
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 19 June 2023
Sec. Gastrointestinal Cancers: Hepato Pancreatic Biliary Cancers
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1169586
This article is part of the Research Topic New Insights into Molecular Mechanisms and Targeted Therapy for Gastrointestinal Tumors View all 9 articles
 Prashanth Ashok Kumar1†*
Prashanth Ashok Kumar1†* Serenella Serinelli2
Serenella Serinelli2 Daniel J. Zaccarini2
Daniel J. Zaccarini2 Richard Huang3
Richard Huang3 Natalie Danziger3Tyler Janovitz3
Natalie Danziger3Tyler Janovitz3 Alina Basnet1Abirami Sivapiragasam1
Alina Basnet1Abirami Sivapiragasam1 Stephen Graziano1Jeffrey S. Ross2,3
Stephen Graziano1Jeffrey S. Ross2,3Introduction: KRAS mutation is a common occurrence in Pancreatic Ductal Adenocarcinoma (PDA) and is a driver mutation for disease development and progression. KRAS wild-type PDA may constitute a distinct molecular and clinical subtype. We used the Foundation one data to analyze the difference in Genomic Alterations (GAs) that occur in KRAS mutated and wild-type PDA.
Methods: Comprehensive genomic profiling (CGP) data, tumor mutational burden (TMB), microsatellite instability (MSI) and PD-L1 by Immunohistochemistry (IHC) were analyzed.
Results and discussion: Our cohort had 9444 cases of advanced PDA. 8723 (92.37%) patients had KRAS mutation. 721 (7.63%) patients were KRAS wild-type. Among potentially targetable mutations, GAs more common in KRAS wild-type included ERBB2 (mutated vs wild-type: 1.7% vs 6.8%, p <0.0001), BRAF (mutated vs wild-type: 0.5% vs 17.9%, p <0.0001), PIK3CA (mutated vs wild-type: 2.3% vs 6.5%, p <0.001), FGFR2 (mutated vs wild-type: 0.1% vs 4.4%, p <0.0001), ATM (mutated vs wild-type: 3.6% vs 6.8%, p <0.0001). On analyzing untargetable GAs, the KRAS mutated group had a significantly higher percentage of TP53 (mutated vs wild-type: 80.2% vs 47.6%, p <0.0001), CDKN2A (mutated vs wild-type: 56.2% vs 34.4%, p <0.0001), CDKN2B (mutated vs wild-type: 28.9% vs 23%, p =0.007), SMAD4 (mutated vs wild-type: 26.8% vs 15.7%, p <0.0001) and MTAP (mutated vs wild-type: 21.7% vs 18%, p =0.02). ARID1A (mutated vs wild-type: 7.7% vs 13.6%, p <0.0001 and RB1(mutated vs wild-type: 2% vs 4%, p =0.01) were more prevalent in the wild-type subgroup. Mean TMB was higher in the KRAS wild-type subgroup (mutated vs wild-type: 2.3 vs 3.6, p <0.0001). High TMB, defined as TMB > 10 mut/mB (mutated vs wild-type: 1% vs 6.3%, p <0.0001) and very-high TMB, defined as TMB >20 mut/mB (mutated vs wild-type: 0.5% vs 2.4%, p <0.0001) favored the wild-type. PD-L1 high expression was similar between the 2 groups (mutated vs wild-type: 5.7% vs 6%,). GA associated with immune checkpoint inhibitors (ICPIs) response including PBRM1 (mutated vs wild-type: 0.7% vs 3.2%, p <0.0001) and MDM2 (mutated vs wild-type: 1.3% vs 4.4%, p <0.0001) were more likely to be seen in KRAS wild-type PDA.
Pancreatic Ductal Adenocarcinoma (PDA) is the most common pancreatic neoplasm comprising 90% of the malignancies arising from the organ (1). The incidence of PDA has been on the rise over the past decade and is projected to be the second leading cause of cancer related mortality in the United States by 2030. Despite the improvement in 5-year survival from 5.26% in 2000 to nearly 10% in 2020, the overall prognosis remains grim (2). The median survival of patients with PDA is only 3-5 months despite all the advances in therapy, necessitating the need for further research (3).
Somatic mutation testing for alterations like BRCA, KRAS, HER2, PALB2 and mismatch repair proteins, for PDA patients is recommended in the NCCN guidelines (4). Around 10% of PDA patients express alterations that are potentially targetable (5). PDA can be highly heterogenous in both biology and clinical behavior (6). KRAS mutation is a common occurrence in PDA and is identified in more than 90% of the sequenced cases. KRAS mutation occurs early in the development of PDA and is a key oncologic driver in this disease (7). KRAS wild-type PDA, although relatively less frequent, appears to have distinctive characteristics, both genetically and clinically. Treatment resistant PDA may feature selection of KRAS wild-type cancer cells that confer a survival advantage, making them more aggressive and resistant to targeted therapy (8). Thus, KRAS mutated and wild-type PDA may represent contrasting molecular subtypes and this distinction has important implications in assessing patients’ clinical course (7, 9). Their response to treatments like immune checkpoint inhibitors (ICPIs) therapy may also be different, as KRAS mutations have shown to affect the immune-microenvironment (10). Modern approaches for the treatment of PDA have moved towards personalized care and identifying therapeutic molecular targets that have the potential to impact the selection of treatment lines (11). This is evident from the results of the recently published results of CodeBreaK 100, that solidified the anti-cancer activity of sotorasib in this disease (12). In this study, we utilized a large database (13), to provide a descriptive analysis of the differences in genomic alterations (GAs) between KRAS mutated and wild-type PDAs (14) and characterize the GAs that occur in the less common KRAS wild-type PDAs.
Approval for this study, including a waiver of informed consent, was obtained from the Western Institutional Review Board (Protocol No. 20152817). A retrospective database search of a CLIA-certified and CAP-accredited reference molecular laboratory was performed for 9,444 PDA tissue samples. All PDA cases were clinically advanced, either inoperable or metastatic. The patient age and gender, routine histology and immunohistochemical staining results and confirmation of the diagnosis, were extracted from medical records and pathology reports. All PDA cases submitted to Foundation Medicine and sequenced using the FoundationOne CDx assay from January 1, 2018, to December 31, 2020, were eligible for inclusion in this study. Only cases with adequate tissue sample size, DNA extraction amounts of 50 ng or greater, tissues with a minimum of 20% tumor nuclear area versus benign nuclear area either before or after pathologist-guided macro-enrichment were included. In addition, cases with low tumor purity on sequencing or inadequate sequencing coverage depth as described in the FoundationOne CDx US FDA approval were excluded from the study (14).
Comprehensive genomic profiling (CGP) of all 9,444 PDA FFPE tissue samples was performed on extracted DNA using hybridization-capture- adaptor ligation–based libraries (FoundationOneCDx, Foundation Medicine, Inc.). All samples forwarded for DNA extraction contained a minimum of 20% tumor nuclei. The samples were assayed using all coding exons from 324 cancer related genes, plus select introns from at least 31 genes frequently rearranged in cancer. The PDA specimens were evaluated for all classes of GAs including base substitutions, insertions, deletions, copy number alterations (amplifications and homozygous deletions), and for select gene fusions/rearrangements, as previously described (14–16). The bioinformatics processes used in this study included Bayesian algorithms to detect base substitutions, local assembly algorithms to detect short insertions and deletions, a comparison with process-matched normal control samples to detect gene copy number alterations and an analysis of chimeric read pairs to identify gene fusions as previously described. To help visualize the sequencing data results, an oncoprint plot was generated with the online tools of the cbio portal as described by Gao et al (17) and Cerami et al (18). Tumor mutational burden was determined on 0.83–1.14 Mb of sequenced, as previously described. In this study, mutational burden scores were defined by mutation/Mb. Assessment of microsatellite instability was performed from DNA sequencing at least 95 loci, as previously described. Each microsatellite locus had repeat length of 7–39 bp. The next-generation sequencing based “microsatellite instability score” was translated into categorical “microsatellite instability high”, “microsatellite instability intermediate”, or “microsatellite stable” by unsupervised clustering of specimens for which microsatellite instability status was previously assessed via gold standard methods (19, 20).
PD-L1 expression was determined on subsets of the tumors using the DAKO 22C3 assay with low-positive tumor cell scoring defined as 1%-49% staining and high-positive tumor cell scoring defined as 50% staining (21). Anti-PD-L1 staining was done using the Dako 22C3 IHC kit, following the instructions provided in the kit protocol. Results were scored using the widely used tumor proportional score system (TPS) (22).
Chi-square test and Mann Whitney U test were used in the statistical comparisons of the 2 groups. Statistical significance was defined as p < 0.05.
8723/9444 (92.37%) of the PDA featured KRAS mutation. 721 (7.63%) patients were KRAS wild-type. Males were more prevalent in both groups [KRAS mutated: 52/48% (M/F), KRAS wild-type 61/39% (M/F)]. The median age was similar in both groups [KRAS mutated: 67 years, KRAS wild-type: 65 years]. The average GA/tumor was 4.88 for KRAS mutated and 4.47 for KRAS wild type (Table 1).
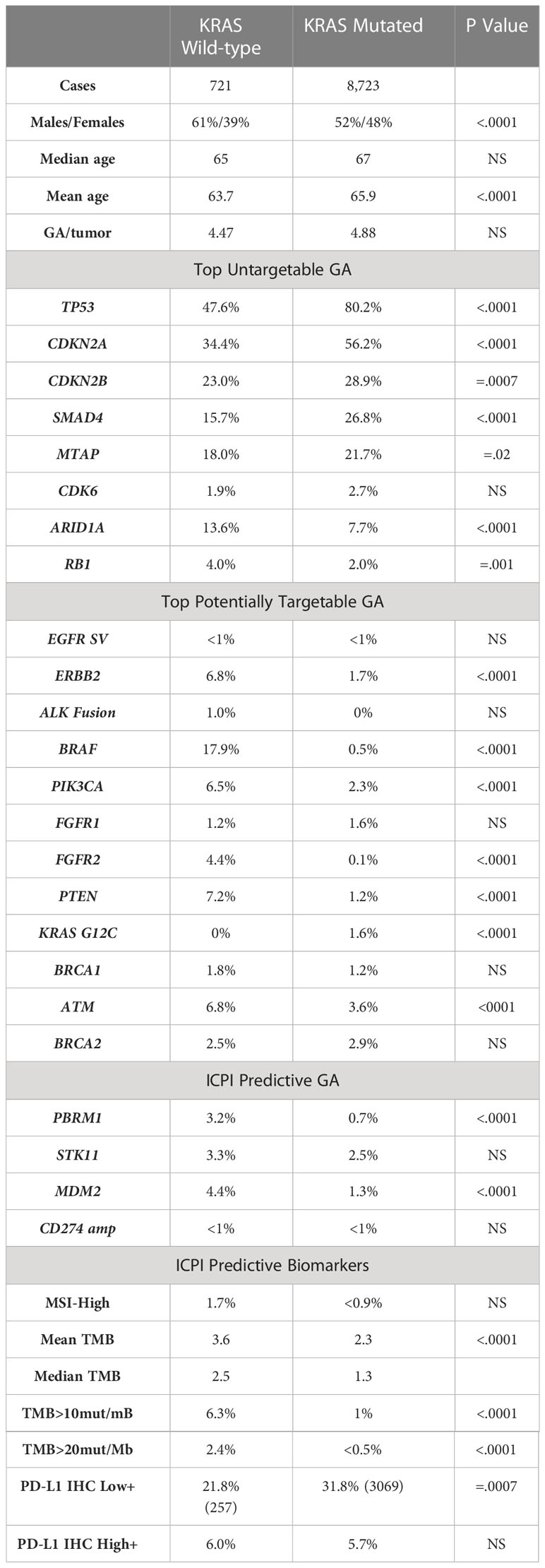
Table 1 Descriptive analysis of the patient characteristics and various GAs.
The descriptive analysis of the various GAs in our cohort, divided into targetable and untargetable GAs is shown in Table 1. Among potentially targetable mutations, GAs more common in KRAS wild-type included ERBB2 (mutated vs wild-type: 1.7% vs 6.8%, p <0.0001), BRAF (mutated vs wild-type: 0.5% vs 17.9%, p <0.0001), PIK3CA (mutated vs wild-type: 2.3% vs 6.5%, p <0.001), FGFR2 (mutated vs wild-type: 0.1% vs 4.4%, p <0.0001), ATM (mutated vs wild-type: 3.6% vs 6.8%, p <0.0001). KRAS G12C comprised 1.6% of the mutated group. On analyzing untargetable GAs, the KRAS mutated group had a significantly higher percentage of TP53 (mutated vs wild-type: 80.2% vs 47.6%, p <0.0001), CDKN2A (mutated vs wild-type: 56.2% vs 34.4%, p <0.0001), CDKN2B (mutated vs wild-type: 28.9% vs 23%, p =0.007), SMAD4 (mutated vs wild-type: 26.8% vs 15.7%, p <0.0001) and MTAP (mutated vs wild-type: 21.7% vs 18%, p =0.02). ARID1A (mutated vs wild-type: 7.7% vs 13.6%, p <0.0001) and RB1 (mutated vs wild-type: 2% vs 4%, p =0.01) were more prevalent in the wild-type subgroup.
Markers predictive of ICPIs therapy response were studied and favored the wild-type subgroup. Mean tumor mutational burden (TMB) was higher in the wild-type subgroup (mutated vs wild-type: 2.3 vs 3.6, p <0.0001). High TMB, defined as TMB > 10 mut/mB (mutated vs wild-type 1% vs 6.3%, p <0.0001) and ultra-high TMB, defined as TMB >20 mut/mB (mutated vs wild-type: 0.5% vs 2.4%, p <0.0001) favored the wild-type. PD-L1 high was similar between the 2 groups (mutated vs wild-type: 5.7% vs 6%). GAs predictive of immune checkpoint therapy (IO) response like PBRM1 (mutated vs wild-type: 0.7% vs 3.2%, p <0.0001) and MDM2 (mutated vs wild-type: 1.3% vs 4.4%, p <0.0001) were more likely to be seen in wild-type subtype.
Figures 1, 2 shows the long tail plots of the various GA in KRAS wild-type and KRAS mutated PDA respectively.
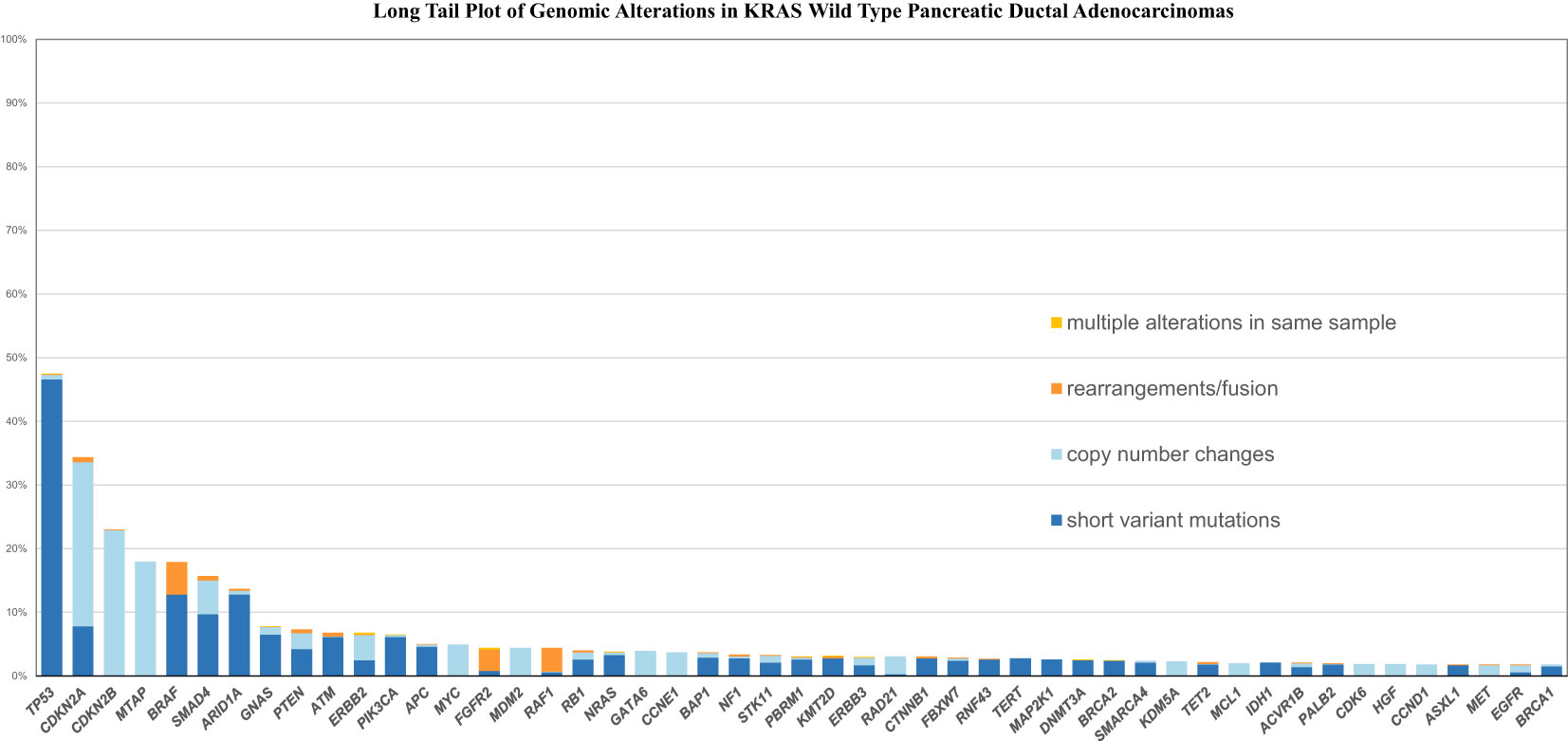
Figure 1 Long tail plot of GA in KRAS Wild-Type PDA.
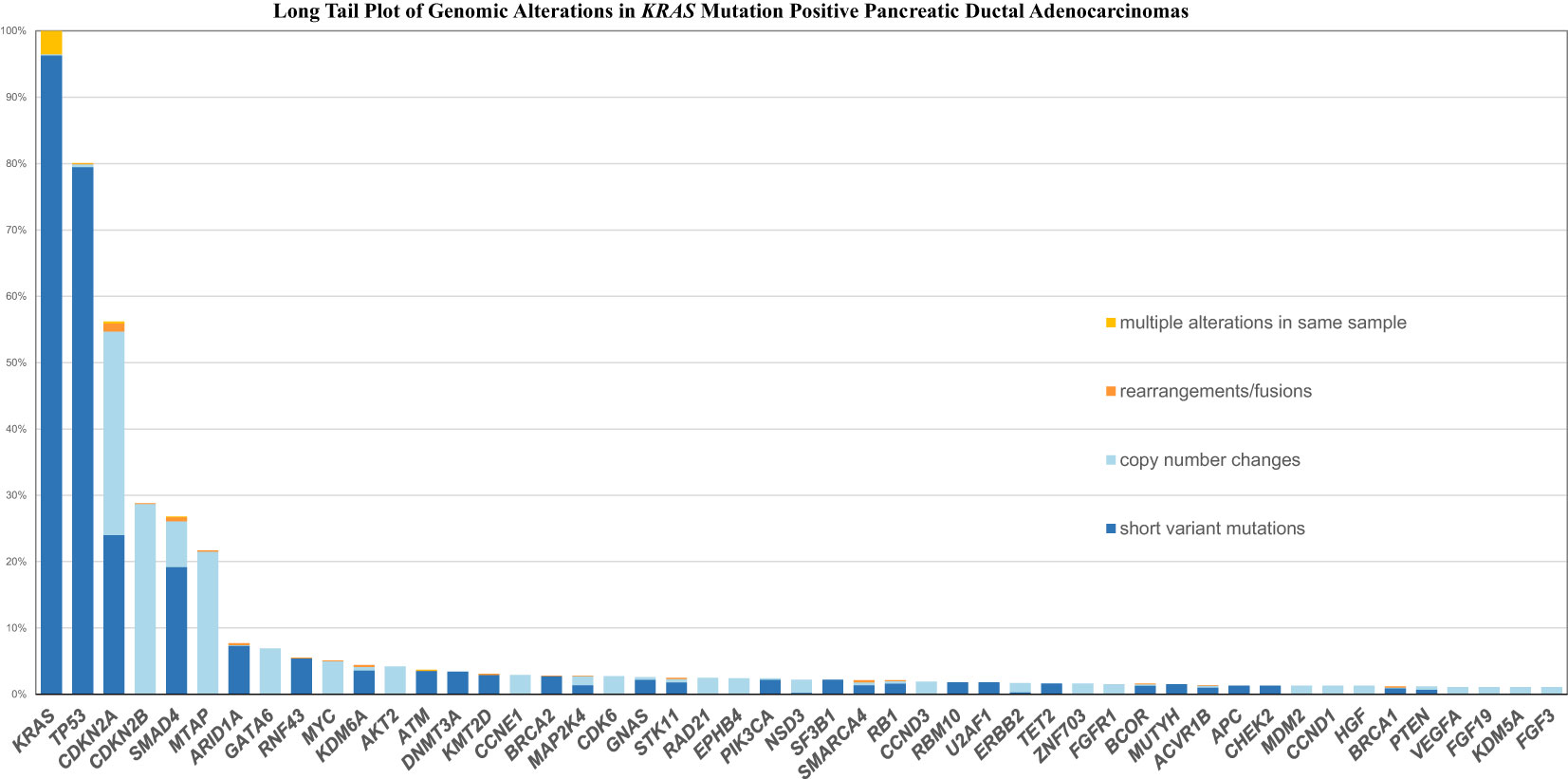
Figure 2 Long Tail Plot of GA in KRAS mutated PDA.
Figure 3 demonstrates the distribution of the various KRAS short variant alterations. Sample size is different from the cohort used in our study and includes earlier versions of the Foundation One CDx test with different/smaller bait sets.
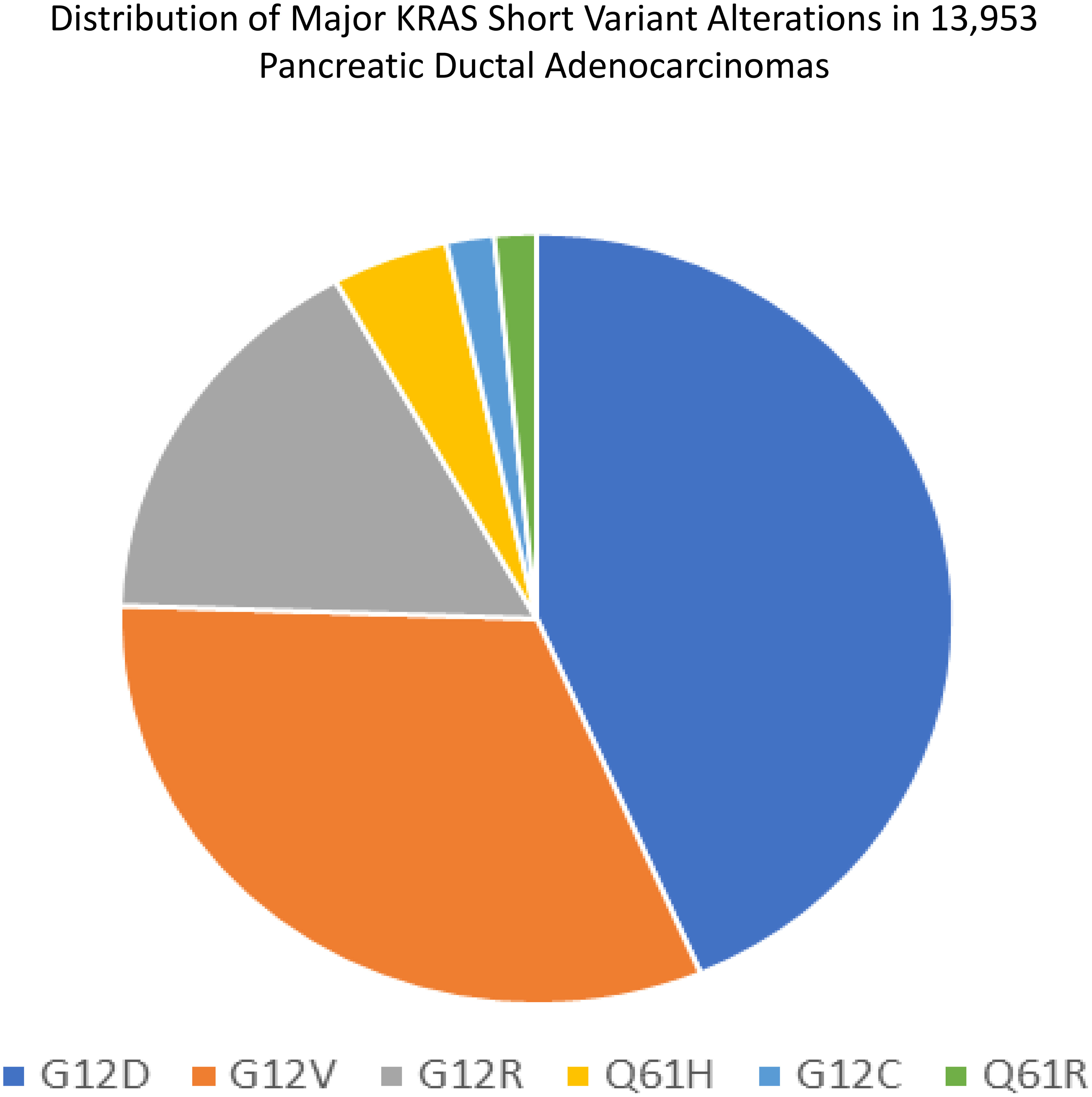
Figure 3 Distribution of major KRAS short variant alterations in 13,953 PDAs. Sample size is different from the cohort used in our study and includes earlier versions of the Foundation One CDx test with different/smaller bait sets.
Figure 4 shows a case of metastatic PDA to the liver in a 51-year-old man. The tumor was positive for CK7 and CA19-9 and negative for CK20, HAS and TTF1. On comprehensive genomic profiling this tumor was microsatellite (MS) stable and featured a TMB of 8 mutations/MB. It had a KRAS G12C mutation along other short variant mutations in CDKN2A/B and TP53 along with MDM2 amplification.
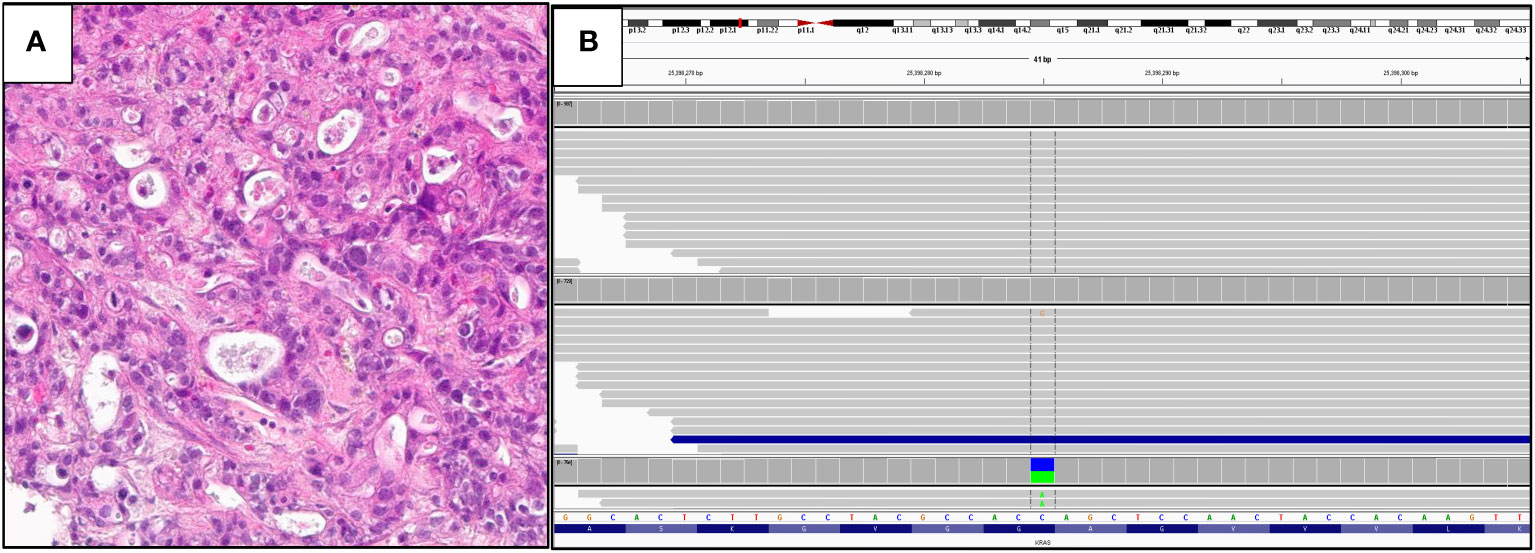
Figure 4 (A) Hematoxylin and eosin: histology of the tumor. (B) IGV View of KRAS G12C mutation.
Figure 5 shows a case of metastatic PDA to the liver in a 75-year-old woman. This tumor stained positively for CK7 and CA19-9 and negatively for CK20 and CDX2. On comprehensive genomic profiling, this tumor was KRAS wild-type, MS stable and featured a low TMB of 1 mutation/Mb. There was major ERBB2 amplification at 148 copies along with APC E468*, MAP2K4 Q316* and TP53 C275F short variant mutations, CCNE1 and CRKL amplification and a CDK12 inversion exons 8-11.
Figures 4B, 5B were generated by the Foundation Medicine “Curation Analysis Tool Interface” linked directly to the Integrative Genomics Viewer (IGV) developed by the Broad Institute of the Massachusetts Institute of Technology (23). All the GA and mutations seen in both the cohorts are available in the supplement (S1: KRAS Wild-Type, S2: KRAS mutated).
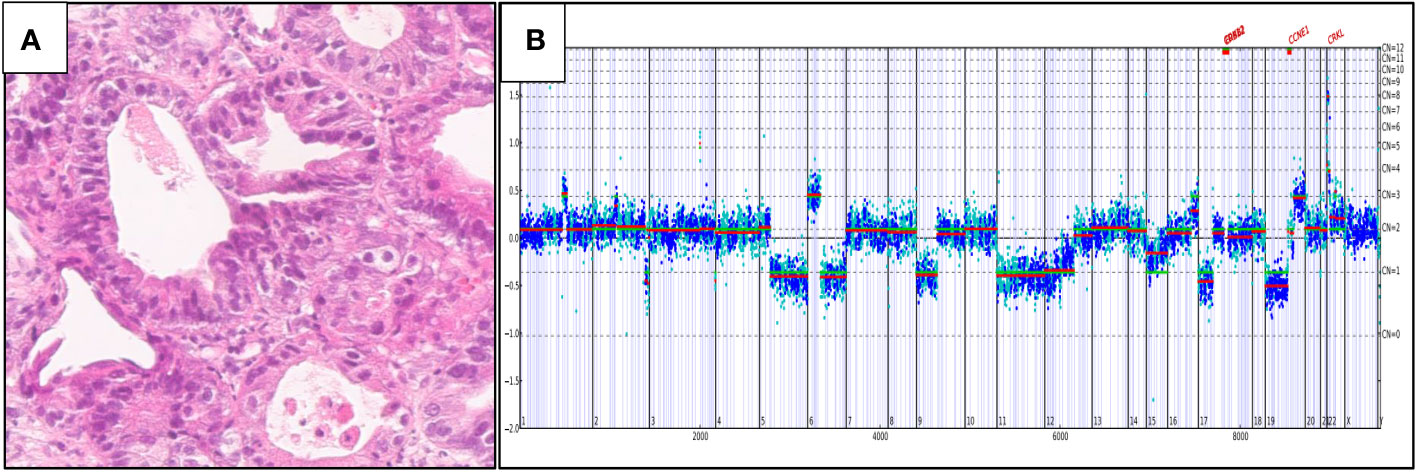
Figure 5 (A) Hematoxylin and eosin: histology of the tumor. (B) ERBB2 amplification at 148 copies.
In our study of PDAs, 93.37% had KRAS mutation, whereas 7.63% were wild-type. This is similar to other studies in literature where KRAS mutation is noted in around in 85-90% of pancreatic cancers (7, 24). The slightly higher frequency of KRAS mutations in the study cohort may reflect that all of the patients had relapsed and refractory disease that was predominantly metastatic at the time of sequencing reflecting the adverse prognostic influence associated with KRAS mutation in PDA. Epidemiologic characteristics of our cohort were consistent with those established for pancreatic cancer, showing male preponderance and older age (25). KRAS mutated PDA has been associated with a worse overall survival and may respond poorly to gemcitabine based therapy (26). Wild-type KRAS is reported in 8-12% of PDAs (27, 28). The possibility that this group may have improved survival, increases the importance to identify potential targets for precision therapies that could potentially increase both the progression-free, and the overall survivals for these patients (28). Voutsadakis et al., used publicly available genomic cohorts (The Cancer Genome Atlas, the MSK-IMPACT study, the pancreatic cancer sub-cohort of a pan-cancer study from China, and the pancreatic cancer cohort from the American Association for Cancer Research project GENIE), along with cBioportal and OncoKB knowledgebase, to characterize the GAs that occur in PDA without commonly seen alterations such as KRAS, TP53, CDKN2A and SMAD4. PDA without these alterations constituted 5-10%. No major differences in GAs were noted. The relatively limited sample size in the wild-type PDA group could be an impediment to adequate analysis in such studies, including our analysis (29).
The treatment of locally advanced, unresectable, and metastatic PDA is centered around using various chemotherapeutic regimes. Olaparib and Rucaparib, which are poly (adenosine diphosphate–ribose) polymerase (PARP) inhibitors, are FDA approved in the 2nd line setting and can also be utilized for maintenance in patients with germline BRCA1 and BRCA2 mutations. There was benefit in terms of progression free survival (PFS), but overall survival remained unchanged (30, 31). Approvals such as pembrolizumab for TMB>10 and Entrectinib and Larotrectinib for NTRK fusions can be utilized in PDA. Overall, the utility of non-chemotherapeutic systemic treatments remains limited (4). PDA is a complex disease in terms of molecular and epigenetic changes, in which KRAS plays a central role from origin to progression (32). Efforts at targeting KRAS in pancreatic cancer have been unsuccessful so far. The mitogen-activated protein kinase (MAPK) pathway is an important pathway interconnected with KRAS. Attempts to target this with MAPK inhibitors either with or without gemcitabine unfortunately did not reveal any survival benefit (33). Blocking MEK and PI3K/AKT using selumetinib was also not useful to improve survival (34). KRAS vaccines like RAS peptide vaccines and those targeting mutant KRAS were unsuccessful (28, 35). Tipifarnib, a drug that targets RAS dependent growth failed to show any benefit in phase 3 trials (36). Several agents have shown promising results in PDA cell lines but the results remain exploratory (28). Sotorasib is a small molecule irreversible inhibitor of KRAS G12C. The CodeBreaK100 showed encouraging activity of this drug in advanced malignancies with KRAS G12C mutation (37). KRAS G12C is seen in 1-2% of patients with KRAS mutated PDA. The results from our cohort were consistent with this (1.6%). The efficacy and safety of sotorosib in pretreated KRAS G12C mutated PDA was reported in ASCO 2022. The overall response rate was 21.1% from a cohort of 38 patients (38).
KRAS wild-type PDA had a higher frequency of ERBB2 genomic alterations (6.8%). Clinical studies of anti-HER2 drugs in PDA have previously focused on ERBB2 amplification. A phase 2 study of traztuzumab, cetuximab and gemcitabine as a first line strategy in metastatic PDA showed disease stabilization in 27% of 33 patients. Cetuximab and traztuzumab combination has also been studied (39). Although the aforementioned studies were small, the durable activity of the antibody drug conjugate, Trastuzumab deruxtecan is well established. It is approved in Non-small cell lung cancer (NSCLC) (55% objective response among 91 patients) based on the DESTINY-Lung01 for patients with ERBB2 kinase domain mutations (40) and in metastatic breast cancer for patients with HER2 low expression by IHC (41). Trials are underway to study the role of Trastuzumab deruxtecan in solid tumors including PDA (42). BRAF mutation was significantly higher in wild-type PDA (17.9%). This can be anticipated as BRAF and KRAS are mutually exclusive in most cancers (43, 44). The FDA has approved the combination of dabrafenib and trametinib in patients with metastatic or unresectable sold tumors with BRAF V600E mutation. They must have progressed through prior lines of treatment and must have no other alternative options. This was based on the Phase 2 ROAR basket study and the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F (45–47). BRAF mutation is historically found in 3% of PDA and individual reports have shown good response with BRAF and MEK inhibitor combination (48). PIK3CA (6.5%), FGFR2 (4.4%) and ATM (6.8%) were the other targetable GAs that were significantly higher in wild-type PDA. Increased Akt signaling is an important effector in the PIK3 pathway. Activating PIK3CA mutations in PDA have been reported in the range of 4% in the past. Several trials evaluating various agents targeting the PIK3 pathway are currently underway, but most of them are early phase (I/II) (49). FGFR alterations are reported in only 4-6% of PDAs. The only approval for FGFR inhibitors are in metastatic urothelial carcinoma (erdafitinib) and in cholangiocarcinoma (pemigatinib) (50, 51). A recent case report showed durable response of >12 months with erdafitinib in a young male with relapsed FGFR rearranged PDA. Incidentally, the patient in that particular case was also KRAS wild-type (52). The authors mention that the pathology revealed poorly differentiated adenocarcinoma, raising the possibility of it being a cholangiocarcinoma, as FGFR GA is more common in biliary tract cancers (52, 53). FGFR2 GAs, which are mostly fusions, are found in 10-15% of intrahepatic cholangiocarcinoma’s (53), compared to 4.4% in our KRAS wild-type PDA cohort. However, the pathologic diagnosis in these cases could be questioned, raising the suspicion that these could be cholangiocarcinoma. ATM mutations are a part of the HRD germline spectrum and may connote sensitivity to PARP inhibitors. Found in 1.7-3.3% of all PDAs, these are significantly more common in KRAS wild-type and the results of early phase clinical trials pertaining to this would have to be observed (54).
On analyzing the data on markers predictive of ICPIs therapy response, several factors (higher mean TMB, higher percentage of high and ultrahigh TMB) suggests that, as a group, the KRAS wild-type PDA may be more responsive to ICPIs therapy than the KRAS mutated PDA. Other predictors of ICPIs response like PBRM1 (55, 56) and MDM2 (57) were also higher, necessitating the need for further research into this aspect.
Limitations of our study include the retrospective nature and the confounding that may arise from it. The lack of clinical outcomes data for our patients is another limitation. Subgroup analysis and stratification of PDA by KRAS mutation status in randomized clinical trials can help overcome this. Some cases like those with FGFR2 alterations could have been intrahepatic cholangiocarcinoma and may have been improperly diagnosed as PDA.
Overall, several targetable GAs and ICPIs positive predictive markers were more frequent in KRAS wild-type PDA. This combined with the fact that TP53 alterations were much lower in KRAS wild-type PDA provides sound evidence that this distinct molecular subtype of PDA has the potential to achieve a much better survival outcome. The presence of TP53 alteration in itself is an adverse prognostic marker in any malignancy (58) and long term PDA survivors have consistently demonstrated absence of KRAS and TP53 mutations (59, 60).
KRAS wild-type PDA has a higher frequency of several targetable GAs and may thus provide more options for targeted treatments. Response to ICPIs therapy may be better, given more patients in this group had high TMB. Overall, lower frequency of TP53 mutation suggests that physicians and academia should strive to achieve longer survivals in KRAS wild-type PDA patients. Lack of prospective clinical outcome data is a limitation of our study.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Western Institutional Review Board (Protocol No. 20152817). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
PA: Conceptualization, Formal analysis, Validation, Visualization, Roles/Writing - original draft, Writing - review and editing. SS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing - review and editing. DZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing - review and editing. RH: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing - review and editing. ND: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing - review and editing. TJ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing - review and editing. AB: Formal analysis, Validation, Visualization, Writing - review and editing. AS: Formal analysis, Validation, Visualization, Writing - review and editing. SG: Formal analysis, Resources, Validation, Visualization, Writing - review and editing. All authors contributed to the article and approved the submitted version.
All Foundation Medicine co-authors disclose that they are employees of Foundation Medicine and own shares in Roche Holdings. Authors affiliated with Upstate have no conflict of interest to disclose.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/CAAC.21654
2. Park W, Rubenstein DM, Chawla A, O’reilly EM. Pancreatic cancer: a review. JAMA (2021) 326(9):851–62. doi: 10.1001/jama.2021.13027
3. Wilkowski R, Wolf M, Heinemann V. Primary advanced unresectable pancreatic cancer. Recent Results Cancer Res (2008) 177:79–93. doi: 10.1007/978-3-540-71279-4_10
4. Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, et al. Pancreatic adenocarcinoma, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw (2021) 19:439–57. doi: 10.6004/JNCCN.2021.0017
5. Zhao Z, Li X, Wang F, Xu Y, Liu S, Han Q, et al. Pathogenic genomic alterations in Chinese pancreatic cancer patients and their therapeutical implications. Cancer Med (2023) 00:1–14. doi: 10.1002/CAM4.5871
6. Panebianco C, Pisati F, Villani A, Andolfo A, Ulaszewska M, Bellini E, et al. Counteracting gemcitabine+nab-paclitaxel induced dysbiosis in KRAS wild type and KRASG12D mutated pancreatic cancer in vivo model. Cell Death Discovery (2023) 9(1):116. doi: 10.1038/S41420-023-01397-Y
7. Luo J. KRAS mutation in pancreatic cancer. Semin Oncol (2021) 48:10. doi: 10.1053/J.SEMINONCOL.2021.02.003
8. Ferguson R, Aughton K, Evans A, Shaw V, Armstrong J, Ware A, et al. Mutant K-ras in pancreatic cancer: an insight on the role of wild-type n-ras and K-Ras-Dependent cell cycle regulation. Curr Issues Mol Biol (2023) 45:2505. doi: 10.3390/CIMB45030164
9. Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol (2019) 16:207–20. doi: 10.1038/S41575-019-0109-Y
10. Gu M, Gao Y, Chang P. KRAS mutation dictates the cancer immune environment in pancreatic ductal adenocarcinoma and other adenocarcinomas. Cancers (Basel) (2021) 13:2429. doi: 10.3390/CANCERS13102429
11. Leroux C, Konstantinidou G. Targeted therapies for pancreatic cancer: overview of current treatments and new opportunities for personalized oncology. Cancers (Basel) (2021) 13:1–28. doi: 10.3390/CANCERS13040799
12. Strickler JH, Satake H, George TJ, Yaeger R, Hollebecque A, Garrido-Laguna I, et al. Sotorasib in KRAS p.G12C–mutated advanced pancreatic cancer. N Engl J Med (2023) 388:33–43. doi: 10.1056/NEJMOA2208470/SUPPL_FILE/NEJMOA2208470_DATA-SHARING.PDF
13. Ashok Kumar P, Graziano SL, Danziger N, Pavlick D, Severson EA, Ramkissoon SH, et al. Genomic landscape of non-small-cell lung cancer with methylthioadenosine phosphorylase (MTAP) deficiency. Cancer Med (2022) 12(2):1157–66. doi: 10.1002/CAM4.4971
14. Milbury CA, Creeden J, Yip WK, Smith DL, Pattani V, Maxwell K, et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PloS One (2022) 17:e0264138. doi: 10.1371/JOURNAL.PONE.0264138
15. Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med (2012) 18:382. doi: 10.1038/NM.2673
16. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol (2013) 311131:1023–31. doi: 10.1038/nbt.2696
17. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):pl1. doi: 10.1126/SCISIGNAL.2004088
18. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
19. Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn (2019) 21:1053–66. doi: 10.1016/J.JMOLDX.2019.06.011
20. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(1):34. doi: 10.1186/S13073-017-0424-2
21. Roach C, Zhang N, Corigliano E, Jansson M, Toland G, Ponto G, et al. Development of a companion diagnostic PD-L1 immunohistochemistry assay for pembrolizumab therapy in non–small-cell lung cancer. Appl Immunohistochem Mol Morphol (2016) 24:392. doi: 10.1097/PAI.0000000000000408
22. Ilie M, Khambata-Ford S, Copie-Bergman C, Huang L, Juco J, Hofman V, et al. Use of the 22C3 anti–PD-L1 antibody to determine PD-L1 expression in multiple automated immunohistochemistry platforms. PloS One (2017) 12(10):e0186537. doi: 10.1371/JOURNAL.PONE.0183023
23. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol (2011) 29:24. doi: 10.1038/NBT.1754
24. Dai M, Chen S, Teng X, Chen K, Cheng W. KRAS as a key oncogene in the clinical precision diagnosis and treatment of pancreatic cancer. J Cancer (2022) 13:3209. doi: 10.7150/JCA.76695
25. Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol (2019) 10:10. doi: 10.14740/WJON1166
26. Kim ST, Lim DH, Jang KT, Lim T, Lee J, La Choi Y, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther (2011) 10:1993–9. doi: 10.1158/1535-7163.MCT-11-0269
27. Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology (2019) 156:2242–2253.e4. doi: 10.1053/J.GASTRO.2019.02.037
28. Luchini C, Paolino G, Mattiolo P, Piredda ML, Cavaliere A, Gaule M, et al. KRAS wild-type pancreatic ductal adenocarcinoma: molecular pathology and therapeutic opportunities. J Exp Clin Cancer Res (2020) 39(1):227. doi: 10.1186/S13046-020-01732-6
29. Voutsadakis IA, Digklia A. Pancreatic adenocarcinomas without KRAS, TP53, CDKN2A and SMAD4 mutations and CDKN2A/CDKN2B copy number alterations: a review of the genomic landscape to unveil therapeutic avenues. Chin Clin Oncol (2023) 12:2–2. doi: 10.21037/CCO-22-108/COIF
30. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA -mutated metastatic pancreatic cancer. N Engl J Med (2019) 381:317–27. doi: 10.1056/NEJMOA1903387/SUPPL_FILE/NEJMOA1903387_DATA-SHARING.PDF
31. Reiss KA, Mick R, O’Hara MH, Teitelbaum U, Karasic TB, Schneider C, et al. Phase II study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin Oncol (2021) 39:2497–505. doi: 10.1200/JCO.21.00003
32. Waters AM, Der CJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med (2018) 8(9):a031435. doi: 10.1101/CSHPERSPECT.A031435
33. Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer (2014) 50:2072–81. doi: 10.1016/j.ejca.2014.04.024
34. Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol (2017) 3:516. doi: 10.1001/JAMAONCOL.2016.5383
35. Palmer DH, Valle JW, Ting Ma Y, Faluyi O, Neoptolemos JP, Jensen Gjertsen T, et al. TG01/GM-CSF and adjuvant gemcitabine in patients with resected RAS-mutant adenocarcinoma of the pancreas (CT TG01-01): a single-arm, phase 1/2 trial. Br J Cancer (2020) 122:971. doi: 10.1038/S41416-020-0752-7
36. Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol (2004) 22:1430–8. doi: 10.1200/JCO.2004.10.112
37. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med (2020) 383:1207. doi: 10.1056/NEJMOA1917239
38. Strickler JH, Satake H, George TJ, Yaeger R, Hollebecque A, Garrido-Laguna I, et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N Engl J Med (2023) 388(1):33–43. doi: 10.1056/NEJMoa220847
39. Jo JH, Jung DE, Lee HS, Park SB, Chung MJ, Park JY, et al. A phase I/II study of ivaltinostat combined with gemcitabine and erlotinib in patients with untreated locally advanced or metastatic pancreatic adenocarcinoma. Int J Cancer (2022) 151:1565–77. doi: 10.1002/IJC.34144
40. Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, et al. Trastuzumab deruxtecan in HER2-mutant non–Small-Cell lung cancer. N Engl J Med (2022) 386:241. doi: 10.1056/NEJMOA2112431
41. Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med (2022) 387:9–20. doi: 10.1056/NEJMOA2203690/SUPPL_FILE/NEJMOA2203690_DATA-SHARING.PDF
42. A phase 2 study of T-DXd in patients with selected HER2 expressing tumors . Available at: https://clinicaltrials.gov/ct2/show/NCT04482309 (Accessed December 8, 2022).
43. Morkel M, Riemer P, Bläker H, Sers C. Similar but different: distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget (2015) 6:20785. doi: 10.18632/ONCOTARGET.4750
44. Guan M, Bender RJ, Pishvaian MJ, Halverson DC, Tuli R, Klempner SJ, et al. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). https://doi.org/101200/JCO2018364_suppl214(2018) 36:214–4. doi: 10.1200/JCO.2018.36.4_SUPPL.214
45. FDA Grants accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation . FDA. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid (Accessed December 9, 2022).
46. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol Off J Eur Soc Med Oncol (2022) 33:406. doi: 10.1016/J.ANNONC.2021.12.014
47. Damodaran S, Zhao F, Deming DA, Mitchell EP, Wright JJ, Gray RJ, et al. Phase II study of copanlisib in patients with tumors with PIK3CA mutations: results from the NCI-MATCH ECOG-ACRIN trial (EAY131) subprotocol Z1F. J Clin Oncol (2022) 40:1552–61. doi: 10.1200/JCO.21.01648
48. Wang Z, He D, Chen C, Liu X, Ke N. Vemurafenib combined with trametinib significantly benefits the survival of a patient with stage IV pancreatic ductal adenocarcinoma with BRAF V600E mutation: a case report. Front Oncol (2022) 11:801320/BIBTEX. doi: 10.3389/FONC.2021.801320/BIBTEX
49. Conway JR, Herrmann D, Evans TJ, Morton JP, Timpson P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut (2019) 68:742. doi: 10.1136/GUTJNL-2018-316822
50. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med (2019) 381:338–48. doi: 10.1056/NEJMOA1817323/SUPPL_FILE/NEJMOA1817323_DATA-SHARING.PDF
51. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol (2020) 21:671. doi: 10.1016/S1470-2045(20)30109-1
52. Ng CF, Glaspy J, Placencio-Hickok VR, Thomassian S, Gong J, Osipov A, et al. Exceptional response to erdafitinib in FGFR2-mutated metastatic pancreatic ductal adenocarcinoma. J Natl Compr Cancer Netw (2022) 20:1076–9. doi: 10.6004/JNCCN.2022.7039
53. Zugman M, Botrus G, Pestana RC, Uson Junior PLS. Precision medicine targeting FGFR2 genomic alterations in advanced cholangiocarcinoma: current state and future perspectives. Front Oncol (2022) 12:860453/BIBTEX. doi: 10.3389/FONC.2022.860453/BIBTEX
54. Lee MS, Pant S. Personalizing medicine with germline and somatic sequencing in advanced pancreatic cancer: current treatments and novel opportunities. Am Soc Clin Oncol Educ book Am Soc Clin Oncol Annu Meet (2021) 41:1–13. doi: 10.1200/EDBK_321255
55. Yang Q, Shen R, Xu H, Shi X, Xu L, Zhang L, et al. Comprehensive analyses of PBRM1 in multiple cancer types and its association with clinical response to immunotherapy and immune infiltrates. Ann Transl Med (2021) 9:465–5. doi: 10.21037/ATM-21-289
56. Zhou H, Liu J, Zhang Y, Huang Y, Shen J, Yang Y, et al. PBRM1 mutation and preliminary response to immune checkpoint blockade treatment in non-small cell lung cancer. NPJ Precis Oncol (2020) 4:1–4. doi: 10.1038/s41698-020-0112-3
57. Fang W, Zhou H, Shen J, Li J, Zhang Y, Hong S, et al. MDM2/4 amplification predicts poor response to immune checkpoint inhibitors: a pan-cancer analysis. ESMO Open (2020) 5:614. doi: 10.1136/ESMOOPEN-2019-000614
58. Overgaard J, Yilmaz M, Guldberg P, Hansen LL, Alsner J. TP53 mutation is an independent prognostic marker for poor outcome in both node-negative and node-positive breast cancer. Acta Oncol (2000) 39:327–33. doi: 10.1080/028418600750013096
59. Voutsadakis IA. Mutations of p53 associated with pancreatic cancer and therapeutic implications. Ann Hepato-Biliary-Pancreatic Surg (2021) 25:315. doi: 10.14701/AHBPS.2021.25.3.315
Keywords: pancreatic ductal adenocarcinoma, KRAS mutation, KRAS wild-type pancreatic cancer, genomic alterations, targeted therapy
Citation: Ashok Kumar P, Serinelli S, Zaccarini DJ, Huang R, Danziger N, Janovitz T, Basnet A, Sivapiragasam A, Graziano S and Ross JS (2023) Genomic landscape of clinically advanced KRAS wild-type pancreatic ductal adenocarcinoma. Front. Oncol. 13:1169586. doi: 10.3389/fonc.2023.1169586
Received: 19 February 2023; Accepted: 07 June 2023;
Published: 19 June 2023.
Edited by:
Tao Zhang, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Laiba Arshad, Forman Christian College, PakistanCopyright © 2023 Ashok Kumar, Serinelli, Zaccarini, Huang, Danziger, Janovitz, Basnet, Sivapiragasam, Graziano and Ross. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prashanth Ashok Kumar, YXNob2trdXBAdXBzdGF0ZS5lZHU=; YWtwamF5YXJhbWFuQGhvdG1haWwuY29t
†ORCID: Prashanth Ashok Kumar, https://orcid.org/0000-0001-6236-2822
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.