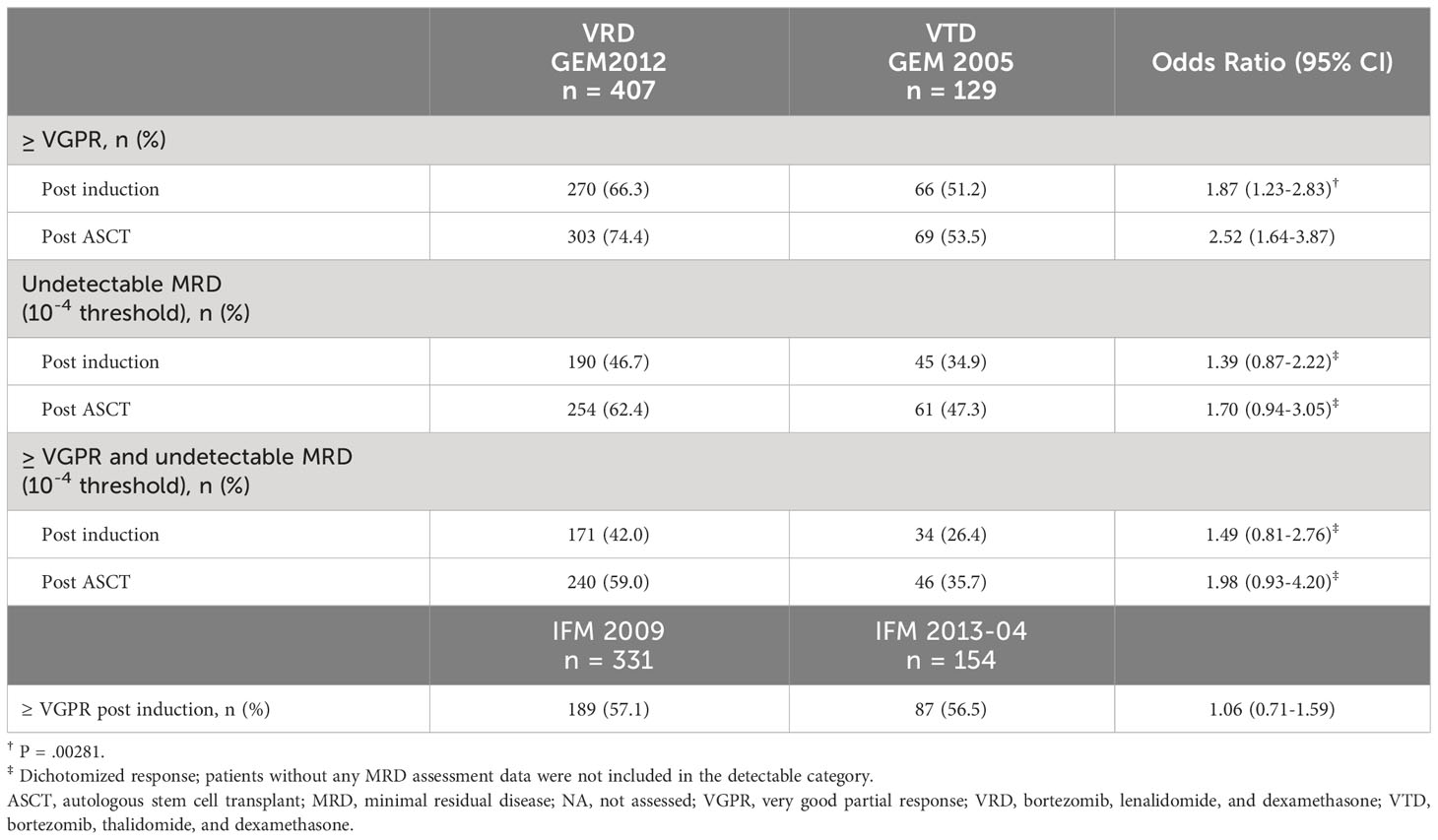
Table 2 Response.

Figure 2 Noninferiority of VRD vs VTD. VGPR, very good partial response; VRD, bortezomib, lenalidomide, and dexamethasone; VTD, bortezomib, thalidomide, and dexamethasone.
In the GEM studies, the ≥ VGPR rate increased over time with both regimens during induction. Among patients in the PS-stratified cohort who initiated cycle six, the ≥ VGPR rate improved from 54.5% at cycle three to 70.1% at cycle six with VRD (n = 378) and from 35.1% to 55.9% with VTD (n = 111) (Figure 3).
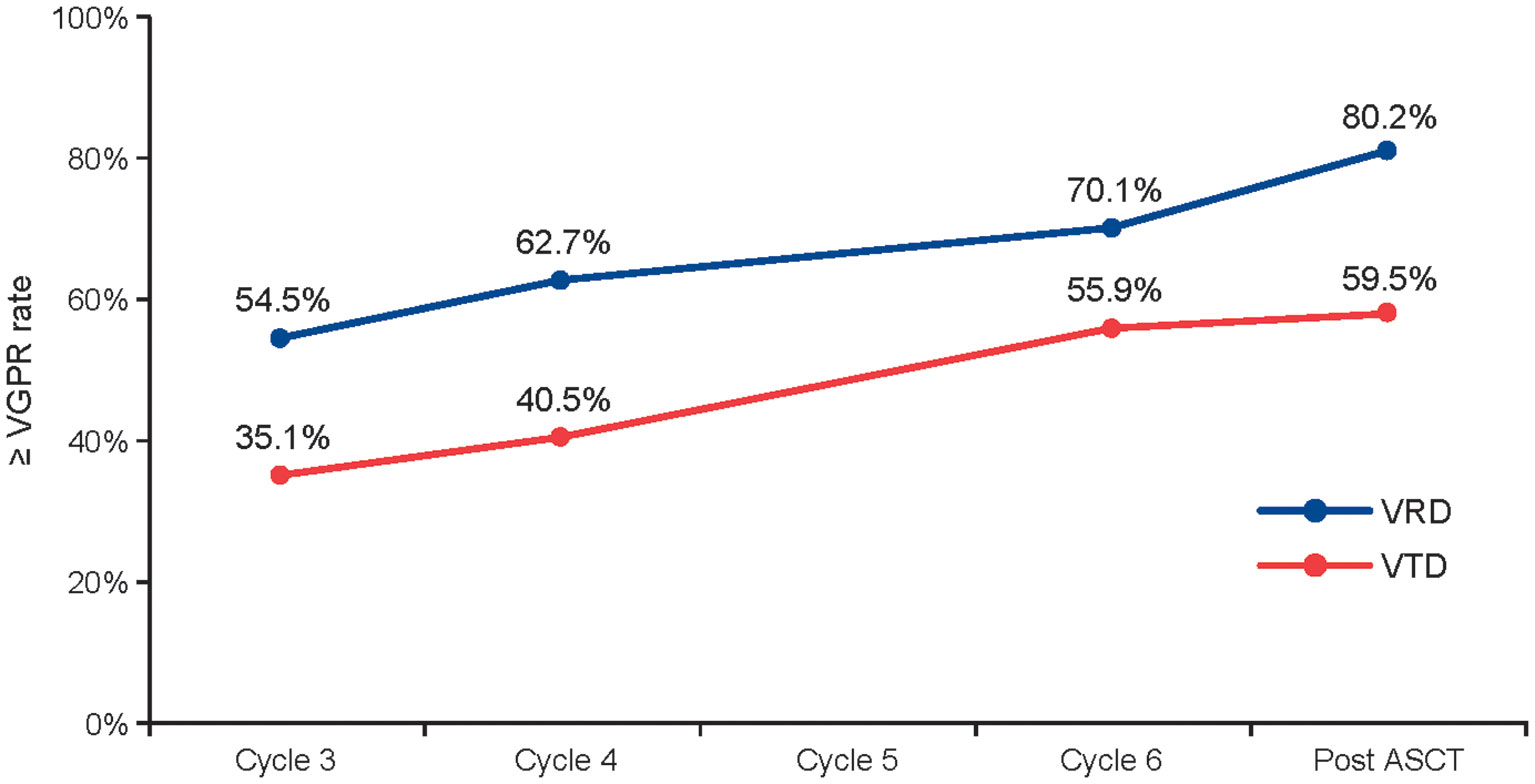
Figure 3 ≥ VGPR rate throughout induction and post ASCT in the GEM studies. Data are based on the 378 patients taking VRD and 111 patients taking VTD who started cycle 6 in the GEM2012 and GEM2005 studies, respectively. ASCT, autologous stem cell transplant; PS, propensity score; VGPR, very good partial response; VRD, bortezomib, lenalidomide, and dexamethasone; VTD, bortezomib, thalidomide, and dexamethasone.
In the overall PS-stratified cohort in the GEM studies, the improvement in ≥ VGPR rate seen for VRD vs VTD post induction was maintained after ASCT (74.4% vs 53.5%). Of note, the ≥ VGPR rate improved more for VRD than for VTD from post induction to post ASCT (8% vs 2%). The undetectable MRD rates (10–4 threshold) post induction (46.7% vs 34.9%) and post ASCT (62.4% vs 47.3%) supported the benefit with VRD vs VTD. Data with a threshold of 10–6 were available for GEM2012, showing an undetectable MRD rate of 28.5% post induction and 41.8% post ASCT. Similar comparisons could not be performed with the IFM studies, as response over time and MRD were not assessed in IFM 2013-04.
Due to differences in median follow-up times and numbers of patients who experienced progression or died between studies, the 2-year event-free rate is a better comparison for the cohorts than median PFS or OS. In the GEM studies, the 2-year PFS rates (ie, those patients who had not experienced progression or died) were 82% and 69%, and the 2-year OS rates (ie, patients who had not died) were 90% and 87% with VRD and VTD, respectively.
In the IFM studies, the 2-year PFS rates were 67% and 71%, and the 2-year OS rates were 93% and 93% with VRD and VTD, respectively. However, differences in the number of cycles of induction received, inclusion of ASCT following induction, and treatment received post ASCT may limit interpretation of these data.
A summary of treatment-emergent adverse events (TEAEs) is provided in Table 3. The most common grade 3/4 (TEAE) in the GEM2012 VRD study was neutropenia (13%), and in the IFM studies, lymphopenia (49% with VRD and 22% with VTD; Table 4). In GEM2012, the rate of peripheral neuropathy was 21% for grade ≥ 2 events and 5% for grade 3/4 events. In the IFM studies, in which intravenous bortezomib was used in VRD and subcutaneous bortezomib was used in VTD, rates of grade ≥ 2 peripheral neuropathy events (30% vs 27% with VRD vs VTD, respectively) and grade 3/4 events (6% vs 8%, respectively) were similar.
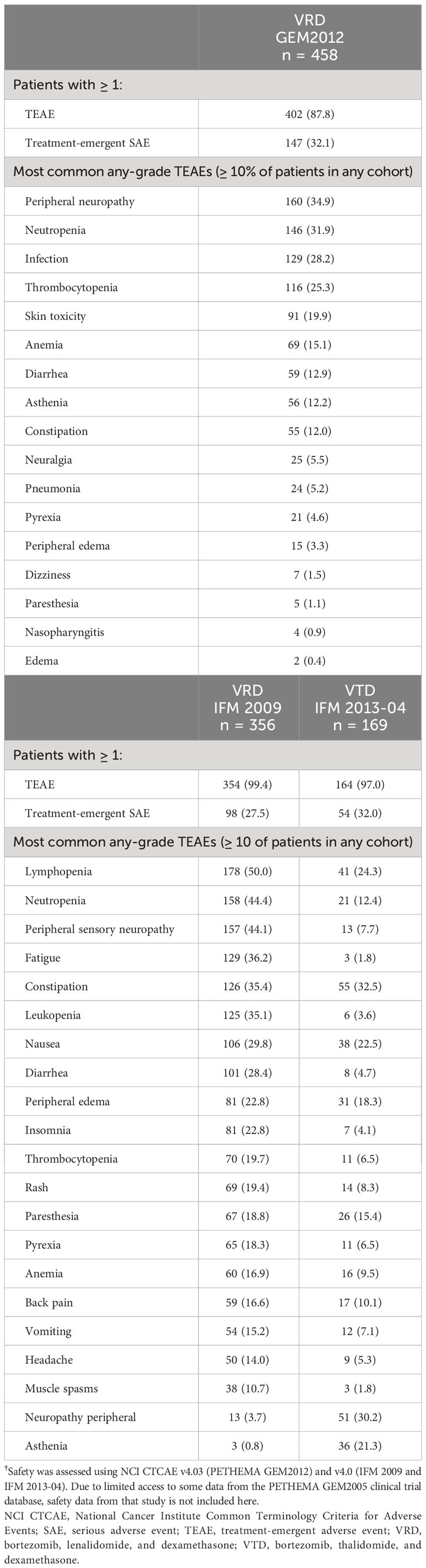
Table 3 Summary of TEAEs†.
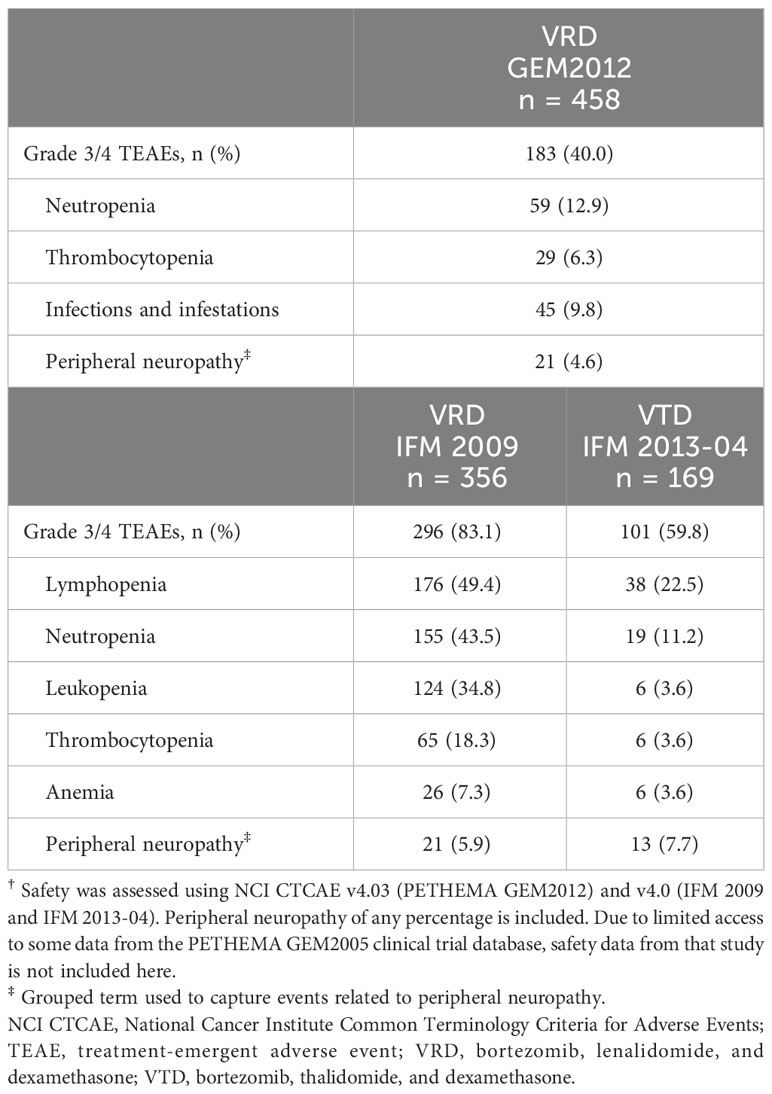
Table 4 Grade 3/4 TEAEs in ≥5% of patients†.
In GEM2012, 22% of patients had at least one TEAE leading to dose reduction, most commonly peripheral neuropathy (17%). At least one TEAE leading to study or treatment discontinuation occurred in 3% of patients with VRD and were most frequently due to infection (1%), septic shock (< 1%), and disease progression (< 1%). In the safety population of the GEM studies, a higher percentage of VTD patients received fewer cycles of therapy compared with VRD. For example, 4.4% vs 6.2% of patients received three or fewer cycles of VRD vs VTD. This trend continued, with 6.1% vs 10.8% of patients receiving four or fewer cycles, and 7.0% vs 13.8% of patients receiving five or fewer cycles of VRD vs VTD, respectively. Thus, more patients receiving VRD vs VTD initiated the protocol-defined sixth cycle of induction.
In the IFM studies, 33% and 18% of patients had at least one TEAE leading to dose reduction with VRD vs VTD, most commonly peripheral neuropathy (26% vs 14%, respectively). At least one TEAE leading to treatment discontinuation occurred in 6% and 11% of patients treated with VRD and VTD. The most common TEAEs leading to discontinuation were peripheral neuropathy (3%) with VRD and peripheral neuropathy (7%) and pulmonary embolism (2%) with VTD. The percentage of patients in the safety population of the IFM studies who received three or fewer cycles was 5.3% for both VRD and VTD.
In the absence of a prospective RCT comparing VRD and VTD, the established methodology of an integrated analysis was used to compare these regimens in TE NDMM. Using patient-level data from the GEM and IFM studies, the analysis met its primary endpoint and demonstrated the noninferiority of VRD vs VTD. Furthermore, VRD induction showed superiority by achieving a statistically significant and clinically relevant improvement in ≥ VGPR rate over VTD when six treatment cycles were compared in the GEM studies (66.3% vs 51.2%; P = .00281). Additionally, the improvement in ≥ VGPR rate from post induction to post ASCT was more notable with VRD than with VTD (rising to 74.4% vs 53.5%). Increasing ≥ VGPR rates over the course of treatment, undetectable MRD, and 2-year PFS rates further supported the benefit of VRD over VTD. The difference in ≥ VGPR rates was more notable in the GEM studies, which featured longer cycles than the IFM studies. It is also important to note that the differences in cycle length and overall treatment duration in the GEM studies may have contributed to a greater ≥ VGPR rate than what might be expected in the clinic. These considerations further highlight the importance of cycle length and treatment duration for achieving a clinically meaningful response during induction. While length of induction therapy in GEM2005 and GEM2012 (6 cycles/24 weeks) was longer than that of some other recent phase 3 trials, such as CASSIOPEIA (34), studies incorporating 6 cycles of induction with VTD have produced similar improvements in CR rates from pre- to posttransplant compared with those using 3 cycles of induction with VTD (18, 19). Further, differences in the length of induction therapy among these trials may reflect variations in regional practices. The current ESMO guidelines recommend 4 to 6 cycles of induction with or without consolidation (4).
Although safety results from the GEM2005 study were not included in this analysis due to limited access, safety data reported in the GEM2005 primary manuscript and safety data reported here for GEM2012 and the IFM studies showed that TEAEs were generally consistent with the known safety profiles of lenalidomide, bortezomib, thalidomide, and dexamethasone. Differences in the most common TEAEs between the regimens were largely consistent with the toxicities of lenalidomide and thalidomide. Overall, TEAEs with the VRD regimen were manageable, and the tolerability profile compared well with that of VTD, with fewer TEAEs leading to discontinuation. Peripheral neuropathy due to thalidomide often increases in frequency with long-term use (16, 17, 35, 36). In addition to the effects of lenalidomide vs thalidomide in the VRD and VTD regimens (including the dose of thalidomide used), TEAEs should be considered in the context of the different bortezomib routes and frequencies of administration used in these studies. Bortezomib was given subcutaneously in GEM2012 and IFM 2013-04 and intravenously in GEM2005 and IFM 2009. Additionally, bortezomib dose intensity was higher when administered in 3-week cycles in the IFM studies vs 4-week cycles in the GEM studies. The efforts made to improve the tolerability of induction regimens were important, as they may have allowed delivery of the full number of planned cycles. This could increase depth of response and lead to improved survival outcomes. One can hypothesize that a weekly subcutaneous bortezomib schedule could further improve tolerability compared with a twice-weekly schedule.
The survival data (PFS and OS) should be interpreted with caution considering the key study design differences. While GEM VRD and VTD trials had largely parallel designs (with six cycles of induction followed by ASCT), post-ASCT treatment differed between the trials. For example, patients in the VRD trial received VRD consolidation (and could enroll in a maintenance study of lenalidomide + dexamethasone ± ixazomib [NCT02406144]), and those in the VTD trial could receive post-ASCT maintenance with thalidomide, thalidomide + bortezomib, or interferon-α2b. Additionally, follow-up times differed between the trials (median 24.0 vs 48.4 months for VRD vs VTD). To address this, we used the event-free rate at 2 years, which mitigates the differences in median follow-up and number of events and is considered a more representative comparison between the two cohorts. The study designs were more divergent in the IFM trials. Arm A of IFM 2009, which was used for the VRD data analyzed, did not include ASCT and instead included a total of eight cycles of VRD. Due to differences in duration of induction between the IFM studies, we chose to include Arm A from the IFM 2009 study (eight 3-week cycles of VRD [24 weeks]) to permit comparison with the IFM 2013-04 study (four 3-week cycles of VTD [12 weeks]). The VTD study used ASCT following four cycles of VTD induction. Follow-up times also differed between the trials (median 35.0 vs 16.6 months for VRD vs VTD). All these factors may have affected the reported survival outcomes.
Another point to consider is that different MRD sensitivity thresholds were used across these studies. In GEM2005, 10−4 was used since the technology for 10−6 was not available at the time the study was conducted. Compared with MRD 10−5 and 10−6, MRD 10−4 is a threshold at which more MRD-positive disease may go undetected, and MRD positivity has been associated with less-durable responses (37). While current response criteria have established an MRD threshold of 10−5, evidence has emerged that 10−6 may be more clinically relevant. Further, achievement of MRD negativity at 10−6 has been associated with longer PFS compared with 10−5 (37).
This analysis has several limitations, the most notable of which is the cross-trial comparison. Although we used PS-based statistical methods, it is possible that differences in baseline factors among the trials could have confounded the comparison between treatment cohorts. In addition, the analyzed trials had relatively short follow-up durations and are older, having been published between 2012 and 2019 with a database closure of 2017. Since this time, regimens based on the anti-CD38 monoclonal antibodies daratumumab and isatuximab have emerged. However, both VRD and VTD are being used in current practice as backbone therapy in modern quadruplet induction regimens incorporating daratumumab or isatuximab. Further, the comparison of VRD and VTD remains relevant as both of these triplet regimens are recommended in current TE NDMM treatment guidelines, including ESMO, EMN, and ASCO/CCO guidelines. VRD is also designated as a category 1 preferred regimen for primary therapy for TE patients by the National Comprehensive Cancer Network Guidelines®, while VTD has a category 1 designation for use in certain circumstances (38).
As with any systematic analysis, the applicability of these results to the real-word setting is of interest, and different practice patterns in the Europe and the US should be acknowledged. In Europe, VTD is a standard of care for patients with TE NDMM (34), whereas in the US, VRD is considered the optimal induction regimen for these patients (39). Therefore, the results of this analysis will have different implications for each respective setting. The inclusion of younger, healthier patients (age <65 years with ECOG performance status of ≤ 3) may not be generally reflective of a real-world patient population. Lastly, the percentage of patients with undetermined cytogenetic risk included in each trial was unbalanced, ranging from 8% to 36%; therefore, it is likely that an analysis of real-world patients may have a more equitable distribution of risk groups.
Although some studies did not meet the criteria for inclusion in the integrated analysis, their results further support the use of VRD. For example, the DSMM XIV, GMMG-HD6, and SWOG S0777 studies showed VRD to be active and well tolerated (26, 27, 40), with the latter supporting the recent European approval of VRD for transplant-ineligible patients. In Myeloma XI, responses to induction were deeper with cyclophosphamide + lenalidomide + dexamethasone vs cyclophosphamide + thalidomide + dexamethasone (≥ VGPR rates, 60.4% vs 52.9%, respectively) (41). Responses also deepened with additional cycles, highlighting the importance of a tolerable regimen to maximize the number of cycles that can be given (42). Of note, although induction was stopped due to toxicity at a similar rate for cyclophosphamide + lenalidomide + dexamethasone vs cyclophosphamide + thalidomide + dexamethasone (5.0% vs 6.7%, respectively), dose modifications of lenalidomide were less frequent than of thalidomide (38.3% vs 73.6%, respectively) (41). Together, these results supported the advantage of lenalidomide- vs thalidomide-containing regimens. Moreover, results of several other studies suggest that there is a role for VRD as consolidation therapy in NDMM (25, 43). For example, in the EMN02/HOVON95 trial, consolidation therapy with VRD followed by maintenance with lenalidomide improved PFS in patients with NDMM vs maintenance alone (43). However, the benefit observed in this trial is possibly due to use of a suboptimal induction regimen—some other trials have not demonstrated the same effect (44).
Given the incurable nature of MM and the fact that patients with MM will ultimately experience relapse, selecting the ideal induction regimen is critical for minimizing disease burden and promoting durable survival outcomes. In lieu of a direct comparison between VRD and VTD, it is our hope that this analysis can be used to help inform these treatment decisions and address clinical questions that are of particular importance to clinicians who treat patients with NDMM. Overall, the results of this integrated analysis provide evidence demonstrating the benefit of VRD over VTD as induction treatment in TE patients with NDMM. These results support the inclusion of VRD as a preferred regimen for primary therapy for transplant candidates (4–6).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to this link: https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
All authors contributed to the concept and design of the work, acquisition, analysis, or interpretation of data for the work; contributed to the drafting of the work; revised the manuscript critically for important intellectual content; approved the final version to be published; and agree to be accountable for all aspects of the work.
This work was funded by Celgene, a Bristol-Myers Squibb Company.
The authors thank Valerie Lauwers-Cances and Laure Devlamynck (Unité de Soutien Méthodologique à la Recherche, Centre Hospitalier et Universitaire de Toulouse, France), Pascale Olivier-Abbal (Service de Pharmacologie Médicale et Clinique, Centre Hospitalier et Universitaire de Toulouse, France), and Catherine Payen and Sandrine Rollet (Service d’Hématologie, Institut Universitaire du Cancer de Toulouse-Oncopole, Toulouse, France) for their assistance with the study. The authors thank Guang Chen for statistical support performed while employed at Celgene, a Bristol-Myers Squibb Company, and thank Peter J. Simon, PhD, CMPP, from MediTech Media, Ltd, and Martin Haschak, PhD from SciMentum, The Nucleus Group Holdings, Inc, for medical writing assistance, which was sponsored by Bristol Myers Squibb. The authors are fully responsible for all content and editorial decisions of this manuscript.
LR and JL report honoraria from Janssen; Celgene, a Bristol-Myers Squibb Company; Takeda; and Amgen. AO reports consultancy and speakers bureau with Amgen; Janssen; Takeda; and Celgene, a Bristol-Myers Squibb Company. RR reports consultancy with Amgen; Celgene, a Bristol-Myers Squibb Company; Janssen; and Takeda. CH reports honoraria from Celgene, a Bristol-Myers Squibb Company; Janssen; Takeda; and Amgen. AS reports honoraria from Bristol Myers Squibb, Takeda, Sanofi, Merck, and Roche; consultancy with Bristol Myers Squibb, Takeda, and Merck; and speakers bureau with Takeda. M-VM reports honoraria and membership on advisory boards with Janssen; Celgene, a Bristol-Myers Squibb Company; Amgen; Takeda; AbbVie; GlaxoSmithKline; EDO; PharmaMar; and Adaptive. MM reports honoraria, membership on advisory boards, and financial support with Celgene, a Bristol-Myers Squibb Company; Janssen; and Takeda and honoraria and membership on advisory boards with Amgen. JS-M reports consultancy with Bristol Myers Squibb; Celgene, a Bristol-Myers Squibb Company; Novartis; Takeda; Amgen; MSD; Janssen; and Sanofi and membership on board of directors or advisory committees with Takeda. KB reports honoraria and consultancy with Celgene, a Bristol-Myers Squibb Company; Amgen; Janssen; and Takeda. MG reports employment with Celgene, a Bristol-Myers Squibb Company. JB reports honoraria from Janssen; Celgene, a Bristol-Myers Squibb Company; and Amgen. PM reports honoraria from Celgene, a Bristol-Myers Squibb Company; Janssen; Takeda; AbbVie; and Amgen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Celgene, a Bristol-Myers Squibb Company. Author MG was employed by the funder and involved in study design, collection, analysis, or interpretation of data and writing the article and deciding to submit the article for publication.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1197340/full#supplementary-material
ASCT, autologous stem cell transplant; MRD, minimal residual disease; NDMM, newly diagnosed multiple myeloma; OS, overall survival; PFS, progression-free survival; PS, propensity score; RCT, randomized controlled trial; TE, transplant eligible; TEAE, treatment-emergent adverse event; VGPR, very good partial response; VRD, bortezomib + lenalidomide + dexamethasone; VTD, bortezomib + thalidomide + dexamethasone; ESMO, European Society for Medical Oncology; EMN, European Myeloma Network; SCO/CCO, American Society of Clinical Oncology/Cancer Care Ontario.
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136(5):E359–86. doi: 10.1002/ijc.29210
2. Sonneveld P, Broijl A. Treatment of relapsed and refractory multiple myeloma. Haematologica (2016) 101(8):995. doi: 10.3324/haematol.2016.148882
3. Surveillance Epidemiology and End Results (SEER): National Cancer Institute (2014). Available at: http://seer.cancer.gov.
4. Dimopoulos MA, Moreau P, Terpos E, Mateos MV, Zweegman S, Cook G, et al. Multiple myeloma: EHA-ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Hemasphere (2021) 5(2):e528. Available at: http://journals.lww.com/hemasphere/fulltext/2021/02000/multiple_myeloma__eha_esmo_clinical_practice.10.aspx.
5. Gay F, Engelhardt M, Terpos E, Wasch R, Giaccone L, Auner HW, et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica (2018) 103(2):197–211. doi: 10.3324/haematol.2017.174573
6. Mikhael J, Ismaila N, Cheung MC, Costello C, Dhodapkar MV, Kumar S, et al. Treatment of multiple myeloma: ASCO and CCO joint clinical practice guideline. J Clin Oncol (2019) 37(14):1228–63. doi: 10.1200/JCO.18.02096
7. Liwing J, Uttervall K, Lund J, Aldrin A, Blimark C, Carlson K, et al. Improved survival in myeloma patients: starting to close in on the gap between elderly patients and a matched normal population. Br J Haematol (2014) 164(5):684–93. doi: 10.1111/bjh.12685
8. Willenbacher E, Weger R, Rochau U, Siebert U, Willenbacher W, Austrian Myeloma Registry. Real-world use of 3rd line therapy for multiple myeloma in Austria: an Austrian Myeloma Registry (AMR) analysis of the therapeutic landscape and clinical outcomes prior to the use of next generation myeloma therapeutics. PloS One (2016) 11(3):e0147381. doi: 10.1371/journal.pone.0147381
9. Yong K, Delforge M, Driessen C, Fink L, Flinois A, Gonzalez-McQuire S, et al. Multiple myeloma: patient outcomes in real-world practice. Br J Haematol (2016) 175(2):252–64. doi: 10.1111/bjh.14213
10. Raab MS, Cavo M, Delforge M, Driessen C, Fink L, Flinois A, et al. Multiple myeloma: practice patterns across Europe. Br J Haematol (2016) 175(1):66–76. doi: 10.1111/bjh.14193
11. Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV, et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc (2004) 79(7):867–74. doi: 10.4065/79.7.867
12. Harousseau JL, Avet-Loiseau H, Attal M, Charbonnel C, Garban F, Hulin C, et al. Achievement of at least very good partial response is a simple and robust prognostic factor in patients with multiple myeloma treated with high-dose therapy: long-term analysis of the IFM 99-02 and 99-04 trials. J Clin Oncol (2009) 27(34):5720–6. doi: 10.1200/JCO.2008.21.1060
13. van de Velde HJ, Liu X, Chen G, Cakana A, Deraedt W, Bayssas M. Complete response correlates with long-term survival and progression-free survival in high-dose therapy in multiple myeloma. Haematologica (2007) 92(10):1399–406. doi: 10.3324/haematol.11534
14. Nadal E, Gine E, Blade J, Esteve J, Rosinol L, Fernandez-Aviles F, et al. High-dose therapy/autologous stem cell transplantation in patients with chemosensitive multiple myeloma: predictors of complete remission. Bone Marrow Transplant (2004) 33(1):61–4. doi: 10.1038/sj.bmt.1704313
15. Lonial S, Anderson KC. Association of response endpoints with survival outcomes in multiple myeloma. Leukemia (2014) 28(2):258–68. doi: 10.1038/leu.2013.220
16. Moreau P, Hulin C, Macro M, Caillot D, Chaleteix C, Roussel M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood (2016) 127(21):2569–74. doi: 10.1182/blood-2016-01-693580
17. Musto P, Montefusco V. Are maintenance and continuous therapies indicated for every patient with multiple myeloma? Expert Rev Hematol (2016) 9(8):743–51. doi: 10.1080/17474086.2016.1196127
18. Rosinol L, Oriol A, Teruel AI, Hernandez D, Lopez-Jimenez J, de la Rubia J, et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood (2012) 120(8):1589–96. doi: 10.1182/blood-2012-02-408922
19. Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as Induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised Phase 3 study. Lancet (2010) 376(9758):2075–85. doi: 10.1016/s0140-6736(10)61424-9
20. Niesvizky R, Flinn IW, Rifkin R, Gabrail N, Charu V, Clowney B, et al. Community-based phase IIIB trial of three UPFRONT bortezomib-based myeloma regimens. J Clin Oncol (2015) 33(33):3921–9. doi: 10.1200/JCO.2014.58.7618
21. Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol (2011) 12(5):431–40. doi: 10.1016/S1470-2045(11)70081-X
22. Wu S, Zheng C, Chen S, Cai X, Shi Y, Lin B, et al. Subcutaneous administration of bortezomib in combination with thalidomide and dexamethasone for treatment of newly diagnosed multiple myeloma patients. BioMed Res Int (2015) 2015(927105):927105. doi: 10.1155/2015/927105
23. Merz M, Salwender H, Haenel M, Mai EK, Bertsch U, Kunz C, et al. Subcutaneous versus intravenous bortezomib in two different induction therapies for newly diagnosed multiple myeloma: an interim analysis from the prospective GMMG-MM5 trial. Haematologica (2015) 100(7):964–9. doi: 10.3324/haematol.2015.124347
24. Rosinol L, Oriol A, Rios R, Sureda A, Blanchard MJ, Hernandez MT, et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood (2019) 134(16):1337–45. doi: 10.1182/blood.2019000241
25. Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med (2017) 376(14):1311–20. doi: 10.1056/NEJMoa1611750
26. Durie BGM, Hoering A, Sexton R, Abidi MH, Epstein J, Rajkumar SV, et al. Longer term follow-up of the randomized phase III trial SWOG S0777: bortezomib, lenalidomide and dexamethasone vs. lenalidomide and dexamethasone in patients (Pts) with previously untreated multiple myeloma without an intent for immediate autologous stem cell transplant (ASCT). Blood Cancer J (2020) 10(5):53. doi: 10.1038/s41408-020-0311-8
27. Knop S, Langer C, Engelhardt MM, Bassermann F, Schreder M, Muegge L-O, et al. Bortezomib, lenalidomide, and dexamethasone (VRD) is superior to lenalidomide, adriamycin, and dexamethasone (RAD) prior to risk-adapted transplant in newly diagnosed myeloma. J Clin Oncol (2020) 38 (15 Supplement). Abstract 8521. doi: 10.1200/JCO.2020.38.15_suppl.8521
28. Goldschmidt H, Mai EK, Bertsch U, Fenk R, Nievergall E, Tichy D, et al. Addition of isatuximab to lenalidomide, bortezomib, and dexamethasone as induction therapy for newly diagnosed, transplantation-eligible patients with multiple myeloma (GMMG-HD7): part 1 of an open-label, multicentre, randomised, active-controlled, phase 3 trial. Lancet Haematol (2022) 9(11):e810–e21. doi: 10.1016/S2352-3026(22)00263-0
29. Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res (2011) 46(3):399–424. doi: 10.1080/00273171.2011.568786
30. D’Agostino R. Tutorial in biostatistics: propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Statist Med (1998) 17:2265–81. doi: 10.1002/(SICI)1097-0258(19981015)17:19<2265::AID-SIM918>3.0.CO;2-B
31. Dimopoulos MA, Orlowski RZ, Facon T, Sonneveld P, Anderson KC, Beksac M, et al. Retrospective matched-pairs analysis of bortezomib plus dexamethasone versus bortezomib monotherapy in relapsed multiple myeloma. Haematologica (2015) 100(1):100–6. doi: 10.3324/haematol.2014.112037
32. Rosenbaum PR, Rubin DB. Reducing bias in observational studies using subclassification on the propensity score. J Am Stat Assoc (1984) 79(387):516–24. doi: 10.1080/01621459.1984.10478078
33. Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, Garcia-Sanchez O, Bottcher S, et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia (2017) 31(10):2094–103. doi: 10.1038/leu.2017.29
34. Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet (2019) 394(10192):29–38. doi: 10.1016/S0140-6736(19)31240-1
35. Tosi P, Zamagni E, Cellini C, Plasmati R, Cangini D, Tacchetti P, et al. Neurological toxicity of long-term (>1 yr) thalidomide therapy in patients with multiple myeloma. Eur J Haematol (2005) 74(3):212–6. doi: 10.1111/j.1600-0609.2004.00382.x
36. Thalidomide. [Summary of Product Characteristics]. Utrecht, Netherlands: Celgene Distribution BV (2018).
37. Kostopoulos IV, Ntanasis-Stathopoulos I, Gavriatopoulou M, Tsitsilonis OE, Terpos E. Minimal residual disease in multiple myeloma: current landscape and future applications with immunotherapeutic approaches. Front Oncol (2020) 10:860. doi: 10.3389/fonc.2020.00860
38. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Multiple Myeloma V.3.2023. (2023).
39. Moreau P, Touzeau C, Vij R, Goldsmith SR, Rosko AE. Newly diagnosed myeloma in 2020. Am Soc Clin Oncol Educ Book (2020) 40:1–15. doi: 10.1200/EDBK_280221
40. Goldschmidt H, Mai EK, Bertsch U, Besemer B, Haenel M, Miah K, et al. Elotuzumab in combination with lenalidomide, bortezomib, dexamethasone and autologous transplantation for newly-diagnosed multiple myeloma: results from the randomized phase III GMMG-HD6 trial. Blood (2021) 138(Supplement 1). Abstract 486. doi: 10.1182/blood-2021-147323
41. Jackson GH, Davies FE, Pawlyn C, Cairns DA, Striha A, Collett C, et al. Lenalidomide before and after autologous stem cell transplantation for transplant-eligible patients of all ages in the randomized, phase III, Myeloma XI trial. Haematologica (2021) 106(7):1957–67. doi: 10.3324/haematol.2020.247130
42. Pawlyn C, Jackson GH, Cairns D, Striha A, Hockaday A, Jacques G, et al. Maximizing pre-transplant response is associated with improved outcome for myeloma patients: exploratory analysis of the Myeloma XI trial. Blood (2018) 132(Supplement 1). Abstract 3280. doi: 10.1182/blood-2018-99-118868
43. Sonneveld P, Dimopoulos MA, Beksac M, van der Holt B, Aquino S, Ludwig H, et al. Consolidation and maintenance in newly diagnosed multiple myeloma. J Clin Oncol (2021) 39(32):3613–22. doi: 10.1200/JCO.21.01045
Keywords: multiple myeloma, bortezomib, lenalidomide, dexamethasone, thalidomide
Citation: Rosiñol L, Hebraud B, Oriol A, Colin A-L, Ríos Tamayo R, Hulin C, Blanchard MJ, Caillot D, Sureda A, Hernández MT, Arnulf B, Mateos M-V, Macro M, San-Miguel J, Belhadj K, Lahuerta JJ, Garelik MB, Bladé J and Moreau P (2023) Integrated analysis of randomized controlled trials evaluating bortezomib + lenalidomide + dexamethasone or bortezomib + thalidomide + dexamethasone induction in transplant-eligible newly diagnosed multiple myeloma. Front. Oncol. 13:1197340. doi: 10.3389/fonc.2023.1197340
Received: 30 March 2023; Accepted: 09 August 2023;
Published: 02 November 2023.
Edited by:
Anja Seckinger, VUB, BelgiumReviewed by:
Monique Hartley-Brown, Dana–Farber Cancer Institute, United StatesCopyright © 2023 Rosiñol, Hebraud, Oriol, Colin, Ríos Tamayo, Hulin, Blanchard, Caillot, Sureda, Hernández, Arnulf, Mateos, Macro, San-Miguel, Belhadj, Lahuerta, Garelik, Bladé and Moreau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Rosiñol, bHJvc2lub2xAY2xpbmljLmNhdA==
†ORCID: Laura Rosiñol, orcid.org/0000-0002-2534-9239
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.