 Fernanda Jardim da Silva1
Fernanda Jardim da Silva1 Juscelino Carvalho de Azevedo Jr.1,2
Juscelino Carvalho de Azevedo Jr.1,2 Ana Carolina Lima Ralph3,4
Ana Carolina Lima Ralph3,4 João de Jesus Viana Pinheiro5
João de Jesus Viana Pinheiro5 Vanessa Morais Freitas4
Vanessa Morais Freitas4 Danielle Queiroz Calcagno1,2,5*
Danielle Queiroz Calcagno1,2,5*- 1Núcleo de Pesquisas em Oncologia, Programa de Pós-Graduação em Oncologia e Ciências Médicas, Universidade Federal do Pará, Belém, Brazil
- 2Hospital Universitário João de Barros Barreto, Programa de Residência Multiprofissional em Saúde (Oncologia), Universidade Federal do Pará, Belém, Brazil
- 3Faculdade de Farmácia, Faculdade Estácio, Carapicuíba, Brazil
- 4Instituto de Ciências da Saúde, Programa de Pós-Graduação em Odontologia, Universidade Federal do Pará, Belém, Brazil
- 5Laboratório de Microambiente Tumoral, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, Brazil
Adenoid cystic carcinoma (ACC) is an aggressive tumor with a high propensity for distant metastasis and perineural invasion. This tumor is more commonly found in regions of the head and neck, mainly the salivary glands. In general, the primary treatment modality for ACC is surgical resection and, in some cases, postoperative radiotherapy. However, no effective systemic treatment is available for patients with advanced disease. Furthermore, this tumor type is characterized by recurrent molecular alterations, especially rearrangements involving the MYB, MYBL1, and NFIB genes. In addition, they also reported copy number alterations (CNAs) that impact genes. One of them is C-KIT, mutations that affect signaling pathways such as NOTCH, PI3KCA, and PTEN, as well as alterations in chromatin remodeling genes. The identification of new molecular targets enables the development of specific therapies. Despite ongoing investigations into immunotherapy, tyrosine kinase inhibitors, and anti-angiogenics, no systemic therapy is approved by the FDA for ACC. In this review, we report the genetic and cytogenetic findings on head and neck ACC, highlighting possible targets for therapeutic interventions.
Background
Adenoid cystic carcinoma (ACC) is a rare and aggressive tumor mainly affecting salivary glands (1). Cytologically, the ACC comprises myoepithelial and luminal cells and is histologically classified into tubular, cribriform, or solid subtypes (2). The solid subtype is the most aggressive and is prone to poor prognosis and a higher frequency of mutations (3, 4).
According to histological patterns, ACC can be classified into high and low grades. Low-grade ACC consists of grades I (predominantly tubular, no solid areas or occasionally solid areas) and II (predominantly cribriform, <30% solid). High-grade ACC is defined as grade III (>30% solid component) (5, 6). It is common for tumors to show transitions between the three histological patterns (5).
The grade III solid histological pattern can be confused with high-grade transformation adenoid cystic carcinoma (ACC-HGT) due to some similarities with cellular atypia, occasional comedoiform necrosis, and frequent mitotic figures. However, the cells show a different aspect (solid-type cells: basaloid, small, hyperchromatic nuclei with scarce cytoplasm. ACC-HGT: larger, more pleomorphic, and vesicular nuclei and balance between nucleus and cytoplasm) (7). Some other aspects differentiate ACC from the solid subtype of ACC-HGT, which is highly prone to lymph node metastases, high rates of mitotic labeling and increased expression of Ki-67, as well as high expression of p53 (7, 8).
ACC is more frequently found in women in their fifth or sixth decade of life (1). Although ACC is usually slow growing, it is a very aggressive tumor with a progressive infiltrative growth pattern and a high propensity for perineural invasion, local recurrence, and distant metastasis, especially in the lung, bones, and liver (9–13). Depending on the clinical stage, different therapeutic approaches can be used for ACC, but surgical intervention with radiotherapy remains the main treatment strategy in the management of ACC. In addition, chemotherapy, targeted therapy, and immunotherapy have also been used to improve the survival and quality of life of patients with ACC (14). Unfortunately, no specific therapies are approved by the food and drug administration (FDA) to treat patients with recurrent or metastatic ACC (15). Knowing the molecular alterations of the tumor, as well as the interaction with the tumor microenvironment, is essential in the identification and development of new therapies (16).
Previous reports have demonstrated that molecular alterations in ACC are characterized mainly by fusions involving the MYB protein family (17). Furthermore, other molecular changes involve copy number alterations (CNAs) and mutations that have a relevant role in the progression of this disease, including RAS, PTEN, PIK3CA, and NOTCH1-4 (3, 16). It is still unclear whether these alterations have any clinical impact.
In the personalized medicine era, it is essential to identify molecular alterations that can play an auxiliary role in therapeutic decision-making, thereby proving their utility as biomarkers for therapy response and clinical management in ACC patients (18). In this review, we focused on reporting the most relevant and recurrent molecular alterations described in ACC of salivary glands and their implications. Moreover, we discussed the clinical trials conducted to evaluate targeted therapies for ACC.
Molecular alterations in ACC
MYB/MYBL1 alterations
In ACC, the most commonly reported molecular alteration is the t(6;9) translocation, which results in the fusion of the 5′-end of MYB to the 3′-end of NFIB (17, 19–21). MYB protein belongs to a family of transcription factors composed of two other members: MYBL1 and MYBL2. This family consists of three functional domains: the C-terminal negative regulatory domain (NRD); the N-terminal DNA binding domain, which is essential for interaction with the MYB binding site; and a centrally located transcription activation domain. The transactivation of MYB is also influenced by interaction with CBP/p300 and p100 coactivators (22). NFIB is a transcription factor involved in various physiological processes, including adipocyte differentiation, maturation of megakaryocytes, and brain and lung development (23).
The frequency of MYB-NFIB fusion is variable (16% to 100%), depending on the approach used for analysis (24). Variability in MYB-NFIB fusion transcripts is frequently observed, mainly due to different breakpoints in both genes (19–21). Generally, most variants involve the fusion of MYB exon 14 to NFIB exon 9 (Figure 1A) (19–21). This diversity can result in full-length and truncated MYB proteins and fusion transcripts. Most truncations occur in the C-terminal domain, and there may also be retained DNA-binding and transactivation domains, suggesting that MYB may act to regulate gene expression in ACC (25).
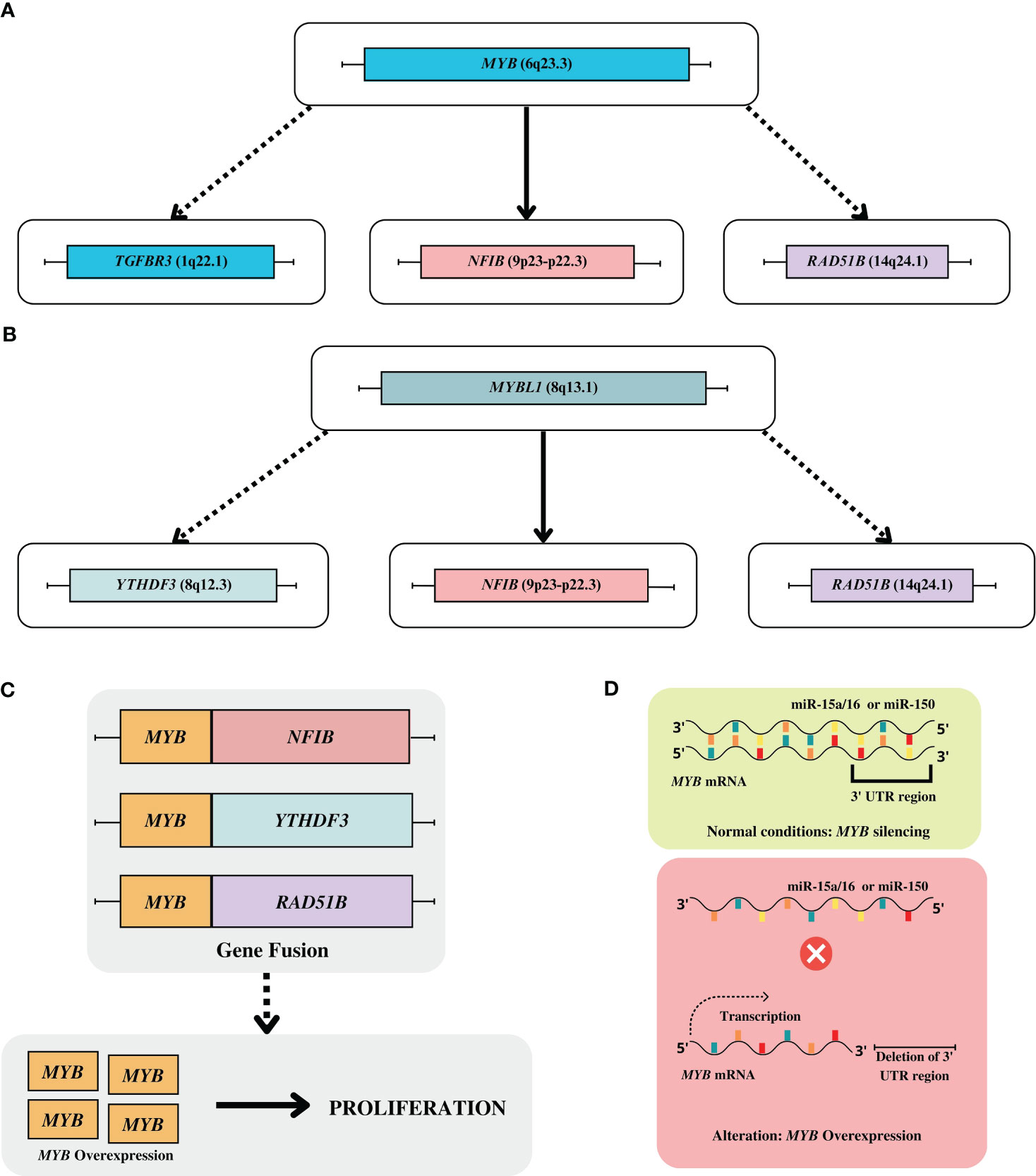
Figure 1 (A) Main gene fusions of MYB gene. (B) Main gene fusions of MYBL1 gene. (C) Gene fusion of MYB with the super enhancers including YTHDF3, RAD51B; (D) Alteration on regulation of MYB expression: in normal conditions the MYB is downregulated by miR-15a/16 or miR-150, however in ACC tumor occur the deletion of 3’ UTR region blocking the ligation of their miRNA, which facilities the overexpression of MYB gene.
These fusions usually result in the overexpression of MYB protein, and its target genes associated with various cellular activities, such apoptosis (API5, BCL2, BIRC3, HSPA8, SET), cell cycle control (CCND1, CCNB1, CDC2, MAD1L1, MET), cell growth/angiogenesis (MYC, KIT, VEGFA, FGF2, CD53, FGFR2, IGF1R), and cell adhesion (CD34) (19, 26–28). Notably, MYC overexpression associated with MYB overexpression is associated with a more advanced clinical stage and shorter disease-free survival (DFS) (29).
Possible downstream effects of MYB overexpression were also investigated. In addition to the target genes already described, other factors or transcription pathways may act on the ACC in a coordinated/mediated means by MYB. One such factor is TP63, which can co-bind with MYB at about 81% of binding sites. Furthermore, at binding sites close to MYB, the EN1, ARID1A, and NOTCH1 genes, and their JAG1 and JAG2 activators are also identified as targets. MYB coordination programs with NOTCH and TP63 also play a role in the transition from grade 2 to grade 3 tumors. While TP63 is strongly expressed in myoepithelial cells from grade 2 tumors, luminal cells showed a high expression of ICN1 (form an active intracellular layer of NOTCH1) in a mutually exclusive manner. In contrast, grade 3 ACCs show strong staining for ICN1 (28). This corroborates the fact that MYB protein is highly expressed in myoepithelial cells (30).
The cell proliferation through MYB-NFIB in the ACC has complex activation mechanisms. Recently, it was demonstrated that cellular stress induced by MYB overexpression leads to a DNA damage response via the ATR pathway (31). Furthermore, MYB-NFIB is regulated by AKT-dependent IGF1R signaling (27).
In addition to the previously reported fusions, other mechanisms that lead to overexpression of MYB have been described. Through whole-genome sequencing (WGS) technology, it was possible to identify alternative mechanisms by which MYB-TGFBR3 and MYB-RAD51B rearrangements can induce MYB overexpression. Such translocations result in the juxtaposition of super enhancers to the MYB locus, increasing their expression levels. In this context, enhancers at the NFIB and TGFBR3 loci are notable since they can interact with the MYB protein, resulting in a positive feedback mechanism that promotes overexpression of MYB (Figure 1C) (28). Subsequently, a fluorescent in situ hybridization (FISH) analysis with wide-ranging probes revealed new breakpoints at the MYB locus that were distant from the MYB gene (~10 Mb), reinforcing the role of distant superenhancing elements in the overexpression of the protein (25). Finally, the deletion of the 3′ untranslated regions (3’UTR) of MYB prevents the silencing of this gene by miRNAs (miR-15a/16 and miR-150) (Figure 1D) (19).
Additionally, another gene from this family of transcription factors also plays a critical role in the development of ACC. It has previously been observed that the MYB protein can be expressed in the ACC regardless of the status of the MYB-NFIB fusion. This led to the consideration that there might be other MYB activation mechanisms (30). Two simultaneous and independently performed studies confirmed this hypothesis and found the presence of translocations involving MYBL1. Novel translocations in the 8q13 and 9p23 regions were identified, resulting in the MYBL1-NFIB fusion and rearrangements resulting in the MYBL1–YTHDF3 fusion. Both cytogenetic alterations can lead to overexpression of MYBL1. An interesting observation is that analysis RT-PCR also detected high levels of MYBL1 mRNA in t(6;9) and t(8;9) negative tumors. Further analysis revealed the presence of truncated MYBL1 proteins due to rearrangements of adjacent intragenic sites on chromosome 8 and a chromosomal translocation between MYBL1 and RAD51B (Figure 1B). The alterations in MYB and MYBL1 appear mutually exclusive (26, 32). Other reports also observed changes in MYBL1, reinforcing the driver gene’s role in ACC (33, 34).
Clinical applicability
The clinical significance of fusions in members of the MYB family still needs to be well established (35–37). However, the MYB-NFIB fusion is specific for ACC (36, 37). Therefore, determining the status of the MYB-NFIB fusion could be useful for the differential diagnosis of ACC and other head and neck neoplasms for the subpopulation of patients with the fusion (38–40). In turn, overexpression of the MYB protein may be useful as a diagnostic biomarker due to its high specificity and sensitivity for ACC (36, 37, 41). It should be noted that the specific cutoff points for MYB immunoreactivity by immunohistochemistry (ICH) and the antibody used, which would be more useful to distinguish ACC, must be carefully defined (30, 36, 37, 41).
Likewise, the diversity of transcripts and multiple breakpoints require that the tool used for identification be carefully chosen (19–21). Issues such as primer design (PCR), probe coverage (FISH) and type of material [fixed in formalin and embedded in paraffin (FFPE) versus frozen] may influence the detection of changes (24). Investigation of MYB-NFIB transcription by RT-PCR showed significant differences in the percentage of positive cases in relation to frozen tissue (86%) and FFPE (44%) (30). Furthermore, most studies rely on RT-PCR to identify fusion transcripts or FISH analysis for the translocation. In this sense, both the primer designs and the cut-off value to determine the positivity of the FISH translocation is variable, which may impact the ability to detect the fusion (19–22). Notably, the high cost and complexity are some of the barriers that make it difficult to include more comprehensive and specific methods in the clinical routine, such as Next Generation Sequencing (NGS) and RNA in situ hybridization (ISH), evaluating the MYB-NFIB fusion routine a standard in the ACC cases a challenge (39, 40).
Critical copy number alterations (CNAs)
In general, the patterns of chromosomal alterations in ACC are heterogeneous due to the multiple technologies used, differences in resolution, tested loci, and rigor of the criteria used to define gains or losses (24, 42).
Notably, CNAs have also been reported in ACC. Among the CNAs, the 6q region gain is the most frequently observed event (3, 43, 44). Furthermore, deletion and loss of heterozygosity (LOH) in this region were associated with locally advanced disease and cribriform histologic subtype, indicating that this event may be related to tumor progression (44, 45). The main candidate tumor suppressor genes in this region are PLAGL1 and LAST1. However, was not observed inactivating mutations or evidence of gene silencing at the RNA and protein levels of these genes in ACC (46). On the other hand, in a recent analysis, it was noticed that the deletion of 6q distal to the MYB breakpoint at 6q23.3 is a concomitant event with the loss of the 3’ MYB region, which results in the subsequent t(6;9) translocation (17)
Gains in the 4q12 region and gene amplifications have been reported, events that may lead to overexpression of C-KIT (47–53). C-KIT is a gene that encodes a Type III tyrosine kinase receptor and plays an essential role in several biological functions, such as survival, metabolism, proliferation, apoptosis, and differentiation (54).
The expression pattern of C-KIT differs according to histological subtype, although some results are controversial. Most reports indicate strong C-KIT staining in the solid histological subtype (49–51). On the other hand, a median staining intensity of the solid phenotype about the cribriform and tubular subtypes was reported. However, cribriform and tubular subtypes may evolve into a solid phenotype during disease progression, suggesting that overexpression of C-KIT may be a key event for tumor progression (52).
This finding is consistent with a higher expression of C-KIT in tumor stages III-IV in patients with perineural invasion, regional local recurrence, and distant metastasis and is related to a poor prognosis (50, 55). Interestingly, C-KIT expression is positively correlated with stem cell factor (SCF) in ACC tumor cells and other tumor microenvironment cells, particularly in nerve cells, which may be associated with the strong trend of perineural invasion for this tumor (55).
Finally, ACC tumors also have a considerable number of cytogenetic deletions affecting genes coding primarily suppressor tumor genes and adhesion molecules, which regulate the cell cycle, resulting in excessive proliferation and contributing to the metastatic process (43, 47, 56). The main findings about the ACC genetic profile are summarized in Table 1.
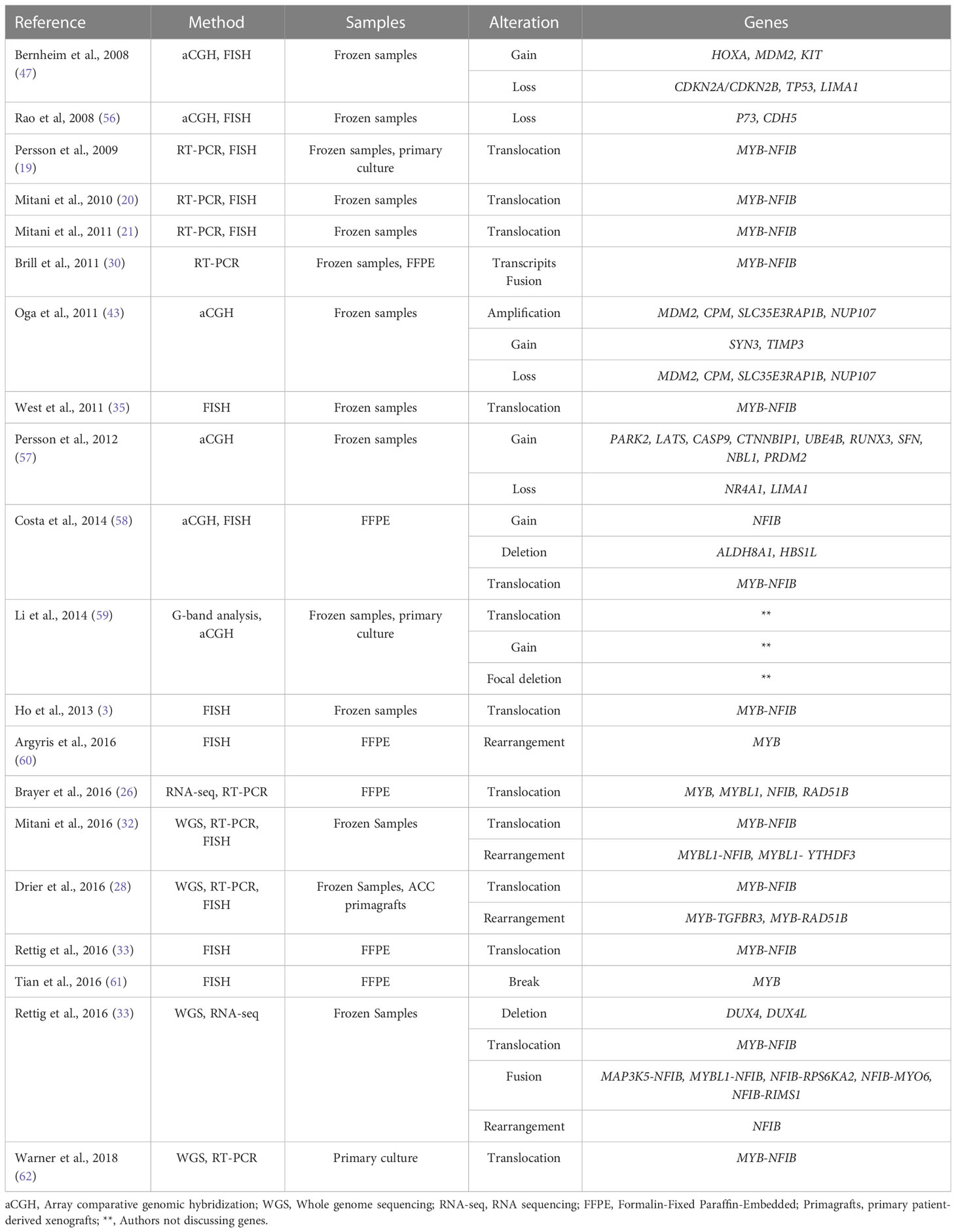
Table 1 Recurrent CNAs in ACC.
Critical mutations
ACC has a low mutational load compared to other types of solid tumors. Despite this, some significant mutations have been reported, mainly in genes associated with cell signaling pathways in DNA damage response, growth factors, and chromatin remodeler genes (56, 63–65).
In relation to clinical-pathological features, missense mutations in RAS and EGFR are considered to be risk factors for DFS and overall survival (OS). In addition, TP53 mutation was associated with expected overall survival in recurrent and metastatic ACC and is more frequent in the solid histological subtype (3). Mutations in PI3KCA, and FGFR2 are also considered frequent, but there was no association with clinical-pathological findings (65, 66). However, both genes are particularly interesting due to their therapeutic potential (66).
Indeed, among the most well-described and important mutations in ACC are those that activate the NOTCH signaling pathway. Studies have shown that NOTCH knockdown is associated with the inhibition of growth, invasion (67) and cell proliferation (68) and that the deregulation of this pathway is associated with invasion and metastasis processes through the modulation of MMPs (metalloproteinases) and the induction of Epithelial-mesenchymal transition (EMT) (69). These findings are consistent with the fact that the NOTCH1 mutation is more frequently observed in the solid histological subtype in metastatic tumors and is associated with a poor prognosis (64, 70, 71). The expression of NOTCH and its receptors may even partially explain the biphasic nature of low-grade and intermediate ACC. A study using single-cell RNA sequencing (scRNA-seq) demonstrated that luminal epithelial cells are more prone to the expression of NOTCH target genes as well as notch receptors, while DLL1, JAG1, and JAG2 ligands are more expressed in myoepithelial cells, suggesting paracrine signaling between these cell types (72).
Activation of the NOTCH pathway in ACC can also occur through mutations in other genes, such as NOTCH2, NOTCH3, NOTCH4, SPEN, FBXW7 and RBPJ (40, 49, 50). Interestingly, ACC tumor samples with mutations in NOTCH1 and SPEN had no alterations in MYB and MYBL1, suggesting that other genetic events than alterations in MYB/MYBL1 may also play a central role in developing the ACC (32).
Strong evidence has suggested that epigenetic processes play an important role in the development of ACC. Mutations in chromatin remodeling genes are frequently reported (SMARC1, SMARCA2, ARID1A, ARID1B, CREBBP, EP300), and this class of genes has also been shown to be downregulated in ACC (SMARCA4, SMARCB1, SMARCC1, SMARCC2, ARID1, and PBRM1) (64, 71, 73, 74).
Chromatin remodeling is an essential process for gene expression, and changes in the chromatin landscape depend directly on the dynamics and density of nucleosomes, which are regulated by genes grouped into four categories: imitation switch (ISWI), chromodomain-helicase-DNA binding (CHD), inositol requiring 80 (INO80) and switching defective/sucrose nonfermenting (SWI/SNF). The activity of the SWI/SNF complex, in turn, depends on the activity of two mutually exclusive catalytic subunits, SMARCA4 and SMARCA2 (75).
The NOTCH signaling pathway can interact with chromatin remodelers through interactions with multiple proteins, mainly associated with the SWI/SNF complex. The catalytic subunit BRM is essential for ensuring the accessibility of chromatin to genes responsive to the NOTCH pathway (53). In this context, the BRM protein was shown to be overexpressed in ACC samples compared to normal salivary gland tissues. Interestingly, the genes encoding the other subunits of the SWI/SNF complex (INI1, BAF155, and BAF170) are downregulated, suggesting that the ACC presents an imbalance between the subunits of the SWI/SNF complex, which culminates in aberrant expression of several genes involved in cell cycle control, cell proliferation and adhesion (76). The simultaneous occurrence of mutations in chromatin remodelers and NOTCH1 suggests that epigenetic mechanisms may promote progression by manipulating the NOTCH pathway through a possible biological synergy (65).
The patterns of gene expression were also described, and these findings culminated in the subdivision of ACC into subgroups according to the expression of MYB/MYBL1. In tumors that normally express these genes, their expression was positively correlated with genes associated with tumor proliferation (EN1, PRAME, SOX4, SOX11, SMO, CDK6, and MYC) (19, 54, 55). It is worth mentioning that MYC overexpression and advanced T stage were associated with lower DFS (29).
The frequency of activating mutations of the NOTCH pathway varies according to the methodology (65). It should be noted that studies that actually distinguish any mutation in NOTCH from activating mutations are limited (64, 71, 77). This distinction is important as a subpopulation of patients with activating mutations in this pathway could benefit from targeted therapies. In this sense, a complete analysis should encompass technologies that map other forms of NOTCH pathway activation and mutations in NOTCH 1/2/3. The combination of DNA-based NGS and NICD1 IHC is efficient in identifying patients with NOTCH pathway activation (64, 71, 77). The main limitation in defining a subgroup of patients who could benefit from targeted treatments remains the scarcity of data on staging classification by ICH, which makes it difficult to distinguish subgroups with activation of the pathway, and the lack of integration between clinical, pathological and genetic data (64, 71, 77).
On the other hand, there are subgroups of patients with ACC that do not express MYB/MYBL1 express KLF4, FOXO1, JUNB, FOSB, VGLL3 or ERBB3, CTNNB1 and SOX4. The expression of ERBB3, CTNNB1 and SOX4 was associated with a survival of less than 10 years in patients with ACC (78).
Targeted therapeutic strategies
Several studies have been developed to evaluate drug efficacy with targets in ACC. Clinical trials have been conducted to analyze patient progress by considering objective response rate (ORR/OR), stable disease (SD), progressive disease (PD), progression-free survival (PFS), or partial response (PR) according to RECIST criteria (66, 79–81). However, preclinical trials are limited due to the number of tumor samples available for experimental studies. In vitro, assays are also insufficient due to the few established cell lines, and some have proven cross-contamination (82). Finally, clinical trials rarely advance to phase 3, mainly due to insufficient objective responses (79–81). Figure 2 and Table 2 summarizes the target agents investigated in clinical trials for patients with ACC.
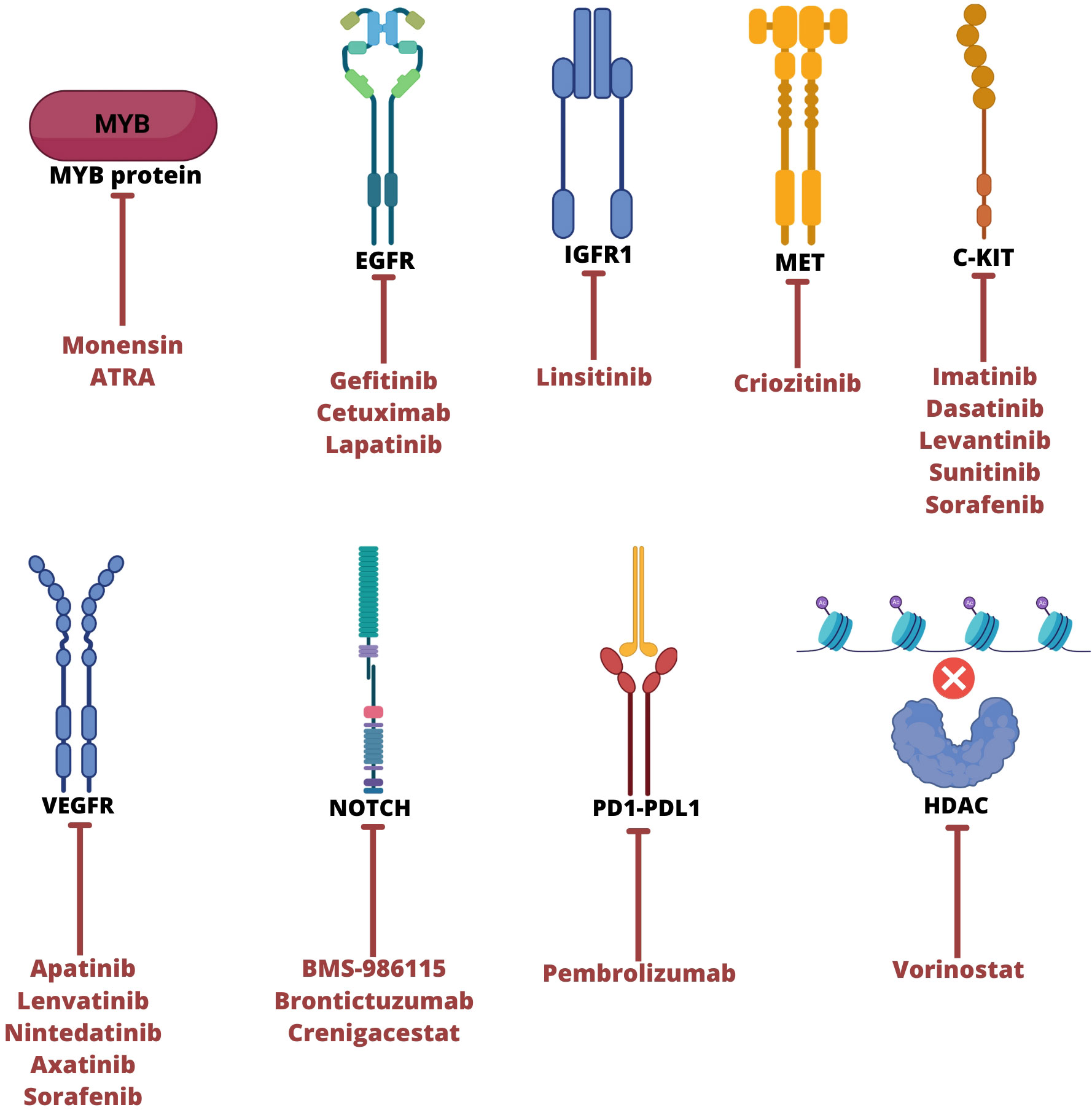
Figure 2 Main therapeutic targets to ACC tumor and their respective therapies. ATRA, all-trans retinoic acid; EGFR, epidermal growth factor receptor; IGFR1, insulin like growth factor 1 receptor; VEGFR, vascular endothelial growth factor; HDAC, Histone deacetylase. Created with BioRender.com.
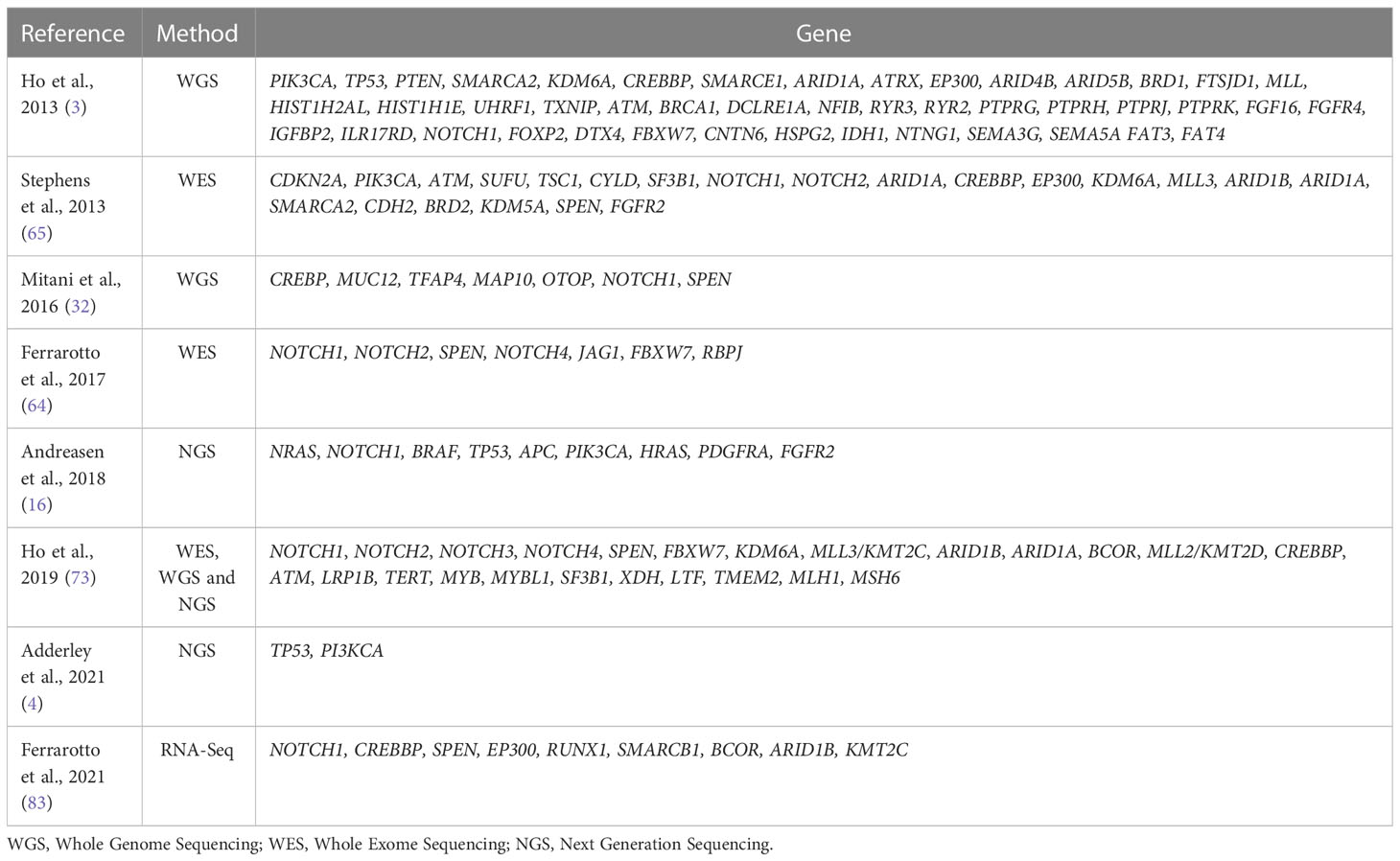
Table 2 Recurrent mutations in ACC.
Inhibitors of MYB
Alterations in the MYB gene are considered a hallmark of ACC and have been a target in investigating new therapies for the disease. Recently, a study evaluated the antiproliferative activity of a small molecule ionophore inhibitor of MYB, monensin, in MYB-reporter cell lines. This study demonstrated a potential interference in ACC cell viability, downregulation of genes activated by MYB-NFIB, and a decrease in MYB-NFIB mRNA levels caused by this drug. These results suggest that monensin acts mainly on MYB-NFIB translocation, which could help develop a suppressor of cell proliferation (84).
Evidence of the frequent MYB-related abnormalities and the inhibitory potential of MYB-NFIB drugs in vitro encourage the development of clinical trials to evaluate this potential in ACC patients. Due all-trans retinoic acid (ATRA) appears capable of interrupting the self-regulatory enhancer loop that occurs with fusion genes involving MYB, decreasing the level of oncogenic fusion protein, a phase II clinical trial using this compound in patients with acute promyelocytic leukemia (APL) It was conducted (85). This is a provisional file, not the final typeset article had no objective response in ACC. On the other hand, a study in ACC population, 61% (11/18) of patients had SD, 28% (5/18) had PD as the best response, and no adverse effects grade ≥ 3 was observed. These results also suggest that the drugs mechanism of action does not appear to be related to MYB overexpression but provided a potential future direction for clinical use with other therapeutic schemes (86).
In general, chromosomal fusion results in overexpression of MYB, but inhibition of their coactivators decreases its activity. A previous study with linsitinib, crizotinib, and gefitinib that target IGFR1, MET, and EGFR decrease mRNA MYB-NFIB levels or even block protein synthesis, which promote cell differentiation and decrease of tumor growth in vivo and in vitro. This evidence implies a possible network between these genes. Recently, studies demonstrated that drugs that inhibit p300 and proteasome coactivators prevent MYB activation leading to the suppression of the proliferation of ACC lines (87, 88).
Inhibitors of tyrosine kinase
The genetic alterations described in ACC impact relevant signaling pathways, including tyrosine kinase activity (48, 49, 63). One of the main targets studied in clinical trials is C-KIT. A relevant group of tyrosine kinase inhibitors (TKIs) evaluated against the protein-encoding this gene involves low molecular weight inhibitors. These types of TKI-specific target cancer cells by ligating transmembrane and intracellular proteins, affecting their enzymatic activity (89). Imatinib is a TKI targeting C-KIT, PDGFR, and ABL and has already demonstrated satisfactory activity in patients with chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST) (90–92).
After promising results of imatinib in GIST and CML, tumors that also overexpress C-KIT, studies have been conducted to investigate their clinical applicability in ACC. Reports using single imatinib therapy demonstrated limited responses, and none of the patients achieved OR, while the SD was not significant (75, 76). Additionally, dasatinib was well tolerated in ACC patients, but only 2.5% (1/20) had OR and 50% (20/40) had SD even with an experimental design that confirmed C-KIT overexpression (78). A phase II study using the combination of imatinib and cisplatin demonstrated that 11% (3/28) of patients evaluated had PR after induction chemotherapy (77). These findings, associated with the absence of mutations in exon 9 or 11 of C-KIT, suggest that only tyrosine kinase inhibitors may not be appropriate for patients with ACC (93–95).
EGFR is usually expressed in salivary glands, and their frequent activation in ACC tumors suggests that it may be a therapeutic target. Gefitinib is a drug capable of inhibiting EGFR in many tumors approved by FDA for treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC). A phase II clinical trial with this drug unfortunately demonstrated no objective response, however, the drug was well tolerated, and there was prolonged SD and OS in some patients (96). Cetuximab, a monoclonal antibody that blocks the EGFR signaling approved for the treatment of colorectal cancer was used in ACC patients. In this phase II clinical trial demonstrated no objective response in ACC, but 87% (20/23) of patients had SD and 52% (12/23) had SD ≥ 6 months (97). Lastly, lapatinib, a double inhibitor of EGFR and erbB2, approved to be used on treatment of breast cancer HER 2+ was used in ACC patients and revealed no objective response, but 52% (15/19) of assessable patients had SD and 60% (9/15) had SD ≥ 6 months. These findings suggest that EGFR signaling is significantly active in ACC tumor and may be used as potential target for treatment of disease (98).
In addition to C-KIT and EGFR inhibitors, multitargeted medications, including other TKIs and inhibitor molecules of FGFR 1-4 signaling, VEGFR-1-3, RET, and PDGFR, have been studied. However, these studies are inconsistent about the best endpoint to evaluate the drug’s effectiveness in metastatic and/or recurrent ACC patients (99, 100). While a few authors defined SD and OR as the best criteria, others indicated that SD could not represent a real drug effect but instead the natural progression of tumor disease that has a long and indolent growth (66).
In this sense, lenvatinib and apatinib that were tested in phase II clinical trials demonstrated clinical efficacy in recurrent/metastatic ACC patients. Both studies established PFS as the primary endpoint, with durations of 9 and 19,8 months, respectively (66, 81). Conversely, other authors defined SD as the primary endpoint. Dovitinib, axitinib, and sunitinib demonstrated a better response in comparison with other TKIs in ACC tumors (SD = 65%, 75,8% and 78%, respectively), but no patient had OR (79, 90, 99).
The presence of FGFR activating mutations appears to be crucial for the satisfactory effect of dovitinib on other types of tumors (100). In ACC, a subpopulation that demonstrates activating mutations may benefit from this treatment, but this would need to be validated in larger cohorts as well as two-arm and randomized studies to observe the real effect of the drug (80).
The use of molecular markers could help to identify patients who may benefit from targeted drugs. For example, a longer median PFS was observed among MYB+/NFIB+ patients than among MYB+/NFIB-, MYB-/NFIB+ and MYB-/NFIB- patients. In addition, 3 cases of 4q12 amplification and, consequently, an increase in the copy number of the PDGFRA/KDR/KIT target genes were identified. In 2 of these 3 patients, axitinib produced tumor regression and SD for ≥ 6 months. Finally, the amplification of NOTCH1 observed in 1 patient correlated with a confirmed partial response (cPR) with a PFS of nearly 1 year. Although these data are not statistically significant, they suggest that molecular investigation should also be considered in clinical trial designs (79).
Cellular signaling pathway suppressors
Multi kinase inhibitors (MKI) and other drug types are currently used in clinical trials aiming to block signaling pathways. A multicenter phase II study evaluated the efficacy of everolimus, a mammalian target of rapamycin (mTOR) inhibitor that is important for proliferation signaling of the Akt pathway, in patients diagnosed with ACC in salivary glands. This study unfortunately demonstrated no complete or partial response (CR and PR) in patients with progressive unresectable ACC, but 79.4% (27/34) had SD, and tumor shrinkage within SD criteria was observed in 44% of these patients, showing a promising effect in advanced ACC patients (101).
Another drug used in clinical trials was nelfinavir, a human immunodeficiency virus (HIV) protease inhibitor that is generally used in acquired immunodeficiency syndrome (AIDS) patients. In cancer, this drug suppresses the PI3K/Akt/mTOR signaling pathway and a previous study evaluated this drug in ACC patients including salivary glands and the best response was SD in 46.7% (7/15) of patients, and no patients had PR or CR (102).
Inhibition of NOTCH pathway
The NOTCH signaling is essential for ACC progression and support the stem cell characteristic maintenance and angiogenesis. A phase I clinical trial testing the BMS-986115 (pan-NOTCH inhibitor) in 36 tumor solid patients including ACC revealed two ACC cases with SD ≥ 6 months, and the author suggest that this drug can interfere actively in ACC progression (103). In addition, the use of brontictuzumab, a monoclonal antibody that blocks NOTCH1 pathway, demonstrate an effective action in ACC patients. A phase I study showed the efficacy of this drug in many cancer patients including ACC with PRs and three prolonged SD occurred in ACC subjects with evidence of NOTCH1 pathway activation (104).
Further, a multicenter phase I clinical trial evaluated the safety and toxicity of crenigacestat (LY3039478) monotherapy, a small molecule inhibitor of NOTCH signaling. In this study, 68% of patients (15/22) had SD and four had SD ≥ 6 months. AE were mild to moderate and some patients had AE grade ≥ 3, including diarrhea 3 (14%), fatigue 1 (5%) and ALT increase 1 (5%) (105).
Antiangiogenic therapies
Angiogenesis develops a crucial role in ACC progression and many genes are required for this process. A multicenter phase II study demonstrated no objective response using nintedatinib, a triple TKi including VEGFR, FGFR and PDGFR. However, this drug was had tolerable toxicity and a high rate of disease control and stabilization in patients with recurrent or metastatic salivary gland cancer with majority of ACC cases. In addition, levantinib demonstrated interesting results in a phase II study: 5 of 33 patients had partial response, 24 had SD and only one had PD. Therefore, 66% (21/32) had tumor regression and 25% (8/32) had 20% or greater of tumor shrinkage, however, AE grade ≥ 3 were reported including hypertension and oral pain (106).
A clinical trial phase II evaluated the safety and efficacy of axitinib in salivary gland tumors including ACC. This drug consists of inhibitor of VEGFR 1, 2 and 3 and it is approved for treatment of advanced renal cell carcinoma. The study showed a tumor shrinkage and SD in ACC patients (107). A European phase II study evaluated sorafenib, a multitarget inhibitor of VEGFR, cKIT, p38a, PDGFRb in ACC population. Majority of patients (68%) had SD and unfortunately 21% had PD. Moreover, the toxicity was frequent (108). Other phase II study using sorafenib in head and neck cancer including ACC patients demonstrate 2 of 19 patients had partial response and 9/19 had stable disease. These findings indicate the limited effect of antiangiogenic therapies (109).
Immune targets
Understanding the tumor immune microenvironment is essential to provides biomarkers to develop novel target therapies. Lymphocytic infiltration supports the immune response against tumor cells and have been associated to favorable prognosis and the expression of molecules associated with immune infiltration are variable and ACC usually presents low immunogenicity (12). Recently, a pioneer phase II clinical trial evaluated the safety and OR of pembrolizumab plus vorinostat in head and neck squamous cells and salivary gland cancer patients (110). Pembrolizumab consists of a humanized mouse- derived PD-1 (programmed cell death protein 1) antibody that interrupt programmed death-ligand 1 (PD-L1) ligation promoting tumor cells apoptosis. FDA approved pembrolizumab to for use in patients with melanoma, head and neck squamous cell carcinoma, classical Hodgkin lymphoma and others solid tumors. On the other hand, vorinostat corresponds to a histone deacetylase (HDAC), that improves cell cycle arrest, apoptosis and angiogenesis inhibition approved to cutaneous T-cell lymphoma treatment (111). In this study with both drugs, 16% (4/25) of patients had PR and 40% (10/25) patients had SD ≥ 6 months. They observed stronger toxicity than studies with only pembrolizumab (110).
Finally, a phase II clinical trial of pembrolizumab, an immunotherapy drug potentially blocking PD-1 was conducted. In this study, the effects of medication accompanied by radiation (Arm A) and single medication therapy (Arm B) were evaluated. The SD was the best response observed among 50% (5/10) of patients in arm A and 70% of patients (7/10) in arm B without statistical significance, and no participants presented OR (112). More details of the current clinical trials involving ACC are summarized in Table 3.

Table 3 Clinical trials for the ACC.
Conclusion
Despite being a rare disease, ACC is a very aggressive tumor, and patients frequently exhibit perineural invasion and distant metastases due to molecular alterations. In this sense, t(6;9) translocation, resulting in MYB-NFIB fusion, is the most frequently reported alteration in ACC. Additionally, translocations involving MYBL1 and other enhancer genes were observed. This alteration results in MYB protein overexpression, which has several activation mechanisms, highlighting the complexity of this tumor.
Chromosomal gains and losses also affect important genes that are responsible for several signaling pathways and cell proliferation, such as NOTCH and C-KIT. Interestingly, the majority of mutations in ACC do not occur simultaneously at t(6;9), suggesting that ACC has a multistep initiation and progression process. Even epigenetic changes are involved in this tumor biology, such as key mutations in chromatin remodeling (MERC1, SMARCA2, ARID1A, ARID1B, SMARCA4) and an imbalance in miRNAs (miR-155, miR-320a, miR-99a, miR-205) that regulate gene expression.
The described genetic alterations have been constantly investigated as possible targets for new therapies. However, no targeted therapy has demonstrated consistent effectiveness, and ACC treatment currently still includes surgery and postoperative radiotherapy, which do not have an effect in metastatic patients. Natural blockers of MYB overexpression have demonstrated promising results in leukemia; however, in ACC patients, preclinical investigation is difficult due to the lack of cell lines and in vivo models.
To try to reverse this situation, studies with significant cohorts are needed, as well as the development of in vitro models. In this sense, 3D cultures would be interesting to analyze the impact of the tumor microenvironment on the ACC. The identification of molecular subtypes with specific alterations would also be interesting in clinical practice for the selection of patients who may benefit from certain therapeutic strategies. But, for that, it is necessary to standardize the methodologies used for the detection of important molecular alterations.
Finally, the authors generally report challenges in evaluating drugs with antitumor activity, since due to the rarity of the disease, it is difficult to establish a homogeneous population regarding histological type, previous therapy and metastatic disease. Apparently, RECIST criteria may not be useful to assess drug efficacy because of slow tumor growth. Therefore, it is recommended that efforts be focused on multicenter studies to improve patient selection (matching according to histological subtype, presence/absence of driver mutations and other relevant clinicopathological criteria) and the design of clinical trials.
Author contributions
Study design: FS and DC. Drafting of the manuscript: FS, JJ, and DC. Tables and Figures: FS and JJ. Manuscript review: AR, JP, and VF. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the Universidade Federal do Pará, Ministério da Educação and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior. Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; to DC, 313127/2020-4) for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACC, Adenoid Cystic Carcinoma; CNAs, Copy number alterations; NRD, Negative regulatory domain; MET, Mesenchymal-epithelial transition; FISH, Fluorescent in situ hybridization; LOH, Loss of heterozygosity; SCF, Stem cell factor; DFS, Disease free survival; OS, Overall survival; MMPs, Metalloproteinasis; ISWI, Imitation switch; CHD, Chromodomain-helicase-DNA binding; INO80, Inositol requiring 80; SWI/SNF, Switching defective/sucrose nonfermenting.; ORR/OR, Objective Response Rate; SD, Stable Disease; PD, Progression Disease; PFS, Progression Free Survival; PR, Partial Response; ATRA, Trial all-trans retinoic acid; APL, Acute promyelocytic leukemia; TKIs, Tyrosine kinase inhibitors; CML, Chronic myeloid leukemia; GIST, Gastrointestinal stromal tumors; cPR, confirmed partial responses; mTOR, Mammalian target of rapamycin; aCGH, Array comparative genomic hybridization; WGS, Whole genome sequencing; RNA-seq, RNA sequencing; FFPE, Formalin-Fixed Paraffin-Embedded; Primagrafts, primary patient-derived xenografts; WES, Whole Exome Sequencing; NGS, Next Generation Sequencing.
References
1. Coca-Pelaz A, Rodrigo JP, Bradley PJ, Vander Poorten V, Triantafyllou A, Hunt JL, et al. Adenoid cystic carcinoma of the head and neck–an update. Oral Oncol (2015) 51(7):652–61. doi: 10.1016/j.oraloncology.2015.04.005
2. Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study 242 cases. Am J Surg (1974) 128(4):512–20. doi: 10.1016/0002-9610(74)90265-7
3. Ho AS, Kannan K, Roy DM, Morris LGT, Ganly I, Katabi N, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet (2013) 45(7):791– 8. doi: 10.1038/ng.2643
4. Adderley H, Rack S, Hapuarachi B, Feeney L, Morgan D, Hussell T, et al. The utility of TP53 and PIK3CA mutations as prognostic biomarkers in salivary adenoid cystic carcinoma. Oral Oncol (2021) 113:105095. doi: 10.1016/j.oraloncology.2020.105095
5. Perzin KH, Gullane P, Conley J. Adenoid cystic carcinoma involving the external auditory canal. clinicopathologic study 16 cases. Cancer (1982) 50(12):2873–83. doi: 10.1002/1097-0142(19821215)50:12<2873::aid-cncr2820501230>3.0.co;2-r
6. Szanto PA, Luna MA, Tortoledo ME, White RA. Histologic grading of adenoid cystic carcinoma of the salivary glands. Cancer (1984) 54(6):1062–9. doi: 10.1002/1097-0142(19840915)54:6<1062::aid-cncr2820540622>3.0.co;2-e
7. Nagao T, Gaffey TA, Serizawa H, Sugano I, Ishida Y, Yamazaki K, et al. Dedifferentiated adenoid cystic carcinoma: a clinicopathologic study of 6 cases. Mod Pathol (2003) 16(12):1265–72. doi: 10.1097/01.MP.0000097366.88165.08
8. Seethala RR, Hunt JL, Baloch ZW, Livolsi VA, Leon Barnes E. Adenoid cystic carcinoma with high-grade transformation: a report of 11 cases and a review of the literature. Am J Surg Pathol (2007) 31(11):1683–94. doi: 10.1097/PAS.0b013e3180dc928c
9. He J-F, Lin Y, Wang B, Zhu W-Y, Wei D, Zhu H-Y. Analysis of the clinicopathological characteristics and prognosis of adenoid cystic carcinoma of the intraoral minor salivary glands: a retrospective study of 40 cases. Int J Oral Maxillofac Surg (2021) 50(4):444–50. doi: 10.1016/j.ijom.2020.07.031
10. Cordesmeyer R, Schliephake H, Kauffmann P, Tröltzsch M, Laskawi R, Ströbel P, et al. Clinical prognostic factors of salivary adenoid cystic carcinoma: a single-center analysis of 61 patients. J Craniomaxillofac Surg (2017) 45(11):1784–7. doi: 10.1016/j.jcms.2017.08.004
11. Jeong IS, Roh J-L, Cho K-J, Choi S-H, Nam SY, Kim SY. Risk factors for survival and distant metastasis in 125 patients with head and neck adenoid cystic carcinoma undergoing primary surgery. J Cancer Res Clin Oncol (2020) 146(5):1343–50. doi: 10.1007/s00432-020-03170-5
12. Nightingale J, Lum B, Ladwa R, Simpson F, Panizza B. Adenoid cystic carcinoma: a review of clinical features, treatment targets and advances in improving the immune response to monoclonal antibody therapy. Biochim Biophys Acta Rev Cancer (2021) 1875(2):188523. doi: 10.1016/j.bbcan.2021.188523
13. Hanna GJ, Bae JE, Lorch JH, Schoenfeld JD, Margalit DN, Tishler RB, et al. Long-term outcomes and clinicogenomic correlates in recurrent, metastatic adenoid cystic carcinoma. Oral Oncol (2020) 106(104690):104690. doi: 10.1016/j.oraloncology.2020.104690
14. Fang Y, Peng Z, Wang Y, Gao K, Liu Y, Fan R, et al. Current opinions on diagnosis and treatment of adenoid cystic carcinoma. Oral Oncol (2022) 130:105945. doi: 10.1016/j.oraloncology.2022.105945
15. Lorini L, Ardighieri L, Bozzola A, Romani C, Bignotti E, Buglione M, et al. Prognosis and management of recurrent and/or metastatic head and neck adenoid cystic carcinoma. Oral Oncol (2021) 115:105213. doi: 10.1016/j.oraloncology.2021.105213
16. Andreasen S. Molecular features of adenoid cystic carcinoma with an emphasis on microRNA expression. APMIS (2018) 126 Suppl 140:7–57. doi: 10.1111/apm.12828
17. Persson M, Andersson MK, Mitani Y, Brandwein-Weber MS, HFJr F, Moskaluk C, et al. Rearrangements, expression, and clinical significance of MYB and MYBL1 in adenoid cystic carcinoma: a multi-institutional study. Cancers (Basel) (2022) 14(15):3691. doi: 10.3390/cancers14153691
18. Su M, Zhang Z, Zhou L, Han C, Huang C, Nice EC. Proteomics, personalized medicine and cancer. Cancers (2021) 13(11):2512. doi: 10.3390/cancers13112512
19. Persson M, Andrén Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA (2009) 106(44):18740–4. doi: 10.1073/pnas.0909114106
20. Mitani Y, Li J, Rao PH, Zhao Y-J, Bell D, Lippman SM, et al. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: incidence, variability, and clinicopathologic significance. Clin Cancer Res (2010) 16(19):4722–31. doi: 10.1158/1078-0432.CCR-10-0463
21. Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao Y-J, et al. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res (2011) 17(22):7003–14. doi: 10.1158/1078-0432.CCR-11-1870
22. Cicirò Y, Sala A. MYB oncoproteins: emerging players and potential therapeutic targets in human cancer. Oncogenesis (2021) 10(2):19. doi: 10.1038/s41389-021-00309-y
23. Becker-Santos DD, Lonergan KM, Gronostajski RM, Lam WL. Nuclear factor I/B: a master regulator of cell differentiation with paradoxical roles in cancer. EBioMedicine (2017) 22:2–9. doi: 10.1016/j.ebiom.2017.05.027
24. De Almeida-Pinto YD, Costa SFDS, de Andrade BAB, Altemani A, Vargas PA, Abreu LG, et al. t(6;9)(MYB-NFIB) in head and neck adenoid cystic carcinoma: a systematic review with meta-analysis. Oral Dis (2019) 25(5):1277–82. doi: 10.1111/odi.12984
25. Togashi Y, Dobashi A, Sakata S, Sato Y, Baba S, Seto A, et al. MYB and MYBL1 in adenoid cystic carcinoma: diversity in the mode of genomic rearrangement and transcripts. Mod Pathol (2018) 31(6):934–46. doi: 10.1038/s41379-018-0008-8
26. Brayer KJ, Frerich CA, Kang H, Ness SA. Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discovery (2016) 6(2):176–87. doi: 10.1158/2159-8290.CD-15-0859
27. Andersson MK, Afshari MK, Andrén Y, Wick MJ, Stenman G. Targeting the oncogenic transcriptional regulator MYB in adenoid cystic carcinoma by inhibition of IGF1R/AKT signaling. J Natl Cancer Inst (2017) 109(9):10. doi: 10.1093/jnci/djx017
28. Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJH, Kluk MJ, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet (2016) 48(3):265–72. doi: 10.1038/ng.3502
29. Fujii K, Murase T, Beppu S, Saida K, Takino H, Masaki A, et al. MYB, MYBL1, MYBL2 and NFIB gene alterations and MYC overexpression in salivary gland adenoid cystic carcinoma. Histopathology (2017) 71(5):823–34. doi: 10.1111/his.13281
30. Brill LB 2nd, Kanner WA, Fehr A, Andrén Y, Moskaluk CA, et al. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol (2011) 24(9):1169–76. doi: 10.1038/modpathol.2011.86
31. Andersson MK, Mangiapane G, Nevado PT, Tsakaneli A, Carlsson T, Corda G, et al. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis (2020) 9(1):5. doi: 10.1038/s41389-020-0194-3
32. Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, et al. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res (2016) 22(3):725–33. doi: 10.1158/1078-0432.CCR-15-2867-T
33. Rettig EM, Talbot CC Jr, Sausen M, Jones S, Bishop JA, Wood LD, et al. Wholegenome sequencing of salivary gland adenoid cystic carcinoma [published correction appears in cancer prev res (Phila). Cancer Prev Res (Phila) (2016) 9(4):265–74. doi: 10.1158/1940-6207.CAPR-15-0316
34. Shibata E, Morita K-I, Kayamori K, Tange S, Shibata H, Harazono Y, et al. Detection of novel fusion genes by next-generation sequencing-based targeted RNA sequencing analysis in adenoid cystic carcinoma of head and neck. Oral Surg Oral Med Oral Pathol Oral Radiol (2021) 132(4):426–33. doi: 10.1016/j.oooo.2021.03.020
35. West RB, Kong C, Clarke N, Gilks T, Lipsick JS, Cao H, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol (2011) 35(1):92–9. doi: 10.1097/PAS.0b013e3182002777
36. Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El-Naggar AK. Clinical significance of myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther (2011) 12(7):569–73. doi: 10.4161/cbt.12.7.17008
37. Pusztaszeri MP, Sadow PM, Ushiku A, Bordignon P, McKee TA, Faquin WC. MYB immunostaining is a useful ancillary test for distinguishing adenoid cystic carcinoma from pleomorphic adenoma in fine-needle aspiration biopsy specimens. Cancer Cytopathol (2014) 122(4):257–65. doi: 10.1002/cncy.21381
38. Han J, Zhang C, Gu T, Yang X, Hu L, Tian Z, et al. Analysis of clinicopathological characteristics, MYB rearrangement and prognostic factors in salivary adenoid cystic carcinoma. Oncol Lett (2019) 17(3):2915–22. doi: 10.3892/ol.2019.9935
39. Ross JS, Wang K, Rand JV, Sheehan CE, Jennings TA, Al-Rohil RN, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol (2014) 38(2):235–8. doi: 10.1097/PAS.0000000000000102
40. Rooper LM, Lombardo KA, Oliai BR, Ha PK, Bishop JA. In situ Hybridization facilitates sensitive and specific diagnosis of adenoid cystic carcinoma regardless of translocation status. Am J Surg Pathol (2021) 45(4):488–97. doi: 10.1097/PAS.0000000000001616
41. Sun T, Akalin A, Dresser K, Fischer AH, Zuo T. The utility of MYB immunohistochemistry (IHC) in fine needle aspiration (FNA) diagnosis of adenoid cystic carcinoma (AdCC). Head Neck Pathol (2021) 15(2):389–94. doi: 10.1007/s12105-020-01202-7
42. Seethala RR, Cieply K, Barnes EL, Dacic S. Progressive genetic alterations of adenoid cystic carcinoma with high-grade transformation. Arch Pathol Lab Med (2011) 135(1):123–30. doi: 10.5858/2010-0048-OAR.1
43. Oga A, Uchida K, Nakao M, Kawauchi S, Furuya T, Chochi Y, et al. Loss of 6q or 8p23 is associated with the total number of DNA copy number aberrations in adenoid cystic carcinoma. Oncol Rep (2011) 26(6):1393–8. doi: 10.3892/or.2011.1446
44. Stallmach I, Zenklusen P, Komminoth P, Schmid S, Perren A, Roos M, et al. Loss of heterozygosity at chromosome 6q23-25 correlates with clinical and histologic parameters in salivary gland adenoid cystic carcinoma. Virchows Arch (2002) 440(1):77–84. doi: 10.1007/s004280100523
45. Cordesmeyer R, Laskawi R, Schliephake H, Kauffmann P, Beck J, Bornemann- Kolatzki K, et al. Shallow whole genome sequencing of adenoid cystic carcinomas of the salivary glands identifies specific chromosomal aberrations related to tumor progression. Oral Oncol (2020) 103(104615):104615. doi: 10.1016/j.oraloncology.2020.104615
46. Rutherford S, Yu Y, Rumpel CA, HFJr F, Moskaluk CA. Chromosome 6 deletion and candidate tumor suppressor genes in adenoid cystic carcinoma. Cancer Lett (2006) 236(2):309–17. doi: 10.1016/j.canlet.2005.05.049
47. Bernheim A, Toujani S, Saulnier P, Robert T, Casiraghi O, Validire P, et al. High- resolution array comparative genomic hybridization analysis of human bronchial and salivary adenoid cystic carcinoma. Lab Invest (2008) 88(5):464–73. doi: 10.1038/labinvest.2008.18
48. Hou H, Jia D, Yan W, Zhang X, Wang C, Li Y, et al. KIT/PDGFRA/KDR amplification defines a novel molecular subtype of adenoid cystic carcinoma patients who may benefit from treatment with tyrosine kinase inhibitors. Transl Cancer Res (2020) 9(8):4703–14. doi: 10.21037/tcr-20-637
49. Edwards PC, Bhuiya T, Kelsch RD. C-kit expression in the salivary gland neoplasms adenoid cystic carcinoma, polymorphous low-grade adenocarcinoma, and monomorphic adenoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2003) 95(5):586–93. doi: 10.1067/moe.2003.31
50. Tang Y, Liang X, Zheng M, Zhu Z, Zhu G, Yang J, et al. Expression of c-kit and slug correlates with invasion and metastasis of salivary adenoid cystic carcinoma. Oral Oncol (2010) 46(4):311–6. doi: 10.1016/j.oraloncology.2010.02.001
51. Penner CR, Folpe AL, Budnick SD. C-kit expression distinguishes salivary gland adenoid cystic carcinoma from polymorphous low-grade adenocarcinoma. Mod Pathol (2002) 15(7):687–91. doi: 10.1097/01.MP.0000018973.17736.F8
52. Freier K, Flechtenmacher C, Walch A, Devens F, Mühling J, Lichter P, et al. Differential KIT expression in histological subtypes of adenoid cystic carcinoma (ACC) of the salivary gland. Oral Oncol (2005) 41(9):934–9. doi: 10.1016/j.oraloncology.2005.05.005
53. Chowsilpa S, An D, Maleki Z. Adenoid cystic carcinoma cytology: salivary gland and nonsalivary gland. Diagn Cytopathol (2020) 48(12):1282–9. doi: 10.1002/dc.24573
54. Marino F, Scalise M, Cianflone E, Mancuso T, Aquila I, Agosti V, et al. Role of ckit in myocardial regeneration and aging. Front Endocrinol (Lausanne) (2019) 10:371. doi: 10.3389/fendo.2019.00371
55. Phuchareon J, van Zante A, Overdevest JB, McCormick F, Eisele DW, Tetsu O. C-kit expression is rate-limiting for stem cell factor-mediated disease progression in adenoid cystic carcinoma of the salivary glands. Transl Oncol (2014) 7(5):537–45. doi: 10.1016/j.tranon.2014.07.006
56. Rao PH, Roberts D, Zhao Y-J, Bell D, Harris CP, Weber RS, et al. Deletion of 1p32-p36 is the most frequent genetic change and poor prognostic marker in adenoid cystic carcinoma of the salivary glands. Clin Cancer Res (2008) 14(16):5181–7. doi: 10.1158/1078-0432.CCR-08-0158
57. Persson M, Andrén Y, Moskaluk CA, Henry F, Susanna C, Futreal PA, et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer (2012) 51(8):805–17. doi: 10.1002/gcc.21965
58. Costa A, Altemani A, Garcı́a-Inclán C, Fresno F, Suárez C, Llorente JL, et al. Analysis of MYB oncogene in transformed adenoid cystic carcinomas reveals distinct pathways of tumor progression. Lab Invest (2014) 94(6):692–702. doi: 10.1038/labinvest.2014.59
59. Li J, Perlaky L, Rao P, Weber RS, El-Naggar AK. Development and characterization of salivary adenoid cystic carcinoma cell line. Oral Oncol (2014) 50(10):991–9. doi: 10.1016/j.oraloncology.2014.06.012
60. Argyris PP, Wetzel SL, Greipp P, Wehrs R, Knutson D, Kloft-Nelsonet S, et al. Clinical utility of myb rearrangement detection and p63/p40 immunophenotyping in the diagnosis of adenoid cystic carcinoma of minor salivary glands: a pilot study. Oral Surg Oral Med Oral Pathol Oral Radiol (2016) 121(3):282–9. doi: 10.1016/j.oooo.2015.10.016
61. Tian Z, Li L, Zhang CY, Gu T, Li J. Differences in MYB expression and gene abnormalities further confirm that salivary cribriform basal cell tumors and adenoid cystic carcinoma are two distinct tumor entities. J Oral Pathol Med (2016) 45(9):698–703. doi: 10.1111/jop.12414
62. Warner KA, Oklejas AE, Pearson AT, Zhang Z, Wu W, Divi V, et al. UMHACC- 2A: MYB-NFIB fusion-positive human adenoid cystic carcinoma cell line. Oral Oncol (2018) 87:21–8. doi: 10.1016/j.oraloncology.2018.10.012
63. Saida K, Murase T, Ito M, Fujii K, Takino H, Masaki A, et al. Mutation analysis of the EGFR pathway genes, EGFR, RAS, PIK3CA, BRAF, and AKT1, in salivary gland adenoid cysticncarcinoma. Oncotarget (2018) 9(24):17043–55. doi: 10.18632/oncotarget.24818
64. Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, et al. Activatingn NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to Notch1inhibitors. J Clin Oncol (2017) 35(3):352–60. doi: 10.1200/JCO.2016.67.5264
65. Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest (2013) 123(7):2965–8. doi: 10.1172/JCI67201
66. Zhu G, Zhang L, Dou S, Li R, Li J, Ye L, et al. Apatinib in patients with recurrent or metastatic adenoid cystic carcinoma of the head and neck: a single- arm, phase II prospective study. Ther Adv Med Oncol (2021) 13:17588359211013626. doi: 10.1177/17588359211013626
67. Chen W, Cao G, Yuan X, Zhang X, Zhang Q, Zhu Y, et al. Notch-1 knockdown suppresses proliferation, migration and metastasis of salivary adenoid cystic carcinoma cells. J Transl Med (2015) 13(1):167. doi: 10.1186/s12967-015-0520-2
68. Su B-H, Qu J, Song M, Huang X-Y, Hu X-M, Xie J, et al. NOTCH1 signaling contributes to cell growth, anti-apoptosis and metastasis in salivary adenoid cystic carcinoma. Oncotarget (2014) 5(16):6885–95. doi: 10.18632/oncotarget.2321
69. Xie J, Lin L-S, Huang X-Y, Gan R-H, Ding L-C, Su B-H, et al. The NOTCH1- HEY1 pathway regulates self-renewal and epithelial-mesenchymal transition of salivary adenoid cystic carcinoma cells. Int J Biol Sci (2020) 16(4):598–610. doi: 10.7150/ijbs.36407
70. Rettig EM, Tan M, Ling S, Yonescu R, Bishop J, Fakhry C, et al. MYB rearrangement and clinicopathologic characteristics in head and neck adenoid cystic carcinoma. Laryngoscope (2015) 125(9):E292–9. doi: 10.1002/lary.25356
71. Sajed DP, Faquin WC, Carey C, Severson EA, H. Afrogheh A, A. Johnson C, et al. Diffuse staining for activated NOTCH1 correlates with NOTCH1 mutation status and is associated with worse outcome in adenoid cystic carcinoma. Am J Surg Pathol (2017) 41(11):1473–82. doi: 10.1097/pas.0000000000000945
72. Parikh AS, Wizel A, Davis D, Lefranc-Torres A, Rodarte-Rascon AI, Miller LE, et al. Single-cell RNA sequencing identifies a paracrine interaction that may drive oncogenic notch signaling in human adenoid cystic carcinoma. Cell Rep (2022) 41(9):111743. doi: 10.1016/j.celrep.2022.111743
73. Ho AS, Ochoa A, Jayakumaran G, Zehir A, Valero Mayor C, Tepe J, et al. Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J Clin Invest (2019) 129(10):4276–89. doi: 10.1172/JCI128227
74. Jagielska B, Sarnowska E, Rusetska N, Jancewicz I, Durzynska M, Kubala S, et al. Advanced adenoid cystic carcinoma (ACC) is featured by SWI/SNF chromatin remodeling complex aberrations. J Cancer Res Clin Oncol (2019) 145(1):201–11. doi: 10.1007/s00432-018-2783-5
75. Centore RC, Sandoval GJ, Soares LMM, Kadoch C, Chan HM. Mammalian SWI/SNF chromatin remodeling complexes: emerging mechanisms and therapeutic strategies. Trends Genet (2020) 36(12):936–50. doi: 10.1016/j.tig.2020.07.011
76. Pillidge Z, Bray SJ. SWI/SNF chromatin remodeling controls notch-responsive enhancer accessibility. EMBO Rep (2019) 20(5):e46944. doi: 10.15252/embr.201846944
77. Feeney L, Hapuarachi B, Adderley H, Rack S, Morgan D, Walker R, et al. Clinical disease course and survival outcomes following disease recurrence in adenoid cystic carcinoma with and without NOTCH signaling pathway activation. Oral Oncol (2022) 133:106028. doi: 10.1016/j.oraloncology.2022.106028
78. Frerich CA, Brayer KJ, Painter BM, Kang H, Mitani Y, El-Naggar AK, et al. Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget (2018) 9(7):7341–58. doi: 10.18632/oncotarget.23641
79. Ho AL, Dunn L, Sherman EJ, Fury MG, Baxi SS, Chandramohan R, et al. A phase II study of axitinib (AG-013736) in patients with incurable adenoid cystic carcinoma. Ann Oncol (2016) 27(10):1902–8. doi: 10.1093/annonc/mdw287
80. Dillon PM, Petroni GR, Horton BJ, Moskaluk CA, Fracasso PM, Douvas MG, et al. A phase II study of dovitinib in patients with recurrent or metastatic adenoid cystic carcinoma. Clin Cancer Res (2017) 23(15):4138–45. doi: 10.1158/1078-0432.CCR-16-2942
81. Locati LD, Galbiati D, Calareso G, Alfieri S, Singer S, Cavalieri S, et al. Patients with adenoid cystic carcinomas of the salivary glands treated with lenvatinib: activity and quality of life. Cancer (2020) 126(9):1888–94. doi: 10.1002/cncr.32754
82. Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PloS One (2009) 4(6):e6040. doi: 10.1371/journal.pone.0006040
83. Ferrarotto R, Mitani Y, McGrail DJ, Li K, Karpinets TV, Bell D, et al. Proteogenomic analysis of salivary adenoid cystic carcinomas defines molecular subtypes and identifies therapeutic targets. Clin Cancer Res (2021) 27(3):852–64. doi: 10.1158/1078-0432.CCR-20-1192
84. Yusenko MV, Trentmann A, Andersson MK, Ghani LA, Jakobs A, Arteaga Paz MF, et al. Monensin, a novel potent MYB inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells. Cancer Lett (2020) 479:61–70. doi: 10.1016/j.canlet.2020.01.039
85. Ni X, Hu G, Cai X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit Rev Food Sci Nutr (2019) 59(sup1):S71–80. doi: 10.1080/10408398.2018.1509201
86. Hanna GJ, ONeill A, Cutler JM, Flynn M, Vijaykumar T, Clark JR, et al. A phase II trial of all-trans retinoic acid (ATRA) in advanced adenoid cystic carcinoma. Oral Oncol (2021) 119(105366):105366. doi: 10.1016/j.oraloncology.2021.105366
87. Yusenko MV, Biyanee A, Andersson MK, Radetzki S, von Kries JP, Stenman G, et al. Proteasome inhibitors suppress MYB oncogenic activity in a p300- dependent manner. Cancer Lett (2021) 520:132–42. doi: 10.1016/j.canlet.2021.07.010
88. Yusenko MV, Biyanee A, Frank D, Köhler LHF, Andersson MK, Khandanpour C, et al. Bcr-TMP, a novel nanomolar-active compound that exhibits both MYB- and microtubule-inhibitory activity. Cancers (2021) 14(1):43. doi: 10.3390/cancers14010043
89. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001-2020). J Hematol Oncol (2020) 13(1):143. doi: 10.1186/s13045-020-00977-0
90. Waller CF. Imatinib mesylate. Recent results Cancer Res Recent Results Cancer Res (2018) 212:1–27. doi: 10.1007/978-3-319-91439-8_1
91. Rausch JL, Boichuk S, Ali AA, Patil SS, Liu L, Lee DM, et al. Opposing roles of KIT and ABL1 in the therapeutic response of gastrointestinal stromal tumor (GIST) cells to imatinib mesylate. Oncotarget (2017) 8(3):4471–83. doi: 10.18632/oncotarget.13882
92. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med (2017) 376(10):917–27. doi: 10.1056/nejmoa1609324
93. Pfeffer MR, Talmi Y, Catane R, Symon Z, Yosepovitch A, Levitt M. A phase II study of imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol (2007) 43(1):33–6. doi: 10.1016/j.oraloncology.2005.12.026
94. Hotte SJ, Winquist EW, Lamont E, MacKenzie M, Vokes E, Chen EX, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a princess Margaret hospital phase II consortium study. J Clin Oncol (2005) 23(3):585–90. doi: 10.1200/JCO.2005.06.125
95. Wong SJ, Karrison T, Hayes DN, Kies MS, Cullen KJ, Tanvetyanon T, et al. Phase II trial of dasatinib for recurrent or metastatic c-KIT expressing adenoid cystic carcinoma and for nonadenoid cystic malignant salivary tumors. Ann Oncol (2016) 27(2):318–23. doi: 10.1093/annonc/mdv537
96. Jakob JA, Kies MS, Glisson BS, Kupferman ME, Liu DD, Lee JJ, et al. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck (2015) 37(5):644–9. doi: 10.1002/hed.23647
97. Locati LD, Bossi P, Perrone F, Potepan P, Crippa F, Mariani L, et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: a phase II study. Oral Oncol (2009) 45(7):574–8. doi: 10.1016/j.oraloncology.2008.07.010
98. Agulnik M, Cohen EW, Cohen RB, Chen EX, Vokes EE, Hotte SJ, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol (2007) 25(25):3978–84. doi: 10.1200/JCO.2007.11.8612
99. Chau NG, Hotte SJ, Chen EX, Chin SF, Turner S, Wang L, et al. A phase II study of sunitinib in recurrent and/or metastatic adenoid cystic carcinoma (ACC) of the salivary glands: current progress and challenges in evaluating molecularly targeted agents in ACC. Ann Oncol (2012) 23(6):1562–70. doi: 10.1093/annonc/mdr522
100. Liang Q, Wang J, Zhao L, Hou J, Hu Y, Shi J. Recent advances of dual FGFR inhibitors as a novel therapy for cancer. Eur J Med Chem (2021) 214(113205):113205. doi: 10.1016/j.ejmech.2021.113205
101. Kim D-W, Oh D-Y, Shin SH, Kang JH, Cho BC, Chung J-S, et al. A multicenter phase II study of everolimus in patients with progressive unresectable adenoid cystic carcinoma. BMC Cancer (2014) 14(1):795. doi: 10.1186/1471-2407-14-795
102. Hoover AC, Milhem MM, Anderson CM, Sun W, Smith BJ, Hoffman HT, et al. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: results of a phase II clinical trial: nelfinavir monotherapy in refractory adenoid cystic carcinoma. Head Neck (2015) 37(5):722–6. doi: 10.1002/hed.23664
103. Aung KL, El-Khoueiry AB, Gelmon K, Tran B, Bajaj G, He B, et al. A multi-arm phase I dose escalating study of an oral NOTCH inhibitor BMS- 986115 in patients with advanced solid tumours. Investigational New Drugs (2018) 36(6):1026–36. doi: 10.1007/s10637-018-0597-6
104. Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol (2018) 29(7):1561–8. doi: 10.1093/annonc/mdy171
105. Even C, Lassen U, Merchan J, Le Tourneau C, Soria JC, Ferte C, et al. Safety and clinical activity of the notch inhibitor, crenigacestat (LY3039478), in an open-label phase I trial expansion cohort of advanced or metastatic adenoid cystic carcinoma. Invest New Drugs (2020) 38(2):402–9. doi: 10.1007/s10637-019-00739-x
106. Tchekmedyian V, Sherman EJ, Dunn L, Tran C, Baxi S, Katabi N, et al. Phase II study of lenvatinib in patients with progressive, recurrent or metastatic adenoid cystic carcinoma. J Clin Oncol (2019) 37(18):1529–37. doi: 10.1200/JCO.18.01859
107. Locati LD, Cavalieri S, Bergamini C, Resteghini C, Alfieri S, Calareso G, et al. Phase II trial with axitinib in recurrent and/or metastatic salivary gland cancers of the upper aerodigestive tract. Head Neck (2019) 41(10):3670–6. doi: 10.1002/hed.25891
108. Thomson DJ, Silva P, Denton K, Bonington S, Mak SK, Swindell R, et al. Phase II trial of sorafenib in advanced salivary adenoid cystic carcinoma of the head and neck. Head Neck (2015) 37(2):182–7. doi: 10.1002/hed.23577
109. Locati LD, Perrone F, Cortelazzi B, Bergamini C, Bossi P, Civelli E, et al. A phase II study of sorafenib in recurrent and/or metastatic salivary gland carcinomas: translational analyses and clinical impact. Eur J Cancer (Oxford Engl 1990) (2016) 69:158–65. doi: 10.1016/j.ejca.2016.09.022
110. Rodriguez CP, Wu QV, Voutsinas J, Fromm JR, Jiang X, Pillarisetty VG, et al. A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res (2020) 26(4):837–45. doi: 10.1158/1078-0432.CCR-19-2214
111. Bubna AK. Vorinostat-an overview. Indian J Dermatol (2015) 60(4):419. doi: 10.4103/0019-5154.160511
Keywords: adenoid cystic carcinoma, oral cancer, molecular profile, target therapies, MYB
Citation: da Silva FJ, Azevedo JC, Ralph ACL, Pinheiro JdJV, Freitas VM and Calcagno DQ (2023) Salivary glands adenoid cystic carcinoma: a molecular profile update and potential implications. Front. Oncol. 13:1191218. doi: 10.3389/fonc.2023.1191218
Received: 23 March 2023; Accepted: 01 June 2023;
Published: 05 July 2023.
Edited by:
Guopei Zhu, Shanghai Jiao Tong University, ChinaReviewed by:
Mattias K. Andersson, University of Gothenburg, SwedenChun-Ye Zhang, Shanghai Jiao Tong University, China
Copyright © 2023 da Silva, Carvalho de Azevedo, Ralph, Pinheiro, Freitas and Calcagno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danielle Queiroz Calcagno, ZGFuaWNhbGNhZ25vQGdtYWlsLmNvbQ==