 Svenja Ebeling
Svenja Ebeling Anita Kowalczyk
Anita Kowalczyk Diego Perez-Vazquez
Diego Perez-Vazquez Irene Mattiola
Irene Mattiola- 1Institute of Microbiology, Infectious Diseases and Immunology (I-MIDI), Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and the Berlin Institute of Health, Berlin, Germany
- 2Laboratory of Mucosal and Developmental Immunology, Deutsches Rheuma-Forschungszentrum (DRFZ), an Institute of the Leibniz Association, Berlin, Germany
Endothelial cells and immune cells are major regulators of cancer progression and prognosis. Endothelial cell proliferation and angiogenesis are required for providing nutrients and oxygen to the nascent tumor and infiltration of immune cells to the tumor is dependent on endothelial cell activation. Myeloid cells and innate lymphocytes have an important role in shaping the tumor microenvironment by crosstalking with cancer cells and structural cells, including endothelial cells. Innate immune cells can modulate the activation and functions of tumor endothelial cells, and, in turn, endothelial cell expression of adhesion molecules can affect immune cell extravasation. However, the mechanisms underlying this bidirectional crosstalk are not fully understood. In this review, we will provide an overview of the current knowledge on the pathways regulating the crosstalk between innate immune cells and endothelial cells during tumor progression and discuss their potential contribution to the development of novel anti-tumor therapeutic approaches.
1 Introduction
The endothelium consists of a monolayer of endothelial cells that lines the inner surface of blood and lymphatic vessels (1). Endothelial cells regulate the function of the circulatory system, such as the blood flow and the blood pressure, as well as vessel permeability and coagulation (1). Blood vessels are formed during embryonic development, in a process called vasculogenesis, and infiltrate every tissue of the organism to provide oxygen and nutrients required for cell survival (1, 2). The mature endothelium is composed of a layer of tightly joint endothelial cells surrounded by a basal muscular layer that regulates vasoconstriction and vasodilation (1). In homeostatic conditions, endothelial cells are in a quiescent state characterized by minimal cell proliferation, and are constantly subjected to autocrine, endocrine, and paracrine signals. Pathways regulated by Wnt, Fibroblast growth factor 2 (FGF-2), Bone Morphogenetic Protein 9 (BMP-9), and Angiopoietin 1 (Ang-1) are required for the maintenance of endothelial cell identity and physical properties, as for preventing the alteration of the blood flow, the vascular permeability and the delivery of nutrients and oxygen (3). Although vascular endothelial growth factor (VEGF) is critical for developmental and pathological angiogenesis, recent evidence showed that autocrine VEGF could also contribute to the maintenance of vascular homeostasis (4).
Beyond its importance for nutrient and oxygen transport, the endothelium is central for the recruitment of immune cells to inflamed tissues (5, 6). Acute inflammation involves the rapid recruitment of leukocytes and requires endothelial cell activation, namely the acquisition of new capacities by resting endothelial cells (6). Endothelial cell activation is characterized by two types of response: a rapid response that is independent of new gene expression, called type I activation, and a slower response that is dependent on new gene expression, called type II activation (6). The latter is triggered by mediators released from activated leukocytes, such as tumor necrosis factor (TNF), interleukin 1 beta (IL-1β) and 6 (IL-6), which induce endothelial cell acquisition of a pro-inflammatory transcriptional program (6, 7). Activated endothelial cells produce Nitric Oxide (NO) that promotes vasodilation and further facilitates immune cell trafficking (8). Upon activation, endothelial cells upregulate the expression of adhesion molecules, such as E-selectin and P-selectin, intercellular adhesion molecule 1 and 2 (ICAM-1 and 2), vascular cell adhesion molecule 1 (VCAM-1), and integrins, and downregulate other intercellular adhesion molecules, such as VE-Cadherin, to enhance leukocyte migration across the endothelial cell layer (9). The process begins with the formation of transient adhesive interactions between free-flowing leukocytes and activated endothelial cells. Chemokines (CC−chemokine ligand 2 (CCL2) and CXC−chemokine ligand 10 (CXCL10)) and pro-inflammatory mediators (TNF and IL-6) secreted either from the activated endothelium (10) or from surrounding immune cells further increase leukocyte adhesion and promote crawling over the luminal endothelial surface (9). The transendothelial migration of immune cells mainly occurs by paracellular diapedesis, with leukocytes passing through the endothelial cell layer where the intercellular contacts are loosening (11). Only in a minority of cases, the migration of leukocytes occurs via transcellular diapedesis, characterized by intraluminal crawling of leukocytes (12), eventually when endothelial cell junctions are too tight (13). Similar receptors and adhesion molecules are involved in both types of diapedeses (14), but the molecular mechanisms that decide the use of one type over the other are still not fully understood. Excessive exposure of endothelial cells to pro-inflammatory mediators, such as TNF and interferon γ (IFN-γ) (15, 16), or bacteria (17) leads to endothelial cell apoptosis and perturbation of vascular permeability. IL-10, an anti-inflammatory cytokine largely described to promote resolution of inflammation and prevent tissue damage (18), was shown to prevent such endothelial cell apoptosis (17), to preserve the expression of endothelial junction proteins, namely occludin and claudin 5 (16), and to maintain proper vascular permeability during inflammation (15).
Depending on the surrounding environment, endothelial cells can acquire different phenotypes and exert different functions. This concept was extended when highly powered single-cell (sc) transcriptomics studies allowed a more detailed characterization of endothelial cell heterogeneity in homeostasis and disease. scRNA sequencing of lung peritumoral and tumoral endothelial cells identified endothelial cell populations endowed with diverse functions correlated with different grades of activation (19). Human pulmonary arterial endothelial cells expressed genes implicated in vascular integrity and homeostasis, whereas postcapillary venous were characterized by the expression of genes involved in leukocyte recruitment, tissue perfusion and blood pressure (19). Interestingly, peritumoral capillary endothelial cells expressed genes associated with semi-professional antigen presentation, including HLA, but not CD80 and CD86, and with scavenging activity, such as MARCO (19). This suggested a role for peritumoral and tumoral endothelial cells in tumor immune surveillance (19). The capability of the endothelium to adapt to changes in the microenvironment highlights its central role in the regulation of tissue homeostasis and points it as a target for novel therapeutic approaches to control inflammation.
The endothelium not only responds to inflammatory signals but constantly communicates with the surrounding cells to provide the required oxygen, nutrients, and macromolecules to every organ in the body (1). Angiogenesis, namely the formation of new vessels from existing ones aimed to provide further nutrients and oxygen, is required from the onset of tumors (1-2 mm of tumor size) (20), and it is widely recognized as an essential hallmark of cancer (21). Tumor cells promote an angiogenic switch in endothelial cells by secreting pro-angiogenic factors such as VEGF, platelet-derived growth factor (PDGF), and angiopoietin 2 (Ang-2), inducing the destabilization of the endothelial cell barrier and promoting endothelial cell migration and proliferation (22). As tumor growth progresses, the high proliferation of cancer cells demands a supply of oxygen (and nutrients) that cannot be satisfied by the existing vasculature (23). Therefore, cancer cells express hypoxia-inducible factor 1 (HIF-1) and secrete VEGF to promote angiogenesis (24). The newly formed vessels within the tumor have an immature phenotype characterized by limited contact between endothelial cells and defective or discontinuous basement membrane. This prevents tumor endothelial cells from properly responding to physiological stimuli (25). Tumor endothelial cells are characterized by a pro-angiogenic phenotype, with high expression of VEGF receptors (VEGFR1 and R2), which pathways support endothelial cell survival and proliferation, and release of matrix metalloproteinases (MMP), involved in the remodeling of the extracellular matrix (ECM) and thus facilitating endothelial cell migration (26, 27). Tumor endothelial cells also show reduced expression of leukocyte binding molecules, including ICAM-1 and 2, VCAM-1, E-selectin and CD34, leading to reduced response to inflammatory signals and defective recruitment of immune cells (28, 29). Moreover, the structural and transcriptional abnormalities in the tumor endothelium facilitate the extravasation of cancer cells and promote metastasis development (30). Altogether, these phenotypical changes in tumor endothelial cells limit immune cell activation and infiltration in the tumor microenvironment (TME), favoring cancer progression. Therefore, a better understanding of the mechanisms regulating endothelial cell manipulation in cancer may improve the efficacy and success of current immunotherapeutic approaches.
Immune cells are part of the TME and have a key role in regulating cancer progression (21). The immune system comprises two branches, consisting of innate and adaptive immunity. Adaptive immune cells are endowed with memory functions and are involved in long-lasting immune responses (31). Conversely, innate immune cells represent the first line of immune defense and promptly respond to environmental challenges mounting appropriate immune responses and activating adaptive immunity (31). Innate immune cells, including myeloid and lymphoid cells, control the early stages of tumor progression by directly interacting with tumor cells and by shaping the TME (31), in a process called cancer immunoediting (21). However, innate immune cell recruitment and activation in the TME is strictly dependent on endothelial cells, which engage a bidirectional crosstalk with innate immune cells (7). The interaction between endothelial cells and innate immune cells represents an important cellular circuit in the regulation of cancer progression. Here, we will provide an overview of the cellular pathways by which myeloid cells and innate lymphocytes interact with endothelial cells in the TME and discuss potential implications of such interactions for anti-cancer treatments.
2 Myeloid cells and angiogenesis in cancer
Myeloid cells are circulating and tissue-resident innate immune cells having an important role in sensing the environment, patrolling tissues, and orchestrating immune responses (32). Myeloid cells constantly crosstalk with non-hematopoietic cells composing tissue niches and with innate and adaptive lymphocytes, promoting their recruitment to sites of inflammation and their activation (33). Research done over two decades provided evidence of myeloid cells being critical players in cancer, by either favoring tumor progression or regression, depending on the regulatory features exerted in the TME (34–36). In the next session, we will give an overview on the pathways regulating the crosstalk between the different subsets of myeloid cells, namely macrophages, neutrophils, dendritic cells and monocytes, and endothelial cells in cancer, with a particular focus on intestinal tumors (Figure 1).
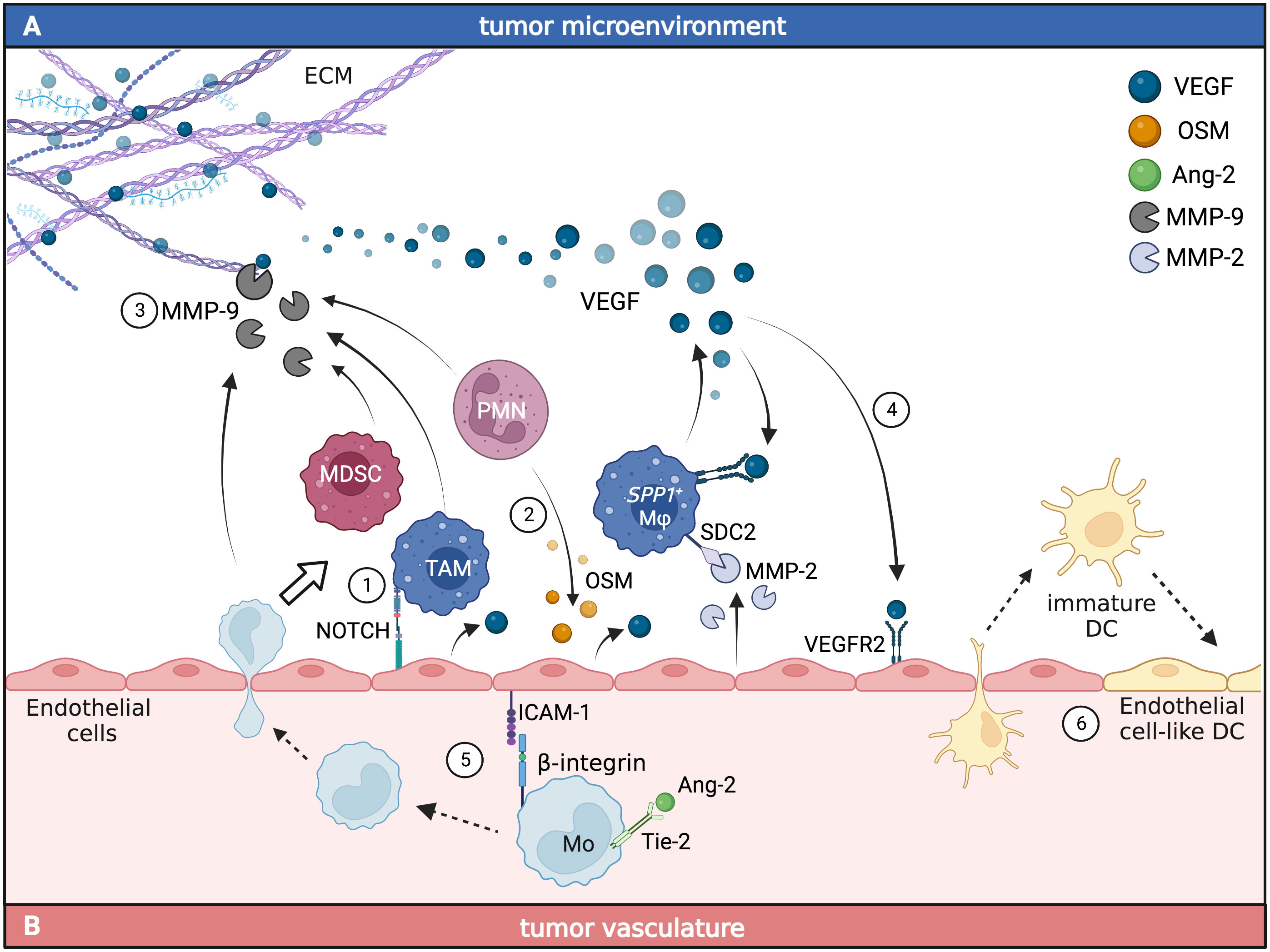
Figure 1 Myeloid and endothelial cell crosstalk promotes angiogenesis in the TME. The interaction between endothelial cells and tumor-infiltrating myeloid cells (A) or circulating myeloid cells (B) leads to the accumulation of pro-angiogenic signals in cancer. In the TME (A), myeloid cells can engage direct or indirect crosstalk with endothelial cells. TAM and polymorphonuclear cells (PMN) can directly induce the production of VEGF in endothelial cells through the activation of Notch1 (1) and OSM-dependent (2) pathways. Together with MDSC, TAM can also indirectly induce the release of VEGF from the ECM through the production of MMP-9 (3). VEGF production from TAM activates VEGFR2 signaling in endothelial cells (4), promoting angiogenesis and leading to increased tumor vascularization. In the tumor vasculature (B), monocytes expressing Tie-2 respond to Ang-2 by increasing the expression of β-integrins that bind ICAM-1 on endothelial cells facilitating monocyte migration to the TME (5), where they differentiate into tumor-associated myeloid cells. Circulating immature DC can support tumor neovasculogenesis by adopting endothelial cell-like functions and being incorporated into newly forming vessels (6). Figure generated with BioRender.
2.1 Tumor-associated macrophages and endothelial cell crosstalk
Tumor-associated macrophages (TAM) are one of the most represented populations of myeloid cells in the TME (36, 37). The initial definition of TAM subsets based on M1 (pro-inflammatory and tumor suppressing) and M2 (anti-inflammatory and promoting tumor progression) (35, 38, 39) was recently overcome by a more heterogenous definition based on transcriptional programs representative of different phenotypes and functions, including activation and cytokine production (40–43). Interestingly, TAM were shown to secrete several pro-angiogenic cytokines and growth factors, such as VEGF (44–46), MMP (46–48), IL-10 (40, 46, 47, 49, 50), IL-8 (45, 46), FGF2 (45, 51, 52) and placental growth factor (PlGF) (52), allowing them to modulate angiogenic pathways.
In relation to VEGF regulation and signaling, macrophages were shown to play a dual role. On the one hand, Stefater and colleagues showed that, during development, the stimulation of non-canonical Wnt-signaling (Wnt11 and Wnt5A) induced retinal CD11b+ F4/80+ macrophages to secrete soluble VEGFR1, sequestering VEGF from the surrounding environment and thus reducing its availability for endothelial cells (53). This prevented uncontrolled outgrow of vessels and suggested a crucial role of macrophages for proper growth of organized vessels (53). On the other hand, in cancer, several studies linked macrophages to increased levels of VEGF in the TME. Hypoxia (54) and HIF-1α-mediated signaling (48) could induce the production of VEGF in TAM, suggesting that they could be a source of VEGF in the TME and could promote tumor angiogenesis. TAM also express VEGFR1 and VEGFR2 (55, 56) having the potential to directly bind and respond to autocrine or paracrine VEGF signaling in the TME. The inhibition of the VEGF/VEGFR2 axis in TAM led to reduced secretion of VEGF, suggesting that TAM could contribute to the accumulation of VEGF in the TME upon activation of VEGFR2 pathways (57). Interestingly, the VEGF expression profile in TAM correlated with the one of MMP-9, a matrix metallopeptidase contributing to the degradation of the ECM and facilitating endothelial cell migration (58). In mouse models of glioblastoma, MMP-9 secreted by F4/80+ macrophages triggered the local activation of sequestered VEGF bound to the ECM, making it available for binding to VEGFR2 and leading to endothelial cell activation (48). In a mouse model of breast cancer (PyMT), the depletion of intratumoral macrophages (colony stimulating factor 1 (CSF-1) null mutant mice) or the conditional knock-out of their secreted VEGF led to a delay of the angiogenic switch of the tumor vasculature (59–61). Altogether, these findings argue for an active role of TAM in promoting tumor angiogenesis through VEGF- and MMP-9-dependent pathways.
The recent development of next-generation sequencing techniques combined with retrospective studies on cancer patients opened new possibilities to better characterize the macrophage subsets in cancer and to further define the pathways involved in their angiogenic signaling. Regarding intestinal cancer, it was thereby possible to identify different macrophage signatures that could correlate with either good or bad prognosis (62). For instance, in colorectal cancer (CRC) patients, high infiltration of CD163+ TAM was linked to poor outcome (63, 64), whereas infiltration of CD206+ TAM correlated with better prognosis (65, 66). Notably, both markers were originally linked to a M2-like profile and therefore a tumor-promoting phenotype (67). Further studies suggested that macrophage transcriptional landscapes in different types of cancer were influenced by the cytokine signature of the respective TME (68). In human CRC, a subset of TAM characterized by SPP1 gene expression was highly enriched in cancerous tissue while being largely absent from healthy tissue (69). Notably, SPP1+ TAM showed an increased expression of VEGF and Syndecan-2 (SDC2) (69), a transmembrane proteoglycan that binds MMP-2, a metalloprotease released by endothelial cells upon hypoxic conditions (70). MMP-2 is involved in the degradation of the ECM and thereby favors the formation of the tumor vasculature (71). These findings suggest that the crosstalk between SPP1+ TAM and tumor endothelial cells promotes tumor angiogenesis. A recent study in mouse and human hepatocellular carcinoma identified a population of TAM and a population of tumor endothelial cells, FOLR2+ macrophages and PLVAP+ endothelial cells respectively, that resembled fetal characteristics (72). Both cell subsets were involved in implementing the onco-fetal features of the TME by interacting via Notch and VEGF signaling and contributing to the formation of an immunosuppressive environment (72). However, further investigations are required to assess a potential crosstalk of such fetal-like TAM and endothelial cell populations in the TME.
Tie-2-expressing macrophages (TEM) represent a macrophage subset characterized by high pro-angiogenic features. Tie-2 is the tyrosine kinase receptor for Ang-1 and Ang-2, molecules that regulate angiogenesis and endothelial cell survival, proliferation and migration (73). TEM were described to be one of the major drivers of angiogenesis in a mouse model of spontaneous pancreatic cancer (RIP-Tag2) and orthotopic brain tumors (74). TEM infiltrated several human cancers (75), and promoted tumor growth and metastases in mouse models of breast (PyMT) and pancreatic (RIP-Tag2) cancer by favoring angiogenesis (76). Notably, Ang-2 induced the upregulation of β2-integrin on monocytes, which interacted with ICAM-1 expressed by endothelial cells and thereby facilitated the migration of monocytes, leading to accumulation of monocyte-derived macrophages in the TME (77). Conversely, in homeostatic conditions, Ang-2-induced Wnt7B production in macrophages promoted apoptosis of endothelial cells, leading to restrained vascular growth (78–80). Thereby, depending on the tissue environment, Ang-2 potentially induces different regulatory pathways in macrophages and has a dual role in the macrophage-endothelial cell crosstalk. However, it remains elusive whether a cancerous environment could trigger either pathway.
Taken together, these findings suggest that a better understanding of TAM and TAM-endothelial cell interactions is required to identify novel pathways and disclose putative targets for innovative therapeutic approaches aimed at restraining tumor angiogenesis.
2.2 Angiogenic features of circulating myeloid cells
In addition to macrophages, other cells of the innate immune compartment such as neutrophils and myeloid-derived suppressor cells (MDSC) regulate angiogenesis through similar signaling pathways. Kujawski and colleagues reported that upon STAT-3-activation MDSC showed pro-angiogenic features mediated by the secretion of VEGF and MMP-9 (81). Interestingly, in a mouse model of spontaneous cervical cancer (K14-HPV/E(2) mice), MMP-9+ neutrophils represented an alternative source to TAM in providing MMP-9 (82) and, in murine models of colorectal cancer (MC26) and lung carcinoma (3LL), MDSC-derived MMP-9 was associated to enhanced tumor vascularization (83). In addition, when co-cultured with human breast cancer cells, neutrophils produced high levels of oncostatin M (OSM) (84), which was shown to act on the endothelium via STAT-3/VEGF signaling in vivo and to enable the remodeling of the TME by activating cancer-associated fibroblasts (CAF) (85, 86). In several murine transplantable models of cancer, MDSC were found to be the predominant source of prokineticin-2 (Bv8), a secreted protein involved in tissue-specific angiogenesis, which promoted vessel formation and tumor growth (87). Interestingly, the migration of these Bv8+ MDSC from the bone marrow into the peripheral blood and TME was mediated by endothelial cell-derived granulocyte colony stimulation factor (G-CSF) (87). This suggested for a bidirectional crosstalk of circulating myeloid cells and endothelial cells in cancer.
Conversely to neutrophils and MDSC, dendritic cells (DC) were described to rather have anti-angiogenic and tumor-suppressing features (38, 88, 89). Conventional DC suppressed tumor angiogenesis through the secretion of pro-inflammatory cytokines and chemokines, including IL-12, IL-18 and CXCL9 (89, 90). In a xenograft model of ovarian carcinoma, high infiltration of plasmacytoid (p)DC and low infiltration of myeloid-derived (m)DC was observed (90). Notably, only mDC were shown to produce IL-12 and to exert anti-angiogenic functions in the TME, whereas pDC were shown to interact with the vasculature by secreting IL-8 and IFN-α (90). In a xenograft model of breast cancer, IFN-α inhibited endothelial cell proliferation and thus impaired tumor vasculature formation (91). Thereby, the TME favored the infiltration of angiogenic subsets of DC and suppressed the anti-angiogenic functions of mDC. In a mouse model of ovarian cancer, immature DC (defined as CD11c+ DEC-205+ CD8α+ CD80-) were also found in the TME, where they were recruited in a β-defensin-dependent manner (92). Those tumor-associated immature DC co-localized with endothelial cells to form new vascular structures and promote neovasculogenesis (92). Therefore, depending on the origin and the differentiation stage, DC could exert either pro- or anti-angiogenic functions.
Monocytes are circulating cells that once recruited to the TME can differentiate into DC, MDSC or macrophages (93), depending on the cytokine and growth factor signature of the TME (94–97). However, monocytes could also contribute to tumor angiogenesis before differentiating into tumor-associated myeloid cell subsets (34, 47). For instance, in a xenograft model of colorectal cancer, a pro-angiogenic monocyte subset was identified, defined by the expression of both CD14 and CD16 and the secretion of MMP-9, which exerted tumor-promoting functions (98). These pro-angiogenic monocytes also expressed Tie-2 and extravasated into the TME in a VEGF-dependent manner (98). In general, CD11b+ myeloid cells, including monocytes, were shown to migrate towards and differentiate upon tumor-specific gradients and factors such as HIF-1α (48) or Ang-2 (47, 74, 76). Nevertheless, due to the heterogeneity of monocyte differentiation stages and subsets, the understanding of the mechanisms by which bona fide monocytes directly interact with endothelial cells in cancer needs further investigation.
3 Innate lymphoid cells and tumor vasculature
Innate lymphoid cells (ILC) are innate lymphocytes residing at barrier tissues that are endowed with immunomodulatory features and have an important role in maintaining tissue homeostasis (99). Based on the expression of master regulator transcription factors and the secretion of peculiar cytokines, ILC can be discerned into three main groups, namely group 1 ILC (ILC1), group 2 ILC (ILC2), and group 3 ILC (ILC3) (100). ILC1 express T-bet and are characterized by the secretion of pro-inflammatory cytokines, including IFN-γ; ILC2 express GATA-3 and secrete type 2 cytokines, including IL-5 and IL-13; and ILC3 express RORγt and are characterized by IL-17 and IL-22 production (99, 100). Due to their cytokine-dependent immunomodulatory features, ILC1, ILC2, and ILC3 are defined as helper-like ILC (101). NK cells represent the first discovered population of ILC (102) and consist of circulating innate lymphocytes characterized by T-bet and Eomes expression and are endowed with cytotoxic functions (102). Therefore, NK cells are defined as cytotoxic ILC (101).
ILC privileged position within tissues allows them to communicate and regulate the homeostatic functions of structural cells (103), including endothelial cells (104). In the next session, we will focus on the pathways regulating the crosstalk between innate lymphocytes and endothelial cells in cancer (Figure 2).
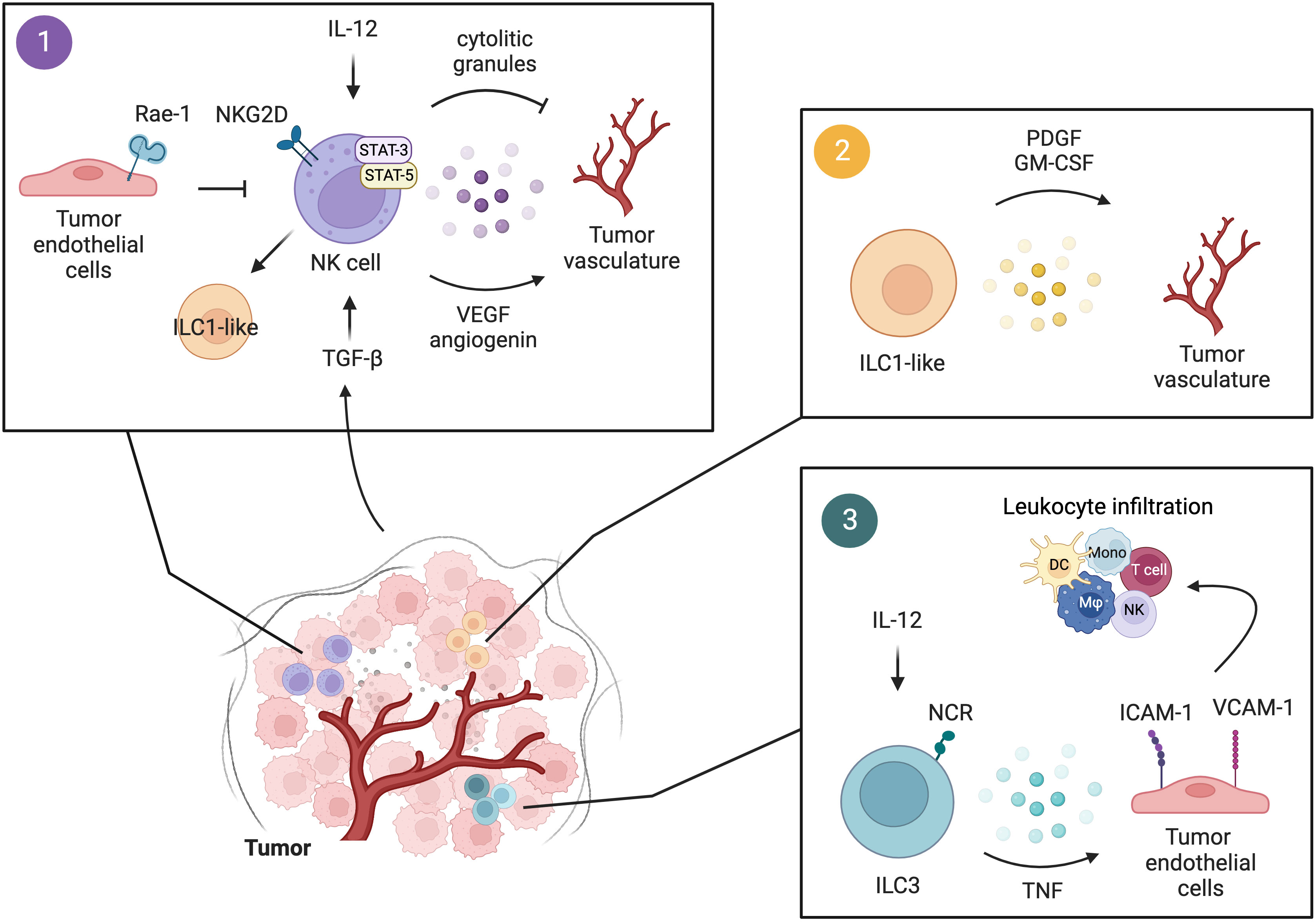
Figure 2 Interactions of innate lymphocytes with the tumor vasculature. 1) Upon IL-12 stimulation, perforin and granzymes released by NK cells act on the tumor vasculature and suppress tumor angiogenesis. Tumor-infiltrating NK cells modulate tumor angiogenesis also through the secretion of VEGF and angiogenin in a STAT-3/5-dependent manner. This pro-angiogenic activity of NK cells can be fostered by TGF-β in the TME, which also induces the conversion of NK cells to a more immature phenotype of ILC1-like cells. Tumor endothelial cells expressing Rae-1 induce constitutive activation of NKG2D+ NK cells leading to reduced NKG2D-dependent responses in NK cells and thus impaired anti-tumor functions. 2) The presence of TGF-β in the TME promotes NK cell conversion to an ILC1-like phenotype, characterized by diminished cytotoxic activity and increased release of pro-angiogenic factors, namely PDGF and GM-CSF. 3) Human and murine NCR+ ILC3 by secreting pro-inflammatory mediators, including TNF, induce the expression of ICAM-1 and VCAM-1 on tumor endothelial cells, favoring leukocyte infiltration and the establishment of a pro-inflammatory environment. Figure generated with BioRender.
3.1 NK cells and tumor angiogenesis
NK cells were initially shown to exert anti-angiogenic functions. In a murine model of Burkitt lymphoma, it was reported that, upon IL-12 treatment, perforin- and granzyme-positive tumor-infiltrating NK cells accumulated in tumor tissues and located close to the vessels (105). This cytotoxic NK cell infiltration correlated with reduced new vessel formation (105). However, more recent evidence reported that NK cells exerted pro-angiogenic functions in humans. Around 90% of human circulating NK cells show a CD56dim phenotype and are characterized by cytotoxic functions, whereas 10% is composed of CD56bright cells, mainly showing immunoregulatory functions (106, 107). Although genetically different, this CD56bright NK cell population shared many similarities with a unique subset of CD56superbrightCD16- decidual NK cells (dNK) deprived of cytotoxic activity, but involved in the modulation of decidual vascular growth during pregnancy through the secretion of angiogenic factors, such as VEGF, PlGF, and IL-8 (108). When transplanted together with human choriocarcinoma-like JEG-3 tumor cells in the subcutis of nude mice, dNK cells markedly supported VEGF and PlGF-dependent tumor vascularization (108). Furthermore, a subset of NK cells resembling dNK cell features was identified in human CRC. These dNK-like cells exhibited increased production of pro-angiogenic factors, such as angiogenin and VEGF, as well as pro-invasive factors, including MMP-9, TIMP-1, and TIMP-2. Of note, the dNK-like cell production of angiogenin and VEGF was dependent on STAT-3 and STAT-5 activation, whereas the production of MMP-9, TIMP-1, and TIMP-2 was STAT-3/5-independent (109). Conversely, Gotthard and colleagues showed that in a model of murine lymphoma (26), STAT-5-deficient NK cells expressed elevated levels of VEGF and were able to induce endothelial cell sprouting ex vivo. Selective deletion of VEGF in NKp46+ innate cells, including NK cells, showed reduced tumor burden correlated with reduced tumor vascularization (110). Therefore, depending on the host species and the origin of the tumor, STAT-5 could have opposite roles in regulating NK cell pro-angiogenic functions. Compared to CD56+CD16+ NK cells, human CD56+CD16- NK cells produce higher levels of pro-angiogenic factors, namely VEGF, IL-8, and PlGF (111). Interestingly, it was shown that such production was enhanced in CD56+CD16- NK cells isolated from squamous cell carcinoma patients and was TGF-β-dependent (111). This suggested a role for TGF-β in modulating NK cell pro-angiogenic functions. TGF-β is a cytokine exerting immunosuppressive functions in the TME (112) and it was shown to modulate the expression of CD16 and thus the NK cell cytotoxic potential (113). In accordance with this, TGF-β in the TME of MCA-induced fibrosarcoma promoted NK cell conversion into a more immature and less cytotoxic phenotype, defined as ILC1-like, characterized by a pro-angiogenic signature (114).
NKG2D is an activating receptor expressed by NK cells that has an important role in NK cell-dependent tumor immunity, including resistance to metastasis (115). Recent work from Thompson and colleagues showed that Rae-1, one of the ligands for NKG2D, was expressed by certain populations of endothelial cells at steady state (116). Rae-1 was detectable in endothelial cells from lymph nodes and spleen but was not detected in endothelial cells from highly vascularized tissues, such as the liver, lung, and heart (116). The constitutive engagement of NK cell NKG2D by endothelial cell Rae-1 led to NKG2D downregulation and diminished NK cell responses (116). Interestingly, Rae-1 expression was induced on endothelial cells from primary melanoma (B16) and correlated with reduced NK cell anti-tumor functions (116). Rae-1 expression was mainly found in endothelial cells of High-Endothelial Venules (HEV), which are blood vessels responsible for lymphocyte migration to lymph nodes (and tumors) (117, 118). These data not only suggest that endothelial cells could control NK cell anti-tumor functions in primary tumors but eventually also at metastatic sites, since NK cell trafficking to the lymph nodes has an important role in NK cell-dependent resistance to metastasis (119).
Taken together, these observations suggest that depending on the host species, the tissue of origin and the cytokine signature of the TME, NK cells can be manipulated to acquire a pro-tumoral and pro-angiogenic phenotype. However, further investigations are needed to better elucidate the molecular pathways responsible for the acquisition of such a pro-angiogenic phenotype in tumor-infiltrating NK cells.
3.2 Helper-like ILC and endothelial cell activation
In recent years, the contribution of ILC to tumor immunity was broadly reported (99, 120, 121). As shown for macrophages, depending on the characteristics of the TME, ILC can acquire diverse activation profiles and can switch from an anti-tumor to a pro-tumor phenotype (99, 122). As potent cytokine producers, ILC can crosstalk with other immune and non-immune cells in the TME, including endothelial cells. In a murine model of melanoma overexpressing IL-12 (B16-IL-12), NKp46+ ILC3 were shown to infiltrate the tumor and mediate the upregulation of ICAM-1 and VCAM-1 adhesion molecules on tumor vessels leading to the generation of a pro-inflammatory microvascular environment (123). This promoted leukocyte recruitment to the tumor bed and suppression of tumor growth (123). Human NCR+ ILC3 were shown to accumulate in human non-small cell lung cancer (NSCLC) where they represented an important source of innate cytokines, including TNF and IL-8 (124). In accordance with the work of Eisering and colleagues, tumor-infiltrating human NCR+ ILC3-derived cytokines promoted the adhesion of in vitro cultured endothelial cells and upregulated their expression of ICAM-1 and VCAM-1 (124). This suggested that both murine and human NCR+ ILC3 could modulate tumor endothelial cell activation, promote leukocyte recruitment, favor the establishment of a pro-inflammatory microenvironment, and thus exert anti-tumoral functions.
A study conducted by Shikhagaie and colleagues identified a novel neuropilin 1 (NRP-1)+ ILC3 subset that is endowed with angiogenic potential (125). NRP-1 is a multifunctional receptor involved in the signal transduction of various angiogenic proteins, including VEGF (126). During pulmonary pathogenesis in chronic obstructive pulmonary disease (COPD), human NRP-1+ ILC3 located in proximity to blood vessels and lung ectopic lymphoid tissues, where they induced VCAM-1 and ICAM-1 expression on mesenchymal stromal cells, and efficiently responded to VEGF-A (125). However, the study did not provide any clear evidence of a direct crosstalk between the NRP-1+ ILC3 and the endothelial compartment. Further studies are therefore warranted to better understand the role of NRP1+ ILC3 in endothelial cell activation and to eventually extend this to cancer.
Since the early 2000s, IL-17 and endothelial cell crosstalk was evaluated in homeostasis and disease, including cancer (127–131). In primary human lung carcinomas, IL-17 was linked to the cancer cell production of a variety of pro-angiogenic factors, such as IL-6, IL-8, and VEGF (132), as well as to the upregulation of VEGF-C, a growth factor involved in lymphangiogenesis (133). Furthermore, it was proposed that murine pulmonary metastatic progression could be facilitated through the IL-17-dependent induction of VCAM-1 expression on lung endothelial cells, which correlated with altered vessel permeability and enhanced transendothelial migration of tumor cells (134). IL-17 was also recognized for its pro-inflammatory role in the development of intestinal malignancies (135). In colorectal and gastric cancers, IL-17 was described to interact with tumor cells to produce angiogenic mediators, including VEGF (129, 136). CRC-infiltrating Th17 cells induced the expression of G-CSF by CAF leading to the recruitment to the TME of CD11b+Gr1+ myeloid cells endowed with pro-angiogenic features, and thus promoting resistance to VEGF blockade therapy (137). In some of those studies, the cellular source of IL-17 was identified and mainly comprised of Th17 cells. However, ILC3 are also important producers of IL-17 (100) and ILC3-derived IL-17 was correlated with cancer progression (138). Therefore, further studies are required to elucidate the potential contribution of IL-17-producing ILC3 to tumor angiogenesis.
IL-22 is a cytokine belonging to the IL-10 family produced by ILC3 to maintain tissue homeostasis (139). The role of IL-22 in cancer is complex since IL-22 was shown to either promote or restrain tumor progression (139). After binding to its receptor (IL-22RA1), which is selectively expressed by non-hematopoietic cells (140), IL-22 induces phosphorylation of STAT-3. STAT-3 is a pleiotropic transcription factor that upon constitutive activation becomes oncogenic (141). High levels of IL-22 were detected in colonic cancer tissues, where IL-22 exerted pro-tumoral functions by promoting cancer cell activation of STAT-3, reducing tumor cell apoptosis, and increasing the levels of VEGF in the TME (142). Importantly, emerging evidence showed that also endothelial cells can express IL-22RA1 and thus can respond to IL-22 (143). Upon IL-22 stimulation, STAT-3 activation led to enhanced endothelial cell proliferation and survival, and promoted the growth of murine T-cell lymphoma (EL4) (143). By regulating the expression of the pro-invasive molecule alanyl aminopeptidase (Anpep) on the surface of endothelial cells, hepatic invariant-NKT (iNKT)-derived IL-22 modulated endothelial cell permeability and favored the seeding of circulating cancer cells in the liver, promoting CRC liver metastases (144). However, the molecular mechanisms by which IL-22 regulates endothelial cell permeability at metastatic sites remain unclear. These findings suggest a role for IL-22 to directly or indirectly shape endothelial cell functions in cancer. Although IL-22+ ILC3 were shown to have an important role in cancer (145, 146), the contribution of ILC3-derived IL-22 to tumor angiogenesis has not been elucidated yet.
ILC3 emerge as potential players in the regulation of tumor vasculature but the contribution of ILC1 and ILC2 to tumor angiogenesis is less understood. NK cell plasticity towards ILC1 was suggested as a strategy employed by tumors to evade immune surveillance (114). However, little is known about the potential of helper-like ILC1 to regulate blood vessel formation in cancer. This might be related to the reduced panel of unique markers that can be used to properly discern between NK cells and ILC1, particularly in cancer, where immune cells acquire mixed phenotypes and show high levels of heterogeneity. When it comes to ILC2, recent evidence showed that murine ILC2 could produce VEGF-A in asthmatic inflammation (147), but there is no evidence that this ILC2 pro-angiogenic function could be observed in cancer. Therefore, further investigations are needed to understand whether ILC1 and ILC2 could modulate cancer angiogenesis.
4 Conclusions and perspectives
In this review, we reported evidence of the crosstalk between endothelial cells and both myeloid cells and innate lymphocytes in the TME and the pathways that until now were described to modulate such a crosstalk. VEGF represents one of the major regulators of the interaction between myeloid cells and endothelial cells in cancer (44–46, 81), but is also involved in the crosstalk with NK cells (108–111), and the interaction between ILC and tumor endothelial cells is regulated by TNF, IL-8 and TGF-β (114, 124) (Figure 3). However, other molecules, including angiopoietins and metalloproteinases, also influence the crosstalk between endothelial cells and innate immune cells in the TME (46–48, 74, 76, 77, 81, 109) (Figure 3). The interaction between innate immune cells and endothelial cells in cancer is complex and sometimes ambiguous, since, depending on the cytokine signature of the TME, it can lead to either tumor regression or progression. Therefore, targeting the pro-angiogenic mediators in the TME with the aim to normalize the tumor vasculature and thus restraining the neoplastic growth might not have an obvious outcome. One of the therapeutic approaches aiming at interfering with tumor angiogenesis consists of anti-VEGF treatment. Anti-VEGF treatment leads to decreased vessel diameter and density, suggesting for a normalization of the tumor vasculature, and increased uptake of chemotherapy agents (148). However, the blockade of VEGF could also impact the migration and function of VEGF responsive immune cells (55, 56, 125, 149) and their progenitors (150), potentially interfering with cancer immune surveillance. Furthermore, clinical data indicated that anti-angiogenic treatments are not always efficient in normalizing the tumor vasculature and could lead to therapy resistance due to the presence of compensatory angiogenic pathways (151). Therefore, a combination approach should be taken into consideration when it comes to anti-angiogenic therapies. A correlation between the angiogenic profile and the response to immune-checkpoint blockade (ICB) was reported (118, 152, 153) and anti-angiogenic treatments were shown to enhance the efficacy of immunotherapies (154–156). Combined treatments with anti-angiogenic agents (anti-VEGF or anti-VEGFR2) and ICB stimulated vessel normalization and the formation of HEV, favoring immune control of tumor progression (154, 155), and induced anti-tumor T cell responses (156). In addition, it was shown that radiotherapy could further enhance the anti-cancer effect of combined anti-angiogenic and ICB treatments (157). These evidences strengthen the concept that indeed the crosstalk between tumor immunity and tumor vasculature has an important role in cancer progression and could be efficiently targeted. Recent studies indicated a contribution of tumor-infiltrating innate lymphocytes in determining the efficacy of ICB (121, 158–160). Importantly, the same stimulator of interferon genes (STING) that was shown to be involved in the suppression of tumor angiogenesis and vessel normalization (161) also contributed to tumor monocyte reprogramming and NK cell-mediated anti-tumor immunity (162). The activation of the STING/IFN I axis in tumors not only improved the innate cell-dependent efficacy of ICB (162), but was also shown to enhance the efficacy of anti-angiogenic therapies, namely VEGFR2 blockade (161). This suggests that combined therapies targeting pro-angiogenic factors and unleashing innate-dependent anti-tumor immunity could not only have therapeutic effects by their selves but could also improve the efficacy of other immunotherapies. Further investigations may extend our understanding of the mechanisms by which endothelial cells crosstalk with myeloid and innate lymphoid cells. Elucidation of the pathways regulating interactions between innate immune cells and the tumor vasculature will allow for a better design of those potential combined therapies that could interfere with tumor angiogenesis, promote normalization of tumor vessels and immune cell trafficking and activation, and enhance the efficacy of immunotherapies.
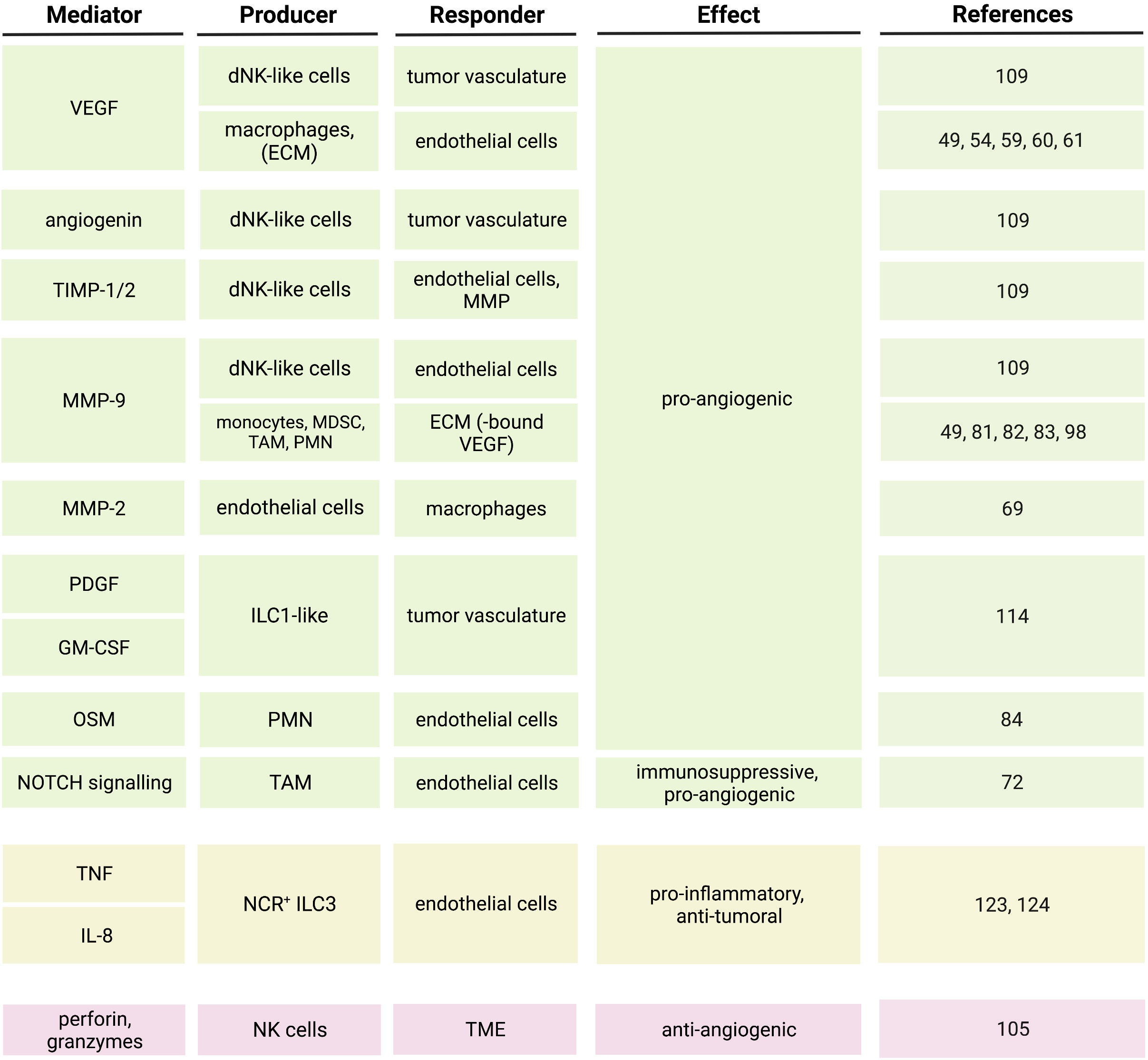
Figure 3 Soluble mediators and pathways regulating the crosstalk between innate immune cells and endothelial cells in the tumor microenvironment.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
We acknowledge financial support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin and the German Research Foundation (DFG).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol (2023) 20:197–210. doi: 10.1038/s41569-022-00770-1
2. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol (2007) 8:464–78. doi: 10.1038/nrm2183
3. Ricard N, Bailly S, Guignabert C, Simons M. The quiescent endothelium: signalling pathways regulating organ-specific endothelial normalcy. Nat Rev Cardiol (2021) 18:565–80. doi: 10.1038/s41569-021-00517-4
4. Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell (2007) 130:691–703. doi: 10.1016/j.cell.2007.06.054
5. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol (2013) 6:61. doi: 10.1186/1756-8722-6-61
6. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi: 10.1038/nri2171
7. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system? Nat Rev Immunol (2022) 22:576–88. doi: 10.1038/s41577-022-00694-4
8. Kuhlencordt PJ, Rosel E, Gerszten RE, Morales-Ruiz M, Dombkowski D, Atkinson WJ, et al. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. Am J Physiol Cell Physiol (2004) 286:C1195–202. doi: 10.1152/ajpcell.00546.2002
9. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15:692–704. doi: 10.1038/nri3908
10. Lee WL, Liles WC. Endothelial activation, dysfunction and permeability during severe infections. Curr Opin Hematol (2011) 18:191–6. doi: 10.1097/MOH.0b013e328345a3d1
11. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-c regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol (2011) 12:761–9. doi: 10.1038/ni.2062
12. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203:2569–75. doi: 10.1084/jem.20060925
13. Filippi MD. Mechanism of diapedesis: importance of the transcellular route. Adv Immunol (2016) 129:25–53. doi: 10.1016/bs.ai.2015.09.001
14. Mamdouh Z, Mikhailov A, Muller WA. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med (2009) 206:2795–808. doi: 10.1084/jem.20082745
15. Lin R, Chen F, Wen S, Teng T, Pan Y, Huang H. Interleukin-10 attenuates impairment of the blood-brain barrier in a severe acute pancreatitis rat model. J Inflammation (Lond) (2018) 15:4. doi: 10.1186/s12950-018-0180-0
16. Oshima T, Laroux FS, Coe LL, Morise Z, Kawachi S, Bauer P, et al. Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res (2001) 61:130–43. doi: 10.1006/mvre.2000.2288
17. Londono D, Carvajal J, Strle K, Kim KS, Cadavid D. IL-10 prevents apoptosis of brain endothelium during bacteremia. J Immunol (2011) 186:7176–86. doi: 10.4049/jimmunol.1100060
18. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
19. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell (2020) 37:21–36 e13. doi: 10.1016/j.ccell.2019.12.001
20. Folkman J. Tumor angiogenesis: a possible control point in tumor growth. Ann Intern Med (1975) 82:96–100. doi: 10.7326/0003-4819-82-1-96
21. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
22. Klein D. The tumor vascular endothelium as decision maker in cancer therapy. Front Oncol (2018) 8:367. doi: 10.3389/fonc.2018.00367
23. Brahimi-Horn C, Berra E, Pouyssegur J. Hypoxia: the tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol (2001) 11:S32–6. doi: 10.1016/S0962-8924(01)02126-2
24. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer (2008) 8:425–37. doi: 10.1038/nrc2397
25. Tilki D, Kilic N, Sevinc S, Zywietz F, Stief CG, Ergun S. Zone-specific remodeling of tumor blood vessels affects tumor growth. Cancer (2007) 110:2347–62. doi: 10.1002/cncr.23024
26. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature (1993) 362:841–4. doi: 10.1038/362841a0
27. Tang Y, Nakada MT, Kesavan P, McCabe F, Millar H, Rafferty P, et al. Extracellular matrix metalloproteinase inducer stimulates tumor angiogenesis by elevating vascular endothelial cell growth factor and matrix metalloproteinases. Cancer Res (2005) 65:3193–9. doi: 10.1158/0008-5472.CAN-04-3605
28. Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med (1995) 181:811–6. doi: 10.1084/jem.181.2.811
29. Zhang J, Lu T, Lu S, Ma S, Han D, Zhang K, et al. Single-cell analysis of multiple cancer types reveals differences in endothelial cells between tumors and normal tissues. Comput Struct Biotechnol J (2023) 21:665–76. doi: 10.1016/j.csbj.2022.12.049
30. Ohga N, Ishikawa S, Maishi N, Akiyama K, Hida Y, Kawamoto T, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol (2012) 180:1294–307. doi: 10.1016/j.ajpath.2011.11.035
31. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi: 10.1146/annurev-immunol-031210-101324
32. Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The myeloid cell compartment-cell by cell. Annu Rev Immunol (2019) 37:269–93. doi: 10.1146/annurev-immunol-042718-041728
33. Gasteiger G, D’Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular innate immunity: an old game with new players. J Innate Immun (2017) 9:111–25. doi: 10.1159/000453397
34. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8:618–31. doi: 10.1038/nrc2444
35. Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol (2015) 36:240–9. doi: 10.1016/j.it.2015.02.005
36. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
37. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell (2006) 124:263–6. doi: 10.1016/j.cell.2006.01.007
38. Kim HJ, Ji YR, Lee YM. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch Pharm Res (2022) 45:401–16. doi: 10.1007/s12272-022-01389-z
39. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
40. Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol (2002) 72:101–6. doi: 10.1189/jlb.72.1.101
41. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity (2021) 54:1883–1900.e5. doi: 10.1016/j.immuni.2021.07.007
42. Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell (2021) 39:1594–1609.e12. doi: 10.1016/j.ccell.2021.10.009
43. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
44. Xiong M, Elson G, Legarda D, Leibovich SJ. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol (1998) 153:587–98. doi: 10.1016/S0002-9440(10)65601-5
45. Pakala R, Watanabe T, Benedict CR. Induction of endothelial cell proliferation by angiogenic factors released by activated monocytes. Cardiovasc Radiat Med (2002) 3:95–101. doi: 10.1016/S1522-1865(02)00159-2
46. Medina RJ, O’Neill CL, O’Doherty TM, Knott H, Guduric-Fuchs J, Gardiner TA, et al. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med (2011) 17:1045–55. doi: 10.2119/molmed.2011.00129
47. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
48. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell (2008) 13:206–20. doi: 10.1016/j.ccr.2008.01.034
49. Huber R, Meier B, Otsuka A, Fenini G, Satoh T, Gehrke S, et al. Tumour hypoxia promotes melanoma growth and metastasis via high mobility group box-1 and M2-like macrophages. Sci Rep (2016) 6:29914. doi: 10.1038/srep29914
50. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res (2010) 20:701–12. doi: 10.1038/cr.2010.52
51. Schulze-Osthoff K, Risau W, Vollmer E, Sorg C. In situ Detection of basic fibroblast growth factor by highly specific antibodies. Am J Pathol (1990) 137:85–92.
52. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
53. Stefater J, Lewkowich I, Rao S, Mariggi G, Carpenter AC, Burr AR, et al. Regulation of angiogenesis by a non-canonical wnt-Flt1 pathway in myeloid cells. Nature (2011) 474:511–5. doi: 10.1038/nature10085
54. Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res (2019) 79:795–806. doi: 10.1158/0008-5472.CAN-18-2545
55. Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res (2008) 68:4340–6. doi: 10.1158/0008-5472.CAN-07-6705
56. Sawano A, Iwai S, Sakurai Y, Ito M, Shitara K, Nakahata T, et al. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood (2001) 97:785–91. doi: 10.1182/blood.V97.3.785
57. Okikawa S, Morine Y, Saito Y, Yamada S, Tokuda K, Teraoku H, et al. Inhibition of the VEGF signaling pathway attenuates tumor−associated macrophage activity in liver cancer. Oncol Rep (2022) 47:1–11. doi: 10.3892/or.2022.8282
58. Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci (2017) 147:1–73. doi: 10.1016/bs.pmbts.2017.02.005
59. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
60. Lin EY, Li JF, Bricard G, Wang W, Deng Y, Sellers R, et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol (2007) 1:288–302. doi: 10.1016/j.molonc.2007.10.003
61. Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature (2008) 456:814–8. doi: 10.1038/nature07445
62. Wang H, Tian T, Zhang J. Tumor-associated macrophages (TAMs) in colorectal cancer (CRC): from mechanism to therapy and prognosis. Int J Mol Sci (2021) 22:8470. doi: 10.3390/ijms22168470
63. Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci (2013) 104:437–44. doi: 10.1111/cas.12096
64. Yang C, Wei C, Wang S, Shi D, Zhang C, Lin X, et al. Elevated CD163(+)/CD68(+) ratio at tumor invasive front is closely associated with aggressive phenotype and poor prognosis in colorectal cancer. Int J Biol Sci (2019) 15:984–98. doi: 10.7150/ijbs.29836
65. Feng Q, Chang W, Mao Y, He G, Zheng P, Tang W, et al. Tumor-associated macrophages as prognostic and predictive biomarkers for postoperative adjuvant chemotherapy in patients with stage II colon cancer. Clin Cancer Res (2019) 25:3896–907. doi: 10.1158/1078-0432.CCR-18-2076
66. Edin S, Wikberg ML, Dahlin AM, Rutegård J, Öberg Å., Oldenborg PA, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PloS One (2012) 7:e47045. doi: 10.1371/journal.pone.0047045
67. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22:6995. doi: 10.3390/ijms22136995
68. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell (2021) 184:792–809.e23. doi: 10.1016/j.cell.2021.01.010
69. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181:442–459.e29. doi: 10.1016/j.cell.2020.03.048
70. Ben-Yosef Y, Miller A, Shapiro S, Lahat N. Hypoxia of endothelial cells leads to MMP-2-dependent survival and death. Am J Physiol Cell Physiol (2005), 289:C1321–31. doi: 10.1152/ajpcell.00079.2005
71. Zucker S, Vacirca J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev (2004) 23:101–17. doi: 10.1023/A:1025867130437
72. Sharma A, Seow JJW, Dutertre CA, Pai R, Blériot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell (2020) 183:377–394.e21. doi: 10.1016/j.cell.2020.08.040
73. Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, et al. Distinct roles of the receptor tyrosine kinases tie-1 and tie-2 in blood vessel formation. Nature (1995) 376:70–4. doi: 10.1038/376070a0
74. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell (2005) 8:211–26. doi: 10.1016/j.ccr.2005.08.002
75. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood (2007) 109:5276–85. doi: 10.1182/blood-2006-10-053504
76. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19:512–26. doi: 10.1016/j.ccr.2011.02.005
77. Scholz A, Lang V, Henschler R, Czabanka M, Vajkoczy P, Chavakis E, et al. Angiopoietin-2 promotes myeloid cell infiltration in a β2-integrin-dependent manner. Blood (2011) 118:5050–9. doi: 10.1182/blood-2011-03-343293
78. Rao S, Lobov IB, Vallance JE, Tsujikawa K, Shiojima I, Akunuru S, et al. Obligatory participation of macrophages in an angiopoietin 2-mediated cell death switch. Development (2007) 134:4449–58. doi: 10.1242/dev.012187
79. Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, et al. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature (2005) 437:417–21. doi: 10.1038/nature03928
80. Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol (2011) 55:495–503. doi: 10.1387/ijdb.103227sn
81. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest (2008) 118:3367–77. doi: 10.1172/JCI35213
82. Pahler JC, Tazzyman S, Erez N, Chen YY, Murdoch C, Nozawa H, et al. Plasticity in tumor-promoting inflammation: impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia (2008) 10:329–40. doi: 10.1593/neo.07871
83. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
84. Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin m: potential implications for tumor progression. Cancer Res (2005) 65:8896–904. doi: 10.1158/0008-5472.CAN-05-1734
85. Araujo AM, Abaurrea A, Azcoaga P, López-Velazco JI, Manzano S, Rodriguez J, et al. Stromal oncostatin m cytokine promotes breast cancer progression by reprogramming the tumor microenvironment. J Clin Invest (2022) 132:e148667. doi: 10.1172/JCI148667
86. Caligiuri A, Gitto S, Lori G, Marra F, Parola M, Cannito S, et al. Oncostatin M: From intracellular signaling to therapeutic targets in liver cancer. Cancers (2022) 14:4211. doi: 10.3390/cancers14174211
87. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature (2007) 450:825–31. doi: 10.1038/nature06348
88. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3:133–46. doi: 10.1038/nri1001
89. Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood (2006) 107:2613–8. doi: 10.1182/blood-2005-07-2965
90. Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res (2004) 64:5535–8. doi: 10.1158/0008-5472.CAN-04-1272
91. Indraccolo S, Gola E, Rosato A, Minuzzo S, Habeler W, Tisato V, et al. Differential effects of angiostatin, endostatin and interferon-alpha(1) gene transfer on in vivo growth of human breast cancer cells. Gene Ther (2002) 9:867–78. doi: 10.1038/sj.gt.3301703
92. Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of vegf-a. Nat Med (2004) 10:950–8. doi: 10.1038/nm1097
93. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
94. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
95. Ugel S, Canè S, De Sanctis F, Bronte V. Monocytes in the tumor microenvironment. Annu Rev Pathol (2021) 16:93–122. doi: 10.1146/annurev-pathmechdis-012418-013058
96. Steube KG, Meyer C, Drexler HG. Secretion of functional hematopoietic growth factors by human carcinoma cell lines. Int J Cancer (1998) 78:120–4. doi: 10.1002/(SICI)1097-0215(19980925)78:1<120::AID-IJC19>3.0.CO;2-F
97. Olingy CE, Dinh HQ, Hedrick CC. Monocyte heterogeneity and functions in cancer. J Leukoc Biol (2019) 106:309–22. doi: 10.1002/JLB.4RI0818-311R
98. Sidibe A, Ropraz P, Jemelin S, Emre Y, Poittevin M, Pocard M, et al. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun (2018) 9:355. doi: 10.1038/s41467-017-02610-0
99. Mattiola I, Diefenbach A. Innate lymphoid cells and cancer at border surfaces with the environment. Semin Immunol (2019) 41:101278. doi: 10.1016/j.smim.2019.06.001
100. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
101. Diefenbach A. Profiling the diversity of innate lymphoid cells. Nat Immunol (2015) 16:222–4. doi: 10.1038/ni.3107
102. Mattiola I. Immune circuits to shape natural killer cells in cancer. Cancers (Basel) (2021) 13:3225. doi: 10.3390/cancers13133225
103. Branzk N, Gronke K, Diefenbach A. Innate lymphoid cells, mediators of tissue homeostasis, adaptation and disease tolerance. Immunol Rev (2018) 286:86–101. doi: 10.1111/imr.12718
104. Lai D, Tang J, Chen L, Fan EK, Scott MJ, Li Y, et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis (2018) 9:369. doi: 10.1038/s41419-018-0412-5
105. Yao L, Sgadari C, Furuke K, Bloom ET, Teruya-Feldstein J, Tosato G. Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood (1999) 93:1612–21. doi: 10.1182/blood.V93.5.1612
106. Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56bright NK cells: an update. J Immunol (2016) 196:2923–31. doi: 10.4049/jimmunol.1502570
107. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
108. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med (2006) 12:1065–74. doi: 10.1038/nm1452
109. Bruno A, Bassani B, D’Urso DG, Pitaku I, Cassinotti E, Pelosi G, et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J (2018) 32:5365–77. doi: 10.1096/fj.201701103R
110. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov (2016) 6:414–29. doi: 10.1158/2159-8290.CD-15-0732
111. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia (2013) 15:133–42. doi: 10.1593/neo.121758
112. Regis S, Dondero A, Caliendo F, Bottino C, Castriconi R. NK cell function regulation by TGF-β-Induced epigenetic mechanisms. Front Immunol (2020) 11:311. doi: 10.3389/fimmu.2020.00311
113. Allan DS, Rybalov B, Awong G, Zúñiga-Pflücker JC, Kopcow HD, Carlyle JR, et al. TGF-β affects development and differentiation of human natural killer cell subsets. Eur J Immunol (2010) 40:2289–95. doi: 10.1002/eji.200939910
114. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol (2017) 18:1004–15. doi: 10.1038/ni.3800
115. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature (2001) 413:165–71. doi: 10.1038/35093109
116. Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, et al. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife (2017) 6:e30881. doi: 10.7554/eLife.30881
117. Veerman K, Tardiveau C, Martins F, Coudert J, Girard JP. Single-cell analysis reveals heterogeneity of high endothelial venules and different regulation of genes controlling lymphocyte entry to lymph nodes. Cell Rep (2019) 26:3116–3131.e5. doi: 10.1016/j.celrep.2019.02.042
118. Asrir A, Tardiveau C, Coudert J, Laffont R, Blanchard L, Bellard E, et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell (2022) 40:318–334.e9. doi: 10.1016/j.ccell.2022.01.002
119. Ran GH, Lin YQ, Tian L, Zhang T, Yan DM, Yu JH, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther (2022) 7:205. doi: 10.1038/s41392-022-01058-z
120. Mattiola I, Diefenbach A. Enabling anti-tumor immunity by unleashing ILC2. Cell Res (2020) 30:461–2. doi: 10.1038/s41422-020-0330-9
121. Mattiola I, Diefenbach A. Regulation of innate immune system function by the microbiome: consequences for tumor immunity and cancer immunotherapy. Semin Immunol (2023) 66:101724. doi: 10.1016/j.smim.2023.101724
122. Heinrich B, Korangy F. Plasticity of innate lymphoid cells in cancer. Front Immunol (2022) 13:886520. doi: 10.3389/fimmu-13-886520
123. Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol (2010) 11:1030–8. doi: 10.1038/ni.1947
124. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun (2015) 6:8280. doi: 10.1038/ncomms9280
125. Shikhagaie MM, Björklund Å K, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 is expressed on lymphoid tissue residing LTi-like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep (2017) 18:1761–73. doi: 10.1016/j.celrep.2017.01.063
126. Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell (1998) 92:735–45. doi: 10.1016/S0092-8674(00)81402-6
127. Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol (2001) 167:4137–40. doi: 10.4049/jimmunol.167.8.4137
128. Li TJ, Jiang YM, Hu YF, Huang L, Yu J, Zhao LY, et al. Interleukin-17-Producing neutrophils link inflammatory stimuli to disease progression by promoting angiogenesis in gastric cancer. Clin Cancer Res (2017) 23:1575–85. doi: 10.1158/1078-0432.CCR-16-0617
129. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun (2011) 407:348–54. doi: 10.1016/j.bbrc.2011.03.021
130. Kato T, Furumoto H, Ogura T, Onishi Y, Irahara M, Yamano S, et al. Expression of IL-17 mRNA in ovarian cancer. Biochem Biophys Res Commun (2001) 282:735–8. doi: 10.1006/bbrc.2001.4618
131. Numasaki M, Lotze MT, Sasaki H. Interleukin-17 augments tumor necrosis factor-alpha-induced elaboration of proangiogenic factors from fibroblasts. Immunol Lett (2004) 93:39–43. doi: 10.1016/j.imlet.2004.01.014
132. Huang Q, Duan L, Qian X, Fan J, Lv Z, Zhang X, et al. IL-17 promotes angiogenic factors IL-6, IL-8, and vegf production via Stat1 in lung adenocarcinoma. Sci Rep (2016) 6:36551. doi: 10.1038/srep36551
133. Chen X, Xie Q, Cheng X, Diao X, Cheng Y, Liu J, et al. Role of interleukin-17 in lymphangiogenesis in non-small-cell lung cancer: enhanced production of vascular endothelial growth factor c in non-small-cell lung carcinoma cells. Cancer Sci (2010) 101:2384–90. doi: 10.1111/j.1349-7006.2010.01684.x
134. Kulig P, Burkhard S, Mikita-Geoffroy J, Croxford AL, Hövelmeyer N, Gyülvészi G, et al. IL17A-mediated endothelial breach promotes metastasis formation. Cancer Immunol Res (2016) 4:26–32. doi: 10.1158/2326-6066.CIR-15-0154
135. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol (2019) 20:1594–602. doi: 10.1038/s41590-019-0514-y
136. Wu X, Yang T, Liu X, Guo JN, Xie T, Ding Y, et al. IL-17 promotes tumor angiogenesis through Stat3 pathway mediated upregulation of VEGF in gastric cancer. Tumour Biol (2016) 37:5493–501. doi: 10.1007/s13277-015-4372-4
137. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med (2013) 19:1114–23. doi: 10.1038/nm.3291
138. Liu Y, Song Y, Lin D, Lei L, Mei Y, Jin Z, et al. NCR(-) group 3 innate lymphoid cells orchestrate IL-23/IL-17 axis to promote hepatocellular carcinoma development. EBioMedicine (2019) 41:333–44. doi: 10.1016/j.ebiom.2019.02.050
139. Hernandez P, Gronke K, Diefenbach A. A catch-22: interleukin-22 and cancer. Eur J Immunol (2018) 48:15–31. doi: 10.1002/eji.201747183
140. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
141. Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated myc expression is required for src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci USA (2001) 98:7319–24. doi: 10.1073/pnas.131568898
142. Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer (2013) 13:59. doi: 10.1186/1471-2407-13-59
143. Protopsaltis NJ, Liang W, Nudleman E, Ferrara N. Interleukin-22 promotes tumor angiogenesis. Angiogenesis (2019) 22:311–23. doi: 10.1007/s10456-018-9658-x
144. Giannou AD, Kempski J, Shiri AM, Lücke J, Zhang T, Zhao L, et al. Tissue resident iNKT17 cells facilitate cancer cell extravasation in liver metastasis via interleukin-22. Immunity (2023) 56:125–142.e12. doi: 10.1016/j.immuni.2022.12.014
145. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210:917–31. doi: 10.1084/jem.20122308
146. Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature (2019) 566:249–53. doi: 10.1038/s41586-019-0899-7
147. Shen X, Pasha MA, Hidde K, Khan A, Liang M, Guan W, et al. Group 2 innate lymphoid cells promote airway hyperresponsiveness through production of VEGFA. J Allergy Clin Immunol (2018) 141:1929–1931.e4. doi: 10.1016/j.jaci.2018.01.005
148. Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res (2005) 65:671–80. doi: 10.1158/0008-5472.671.65.3
149. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother (2008) 57:1115–24. doi: 10.1007/s00262-007-0441-x
150. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
151. Bueno MJ, Mouron S, Quintela-Fandino M. Personalising and targeting antiangiogenic resistance: a complex and multifactorial approach. Br J Cancer (2017) 116:1119–25. doi: 10.1038/bjc.2017.69
152. Subramanian M, Kabir AU, Barisas D, Krchma K, Choi K. Conserved angio-immune subtypes of the tumor microenvironment predict response to immune checkpoint blockade therapy. Cell Rep Med (2023) 4:100896. doi: 10.1016/j.xcrm.2022.100896
153. Hua Y, Vella G, Rambow F, Allen E, Antoranz Martinez A, Duhamel M, et al. Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1(+) T lymphocyte niches through a feed-forward loop. Cancer Cell (2022) 40:1600–1618 e10. doi: 10.1016/j.ccell.2022.11.002
154. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med (2017) 9:eaak9679. doi: 10.1126/scitranslmed.aak9679
155. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (2020) 71:1247–61. doi: 10.1002/hep.30889
156. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-a drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol (2019) 4:eaay0555. doi: 10.1126/sciimmunol.aay0555
157. Zhu L, Yu X, Wang L, Liu J, Qu Z, Zhang H, et al. Angiogenesis and immune checkpoint dual blockade in combination with radiotherapy for treatment of solid cancers: opportunities and challenges. Oncogenesis (2021) 10:47. doi: 10.1038/s41389-021-00335-w
158. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
159. Jacquelot N, Seillet C, Wang M, Pizzolla A, Liao Y, Hediyeh-Zadeh S, et al. Blockade of the co-inhibitory molecule PD-1 unleashes ILC2-dependent antitumor immunity in melanoma. Nat Immunol (2021) 22:851–64. doi: 10.1038/s41590-021-00943-z
160. Goc J, Lv M, Bessman NJ, Flamar AL, Sahota S, Suzuki H, et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell (2021) 184:5015–5030.e16. doi: 10.1016/j.cell.2021.07.029
161. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest (2019) 129:4350–64. doi: 10.1172/JCI125413
Keywords: myeloid cells, innate lymphocytes, endothelial cells, cancer, angiogenesis
Citation: Ebeling S, Kowalczyk A, Perez-Vazquez D and Mattiola I (2023) Regulation of tumor angiogenesis by the crosstalk between innate immunity and endothelial cells. Front. Oncol. 13:1171794. doi: 10.3389/fonc.2023.1171794
Received: 22 February 2023; Accepted: 10 April 2023;
Published: 10 May 2023.
Edited by:
Pina Ziranu, University of Cagliari, ItalyReviewed by:
Stefano Mariani, University of Cagliari, ItalyCopyright © 2023 Ebeling, Kowalczyk, Perez-Vazquez and Mattiola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Mattiola, aXJlbmUubWF0dGlvbGFAY2hhcml0ZS5kZQ==
†These authors have contributed equally to this work