 Ilaria Bertacca
Ilaria Bertacca Francesco Pegoraro
Francesco Pegoraro Annalisa Tondo
Annalisa Tondo Claudio Favre
Claudio Favre- 1Paediatric Hematology/Oncology Department, Meyer Children’s Hospital, Firenze, Italy
- 2Department of Health Sciences , University of Firenze, Firenze, Italy
The treatment of childhood solid cancer has markedly evolved in recent years following a refined molecular characterization and the introduction of novel targeted drugs. On one hand, larger sequencing studies have revealed a spectrum of mutations in pediatric tumors different from adults. On the other hand, specific mutations or immune dysregulated pathways have been targeted in preclinical and clinical studies, with heterogeneous results. Of note, the development of national platforms for tumor molecular profiling and, in less measure, for targeted treatment, has been essential in the process. However, many of the available molecules have been tested only in relapsed or refractory patients, and have proven poorly effective, at least in monotherapy. Our future approaches should certainly aim at improving the access to molecular characterization, to obtain a deeper picture of the distinctive phenotype of childhood cancer. In parallel, the implementation of access to novel drugs should not only be limited to basket or umbrella studies but also to larger, multi-drug international studies. In this paper we reviewed the molecular features and the main available therapeutic options in pediatric solid cancer, focusing on available targeted drugs and ongoing investigations, aiming at providing a useful tool to navigate the heterogeneity of this promising but complex field.
Introduction
The treatment of solid and central nervous system (CNS) cancers in children has dramatically evolved in the last decades. The development of intensified cytotoxic chemotherapy and multimodal approaches has led to a significant improvement in survival. In parallel, molecular and diagnostic advances have resulted in more accurate stratification protocols, allowing the selection of those patients who require intensified treatments and reducing long-term toxicities. Nevertheless, pediatric oncologists still have to deal with poorly addressable tumors and severe chemotherapy burdens in cancer survivors (1).
Recently, the implementation of widespread genome-wide profiling programs has contributed to unveiling the genetic heterogeneities and specific nature of childhood solid cancers, as well as their dissimilarity from adult-onset tumors. Moreover, these studies have revealed the contribution of genetic predisposition to pediatric neoplasms and have driven the implementation of targeted approaches (2). Many trials are evaluating pediatric cancer molecular stratification and targeted treatment: the INdividualized therapy FOor Relapsed Malignancies in childhood (INFORM); the individualized THERapy (iTHER) program for children with relapsed or refractory cancer; the MoleculAr Profiling for Pediatric and Young Adult Cancer Treatment Stratification (MAPPYACTS); the pediatric Molecular Analysis for Therapy CHoice (pediatric MATCH); the individualized CAncer Therapy (iCAT) program; the Genomic Assessment Improves Novel Therapy (GAIN) project; the PRecision Oncology For Young peopLE (PROFYLE) program; the Stratified Medicine Paediatrics (SMPaeds) study; and the ZERO childhood cancer program (Figure 1). However, among pediatric solid cancers, many entities still lack effective therapeutic strategies, in most cases resulting in poor survival and long-term outcome.
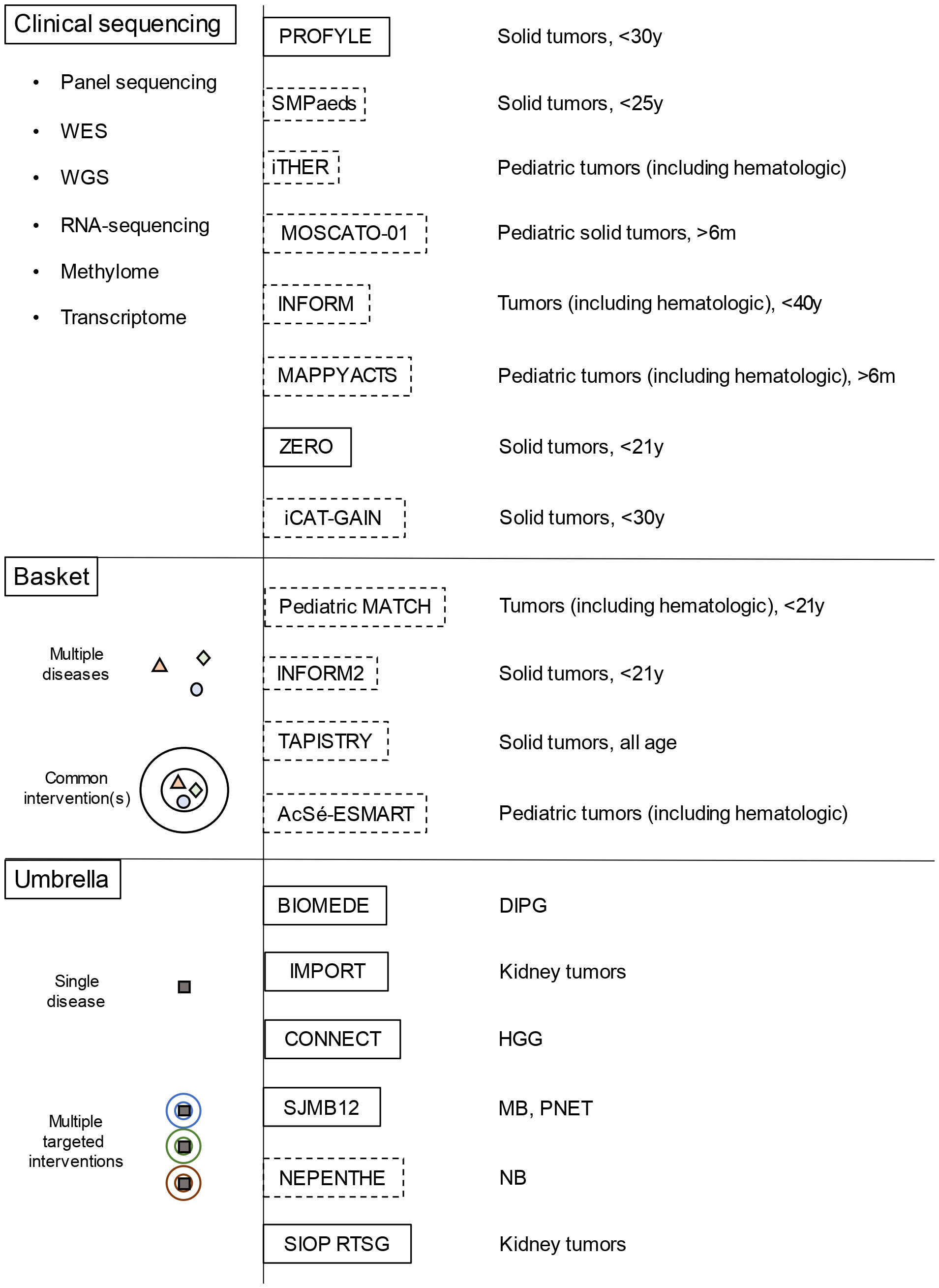
Figure 1 Overview of the main molecular profiling platforms and of basket/umbrella trials for childhood solid cancer (dotted lines represent platforms/trials for relapsed/refractory tumors, while the continuous lines those for newly diagnosed tumors) WES, whole-exome sequencing; WGS, whole-genome sequencing; DIPG, diffuse intrinsic pontine glioma; MB, medulloblastoma; NB, neuroblastoma; HGG, high grade gliomas; PNET, primitive neuroectodermal tumors.
In this review, we will discuss the main genetic abnormalities displayed by pediatric solid tumors, and describe how these abnormalities can be targeted by innovative treatments. Data on already published studies will be provided, as well as available preliminary data on molecules that are still being investigated, with the ultimate goal of providing clinicians with an updated tool for everyday clinical management.
Neuroblastoma
Neuroblastoma is a neuroendocrine tumor of the developing sympathetic nervous system and the most common malignancy diagnosed in the first year of life. The most common genetic alterations in neuroblastoma are MYCN amplification, anaplastic lymphoma kinase (ALK) mutations, segmental chromosomal alterations, and DNA copy number alterations (2, 3). MYCN amplification is found in around 20% of cases, typically coexisting with a segmental chromosomal loss of chromosome 1p (4). ALK mutations are found in around 10-15% of sporadic neuroblastomas but are also typically responsible for familial forms (5, 6). Rarely, mutations are found in genes of the mitogen-activated protein kinase (MAPK) pathway (e.g., RAS, BRAF, PTPN11, FGFR), which are targetable by specific molecules (3) (Figure 2). MYCN amplification is associated with an aggressive subtype and poor survival, as well as chromosome 11q deletion (7). The activation of telomere maintenance mechanisms (TMMs) by multiple genetic alterations, such as TERT rearrangements, MYCN amplification, and ATRX mutations, is emphasized in relapsed neuroblastoma and has a markedly poor prognosis, especially when associated with MAPK or p53 pathway mutations (8) (Table 1).
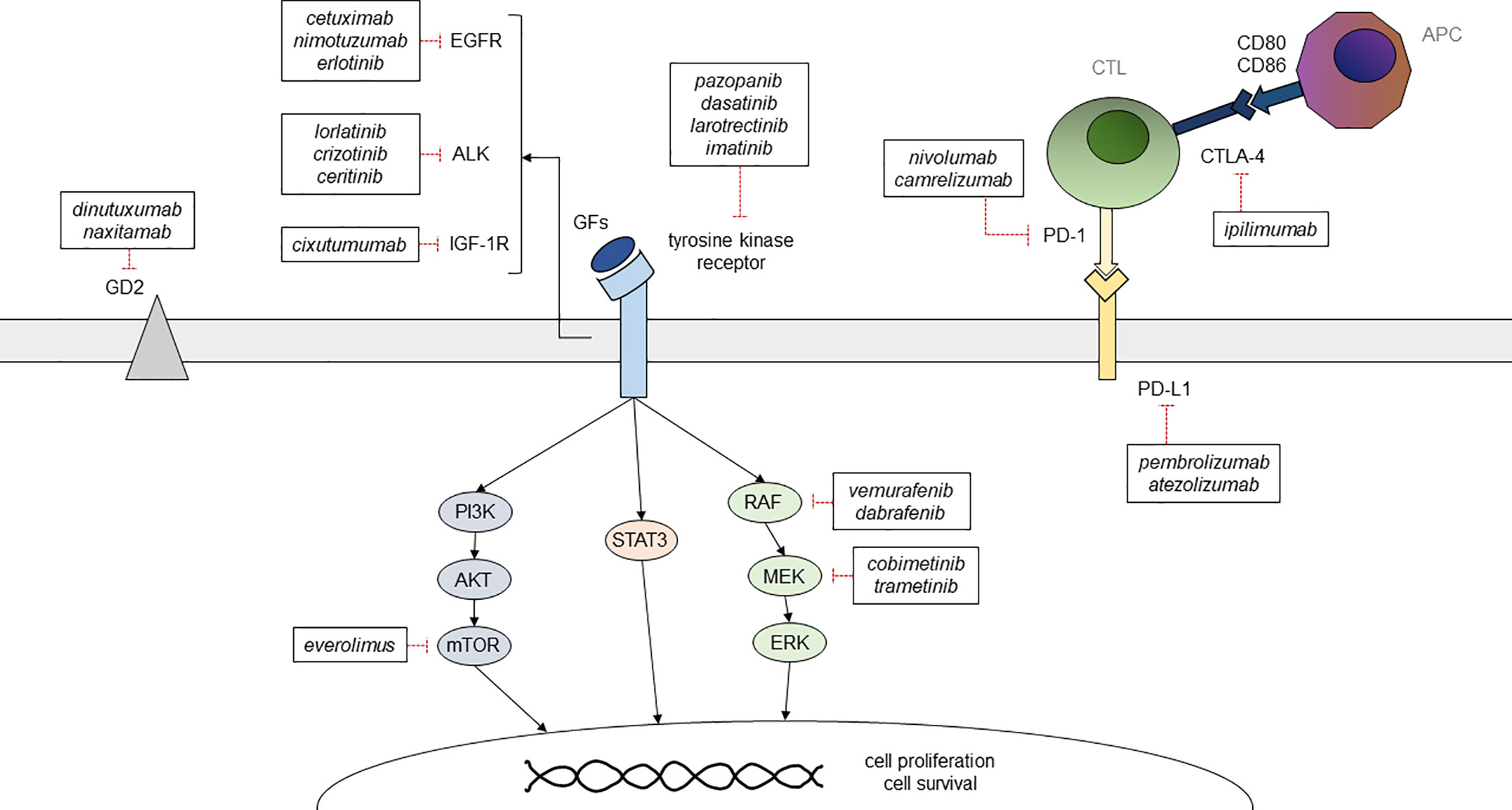
Figure 2 Summary of the main targetable pathways in childhood solid cancer and of the available therapeutic options GFs, growth factors; CTL, cytotoxic T lymphocyte; APC, antigen presenting cell.
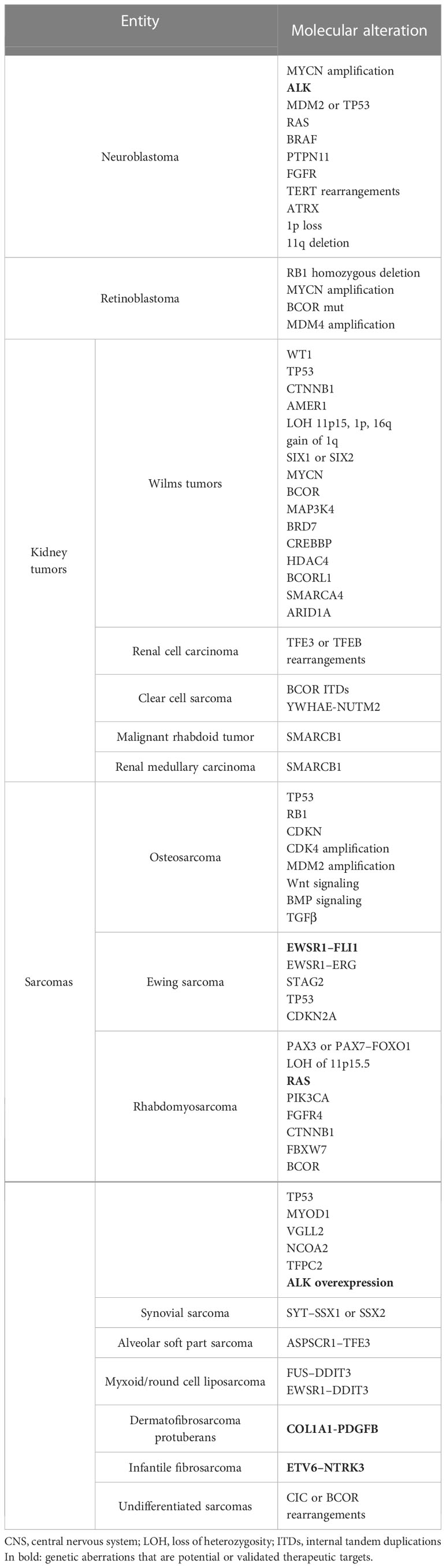
Table 1 Common genetic alterations in pediatric non-CNS solid tumors.
First-line approaches to neuroblastoma consist of chemotherapy, radiotherapy, and autologous hematopoietic stem cell transplantation (HSCT), while the use of anti-GD2 chimeric antibody as maintenance therapy has proven effective in reducing relapse rates (9) (Figure 2). First-line chemotherapy combines multiple drugs such as etoposide, vincristine, carboplatin, cisplatin, and cyclophosphamide +/- doxorubicin, depending on the patient’s risk category. Such regimens are followed by myeloablative therapy with busulfan and melphalan in high-risk patients (NCT01728155, NCT01704716). The ALK inhibitor crizotinib has already been used with variable efficacy in pediatric solid cancer, including neuroblastoma (10) (Figure 2). At the moment, a phase-III trial comparing iobenguane I-131 meta-iodobenzylguanidine (MIBG) or crizotinib plus standard therapy in high-risk neuroblastoma is ongoing (NCT03126916). Crizotinib is also being evaluated in association with chemotherapy for relapsed neuroblastoma (NCT01606878), while it did not reach adequate response rates in monotherapy (11). Phase-I trials are ongoing using other ALK inhibitors, such as lorlatinib and ceritinib (NCT03107988, NCT01742286) (Table 2). Immune checkpoint inhibitors, such as the anti-PD-1 nivolumab, did not prove effective in monotherapy for relapsed neuroblastoma (35, 36), but their association with both targeted molecules and conventional therapies is being tested in newly diagnosed and relapsed neuroblastoma. Also, trials on chimeric antigen receptor T-cell (CAR-T) based therapies targeting CD171 (NCT02311621) and GD2 (NCT02765243) on relapsed neuroblastoma are ongoing (Table 3). Nowadays, children with neuroblastoma can also benefit from precision diagnostic and therapeutic trials such as the PEDS-PLAN (NCT02559778) and the NEPENTHE (NCT02780128) studies (Figure 1).
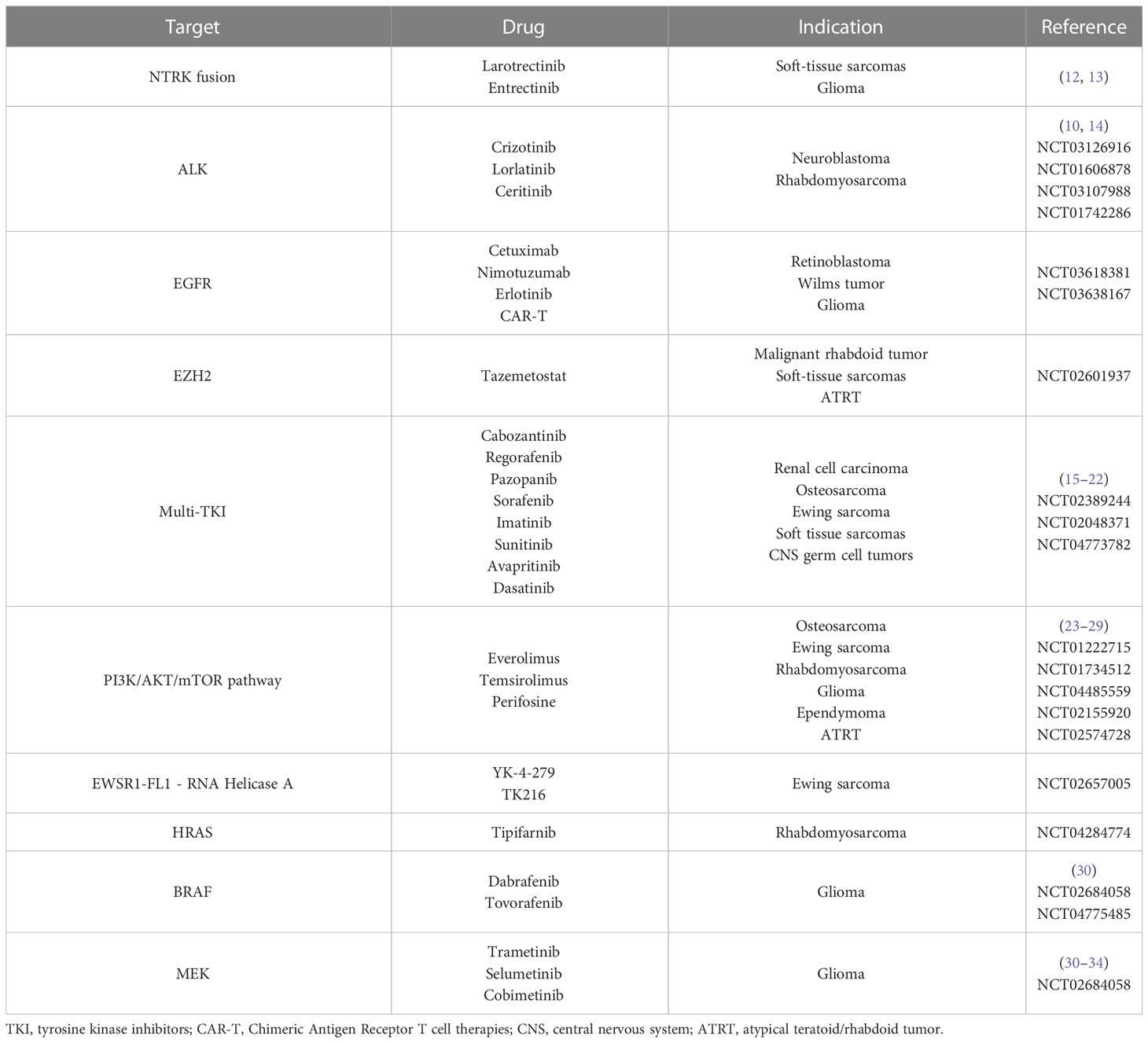
Table 2 Targeted therapies towards specific mutations.
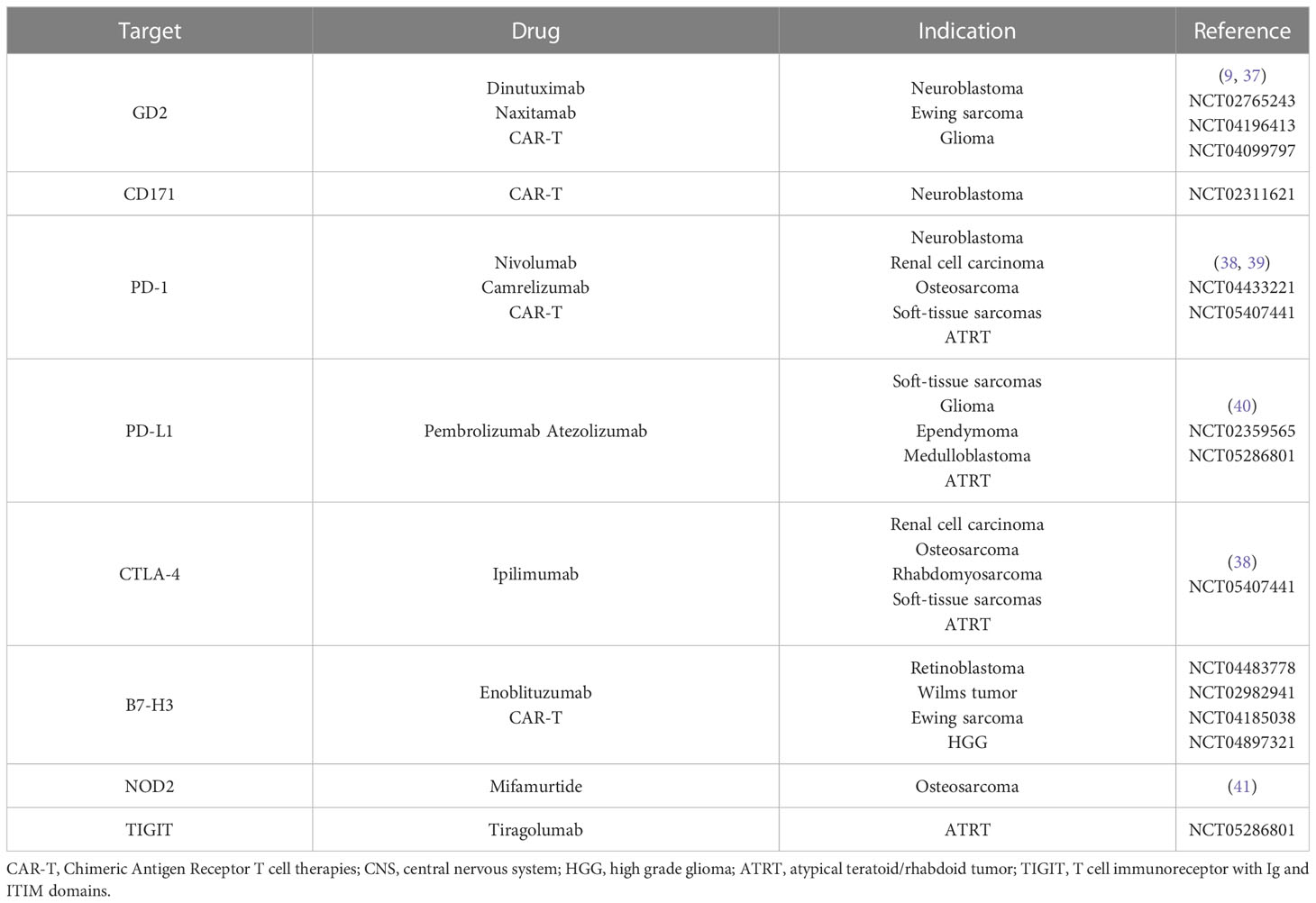
Table 3 Immune-based therapies.
Retinoblastoma
Retinoblastoma is a rare tumor of retinal progenitor cells that accounts for around 2-3% of childhood cancer (42). Biallelic mutations inactivating RB1 are the most common drivers of both sporadic and familial retinoblastoma, but also MYCN amplification and BCOR mutation can be involved (43); less frequently, copy number alterations (e.g., involving MDM4) are found (44) (Table 1).
The treatment of retinoblastoma relies on a multimodal approach based on risk stratification (depending on disease staging, extraocular involvement, and germline mutations) and available institutional resources (45). Adjuvant intravenous chemotherapy with vincristine, etoposide, and carboplatin can be administered in patients with high-risk features (46), and new approaches include intra-arterial delivery of chemotherapy agents such as melphalan, topotecan, or carboplatin (47). However, despite the thorough molecular profiling of retinoblastoma, targeted approaches have not been investigated, except for CAR-T-based therapies directed against EGFR806 (NCT03618381) or B7H3 (NCT04483778) in the context of large-spectrum phase-I trials (Tables 2, 3).
Kidney tumors
Wilms tumor (WT), also known as nephroblastoma, is the most common kidney tumor in childhood (90%). Less common pediatric kidney cancers include renal cell carcinoma (RCC) (5%), clear-cell sarcoma of the kidney (CCSK) (3.5%), congenital mesoblastic nephroma (4%), malignant rhabdoid tumor (MRT) (1.5%), and other rare cancers such as cystic nephroma and metanephric tumors (2%) (48).
Wilms tumor
Aberrations of WT1 and TP53, Wnt pathway activating mutations involving CTNNB1 and AMER1, and loss of heterozygosity (LOH) of 11p15 resulting in overexpression of IGF2 are known to be associated with WT. Abnormalities of 11p15 methylation, as well as 1q gain, LOH of 1p, and 16q were shown to be prognostic biomarkers for inferior survival (49). Recently, whole-exome sequencing analyses identified novel mutations involving microRNA processing genes, renal developmental genes SIX-1 and SIX-2, MYCN, histone modification mediators such as BCOR, MAP3K4, BRD7, CREBBP, and HDAC4, transcriptional repressors such as BCORL1 and epigenetic remodelers SMARCA4 and ARID1A (50, 51) (Table 1).
Standard treatment of WT includes surgery, pre- and post-operative chemotherapy, and radiation in advanced-stage disease and in intermediate or high-risk histology. According to the International Society of Paediatric Oncology (SIOP) protocol, first-line chemotherapy consists of a combination of vincristine, dactinomycin, and doxorubicin; etoposide and cyclophosphamide are used in the Children’s Oncology Group (COG) protocol (52). Several molecules, such as the IGF-1R inhibitor cixutumumab, the multi-tyrosine kinase inhibitors (TKIs) sorafenib and cabozantinib, and the aurora A kinase inhibitor alisertib, were used in children and young adults with refractory solid tumors including WT, but no relevant clinical activity was demonstrated (53–55). A phase I trial is ongoing to evaluate the efficacy of vorinostat, a histone deacetylase inhibitor, in combination with standard chemotherapy in patients with recurrent and refractory solid tumors such as WT (NCT04308330). Also, the combination of antibodies directed against tumor cells antigens and an anti-cancer drug is being studied in WT. A phase II trial of the anti-CD56 antibody lorvotuzumab linked with the anti-mitotic agent mertansine showed good tolerability, but results on efficacy are still expected (NCT02452554) (Table 4). Recently, some interest has been addressed regarding B7-H3 (CD276), a checkpoint molecule that was found to be overexpressed in WT and probably related to unfavorable prognosis; promising preclinical data on the anti-B7-H3 antibody-drug conjugate are available in xenografts models of different pediatric solid tumors including WT (60). Moreover, enoblituzumab, a monoclonal antibody directed against CD276 in children with B7-H3-expressing WT (NCT02982941), and CAR-T cell immunotherapy targeting B7-H3 (NCT04483778) and EGFR (NCT03618381) are being tested in WT (Tables 2, 3).
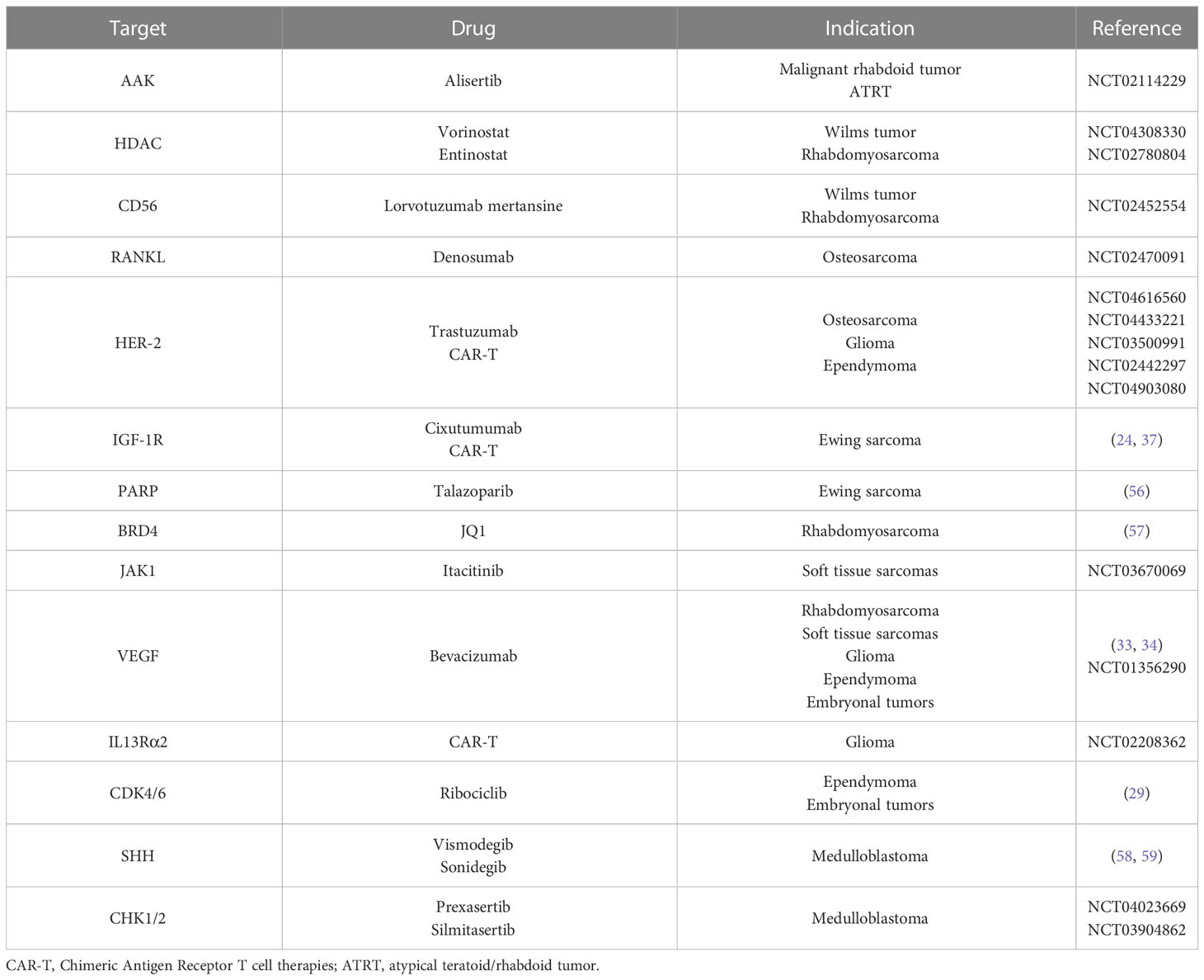
Table 4 Other targeted therapies for pediatric solid tumors.
Other kidney tumors
RCC in children and adolescents is characterized by translocations involving the TFE3 gene, located on chromosome X, or, less frequently, the TFEB gene. TFE3 fusion partners include ASPL, PRCC, SFPQ, and others. The TFEB gene is most commonly fused with the MALAT1 gene (61) (Table 1). RCC treatment consists of surgery and adjuvant therapy. TKIs such as sunitinib and axitinib have been tested in pediatric RCC with promising results (15) (Table 2). The combination of TKIs with immune-checkpoint inhibitors (CPIs) (anti-CTLA-4, anti-PD-1/PD-L1) achieved higher response rates, and it is now recommended as first-line therapy in adults with metastatic RCC. In childhood, CPIs have only been evaluated in early-phase trials showing safety, tolerability, and variable clinical efficacy (38) (Table 3).
CCSK is the third most common pediatric kidney tumor. Internal tandem duplications (ITDs) in the BCOR gene are the prevalent genetic aberrations (70%) in this entity and are mutually exclusive with the less common chromosomal translocation t(10;17), which results in the YWHAE-NUTM2 gene fusion (62). So far, no targeted therapies directed against these molecular features have been developed. MRT is primarily driven by the loss of the SMARCB1 gene (63) (Table 1). Phase I/II studies on the EZH2 inhibitor tazemetostat in children with SMARCB1-deficient solid tumors are underway with promising results (NCT02601937) (Table 2), and preclinical trials are evaluating other potential therapeutic agents, such as aurora A kinase inhibitors, MDM2/4 inhibitors and proteasome inhibitors (48). Renal Medullary Carcinoma (RMC), a non-clear-cell RCC, has been associated with SMARCB1 deficiency (Table 1). Novel therapies effective against MRT may also be useful for this subgroup of RCC.
Sarcomas
Sarcomas are a group of solid tumors developing from mesenchymal cells that can affect bone and soft tissues. Each subtype has a different phenotype and distinct genetic features.
Osteosarcoma
Osteosarcoma is the most common primary malignant bone tumor occurring in children and adolescents. It is characterized by a high level of genomic instability, probably consequent to mutations in genes that are essential for mitotic checkpoints, such as the inactivation of TP53 and the RB1 tumor suppressor genes. Less frequently, loss of CDKN genes and amplification of CDK4 have been reported. In some cases, p53 inactivation indirectly results from MDM2 amplification (64) (Table 1). Gain-of-function mutations in the effectors of PI3K/Akt pathway can be found in a high percentage of osteosarcomas, especially in advanced stages (65). Aberrant expression of genes involved in bone cell differentiation, such as Wnt family and BMP/TGFβ family members, has also been associated with osteosarcomagenesis (66). There is no evidence of reliable molecular prognostic factors for osteosarcoma, but the expression of P‐glycoprotein (Pgp), an efflux pump that removes chemotherapeutic drugs from cells, has been associated with poorer survival in patients affected by osteosarcoma (41).
The standard treatment of osteosarcoma consists of a combination of chemotherapy and surgery, with a poor prognosis for patients with metastatic (usually to the lung) or relapsed disease (66). Methotrexate, doxorubicin, and cisplatin represent the backbone of the medical treatment, and poor responders also receive high-dose ifosfamide (41). In recent years, TKIs have had an increasing role in the treatment of osteosarcoma: cabozantinib in patients with advanced or recurrent osteosarcoma and Ewing sarcoma (16); anlotinib in unresectable or metastatic bone sarcomas (17); regorafenib in recurrent, progressive and metastatic bone sarcomas (NCT02389244); and pazopanib in recurrent osteosarcoma metastatic to the lung (18) (Table 2) (Figure 2). A phase II randomized study is still ongoing to evaluate the efficacy and safety of lenvatinib in combination with chemotherapy in relapsed and refractory osteosarcoma (NCT04154189). mTOR inhibitors have been used in osteosarcomas, showing poor antineoplastic activity as monotherapy (67), probably due to the presence of many feedback loops in the IGF/PI3K/mTOR pathway. Combination strategies co-targeting two or more proteins are being evaluated in order to avoid drug resistance. The multitargeted TKI sorafenib in combination with the mTOR inhibitor everolimus (Figure 2) proved to be effective in unresectable osteosarcoma progressing after standard treatment, but it did not reach the prespecified target of 6-month progression-free survival (PFS) of 50% (23) (Table 2). Similarly, immune checkpoint inhibitors such as anti-PD-1 and anti-CTLA-4 antibodies have been investigated with limited activity when used as a single agent (NCT02406781; ADVL1412) (68), but the combination of the anti-VEGFR apatinib and the PD-1 inhibitor camrelizumab seemed to prolong PFS in comparison to apatinib alone in advanced osteosarcoma but did not achieve the prespecified target of 6-month PFS of 60% (39). Concerning immunotherapies, the Italian Sarcoma Group led a phase II trial showing the benefit of adjuvant mifamurtide – an immune-stimulating compound that promotes macrophage and monocyte antitumor activity – in patients with non-metastatic osteosarcoma expressing P-glycoprotein (Pgp+) (41) (Table 3). Denosumab, a monoclonal antibody directed against RANKL – of which overexpression has been related to poorer outcomes – is being investigated in patients with recurrent or refractory osteosarcoma (NCT02470091). Another phase II trial is evaluating the efficacy of the anti-HER-2 monoclonal antibody trastuzumab linked to the topoisomerase-I inhibitor deruxtecan (NCT04616560) (Table 4). Finally, innovative CAR-T cell based approaches are being evaluated in multiple sarcomas (NCT04433221) (Tables 3, 4). Potential targets include surface antigens overexpressed by osteosarcoma cell lines, such as HER-2 and PD-1.
Ewing sarcoma
Ewing sarcoma (EWS) is the second most common pediatric malignant bone tumor, but it can also occur in soft tissues. The translocation between the EWSR1 gene and the FLI1 gene is the most common (85%), resulting in a fusion product that functions as an oncoprotein. Less frequently, the translocation of EWSR1 involves other members of ETS family transcription factors, such as ERG (10%), or non-ETS family genes, such as NFACT2. A few additional pathogenic alterations have been observed, like loss-of-function mutations involving STAG2, TP53, and CDKN2A genes (69, 70) (Table 1). STAG2 and TP53 mutations have been associated with a dismal prognosis, especially when coexisting, as for the loss of CDKN2A (69). Rearrangement of the CIC and BCOR genes have been implicated in some cases of small round cell sarcomas, also defined as “Ewing-like” because of their clinical and morphological similarities with EWS, but sometimes with a worse prognosis due to a poorer response to treatments (71, 72).
Standard treatment for Ewing sarcoma relies on combined chemotherapy (e.g., vincristine, doxorubicin, ifosfamide, cyclophosphamide, etoposide, and dactinomycin), radiotherapy, and surgery approach, less frequently including autologous HSCT in cases requiring high-dose chemotherapy with busulfan and melphalan (73). Targeting the EWSR1-FLI1 fusion protein is not easy, due to its structure and the lack of enzymatic activity. However, molecules inhibiting the interaction between EWSR1-FLI1 and the RNA Helicase A might be effective: YK-4-279 has shown promising results in preclinical studies (74, 75), and a phase I clinical trial is ongoing to evaluate the efficacy of TK216 in combination with vincristine (NCT02657005) (Table 2). IGF-1R targeted antibodies have been evaluated in advanced-stage EWS, inducing a short-term response when used as a single agent, but an improvement of PFS when combined with mTOR inhibitors (cixutumumab/temsirolimus) (24, 76). A phase I trial of the PARP inhibitor talazoparib in combination with irinotecan +/- temozolomide showed promising results in recurrent and refractory solid tumors, including EWS (56) (Table 4). The multitargeted TKI cabozantinib demonstrated antitumor activity in patients with advanced or recurrent EWS and osteosarcoma (16), while a phase II trial using regorafenib is still ongoing with promising early results (NCT02048371) (Table 2). Finally, CAR-T cell based therapy is currently being investigated in preclinical trials using in vitro and in vivo xenograft models, showing promising antitumor activity. Potential targets include VEGFR2, IGF1R, ROR1, GD2, B7-H3, EphA2, and NKG2D (37) (Tables 3, 4).
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma in childhood and adolescence. It is classified on the basis of genetic and morphologic features into embryonal, alveolar, spindle cell, and pleomorphic RMS (77). Alveolar RMS (ARMS) is the second most common subtype (20%) and it is usually characterized by the translocation between PAX and FOXO1 genes: PAX3-FOXO1 is the most common (75%), while PAX7-FOXO1 occurs in 10% of cases. The alveolar histologic subtype is an unfavorable prognostic factor that classifies the patient within the very high-risk group. Embryonal rhabdomyosarcoma (ERMS), the most frequent subtype (70-80%), has a wider range of genetic aberrations and a higher mutation burden compared to ARMS. The most common chromosomal aberration – in up to 50% of cases – is the loss of heterozygosity at 11p15.5. Various mutations involve the RTK/RAS/PIK3CA pathway, including RAS (approximately 25% of cases of fusion-negative RMS), PIK3CA, and FGFR4. Also, cell cycle regulatory genes and tumor suppressors were found to be altered, including CTNNB1, FBXW7, BCOR and TP53 (78). Spindle cell RMS (ssRMS) often harbors MYOD1 mutation, which is associated with poor prognosis (79). Other recurrent aberrations include gene fusions involving VGLL2, NCOA2, and TFPC2 (80, 81). Furthermore, ALK overexpression has been reported, especially in ARMS (82) (Table 1).
The standard treatment of RMS includes surgery, chemotherapy, and radiation therapy (83). The first-line chemotherapy regimens often include vincristine, dactinomycin, and ifosfamide, with or without doxorubicin. In high-risk groups, maintenance chemotherapy consisting of vinorelbine and cyclophosphamide showed an improvement in overall survival (84). Therapies that directly inhibit PAX-FOXO1 fusion protein with good specificity and affinity are yet to be designed. A promising strategy is to target BRD4, an epigenetic reader that mediates PAX-FOXO1 transcription through the novel molecule JQ1, which reduces the expression of the oncogenic fusion protein (57) (Table 4). Various receptor tyrosine kinase (RTK) inhibitors have been tested in RMS. An ongoing phase II trial is evaluating the efficacy of tipifarnib, an indirect HRAS inhibitor, in pediatric patients with advanced or recurrent HRAS mutated solid tumors (NCT04284774). The mTOR inhibitor temsirolimus has been tested in relapsed RMS with a satisfying antitumor response, achieving superior event-free survival rates compared with bevacizumab (NCT01222715). A phase II clinical trial is ongoing exploring the ALK inhibitor crizotinib in patients with advanced tumors induced by causal alterations of either ALK or MET (2011-001988-52), while there are promising preclinical data on the ALK inhibitor ceritinib combined with dasatinib, an Src family kinase inhibitor (14) (Table 2). Conversely, no sustained response has been achieved by targeting IGF-1R. The addition of the anti-IGF-1R cixutumumab to multiagent chemotherapy for metastatic RMS did not improve survival (85), while a phase II trial studying the efficacy of the IGF-1R monoclonal antibody ganitumab in combination with dasatinib in relapsed and refractory RMS (NCT03041701) was closed early due to lack of the study drug. As discussed for WT, since CD56 is expressed on several tumors cells, the antibody-drug conjugate lorvotuzumab mertansine was evaluated in recurrent solid cancers including RMS but results on efficacy are pending (NCT02452554), as is for entinostat, an oral histone deacetylase inhibitor that has been evaluated in a phase I trial in pediatric patients with recurrent or refractory solid tumors (NCT02780804) (Table 4).
Non-rhabdomyosarcoma soft tissue sarcomas
The non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) group includes multiple histological variants, and pathognomonic chromosomal aberrations have been identified in certain subtypes. Synovial sarcoma has been associated with SYT-SSX1/2 translocation (86). Alveolar soft part sarcoma (ASPS) often carries a translocation between ASPSCR1 and TFE3 genes, resulting in a fusion protein that transcriptionally upregulates MET (87). Myxoid/Round cell Liposarcoma (MRLPS) represents 20-30% of LPS and it is the only subtype described in childhood and adolescence. Round cell LPS is defined as having more than 5% of small round cells in a myxoid LPS. Most MRLPSs carry a pathognomonic translocation between the FUS gene and the DDIT3 gene (also known as CHOP), whereas a smaller proportion is associated with EWSR1-DDIT3 translocation. Overexpression of p53 in myxoid LPS has been associated with poor prognosis (88). Dermatofibrosarcoma protuberans is characterized by a COL1A1-PDGFβ translocation in up to 90% of cases, resulting in autocrine stimulation of the PDGF receptor (89). The ETV6-NTRK3 gene fusion is pathognomonic for infantile fibrosarcoma (70-100% of cases) (90) (Table 1).
Surgery remains the mainstay of treatment for NRSTS, while radiation and chemotherapy with doxorubicin and ifosfamide can be administered as a neoadjuvant or adjuvant treatment to improve the efficacy of surgery or in patients deemed at high risk for metastasis (91). Although multi-TKI pazopanib (Figure 2) is not approved for many STSs such as LPS, RMS apart from alveolar and pleomorphic subtypes, and dermatofibrosarcoma protuberans, it is occasionally used off-label based on published studies on its antitumor activity (19). Other TKIs such as sunitinib and cediranib also achieved tumor responses or disease stabilization in ASPS (20) (Table 2). Unlike for other NRSTSs, the anti-PD-L1 antibody pembrolizumab (Figure 2) has shown some efficacy in ASPS. Indeed, the combination of immune checkpoint inhibitors with anti-angiogenic therapies has achieved significant improvements in response rates (40) (Table 3). Similarly, the TRK inhibitor larotrectinib (Figure 2) showed encouraging antitumor activity in pediatric patients with TRK fusion-positive tumors, including STSs (12). Moreover, the PDGFR inhibitor imatinib mesylate (Figure 2) proved to be effective in recurrent, unresectable, and metastatic dermatofibrosarcoma protuberans, mainly in patients with a t(17;22) translocation (21). Also, interim results from a phase I study of the EZH2 inhibitor tazemetostat in relapsed or refractory INI1-negative tumors (e.g., epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, dedifferentiated chordoma) or synovial sarcoma showed promising anti-tumor activity (NCT02601937) (Table 2). An ongoing phase I trial is evaluating the efficacy of the JAK1-selective inhibitor itacitinib in patients with refractory advanced or metastatic sarcomas (NCT03670069) (Table 4). Finally, a modified T cell receptor (TCR) based immunotherapy directed against NY-ESO-1, which is expressed in 90% of MRLPS tumors, has shown promising preliminary results in adults (NCT02992743).
Central nervous system
As a group, central nervous system (CNS) tumors are the most common solid neoplasm during childhood and the leading cause of cancer-related mortality in this age group. Among CNS tumors, gliomas account for approximately 50% of cases in children aged 0-14 years (92); they include several histological variants such as low and high-grade gliomas, other astrocytomas, ependymomas, and oligodendrogliomas.
Low-grade gliomas
Low-grade gliomas (LGGs) are the most common pediatric brain tumors. LGGs usually occur sporadically, but they can be associated with cancer-predisposition syndromes such as Neurofibromatosis type 1 (NF1) and Tuberous Sclerosis (TS) (93). The presence of NF1 has been reported as a favorable prognostic factor in optic pathway glioma (33). LGGs often carry BRAF gene fusions (e.g., KIAA1549-BRAF) or activating mutations (e.g., BRAF V600E), NF1 mutations, RAF fusions, FGFR1 mutation or rearrangement, impacting both the RAS/MAPK and PI3K/AKT/mTOR pathways. The BRAF V600E mutation seems to correlate with a poorer prognosis across a broad spectrum of pediatric LGG (34). Gangliogliomas, a subset of glioneuronal tumors, often harbor the activating BRAF V600E mutation as well (94). Rearrangements of MYB or MYBL1 occur most frequently in diffuse LGGs (95, 96) (Table 5).
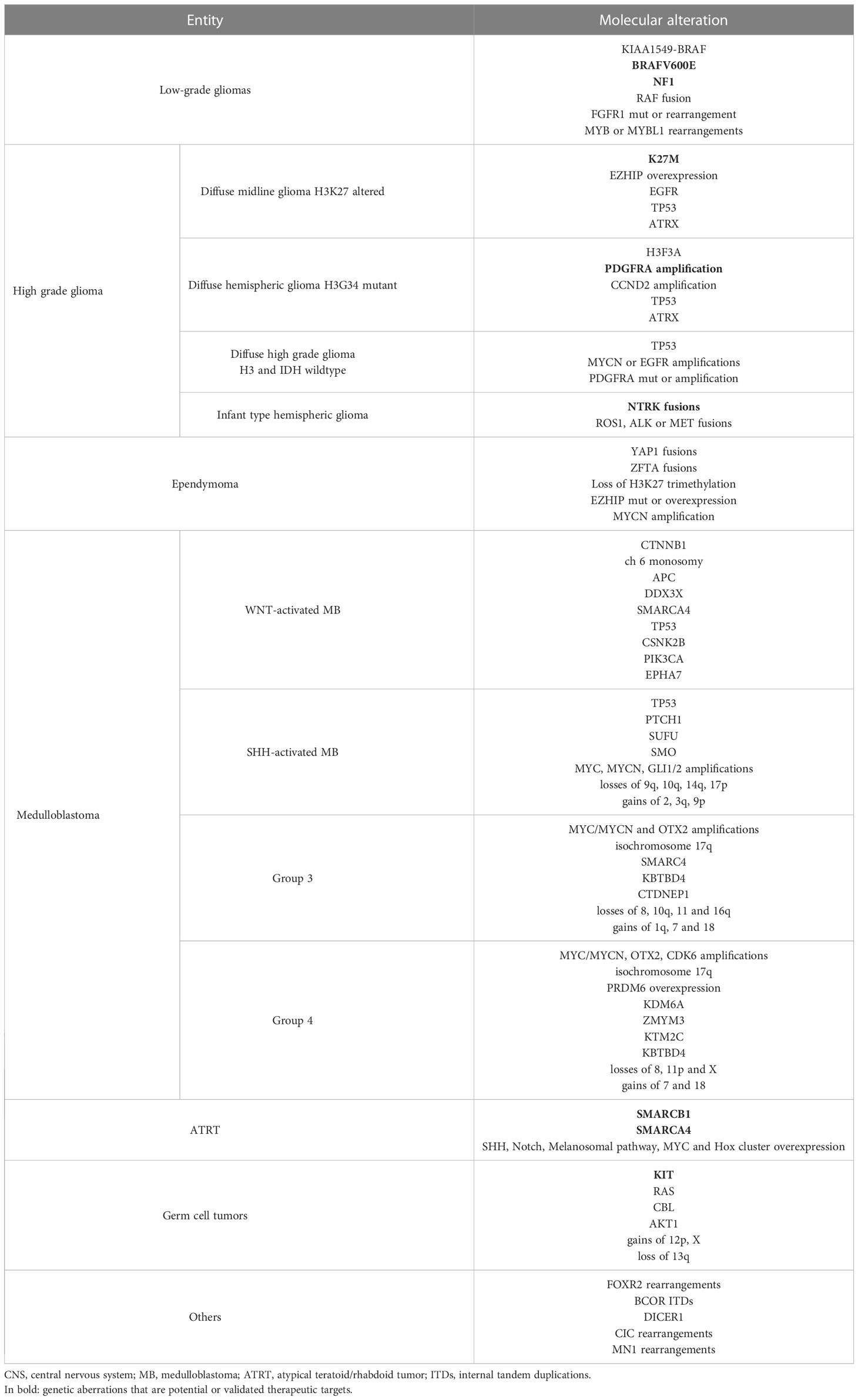
Table 5 Common genetic alterations in pediatric CNS solid tumors.
The mainstay of LGGs treatment is complete surgical resection. When radical surgery is not feasible, chemotherapy or radiotherapy may be used to treat the residual lesions. The standard chemotherapy regimens recommended for pLGGs include carboplatin and vincristine, or vinblastine monotherapy (97). Recently, the Food and Drug Administration (FDA) approved the use of the BRAF inhibitor dabrafenib in combination with the MEK inhibitor trametinib (Figure 2) for pediatric patients with BRAF V600E mutated LGG, based on a large phase II open label trial (NCT02684058). A phase II multicentre trial showed some efficacy of the MEK inhibitor selumetinib in recurrent, refractory, and progressive pLGG carrying BRAF aberrations and NF1 mutations (31). Cobimetinib, another MEK inhibitor (Figure 2), proved to be safe and effective in LGGs with MAPK pathway activation (32). Recently, the pan-RAF inhibitor tovorafenib (DAY101), provided encouraging response data in pediatric and young adult pretreated patients with recurrent or progressive low-grade glioma or advanced solid tumors harboring a known activating BRAF alteration (NCT04775485). The selective mTOR inhibitor everolimus (Figure 2) is approved for TS-associated subependymal giant cell astrocytoma and is also tolerable and effective in terms of disease stabilization in sporadic pLGGs (NCT01734512) (25) (Table 2). Finally, the treatment with bevacizumab, an antibody directed against VEGF, has shown good short-term disease control, even if several patients progressed after the drug discontinuation (98, 99) (Table 4).
High-grade gliomas
High-grade gliomas (HGGs) are grade III-IV tumors that still have a very poor prognosis. According to the 2021 World Health Organization (WHO) classification of CNS tumors, pediatric diffuse high-grade gliomas include several categories of which the two major entities are diffuse midline glioma (DMG) H3K27 altered and diffuse hemispheric glioma H3G34 mutant (100).
Concerning the first subgroup, the loss of H3K27 trimethylation can result from K27M mutations, EZHIP overexpression, or EGFR mutations (101). Of note, H3K27 mutations have been associated with poor prognosis (102). Of note, the DMG entity now includes diffuse intrinsic pontine gliomas in addition to diffuse gliomas arising in other midline locations. The second main entity, diffuse hemispheric glioma, is typically characterized by a missense mutation in the H3F3A gene. PDGFRA and CCND2 amplifications are less common and are both associated with poor outcomes. TP53 and ATRX mutations are detected in almost all cases of diffuse hemispheric glioma H3G34 mutant, but they can also occur in H3K27-mutated diffuse midline gliomas (103). The H3- and IDH-wildtype diffuse HGG represents an additional entity, which by definition lacks alterations in histone H3, IDH1, and IDH2 genes. This group exhibits great molecular heterogeneity, including TP53 mutations, MYCN and EGFR amplifications, and PDGFRA mutation or amplification (103). MYCN-mutated high-grade gliomas are associated with a poor prognosis. Finally, the infant-type hemispheric glioma category often carries RTK fusions involving the NTRK, ROS1, ALK, and MET genes, while BRAF V600E mutations are observed in 10-15% of pediatric HGGs (102, 103) (Table 5).
The standard treatment of HGG Is based on surgical resection followed by radiation therapy +/- concurrent chemotherapy (104). Temozolomide is the most commonly used conventional drug for newly diagnosed pHGGs (102). Many TKIs have been investigated in HGGs but in most cases did not provide sufficient disease control when investigated in monotherapy (105, 106). However, the multi-TKI entrectinib showed durable responses in children with solid tumors with NTRK1/2/3 or ROS1 fusions, including primary brain tumors (13). A phase I/II multicenter trial of avapritinib in pediatric relapsed and refractory solid tumors harboring mutations in KIT or PDGFRA and H3K27 altered gliomas is recruiting (NCT04773782). Moreover, preliminary results of recent ongoing trials suggest promising results when TKIs are used in combination with other targeted therapies, such as the mTOR inhibitor everolimus (26) (NCT04485559). In addition, a phase I study demonstrated the feasibility of the combination therapy with temsirolimus and the AKT inhibitor perifosine in recurrent and refractory pediatric solid tumors, including HGGs (27) (Table 2). On the other hand, the EGFR inhibitors cetuximab and nimotuzumab (Figure 2) were evaluated in addition to standard treatment in pediatric HGGs, but both were not able to markedly improve overall survival compared to controls (107, 108). A phase II trial has evaluated the use of the BRAF inhibitor dabrafenib in combination with the MEK inhibitor trametinib (Figure 2) in children and adolescents with relapsed and refractory BRAF-mutated high-grade glioma (NCT02684058). The efficacy and safety of this combination therapy in these patients have already been reported in some case series (30) (Table 2). Finally, multiple trials are evaluating CAR-T cell immunotherapy targeting B7-H3-expressing pontine DMG (NCT04185038) or solid tumors (NCT04897321), HER-2 in recurrent or refractory HER2-positive CNS tumors (NCT03500991, NCT02442297), IL13Rα2 in recurrent or refractory malignant gliomas (NCT02208362), EGFR in pediatric recurrent or refractory EGFR positive CNS tumors (NCT03638167), and GD2-expressing CNS tumors including DMG (NCT04196413, NCT04099797) (Tables 2–4).
Ependymoma
Based on its anatomic location, ependymoma can be classified into three main groups: supratentorial, posterior fossa, and spinal cord (109). Each of these groups consists of different clinical, genetic, and histopathological subtypes. Supratentorial ependymomas generally have a more favorable prognosis. According to the 2021 WHO classification of CNS tumors, there are two main subsets of supratentorial ependymoma, namely the ZFTA (also called C11orf95) and the YAP1 fusion positive supratentorial ependymomas. Regarding the first entity, the ZFTA-RELA fusion protein is the most frequently identified alteration. Those that have neither the ZFTA nor the YAP1 fusion constitute a different supratentorial ependymoma subset (100). Posterior fossa ependymomas are distinguished into PFA (pediatric type) and PFB, which mainly occur in older children and in adults, and have a better prognosis. Posterior fossa ependymomas lack recurrent mutations. However, PFA often exhibits loss of H3K27 trimethylation and overexpression of EZHIP. Among spinal cord ependymomas, MYCN amplification defines a distinctive subtype with poorer outcomes (109, 110) (Table 5).
Surgical resection followed by radiation therapy to the tumor bed is the pivotal treatment of ependymomas and often guarantees a good long-term prognosis. Current treatment approaches do not include chemotherapy in most cases. However, there are few treatment options for recurrent disease besides re-irradiation (111). Only a few targeted therapies have been investigated in ependymoma. The role of antiangiogenic agents in pediatric brain tumors remains controversial, due to conflicting results (112, 113). A phase II study, aiming to evaluate the efficacy of the anti-VEGF antibody bevacizumab in children with recurrent and progressive medulloblastoma, ependymoma, or the atypical teratoid rhabdoid tumor (ATRT) is recruiting (NCT01356290) (Table 4). The use of mTOR inhibitors in combination proved to be safe and was able to stabilize disease progression in some children with recurrent or refractory brain ependymoma (28, 29). Further phase II trials are ongoing to assess their anti-tumor activity (NCT02155920; NCT02574728) (Table 2). The EGFR inhibitor erlotinib was compared with etoposide in a phase II study in pediatric patients with recurrent ependymoma, but its efficacy was limited (114). A phase I trial is evaluating the safety profile and efficacy of HER2-targeting CAR-T cell therapy in recurrent and progressive ependymoma (NCT04903080) (Table 4). Similarly, a phase I study of the anti-PD-L1 pembrolizumab (Figure 2) in younger patients with recurrent and refractory HGG, ependymoma, and medulloblastoma is recruiting (NCT02359565) (Table 3). Other interesting targets for specific molecules include EZHIP, PARP, HDAC, the chemokine receptor CXCR4, the RAF/MEK/ERK pathway for NF2-associated tumors, and the Wnt-β-catenin (whose activation seems to be promoted by YAP1) and RELA pathways (since RELA mediates the activation of the NFkB pathway) (115).
Embryonal tumors
Embryonal tumors account for 10-15% of primary CNS tumors in children and adolescents. Medulloblastoma (MB) is the most common, representing about 70% of embryonal tumors (92). The WHO 2021 classification distinguishes various molecular and histological subgroups. Among them, WNT-activated MB has a good long-term prognosis, since it is usually responsive to the currently available treatments (100). It harbors CTNNB1 somatic mutations and chromosome 6 monosomy in almost all cases (80-90%) (116). APC pathogenic variants are generally identified in CTNNB1 wild-type tumors, explaining the WNT pathway activation (117). Other recurrent mutations can affect the DDX3X, SMARCA4, TP53, CSNK2B, PIK3CA, and EPHA7 genes (118). SHH-activated MB is more common in infants and adults and has an intermediate prognosis. The WHO 2021 classification distinguishes SHH-driven MB into TP53-mutant or wildtype. The activation of the SHH signaling pathway represents the most common genetic event, caused by mutations or deletions in PTCH1 and SUFU genes, SMO activating mutations, MYC/MYCN or GLI1/GLI2 amplifications. In addition, alterations in p53 and PI3K pathways can drive tumorigenesis. MYC/MYCN amplification and TP53 mutations have been related to poor prognosis (119). Other frequent chromosomal alterations include the loss of chromosomes 9q (causing loss of heterozygosity of PTCH1), 10q, 14q, and 17p, and gains of chromosomes 2, 3q, and 9p (120, 121). A common driver pathway that defines group-3 and -4 MBs has not yet been identified. However, these subtypes share some genetic aberrations with the WNT and SHH subtypes. Distinctive features include OTX2 and CDK6 amplifications, SMARC4, KBTBD4, CTDNEP1, KDM6A, ZMYM3 and KTM2C mutations, PRDM6 overexpression (group 4), isochromosome 17q (present in about 50% of cases in both subgroups), loss of chromosomes 8, 10q, 11p, 16q and X and gain of chromosomes 1q, 7, and 18 (118, 121) (Table 5).
Standard treatment of MB consists of surgical tumor resection with craniospinal irradiation (except in infants) and chemotherapy, depending on risk stratification. The first-line chemotherapy consists of cisplatin, vincristine, and cyclophosphamide, while in MB metastatic at diagnosis, the combination of cyclophosphamide, vincristine, methotrexate, carboplatin, etoposide, and concomitant intraventricular methotrexate allowed achieving acceptable survival rates (122). The use of targeted approaches is rapidly evolving. Smoothened inhibitors (SMOi), such as vismodegib and sonidegib, have shown temporary activity in SHH-activated MB, but they have been associated with severe growth deceleration due to premature growth plate fusion, restricting their use to older adolescents and young adults (58, 59). Prexasertib, a CHK1/2 inhibitor, is being investigated in combination with chemotherapy in pediatric refractory or recurrent group 3, group 4, and SHH-activated MB (NCT04023669). A phase I/II trial to evaluate the CHK2 inhibitor silmitasertib in children with recurrent, progressive, or refractory SHH-activated MB is recruiting (NCT03904862) (Table 4).
ATRT is a rare CNS embryonal tumor that usually affects children in the first years of life. It has a poor prognosis since it usually grows fast and spreads through the cerebrospinal fluid (123). The main recurrent molecular aberration of ATRT is biallelic loss of function of SMARCB1, resulting from pathogenic variants, mutations, or partial or whole loss of chromosome 22. Rare cases (<5%) of SMARCB1-wildtype ATRT generally harbor SMARCA4 mutations (124) (Table 5). Based on DNA methylation profiling and gene expression, other recurrent molecular features have been identified and related to three ATRT subtypes, namely the overexpression of the SHH and Notch pathways (ATRT-SHH), the upregulation of the melanosomal pathway (ATRT-TYR), and the overexpression of the MYC oncogene and the Hox cluster (ATRT-MYC) (125).
The most common approach to ATRT is an aggressive multimodal treatment consisting of maximal safe surgical resection, followed by chemotherapy +/- radiotherapy. Considering its toxicity, various trials have been carried out with the goal of avoiding radiation therapy, especially in children <3 years (126). Standard chemotherapy is mainly based on two regimens, the first including etoposide, vincristine, cisplatin and cyclophosphamide, the second including etoposide, vincristine, carboplatin and ifosfamide. Additional high-dose chemotherapy (carboplatin + thiotepa + etoposide and cyclophosphamide + melphalan) followed by autologous HSCT is usually administered in high-risk patients (127). A phase I study of tazemetostat, a selective EZH2 inhibitor, in children with relapsed or refractory SMARCB1-negative tumors has provided promising interim results (NCT02601937) (Table 2). Another phase I/II trial of a combination regimen (i.e., tazemetostat, the anti-PD-1 nivolumab, and the anti-CTLA-4 ipilimumab) in SMARCB1 or SMARCA4-deficient neoplasms has just been designed (NCT05407441) (Table 3). The CDK4/6 inhibitor ribociclib was evaluated in combination with the mTOR inhibitor everolimus in children with recurrent, progressive or refractory brain cancers and was shown to be well tolerated (29), while a phase II trial of alisertib, an aurora A kinase inhibitor, is recruiting (NCT02114229) (Table 4). Furthermore, a phase I/II study will evaluate the efficacy of a combination regimen with the immune checkpoint inhibitors atezolizumab (anti-PD-L1) and tiragolumab (a novel anti-T-cell immunoreceptor with Ig and ITIM domains, TIGIT) in relapsed and refractory SMARCB1 or SMARCA4-deficient tumors (NCT05286801) (Table 3).
Since recurrent CNS neoplasms are often genetically distinct from the primary one and resistant to treatments, a phase I trial is ongoing to evaluate rational combination therapies in refractory, relapsed, or recurrent brain tumors, based on tumor type and molecular characteristics (NCT03434262).
Germ cell tumors
Germ cell tumors (GCTs) represent approximately 3% of pediatric primary CNS tumors worldwide but have higher incidence rates in East Asia, especially Japan and South Korea (92). They have traditionally been classified into germinoma, which accounts for 50-70% of cases, and non-germinomatous GCTs, which include many entities with a variable prognosis. The histopathological, molecular, and therapeutic features of intracranial GCTs are similar to the extracranial ones (128). The KIT/RAS and AKT/mTOR pathways are commonly involved. KIT mutations represent the most frequent molecular feature, followed by KRAS, NRAS, and CBL mutations, all resulting in KIT overexpression (129). Some studies reported that KIT/RAS pathway mutations were significantly more frequent in germinomas and in male patients (130). AKT1 copy number gains were found in a high percentage of tumors with wildtype KIT, KRAS, and NRAS (129). Chromosomal instability is also common in intracranial GCTs. Gains of chromosomes 12p or X, and loss of 13q have been also seldom described and were found to significantly worsen prognosis (131) (Table 5).
The treatment of CNS GCTs combines the use of multiple chemotherapy agents, including carboplatin, etoposide, ifosfamide, and cyclophosphamide, and radiation therapy, while surgery plays a less established role, apart from non germinomatous tumors (132). So far, the field of targeted therapies has been poorly explored in GCTs. The TKI imatinib (Figure 2) was evaluated in children with recurrent or refractory CNS tumors expressing KIT and/or PDGFRA, proving to be safe but not particularly effective (133). A retrospective study analyzed the feasibility and tolerability of dasatinib (Figure 2), another TKI with improved CNS penetration, in patients with newly diagnosed or recurrent CNS germinoma (22), suggesting a potential role in future treatment strategies (Table 2).
Other tumors
In recent years, the use of the term primitive neuroectodermal tumors (PNETs) has been questioned. Advanced molecular analyses revealed that most of PNETs can either be classified into other known CNS tumors (e.g., HGG, ependymoma, embryonal tumors) or in new molecularly defined entities (134). In the 2021 WHO classification, two embryonal tumor subtypes were introduced: CNS neuroblastoma FOXR2-activated and CNS tumor with BCOR internal tandem duplication (100). The primary intracranial sarcoma DICER1-mutant and the CIC-rearranged sarcoma are now included in the mesenchymal tumors group. Another genetically defined new entity is CNS high-grade neuroepithelial tumor with MN1 alteration (135) (Table 5). So far, no specific therapeutic protocols have been developed for these rare CNS tumor subtypes. The presence of distinct molecular features is attractive for the use of targeted drugs, but it requires further evaluation (134).
Conclusions
In the last few decades, we have witnessed a rapid evolution of available options for treating childhood cancer, following the development of multiple molecules targeting specific mutations and pathomechanisms. Novel, molecular alterations have been identified, as well as the role of germline variants in childhood cancer development (up to 10-15% of cases). The identified molecular abnormalities resulted in multiple investigations of new targeted treatments. These drugs were often investigated in monotherapy, which probably limited their efficacy, and combination therapies should be rapidly introduced in clinical investigations. However, some of these agents have already proven useful as add-on therapy and are now included in the standard of care. Nevertheless, we have to acknowledge that many childhood solid cancers remain burdened by high mortality rates and severe sequelae. In the following years, our efforts should be oriented in multiple directions. On one hand, improving patients’ access to tumor profiling, in both high- and low-income countries, will guarantee a deeper understanding of the molecular landscape of childhood cancer. On the other hand, therapeutic efforts should be directed to the validation of available options within structured protocols and to the constant development of new molecules. Finally, a deeper cross-talk among clinicians by implementing multidisciplinary tumor boards (136), and between clinicians and caregivers, would certainly be beneficial.
Author contributions
IB and FP performed the literature review and wrote the manuscript, while AT and CF critically revised it. All authors have read and approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Blattner-Johnson M, Jones DTW, Pfaff E. Precision medicine in pediatric solid cancers. Semin Cancer Biol (2022) 84:214–27. doi: 10.1016/j.semcancer.2021.06.008
2. Jones DTW, Banito A, Grünewald TGP, Haber M, Jäger N, Kool M, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer (2019) 19(8):420–38. doi: 10.1038/s41568-019-0169-x
3. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers (2016) 2:16078. doi: 10.1038/nrdp.2016.78
4. Thompson D, Vo KT, London WB, Fischer M, Ambros PF, Nakagawara A, et al. Identification of patient subgroups with markedly disparate rates of MYCN amplification in neuroblastoma: a report from the international neuroblastoma risk group project. Cancer (2016) 122(6):935–45. doi: 10.1002/cncr.29848
5. Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature (2008) 455(7215):967–70. doi: 10.1038/nature07398
6. Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature (2008) 455(7215):930–5. doi: 10.1038/nature07261
7. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45:279–84. doi: 10.1038/ng.2529
8. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature (2015) 526:700–4. doi: 10.1038/nature14980
9. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi: 10.1056/NEJMoa0911123
10. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a children's oncology group phase 1 consortium study. Lancet Oncol (2013) 14(6):472–80. doi: 10.1016/S1470-2045(13)70095-0
11. Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of crizotinib in patients with ALK-aberrant Relapsed/Refractory neuroblastoma: a children's oncology group study (ADVL0912). Clin Cancer Res (2021) 27(13):3543–8. doi: 10.1158/1078-0432.CCR-20-4224
12. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for pediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicenter, open-label, phase 1/2 study. Lancet Oncol (2018) 19(5):705–14. doi: 10.1016/S1470-2045(18)30119-0
13. Desai AV, Robinson GW, Gauvain K, Basu EM, Macy ME, Maese L, et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro Oncol (2022) 24(10):1776–89. doi: 10.1093/neuonc/noac087
14. van Erp AEM, Hillebrandt-Roeffen MHS, van Houdt L, Fleuren EDG, van der Graaf WTA, Versleijen-Jonkers YMH. Targeting anaplastic lymphoma kinase (ALK) in rhabdomyosarcoma (RMS) with the second-generation ALK inhibitor ceritinib. Target Oncol (2017) 12(6):815–26. doi: 10.1007/s11523-017-0528-z
15. Ambalavanan M, Geller JI. Treatment of advanced pediatric renal cell carcinoma. Pediatr Blood Cancer (2019) 66(8):e27766. doi: 10.1002/pbc.27766
16. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol (2020) 21(3):446–55. doi: 10.1016/S1470-2045(19)30825-3
17. Liu Z, Gao S, Zhu L, Wang J, Zhang P, Li P, et al. Efficacy and safety of anlotinib in patients with unresectable or metastatic bone sarcoma: a retrospective multiple institution study. Cancer Med (2021) 10(21):7593–600. doi: 10.1002/cam4.4286
18. Frankel P, Ruel C, Uche A, Choy E, Okuno S, Somiah N, et al. Pazopanib in patients with osteosarcoma metastatic to the lung: phase 2 study results and the lessons for tumor measurement. J Oncol (2022) 2022:3691025. doi: 10.1155/2022/3691025
19. Thirasastr P, Somaiah N. Overview of systemic therapy options in liposarcoma, with a focus on the activity of selinexor, a selective inhibitor of nuclear export in dedifferentiated liposarcoma. Ther Adv Med Oncol (2022) 14:1–13. doi: 10.1177/17588359221081073
20. Paoluzzi L, Maki RG. Diagnosis, prognosis, and treatment of alveolar soft-part sarcoma: a review. JAMA Oncol (2019) 5(2):254–60. doi: 10.1001/jamaoncol.2018.4490
21. Shah A, Tassavor M, Sharma S, Torbeck R. The various treatment modalities of dermatofibrosarcoma protuberans. Dermatol Online J (2021) 27(6):20. doi: 10.5070/D327654070
22. Osorio DS, Finlay JL, Dhall G, Goldman S, Eisenstat D, Brown RJ. Feasibility of dasatinib in children and adolescents with new or recurrent central nervous system germinoma. Pediatr Blood Cancer (2013) 60(9):E100–2. doi: 10.1002/pbc.24567
23. Grignani G, Palmerini E, Ferraresi V, D'Ambrosio L, Bertulli R, Asaftei SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol (2015) 16(1):98–107. doi: 10.1016/S1470-2045(14)71136-2
24. Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory ewing's sarcoma family tumors. Clin Cancer Res (2012) 18(9):2625–31. doi: 10.1158/1078-0432.CCR-12-0061
25. Wright KD, Yao X, London WB, Kao PC, Gore L, Hunger S, et al. A POETIC phase II study of continuous oral everolimus in recurrent, radiographically progressive pediatric low-grade glioma. Pediatr Blood Cancer (2021) 68(2):e28787. doi: 10.1002/pbc.28787
26. Miklja Z, Yadav VN, Cartaxo RT, Siada R, Thomas CC, Cummings JR, et al. Everolimus improves the efficacy of dasatinib in PDGFRα-driven glioma. J Clin Invest (2020) 130(10):5313–25. doi: 10.1172/JCI133310
27. Becher OJ, Gilheeney SW, Khakoo Y, Lyden DC, Haque S, De Braganca KC, et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr Blood Cancer (2017) 64(7):1–9. doi: 10.1002/pbc.26409
28. Qayed M, Cash T, Tighiouart M, MacDonald TJ, Goldsmith KC, Tanos R, et al. A phase I study of sirolimus in combination with metronomic therapy (CHOAnome) in children with recurrent or refractory solid and brain tumors. Pediatr Blood Cancer (2020) 67(4):e28134. doi: 10.1002/pbc.28134
29. DeWire MD, Fuller C, Campagne O, Lin T, Pan H, Young Poussaint T, et al. Dunkel IJ. A Phase I Surg Study Ribociclib Everolimus Children Recurrent Refractory Malignant Brain Tumors: A Pediatr Brain Tumor Consortium Study Clin Cancer Res (2021) 27(9):2442–51. doi: 10.1158/1078-0432.CCR-20-4078
30. Toll SA, Tran HN, Cotter J, Judkins AR, Tamrazi B, Biegel JA, et al. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget (2019) 10(4):551–7. doi: 10.18632/oncotarget.26560
31. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol (2019) 20(7):1011–22. doi: 10.1016/S1470-2045(19)30277-3
32. Trippett T, Toledano H, Campbell Hewson Q, Verschuur A, Langevin AM, Aerts I, et al. Cobimetinib in pediatric and young adult patients with relapsed or refractory solid tumors (iMATRIX-cobi): a multicenter, phase I/II study. Target Oncol (2022) 17(3):283–93. doi: 10.1007/s11523-022-00888-9
33. Stokland T, Liu JF, Ironside JW, Ellison DW, Taylor R, Robinson KJ, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol (2010) 12(12):1257–68. doi: 10.1093/neuonc/noq092
34. Colli SL, Cardoso N, Massone CA, Cores M, García Lombardi M, De Matteo EN, et al. Molecular alterations in the integrated diagnosis of pediatric glial and glioneuronal tumors: a single center experience. PLoS One (2022) 17(4):e0266466. doi: 10.1371/journal.pone.0266466
35. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol (2020) 21(4):541–50. doi: 10.1016/S1470-2045(20)30023-1
36. Pasqualini C, Rubino J, Brard C, Cassard L, André N, Rondof W, et al. Phase II and biomarker study of programmed cell death protein 1 inhibitor nivolumab and metronomic cyclophosphamide in paediatric relapsed/refractory solid tumours: arm G of AcSé-ESMART, a trial of the European innovative therapies for children with cancer consortium. Eur J Cancer (2021) 150:53–62. doi: 10.1016/j.ejca.2021.03.032
37. Lin Z, Wu Z, Luo W. A novel treatment for ewing's sarcoma: chimeric antigen receptor-T cell therapy. Front Immunol (2021) 12:707211. doi: 10.3389/fimmu.2021.707211
38. Ray S, Jones R, Pritchard-Jones K, Dzhuma K, van den Heuvel-Eibrink M, Tytgat G, et al. Pediatric and young adult renal cell carcinoma. Pediatr Blood Cancer (2020) 67(11):e28675. doi: 10.1002/pbc.28675
39. Xie L, Xu J, Sun X, Guo W, Gu J, Liu K, et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: a single-arm, open-label, phase 2 trial. J Immunother Cancer (2020) 8(1):e000798. doi: 10.1136/jitc-2020-000798
40. You Y, Guo X, Zhuang R, Zhang C, Wang Z, Shen F, et al. Activity of PD-1 inhibitor combined with anti-angiogenic therapy in advanced sarcoma: a single-center retrospective analysis. Front Mol Biosci (2021) 8:747650. doi: 10.3389/fmolb.2021.747650
41. Palmerini E, Meazza C, Tamburini A, Bisogno G, Ferraresi V, Asaftei SD, et al. Phase 2 study for nonmetastatic extremity high-grade osteosarcoma in pediatric and adolescent and young adult patients with a risk-adapted strategy based on ABCB1/P-glycoprotein expression: an Italian sarcoma group trial (ISG/OS-2). Cancer (2022) 128(10):1958–66. doi: 10.1002/cncr.34131
42. Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am (2015) 62(1):201–23. doi: 10.1016/j.pcl.2014.09.014
43. Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol (2013) 14(4):327–34. doi: 10.1016/S1470-2045(13)70045-7
44. Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature (2006) 444(7115):61–6. doi: 10.1038/nature05194
45. Ancona-Lezama D, Dalvin LA, Shields CL. Modern treatment of retinoblastoma: a 2020 review. Indian J Ophthalmol (2020) 68(11):2356–65. doi: 10.4103/ijo.IJO_721_20
46. hévez-Barrios P, Eagle RC Jr, Krailo M, Piao J, DM A, Gao Y, et al. Study of unilateral retinoblastoma with and without histopathologic high-risk features and the role of adjuvant chemotherapy: a children's oncology group study. J Clin Oncol (2019) 37(31):2883–91. doi: 10.1200/JCO.18.01808
47. Schaiquevich P, Francis JH, Cancela MB, Carcaboso AM, Chantada GL, Abramson DH. Treatment of retinoblastoma: what is the latest and what is the future. Front Oncol (2022) 12:822330. doi: 10.3389/fonc.2022.822330
48. Jain J, Sutton KS, Hong AL. Progress update in pediatric renal tumors. Curr Oncol Rep (2021) 23(3):33. doi: 10.1007/s11912-021-01016-y
49. Pater L, Melchior P, Rübe C, Cooper BT, McAleer MF, Kalapurakal JA, et al. Wilms tumor. Pediatr Blood Cancer (2021) 68 Suppl 2:e28257. doi: 10.1002/pbc.28257
50. Gadd S, Huff V, Walz AL, Ooms AHAG, Armstrong AE, Gerhard DS, et al. A children's oncology group and TARGET initiative exploring the genetic landscape of wilms tumor. Nat Genet (2017) 49(10):1487–94. doi: 10.1038/ng.3940
51. Treger TD, Chowdhury T, Pritchard-Jones K, Behjati S. The genetic changes of wilms tumour. Nat Rev Nephrol (2019) 15(4):240–51. doi: 10.1038/s41581-019-0112-0
52. Wang J, Li M, Tang D, Gu W, Mao J, Shu Q. Current treatment for wilms tumor: COG and SIOP standards. World J Pediatr Surg (2019) 2:e000038. doi: 10.1136/wjps-2019-000038
53. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the children's oncology group. Pediatr Blood Cancer (2014) 61(3):452–6. doi: 10.1002/pbc.24605
54. Kim A, Widemann BC, Krailo M, Jayaprakash N, Fox E, Weigel B, et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: a report from the children's oncology group. Pediatr Blood Cancer (2015) 62(9):1562–6. doi: 10.1002/pbc.25548
55. Mossé YP, Fox E, Teachey DT, Reid JM, Safgren SL, Carol H, et al. A phase II study of alisertib in children with Recurrent/Refractory solid tumors or leukemia: children's oncology group phase I and pilot consortium (ADVL0921). Clin Cancer Res (2019) 25(11):3229–38. doi: 10.1158/1078-0432.CCR-18-2675
56. Federico SM, Pappo AS, Sahr N, Sykes A, Campagne O, Stewart CF, et al. A phase I trial of talazoparib and irinotecan with and without temozolomide in children and young adults with recurrent or refractory solid malignancies. Eur J Cancer (2020) 137:204–13. doi: 10.1016/j.ejca.2020.06.014
57. Chang WI, Lin C, Liguori N, Honeyman JN, DeNardo B, El-Deiry W. Molecular targets for novel therapeutics in pediatric fusion-positive non-CNS solid tumors. Front Pharmacol (2022) 12:747895. doi: 10.3389/fphar.2021.747895
58. Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget (2017) 8(41):69295–302. doi: 10.18632/oncotarget.20619
59. Kieran MW, Chisholm J, Casanova M, Brandes AA, Aerts I, Bouffet E, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol (2017) 19(11):1542–52. doi: 10.1093/neuonc/nox109
60. Kendsersky NM, Lindsay J, Kolb EA, Smith MA, Teicher BA, Erickson SW, et al. The B7-H3-Targeting antibody-drug conjugate m276-SL-PBD is potently effective against pediatric cancer preclinical solid tumor models. Clin Cancer Res (2021) 27(10):2938–46. doi: 10.1158/1078-0432.CCR-20-4221
61. Caliò A, Marletta S, Brunelli M, Pedron S, Portillo SC, Segala D, et al. TFE3 and TFEB-rearranged renal cell carcinomas: an immunohistochemical panel to differentiate from common renal cell neoplasms. Virchows Arch (2022) 481(6):877–91. doi: 10.1007/s00428-022-03380-x
62. Aldera AP, Pillay K. Clear cell sarcoma of the kidney. Arch Pathol Lab Med (2020) 144(1):119–23. doi: 10.5858/arpa.2018-0353-RS
63. Pawel BR. SMARCB1-deficient tumors of childhood: a practical guide. Pediatr Dev Pathol (2018) 21(1):6–28. doi: 10.1177/1093526617749671
64. Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma (2012) 2012:627254. doi: 10.1155/2012/627254
65. Zhang J, Yu XH, Yan YG, Wang C, Wang WJ. PI3K/Akt signaling in osteosarcoma. Clin Chim Acta (2015) 444:182–92. doi: 10.1016/j.cca.2014.12.041
66. Rathore R, Van Tine BA. Pathogenesis and current treatment of osteosarcoma: perspectives for future therapies. J Clin Med (2021) 10(6):1182. doi: 10.3390/jcm10061182
67. Truong DD, Lamhamedi-Cherradi SE, Ludwig JA. Targeting the IGF/PI3K/mTOR pathway and AXL/YAP1/TAZ pathways in primary bone cancer. J Bone Oncol (2022) 33:100419. doi: 10.1016/j.jbo.2022.100419
68. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res (2016) 22(6):1364–70. doi: 10.1158/1078-0432.CCR-15-0491
69. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discovery (2014) 4(11):1342–53. doi: 10.1158/2159-8290.CD-14-0622
70. Grünewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, et al. Ewing Sarcoma. Nat Rev Dis Primers (2018) 4(1):5. doi: 10.1038/s41572-018-0003-x
71. Song MJ, Cho KJ, Kim TI, Kim W, Song JS. Distinct histologic and genetic characteristics of round cell sarcoma with CIC-DUX4 fusion and comparison with Ewing sarcoma. Pathol Res Pract (2022) 231:153779. doi: 10.1016/j.prp.2022.153779
72. Yoshida A. Ewing And Ewing-like sarcomas: a morphological guide through genetically-defined entities. Pathol Int (2023) 73(1):12–26. doi: 10.1111/pin.13293
73. Zöllner SK, Amatruda JF, Bauer S, et al. Ewing Sarcoma-diagnosis, treatment, clinical challenges and future perspectives. J Clin Med (2021) 10(8):1685. doi: 10.3390/jcm10081685
74. Lamhamedi-Cherradi SE, Menegaz BA, Ramamoorthy V, Aiyer RA, Maywald RL, Buford AS, et al. An oral formulation of YK-4-279: preclinical efficacy and acquired resistance patterns in Ewing sarcoma. Mol Cancer Ther (2015) 14(7):1591–604. doi: 10.1158/1535-7163.MCT-14-0334
75. Zöllner SK, Selvanathan SP, Graham GT, Commins RMT, Hong SH, Moseley E, et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-m cell cycle arrest and enhanced vincristine sensitivity in ewing's sarcoma. Sci Signal (2017) 10(499):eaam8429. doi: 10.1126/scisignal.aam8429
76. Amin HM, Morani AC, Daw NC, Lamhamedi-Cherradi SE, Subbiah V, Menegaz BA, et al. IGF-1R/mTOR targeted therapy for Ewing sarcoma: a meta-analysis of five IGF-1R-Related trials matched to proteomic and radiologic predictive biomarkers. Cancers (2020) 12(7):1768. doi: 10.3390/cancers12071768
77. Rudzinski ER, Kelsey A, Vokuhl C, Linardic CM, Shipley J, Hettmer S, et al. Pathology of childhood rhabdomyosarcoma: a consensus opinion document from the children's oncology group, European paediatric soft tissue sarcoma study group, and the cooperative weichteilsarkom studiengruppe. Pediatr Blood Cancer (2021) 68(3):e28798. doi: 10.1002/pbc.28798
78. Shern JF, Selfe J, Izquierdo E, Patidar R, Chou HC, Song YK, et al. Genomic classification and clinical outcome in rhabdomyosarcoma: a report from an international consortium. J Clin Oncol (2021) 39(26):2859–71. doi: 10.1200/JCO.20.03060
79. Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer (2014) 53(9):779–87. doi: 10.1002/gcc.22187
80. Chen S, Rudzinski ER, Arnold MA. Challenges in the diagnosis of pediatric spindle Cell/Sclerosing rhabdomyosarcoma. Surg Pathol Clin (2020) 13(4):729–38. doi: 10.1016/j.path.2020.08.010
81. Le Loarer F, Cleven AHG, Bouvier C, Castex MP, Romagosa C, Moreau A, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol (2020) 33(3):404–19. doi: 10.1038/s41379-019-0323-8
82. Corao DA, Biegel JA, Coffin CM, Barr FG, Wainwright LM, Ernst LM, et al. ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr Dev Pathol (2009) 12(4):275–83. doi: 10.2350/08-03-0434.1
83. Gurria JP, Dasgupta R. Rhabdomyosarcoma and extraosseous Ewing sarcoma. Children (2018) 5(12):165. doi: 10.3390/children5120165
84. Bisogno G, De Salvo GL, Bergeron C, Gallego Melcón S, Merks JH, Kelsey A, et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2019) 20(11):1566–75. doi: 10.1016/S1470-2045(19)30617-5
85. Malempati S, Weigel BJ, Chi YY, Tian J, Anderson JR, Parham DM, et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: a report from the children's oncology group. Cancer (2019) 125(2):290–7. doi: 10.1002/cncr.31770
86. Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med (1998) 338(3):153–60. doi: 10.1056/NEJM199801153380303
87. Lee CJ, Modave E, Boeckx B, Kasper B, Aamdal S, Leahy MG, et al. Correlation of immunological and molecular profiles with response to crizotinib in alveolar soft part sarcoma: an exploratory study related to the EORTC 90101 "CREATE" trial. Int J Mol Sci (2022) 23(10):5689. doi: 10.3390/ijms23105689
88. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol (2018) 36(2):151–9. doi: 10.1200/JCO.2017.74.9598
89. Thway K, Noujaim J, Jones RL, Fisher C. Dermatofibrosarcoma protuberans: pathology, genetics, and potential therapeutic strategies. Ann Diagn Pathol (2016) 25:64–71. doi: 10.1016/j.anndiagpath.2016.09.013
90. Perreault S, Chami R, Deyell RJ, El Demellawy D, Ellezam B, Jabado N, et al. Canadian Consensus for biomarker testing and treatment of TRK fusion cancer in pediatric patients. Curr Oncol (2021) 28(1):346–66. doi: 10.3390/curroncol28010038
91. Loeb DM, Thornton K, Shokek O. Pediatric soft tissue sarcomas. Surg Clin North Am (2008) 88(3):615–27. doi: 10.1016/j.suc.2008.03.008
92. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noac202
93. Pollack IF, Agnihotri S, Broniscer A. Childhood brain tumors: current management, biological insights, and future directions. J Neurosurg Pediatr (2019) 23(3):261–73. doi: 10.3171/2018.10.PEDS18377
94. Alturkustani M. Classification of pediatric gangliogliomas based on the histological infiltration. Curr Oncol (2022) 29(10):6764–75. doi: 10.3390/curroncol29100532
95. Raabe E, Kieran MW, Cohen KJ. New strategies in pediatric gliomas: molecular advances in pediatric low-grade gliomas as a model. Clin Cancer Res (2013) 19(17):4553–8. doi: 10.1158/1078-0432.CCR-13-0662
96. Chalil A, Ramaswamy V. Low grade gliomas in children. J Child Neurol (2016) 31(4):517–22. doi: 10.1177/0883073815599259
97. Lim YJ. Medical treatment of pediatric low-grade glioma. Brain Tumor Res Treat (2022) 10(4):221–5. doi: 10.14791/btrt.2022.0039
98. Green K, Panagopoulou P, D'Arco F, O'Hare P, Bowman R, Walters B, et al. A nationwide evaluation of bevacizumab-based treatments in paediatric low-grade glioma in the UK: safety. efficacy, visual morbidity and outcomes. Neuro Oncol (2022) 25(4):774–85. doi: 10.1093/neuonc/noac223
99. Kalra M, Heath JA, Kellie SJ, Dalla Pozza L, Stevens MM, Swamy S, et al. Confirmation of bevacizumab activity, and maintenance of efficacy in retreatment after subsequent relapse, in pediatric low-grade glioma. J Pediatr Hematol Oncol (2015) 37(6):e341–6. doi: 10.1097/MPH.0000000000000371
100. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
101. Persson ML, Douglas AM, Alvaro F, Faridi P, Larsen MR, Alonso MM, et al. The intrinsic and microenvironmental features of diffuse midline glioma: implications for the development of effective immunotherapeutic treatment strategies. Neuro Oncol (2022) 24(9):1408–22. doi: 10.1093/neuonc/noac117
102. Rallis KS, George AM, Wozniak AM, Bigogno CM, Chow B, Hanrahan JG, et al. Molecular genetics and targeted therapies for paediatric high-grade glioma. Cancer Genomics Proteomics (2022) 19(4):390–414. doi: 10.21873/cgp.20328
103. Thomas DL. 2021 updates to the world health organization classification of adult-type and pediatric-type diffuse gliomas: a clinical practice review. Chin Clin Oncol (2023) 12(1):7. doi: 10.21037/cco-22-120
104. Franson A, McClellan BL, Varela ML, Comba A, Syed MF, Banerjee K, et al. Development of immunotherapy for high-grade gliomas: overcoming the immunosuppressive tumor microenvironment. Front Med (Lausanne) (2022) 9:966458. doi: 10.3389/fmed.2022.966458
105. Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol (2011) 103(3):491–501. doi: 10.1007/s11060-010-0402-7
106. Qaddoumi I, Kocak M, Pai Panandiker AS, Armstrong GT, Wetmore C, Crawford JR, et al. Phase II trial of erlotinib during and after radiotherapy in children with newly diagnosed high-grade gliomas. Front Oncol (2014) 4:67. doi: 10.3389/fonc.2014.00067
107. Macy ME, Kieran MW, Chi SN, Cohen KJ, MacDonald TJ, Smith AA, et al. A pediatric trial of radiation/cetuximab followed by irinotecan/cetuximab in newly diagnosed diffuse pontine gliomas and high-grade astrocytomas: a pediatric oncology experimental therapeutics investigators' consortium study. Pediatr Blood Cancer (2017) 64(11):e26621. doi: 10.1002/pbc.26621
108. Parenrengi MA, Suryaningtyas W, Al Fauzi A, Hafid Bajamal A, Kusumastuti K, Utomo B, et al. Nimotuzumab as additional therapy for GLIOMA in pediatric and adolescent: a systematic review. Cancer Control (2022) 29:1–10. doi: 10.1177/10732748211053927
109. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol (2020) 30(5):863–6. doi: 10.1111/bpa.12866
110. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell (2015) 27(5):728–43. doi: 10.1016/j.ccell.2015.04.002
111. Cage TA, Clark AJ, Aranda D, Gupta N, Sun PP, Parsa AT, et al. A systematic review of treatment outcomes in pediatric patients with intracranial ependymomas. J Neurosurg Pediatr (2013) 11(6):673–81. doi: 10.3171/2013.2.PEDS12345
112. Barone A, Rubin JB. Opportunities and challenges for successful use of bevacizumab in pediatrics. Front Oncol (2013) 3:92. doi: 10.3389/fonc.2013.00092
113. DeWire M, Fouladi M, Turner DC, Wetmore C, Hawkins C, Jacobs C, et al. An open-label, two-stage, phase II study of bevacizumab and lapatinib in children with recurrent or refractory ependymoma: a collaborative ependymoma research network study (CERN). J Neurooncol (2015) 123(1):85–91. doi: 10.1007/s11060-015-1764-7
114. Jakacki RI, Foley MA, Horan J, Wang J, Kieran MW, Bowers DC, et al. Single-agent erlotinib versus oral etoposide in patients with recurrent or refractory pediatric ependymoma: a randomized open-label study. J Neurooncol (2016) 129(1):131–8. doi: 10.1007/s11060-016-2155-4
115. Larrew T, Saway BF, Lowe SR, Olar A. Molecular classification and therapeutic targets in ependymoma. Cancers (Basel) (2021) 13(24):6218. doi: 10.3390/cancers13246218
116. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol (2016) 131(6):821–31. doi: 10.1007/s00401-016-1569-6
117. Northcott P, Buchhalter I, Morrissy A, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature (2017) 547:311–7. doi: 10.1038/nature22973
118. Juraschka K, Taylor MD. Medulloblastoma in the age of molecular subgroups: a review. J Neurosurg Pediatr (2019) 24(4):353–63. doi: 10.3171/2019.5.PEDS18381
119. Kim JW, Park SH, Choi SA, Kim SK, Koh EJ, Won JK, et al. Molecular subgrouping of medulloblastoma in pediatric population using the NanoString assay and comparison with immunohistochemistry methods. BMC Cancer (2022) 22(1):1221. doi: 10.1186/s12885-022-10328-6
120. Menyhárt O, Győrffy B. Principles of tumorigenesis and emerging molecular drivers of SHH-activated medulloblastomas. Ann Clin Transl Neurol (2019) 6(5):990–1005. doi: 10.1002/acn3.762
121. Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer (2020) 20(1):42–56. doi: 10.1038/s41568-019-0223-8
122. Thomas A, Noël G. Medulloblastoma: optimizing care with a multidisciplinary approach. J Multidiscip Healthc (2019) 12:335–47. doi: 10.2147/JMDH.S167808
123. Karim A, Shaikhyzada K, Suleimenova A, Ibraimov B, Nurgaliev D, Poddighe D. Case report: atypical teratoid/rhabdoid tumor of the lateral ventricle in a male adolescent (case-based review and diagnostic challenges in developing countries). Front Oncol (2022) 12:985862. doi: 10.3389/fonc.2022.985862
124. Holdhof D, Johann PD, Spohn M, Bockmayr M, Safaei S, Joshi P, et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol (2021) 141(2):291–301. doi: 10.1007/s00401-020-02250-7
125. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald M, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol (2020) 22(5):613–24. doi: 10.1093/neuonc/noz235
126. Alva E, Rubens J, Chi S, Rosenberg T, Reddy A, Raabe EH, et al. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia (2023) 37:100880. doi: 10.1016/j.neo.2023.100880
127. Guo G, Zhuang J, Zhang K, Zhou Z, Wang Y, Zhang Z. Atypical Teratoid/Rhabdoid tumor of the central nervous system in children: case reports and literature review. Front Surg (2022) 9:864518. doi: 10.3389/fsurg.2022.864518
128. Echevarría ME, Fangusaro J, Goldman S. Pediatric central nervous system germ cell tumors: a review. Oncologist (2008) 13(6):690–9. doi: 10.1634/theoncologist.2008-0037
129. Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature (2014) 511(7508):241–5. doi: 10.1038/nature13296
130. Takami H, Fukuoka K, Fukushima S, Nakamura T, Mukasa A, Saito N, et al. Integrated clinical, histopathological, and molecular data analysis of 190 central nervous system germ cell tumors from the iGCT consortium. Neuro Oncol (2019) 21(12):1565–77. doi: 10.1093/neuonc/noz139
131. Satomi K, Takami H, Fukushima S, Yamashita S, Matsushita Y, Nakazato Y, et al. 12p gain is predominantly observed in non-germinomatous germ cell tumors and identifies an unfavorable subgroup of central nervous system germ cell tumors. Neuro Oncol (2022) 24(5):834–46. doi: 10.1093/neuonc/noab246
132. Fetcko K, Dey M. Primary central nervous system germ cell tumors: a review and update. Med Res Arch (2018) 6(3):1719. doi: 10.18103/mra.v6i3.1719
133. Baruchel S, Sharp JR, Bartels U, Hukin J, Odame I, Portwine C, et al. A Canadian paediatric brain tumour consortium (CPBTC) phase II molecularly targeted study of imatinib in recurrent and refractory paediatric central nervous system tumours. Eur J Cancer (2009) 45(13):2352–9. doi: 10.1016/j.ejca.2009.05.008
134. Gojo J, Kjaersgaard M, Zezschwitz BV, Capper D, Tietze A, Kool M, et al. Rare embryonal and sarcomatous central nervous system tumours: state-of-the art and future directions. Eur J Med Genet (2022) 66(1):104660. doi: 10.1016/j.ejmg.2022.104660
135. Łastowska M, Trubicka J, Sobocińska A, Wojtas B, Niemira M, Szałkowska A, et al. Molecular identification of CNS NB-FOXR2, CNS EFT-CIC, CNS HGNET-MN1 and CNS HGNET-BCOR pediatric brain tumors using tumor-specific signature genes. Acta Neuropathol Commun (2020) 8(1):105. doi: 10.1186/s40478-020-00984-9
Keywords: targeted, childhood, cancer, tumors, precision oncology
Citation: Bertacca I, Pegoraro F, Tondo A and Favre C (2023) Targeted treatment of solid tumors in pediatric precision oncology. Front. Oncol. 13:1176790. doi: 10.3389/fonc.2023.1176790
Received: 28 February 2023; Accepted: 24 April 2023;
Published: 05 May 2023.
Edited by:
Oleg Tsodikov, University of Kentucky, United StatesReviewed by:
Markos Leggas, St. Jude Children’s Research Hospital, United StatesAlessandro Laganà, Icahn School of Medicine at Mount Sinai, United States
David D. Eisenstat, Royal Children’s Hospital, Australia
Copyright © 2023 Bertacca, Pegoraro, Tondo and Favre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Favre, Y2xhdWRpby5mYXZyZUBtZXllci5pdA==