
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 17 August 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1195814
 Firas Akrout1,2*†
Firas Akrout1,2*† Ahlem Achour3,4,5†
Ahlem Achour3,4,5† Carli M. J. Tops4
Carli M. J. Tops4 Richard Gallon6Rym Meddeb3,5Sameh Achoura1,2Mariem Ben Rekaya7,8Emna Hamdeni7Soumaya Rammeh7,8Ridha Chkili1,2Nada Mansouri2,9Neila Belguith3,10Ridha Mrad3,5
Richard Gallon6Rym Meddeb3,5Sameh Achoura1,2Mariem Ben Rekaya7,8Emna Hamdeni7Soumaya Rammeh7,8Ridha Chkili1,2Nada Mansouri2,9Neila Belguith3,10Ridha Mrad3,5Constitutional mismatch repair deficiency (CMMRD) syndrome is a rare autosomal recessive genetic disorder caused by biallelic germline mutations in one of the mismatch repair genes. Carriers are at exceptionally high risk for developing, typically in early life, hematological and brain malignancies, as well as cancers observed in Lynch syndrome. We report a homozygous MLH1 missense variant (c.1918C>A p.(Pro640Thr)) in a Tunisian patient with CMMRD syndrome and a family history of early-age colorectal cancer. The proband presented initially with colonic oligopolyposis and adenosquamous carcinoma of the caecum. He later developed several malignancies, including undifferentiated carcinoma of the parotid, grade 4 IDH-mutant astrocytoma, and ampulla of Vater adenocarcinoma. The patient was older than typical for this disease and had a remarkably prolonged survival despite developing four distinct aggressive malignancies. The current report highlights the challenges in assessing the pathogenicity of the identified variant and the remarkable phenotypic diversity in CMMRD.
Constitutional mismatch repair deficiency (CMMRD; MIM 276300) is a recessive childhood cancer syndrome caused by pathogenic variants in both alleles of one of the mismatch repair (MMR) genes (MLH1, MIM *120436; MSH2, MIM *609309; MSH6, MIM *600678 and PMS2, MIM *600259) (1). Unlike Lynch syndrome (LS), which is due to monoallelic variants in one of the same genes and leads mainly to colorectal and endometrial cancer development, CMMRD predisposes to a broad tumor spectrum. These malignancies include most frequently hematological, brain, and LS-associated neoplasms. CMMRD is often associated with features suggestive of neurofibromatosis type 1, specifically café-au-lait macules. Consensus reports suggest use of an indication score to identify at-risk patients, genetic testing, and ancillary molecular assays to confirm the diagnosis of CMMRD, and to implement cancer surveillance programs (2, 3). We herein describe a family in which three of the four siblings met the clinical criteria of CMMRD testing. One of them survived long-term, underwent genetic testing, and was found to have a homozygous germline missense variant (c.1918C>A p.(Pro640Thr)) in the MLH1 gene. We describe the clinical, pathological, and genetic findings of the patient harboring this variant, who exhibits strong evidence supporting the diagnosis of CMMRD and whose phenotype exhibits several original features.
The proband, a Tunisian male and the child of first cousins, presented at age 18 with a right-sided malignant colonic obstruction for which he underwent a total colectomy. Histopathology classified the tumor as an invasive adenosquamous carcinoma (pT4a, pN0, R0). Four tubulo-villous and tubular adenomatous polyps were also found in the removed specimen. Two of them showed high-grade dysplasia. Chemotherapy was administered according to the FUFOL regimen. Synchronous liver metastasis was identified during the postoperative investigation, and right hepatectomy was performed. At age 32, an undifferentiated carcinoma of the parotid was diagnosed, which was radically resected and locally treated with radiotherapy. One year later, about 20 synchronous rectal adenomas were detected on screening colonoscopy. One of them was severely dysplastic and a proctectomy was performed. At age 37, the patient exhibited headache, vomiting, and left-sided weakness. Imaging revealed a right frontal mass involving the corpus callosum with a marked ring-like gadolinium enhancement, central necrosis, and peritumoral edema (Figure 1A). Craniotomy and gross total resection of the tumor were done, and it was diagnosed as grade 4, isocitrate dehydrogenase (IDH)-mutant astrocytoma. He received radiochemotherapy according to Stupp’s protocol. At age 38, he developed obstructive jaundice due to a mass in the head of the pancreas. He underwent endoscopic retrograde cholangiopancreatography followed by papillotomy and placement of a plastic stent in the common bile duct. Histopathological examination revealed an adenocarcinoma of the ampulla of Vater. Three months later, the patient passed away from aggressive local and distant recurrence of his high-grade astrocytoma. A timeline depicting patient care is shown in Figure 1B.
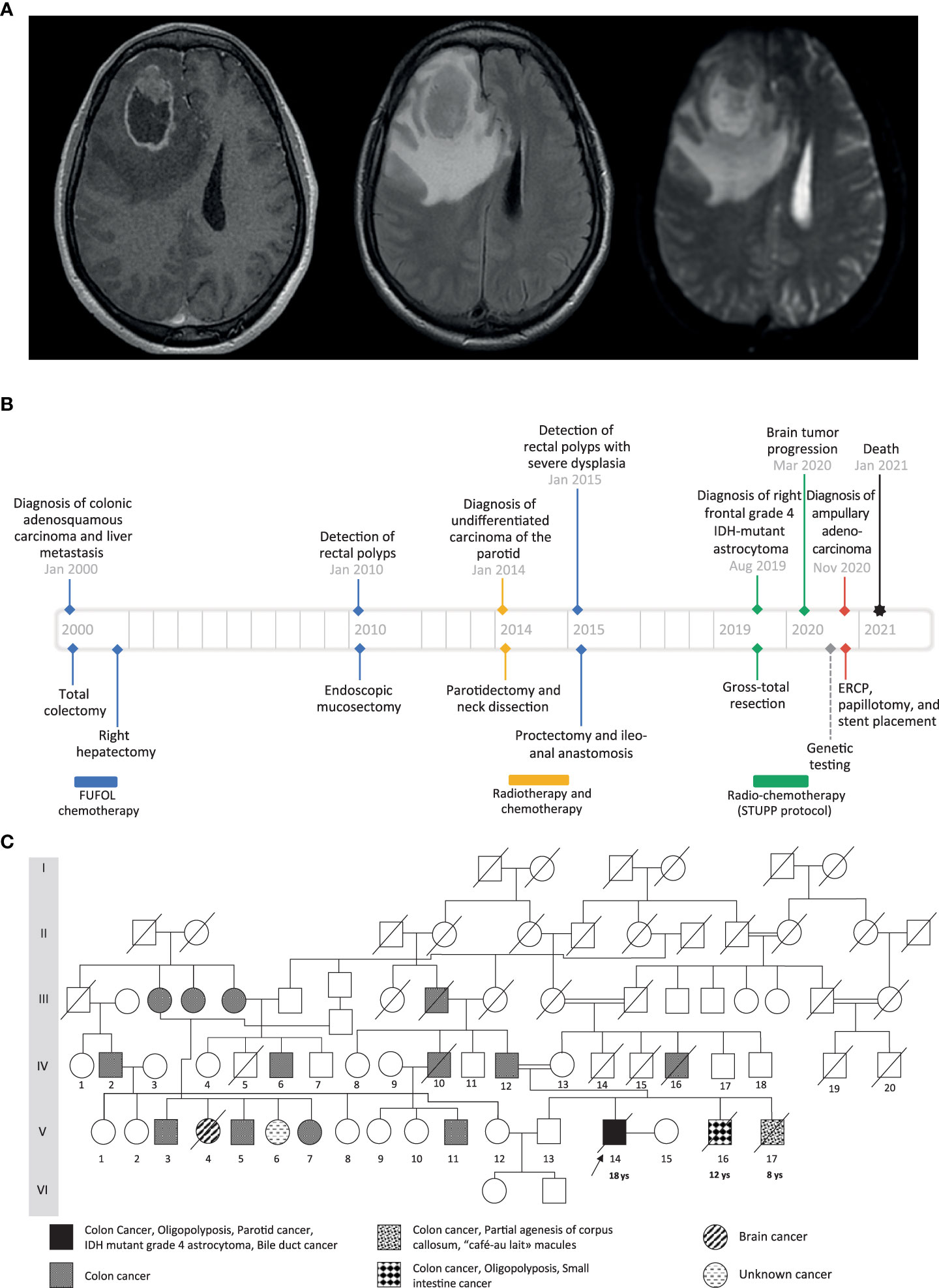
Figure 1 (A) Preoperative cranial MRI of the proband (from left to right: axial T1-weighted with gadolinium injection, axial T2 FLAIR-weighted, axial apparent diffusion coefficient). There is a solid intra-axial mass within the right frontal lobe with marked ring-like enhancement and an internal necrotic area. The enhancing component of the lesion shows diffusion restriction. (B) Timeline showing the clinical history of the patient. (C) Family pedigree. The black arrow indicates the proband case. The age of the first tumor is given below the proband and the two affected brothers.
Because of the history of metachronous malignancies and consanguinity, a cancer predisposition syndrome was suspected. The proband and his family were referred to genetic consultation, and a six-generation pedigree was established (Figure 1C). Physical examination revealed neither café-au-lait spots nor other features of neurofibromatosis type 1. Family history was significant for early-onset colorectal cancer (CRC) in two brothers: One (V.16) had undergone a screening colonoscopy at age 12, and was found to have several dysplastic polyps involving the colon and the small intestine. One of them showed invasive adenocarcinoma requiring a total colectomy. The youngest brother (V.17) was diagnosed with adenocarcinoma of the sigmoid at age 8. He also had a history of partial agenesis of the corpus callosum and café-au-lait macules. Both died from their diseases at ages 18 and 9, respectively. The proband’s father (IV.12) was diagnosed, at age 58, with several highly dysplastic polyps on screening colonoscopy, which led to performing a preventive total colectomy. The eldest brother (V.13) and the mother (IV.13) didn’t present any tumors at ages 41 and 63, respectively. Family history and clinical phenotype were consistent with CMMRD and the proband received a score of 11, meeting the indication criteria for CMMRD testing (2).
Genomic DNA was extracted from the peripheral blood leukocytes (PBLs) of the proband, unaffected brother, and parents. A CRC predisposition gene panel, including the MMR genes, was analyzed. Next-generation sequencing was performed on an Illumina platform with the Agilent enrichment kit SureSelectXT Clearseq inherited disease Panel. Data analysis was processed for all coding exons, including 20 nucleotides in the flanking intron sequences, with in-house pipelines. Sanger sequencing was performed using Big Dye terminator cycle sequencing Kit v.3.1 on a Seq Studio Applied Biosystems Sequencing platform. Genetic analysis revealed a homozygous variant (c.1918C>A; p.(Pro640Thr)) in the MLH1 gene: a substitution of cytosine by adenine in exon 17 leading to a missense mutation of a proline to a threonine. The variant was also identified in a heterozygous state in the father, the mother, and the eldest brother (Figure 2A). Unfortunately, the two deceased brothers have not been tested because DNA and tissue samples were unavailable.
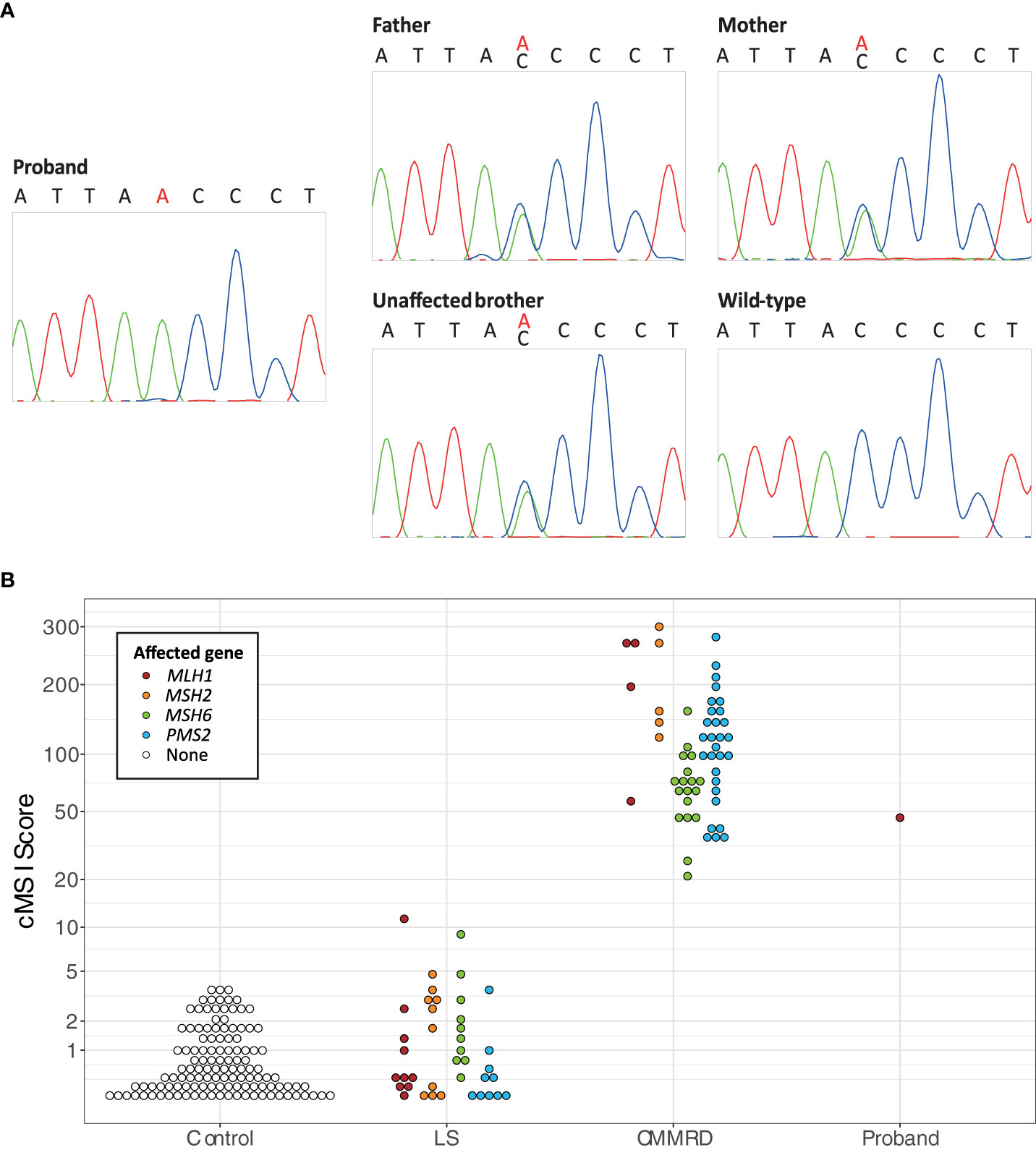
Figure 2 (A) Sanger sequencing results of the segregation study of the MLH1 c.1918C>A variant, including a chromatogram of the proband showing the homozygous MLH1 c.1918C>A variant, chromatograms of the father, the mother, and the unaffected brother showing the heterozygous MLH1 c.1918C>A variant, and a chromatogram of the wild type sequence. (B) The constitutional MSI (cMSI) scores of 123 control, 40 Lynch syndrome (LS; MLH1 n = 10, MSH2 n = 10, MSH6 n = 10, PMS2 n = 10), 56 constitutional mismatch repair deficiency syndrome (CMMRD; MLH1 n = 4, MSH2 n = 5, MSH6 n = 18, PMS2 n = 29), and 43 control patients using data from Gallon et al. (4), compared to the cMSI score of the proband. The y-axis is scaled based on a logit transformation.
The MLH1 c.1918C>A variant was not found in gnomAD (https://gnomad.broadinstitute.org/) and was predicted to be damaging by in silico software, including Polyphen2 (HumVar score 0.999) (http://genetics.bwh.harvard.edu/pph2/), SIFT (Score 0.00) (https://sift.bii.a-star.edu.sg/), AlignGVGD (Class C35) (http://agcgd.hci.utah.edu), and CADD (Phred score 26.7, Raw score 3.86) (http://cadd.gs.washington.edu/). The MAPP-MMR + Polyphen-2 prior probability for pathogenicity was 0.83 (http://priors.hci.utah.edu/PRIORS/) (5). MLH1 c.1918C>A has recently been observed as a heterozygous variant in several families with an LS phenotype. It was also shown to disrupt protein expression in a transfected HEK293T cell line model, but had minimal effect on MMR activity in vitro (6). In contrast, Takahashi et al. (7) had previously found >75% expression of MLH1 protein containing the variant in a transfected HCT116 cell line model, but an approximate 50% reduction in repair activity in vitro. These data suggest homozygosity of MLH1 c.1918C>A may cause CMMRD in the proband, but further investigations were pursued to confirm the diagnosis given the conflicting functional test results.
Immunohistochemistry staining for all four MMR proteins was assessed for all tumor specimens of the proband. We used an external positive control tissue from another individual for each antibody stain. The microscopic examination showed loss of MLH1 and PMS2 expression in neoplastic cells and surrounding normal cells, which is consistent with but not confirmatory of a CMMRD diagnosis caused by variants in MLH1. Immunostaining was retained for MSH2 and, to a lesser extent, for MSH6 (Figure 3).
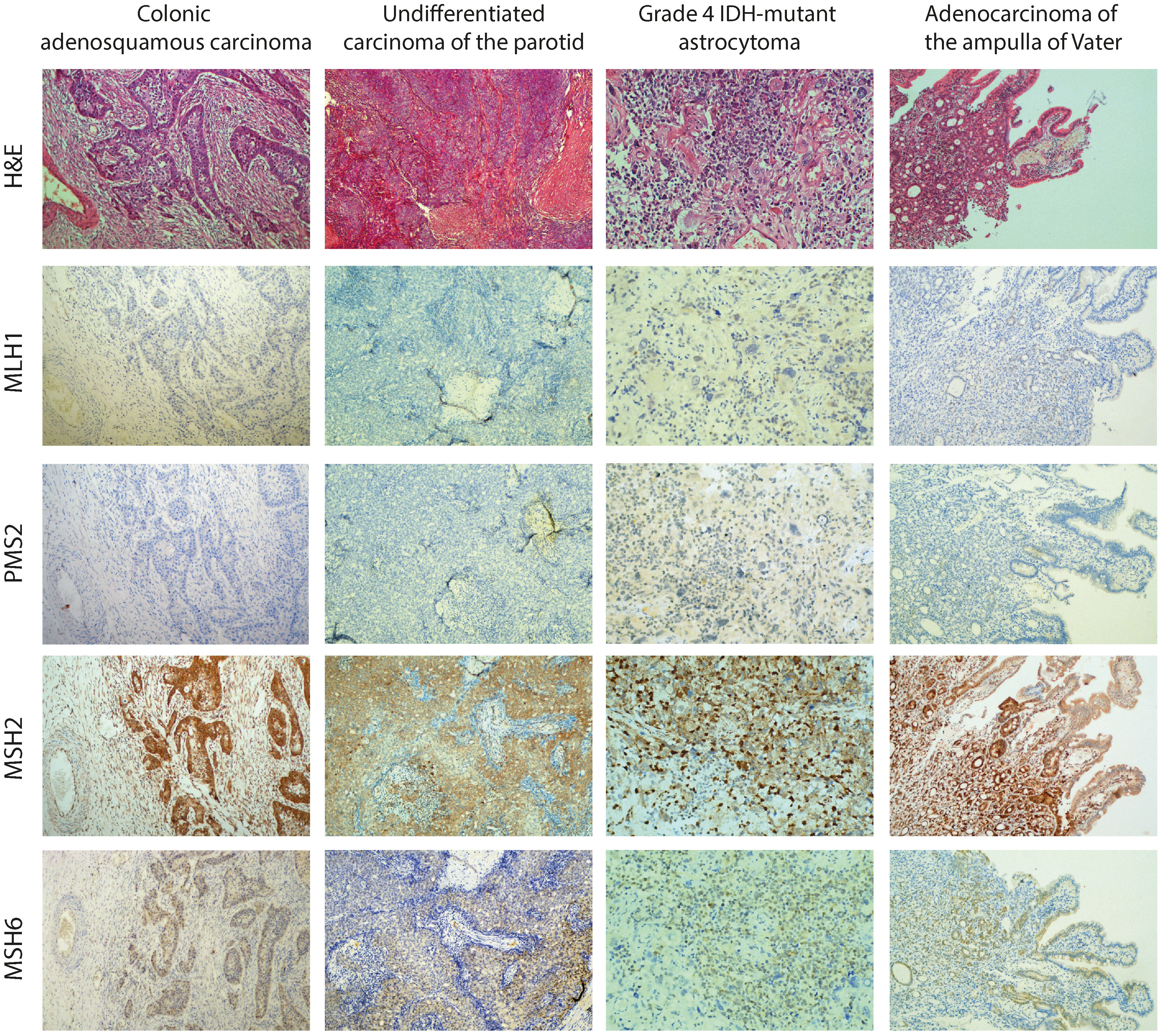
Figure 3 Histology (hematoxylin and eosin (H&E)) and immunohistochemical staining for all four MMR proteins (MLH1, PMS2, MSH2, and MSH6) in all tumor specimens of the proband (colonic and parotid tumor at 40× magnification, brain tumor and pancreatic tumor at 100× magnification). As the representative images show, immunostaining for MLH1 and PMS2 revealed a loss of expression in neoplastic cells and surrounding normal cells.
Microsatellite instability (MSI) analysis was performed on DNA samples extracted from formalin fixed paraffin embedded tissue of the colorectal, cerebral, parotid, and ampulla of Vater tumors, and non-tumoral colorectal mucosa. Six monomorphic microsatellite markers (NR21, NR24, NR27, BAT25, BAT26 and HSP110) were amplified and the amplicons were separated and visualized using the ABI Seq Studio Genetic Analyzer (Applied Biosystem). Fragment length analysis of microsatellite amplicons showed instability in both tumoral and non-tumoral colorectal tissues, as well as in the cerebral, parotid, and ampulla of Vater tumors. However, in blood, all MSI markers showed a stable profile (Supplementary Table 1).
Along with tumor analyses, the impact of MLH1 c.1918C>A; p.(Pro640Thr) on MMR function was further assessed using the cell-free in vitro MMR activity (CIMRA) assay (8) and was shown to have 17% relative MMR activity. Together, these results provide strong evidence for the pathogenicity of MLH1 c.1918C>A (Table 1) and support a diagnosis of CMMRD in the proband.

Table 1 MLH1 c.1918C>A; p.(Pro640Thr) variant interpretation according to ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Variant Curation Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1.
As a final confirmation of the diagnosis, PBL DNA of the proband was analyzed for constitutional MSI (cMSI), MSI in non-neoplastic tissues that is a highly specific molecular feature of CMMRD. cMSI is not detectable by methods designed for tumor MSI analysis and requires specialist assays (4). The cMSI assay amplifies and sequences 32 highly sensitive MSI markers, and uses the frequency of microsatellite insertion and deletion variants to calculate a cMSI score for each sample. The higher the cMSI score the higher the constitutional burden of MSI. The proband had a cMSI score of 44.3, which falls within the published cMSI score range of known CMMRD patients (20.9-300.0) and is much greater than the cMSI scores of LS carriers (0.0-11.3) and controls (0.0-3.6) (4). cMSI analysis, therefore, confirmed the CMMRD diagnosis (Figure 2B).
Biallelic germline variants in MMR genes are known to cause the recessive CMMRD syndrome. The latter was recognized as a distinct childhood cancer predisposition syndrome in 1999, although it was initially named Turcot syndrome (9, 10). After its first description, more than a hundred cases of biallelic germline variants in one of the MMR genes, including PMS2, MSH2, MSH6, and MLH1, have been identified (11). The c.1918C>A; p.(Pro640Thr) variant of the MLH1 gene identified in our patient has been previously detected in LS (6). However, to our knowledge, a biallelic case of MLH1 c.1918C>A; p.(Pro640Thr) has never been reported in CMMRD syndrome. Many pieces of evidence were suggestive of the pathogenicity of this variant and its involvement in our patient’s phenotype. Apart from its absence from population databases (PM2_Supporting), it is predicted to be damaging (Prior probability of pathogenicity 0.83) (PP3_Moderate) and is located in a conserved interaction domain between MLH1 and PMS2 506-675 (12). Recently, Mahdouani et al. reported MLH1 c.1918C>A; p.(Pro640Thr) heterozygotes in three Tunisian families with an LS phenotype from the same geographical area of our proband. They compared protein stability and catalytic activity between the MLH1 p.(Pro640Thr) variant and a validated reference pathogenic variant MLH1 p.(Ala681Thr). The p.(Pro640Thr) variant results in severe defects in protein stability and minimal reduction in catalytic activity (6) (PS3_Strong). However, due to conflicting functional results from Takahashi et al., we conducted further assessment of the functional impact of the variant.
IHC showed loss of MLH1 and PMS2 expression in all tumor and normal cells within tumor tissue sections, consistent with a CMMRD diagnosis. MLH1 and PMS2 are known to act as partners in the repair process, and so functional defects in MLH1 can lead to the instability of both MLH1 and PMS2. Therefore, the IHC results were consistent with the functional analyses of Mahdouani et al., who found the missense variant p.(Pro640Thr) reduced MLH1 protein expression (6). MSI was identified in both non-neoplastic and neoplastic colorectal tissue, and found MLH1 p.(Pro640Thr) caused reduced MMR activity by CIMRA (8). According to the ACMG-adapted InSiGHT criteria (under review, Draft InSiGHT ACMG MMR gene variant classification criteria version 1; https://www.insight-group.org/criteria/), these findings allowed us to classify the variant as pathogenic (Table 1).
The variant was also identified in the heterozygous state in the father, who had a history of CRC consistent with LS, and in the unaffected mother and brother, whose lack of cancer history can be explained by the incomplete penetrance of the LS phenotype. The two brothers who died from their disease could not be tested. However, we speculate that they were homozygous for the MLH1 c.1918C>A variant given their ages at diagnosis and the severity of their phenotypes.
MSI was observed in tumor tissues and in colorectal mucosa of the proband but not in their PBLs using PCR and fragment length analysis of six mononucleotide repeat MSI markers. Several parameters influence MSI assay sensitivity, such as the structure of the repetitive motif (di- or mono-nucleotide repeat), the number of used markers, and the analysis method. Moreover, MSI is the consequence of MMR deficiency during DNA replication. We, therefore, hypothesize that MSI detection depends on the rate of cell division and the lifespan of the cells in each tissue, which could explain the MSS profile detected in our patient’s non-neoplastic blood cells as compared to the MSI profile of the colorectal mucosa. The MSI score was the lowest in the cerebral tumor and highest in the colorectal mucosa and tumor tissues, as well as parotid and ampulla of Vater tumors (Supplementary Table 1). Previous studies reported that MSI often fails in brain malignancies and it has been suggested that IHC in all malignant brain tumors younger than 25 years old could be used to screen for CMMRD (13).
Detection of MSI in the constitutional tissues (cMSI) of CMMRD patients requires specialist techniques. For example, Bodo et al. found that PCR and fragment length analysis of mononucleotide repeat MSI markers could not detect MSI in the PBLs but could detect MSI in the immortalized lymphoblastoid cell lines (cultured to develop the MSI signal) of CMMRD patients – a technique named ex vivo MSI analysis (14). More recently, several studies have shown the utility of next-generation sequencing for the detection of cMSI directly from CMMRD PBLs, using either targeted amplicon sequencing or whole genome sequencing approaches (15–17). Here, we used a published amplicon sequencing-based cMSI assay of 32 mononucleotide repeat MSI markers that had been selected from genome sequencing of CMMRD PBL samples specifically for cMSI analysis, which is highly sensitive and specific for CMMRD detection (4). The proband’s cMSI score of 44.3 corroborates a diagnosis of CMMRD and classification of MLH1 c.1918C>A; p.(Pro640Thr) as pathogenic. Interestingly, a cMSI score of 44.3 is the lowest observed for CMMRD caused by MLH1 deficiency but is consistent with previous observations that missense MMR gene variants are associated with lower cMSI scores (4). Despite observed correlations between MMR genotype and cMSI phenotype, no significant association of cMSI score with age at first cancer was previously found (4). Here, it would have been of interest to compare cMSI scores of the proband and their two affected brothers, who we assume shared a homozygous MLH1 c.1918C>A genotype, given their very different ages at first cancer, but DNA was not available from the brothers for genetic or cMSI analyses.
CMMRD is poorly recognized by clinicians, and its diagnosis tends to be delayed due to the lack of specific clinical features and the broad tumor spectrum. To address this problem, the European consortium “Care for CMMRD” suggested a scoring system to help physicians to diagnose this disease at the time of the first malignancy (2). According to this scoring system, our patient would have reached a high enough score at the time of his first tumor to initiate CMMRD testing. However, he was suspected of suffering from CMMRD only when he developed his third malignancy. Hence, clinicians should be aware of this cancer predisposition syndrome as a rare cause of early-onset malignancy to achieve timely diagnosis and thus provide an appropriate surveillance program and genetic counseling.
Our case supplements the existing literature by illustrating several atypical features. First, the patient had an atypical course of his disease, with the development of initial cancer at a relatively older age and a remarkably prolonged survival despite developing four distinct aggressive malignancies. In 2014, Wimmer and Kratz reviewed all reported cases, including 146 individuals with CMMRD syndrome, and 82% were younger than 18 years of age at the time of diagnosis of their first malignancy (2). Hematological malignancies are reported to occur in early childhood, around age 5 years, whereas brain cancers develop later with a median age at diagnosis of 9 years, and LS-associated tumors, mostly CRC, develop even later with a median age at diagnosis of 17 years. In our case, the patient presented initially at 18 years of age with colonic cancer. This suggests that CMMRD syndrome is not confined to children and may also manifest in early adulthood. Long-term survival in CMMRD syndrome is also uncommon due to a particularly poor prognosis, with median overall survival of 27 months after the diagnosis of the first malignancy (18). Studies suggest that prognosis varies depending on which MMR gene is affected. Indeed, individuals with biallelic MLH1 or MSH2 variants show a more severe phenotype and a lower chance of surviving the first tumor than those with biallelic MSH6 or PMS2 variants (1). Surprisingly, the proband in the present study lived 20 years after his first cancer diagnosis, and developed three other malignancies. His relatively long lifespan contrasts with the variant occurring in the MLH1 gene rather than other MMR genes. Previous cases of attenuated CMMRD have been reported, characterized most frequently by CRC in early adulthood rather than hematological malignancies or brain tumors, caused by a hypomorphic splice site variant in PMS2 (19). These cases have also been analyzed by the cMSI assay used here and, similar to the proband, had relatively low cMSI scores (4). This, the functional results of Mahdouani et al. (6), and the atypical phenotype of the proband suggest the MLH1 c.1918C>A; p.(Pro640Thr) could also be considered hypomorphic. Alternatively, the relatively mild phenotype of the proband may be explained by other genetic or environmental factors, and the effective management of each tumor, including radical treatment of his cancers and prophylactic proctectomy to avoid additional rectal malignancies.
Second, the vast majority of high-grade astrocytomas in CMMRD patients are IDH-wildtype (glioblastoma), and IDH-mutant astrocytoma, as diagnosed in the proband, rarely occurs. IDH-wildtype and -mutant tumors are two distinct subsets of high-grade astrocytomas that develop through different genetic pathways and exhibit dissimilar prognoses (20). In a histological review of 26 grade 4 astrocytomas in the setting of CMMRD syndrome, all tumors were identified as IDH-wildtype except for one (21). We report another rare case with grade 4 IDH-mutant astrocytoma. The current standard treatment for grade 4 astrocytoma involves maximal surgical resection, followed by radiotherapy and temozolomide chemotherapy (22). However, MMR-deficient high-grade astrocytomas are known to be resistant to temozolomide but immunotherapy has shown promising results (21, 23). Unfortunately, our index patient was diagnosed with CMMRD syndrome after undergoing temozolomide treatment, which could explain the observed tumor resistance to this methylating drug. Thus, timely diagnosis of CMMRD syndrome is necessary not only for pre-emptive cancer surveillance but also for choosing the most effective therapeutic regimen.
Third, our patient developed an undifferentiated carcinoma of the parotid and an adenocarcinoma of the ampulla of Vater, two unusual malignancies that have been reported only once or twice in CMMRD syndrome. The European Consortium Care for CMMRD divided CMMRD tumors into four groups: hematological malignancies, central nervous system tumors, LS-associated tumors, and other unusual neoplasms. Several tumors belonging to the latter group were described in isolated CMMRD cases, including neuroblastoma, Wilms tumors, rhabdomyosarcoma, ovarian neuroectodermal tumor, infantile myofibromatosis, breast cancer, sarcoma, and pilomatricoma (2). Parotid cancer was not listed but had been separately reported by Baas et al. in a child carrying a biallelic PMS2 variant, who had a history of B-cell Hodgkin lymphoma, agenesis of the corpus callosum, and developed at age 11 a mucoepidermoid carcinoma of the parotid gland (24). To our knowledge, our case is the second report of a malignant salivary gland tumor in CMMRD. Pancreaticobiliary tumors are also unusual in CMMRD syndrome, although these are included in the spectrum of LS-associated malignancies (25). A carcinoma of the papilla, diagnosed at age 22, has been described in one CMMRD case carrying a biallelic PMS2 variant (26), and an ampullary adenocarcinoma, diagnosed at age 12, has been described in another carrying a biallelic MSH6 variant (27). To our knowledge, our proband is the third reported case of pancreaticobiliary cancer in a CMMRD patient. Small intestine cancer is often observed in individuals with CMMRD syndrome and belongs to the LS-associated tumors group (28, 29). This was the case in the proband’s brother, who developed small bowel cancer at age 17 and was likely homozygous for the MLH1 c.1918C>A variant.
Finally, the proband presented with colonic oligopolyposis reminiscent of attenuated familial adenomatous polyposis or MUTYH-associated polyposis. Colonic polyposis is absent in LS, and polyps in this “hereditary non-polyposis syndrome” are only slightly more prevalent than in the general population (30). However, oligopolyposis is a common finding in CMMRD syndrome and can cause misdiagnosis of CMMRD patients as having attenuated familial adenomatous polyposis (26). Therefore, CMMRD syndrome should be considered in patients with childhood-onset adenomatous polyposis in the absence of proven APC or MUTYH germline variants, as suggested by Herkert et al. and argued by expert opinion from the international CMMRD Consortium (2, 31).
In conclusion, the CMMRD case described here contributes to the continuing evolution of our knowledge of the CMMRD syndrome. Molecular diagnosis allowed accurate genetic counseling for this family, with predictive genetic testing being proposed to facilitate targeted preventive surveillance. Additional assessment of the functional impact of MLH1 c.1918C>A; p.(Pro640Thr) and the detection of increased cMSI in the proband has confirmed its pathogenicity. The incidence of the MLH1 c.1918C>A variant in Tunisian families with LS and CMMRD could be consistent with a founder effect. However, more studies and enlarging our cohort are needed to confirm this hypothesis. Our report of a CMMRD patient, diagnosed with parotid and pancreaticobiliary malignancies, provides an unusual phenotypic expression of biallelic MMR gene variant carriers and broadens the tumor spectrum observed in this condition. Even though CMMRD syndrome is exceptionally rare, increasing awareness of this disease is crucial, not only to implement cancer surveillance and prophylaxis but also to deliver accurate and effective cancer therapy.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Ethics Committee of Charles Nicolle Hospital (FWA00032748). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FA and AA conceived and drafted the manuscript. CT, NB, and AA contributed to the genomic data analysis and interpretation. RG performed cMSI analysis and contributed sections to the manuscript. NB contributed to genetic counseling. SR, MB, and EH performed MSI testing and contributed sections to the manuscript. CT, NB, RyM, and RiM critically revised the manuscript for important intellectual content. FA, SA and RC participated in the patient’s clinical care and contributed intellectually. SR and NM interpreted immunohistochemistry data and provided a pathological assessment of the case. FA and AA authors contributed equally to this work. All authors read and approved the final manuscript.
The authors thank all members of this family for their cooperation, which was essential for completing this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1195814/full#supplementary-material
1. Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet (2008) 124(2):105–22. doi: 10.1007/s00439-008-0542-4
2. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet (2014) 51(6):355–65. doi: 10.1136/jmedgenet-2014-102284
3. Aronson M, Colas C, Shuen A, Hampel H, Foulkes WD, Baris Feldman H, et al. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): recommendations from the international consensus working group. J Med Genet (2021) 59:318–327. doi: 10.1136/jmedgenet-2020-107627
4. Gallon R, Phelps R, Hayes C, Brugieres L, Guerrini-Rousseau L, Colas C, et al. Constitutional microsatellite instability, genotype, and phenotype correlations in constitutional mismatch repair deficiency. Gastroenterology (2023) 164(4):579–92 e8. doi: 10.1053/j.gastro.2022.12.017
5. Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat (2013) 34(1):255–65. doi: 10.1002/humu.22214
6. Mahdouani M, Ben Ahmed S, Hmila F, Rais H, Ben Sghaier R, Saad H, et al. Functional characterization of MLH1 missense variants unveils mechanisms of pathogenicity and clarifies role in cancer. PLoS One (2022) 17(12):e0278283. doi: 10.1371/journal.pone.0278283
7. Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res (2007) 67(10):4595–604. doi: 10.1158/0008-5472.CAN-06-3509
8. Drost M, Tiersma Y, Thompson BA, Frederiksen JH, Keijzers G, Glubb D, et al. A functional assay-based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet Med (2019) 21(7):1486–96. doi: 10.1038/s41436-018-0372-2
9. Ricciardone MD, Ozcelik T, Cevher B, Ozdag H, Tuncer M, Gurgey A, et al. Human MLH1 deficiency predisposes to hematological Malignancy and neurofibromatosis type 1. Cancer Res (1999) 59(2):290–3.
10. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The molecular basis of Turcot’s syndrome. N Engl J Med (1995) 332(13):839–47. doi: 10.1056/NEJM199503303321302
11. Citak EC, Sagcan F, Gundugan BD, Bozdogan ST, Yilmaz EB, Avci E, et al. Metachronous Wilms Tumor, Glioblastoma, and T-cell Leukemia in an Child With Constitutional Mismatch Repair Deficiency syndrome due to Novel Mutation in MSH6 (c.2590G>T). J Pediatr Hematol Oncol (2021) 43(2):e198–202. doi: 10.1097/MPH.0000000000001687
12. Guerrette S, Acharya S, Fishel R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem (1999) 274(10):6336–41. doi: 10.1074/jbc.274.10.6336
13. Carrato C, Sanz C, Munoz-Marmol AM, Blanco I, Pineda M, Del Valle J, et al. The challenge of diagnosing constitutional mismatch repair deficiency syndrome in brain Malignancies from young individuals. Int J Mol Sci (2021) 22(9):4629. doi: 10.3390/ijms22094629
14. Bodo S, Colas C, Buhard O, Collura A, Tinat J, Lavoine N, et al. Diagnosis of constitutional mismatch repair-deficiency syndrome based on microsatellite instability and lymphocyte tolerance to methylating agents. Gastroenterology. (2015) 149(4):1017–29.e3. doi: 10.1053/j.gastro.2015.06.013
15. Chung J, Negm L, Bianchi V, Stengs L, Das A, Liu ZA, et al. Genomic microsatellite signatures identify germline mismatch repair deficiency and risk of cancer onset. J Clin Oncol (2023) 41(4):766–77. doi: 10.1200/JCO.21.02873
16. Gallon R, Muhlegger B, Wenzel SS, Sheth H, Hayes C, Aretz S, et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum Mutat (2019) 40(5):649–55. doi: 10.1002/humu.23721
17. Gonzalez-Acosta M, Marin F, Puliafito B, Bonifaci N, Fernandez A, Navarro M, et al. High-sensitivity microsatellite instability assessment for the detection of mismatch repair defects in normal tissue of biallelic germline mismatch repair mutation carriers. J Med Genet (2020) 57(4):269–73. doi: 10.1136/jmedgenet-2019-106272
18. Lavoine N, Colas C, Muleris M, Bodo S, Duval A, Entz-Werle N, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet (2015) 52(11):770–8. doi: 10.1136/jmedgenet-2015-103299
19. Li L, Hamel N, Baker K, McGuffin MJ, Couillard M, Gologan A, et al. A homozygous PMS2 founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J Med Genet (2015) 52(5):348–52. doi: 10.1136/jmedgenet-2014-102934
20. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
21. Guerrini-Rousseau L, Varlet P, Colas C, Andreiuolo F, Bourdeaut F, Dahan K, et al. Constitutional mismatch repair deficiency-associated brain tumors: report from the European C4CMMRD consortium. Neurooncol Adv (2019) 1(1):vdz033. doi: 10.1093/noajnl/vdz033
22. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
23. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol (2016) 34(19):2206–11. doi: 10.1200/JCO.2016.66.6552
24. Baas AF, Gabbett M, Rimac M, Kansikas M, Raphael M, Nievelstein RA, et al. Agenesis of the corpus callosum and gray matter heterotopia in three patients with constitutional mismatch repair deficiency syndrome. Eur J Hum Genet (2013) 21(1):55–61. doi: 10.1038/ejhg.2012.117
25. Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA (2009) 302(16):1790–5. doi: 10.1001/jama.2009.1529
26. Levi Z, Kariv R, Barnes-Kedar I, Goldberg Y, Half E, Morgentern S, et al. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: from a single adenoma to polyposis-like phenotype and early onset cancer. Clin Genet (2015) 88(5):474–8. doi: 10.1111/cge.12518
27. Shimamura Y, Walsh CM, Cohen S, Aronson M, Tabori U, Kortan PP, et al. Role of video capsule endoscopy in patients with constitutional mismatch repair deficiency (CMMRD) syndrome: report from the International CMMRD Consortium. Endosc Int Open (2018) 6(8):E1037–43. doi: 10.1055/a-0591-9054
28. Roy S, Raskin L, Raymond VM, Thibodeau SN, Mody RJ, Gruber SB. Pediatric duodenal cancer and biallelic mismatch repair gene mutations. Pediatr Blood Cancer. (2009) 53(1):116–20. doi: 10.1002/pbc.21957
29. Suerink M, Kilinc G, Terlouw D, Hristova H, Sensuk L, van Egmond D, et al. Prevalence of mismatch repair deficiency and Lynch syndrome in a cohort of unselected small bowel adenocarcinomas. J Clin Pathol (2021) 74(11):724–9. doi: 10.1136/jclinpath-2020-207040
30. Kalady MF, Kravochuck SE, Heald B, Burke CA, Church JM. Defining the adenoma burden in lynch syndrome. Dis Colon Rectum (2015) 58(4):388–92. doi: 10.1097/DCR.0000000000000333
Keywords: CMMRD, hereditary cancer syndromes, next-generation sequencing, MLH1 gene, Lynch syndrome, cMSI, case report
Citation: Akrout F, Achour A, Tops CMJ, Gallon R, Meddeb R, Achoura S, Ben Rekaya M, Hamdeni E, Rammeh S, Chkili R, Mansouri N, Belguith N and Mrad R (2023) Constitutional mismatch repair deficiency syndrome with atypical features caused by a homozygous MLH1 missense variant (c.1918C>A, p.(Pro640Thr)): a case report. Front. Oncol. 13:1195814. doi: 10.3389/fonc.2023.1195814
Received: 29 March 2023; Accepted: 24 July 2023;
Published: 17 August 2023.
Edited by:
Leonhard Müllauer, Medical University of Vienna, AustriaReviewed by:
Dongliang Wang, ChosenMed Technology (Beijing) Co., Ltd., ChinaCopyright © 2023 Akrout, Achour, Tops, Gallon, Meddeb, Achoura, Ben Rekaya, Hamdeni, Rammeh, Chkili, Mansouri, Belguith and Mrad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Firas Akrout, YWtyb3V0LmZpcmFzQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.