 Kleoniki Roka
Kleoniki Roka Elena E. Solomou
Elena E. Solomou Antonis Kattamis
Antonis Kattamis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 19 May 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1167848
This article is part of the Research TopicBiological Aspects of Bone Marrow FailureView all 9 articles
Variations in the length of telomeres and pathogenic variants involved in telomere length maintenance have been correlated with several human diseases. Recent breakthroughs in telomere biology knowledge have contributed to the identification of illnesses named “telomeropathies” and revealed an association between telomere length and disease outcome. This review emphasizes the biology and physiology aspects of telomeres and describes prototype diseases in which telomeres are implicated in their pathophysiology. We also provide information on the role of telomeres in hematological diseases ranging from bone marrow failure syndromes to acute and chronic leukemias.
In the 1930s, H. Muller and B. McClintock reported that while the chromosomal ends were protected from rearrangements and fusion, damaged chromosomes were unstable and susceptible to these events (1–4). Muller used the Hellenic words “telos” and “meros”, interpreted as end and part accordingly; hence ‘end-parts, and proposed the term telomere (3–5). However, the molecular nature of telomeres was unknown at the time.
Hayflick demonstrated in 1961 that human fetal cells possessed a maximum number of potential cellular divisions of 50 to 60 doublings. The cells that reached this limit stopped dividing and became senescent. Hayflick also proved that cells reconstituted from the frozen state remembered the population division level at which they were frozen, and underwent further doubling only up to a predetermined maximum. Hayflick’s findings reversed Carrel’s concept since the early 1900s, regarding the cellular ability to replicate indefinitely. The limited number that a physiological cell population divides before subsequently becoming senescent was named the “Hayflick limit” or replicative senescence (4, 6, 7).
In the early 1970s, Watson and Olovnikow observed asymmetry in linear DNA replication and hypothesized that chromosomal DNA was lost from the ends of the lagging strand with each cellular division while the discharge of the terminal RNA primer led to progressing shortening of chromosomes (4, 8, 9).
Blackburn and Gall were the first to sequence telomeric DNA in the protozoan Tetrahymena thermophila in 1978, and a few years later Backburn and Greider discovered the existence of a terminal transferase activity leading to the addition of repeated DNA sequences to the chromosomal ends, named telomerase (10–12).
Since then, more than 35000 peer-reviewed research articles have been published on telomeres, their biology, their relationship with aging, and practically every human disease.
Telomeres are non-coding nucleoprotein structures that protect the ends of eukaryotic chromosomes by discriminating natural ends from regular incidental DNA breaks (Figure 1). In this manner, telomeres prevent undesirable end-to-end fusion, nucleolytic degradation, and genomic instability. Without telomeres, every mitosis would result in the loss of genetic information from the leading strand (13).
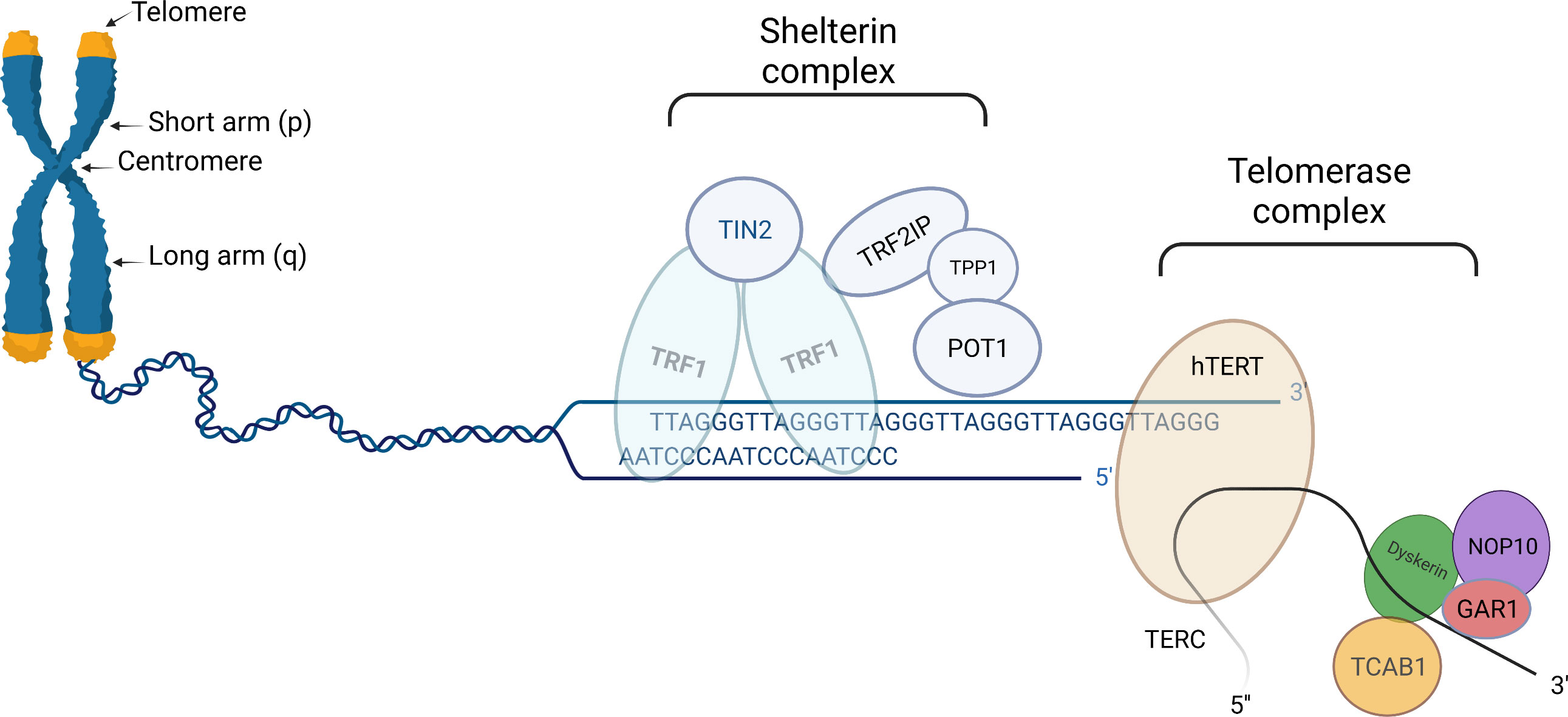
Figure 1 Schematic presentation of telomere (left part), shelterin complex (middle), and telomerase complex (right part). Telomeres are protective structures of tandemly repeated DNA sequence at the ends of eukaryotic chromosomes. In humans, the telomeric DNA sequence consists of repeats of TTAGGG. Shelterin complex is linked to telomeres, helps to stabilize chromosomal ends, and suppresses the DNA damage response system from activating in response to DNA breaks. Human shelterin is specific and consists of six distinct proteinic factors: Telomeric Repeat Binding Factor1(TRF1), TRF2, Repressor/Activator Protein 1 (RAP1), Interacting Nuclear Factor 2 (TIN2), Protector of Telomeres 1 (POT1) as well as POT1-TIN2 organizing protein (TPP1). Telomerase complex preserves critically short telomeres by adding TTAGGG sequences onto the telomeric chromosomal ends; thus contributing by slowing down telomere attrition, although not completely preventing it. Telomerase complex is comprised of a telomerase reverse transcriptase (hTERT) catalytic component, an RNA component (TERC), and several species-specific accessory proteins. In humans, these proteins include dyskerin (DKC1), Nuclear Protein Family A, member 2 (NHP2), Nuclear Protein Family A, member 3 (NOP10), Pontin/Reptin, TCAB1, and Nuclear Protein Family A, member 1(GAR1). “Created with Biorender.com”.
Telomere length (TL) differs among diverse species (14). Human telomeres are comprised of thousands of tandem repeats of the 5’-TTAGGG-3’ sequence and their length typically ranges between 10 and 15 kilobase pairs (kbp) (13, 14). Although human telomere length has a broad distribution, there are finite upper and lower borders (15).
TL is maintained by a dynamic balance that involves processes of shortening telomeres, such as non-sufficient synthesis of the lagging strand of DNA and end-processing events, as well as processes of telomere lengthening. Only a subset of cell types (germinal, stem, and tumor cells) can avoid progressive telomere shortening through the activation of mechanisms known as “Telomere maintenance mechanisms (TMMs)”. Currently, only two TMMs are known: telomerase-mediated telomere maintenance which is considered the primary TMM, and alternative lengthening of telomeres (ALT) (16–19).
Regardless of the maintenance mechanisms, telomeres shorten by 50-200bp with each cell division (16, 20). Ultimately, repeats loss results in extremely short, dysfunctional telomeres with a loss of capability to adequately cap ends of chromosomes, thus causing impaired cell proliferation, cell senescence, and apoptosis (21).
The time limit at which a chromosome end loses its cap as a result of telomere shortening alters according to a specific rate of each cell type and tissue. Therefore, the shortest telomere is extremely important for cell viability and chromosomal stability (22).
Progressive shortening of TL through loss of telomeric DNA manifests also in the hematopoietic system’s cells as a result of the natural replicative aging process. Since 1994, Vaziri et al. demonstrated that shorter TL was found in human candidate stem cells of adult bone marrow (BM) compared to cells isolated from fetal liver or umbilical cord blood, thus confirming the progressive decline in telomere length with age in hematopoietic stem cells (HSCs) (23, 24).
Telomerase, a reverse transcriptase enzyme, preserves critically short telomeres by elongating chromosome ends de novo (Figure 1) (25). The increase in TL is achieved through the addition of TTAGGG sequences to the telomeric chromosomal ends. It should be noted, that telomerase although contributing to slowing down this process does not completely prevent telomere attrition (16).
Telomerase is comprised of a telomerase reverse transcriptase (hTERT) catalytic component, an RNA component (TERC), and several species-specific accessory proteins, serving as primers and templates for extending telomeric nucleotide repeats (26–28). The protein known as hTERT functions as a specific reverse transcriptase with conserved catalytic domains. The fundamental non-coding RNA for telomere synthesis, TERC, acts as a template for elongation of the 3′ overhang of the telomeric G-rich strand (18). In humans, the accessory proteins include dyskerin (DKC1), Nuclear Protein Family A, member 2 (NHP2), Nuclear Protein Family A, member 3 (NOP10), Pontin/Reptin, TCAB1, and Nuclear Protein Family A, member 1(GAR1). These proteins have been reported to assist in telomere assembly, trafficking, recruitment, and stability (29).
Telomerase activity (TA) varies throughout human life. The majority of human tissues show a peak in TA expression within the first few weeks of embryogenesis (30). This specific enzyme’s activity is largely repressed starting from the neonatal period, although this is not seen in certain highly proliferative tissues like the skin, intestine, and bone marrow, which are thought to contain stem cell-like subpopulations, or in dividing lymphocytes, ovaries, and testes (31). In mammalian somatic cells and cells with poor proliferative ability, TA is completely absent. Telomerase is also upregulated in the majority of neoplastic cells, which illustrates the importance of telomere maintenance for proliferative potential (32–34).
The protein complex known as shelterin, which is linked to telomeres, helps to stabilize chromosomal ends and suppresses the DNA damage response (DDR) system from activating in response to DNA breaks (Figure 1). Shelterin’s structure is specific for binding to the structure and sequence of mammalian telomeres. Human shelterin is composed of six distinct protein factors: Telomeric Repeat Binding Factor1 (TRF1), TRF2, Repressor/Activator Protein 1 (RAP1), Interacting Nuclear Factor 2 (TIN2), Protector of Telomeres 1 (POT1) as well as POT1-TIN2 organizing protein (TPP1) (4, 16, 35). Shelterin complex through interacting with TERT binds particularly to telomeric DNA and promotes the telomerase complex to be recruited to chromosomal ends (5, 35).
Another relevant complex is the CST. It consists of three elements: conserved telomere protection component 1 (CTC1), suppressor of cdc thirteen 1 (STN1), and telomeric pathway with STN1 (TEN1). The CST complex partially functions in the synthesis of the telomere-lagging strand (35).
A recently recognized element of the telomere complex is TERRA, a long non-coding telomeric repeat-containing RNA (TERRA) that is transcribed by RNA polymerase II from intrachromosomal telomeric repeats. Although the function of TERRA is not yet fully understood, it is likely to play a role in telomerase regulation, heterochromatin organization in telomeres, gene expression regulation, as well as DDR induced by dysfunctional telomeres (36, 37).
TL is considered a dynamic marker reflecting genetic predisposition alongside an individual’s environmental conditions (38) (Figure 2).

Figure 2 Factors influencing telomere length (TL). Generally, TL is considered a dynamic marker reflecting genetic predisposition as well as individual environmental conditions. TL gradually decreases with age. Additional factors that have been correlated with TL decrease are stress, smoking, alcohol, obesity, chronic inflammation, viral infections as well as hormone imbalance. On the other hand, factors that have been shown to increase telomere length apart from telomerase activity are regular physical activity, low oxidative stress, antioxidants, and low stress daily life.
Since the early 1990s, TL has been shown to gradually decrease with age and is considered a sign of human aging (39–41). TL shows a narrow distribution across human populations, ranging from 8 kb to 13 kb in leukocytes from newborns. Telomeres eventually shorten with age normally at a constant rate (5). In contrast to later life, the rate of telomere loss is more pronounced during the first two years of life than during later life (42, 43).
Furthermore, TL is longer in the genomes of African vs. European ancestry (41), females vs. males (44), preterm vs. full-term babies (38), and offspring of older fathers (45–47). Recently, it has been denoted that offspring of older mothers are associated with longer TL (47, 48), thus implying that a joint effect of parental age on TL exists (47, 49, 50). TL also varied among twins and family members, suggesting that extrinsic variables may have an additional impact on TL and attrition (50).
Numerous other factors are correlated with telomere length, named physical activity, smoking, obesity, alcohol use, vitamins, trace elements, chronic inflammation, hormonal replacement therapy, dietary antioxidants, socio-economic status, as well as perceived stress levels (4, 41, 51–55).
Genetic disorders have also been linked to abnormal telomere shortening, involving genes whose dysregulation disrupts the maintenance of telomeres and leads to clinically relevant diseases, named telomeropathies (5).
Recently, Schneider et al. showed that reduced TL was linked to a minor increase in overall mortality risk, although a higher risk of mortality was linked to particular diseases and organs. Malignant tumors, cancer of the esophagus, and lymphoid and myeloid leukemia were considerably more frequent in individuals with shorter TL, despite the lack of an increase in cancer-related deaths (56).
Telomere biology disorders (TBDs) or telomeropathies encompass a broad variety of infrequent disorders that result from inherited defects in telomerase processes or the DDR system (29, 35, 57).
Currently, short telomere syndromes (STS) are considered the best-studied telomere disorder (Figure 3). They are accelerated premature aging syndromes that show genetic anticipation and multisystem manifestations (35, 58, 59). Of notice, ancestral generations may present with lung disease whereas their children with bone marrow failure (BMF). Therefore, the entire spectrum of clinical features may appear in a single family.
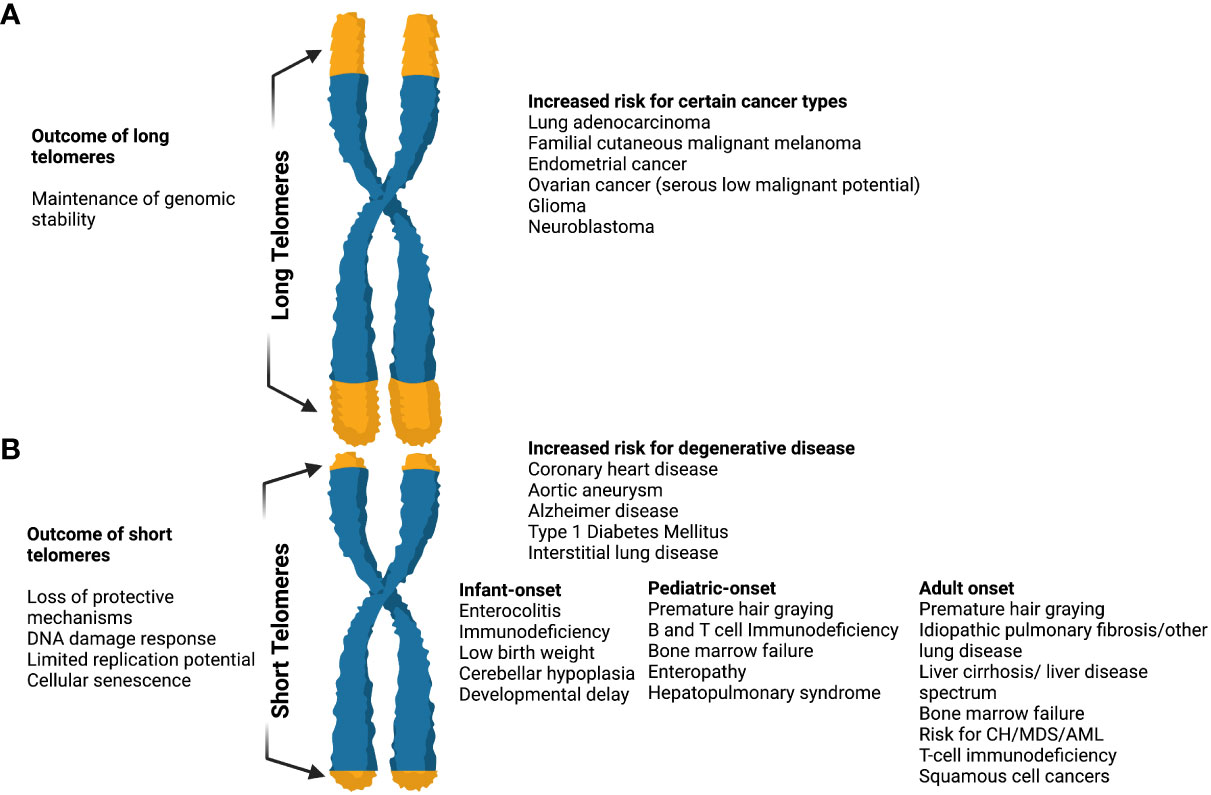
Figure 3 The effects of long and short telomeres. (A) Long telomeres are found in some cancer types (i.e. lung adenocarcinoma, familial cutaneous malignant melanoma, endometrial cancer, glioma, etc). They result in the maintenance of genomic instability, although there is increasing evidence that the price of long telomeres is an increased risk of melanoma and glioma. The complete long telomere syndrome spectrum remains incompletely understood. (B) Short telomeres result in DNA damage response, loss of protective mechanisms, limited replication potential, and cellular senescence. When present they result in an increased risk of degenerative diseases like coronary heart disease, diabetes mellitus type I, interstitial lung diseases, etc. In short telomere syndromes, different phenotypes are revealed depending on age. The cancer spectrum is notably different when compared to long telomere syndromes and usually involves squamous cell and hematologic malignancies. In general, short telomere syndrome phenotypes can be subclassified in infant, pediatric, and adult onset as shown. “Created with Biorender.com”.
The severity of telomere shortening determines the type, age of onset, as well as degree of telomere-driven disorders. Thus, STS may present with different manifestations across the age spectrum (58).
The main characteristic that differentiates short telomere syndromes from other Mendelian disorders is the likelihood of unique clinical phenotypes even though all affected individuals share the same genetic abnormality. In general, adults typically develop the disease in slow-turnover tissues, mostly the lung and liver, whereas children and young individuals develop it in high-turnover tissues, i.e. bone marrow (5).
The first disorder to be linked to a pathogenic variant in telomere gene is Dyskeratosis Congenita (DC) (60). It was first described by Zinnser in 1906 in a case report of two brothers with leukoplakia, abnormal skin color, as well as nail dystrophy. Primarily the disorder was thought to be inherited as an X-linked recessive type (XLR). To date, the detection of pathogenic variants in dyskerin (DKC1) on the X chromosome leading to reduced TA and shorter TL is the first connection between telomere biology and human illnesses (61).
It has been demonstrated that females are also affected, that the manifestation spectrum is wide and that at least 18 different genes with all inheritance patterns — XLR, autosomal dominant (AD), and autosomal recessive (AR)— are etiologically related (61).
DC is genetically heterogeneous. Currently, directly related pathogenic variants in DC include DKC1, TERC, TERT, NOP10, NHP2, PARN, RTEL1, TPP1, TCAB1, CTC1, POT1, STN1, and TINF2 genes (15, 62).
Patients with TBDs can be diagnosed using flow FISH lymphocyte telomere lengths that are smaller than the age-specific 1st percentile (Figure 4). The ability to distinguish patients with DC from their unaffected relatives using total lymphocyte flow FISH telomere lengths less than the 1st percentile for age had a sensitivity and specificity of 97% and 91%, respectively (61).
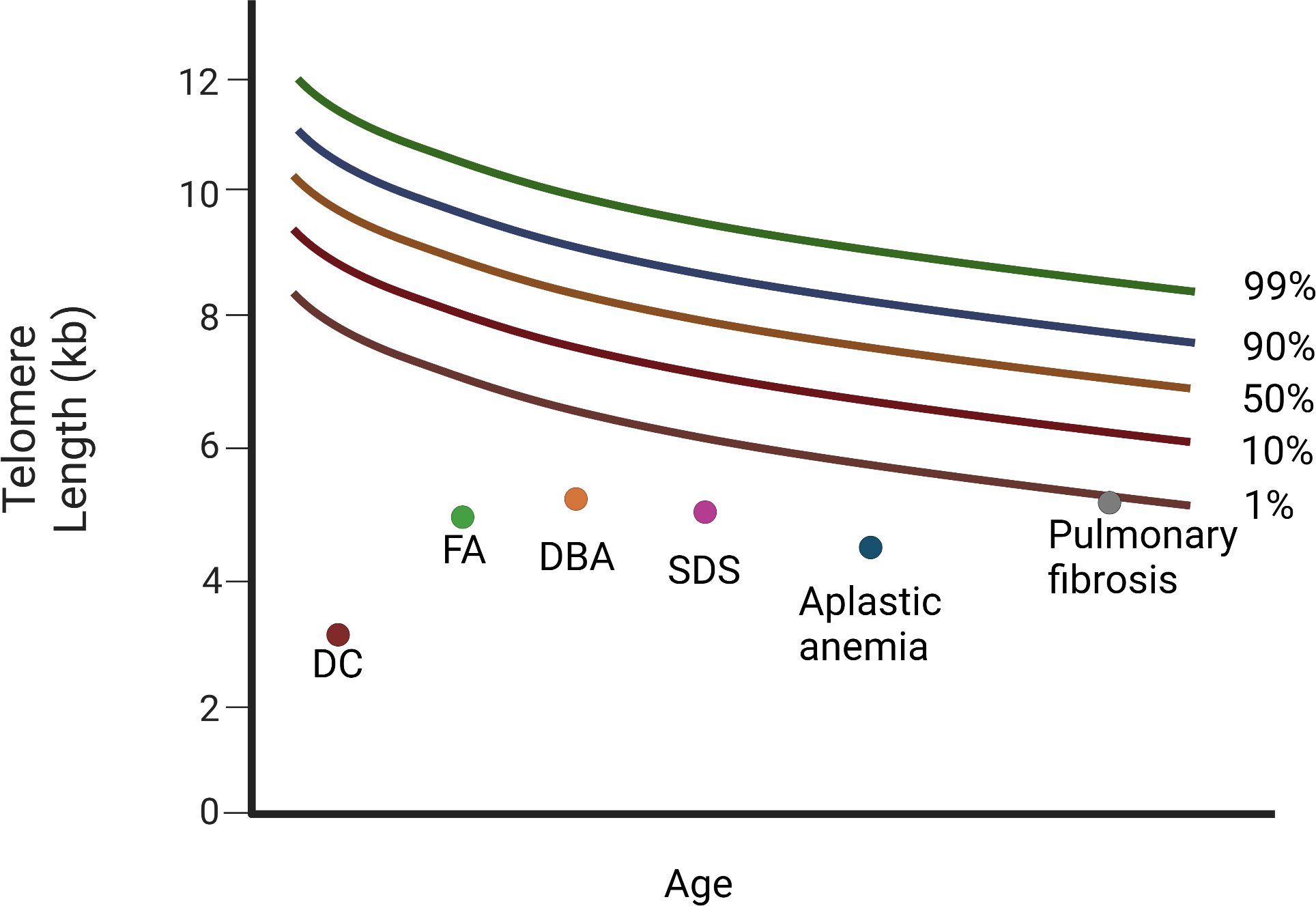
Figure 4 Diagram of relative telomere length (TL) in patients with Dyskeratosis congenita, bone marrow failure syndromes, and the cryptic form of DC, idiopathic pulmonary fibrosis. Different diseases have variable telomere length. Dyskeratosis congenita (DC) is characterised by the shortest telomeres. FA, DBA, and SDS, as well as aplastic anemia (AA), display a mean telomere length below the 1st percentile. Patients with pulmonary fibrosis have telomere length around the 1st percentile (DC, dyskeratosis congenita; FA, Fanconi anemia; DBA, Diamond-Blackfan anemia; SDS, Schwachman-Diamond Syndrome). “Created with Biorender.com”.
The severity of telomere shortening directly affects the severity of symptoms in DC. DC manifests between 15 and 25 years of age in the less severe cases of telomere shortening (TL less than 10% percentile), whereas in the most severe cases (i.e., those with greater telomere shortening and TL less than 1% compared to healthy controls), the age of onset of symptoms is within the first 10 years of life or even during pregnancy. Depending on the mutated gene, DC may appear in a less typical way, that is, bone marrow aplasia or pulmonary fibrosis only (63).
Mucocutaneous triad-nail dystrophy, mucosal leukoplakia, and abnormal skin pigmentation- characterize the classic form of DC (64). Currently, minimal clinical criteria have been established for DC diagnosis. They consist of at least two of the four major characteristics, (abnormal skin pigmentation, nail dystrophy, leukoplakia, and BMF), and at least two of the other extra-hematopoietic manifestations known to occur in DC (Table 1) (64).
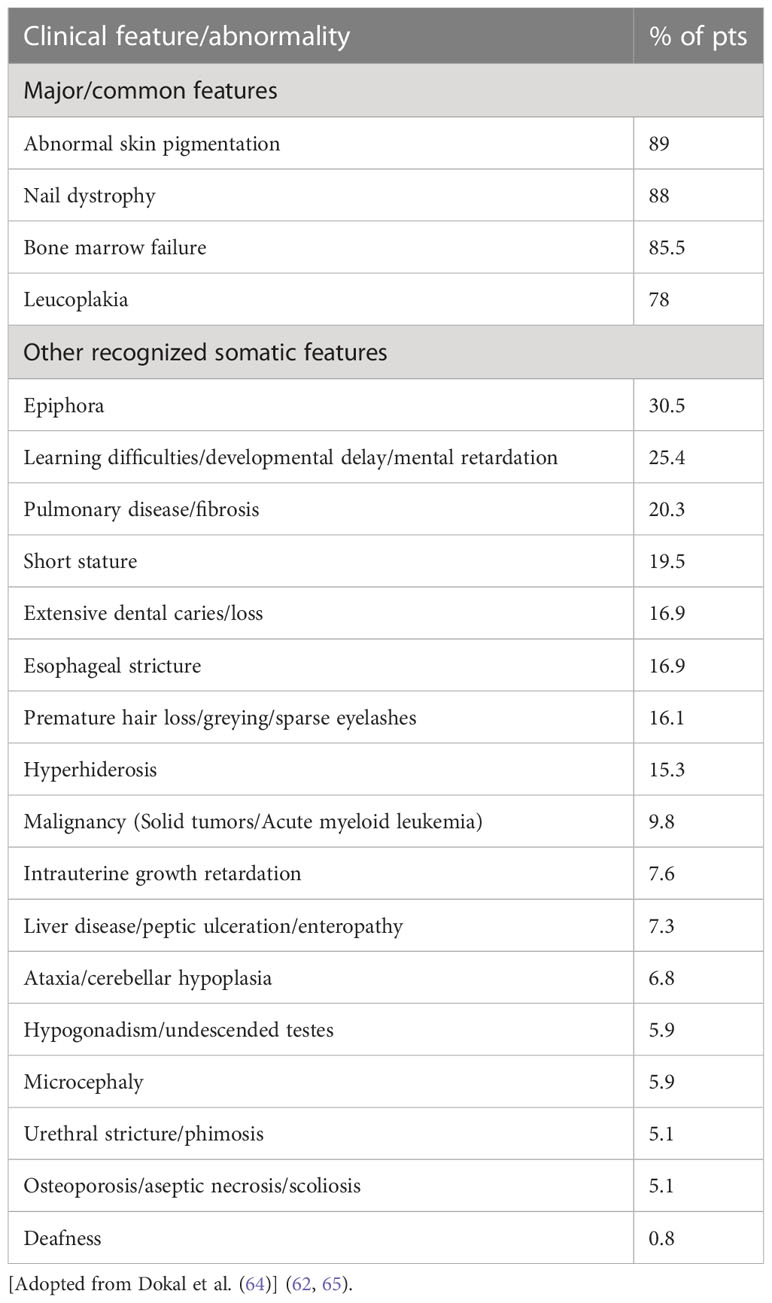
Table 1 Major and minimal clinical criteria for diagnosis of Dyskeratosis Congenita.
Apart from the classical forms of DC, severe forms have been described. Patients with Hoyeraal-Hreidarsson syndrome (HH; OMIM#305000)) present with intrauterine growth retardation, microcephaly, cerebellar hypoplasia, developmental delay, immunodeficiency, and BMF. Patients with Revesz syndrome (RS; OMIM#268130) additionally present with bilateral exudative retinopathy. Finally, patients with Coats plus syndrome (CRMCC; OMIM#612199) may additionally present with intracranial calcifications, brain cysts, leukodystrophy, cerebroretinal microangiopathy, osteopenia, bone fractures, and poor bone healing. These patients may also present gastrointestinal bleeding (62, 66).
Furthermore, it has been demonstrated that some individuals experience a cryptic form of DC marked by delayed symptoms in adulthood (67). This cryptic telomeropathy (cryptic DC) is a late-onset variant that manifests in early adulthood with diverging oligosymptomatic characteristics (62). Isolated aplastic anemia (AA), myelodysplastic syndrome (MDS), acute myelogenous leukemia (AML), cirrhosis of the liver, interstitial lung disease, or a combination of these are common manifestations in patients with cryptic DC (68, 69). The predominant adult-onset presentation of STS, idiopathic pulmonary fibrosis (IPF), is an age-related lung scarring disorder, often identified after the sixth decade (70). In the last few years, it is obvious that the number of individuals that might be considered to have DC has grown considerably. Thus, terms such as “ telomeropathies,” “short telomere syndromes,” and “telomere biology disorders “have been introduced to describe these patients (29). Up to date, individuals with features that fall into the following categories can be considered DC patients:
1. Patients carrying the three defining mucocutaneous characteristics,
2. Patients who exhibit one out of three mucocutaneous characteristics, BMF, or other somatic characteristics of DC,
3. Patients presenting AA or MDS or pulmonary fibrosis linked with a pathogenic telomerase variant,
4. Patients who exhibit four or more features of the Hoyeraal– Hreidarsson syndrome (growth retardation, developmental delay, microcephaly, BMF, immunodeficiency, and cerebellar hypoplasia), or
5. Patients who exhibit two or more DC symptoms, linked to very short telomeres (less than 1st percentile compared to healthy controls) (64).
A common manifestation of TBD is BMF. Individuals with classical DC are almost 80% likely to have at least one single lineage of cytopenia by the age of 30. One cell lineage may initially be altered, but it can subsequently turn into severe pancytopenia or MDS (68). A prospective study showed that by the age of 50 years, 50% of individuals had clinically relevant BMF, and 20% had evident MDS. In some individuals, aplastic anemia may develop, even in the absence of DC-associated symptoms (65, 68).
Apart from genome instability, short telomeres have been shown to promote clonal hematopoiesis (CH), thus providing another paradigm of leukemogenesis (5). It is well-established that human DNA changes with age. Somatic mutations in the same genes associated with MDS and AML are also associated with advanced age. These mutations define CH. Clonal hematopoiesis of indeterminate potential (CHIP) is characterized by the presence of somatic variants in the peripheral blood with a variant allele frequency (VAF) ≥2% in genes associated with hematological malignancies (71). These variants occur more frequently in advanced age (~ 10% at age> 65 years). That said, recent data show that young patients with CHIP are at an increased risk of developing not only hematological malignancies but also coronary heart disease, cardiovascular events, and stroke (72, 73).
Short telomeres are also connected with CHIP; adults with short telomere syndromes have an increased possibility of mutations in the same genes implicated in CHIP. Thus far, data suggest that telomere shortening associated with pathogenic variants in telomerase and telomere-related genes can give rise to CHIP in these patients (74, 75). Schratz et al, reported that 12.8% of patients with short telomere syndromes had cancer, of whom 75% were diagnosed with MDS/AML whereas 30% of adult patients without MDS/AML had age-related CH (74). This percentage was significantly higher than that in patients without TBD, even at older ages (6-10%) (74). Furthermore, in a larger study of 120 patients with TBD, CH-related mutations were found in 48% and specific pathogenic variants correlated with specific somatic mutations. Indeed, it was shown that somatic POT1 mutations were strongly associated with the TINF2 pathogenic variant, whereas both TERTp and PPM1D, were mostly detected in TERT/TERC patients with multi-organ disease (76, 77).
The spectrum of solid malignancies associated with STS overlaps with tumors that develop in individuals with T-cell immunodeficiency, including those with acquired immunodeficiency syndrome (AIDS) and those who have received solid organ transplants. To date, every aspect of T-cell development is limited by short telomeres, both in individuals with short telomere syndromes and in mouse models (5).
Alter et al. reported a 4.2 O/E ratio for any malignancy in non-transplanted DC patients. Notably, the O/E ratio for the development of head and neck squamous cell carcinoma (HNSCC), specifically in the tongue, was extremely high (74 and 216, respectively). Concerning hematological disorders, these patients had O/E ratios of 578 and 73 to develop MDS and AML, respectively. Moreover, in DC patients following transplantation the O/E ratio for the development of any malignancy, HNSCC in general, and HNSCC in the tongue was even higher (30, 432, and 1561, respectively). Transplanted DC patients also had an increased possibility of developing Hodgkin (HD) or non-Hodgkin lymphoma (NHL) (78).
Considering the significant risk of cancer, particularly as individuals reach adulthood, it is advised to avoid smoking, exposure to the sun, and alcohol use. Additionally, routine screenings for both hematological and non-hematological malignancies should be performed. Although cancer treatment depends on the specific type, it should take into account the underlying genetic defects (i.e., the need for increased supportive care and reduction in drug dosages) (79).
Early mortality of DC is mainly caused by BMF. It should be noted that patients with DC and BMF showed no response to immunosuppressives (80, 81). According to recent research, sex hormones may increase TA, by acting on the TERT gene. Thus, the anabolic steroids danazol and oxymetholone can improve hematopoietic function. The wide phenotypic spectrum of DC, especially silent carriers, may complicate the selection of related stem cell donors (82). Currently, the only long-term treatment for hematopoietic manifestations linked to DC is allogeneic hematopoietic stem cell transplantation (HSCT) although there is an increased risk of pulmonary/vascular and liver complications (83). Finally, non-myeloablative fludarabine-based protocols have decreased the frequent and severe adverse effects observed with standard myeloablative conditioning regimens (65, 79, 83).
Studies of primary cultured fibroblasts, in which cells with longer telomeres showed longer replicative potential, supported the evidence that longer TL provides a longevity advantage (54). Furthermore, exogenous TERT expression was sufficient to bypass cellular senescence and immortalize primary cells (55). Recent evidence has supported a greater role for long telomeres in promoting age-related cancer risk, in contrast to observations supporting a low penetrance and narrow spectrum of cancer associated with short telomeres (5) (Figure 3).
In humans, upregulation of telomerase was initially related to an increased risk of familial cancer in a five-generation autosomal dominant family with cutaneous malignant melanoma (CMM) which was found to carry a pathogenic variant in the TERT promoter (15).
Additionally, pathogenic variants in five telomere-related genes (TERT promoter and shelterin genes POT1, TPP1, TERF2IP, and TINF2) showing autosomal dominant inheritance have been linked to the risk of familial melanoma, glioma, and chronic lymphocytic leukemia, explaining 1-10% of families with such cancer types (5, 84, 85). Haycock et al, in the Mendelian Randomization study, reported that increased TL due to germline genetic variation was widely correlated with elevated risk for site-specific cancers. Notably, the observed associations were stronger for rare cancers and tissue sites characterized by lower rates of stem cell division, such as glioma, serous low-malignant potential ovarian cancer, lung adenocarcinoma, neuroblastoma, bladder cancer, and melanoma (84). In the same study, the risk of non-neoplastic or degenerative diseases was reduced (84).
IBMFs comprise a set of diverse diseases, in which the production of one or more blood cell lines fails (86, 87). Clinical manifestations vary depending on the type and number of blood cell lines involved and may include various combinations of anemia, leukopenia, and/or thrombocytopenia (86–88). Despite having heterogeneous phenotypes and various underlying pathogenetic mechanisms, all IBMFs have a higher risk of MDS and/or AML development. Among all patients with IBMF, those with DC and Fanconi anemia (FA, OMIM#227650) presented the highest risk (Figure 4) (86).
Several investigators have demonstrated that apart from DC patients, TL is extremely short in subjects with AA (81) and non-DC IBMFS, such as FA, Schwachman-Diamond syndrome (SDS, OMIM#260400), as well as Diamond-Blackfan anemia (DBA, OMIM#105650) (89, 90).
FA, the most prevalent form of IBMFs, is a rare, multisystemic, hereditary disease characterized by a high frequency of spontaneous chromosomal breakage. This results in genomic instability, which impairs DNA repair and cell-cycle regulation (82, 91, 92). Individuals with FA show hypersensitivity to DNA cross-linking agents (92, 93).
FA is caused by germline pathogenic variants in genes involved in the FA/Breast Cancer Susceptibility (BRCA) DNA repair pathway. Currently, pathogenic variants in 22 genes whose protein products collaborate in the FA/BRCA pathway have been detected in individuals with FA, the most commonly found of which are FANCA, FANCC, and FANCG (94–96).
Shorter telomeres, telomere loss/breaks, and high levels of telomerase activity are typically found in lymphocytes of FA patients (97). To date, it has been suggested that telomere shortening in patients with FA correlates with the progression of bone marrow aplasia (98, 99). In fact, Li et al. demonstrated that the vast majority of patients with FA and severe BMF had higher telomere shortening rates compared to those with FA and on severe BMF (99).
Increased chromosomal breakage, excessive free radical production, and/or abnormal regulation of cell proliferation-related to the deregulation of apoptosis are some of the hypothesized causes of rapid telomere shortening observed in FA (82, 97, 100). Shah et al. recently reported different TL shortening among different complementation groups, with the FANCL group showing the most severe (97). Finally, it has been proposed that the very short telomeres observed in individuals with FA might be the result of hematopoietic failure, stress, and/or treatment (82).
SDS is an autosomal recessive disease characterized by neutropenia, insufficient pancreatic exocrine function, and extra-hematopoietic manifestations, particularly metaphyseal dysostosis (79, 101). The vast majority of individuals with SDS, although not all have been found to harbor pathogenic variants only in the SBDS gene (102, 103).
Thornley et al, reported that SDS patients had a considerably reduced mean telomere length adjusted to age compared to healthy controls (p<0.0001) and concluded that stem cell hyperproliferation is implicated for the shorter TL (104). It should be noted that TL in patients with SDS is not as short as that in DC patients (90, 104). In 2018 Liu et al, showed that the underlying mechanism of telomere shortening found in SDS patients is a cell cycle-dependent telomere-protecting protein named SBDS which enables TPP1-mediated telomerase recruitment (102).
DBA is the most common cause of inherited isolated failure in red blood cell production. Most patients show transfusion-dependent anemia in the first year of life, and approximately half of the patients have congenital deformities such as dysmorphic features, short stature, and abnormalities of the eyes, kidneys, and hands (101, 105).
DBA has an autosomal dominant mode of inheritance. Pathogenic variants in genes encoding essential parts of the small 40S or large 60S ribosomal subunits (RPS19, most often, RPS24, RPS1, RPS15, RPS27A, RPS10, RPS29, RPS26, RPL5, RPL11, RPL35A, as well as RPL15) result to a defect in the ribosomal RNA maturation (101). Currently, pathogenic variants in 20 ribosomal protein (RP) genes have been linked to DBA (105).
TL in patients with DBA is not as short as that in patients with DC and seems to be closer to that in healthy controls (90, 106).
AA was initially described by Ehrlich in 1888, and almost all sporadic cases implicate immune pathophysiology. The improvement in blood counts following immunosuppressive treatment (IST) observed in AA patients is the strongest and most pertinent evidence for the involvement of an immune mechanism (107).
According to different studies, the telomere length of leukocytes from patients with AA shows a wide variation, with a greater percentage of patients having shorter telomeres than healthy controls (108, 109).
Short telomeres in patients with acquired severe aplastic anemia were initially thought to result from hematopoietic stress (110). Currently, it is believed that the destruction of HSPCs by the patient’s immune system results in increased mitotic demand on a limited pool of stem cells, and subsequently a probable reduction in TL (107, 111).
Although telomere shortening in marrow failure is caused by loss-of-function mutations in the telomerase complex (TERC and TERT) genes, it was later found that these mutations were also observed in patients without DC. A minority of patients with acquired aplastic anemia harbor telomerase mutations and show short telomeres (112, 113).
It is noteworthy that Calado et al. found that although the TL of individuals at diagnosis of AA was comparable to that of age-matched subjects, it was conversely associated with the likelihood of developing a cytogenetically aberrant clone. Furthermore, it was shown that the percentage of monosomy-7 cells in the BM at diagnosis of AA was negatively associated with telomere length years before the development of an autonomous and clinically detectable abnormal clone (111).
Ball et al. first reported and confirmed by others that patients with AA had considerably shorter telomeres than controls and that in patients who had a full hematological recovery, the rate of telomere shortening returned to normal for age-related loss (108, 114).
Multivariate analysis in a single institution of 183 patients with severe aplastic anemia (SAA) demonstrated that TL was associated with relapse, clonal evolution, and mortality (110). A major unfavorable event in SAA is the clonal evolution to AML (115). Very short telomeres are the main risk factors for malignant clonal evolution. In fact, patients with shorter telomeres in the SAA cohort evolved to monosomy 7 at a rate that was approximately five to seven-fold higher rate than those with longer telomeres (95% CI, 8.7–37.5%) (110). According to these findings, patients undergoing clonal evolution have accelerated telomere attrition; thus, telomere length measurement can be used to identify patients with SAA at risk (110). Moreover, such findings suggest that in the near future risk stratification and eventually, management of medical treatment may depend on the ability to prognosticate severe disease-related complications (110).
Notably, AA subjects responding to immunosuppressive treatment had TL similar to that of healthy controls, whereas non-responders had considerably shorter telomeres (23). The TL and the number of reticulocytes at diagnosis can predict response to IST (110).
Children with AA also have significantly shorter telomeres compared to healthy controls. Telomere length appeared to be a significant predictive variable in multivariate analysis for response to IST (81, 116).
MDS consist of numerous clonal disorders deriving from multipotent hematopoietic stem cells. Ineffective hematopoiesis associated with peripheral blood cytopenia and bone marrow morphologic abnormalities are the main characteristics of MDS. Approximately 1/3 of patients with MDS may progress to acute leukemia (117). Nowadays, the only potentially curative therapy for MDS patients is allogeneic HSCT, although long-term survival is limited by transplant-related risk for complications (118, 119).
Telomere length showed heterogeneity in MDS. Reduced TL was observed in approximately 50% of patients with MDS versus age-matched controls (16, 120, 121). Additionally, reduction in TL seems to be more marked in MDS in transformation (120, 122), in patients with complex abnormalities in the karyotype (120), in patients with more severe pretransplant cytopenias (118), and also in patients with a greater risk international prognostic scoring system (IPSS) or MDS WHO classification-based prognostic scoring system (WPSS) vs those with lower risk (121, 123). Telomere shortening may also be indicative of a higher risk for leukemic transformation (124), and poor prognosis (125, 126).
Regarding telomerase activity, a study showed that upregulation of telomerase is unusual in MDS patients, despite telomere shortening that occurs in almost 40% of patients with MDS at diagnosis (122). According to Park et al. increased TA may be a poor prognostic indicator in MDS, as individuals with a high TA had a statistically significant shorter interval before evolving to acute leukemia (p = 0.010) (127).
Recently, Myllymäki et al. reported that recipient blood TL proved to be a significant predictor of survival and non-relapse mortality (NRM) after HSCT in MDS patients aged>40 years, regardless of established clinical parameters like age, somatic mutations, hematologic factors, treatment history for MDS, performance status, comorbidity index, as well as donor-recipient HLA matching. In the same study, mutations in three genes, SRSF2 (P=0.001), DNA methyltransferase 3A (DNMT3A; P=0.001), and STAG2 (P=0.04) were more frequently found in individuals with the longest quartile blood TL versus those with the shortest quartile TL. In contrast, three different gene mutations were considerably more frequent in individuals with the shortest telomere length: PPM1D (P=0.01), WT1 (P=0.03), and ATM (P=0.04). Furthermore, it was shown that individuals with severe acute graft-versus-host disease (GVHD) had the greatest effects of TL on NRM and were more prominent after high-intensity conditioning (118).
Interestingly, 2.7% of MDS patients who underwent allogeneic transplantation and did not have a clinical diagnosis of TBD had uncommon TERT variants according to a large study by Reilly et al. These polymorphisms were related to shorter TL, younger age at MDS diagnosis, and a higher risk of NRM in MDS patients following allogeneic SCT (128). Multivariate analysis revealed that these uncommon TERT uncommon variants were linked to poorer overall survival (OS) (P=0.034), which was caused by a higher incidence of NRM (P= 0.015). These results imply that uncommon TERT variants may help in the identification of a subset of MDS patients with undiagnosed telomere biology disorders (128).
Children with MDS tend to exhibit short telomeres (129). Children show considerable differences in terms of morphology, cytogenetics, and therapeutic approaches compared with adults. The mutational landscape is also different between children and adults with MDS. The implementation of specific molecular testing and NGS in daily practice is expected to identify more children with MDS and an underlying genetic predisposition syndrome (130).
Since the early 2000s, several researchers have studied differences in both TL and TA in patients with leukemia (131, 132).
Leukemic cells have shorter TL compared to bone marrow mononuclear cells and in subjects with B-cell acute lymphoblastic leukemia (B-ALL) vs T-cell acute lymphoblastic leukemia (T-ALL) vs acute myelogenous leukemia (AML). Telomerase activity was found to be higher in B-ALL vs T-ALL vs AML patients (133). Moreover cytogenetic abnormalities were associated with shorter telomeres in both ALL and AML (133, 134).
Shorter TL is found in adults with AML vs matched controls (median: -2.5TFU; p<0.001) (134), with aberrant karyotype vs normal karyotype (median: -3.0vs -2.3TFU; p<0.03) (134), with multiple aberrations vs less than two aberrations (median: -3.7 vs -2.9 TFU; p<0.03), and younger than 60 years vs older than 60 years (median: -4.3 vs-1.7 TFU; p<0.001) (134, 135).
Noteworthy, individuals diagnosed with AML monocytic subtype M5 according to the FAB classification system, showed the shortest telomeres (-3.8 TFU); versus individuals with M1 (-1.9 TFU, p<0.001) (134). Gaffari et al. reported that patients with acute promyelocytic leukemia (M3) have a significantly shorter TL compared to the control group (median 3.5 versus 11.37 kbp; p < 0.001). The same study demonstrated that all patients had higher TA and that TA was noticeably higher in relapsed group vs the group that had just been diagnosed (136).
It has also been reported that activating mutations like FLT3-ITD have been linked to shorter TL in patients (135–137), whereas mutations in epigenetic modifying enzymes, mainly IDH1 and IDH2, have been linked to longer TL in patients (137). Dratwa et al. observed that patients in various risk stratification categories for FLT3-ITD and NPM1 showed different TL and OS rates. The shortest telomeres and noticeably worse OS were seen in AML individuals with FLT3-ITD mutations and without NPM1 mutation (135). Moreover, shorter pre-treatment TL was associated with an inferior relapse-free, event-free, and OS in intermediate-risk AML (138).
Of interest, in a recent study of 5207 patients with AML from the German Study Alliance Leukemia registry, patients younger than 36 years with aberrant karyotype were screened for underlying germline predisposition syndromes named “Myeloid neoplasms with germ line predisposition (MNGLP)” which was detected in 34.5% of them. Patients harboring pathogenic variants in MNGLP-associated genes displayed a markedly decreased OS. Thus, researchers proposed genetic screening for underlying hereditary cancer predisposition syndromes in young patients with AML and aberrant karyotype, as well as the implementation of different treatment protocols to reduce toxicity and improve response to therapy (139).
Children with AML have very short TL compared to healthy controls (134, 140). Variants in the telomerase complex genes TERT and TERC do not appear to increase the risk of developing pediatric AML, and these mutations are rather uncommon in pediatric AML (140).
It should be noted that in comparison to CEBPA-double mutants and NPM1 mutants, children with AML who harbored FLT3/ITD or a WT1 mutation and FLT3/ITD exhibited shorter telomeres. Finally, the presence of the high-risk molecular aberration FLT3/ITD was linked to shorter TL, but not to OS (140).
Several studies have reported shorter TL in blood samples of both adult and pediatric patients with newly diagnosed ALL compared to controls (141–143). Furthermore, it seems that TL is shorter to relapse patients vs newly diagnosed ALL patients vs healthy controls (143). TA is significantly elevated in ALL patients when compared to normal controls. The average TA level is reported higher in relapsed patients vs newly diagnosed vs control group (143).
TL is shorter in T-ALL vs B-ALL patients (133), and in adult vs pediatric ALL patients (141). Importantly, across all the AL subtypes, B-ALL cells showed the shortest telomeres and the highest TA (133). Male individuals have been reported to have higher TA levels than female individuals (144).
According to Wang et al, patients with high TA had a higher ratio of unfavorable cytogenetic abnormalities in ALL patients such as t(9;22), t(4;11), and t(11;14), whereas patients with low TA had a higher ratio of normal and other abnormalities (p < 0.05). In the same study, it was demonstrated that ALL subjects with high TA showed a significantly worse OS (31.65%) versus ALL subjects with low TA (58.82%, p < 0.01) (143).
Borseen et al. reported that TL is significantly shorter in pediatric ALL at diagnosis compared to TL and end of therapy. Furthermore, TL was shorter in patients with t(9;22)(q11;q34)[BCR/ABL1] compared to high-hyperdiploid (51-61 chromosomes) or t(12;21) subjects and also compared to all B-cell precursor-ALL (BCP). Moreover, in comparison to BCP-ALL cases, hTERT methylation was more prevalent in T-ALL ones (18% compared to 72%). Moreover, BCP individuals with t(12;21) showed a significantly higher methylation rate (63%) compared to high-hyperdiploid BCP cases (7%). These findings are a strong indication that hTERT promoter methylation status may define certain ALL subgroups and have functional relevance. Finally, patients with BCP-ALL and short telomeres had a better prognosis (145).
The main characteristic of the myeloproliferative neoplasm named chronic myeloid leukemia (CML) is an uncontrolled malignant proliferation of myeloid cells in peripheral blood and bone marrow. The indolent chronic phase (CP), which lasts almost 3-5 years and has a notable increase in myeloid precursors and mature cells, is the first phase of CML’s natural triphasic course. CP proceeds to an accelerated phase (AP) if left untreated within a median time of 3-15 months, and then further progresses to a highly aggressive blast phase (BP). BP is characterized by a rapid expansion of primitive cells in BM that escape into the circulation. Philadelphia chromosome (Ph), which results from the reciprocal translocation t(9;22)(q34.1;q11.2) is a hallmark of CML (146).
TL in CML patients is shorter when compared to healthy individuals (146–149). In retrospective studies, it was demonstrated that accelerated telomere shortening was associated with the stage of disease, cytogenetic remission status, progression to accelerated phase, blast crisis, as well as clinical risk score at diagnosis (150).
Furthermore, average TL has been shown to be adversely associated with the progression of the disease, thus individuals in AP and BP present considerably shorter telomeres compared to patients in CP (147). Since 2000, it was shown that subjects in CP who eventually developed BP within 2 years had considerably shorter telomeres versus those who did not experience BP development for at least 2 years (PF 0.05) (148).
Increased expression of telomerase has been found to associate with an accumulation of additional cytogenetic abnormalities (151). Also, it has been suggested that patients with a more favorable prognosis may have longer telomeres (148).
Wenn et al. reported that CML patients with the longest telomeres showed a lower clinical risk profile compared to patients with shorter telomeres, thus implying that TL analysis may predict response to initial treatment with TKIs after 12 and 18 months according to ELN criteria (152).
In a recent study of 96 individuals with CP-CML, Estrada et al. found that patients with longer age-adjusted telomeres at diagnosis were more likely to achieve deep molecular response with imatinib when compared to individuals with shortened telomeres. Furthermore, major molecular responses were achieved remarkably earlier in subjects with long telomeres (p=0.012) (153).
High telomerase expression and activity are found in CLL subjects with short telomeres, presumably to preserve the critical telomere length for cell survival (154).
The majority of CLL cases were found to keep a rather stable telomere length, nevertheless a subset of follow-up samples, especially those undergoing clonal evolution showed changes in TL (155).
Moreover, markers of worse prognosis like unmutated IGHV genes, increased genomic complexity, 11q deletion/mutated ATM gene, 17p deletion/mutated TP53 gene (155–157), as well as short time to first treatment (157) and short OS have all been linked to short telomeres (155, 157). According to Rambazzo et al, CLL subjects with 11q, 17p deletion, or 12 trisomy had considerably higher levels of telomerase and shorter telomeres, compared to those with no chromosomal abnormalities or only 13q deletion (P<0.001). In the same study, TL/telomerase level profiles were able to identify subgroups of patients with distinct clinical outcomes (P<0.0001) (158).
Of notice, Olbertova et al. reported substantially longer RTLs in individuals at Rai and Binet early disease stages at diagnosis (155). Longer telomeres in CLL patients were associated with improved outcomes compared to short telomeres (159), and significantly better prognosis in those with also mutated IGVH compared to CLL patients with short TL and unmutated IGVH (158).
The discovery of telomeres and telomerase has provided only a beginning in understanding the basic elements of the cell cycle and of several diseases with either shorter or longer telomeres that were previously considered idiopathic. Nowadays, studies have shown that multiple genetic and nongenetic factors might impact telomere length, thus it is not yet fully understood how these factors may interact. While genetic mutations involved in telomere length maintenance mechanisms are still being identified, it is more than obvious that additional studies are needed. One of the most important challenges is determining whether the combination of different factors-clinical, genetic, diseases specific- with telomere length may aid in identifying specific disease groups with different characteristics in clinical behavior, response to treatment, progression risk, and outcome.
Finally, while mechanisms of aging and cancer remain challenging in understanding, insights into telomeres and telomerase activity lead researchers to examine the possibility of potential drugs targeting those mechanisms (160). The assumption that inhibition of telomerase could serve as a promising therapeutic approach remains a challenge with several factors currently under investigation.
KR: Conceptualization, Data curation, Formal analysis, Writing: review and editing. ES: Conceptualization, Writing: review and editing, Supervision, Funding acquisition, AK: Conceptualization, Writing: review and editing, Supervision, Funding acquisition. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. McClintock B. The stability of broken ends of chromosomes in zea mays. Genetics (1941) 26(2):234–82. doi: 10.1093/genetics/26.2.234
3. Chakravarti D, LaBella KA, DePinho RA. Telomeres: history, health, and hallmarks of aging. Cell (2021) 184(2):306–22. doi: 10.1016/j.cell.2020.12.028
4. Herrmann M, Pusceddu I, März W, Herrmann W. Telomere biology and age-related diseases. Clin Chem Lab Med (2018) 56(8):1210–22. doi: 10.1515/cclm-2017-0870
5. Armanios M. The role of telomeres in human disease. Annu Rev Genomics Hum Genet (2022) 23:363–81. doi: 10.1146/annurev-genom-010422-091101
6. Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
7. Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol (2000) 1(1):72–6. doi: 10.1038/35036093
8. Watson JD. Origin of concatemeric T7 DNA. Nat New Biol (1972) 239(94):197–201. doi: 10.1038/newbio239197a0
9. Olovnikov AM. A theory of marginotomy. the incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol (1973) 41(1):181–90. doi: 10.1016/0022-5193(73)90198-7
10. Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in tetrahymena. J Mol Biol (1978) 120(1):33–53. doi: 10.1016/0022-2836(78)90294-2
11. Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell (1985) 43(2 Pt 1):405–13. doi: 10.1016/0092-8674(85)90170-9
12. Greider CW, Blackburn EH. A telomeric sequence in the RNA of tetrahymena telomerase required for telomere repeat synthesis. Nature (1989) 337(6205):331–7. doi: 10.1038/337331a0
13. Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science (2015) 350(6265):1193–8. doi: 10.1126/science.aab3389
14. Srinivas N, Rachakonda S, Kumar R. Telomeres and telomere length: a general overview. Cancers (Basel) (2020) 12(3):1–29. doi: 10.3390/cancers12030558
15. McNally EJ, Luncsford PJ, Armanios M. Long telomeres and cancer risk: the price of cellular immortality. J Clin Invest (2019) 129(9):3474–81. doi: 10.1172/JCI120851
16. Allegra A, Innao V, Penna G, Gerace D, Allegra AG, Musolino C. Telomerase and telomere biology in hematological diseases: a new therapeutic target. Leuk Res (2017) 56:60–74. doi: 10.1016/j.leukres.2017.02.002
17. Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discovery (2006) 5(7):577–84. doi: 10.1038/nrd2081
18. Giardini MA, Segatto M, da Silva MS, Nunes VS, Cano MI. Telomere and telomerase biology. Prog Mol Biol Transl Sci (2014) 125:1–40. doi: 10.1016/B978-0-12-397898-1.00001-3
19. De Vitis M, Berardinelli F, Sgura A. Telomere length maintenance in cancer: At the crossroad between telomerase and alternative lengthening of telomeres (ALT). Int J Mol Sci (2018) 19(2):1–15. doi: 10.3390/ijms19020606
20. Nogueira BMD, Machado CB, Montenegro RC, MEA DEM, Moreira-Nunes CA. Telomere length and hematological disorders: a review. In Vivo (2020) 34(6):3093–101. doi: 10.21873/invivo.12142
21. Saldanha SN, Andrews LG, Tollefsbol TO. Assessment of telomere length and factors that contribute to its stability. Eur J Biochem (2003) 270(3):389–403. doi: 10.1046/j.1432-1033.2003.03410.x
22. Bourgeron T, Xu Z, Doumic M, Teixeira MT. The asymmetry of telomere replication contributes to replicative senescence heterogeneity. Sci Rep (2015) 5:15326. doi: 10.1038/srep15326
23. Greenwood MJ, Lansdorp PM. Telomeres, telomerase, and hematopoietic stem cell biology. Arch Med Res (2003) 34(6):489–95. doi: 10.1016/j.arcmed.2003.07.003
24. Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM, et al. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci U.S.A. (1994) 91(21):9857–60. doi: 10.1073/pnas.91.21.9857
25. Vaiserman A, Krasnienkov D. Telomere length as a marker of biological age: state-of-the-Art, open issues, and future perspectives. Front Genet (2020) 11:630186. doi: 10.3389/fgene.2020.630186
26. Laberthonnière C, Magdinier F, Robin JD. Bring it to an end: does telomeres size matter? Cells (2019) 8(1):1–17. doi: 10.3390/cells8010030
27. Wyatt HD, West SC, Beattie TL. InTERTpreting telomerase structure and function. Nucleic Acids Res (2010) 38(17):5609–22. doi: 10.1093/nar/gkq370
28. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet (2019) 20(5):299–309. doi: 10.1038/s41576-019-0099-1
29. Armando RG, Mengual Gomez DL, Maggio J, Sanmartin MC, Gomez DE. Telomeropathies: etiology, diagnosis, treatment and follow-up. ethical and legal considerations. Clin Genet (2019) 96(1):3–16. doi: 10.1111/cge.13526
30. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR, et al. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res (1998) 58(18):4168–72.
31. Shalaby T, Hiyama E, Grotzer MA. Telomere maintenance as therapeutic target in embryonal tumours. Anticancer Agents Med Chem (2010) 10(3):196–212. doi: 10.2174/1871520611009030196
32. Wong JM, Collins K. Telomere maintenance and disease. Lancet (2003) 362(9388):983–8. doi: 10.1016/S0140-6736(03)14369-3
33. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer (1997) 33(5):787–91. doi: 10.1016/S0959-8049(97)00062-2
34. Bernardes de Jesus B, Blasco MA. Telomerase at the intersection of cancer and aging. Trends Genet (2013) 29(9):513–20. doi: 10.1016/j.tig.2013.06.007
35. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet (2012) 13(10):693–704. doi: 10.1038/nrg3246
36. Diman A, Decottignies A. Genomic origin and nuclear localization of TERRA telomeric repeat-containing RNA: from darkness to dawn. FEBS J (2018) 285(8):1389–98. doi: 10.1111/febs.14363
37. Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med (2016) 8(1):69. doi: 10.1186/s13073-016-0324-x
38. Gorenjak V, Petrelis AM, Stathopoulou MG, Visvikis-Siest S. Telomere length determinants in childhood. Clin Chem Lab Med (CCLM) (2020) 58(2):162–77. doi: 10.1515/cclm-2019-0235
39. Lindsey J, McGill NI, Lindsey LA, Green DK, Cooke HJ. In vivo loss of telomeric repeats with age in humans. Mutat Res (1991) 256(1):45–8. doi: 10.1016/0921-8734(91)90032-7
40. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
41. Demanelis K, Jasmine F, Chen LS, Chernoff M, Tong L, Delgado D, et al. Determinants of telomere length across human tissues. Science (2020) 369(6509):1–12. doi: 10.1101/793406
42. Frenck RW Jr., Blackburn EH, Shannon KM. The rate of telomere sequence loss in human leukocytes varies with age. Proc Natl Acad Sci U.S.A. (1998) 95(10):5607–10. doi: 10.1073/pnas.95.10.5607
43. Zeichner SL, Palumbo P, Feng Y, Xiao X, Gee D, Sleasman J, et al. Rapid telomere shortening in children. Blood (1999) 93(9):2824–30. doi: 10.1182/blood.V93.9.2824
44. Gardner M, Bann D, Wiley L, Cooper R, Hardy R, Nitsch D, et al. Gender and telomere length: systematic review and meta-analysis. Exp Gerontol (2014) 51:15–27. doi: 10.1016/j.exger.2013.12.004
45. Broer L, Codd V, Nyholt DR, Deelen J, Mangino M, Willemsen G, et al. Meta-analysis of telomere length in 19 713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet (2013) 21(10):1163–8. doi: 10.1038/ejhg.2012.303
46. Unryn BM, Cook LS, Riabowol KT. Paternal age is positively linked to telomere length of children. Aging Cell (2005) 4(2):97–101. doi: 10.1111/j.1474-9728.2005.00144.x
47. Wulaningsih W, Hardy R, Wong A, Kuh D. Parental age and offspring leukocyte telomere length and attrition in midlife: evidence from the 1946 British birth cohort. Exp Gerontol (2018) 112:92–6. doi: 10.1016/j.exger.2018.09.008
48. Latour CD, O'Connell K, Romano ME, Kantor ED, Du M. Maternal age at last birth and leukocyte telomere length in a nationally representative population of perimenopausal and postmenopausal women. Menopause (2020) 27(11):1242–50. doi: 10.1097/GME.0000000000001669
49. Andreu-Sánchez S, Aubert G, Ripoll-Cladellas A, Henkelman S, Zhernakova DV, Sinha T, et al. Genetic, parental and lifestyle factors influence telomere length. Commun Biol (2022) 5(1):565. doi: 10.1038/s42003-022-03521-7
50. Broer L, Codd V, Nyholt DR, Deelen J, Mangino M, Willemsen G, et al. Meta-analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet (2013) 21(10):1163–8. doi: 10.1038/ejhg.2012.303
51. Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet (2003) 361(9355):393–5. doi: 10.1016/S0140-6736(03)12384-7
52. Arsenis NC, You T, Ogawa EF, Tinsley GM, Zuo L. Physical activity and telomere length: impact of aging and potential mechanisms of action. Oncotarget (2017) 8(27):45008–19. doi: 10.18632/oncotarget.16726
53. Astuti Y, Wardhana A, Watkins J, Wulaningsih W. Cigarette smoking and telomere length: a systematic review of 84 studies and meta-analysis. Environ Res (2017) 158:480–9. doi: 10.1016/j.envres.2017.06.038
54. Lin J, Epel E. Stress and telomere shortening: insights from cellular mechanisms. Ageing Res Rev (2022) 73:101507. doi: 10.1016/j.arr.2021.101507
55. Mathur MB, Epel E, Kind S, Desai M, Parks CG, Sandler DP, et al. Perceived stress and telomere length: a systematic review, meta-analysis, and methodologic considerations for advancing the field. Brain Behav Immun (2016) 54:158–69. doi: 10.1016/j.bbi.2016.02.002
56. Schneider CV, Schneider KM, Teumer A, Rudolph KL, Hartmann D, Rader DJ, et al. Association of telomere length with risk of disease and mortality. JAMA Intern Med (2022) 182(3):291–300. doi: 10.1001/jamainternmed.2021.7804
57. Grill S, Nandakumar J. Molecular mechanisms of telomere biology disorders. J Biol Chem (2021) 296:100064. doi: 10.1074/jbc.REV120.014017
58. Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest (2013) 123(3):996–1002. doi: 10.1172/JCI66370
59. Mangaonkar AA, Patnaik MM. Short telomere syndromes in clinical practice: bridging bench and bedside. Mayo Clin Proc (2018) 93(7):904–16. doi: 10.1016/j.mayocp.2018.03.020
60. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-Linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet (1998) 19(1):32–8. doi: 10.1038/ng0598-32
61. Savage SA. Dyskeratosis congenita and telomere biology disorders. Hematol Am Soc Hematol Educ Program (2022) 2022(1):637–48. doi: 10.1182/hematology.2022000394
62. Dorgaleleh S, Naghipoor K, Hajimohammadi Z, Dastaviz F, Oladnabi M. Molecular insight of dyskeratosis congenita: defects in telomere length homeostasis. J Clin Transl Res (2022) 8(1):20–30.
63. Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica (2012) 97(3):353–9. doi: 10.3324/haematol.2011.055269
64. Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita - update 2015. Eur J Hum Genet (2015) 23(4):e1–e4. doi: 10.1038/ejhg.2014.170
65. Dokal I. Dyskeratosis congenita. Hematol Am Soc Hematol Educ Program 2011 (2011) p:480–6. doi: 10.1182/asheducation-2011.1.480
66. Callea M, Martinelli D, Cammarata-Scalisi F, Grimaldi C, Jilani H, Grimaldi P, et al. Multisystemic manifestations in rare diseases: the experience of dyskeratosis congenita. Genes (Basel) (2022) 13(3):1–11. doi: 10.3390/genes13030496
67. Calado RT, Young NS. Telomere diseases. N Engl J Med (2009) 361(24):2353–65. doi: 10.1056/NEJMra0903373
68. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol (2019) 12(12):1037–52. doi: 10.1080/17474086.2019.1662720
69. Vieri M, Brümmendorf TH, Beier F. Treatment of telomeropathies. Best Pract Res Clin Haematol (2021) 34(2):101282. doi: 10.1016/j.beha.2021.101282
70. Kam MLW, Nguyen TTT, Ngeow JYY. Telomere biology disorders. NPJ Genom Med (2021) 6(1):36. doi: 10.1038/s41525-021-00198-5
71. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126(1):9–16. doi: 10.1182/blood-2015-03-631747
72. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med (2017) 377(2):111–21. doi: 10.1056/NEJMoa1701719
73. Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke (2022) 53(3):788–97. doi: 10.1161/STROKEAHA.121.037388
74. Schratz KE, Haley L, Danoff SK, Blackford AL, DeZern AE, Gocke CD, et al. Cancer spectrum and outcomes in the mendelian short telomere syndromes. Blood (2020) 135(22):1946–56. doi: 10.1182/blood.2019003264
75. Schratz KE, Armanios M. Cancer and myeloid clonal evolution in the short telomere syndromes. Curr Opin Genet Dev (2020) 60:112–8. doi: 10.1016/j.gde.2020.02.019
76. Gutierrez-Rodrigues F, Groarke EM, Clé DV, Patel BA, Donaires FS, Spitofsky N, et al. Clonal hematopoiesis in telomere biology disorders associates with the underlying germline defect and somatic mutations in POT1, PPM1D, and TERT promoter. Blood (2021) 138(Supplement 1):1111–1. doi: 10.1182/blood-2021-151199
77. Schratz KE, Gaysinskaya V, Cosner ZL, DeBoy EA, Xiang Z, Kasch-Semenza L, et al. Somatic reversion impacts myelodysplastic syndromes and acute myeloid leukemia evolution in the short telomere disorders. J Clin Invest (2021) 131(18):1–9. doi: 10.1172/JCI147598
78. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica (2018) 103(1):30–9. doi: 10.3324/haematol.2017.178111
79. Dokal I, Tummala H, Vulliamy T. Inherited bone marrow failure in the pediatric patient. Blood (2022) 140(6):556–70. doi: 10.1182/blood.2020006481
80. Al-Rahawan MM, Giri N, Alter BP. Intensive immunosuppression therapy for aplastic anemia associated with dyskeratosis congenita. Int J Hematol (2006) 83(3):275–6. doi: 10.1532/IJH97.06030
81. Sakaguchi H, Nishio N, Hama A, Kawashima N, Wang X, Narita A, et al. Peripheral blood lymphocyte telomere length as a predictor of response to immunosuppressive therapy in childhood aplastic anemia. Haematologica (2014) 99(8):1312–6. doi: 10.3324/haematol.2013.091165
82. Savage SA, Alter BP. The role of telomere biology in bone marrow failure and other disorders. Mech Ageing Dev (2008) 129(1-2):35–47. doi: 10.1016/j.mad.2007.11.002
83. Gadalla SM, Sales-Bonfim C, Carreras J, Alter BP, Antin JH, Ayas M, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant (2013) 19(8):1238–43. doi: 10.1016/j.bbmt.2013.05.021
84. Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, et al. Association between telomere length and risk of cancer and non-neoplastic diseases: a mendelian randomization study. JAMA Oncol (2017) 3(5):636–51. doi: 10.1001/jamaoncol.2016.5945
85. Rode L, Nordestgaard BG, Bojesen SE. Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol (2016) 45(5):1634–43. doi: 10.1093/ije/dyw179
86. Savage SA, Dufour C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol (2017) 54(2):105–14. doi: 10.1053/j.seminhematol.2017.04.004
87. Alter BP. Bone marrow failure syndromes in children. Pediatr Clin North Am (2002) 49(5):973–88. doi: 10.1016/S0031-3955(02)00031-7
88. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol (2017) 177(4):526–42. doi: 10.1111/bjh.14535
89. Miwata S, Narita A, Okuno Y, Suzuki K, Hamada M, Yoshida T, et al. Clinical diagnostic value of telomere length measurement in inherited bone marrow failure syndromes. Haematologica (2021) 106(9):2511–5. doi: 10.3324/haematol.2021.278334
90. Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere length in inherited bone marrow failure syndromes. Haematologica (2015) 100(1):49–54. doi: 10.3324/haematol.2014.114389
91. Soulier J. Fanconi anemia. Hematology (2011) 2011(1):492–7. doi: 10.1182/asheducation-2011.1.492
92. Dokal I. Inherited bone marrow failure syndromes. J Hematopathol (2011) 4(2):53–60. doi: 10.1007/s12308-011-0089-5
93. Dufour C, Pierri F. Modern management of fanconi anemia. Hematology (2022) 2022(1):649–57. doi: 10.1182/hematology.2022000393
94. Fiesco-Roa M, García-de Teresa B, Leal-Anaya P, van 't Hek R, Wegman-Ostrosky T, Frías S, et al. Fanconi anemia and dyskeratosis congenita/telomere biology disorders: two inherited bone marrow failure syndromes with genomic instability. Front Oncol (2022) 12:949435. doi: 10.3389/fonc.2022.949435
95. Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol (2014) 27(3-4):214–21. doi: 10.1016/j.beha.2014.10.002
96. Chan SH, Ni Y, Li ST, Teo JX, Ishak NDB, Lim WK, et al. Spectrum of germline mutations within fanconi anemia-associated genes across populations of varying ancestry. JNCI Cancer Spectr (2021) 5(1):1–12. doi: 10.1093/jncics/pkaa117
97. Shah A, George M, Dhangar S, Rajendran A, Mohan S, Vundinti BR. Severe telomere shortening in fanconi anemia complementation group l. Mol Biol Rep (2021) 48(1):585–93. doi: 10.1007/s11033-020-06101-2
98. Leteurtre F, Li X, Guardiola P, Le Roux G, Sergère JC, Richard P, et al. Accelerated telomere shortening and telomerase activation in fanconi's anaemia. Br J Haematol (1999) 105(4):883–93. doi: 10.1046/j.1365-2141.1999.01445.x
99. Li X, Leteurtre F, Rocha V, Guardiola P, Berger R, Daniel M-T, et al. Abnormal telomere metabolism in fanconi's anaemia correlates with genomic instability and the probability of developing severe aplastic anaemia. Br J Haematol (2003) 120(5):836–45. doi: 10.1046/j.1365-2141.2003.04225.x
100. Merfort LW, Lisboa MO, Cavalli LR, Bonfim CMS. Cytogenetics in fanconi anemia: the importance of follow-up and the search for new biomarkers of genomic instability. Int J Mol Sci (2022) 23(22):1–15. doi: 10.3390/ijms232214119
101. Cantu C, Proytcheva MA. Bone marrow failure syndromes, a practical approach to diagnosis. J Hematopathol (2015) 8(3):101–12. doi: 10.1007/s12308-015-0252-5
102. Liu Y, Liu F, Cao Y, Xu H, Wu Y, Wu S, et al. Shwachman-diamond syndrome protein SBDS maintains human telomeres by regulating telomerase recruitment. Cell Rep (2018) 22(7):1849–60. doi: 10.1016/j.celrep.2018.01.057
103. Myers KC, Bolyard AA, Otto B, Wong TE, Jones AT, Harris RE, et al. Variable clinical presentation of shwachman-diamond syndrome: update from the north American shwachman-diamond syndrome registry. J Pediatr (2014) 164(4):866–70. doi: 10.1016/j.jpeds.2013.11.039
104. Thornley I, Dror Y, Sung L, Wynn RF, Freedman MH. Abnormal telomere shortening in leucocytes of children with shwachman-diamond syndrome. Br J Haematol (2002) 117(1):189–92. doi: 10.1046/j.1365-2141.2002.03371.x
105. Da Costa L, Leblanc T, Mohandas N. Diamond-blackfan anemia. Blood (2020) 136(11):1262–73. doi: 10.1182/blood.2019000947
106. Pavesi E, Avondo F, Aspesi A, Quarello P, Rocci A, Vimercati C, et al. Analysis of telomeres in peripheral blood cells from patients with bone marrow failure. Pediatr Blood Cancer (2009) 53(3):411–6. doi: 10.1002/pbc.22107
108. Ball SE, Gibson FM, Rizzo S, Tooze JA, Marsh JC, Gordon-Smith EC. Progressive telomere shortening in aplastic anemia. Blood (1998) 91(10):3582–92. doi: 10.1182/blood.V91.10.3582
109. Brümmendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood (2001) 97(4):895–900. doi: 10.1182/blood.V97.4.895
110. Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. Jama (2010) 304(12):1358–64. doi: 10.1001/jama.2010.1376
111. Calado RT, Cooper JN, Padilla-Nash HM, Sloand EM, Wu CO, Scheinberg P, et al. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia (2012) 26(4):700–7. doi: 10.1038/leu.2011.272
112. Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood (2003) 102(3):916–8. doi: 10.1182/blood-2003-01-0335
113. Du HY, Pumbo E, Ivanovich J, An P, Maziarz RT, Reiss UM, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood (2009) 113(2):309–16. doi: 10.1182/blood-2008-07-166421
114. Brümmendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length dynamics in normal individuals and in patients with hematopoietic stem cell-associated disorders. Ann N Y Acad Sci (2001) 938:293–303; discussion 303-4. doi: 10.1111/j.1749-6632.2001.tb03598.x
115. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood (2006) 108(8):2509–19. doi: 10.1182/blood-2006-03-010777
116. Narita A, Muramatsu H, Sekiya Y, Okuno Y, Sakaguchi H, Nishio N, et al. Paroxysmal nocturnal hemoglobinuria and telomere length predicts response to immunosuppressive therapy in pediatric aplastic anemia. Haematologica (2015) 100(12):1546–52. doi: 10.3324/haematol.2015.132530
117. Sieglová Z, Zilovcová S, Cermák J, Ríhová H, Brezinová D, Dvoráková R, et al. Dynamics of telomere erosion and its association with genome instability in myelodysplastic syndromes (MDS) and acute myelogenous leukemia arising from MDS: a marker of disease prognosis? Leuk Res (2004) 28(10):1013–21. doi: 10.1016/j.leukres.2003.11.020
118. Myllymäki M, Redd R, Reilly CR, Saber W, Spellman SR, Gibson CJ, et al. Short telomere length predicts nonrelapse mortality after stem cell transplantation for myelodysplastic syndrome. Blood (2020) 136(26):3070–81. doi: 10.1182/blood.2020005397
119. de Witte T, Bowen D, Robin M, Malcovati L, Niederwieser D, Yakoub-Agha I, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood (2017) 129(13):1753–62. doi: 10.1182/blood-2016-06-724500
120. Boultwood J, Fidler C, Kusec R, Rack K, Elliott PJ, Atoyebi O, et al. Telomere length in myelodysplastic syndromes. Am J Hematol (1997) 56(4):266–71. doi: 10.1002/(SICI)1096-8652(199712)56:4<266::AID-AJH12>3.0.CO;2-7
121. Yan S, Han B, Li H, Wu Y, Zhou D, Zhao Y. Telomerase gene screening and telomere overhang detection in Chinese patients with myelodysplastic syndrome. Leuk Res (2013) 37(10):1359–62. doi: 10.1016/j.leukres.2013.06.011
122. Ohyashiki K, Iwama H, Yahata N, Tauchi T, Kawakubo K, Shimamoto T, et al. Telomere dynamics in myelodysplastic syndromes and acute leukemic transformation. Leuk Lymphoma (2001) 42(3):291–9. doi: 10.3109/10428190109064585
123. Poloni A, Serrani F, Berardinelli E, Maurizi G, Mariani M, Costantini B, et al. Telomere length, c-myc and mad-1 expression could represent prognosis markers of myelodysplastic syndrome. Leuk Res (2013) 37(11):1538–44. doi: 10.1016/j.leukres.2013.07.022
124. Göhring G, Hofmann W, Hofmann W, Hellström-Lindberg E, Roy L, Morgan M, et al. Telomere shortening, clonal evolution and disease progression in myelodysplastic syndrome patients with 5q deletion treated with lenalidomide. Leukemia (2012) 26(2):356–8. doi: 10.1038/leu.2011.193
125. Menshawy NE, Ashwah SE, Ebrahim MA. Short dysfunctional telomere is highly predictive of dismal outcome in MDS but not in AML patients. Int J Hematol Oncol Stem Cell Res (2020) 14(3):188–99. doi: 10.18502/ijhoscr.v14i3.3728
126. Hwang SM, et al. Short telomere length and its correlation with gene mutations in myelodysplastic syndrome. J Hematol Oncol (2016) 9(1):62. doi: 10.1186/s13045-016-0287-9
127. Park HS, et al. Dysregulation of telomere lengths and telomerase activity in myelodysplastic syndrome. Ann Lab Med (2017) 37(3):195–203. doi: 10.3343/alm.2017.37.3.195
128. Reilly CR, et al. The clinical and functional effects of TERT variants in myelodysplastic syndrome. Blood (2021) 138(10):898–911. doi: 10.1182/blood.2021011075
129. Im K, et al. Profiles of telomere length adult and childhood myelodysplastic syndromes: a comparison with hematologic diseases. Blood (2015) 126(23):5231–1. doi: 10.1182/blood.V126.23.5231.5231
130. Locatelli F, Strahm B. How I treat myelodysplastic syndromes of childhood. Blood (2018) 131(13):1406–14. doi: 10.1182/blood-2017-09-765214
131. Engelhardt M, et al. Telomerase activity and telomere length in acute and chronic leukemia, pre- and post-ex vivo culture. Cancer Res (2000) 60(3):610–7.
132. Kawauchi K, Akiyama M, Yamada O. The mechanisms of telomere and telomerase regulation in hematologic malignancies. Front Clin Drug Res - Anti-Cancer Agents (2013) p:115–83. doi: 10.2174/9781608057986113010005
133. Capraro V, et al. Telomere deregulations possess cytogenetic, phenotype, and prognostic specificities in acute leukemias. Exp Hematol (2011) 39(2):195–202.e2. doi: 10.1016/j.exphem.2010.10.008
134. Hartmann U, et al. Telomere length and hTERT expression in patients with acute myeloid leukemia correlates with chromosomal abnormalities. Haematologica (2005) 90(3):307–16.
135. Dratwa M, et al. TERT genetic variability and telomere length as factors affecting survival and risk in acute myeloid leukaemia. Sci Rep (2021) 11(1):23301. doi: 10.1038/s41598-021-02767-1
136. Ghaffari SH, et al. Telomerase activity and telomere length in patients with acute promyelocytic leukemia: indicative of proliferative activity, disease progression, and overall survival. Ann Oncol (2008) 19(11):1927–34. doi: 10.1093/annonc/mdn394
137. Watts JM, et al. Telomere length and associations with somatic mutations and clinical outcomes in acute myeloid leukemia. Leuk Res (2016) 49:62–5. doi: 10.1016/j.leukres.2016.07.013
138. Gu R, et al. Evaluation of pretreatment telomere length as a prognostic marker in intermediate-risk acute myeloid leukemia. Int J Lab Hematol (2021) 43(6):1510–5. doi: 10.1111/ijlh.13665
139. Kirschner M, et al. Impaired overall survival in young patients with acute myeloid leukemia and variants in genes predisposing for myeloid malignancies. Hemasphere (2022) 6(11):e787. doi: 10.1097/HS9.0000000000000787
140. Aalbers AM, et al. Telomere length and telomerase complex mutations in pediatric acute myeloid leukemia. Leukemia (2013) 27(8):1786–9. doi: 10.1038/leu.2013.57
141. Karow A, et al. Targeting telomere biology in acute lymphoblastic leukemia. Int J Mol Sci (2021) 22(13):1–11. doi: 10.3390/ijms22136653
142. Engelhardt M, et al. Telomerase activity and telomere length in pediatric patients with malignancies undergoing chemotherapy. Leukemia (1998) 12(1):13–24. doi: 10.1038/sj.leu.2400889
143. Wang Y, et al. Telomerase activity and telomere length in acute leukemia: correlations with disease progression, subtypes and overall survival. Int J Lab Hematol (2010) 32(2):230–8. doi: 10.1111/j.1751-553X.2009.01178.x
144. Asfour IA, et al. Correlation of telomerase activity to apoptosis and survival in adult acute lymphoblastic leukemia: an Egyptian single-center study. Ann Hematol (2008) 87(3):213–21. doi: 10.1007/s00277-007-0395-2
145. Borssén M, et al. hTERT promoter methylation and telomere length in childhood acute lymphoblastic leukemia: associations with immunophenotype and cytogenetic subgroup. Exp Hematol (2011) 39(12):1144–51. doi: 10.1016/j.exphem.2011.08.014
146. Wang L, et al. Understanding and monitoring chronic myeloid leukemia blast crisis: how to better manage patients. Cancer Manag Res (2021) 13:4987–5000. doi: 10.2147/CMAR.S314343
147. Samassekou O. Dynamic length changes of telomeres and their nuclear organization in chronic myeloid leukemia. Cancers (Basel) (2013) 5(3):1086–102. doi: 10.3390/cancers5031086
148. Brümmendorf TH, et al. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood (2000) 95(6):1883–90. doi: 10.1182/blood.V95.6.1883
149. Samassekou O, et al. Individual telomere lengths in chronic myeloid leukemia. Neoplasia (2009) 11(11):1146–54. doi: 10.1593/neo.09836
150. Drummond MW, et al. Concise review: telomere biology in normal and leukemic hematopoietic stem cells. Stem Cells (2007) 25(8):1853–61. doi: 10.1634/stemcells.2007-0057
151. Ohyashiki K, et al. Telomerase activity and cytogenetic changes in chronic myeloid leukemia with disease progression. Leukemia (1997) 11(2):190–4. doi: 10.1038/sj.leu.2400560
152. Wenn K, et al. Telomere length at diagnosis of chronic phase chronic myeloid leukemia (CML-CP) identifies a subgroup with favourable prognostic parameters and molecular response according to the ELN criteria after 12 months of treatment with nilotinib. Leukemia (2015) 29(12):2402–4. doi: 10.1038/leu.2015.245
153. Estrada N, et al. Influence of telomere length on the achievement of deep molecular response with imatinib in chronic myeloid leukemia patients. Hemasphere (2021) 5(12):e657. doi: 10.1097/HS9.0000000000000657
154. Jebaraj BMC, et al. Short telomeres are associated with inferior outcome, genomic complexity, and clonal evolution in chronic lymphocytic leukemia. Leukemia (2019) 33(9):2183–94. doi: 10.1038/s41375-019-0446-4
155. Olbertova H, et al. Evolution of TP53 abnormalities during CLL disease course is associated with telomere length changes. BMC Cancer (2022) 22(1):137. doi: 10.1186/s12885-022-09221-z
156. Robbe P, et al. Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features. Nat Genet (2022) 54(11):1675–89. doi: 10.1038/s41588-022-01211-y
157. Strefford JC, et al. Telomere length predicts progression and overall survival in chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Leukemia (2015) 29(12):2411–4. doi: 10.1038/leu.2015.217
158. Rampazzo E, et al. Telomere length and telomerase levels delineate subgroups of b-cell chronic lymphocytic leukemia with different biological characteristics and clinical outcomes. Haematologica (2012) 97(1):56–63. doi: 10.3324/haematol.2011.049874
159. Adam R, et al. Prognostic role of telomere length in malignancies: a meta-analysis and meta-regression. Exp Mol Pathol (2017) 102(3):455–74. doi: 10.1016/j.yexmp.2017.05.010
Keywords: telomeres, telomere biology disorders, hematological malignancies, pediatric, adult
Citation: Roka K, Solomou EE and Kattamis A (2023) Telomere biology: from disorders to hematological diseases. Front. Oncol. 13:1167848. doi: 10.3389/fonc.2023.1167848
Received: 16 February 2023; Accepted: 02 May 2023;
Published: 19 May 2023.
Edited by:
Carlo Finelli, Sant’Orsola-Malpighi Polyclinic, ItalyReviewed by:
Alejandro Ferrer, Mayo Clinic, United StatesCopyright © 2023 Roka, Solomou and Kattamis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kleoniki Roka, bmlraXJva2FAbWVkLnVvYS5ncg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.