 Yu-Xiang Cai
Yu-Xiang Cai Jin-Sheng Liu
Jin-Sheng Liu Jian Xu1†
Jian Xu1† Yong-Ze He
Yong-Ze He Zhi-Qiang Li
Zhi-Qiang Li- 1Department of Pathology, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Department of Neurosurgery, Zhongnan Hospital of Wuhan University, Wuhan, China
Background: The purpose of this study is to present a series of primary intracranial sarcomas (PIS), a rare type of tumor of the central nervous system, in order to improve our understanding of the disease. These tumors are heterogeneous and prone to recurrence after resection, exhibiting a high mortality rate. As PIS has yet to be understood and studied on a large scale, it is vital for further evaluation and research.
Methods: Our study included 14 cases of PIS. The patients’ clinical, pathological, and imaging features were retrospectively analyzed. Additionally, targeted DNA next-generation sequencing (NGS) was applied for the 481-gene panel to detect gene mutations.
Results: The average age for PIS patients was 31.4 years. Headache (7, 50.0%) was the most common symptom leading to the hospital visit. Twelve cases had PIS located in the supratentorial area and two in the cerebellopontine angle region. The maximum tumor diameter ranged from 19.0 mm to 130.0 mm, with an average diameter of 50.3 mm. Pathological types of tumors were heterogeneous, with chondrosarcoma being the most common, followed by fibrosarcoma. Eight of the 10 PIS cases that underwent MRI scanning showed gadolinium enhancement; 7 of these cases were heterogeneous, and 1 of them was garland-like. Targeted sequencing was performed in two cases and identified mutations in genes such as NRAS, PIK3CA, BAP1, KDR, BLM, PBRM1, TOP2A, DUSP2, and CNV deletions of SMARCB1. Additionally, the SH3BP5::RAF1 fusion gene was also detected. Of the 14 patients, 9 underwent a gross total resection (GTR), and 5 chose subtotal resection. Patients who underwent GTR displayed a trend toward superior survival. Among the 11 patients with available follow-up information, one had developed lung metastases, three had died, and eight were alive.
Conclusion: PIS is extremely rare compared to extracranial soft sarcomas. The most common histological type of intracranial sarcoma (IS) is chondrosarcoma. Patients who underwent GTR of these lesions showed improved survival rates. Recent advancements in NGS aided in the identification of diagnostic and therapeutic PIS-relevant targets.
1 Introduction
Primary intracranial sarcomas (PIS) are a rare group of heterogeneous tumors (1), with their prevalence ranging from 0.1% to 4.3% across the literature (2). These tumors probably have the origin of multipotent primitive mesenchymal cells within leptomeninges or dura (1, 3–5). The ionizing radiation is the only evident associated risk factor (4, 5). Combining clinical and imaging data with a histopathological examination of PIS aids in its diagnosis (1).
The underlying mechanisms of PIS remain unclear due to its rarity and heterogeneity. In pediatric sarcomas, there is significant activation of signaling pathways, such as angiogenesis as well as receptor tyrosine kinases (RTKs), associated with tumor survival (6–9). The platelet growth factor receptor (PDGFR), as an RTK, shows overexpression within Ewing sarcoma, rhabdomyosarcoma, and osteosarcoma (OS) (9–12). Its involvement in tumorigenesis bears considerable implications in terms of prognosis and treatment. PIS does not have an accurate approach to its treatment established to date. A combination of surgical and adjuvant therapies is typically utilized in PIS management, whereas radiation therapy is a common approach for local disease control (9, 13, 14). Physicians can better comprehend the molecular characteristics of the disease and identify potential treatments when next-generation sequencing (NGS) and other molecular biology techniques are applied in clinical practice. Moreover, this high-throughput approach has the potential to identify critical information that could lead to changes in the diagnosis (15).
The knowledge concerning clinicopathological features of PIS is deficient as fewer studies have been conducted on PIS. Thus, an investigation of 14 PIS cases was conducted to comprehend their behavior and prognosis better. Furthermore, targeted sequencing was carried out on some of the PIS cases to obtain diagnostic and therapeutic insight.
2 Materials and methods
2.1 Selection of patients
In this study, 14 cases of PIS were retrospectively collected from the archives of Zhongnan Hospital of Wuhan University. Cases of malignant meningioma, gliosarcoma, malignant solitary fibrous tumor, and intracranial metastatic sarcoma were excluded from the study. Clinical data obtained from the patients included their sex, age, location, clinical manifestations, and radiologic appearances. Clinical courses of patients were tracked retrospectively. All cases were reviewed by two pathologists (ST, YC) who assessed their pathological features. This study was approved by the Institutional Review Board at the Zhongnan Hospital of Wuhan University (approval code: 2022007K).
2.2 Histopathology and immunohistochemistry
The specimens were fixed with 10% neutral formalin solution, routinely embedded in paraffin, 4μm thick sections were stained with haematoxylin and eosin-stain and immunohistochemistry. The following antibodies were performed using automated instruments CK (AE1/AE3, ZSGB-BIO, prediluted), EMA (GP14, ZSGB-BIO, prediluted), Bcl-2 (EP36, ZSGB-BIO, prediluted), SMA (UMAB237, ZSGB-BIO, prediluted), Dsmin (OTI4A8, ZSGB-BIO, prediluted), CD34 (QBEnd10, DAKO, prediluted), MDM2 (1E6&17B3, ZSGB-BIO, prediluted), CD99 (12E7, DAKO, prediluted), SATB2 (OTI5H7, ZSGB-BIO, prediluted), TLE1 (UMAB253, ZSGB-BIO, prediluted), SSTR-2 (EP149, ZSGB-BIO, prediluted), ALK (OTI1H7, ZSGB-BIO, prediluted), S-100 (15E2E2 + 4C4.9, ZSGB-BIO, prediluted), IDH1 (H09, ZSGB-BIO, prediluted), Myogenin (EP162, ZSGB-BIO, prediluted), MyoD1(EP212, ZSGB-BIO, prediluted), CyclinD1(SA38-08, ZSGB-BIO, prediluted), CD68(KP1, ZSGB-BIO, prediluted), SMARCB1 (OTIR4G9, ZSGB-BIO, prediluted), STAT6 (EP325, ZSGB-BIO, prediluted), GFAP (EP13, ZSGB-BIO, prediluted), Olig-2 (EP112, ZSGB-BIO, prediluted), Ki-67 (MIB-1, ZSGB-BIO, prediluted).
2.3 Targeted next-generation DNA sequencing analysis
DNA was extracted from five 10-µm sections from formalin-fixed paraffin-embedded (FFPE) tissue, with the help of QIAamp DNA FFPE Kit (QIAGEN, Valencia, CA, USA) in line with specific protocols. DNA quality was assessed through spectrophotometry, and optical density (OD) values were detected at 230, 260, and 280 nm. Qubit 3.0 software was employed for DNA quantification. Library preparation was carried out according to the previous description (16). Through the adoption of xGen Lockdown Hybridization and Wash Reagents Kit (Integrated DNA Technologies), GeneseeqOne™ bone tumor gene panel (481-cancer-relevant genes, Geneseeq Technology Inc.) was utilized for the enrichment of target based on hybridization. Libraries were captured through Dynabeads M-270 (Life Technologies), which were later subjected to amplification within the KAPA HiFi HotStart ReadyMix (KAPA Biosystems) as well as quantified with KAPA Library Quantification kit (KAPA Biosystems) employing the qPCR. Later, the target enriched libraries were sequenced with the help of the NovaSeq4000 platform (Illumina), with pair-end reads of 2 × 150bp. The sequencing data were demultiplexed by utilizing software bcl2fastq (v2.19), whereas Trimmomatic was utilized for the analysis (17). For removing N-bases or those bases with low-quality (quality<15), Burrows-Wheeler Aligner was used to map sequencing data to reference the hg19 genome (Human Genome version 19) (18). Picard (https://broadinstitute.github.io/picard/) was utilized to remove PCR duplicates. Indels-surrounding local realignments along with base-quality reassurance were conducted using Genome Analysis Toolkit (GATK) (19). At least three distinct mutant reads and a mutant allele frequency (MAF) threshold of 0.5% were used in HaplotypeCaller/UnifiedGenotyper in GATK and VarScan2 (20) that were later applied in calling indels and SNPs. Common variants were removed using dbSNP as well as the 1000 Genome project. As for germline mutations, they were compared against whole blood controls in the cases of filtration. The in-house list of relapsed sequencing errors was obtained, based on more than 10,000 healthy subjects using an identical sequencing platform, and was used to filter the obtained somatic variants. For identifying gene fusions, FACTERA (6) was adopted, whereas ADTEx (21) was utilized for analyzing copy number variations (CNVs). The threshold log2 ratio was deemed to be 2.0 for copy number gain in the tissues, while in the whole samples, it was deemed to be 0.67 for copy number loss. Thresholds based on prior assay verification were determined through the droplet digital PCR (ddPCR)-detected absolute CNVs. FACETS (22) was employed for analyzing allele-specific CNVs, and upon the drift threshold of 0.2, unstable joint segments were determined. To determine the chromosomal instability (CIN) proportion, the drifted segment size was divided by the overall segment size.
3 Results
3.1 Clinical features and demographic information of the patients
The clinical features of the cases are described in Table 1. There were 14 patients included in the study, 8 male and 6 female, with a mean age of 31.4 years (median 28, range 3–71). The most common symptom of a hospital visit was headache (7), (50.0%), dizziness ranks the second most common symptom (4), (28.6%). Other presenting symptoms inclued twitching of extremities, blindness, numbness, and hearing loss. The tumor was located supratentorial are in 12 cases (85.1%), and 2 case situated in cerebellopontine angle. The maximum diameter tumor calculated based on radiographic examination ranged from 19.0 to 130.0 mm, with a average of 50.3 mm.
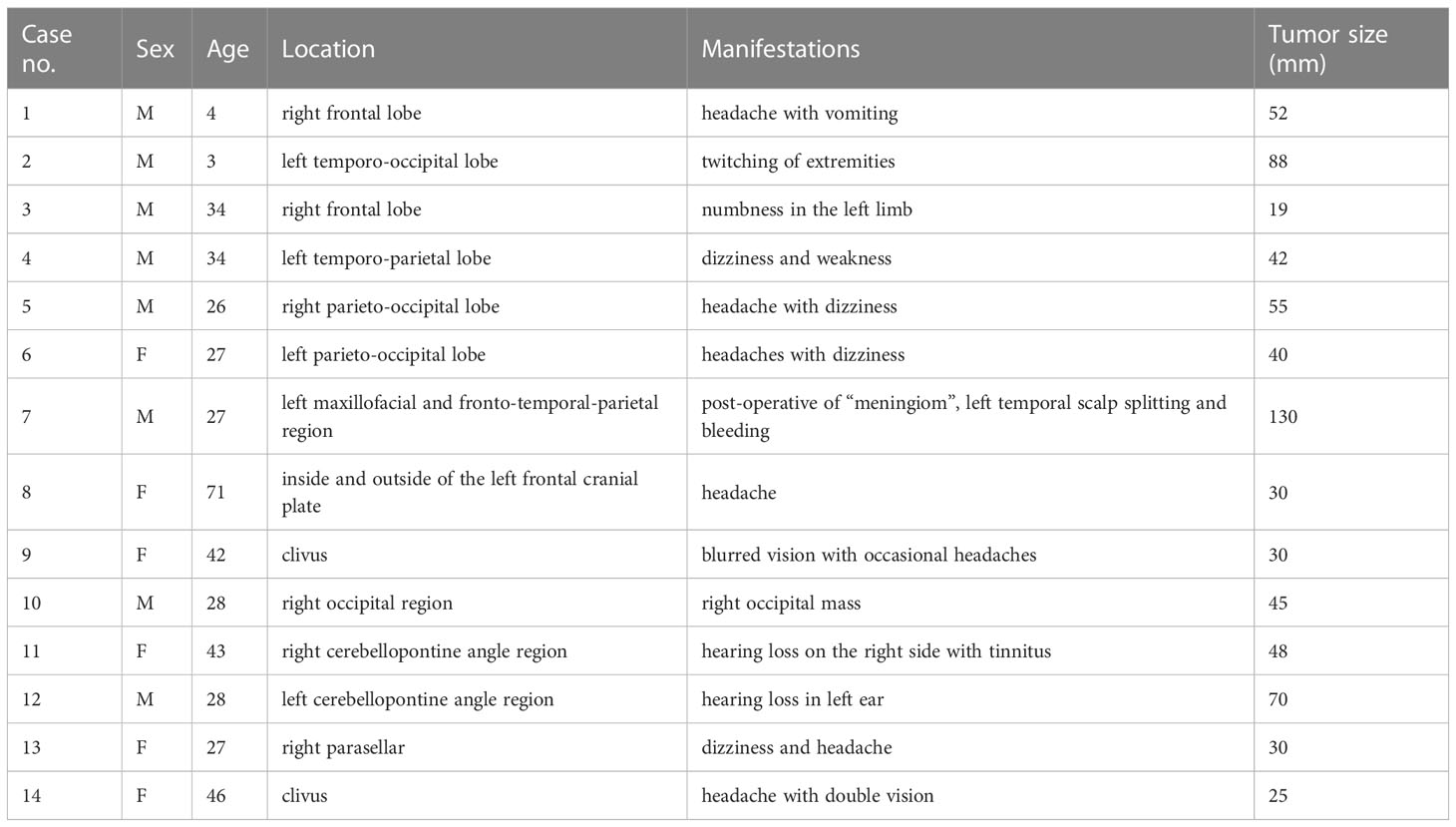
Table 1 Clinical characteristics of PIS patients.
3.2 Pathological findings and genomic features
AS showed in Table 2, the histological heterogeneity is evident. According to the WHO classification of soft tissue tumors, histologic types were grouped as follows: adult fibrosarcoma [case 1, 2 (Figure 1)], myxofibrosarcoma [case 3 (Figure 2)], undifferentiated pleomorphic sarcoma (case 4), undifferentiated round cell sarcoma (case 5), leiomyosarcoma (case 6), extraskeletal myxoid chondrosarcoma (case 7), and angiosarcoma (case 8), mesenchymal chondrosarcoma (case 9), undifferentiated spindle cell sarcoma [case 10 (Figure 3)], and chondrosarcoma (case 11-14). And there is 10 low-grade lesion, which corresponded to the French Federation of Cancer Centers Sarcoma Group (FNCLCC) system grades 1 and 2. And 4 high-grade lesions, which corresponded to the FNCLCC system grades 3 (2, 23). NGS sequencing was applied in 2 cases (case 3, 10). In case 3, tumor cells are fusiform, moderately dense, with obviously myxoid stroma and elongated, curvilinear, thin-walled blood vessels, tumor cell with ill-defined, slightly eosinophilic cytoplasm and atypical, enlarged, hyperchromatic nuclei. Immunohistochemical revealed MDM-2, SATB2 were positive, GFAP, Olig-2, CD34, Desmin, SMA, S-100, SSTR2, EMA, Myogenin, MyoD1 were negative, and the Ki-67 LI (labeling index) was approximately 20%. Targeted NGS was performed with the resected specimen of case 3, and several key gene mutations were identified including NRAS p.Q61K missense mutation (exon 3), PIK3CA p.H1047L missense mutation (exon 20), BAP1 p.S325F missense mutation (exon 11), KDR p.D1046E missense mutation (exon 23), and BLM p.H651Qfs*4 frame shift mutation (exon 8). In case 10, the tumor cells were short spindle-shaped with round or ovoid nuclei and pale eosinophilic cytoplasm. The tumor was arranged in fascicular and myxoid stroma was seen between the tumor cells, and dilated blood vessels were seen inside the tumor. The tumor cells expressed SATB2, Bcl-2, TLE1, SMARCB1, but not CK, EMA, CD34, SMA, Desmin, S-100, CD99, STAT6, MyoD1, Myogenin, and Ki-67 LI is about 10%. NGS identified PBRM1 p.R876S missense mutation (exon 18), TOP2A p.P371L missense mutation (exon 10), DUSP2 p.C39* nonsense mutation (exon 1), CNV deletions of SMARCB1, and SH3BP5::RAF1 fusion gene. The immunohistochemical expression of PIS is shown in Table 2.
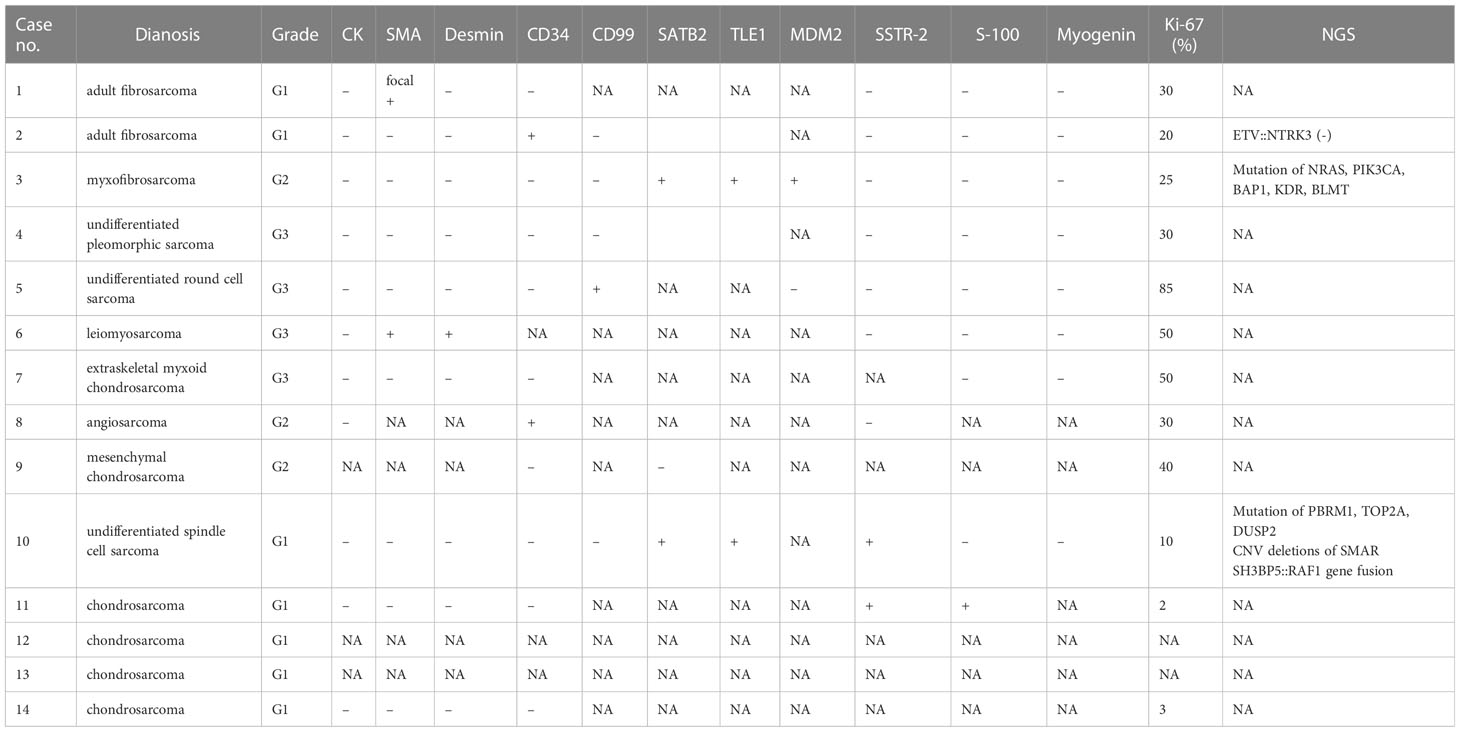
Table 2 The pathological characteristics of PIS patients.
Figure 1 In Case 2, the tumor cells are long spindles, relatively uniform in size, and arranged in bundles (A, B). The IHC analysis displays positiveness for CD34 (C), and the Ki-67 LI is about 30% (D). MRI shows that the tumor is located in the left temporo-occipital lobe, a solid-cystic irregular appearance, T1WI (E), T2WI (F), and T2 FLAIR (G) shows heterogeneous intensity, contrast-enhanced T1WI (H) shows garland-like enhancement.

Figure 2 In Case 3, the tumor cells are fusiform, moderately dense, with obviously myxoid stroma and lengthened, curvilinear, thin-walled blood vessels (A, B). Target NGS sequencing finds the mutation of NRAS (C) and PIK3CA (D). MRI reveals that a sub-round solid lesion is located in the right frontal lobe, T1-weight imaging (T1WI) (E) shows hypointensity, T2-weight imaging (T2WI) (F) and T2 Fluid Attenuated Inversion Recovery (T2 FLAIR) (G) shows hyperintensity, and contrast-enhanced T1WI (H) shows inhomogeneous enhancement.
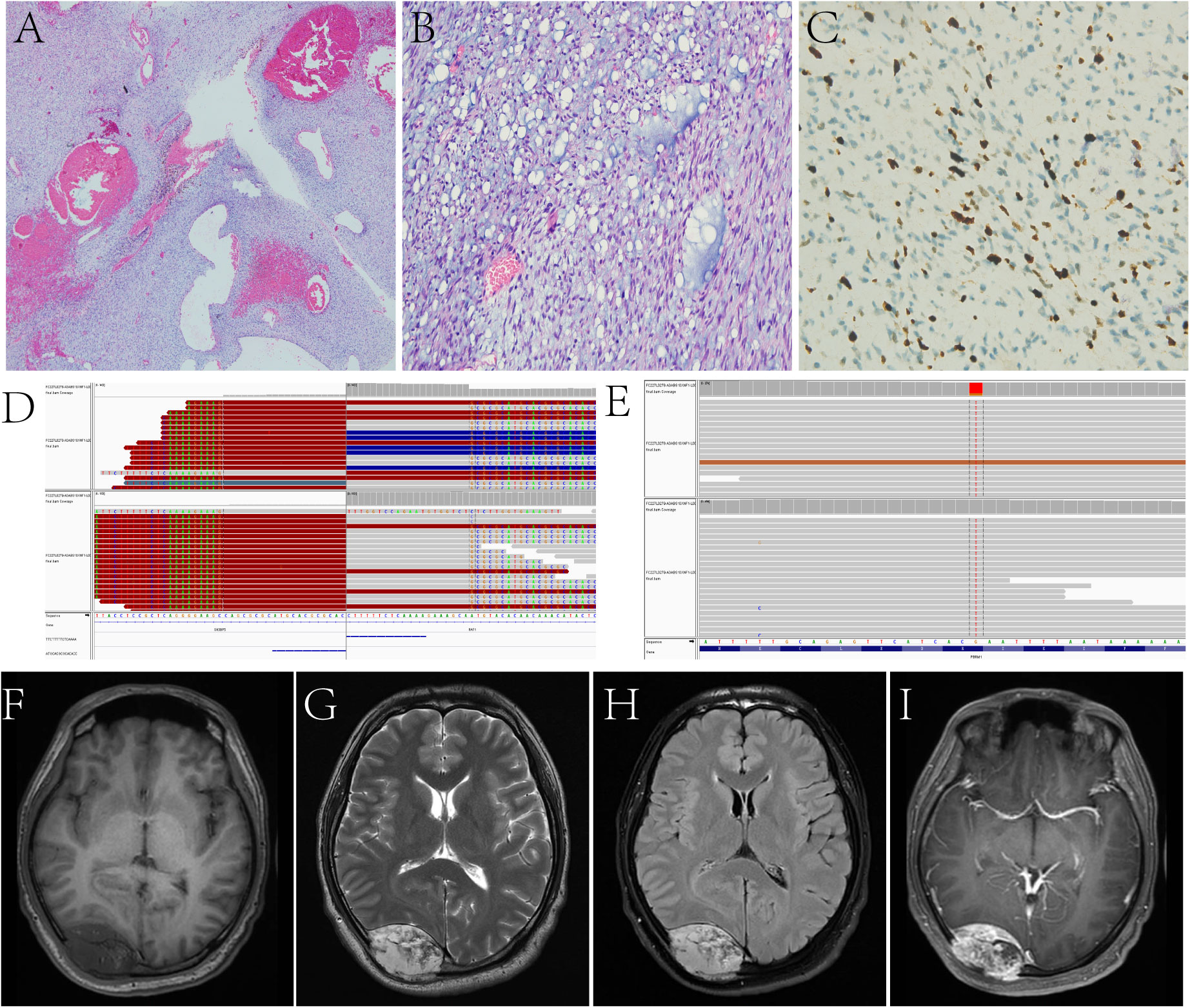
Figure 3 In Case 10, the tumor is arranged in bundles, the myxoid stroma is seen between the tumor cells, and dilated blood vessels are seen inside the tumor (A, B). Ki-67 LI is about 10% (C). Target NGS sequencing finds the SH3BP5::RAF1 gene fusion (D) and the mutation of PBRM1 (E). A fusiform solid lesion is sited in the right occipital region and can be seen in the extradural area and invades the skull, T1WI (F) shows hypointensity, T2WI (G) and T2 FLAIR (H) shows hyperintensity, and contrast-enhanced T1WI (I) shows inhomogeneous enhancement.
3.3 Radiological findings
In the present retrospective study, radiological data were missing for some patients. Among the 10 patients who underwent an MRI scan, the lesions showed hypointensity signals in 6 cases (60.0%), heterogeneous intensity signals in 3 cases (30.0%), and an isointensity signal in 1 case (10.0%) in T1WI. The lesions in 6 cases (60.0%) showed hyperintensity signals, 3 cases (30.0%) showed heterogeneous intensity signals, and 1 case (10.0%) showed an isointensity signal in T2WI. Moreover, 6 cases (60.0%) showed hyperintensity signals, 3 cases (30.0%) showed heterogeneous intensity signals, and 1 case (10.0%) showed an isointensity signal in T2FLAIR. Ten cases accepted the examination of enhanced MRI and showed gadolinium enhancement of the lesion in 8 cases, including 7 heterogeneous enhancements and 1 ring-like enhancement, and they were non-enhanced in 2 cases. In two patients, there was a large band of edema surrounding the lesion (Cases 2, 6). Three out of five patients with brain parenchymal tumors were examined by magnetic resonance spectroscopy (MRS). Two of them (Cases 2, 5) exhibited an elevated Cho peak whereas a reduced NAA peak, and one (Case 9) showed a normal Cho peak whereas a reduced NAA peak in the tumor region. This suggests signs of malignancy in the tumors. The remaining radiological features are detailed in Table 3.
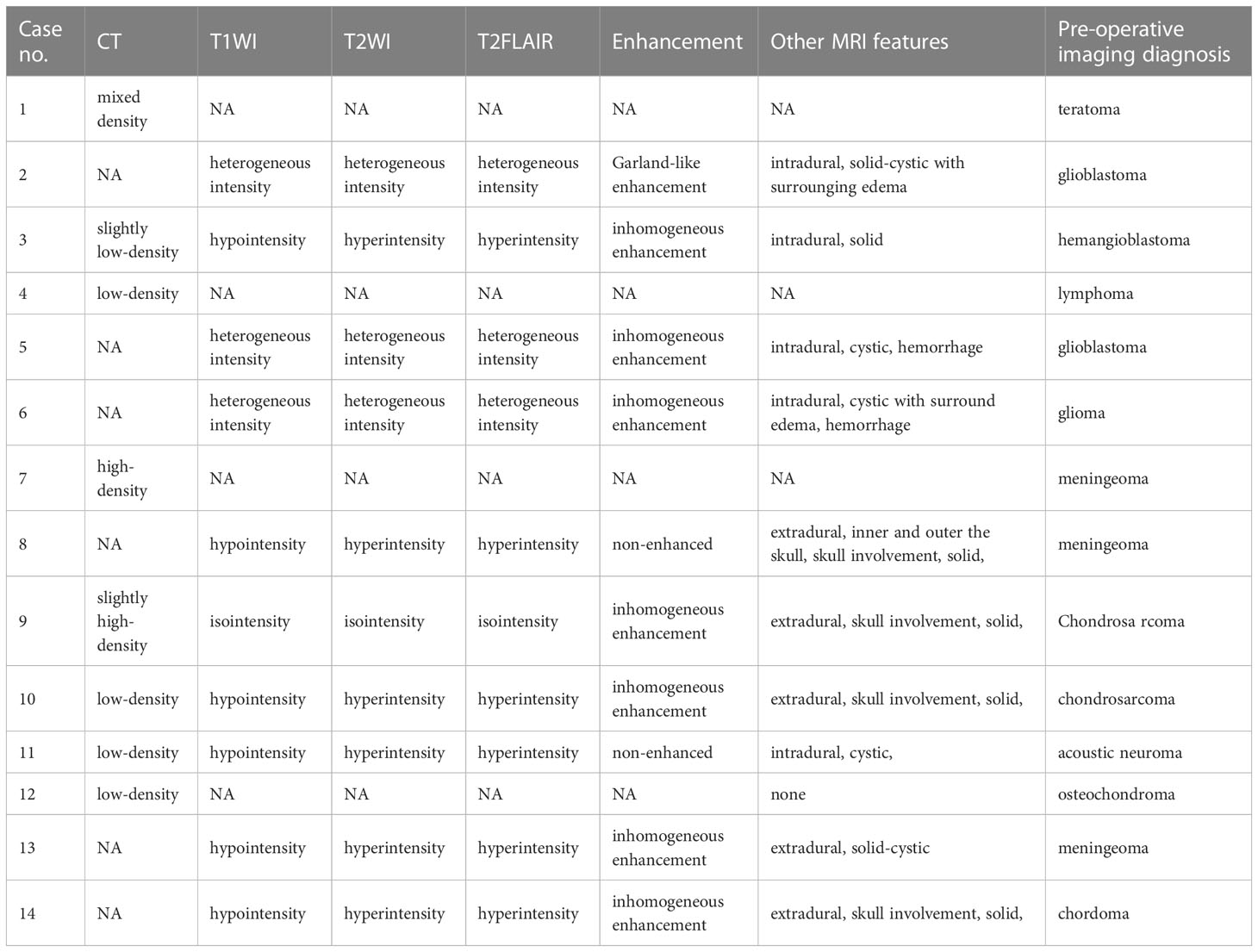
Table 3 Radiological features of the PIS patients.
3.4 Surgical information
According to tumor size and location, different incisions as well as surgical approaches were used, such as horseshoe-shaped incision, pterional approach, suboccipital retrosigmoid approach, infratemporal approach, transsphenoidal approach, etc. The character and color of the tumors were different; most of them were fish-meat-like with abundant blood supply. The tumor resection extent was assessed in line with surgical records as well as post-surgery imaging examinations. The details have been described in Table 4.
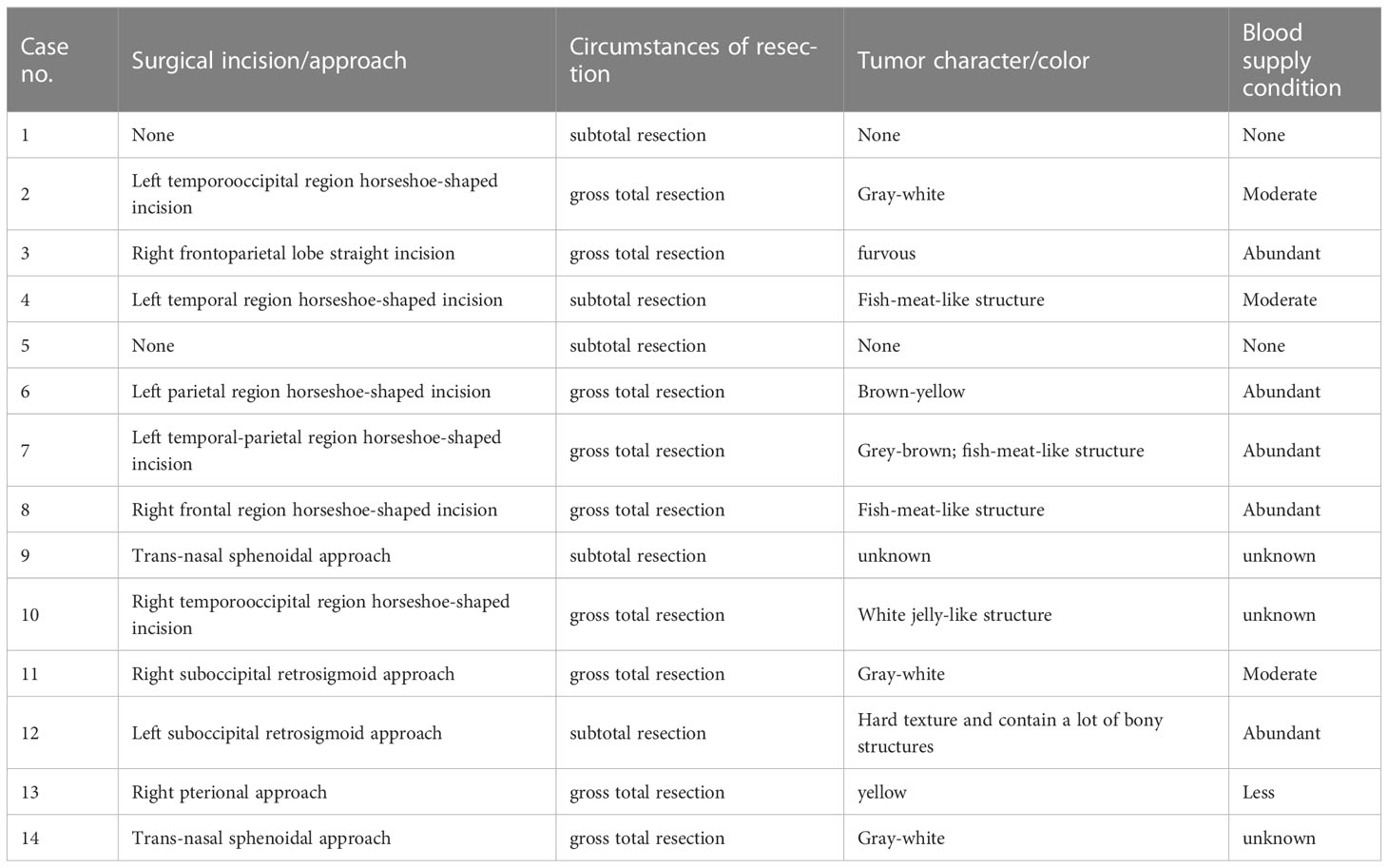
Table 4 Surgical information of the PIS patients.
3.5 Adjutant therapy and follow-up
A total of 9 patients received radiotherapy out of the 11 patients with available follow-up information. Only three patients received postoperative chemotherapy. In four patients, tumor recurrence was observed, 1 patient (Case 2) received reoperation, 1 patient (Case 9) received palliative care due to cachexy, and the other patient (Case 7) chose radiotherapy. One patient had lung metastases based on imaging (Case 9). After follow-up, 3 patients died of tumor between 3 months and 15 months, 8 patients were alive. The details can be found in Table 5.
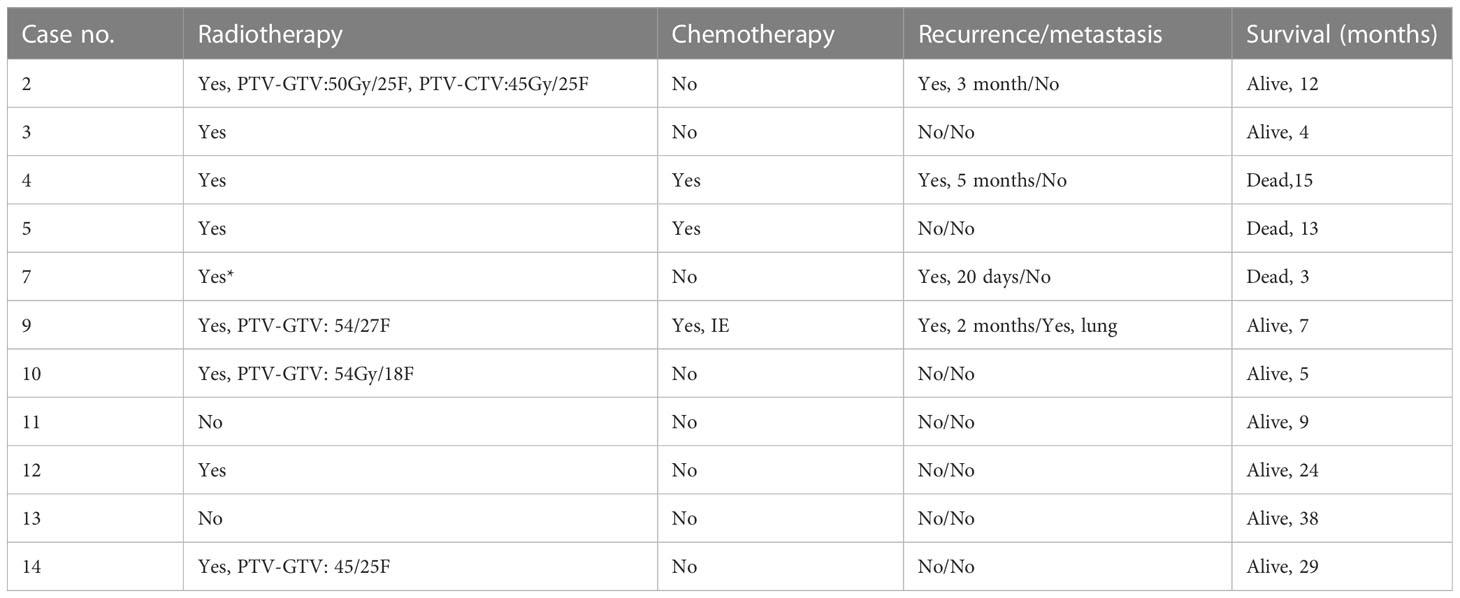
Table 5 Adjuvant therapy and follow-up information for the PIS patients whose follow-up information was available.
4 Discussion
Owing to the rarity and fewer studies conducted on primary intracranial sarcomas (PIS), there exists a lack of understanding concerning their etiology, pathogenesis, and susceptibility factors leading to potential diagnostic challenges and mistreatment (2). Hence, to shed light on the same, the present study was conducted on 14 PIS cases to summarize their clinical and pathological information on PIS.
PIS can affect individuals of all ages; a PIS study based on all age groups indicated that the age at diagnosis ranged from 3 to 63 years (median, 28 years) (2). In the present study, 2 juvenile patients and 12 adult patients were investigated, with an average age of 31 years (median 28 years, range 3–71 years). The majority of the study cases were younger than 50 years, suggesting that PIS might probably take place among young patients, thus, indicating that more attention should be paid to the trend of incidence age. A total of 8 originated from the brain parenchyma, while the remaining 4 originated in the skull or dura mater in our cases. Tumor located supratentorial in 12 cases (85.1%), and other 2 case situated in cerebellopontine angle. Previous reports have shown PIS can be found in the parietal (17.9%), frontal (14.1%), and temporal (14.1%) lobes most common (1, 2, 24). In the present study, the frontal and parietal lobes were the most frequently affected areas; other locations included the temporal lobe, occipital lobe, clivus, cerebellopontine angle region, and parasellar region.
The location of the tumor and its occupying effect are the primary determinants for the clinical symptoms of PIS, often resulting in nonspecific symptoms. In this study, the most common symptoms consisted of headache, dizziness, twitching of extremities, blindness, numbness, and hearing loss. Other symptoms, including memory loss, progressive dementia, sensorimotor dysfunction, and seizures, were also reported (2). In contrast to glial tumors, which tend to stay within the central nervous system, PIS tend to spread beyond it. As a result, up to 40% of individuals with PIS may experience metastatic disease, which is associated with a poor prognosis (13, 14). In the present study, one case displayed imaging evidence of metastasis, thus, highlighting the importance of conducting thorough imaging inspections during the perioperative and follow-up periods.
The PIS management options are akin to those for other CNS tumors and include surgical resection, chemotherapy, and radiotherapy (19–22). Surgery plays a crucial role in both diagnosis and treatment due to its ability to identify molecular and histological changes that are significant in guiding adjuvant therapies (25). In the study, 9 patients achieved gross total resection, and 5 patients underwent subtotal resection. Since PIS tend to be highly aggressive, complete removal of the affected tissue may not be attainable. Therefore, postoperative radiotherapy and/or systemic chemotherapy are necessary to ensure effective treatment (2, 24). In patients with metastatic disease, craniospinal irradiation is deemed a beneficial therapeutic choice (9).
Although various therapies are administered, PIS tends to exhibit lower rates of local tumor control and overall survival in comparison to extracranial soft tissue sarcomas (4, 26). In our study, 4 patients relapsed, 1 patient developed pulmonary metastasis, 3 patients died from the tumor, and 8 patients survived. Previous literature regarding CNS sarcoma recurrence and mortality rates after initial management revealed discouraging results, with 26.9% of patients encountering disease recurrence and 76.9% dying due to disease progression (1). Compared with the literature, we reported a lower recurrence and mortality rate of PIS, which may be related to the shorter follow-up period of some cases. Various pathological subtypes of PIS may display varied prognosis. El Ghatany et al. observed that the survival duration was reduced among patients without GTR, those with metastatic disease, and individuals diagnosed before 1 year of age (13, 14). In our series, patients who had GTR of the tumor had a trend towards superior survival. Tables 4 and 5 summarizes the clinical characteristics and outcomes.
Based on an analysis conducted by Haider et al. on PIS, fibrosarcoma (16.7%), synovial sarcoma (12.8%), and extraosseous mesenchymal chondrosarcoma (11.5%) were identified as the most frequent histological types (1). The present study, however, revealed chondrosarcoma to be the most prevalent, which was possibly affected by the inclusion criteria of the study; moreover, Haider et al. did not include intracranial skull-based chondrosarcoma in their analysis.
The present research identified interesting molecular alterations in two patients through target next-generation sequencing (NGS). Mutation of PIK3CA and NRAS, the key molecules in the common abnormal pathways (MAPK and PI3K/AKT/mTOR pathways) within cancers, were found in case 3. PIK3CA gene mutations lead to PI3K pathway kinase hyperactivity, causing abnormal cell growth (27–29). Furthermore, these mutations are related to anti-cancer therapeutic efficacy and patient survival (30). The NRAS gene mutations are mostly related to hematopoietic cancers, bladder cancers, and melanomas (31). The activation of the PI3K/Akt/mTOR pathway has been found to be associated with the histologic malignancy and tumor progression of myxofibrosarcoma, and the PI3K/Akt/mTOR pathway has the potential to be a therapeutic target for primary and recurrent myxofibrosarcomas, but no PIK3CA mutation was found in their study (32). NGS identified alterations in genes associated with the SWI/SNF chromatin-remodeling complexes, specifically PBRM1 mutation and CNV deletions of SMARCB1 were found in case 3. The PBRM1 gene activates p21 expression and suppresses cell growth (33). This gene is also necessary for cell aging and DNA double-strand breaks-mediated transcriptional silencing, which enhances DNA damage repair; the inability to perform these functions may be due to mutants of PBRM1 (34). As the tumor suppressor, the SMARCB1 gene is related to chromatin remodeling and is an important part of the SWI/SNF chromatin-remodeling complex (35). Upon SMARCB1 deficiency, SWI/SNF complex cannot bind to the target, causing extensive transcriptional dysregulation. In case 10, the copy number of SMARCB1 was found to be 1.0, suggesting the possibility of heterozygous deletion. IHC also revealed that SMARCB1 expression was not defective, suggesting that it may not be the driver gene for this tumorigenesis. SH3BP5::RAF1, a new fusion gene, was first reported in the present study. Since there is only one case, its function and diagnostic value for this tumor still needs further study.
5 Conclusion
In conclusion, PIS is extremely rare compared to extracranially occurring soft sarcomas. Intracranial chondrosarcoma usually originates from the skull, different inclusion criteria led to differences in their incidence. Other type of PIS, including adult fibrosarcoma, myxofibrosarcoma, and undifferentiated sarcoma, which have no exact direction of differentiation. Patients with GTR have a higher survival rate. NGS assay can help identify diagnostic and therapeutically relevant targets, but further clinical studies are needed for personalized therapies and target treatment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA978484.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board at the Zhongnan Hospital of Wuhan University. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. Written informed consent was not obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZL, ST and YC, designed the experiments; JL, YH, JX, HZ is responsible for information collection and data statistics; YC is responsible for writing the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Discipline Cultivation Project of Zhongnan Hospital of Wuhan University (No. ZNXKPY2022001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Haider AS, Palmisciano P, Sagoo NS, Bin Alamer O, El Ahmadieh TY, Pan E, et al. Primary central nervous system sarcomas in adults: a systematic review. Clin Neurol Neurosurg (2022) 214:107127. doi: 10.1016/j.clineuro.2022.107127
2. Oliveira AM, Scheithauer BW, Salomao DR, Parisi JE, Burger PC, Nascimento AG. Primary sarcomas of the brain and spinal cord: a study of 18 cases. Am J Surg Pathol (2002) 26(8):1056–63. doi: 10.1097/00000478-200208000-00011
3. Stiller CA, Trama A, Serraino D, Rossi S, Navarro C, Chirlaque MD, et al. Descriptive epidemiology of sarcomas in Europe: report from the rarecare project. Eur J Cancer (2013) 49(3):684–95. doi: 10.1016/j.ejca.2012.09.011
4. Grünewald TG, Alonso M, Avnet S, Banito A, Burdach S, Cidre-Aranaz F, et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol Med (2020) 12(11):e11131. doi: 10.15252/emmm.201911131
5. Zakaria WK, Taha AN. Primary central nervous system sarcomas: clinical, radiological and pathological features in our institution. Romanian Neurosurg (2019) 33:287–94. doi: 10.33962/roneuro-2019-048
6. Anderson JL, Denny CT, Tap WD, Federman N. Pediatric sarcomas: translating molecular pathogenesis of disease to novel therapeutic possibilities. Pediatr Res (2012) 72(2):112–21. doi: 10.1038/pr.2012.54
7. Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res (2006) 66(9):4742–9. doi: 10.1158/0008-5472.Can-05-4292
8. Abraham J, Prajapati SI, Nishijo K, Schaffer BS, Taniguchi E, Kilcoyne A, et al. Evasion mechanisms to Igf1r inhibition in rhabdomyosarcoma. Mol Cancer Ther (2011) 10(4):697–707. doi: 10.1158/1535-7163.Mct-10-0695
9. Maher OM, Khatua S, Mukherjee D, Olar A, Lazar A, Luthra R, et al. Primary intracranial soft tissue sarcomas in children, adolescents, and young adults: single institution experience and review of the literature. J Neurooncol (2016) 127(1):155–63. doi: 10.1007/s11060-015-2027-3
10. Ho AL, Vasudeva SD, Laé M, Saito T, Barbashina V, Antonescu CR, et al. Pdgf receptor alpha is an alternative mediator of rapamycin-induced akt activation: implications for combination targeted therapy of synovial sarcoma. Cancer Res (2012) 72(17):4515–25. doi: 10.1158/0008-5472.Can-12-1319
11. Kubo T, Piperdi S, Rosenblum J, Antonescu CR, Chen W, Kim HS, et al. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer (2008) 112(10):2119–29. doi: 10.1002/cncr.23437
12. Zwerner JP, May WA. Dominant negative pdgf-c inhibits growth of Ewing family tumor cell lines. Oncogene (2002) 21(24):3847–54. doi: 10.1038/sj.onc.1205486
13. Benesch M, von Bueren AO, Dantonello T, von Hoff K, Pietsch T, Leuschner I, et al. Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European cws and hit study groups. J Neurooncol (2013) 111(3):337–45. doi: 10.1007/s11060-012-1020-3
14. Al-Gahtany M, Shroff M, Bouffet E, Dirks P, Drake J, Humphreys R, et al. Primary central nervous system sarcomas in children: clinical, radiological, and pathological features. Childs Nerv Syst (2003) 19(12):808–17. doi: 10.1007/s00381-003-0839-5
15. Nejo T, Takayanagi S, Tanaka S, Shinozaki-Ushiku A, Kohsaka S, Nagata K, et al. Primary intracranial spindle cell sarcoma, Dicer1-mutant, with Mdm2 amplification diagnosed on the basis of extensive molecular profiling. Clin Med Insights Case Rep (2022) 15:11795476221131189. doi: 10.1177/11795476221131189
16. Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating novel resistance mechanisms to third-generation egfr tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res (2018) 24(13):3097–107. doi: 10.1158/1078-0432.Ccr-17-2310
17. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
18. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
19. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet (2011) 43(5):491–8. doi: 10.1038/ng.806
20. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. Varscan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res (2012) 22(3):568–76. doi: 10.1101/gr.129684.111
21. Amarasinghe KC, Li J, Halgamuge SK. Convex: copy number variation estimation in exome sequencing data using hmm. BMC Bioinf (2013) 14 Suppl 2(Suppl 2):S2. doi: 10.1186/1471-2105-14-s2-s2
22. Shen R, Seshan VE. Facets: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res (2016) 44(16):e131. doi: 10.1093/nar/gkw520
23. Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med (2006) 130(10):1448–53. doi: 10.5858/2006-130-1448-gostsr
24. Zhang G, Xiao B, Huang H, Zhang Y, Zhang X, Zhang J, et al. Intracranial synovial sarcoma: a clinical, radiological and pathological study of 16 cases. Eur J Surg Oncol (2019) 45(12):2379–85. doi: 10.1016/j.ejso.2019.08.015
25. Brown HK, Schiavone K, Gouin F, Heymann MF, Heymann D. Biology of bone sarcomas and new therapeutic developments. Calcif Tissue Int (2018) 102(2):174–95. doi: 10.1007/s00223-017-0372-2
26. Ozturk AK, Gokaslan ZL, Wolinsky JP. Surgical treatment of sarcomas of the spine. Curr Treat Options Oncol (2014) 15(3):482–92. doi: 10.1007/s11864-014-0290-8
27. Tang C, Ma J, Liu X, Liu Z. Identification of four immune subtypes in bladder cancer based on immune gene sets. Front Oncol (2020) 10:544610. doi: 10.3389/fonc.2020.544610
28. Fleming GF, Ma CX, Huo D, Sattar H, Tretiakova M, Lin L, et al. Phase ii trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat (2012) 136(2):355–63. doi: 10.1007/s10549-011-1910-7
29. Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic functional characterization of resistance to Pi3k inhibition in breast cancer. Cancer Discovery (2016) 6(10):1134–47. doi: 10.1158/2159-8290.Cd-16-0305
30. Alqahtani A, Ayesh HSK, Halawani H. Pik3ca gene mutations in solid malignancies: association with clinicopathological parameters and prognosis. Cancers (Basel) (2019) 12(1):93–110. doi: 10.3390/cancers12010093
31. Li Y, Sun Z, Cunningham JM, Aubry MC, Wampfler JA, Croghan GA, et al. Genetic variations in multiple drug action pathways and survival in advanced stage non-small cell lung cancer treated with chemotherapy. Clin Cancer Res (2011) 17(11):3830–40. doi: 10.1158/1078-0432.Ccr-10-2877
32. Takahashi Y, Kohashi K, Yamada Y, Endo M, Setsu N, Ishii T, et al. Activation of the Akt/Mammalian target of rapamycin pathway in myxofibrosarcomas. Hum Pathol (2014) 45(5):984–93. doi: 10.1016/j.humpath.2013.12.012
33. Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, et al. Baf180 is a critical regulator of P21 induction and a tumor suppressor mutated in breast cancer. Cancer Res (2008) 68(6):1667–74. doi: 10.1158/0008-5472.Can-07-5276
34. Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, et al. Pbrm1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Res (2021) 81(11):2888–902. doi: 10.1158/0008-5472.Can-21-0628
35. Schweiger T, Liebmann-Reindl S, Glueck O, Starlinger P, Laengle J, Birner P, et al. Mutational profile of colorectal cancer lung metastases and paired primary tumors by targeted next generation sequencing: implications on clinical outcome after surgery. J Thorac Dis (2018) 10(11):6147–57. doi: 10.21037/jtd.2018.10.72
Keywords: primary intracranial sarcomas, case series, pathology, MRI, next-generation sequencing
Citation: Cai Y-X, Liu J-S, Xu J, He Y-Z, Zhang H-N, Tian S-F and Li Z-Q (2023) Primary intracranial sarcomas: a clinicopathological investigation. Front. Oncol. 13:1195467. doi: 10.3389/fonc.2023.1195467
Received: 28 March 2023; Accepted: 18 May 2023;
Published: 09 June 2023.
Edited by:
Pankaj Pathak, National Institutes of Health (NIH), United StatesReviewed by:
Pankaj Jha, Medgenome Pvt. Ltd, IndiaJohn Chrisinger, Washington University in St. Louis, United States
Copyright © 2023 Cai, Liu, Xu, He, Zhang, Tian and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Su-Fang Tian, c2Z0aWFuQHdodS5lZHUuY24=; Zhi-Qiang Li, bGl6aGlxaWFuZ0B3aHUuZWR1LmNu
†These authors have contributed equally to this work