 Ying Li
Ying Li Shuai Xiang2
Shuai Xiang2 Wenjun Pan
Wenjun Pan Jing Wang
Jing Wang Hanxiang Zhan
Hanxiang Zhan Shanglong Liu
Shanglong Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 29 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1166860
This article is part of the Research Topic Cancer Cell-intrinsic and -extrinsic Factors Affecting Tumor Immune Evasion View all 10 articles
Pancreatic cancer is one of the most malignant tumors with increased incidence rate. The effect of surgery combined with chemoradiotherapy on survival of patients is unsatisfactory. New treatment strategy such as immunotherapy need to be investigated. The accumulation of desmoplastic stroma, infiltration of immunosuppressive cells including myeloid derived suppressor cells (MDSCs), tumor associated macrophages (TAMs), cancer‐associated fibroblasts (CAFs), and regulatory T cells (Tregs), as well as tumor associated cytokine such as TGF-β, IL-10, IL-35, CCL5 and CXCL12 construct an immunosuppressive microenvironment of pancreatic cancer, which presents challenges for immunotherapy. In this review article, we explore the roles and mechanism of immunosuppressive cells and lymphocytes in establishing an immunosuppressive tumor microenvironment in pancreatic cancer. In addition, immunotherapy strategies for pancreatic cancer based on tumor microenvironment including immune checkpoint inhibitors, targeting extracellular matrix (ECM), interfering with stromal cells or cytokines in TME, cancer vaccines and extracellular vesicles (EVs) are also discussed. It is necessary to identify an approach of immunotherapy in combination with other modalities to produce a synergistic effect with increased response rates in pancreatic cancer therapy.
Pancreatic cancer is one of the most malignant digestive system tumors, ranking the fourth most common cause of cancer death with increased incidence rate (1). Surgical treatment is one of the most effective way, but the rate of radical resection is less than 20%. The main causes of death following operation are the recurrence and metastasis. Moreover, pancreatic cancer is striking resistance to radiotherapy and chemotherapy (2). It is necessary to explore a new therapeutic approach. Therefore, immunotherapy has been given high hopes and now become a key pillar of cancer treatment alongside chemoradiotherapy. In pancreatic cancer, cytotoxic T lymphocytes are suppressed by immunosuppressive interaction between cancer cells with stromal cells in tumor microenvironment (TME) like cancer‐associated fibroblasts (CAFs), myeloid derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor associated macrophages (TAMs). Although various immunotherapy methods are different, it is suggested to use a combination of immunotherapeutic approaches for helping active cytotoxic T lymphocytes (CTLs) infiltrate the pancreatic TME or rescuing the exhausted CTLs (3). Therefore, maintaining the activated state of CTLs in TME is the key to tumor immunotherapy. The TME of pancreatic cancer is characterized by low oxygen, acid, high permeability, a large number of growth factors and hydrolases, resulting in insensitivity to apoptosis, resistance to growth inhibition, immune tolerance and immune escape (4). Immune microenvironment remodeling in pancreatic cancer plays an important role in the development of malignant tumors, which poses a challenge to the immunotherapy of pancreatic cancer. First, pancreatic cancer lacks the expression of immunogenic antigens, and their major histocompatibility antigens (MHC) I molecules are significantly downregulated. Secondly, the activities of effector T cells including natural killer (NK) cells, dendritic cells (DCs) and macrophages are significantly decreased, while inhibitory immune cells including Tregs, MDSCs and M2-TAMs aggregate in TME. Meanwhile, TGF-β, IL-10, IL-35, CCL5, CXCL12 and other immune regulatory factors together form an immunosuppressive microenvironment (5). It is obvious that TME, including a heterogeneous composition of immunosuppressive cells and cytokines, is the critical factor that limits the efficacy of immunotherapy (6). Therefore, exploring the formation of tumor immunosuppressive microenvironment and the underlying molecular mechanisms are of great significance in elucidating the immune tolerance of pancreatic cancer and developing new methods of immunotherapy. In this review, we will focus on the role and the mechanisms of stromal cells in the formation of immunosuppressive TME, as well as the potential clinical application of immunotherapy strategies currently under investigation in targeting tumor microenvironment, aiming to explore new research strategies for pancreatic cancer immunotherapy.
Various types of immune cells exist in the TME of pancreatic cancer, but these immune cells present abnormality quantity and function, leading to the disorder of the immune function and decrease of anti-tumor ability. The number of CD4+, CD8+ effector T cells, NK cells and DCs with anti-tumor effects are reduced, and they present nonfunctional status or immature phenotypes. Tregs, CAFs, MDSCs and TAMs with immunosuppressive ability are active and present in large quantities (7). Immunosuppressive cells, cytokines and effector cells’ disability form the immunosuppressive microenvironment of pancreatic cancer, which is an important reason for the insensitivity of pancreatic cancer to immunotherapy. Pancreatic cancer cells secret of various cytokines (such as IL-10, TGF-B and IL-23) and chemokines (such as CXCL1-3, CXCL5, CXCL12, CCI2 and VEGF) to enhance the activation of stromal cells and the accumulation of immunosuppressive cells (8). Activated stromal cells then produce a large number of extracellular matrix and form fibrous basement around pancreatic cancer cells, which hinders the infiltration of effector T cells into tumor (9). Immunosuppressive cells including Tregs, MDSCs and TAMs, up regulate immune checkpoint mediators like programmed death‐1 receptor (PD‐1)–ligand (PD‐L1) and CTL‐associated antigen 4 (CTLA‐4), conferring an inhibitory effect on T cells and NK cells which contribute to form a unique immune suppression tumor microenvironment of pancreatic cancer (10) (Table 1). TME become a chief mediator of tumor immune tolerance and immunotherapy resistance (Figure 1).
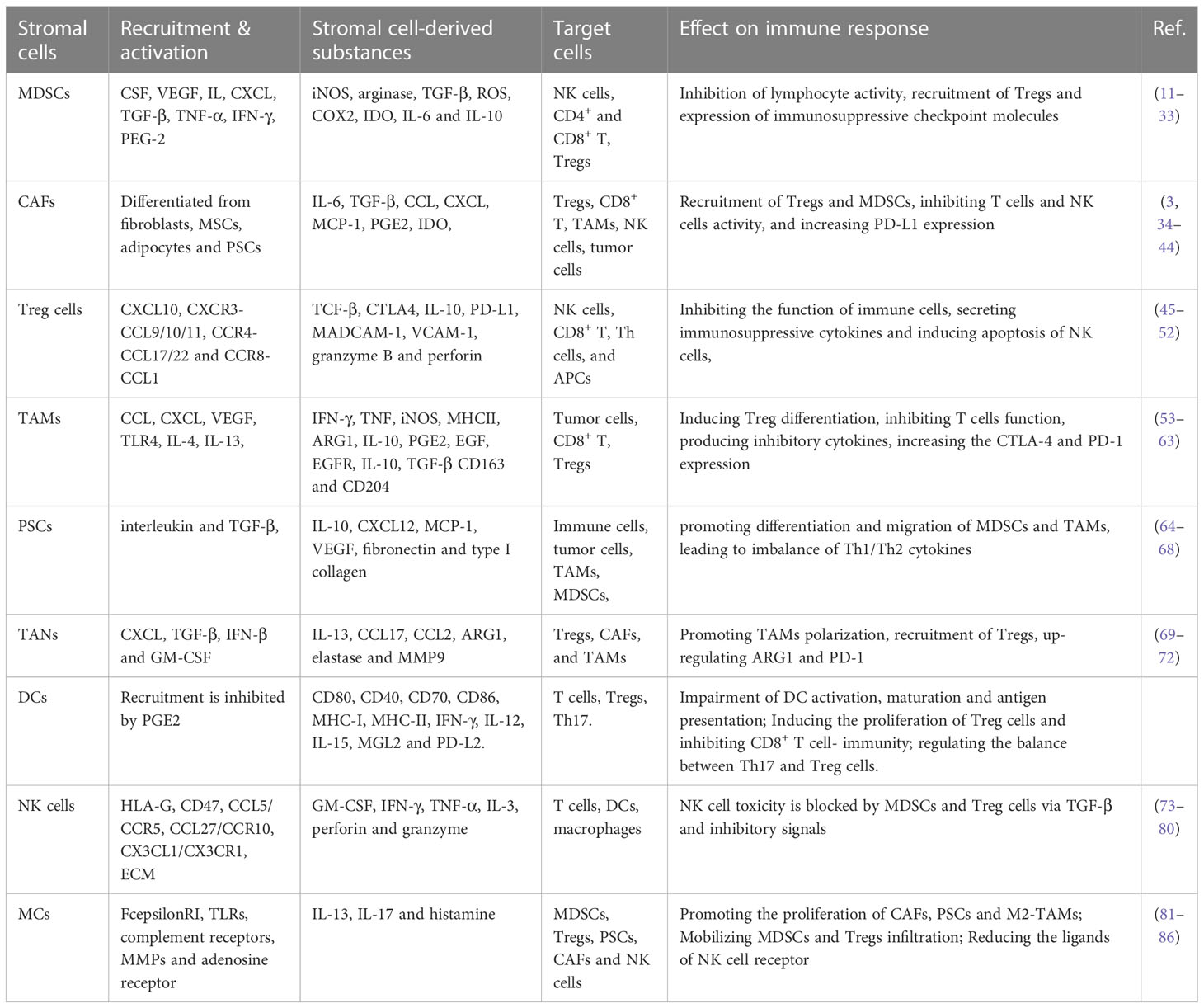
Table 1 The effect of stromal components in pancreatic TME on cancer immunosuppression.
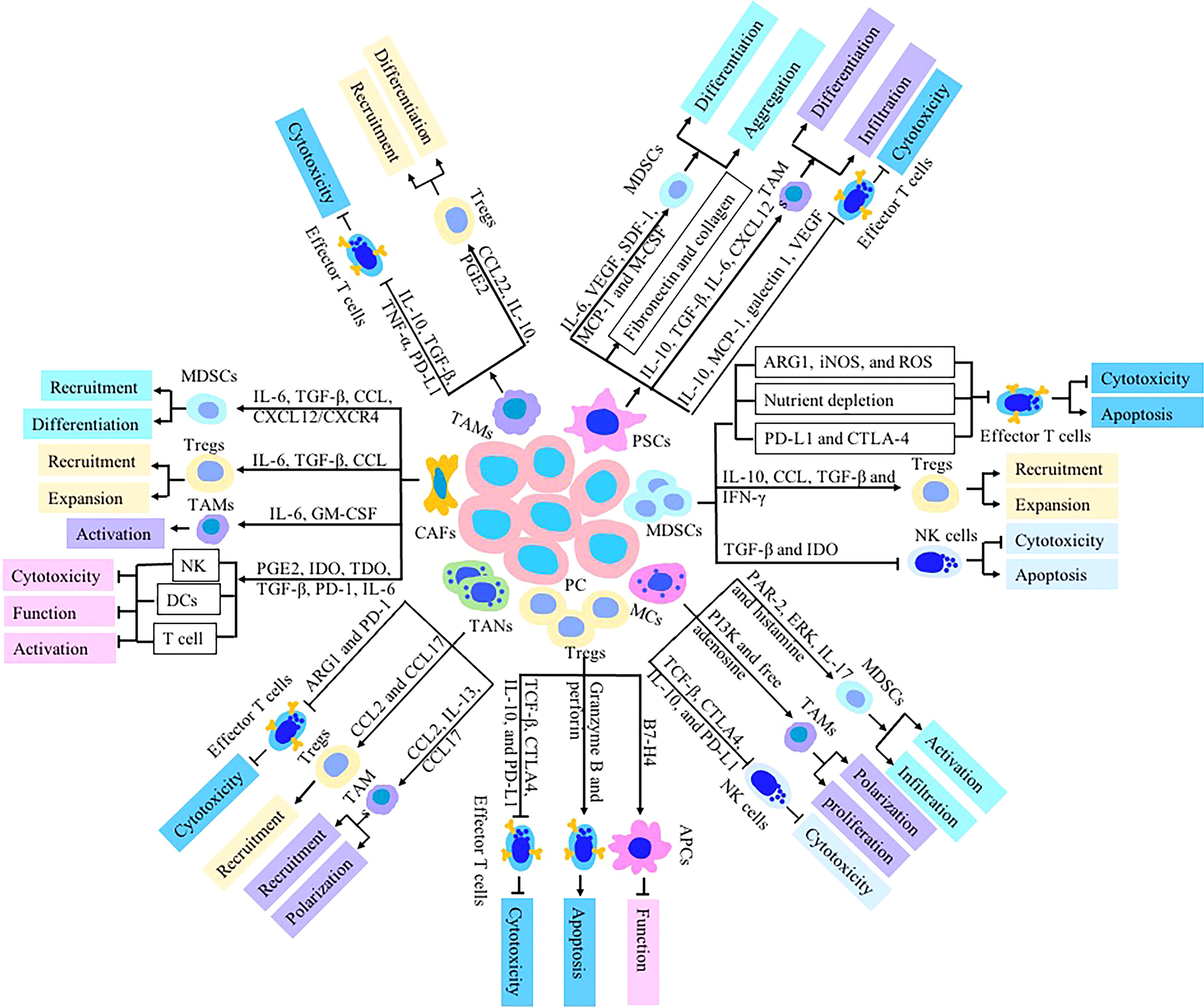
Figure 1 Immunologic effects of stromal cells including MDSCs, CAFs, TAMs, PSCs, Tregs, TANs and MCs on pancreatic TME. These stromal cells dysregulate the function of immune cells-mediated antitumor immunity to establish an immunosuppressive TME via secretion of chemokines and cytokines such as TGF-β, IL-10, IL-35, CCL5, CXCL12, GM-CSF, PGE2, IDO, PD-L1 and CTLA-4. The cytotoxic activity and cytokines production of NK cells, DCs and CTLs are inhibited in pancreatic TME.
MDSCs are heterogeneous cell populations which mainly composed of immature myeloid cells that are found in pathological settings (87). They are precursors of immature monocytes, macrophages, granulocytes and dendritic cells. In patients with pancreatic cancer, the differentiation of monocytes and granulocyte precursors into mature myeloid cells is blocked and they are stimulated by tumor derived factors to differentiate into MDSCs. Studies have shown that MDSCs are mainly distributed in bone marrow, spleen, lymph nodes, peripheral blood and tumor tissues, and gradually accumulate in TME (88). Tumor associated inflammation can stimulate the expansion of MDSCs. Growth factors produced by chronic inflammation (GM-CSF, G-CSF, M-CSF, VEGF and TGF-β), proinflammatory cytokine (IL-1, IL-4, IL-6, TNF-α and IFN-γ) and chemokines (CCL2, CCL4, CXCL1, and CXCL12) can stimulate the expansion of MDSCs (11). These factors induce the amplification of MDSCs via JAK/STAT, PI3K/Akt, Notch, NF-κB, NLRP3, Wnt and adenosine receptors A2b signaling pathway, and then MDSCs are recruited to the tumor site by PEG-2, resulting in promoting production of iNOS, arginase, IL-10 and TGF-β (12). Signals that are responsible for converting immature myeloid cells to MDSCs include tumor stroma (proinflammatory cytokines, HMGB1), NF-κB, STAT1, STAT6, COX2 and PGE2 (13). In pancreatic microenvironment, pancreatic stellate cells (PSCs), a subset of pancreatic cancer-associated fibroblasts, promote myeloid cells to differentiate into MDSCs and induce MDSCs to aggregate into TME by producing IL-6, vascular endothelial growth factor (VEGF), macrophage colony-stimulating factor (M-CSF), SDF-1 and MCP-1 in a STAT3-dependent manner (14). Other mediators that are major contributors to induce MDSCs include S100A8 and S100A9 which are regulated by STAT3 and NF-κB. TNF-α also drives the suppressive activity of MDSC by enhancing the binding of S100A8/A9 to the N-glycan motif of receptor for advanced glycation end products (RAGE) (15).
As an important contributor of the immunosuppressive microenvironment in pancreatic cancer, MDSCs induce the inactivation of NK cells, CD4+ and CD8+ T cells by inhibition of lymphocyte activity, recruitment of Tregs and expression of immunosuppressive checkpoint molecules (16). (1) Direct or indirect inhibition of lymphocyte activity. These mechanisms include elimination of key nutrition needed for T cell proliferation by depleting sequestering L-cysteine and L-arginine, and production of NO and ROS. In the pancreatic TME, MDSCs can compete with T cells to use cysteine which is very important for T cell activation and proliferation, resulting in T cells unable to synthesize de novo and hindering the production of anti-tumor T cells (17). In addition, MDSCs also decompose L-arginine and L-tryptophan, further consume essential amino acids and prevent the acquisition of anti-tumor immune cells, resulting in inhibition of anti-tumor immune response. Studies have shown that the expression of arginase 1 (ARG1) and inducible nitric oxide synthase (iNOS) can promote arginine metabolism (18). The high expression of ARG1 and iNOS in MDSCs consumes L-arginine by decomposing L-arginine into L-ornithine and urea. The reduction of L-arginine and the production of nitric oxide in tumor microcirculation can down regulate TCR ζ and MHC II expression, and inhibit T cell activity (19). Release of ROS molecules, which is a critical molecule to maintain the MDSCs in undifferentiated state, is another major mechanism that MDSCs use to suppress T cells by enabling CD8+ T cells to lose ability to bind phosphorylated MHC and inducing antigen-specific tolerance of CD8+ T cells (20). ROS is formed by superoxide reacting with H2O2, hypochlorous acid and hydroxyl radical released by MDSCs. H2O2 inactivate T cells by decreasing T cellular CD3ζ expression and reducing their expression of IFN-γ (21, 22). ROS inhibitors is found to reverse the suppressive effect of MDSCs on T cells to some degree (23). MDSCs also down regulate B cell-mediated immune responses and suppress antibody production by means of arginase, NO and ROS production (24). Under normal physiological conditions, L-tryptophan can produce kynurenine by up regulating the decomposition of indoleamine-2,3 dioxygenase through a STAT3 dependent mechanism. The reduction of L-tryptophan and the production of kynurenine induce the apoptosis of T cells and NK cells, and promote the differentiation of CD4+ T cells into Tregs (25). MDSCs just stand at the top of this regulatory chain and consume L-tryptophan, which promotes the accumulation of canine urinary ammonia, and finally leads to the promotion of immunosuppression of tumor microenvironment. (2) Stimulatory effect on Tregs, which is one important mechanism of MDSC-mediated T cell inhibition. Immunosuppressive cytokines (IL-10), chemokine ligand (CCL4 and CCL5) and TGF-β produced by MDSCs can induce Tregs into the TME. MDSCs also promote the expansion of induced Treg cells through TGF-β, CD40/CD40L, IL-10 and IFN-γ (26). CD40/CD40L signal pathway plays a role in the homeostasis of Tregs and the homeostasis is disrupted under pathologic conditions. It has been proven that expression of CD40 on MDSCs is critically important for MDSCs-induced Treg proliferation. The involvement of CD40/CD40L in the interplay between MDSCs and Treg in the presence of TGF-β and IL-10 promotes activation and expansion of Treg. Interference with CD40/CD40L interactions reverse the suppression of T-cell responses and inhibits Tregs activity (27, 28). (3) Expression of immunosuppressive checkpoint molecules. PD-1/PD-L1 and CTLA-4 are the best-known co-inhibitory pathways. MDSCs express a high level of PD-L1, which binds to PD-1 expressed on T cells, induces conformational changes, and then makes T cells inactive and dysfunctional (29). Under hypoxia conditions in TME, hypoxia inducible factor 1-α (HIF1-α) selectively up-regulate the expression of PD-L1 on the surface of MDSCs by binding to the hypoxia-response element (HRE) in the PD-L1 proximal promoter (30). Blockade of PD-L1 expression abrogate T cell suppression regulated by MDSCs via modulating production of IL-6 and IL-10 (30). In addition, the ARG1 secreted from MDSCs promote the binding of inhibitory checkpoint ligands with receptors expressed on T cells, accelerating the depletion of T cells, and this effect is abrogated by arginase inhibitor (31). (3) Immunosuppressive cytokines secreted by MDSCs are important factors to inhibit antitumor immune response in pancreatic cancer. The factors involved in MDSC-mediated immune suppression include TGF-β, COX2, indoleamine 2,3-dioxygenase (IDO), IL-6 and IL-10 via NF-κB and STAT3 signaling pathways (32). TGF-β derived from MDSCs plays a key role in inhibiting antitumor immune response. As immunosuppressive cytokines, TGF-β inhibit the activity of cytotoxic T cells and NK cells by reducing IFN-γ. The anti-TGF-β monoclonal antibody restores T-cell anti-tumor response and IFN-γ production to a certain degree (33). Intimate interactions exist between MDSCs and other components such as TAMs, NK cells, CAFs, and T cells in the TME. Due to suppressive activities against effector lymphocytes in TME, MDSCs act as a major barrier to pancreatic cancer immunotherapy. Various therapeutic approaches targeting MDSCs including immunostimulatory adjuvants, therapeutic blockade of the mobilization and survival of MDSCs, and anti-inflammatory agents are being tested in preclinical and clinical studies.
CAFs in TME are heterogeneous cell populations with different origins. Bone marrow-derived fibroblasts and locally infiltrated fibroblasts are considered to be the major sources of CAFs. Mesenchymal stem cells, adipocytes and PSCs can also be transformed into CAFs (34). Various cellular precursors of CAFs result in fibroblast heterogeneity. Four different CAFs populations (referred as CAF-S1 to CAF-S4) have been identified recently by combining the analysis of six CAFs markers including smooth-muscle α-actin (SMA), fibroblast activated protein (FAP), fibroblast specific protein-1 (FSP1, also known as S100A4), integrin β1 (CD29), platelet derived growth factor receptor (PDGFR) and podoplanin (PDPN) (35). As an important part of pancreatic TME, CAFs actively affect nearly all aspects of cancer biology including tumor growth, immunosuppression, invasion and angiogenesis. CAFs promote tumor immune escape through secretion of cytokines, chemokines, metabolites and enzymes, and recruitment of inhibitory immune cells. Cytokine (e.g., IL-6 and TGF-β) and chemokine (e.g., CCL2, CCL5, CXCL1 and CXCL12) released from CAFs attract suppressive immune subsets including MDSCs and Tregs into TME (36). Inducing monocytes to differentiate into M2 macrophages by CAFs is mainly mediated via CXCL12/stromal cell-derived factor 1 (SDF1), M-CSF/CSF-1, IL-6, IL-8 and CCL2/monocyte chemotactic protein-1 (MCP-1). CAFs enhance pro-tumoral M2-TAMs activation via increased secretion of IL-6 and GM-CSF in response to cancer cell stimulation (37). Then CAFs coordinate and synergize with M2-TAMs to inhibit the activity of T cells. The proportion of TAMs and polymorphonuclear MDSCs (PMN-MDSCs) in pancreatic TME is decreased significantly through inhibiting colony stimulating factor 1 receptor (CSF-1R) and CXC chemokine receptor 2 (CXCR2). CAFs recruit monocytes into TME through the CXCL12/CXCR4 and CCL2 pathway, and induce their differentiation into MDSCs through IL-6 -STAT3 signal axis (38, 39). CAFs attract Tregs and decrease CD8+ T cells infiltration into TME by secreting TGF-β and IL-6, thus interfering with tumor-directed immune response (40). In pancreatic cancer, CAFs down regulate NKp30, NKp44, NKG2D, perforin and granzyme B expressed on NK cells by secreting prostaglandin E2 (PGE2), IDO and TGF-β, thereby decreasing NK cell cytotoxic activity (41, 42). Another mechanism by which CAFs trigger an immunosuppressive response in TME is by inhibiting the function of DCs. TGF-β secreted by CAFs induces DCs to down regulate the major histocompatibility complex (MHC) class II molecules and costimulatory molecules such as CD40, CD80 and CD86, thus decreasing their ability to activate cytotoxic T cell responses and their antigen presenting function (35). Activated CAFs express tryptophan-2,3-dioxygenase (TDO), promote the degradation of tryptophan and inhibit the function of DCs. CAFs kill directly CD8+ T cells or reduce CD8+ T population through secreting PD-1, Fas ligand (FasL) and IL-6, and inhibiting the expression of B cell lymphoma 2 (Bcl-2) (3, 40). CAFs have immune regulatory effects on tumor cells, such as up regulating the expression of immune markers on tumor cells. CAFs increase the expression of PD-L1 in pancreatic cancer by secreting CXCL5 and CXCL12 via phosphatidylinositol-3-kinase/protein kinase B signaling pathway, thus promoting the formation of immunosuppressive microenvironment (43, 44). However, Costa et al. revealed the fibroblast heterogeneity and showed that CAF-S1 promotes an immunosuppressive microenvironment by recruiting CD4+CD25+ T cells, while another subtype, CAF-S4, do not exhibit the properties. CAF-S1 is associated with accumulation of FOXP3+ T Lymphocytes and enhances regulatory T cell differentiation through B7H3, CD73, and DPP4 (89). Both CAF-S1 and CAF-S4 promote metastases through complementary mechanisms (90). FAPHigh CAFs attract CD4+ CD25+ T lymphocytes and retain them through high expression of OX40 L/TNFSF4, PD-L2/PDCD1LG2 and JAM2 (91). FAPHigh CAFs secrete high levels of TGF-β ligands to promote cell death of effector CD8+ T lymphocytes by inhibiting the expression of the pro-survival protein BCL2 (92). α-SMA+ FAP+ CAFs decrease the proliferation of CD8+ T cells and induce the recruitment of CD4+ CD25+ T cells via secreting TGF-β and IL-6 (93). Both ecm-myCAF and TGF-ß-my CAF exhibit an immunosuppressive function and contribute to resistance to immunotherapy via recruiting FOXP3high Tregs and increasing PD-1 and CTLA-4 protein expression (94). α-SMA+ CAFs are positively correlated with PD-L1 expression by abolishing T cell anti-tumor immune response (95). The amounting evidence confirms that specific CAF subpopulation functions is complex with the different specific markers, which is one of the most challenges in the study of CAFs. The immunosuppressive effect of CAFs on tumor cells reflects that CAFs can become a therapeutic target in pancreatic cancer immunotherapy. Many drugs targeting key regulator of CAFs are undergoing clinical and/or preclinical evaluation. At present, anti CAFs treatment mainly focuses on fibroblast activation protein (FAP). As a specific marker of CAFs, FAP is a transmembrane serine protease, which is highly expressed on CAFs in most epithelial tumors. In the pancreatic cancer, the deletion of FAP gene can significantly reduce the infiltration of FAP+ CAFs and induce tumor hypoxic necrosis (96). Elimination of FAP+ CAFs by DNA vaccination and chimeric antigen receptor (CAR)-T cells has become an important immunotherapy strategy (97). A study showed that oral FAP-DNA vaccine could induce both CD8+ and CD4+ immune responses, and antitumor immunity was enhanced by combing FAP DNA vaccine with other tumor antigen-specific DNA vaccines (98). However, it should not be ignored that bone marrow mesenchymal stem cells and skeletal muscle expressing FAP may also be recognized and impaired by FAP specific CAR-T cells, leading to fatal bone marrow toxicity and cachexia. In addition to directly eliminating and inhibiting CAFs, it is also effective to restore CAFs to normal state. Angiotensin receptor blockers can reprogram CAFs into to a quiescent state, alleviate immunosuppression and improve T lymphocyte activity (99). but its clinical application is limited because of systemic adverse reactions. Resetting activated CAFs into the static state through vitamin A and vitamin D has attracted extensive attention. Administration of all-trans-retinol can inhibit activated CAFs that promote tumor signal pathway, significantly increase CD8+ T cell infiltration, and improve the efficacy of cancer chemotherapy (100–102). A randomized, double-blind, placebo- controlled trial performed by Morita et al. reported that Vitamin D supplementation decreased serum levels of PD-L1 in digestive tract cancer and significantly reduced the risk of relapse/death in the highest level of PD-L1 (103). Due to the toxic effects of directly targeting CAFs, researchers begin to focus on the effector molecules that inhibit the secretion of CAFs. Targeting CAFs related signaling pathways with immunotherapy is a possible approach for improving the anti-tumor efficiency. Blocking cytokines secreted by CAFs (such as IL-6, IL-8, IL-10 and TGF-β) combined with PD-1/PD-L1 can improve the anti-tumor immune response through increasing T cell infiltration and alleviating the role of PD-1 in inhibiting T cell activity (3). Thus, targeting CAFs related signaling pathways may be a new strategy for the treatment of pancreatic cancer.
Treg cells are a kind of T cell subsets that regulate a variety of immune cell functions. They play a critical role in suppressing anti-tumor immune response. Treg cells maintain the balance of the immune system and avoiding autoimmune diseases, allergic reactions and transplantation rejection under physiological conditions (104). In patients with pancreatic cancer, Treg cells are an important component involved in the inhibition of tumor immune response by disordering the functions of CD4+ T and CD8+ T cells, macrophages, NK cells and DC cells (105, 106). The number of Treg cells in pancreatic TME increases significantly, and is correlated with tumor metastasis and poor prognosis (45). The infiltrated Treg cells were directly proportional to the infiltrated CD4+ T cells and inversely proportional to the infiltrated CD8+ T cells. Treg cells reduce the infiltration of antitumor effector lymphocytes in peritumoral tissues by secreting IL-35. Treg cells are chemoattracted to TME by chemokines including CXCR3-CCL9/10/11, CCR4-CCL17/22 and CCR8-CCL1. Tumor cells secrete cytokines such as CCL5, CCL17 and CCL22 which Treg are chemotactic to (46). In addition, studies have shown that pancreatic cancer cells induce PSCs to express and secrete CXCL10 which acts as a chemoattractant for Treg cells. CXCL10 mediate the recruitment of CXCR3+Treg cells to tumors via its cognate receptor CXCR3 (47). Treg cells secrete inhibitory cytokines such as TCF-β, CTLA4, IL-10, and PD-L1 to mediate the immune tolerance of the effective cell to pancreatic cancer associated antigen, thereby avoiding the tumor cells from immune surveillance. Pancreatic cancer with higher Treg and lower Th17 cells alter cytokine IL-10, IL-17, IL-23, INF-γ and TGF-β expression by regulating transcription factors such as CTLA-4, RORγt, ROR-α, and FoxP3 (48). Other studies have found that tumor derived endothelial cells highly express mucosal epithelial cell adhesion molecule-1 (MADCAM-1), vascular cell adhesion molecule-1 (VCAM-1), and the ligands of these adhesion molecules are specifically expressed on Treg cells. Specifically blocking the adhesion molecules reduce the adhesion of Treg cells to tumor derived vascular endothelial cells and reduce the infiltration of Treg cells into TME (49). Cytokines secreted by cancer cells and other stromal cells in pancreatic tumor microenvironment, such as TGF-β1 induce the differentiation of CD4+ CD25- T cells into CD4+ CD25+ Foxp3+ Treg cells. Application of TGF-β neutralizing antibodies can block this differentiation process (50). Treg cells inhibit the function of immune cells such as CD8 cytotoxic T lymphocytes, CD4 helper T cells and NK cells. However, the mechanism is still controversial. Experimental data supports that it mediates the inhibitory effect on immune response mainly by secreting immunosuppressive cytokines such as TGF- β and IL-10. Treg cells induce antigen presenting cells (APCs) to express B7-H4 by triggering high levels of IL-10 production, and then these APCs expressing B7-H4 inhibit the proliferation of T cells by interacting with the corresponding receptors (51). Activated Treg cells directly induce apoptosis of NK cells and CD8+ T cells by secreting granzyme B and perforin. In addition, Treg cells combine with APCs to induce the expression of IDO, so as to inhibit the activation of T cells (52). In the pancreatic TME, tumor cells are conferred with the biological functions of the immune cells. They cooperate with infiltrating Treg cells and macrophages to play an immunosuppressive function and promote tumor progression.
TAMs are important inflammatory cells in the pancreatic TME. Tumor cells secrete chemokines (CCL2, CCL7, and CXCL12) and cytokines (VEGF) to recruit peripheral blood monocytes into tumor tissues and induce them to differentiate into TAMs. Macrophages can be divided into M1 type and M2 type. The former, which is induced by Toll-like receptor 4 (TLR4) and IFN-γ, secrete tumor necrosis factor (TNF), iNOS and MHC class II molecules, and has the ability to coordinate the anti-tumor immune response of Th cells; M2-macrophages are induced by IL-4 and IL-13 which secreted by CD4+ T cells. M2-macrohphages with high level of ARG1, IL-10, CD163 and CD204 expression have anti-inflammatory responses and pro-repair, pro-tumoral and anti-parasitic ability (53). Macrophages in a state of constant transition between the two forms of M1 and M2 phenotype adopt context-dependent phenotypes when stimulated. They are differentiated and activated mainly by IRF/STAT (interferon-regulatory factor/signal transducer and activator of transcription) signaling pathway (54). Tumor associated macrophages (TAMs) which are broadly considered M2-like secrete a large number of cytokines including PGE2, epithelial growth factor (EGF), epithelial growth ligands of the factor receptor (EGFR), IL-10 and TGF-β, which stimulate tumor cell proliferation and survival (55). C5aR1 which is generally considered to be proinflammatory can induce macrophages to differentiate into M2 type via activation of NF-κB pathway (56). C1q characterizes macrophages with a tolerant or immunosuppressive phenotype, which lead to the up regulation of PD-L1 and PD-L2 and the inhibition of T cell proliferation. C1q also has a direct effect on T cells by regulating the mitochondrial metabolism of CD8+ T cells (57). TSC-mTOR pathway which acts as central interchange for cell proliferation switches macrophages into M2-like TAMs to promote immune-suppressive TME (58).
TAMs take precedence in the pancreatic TME leading to immune evasion through production of inhibitory cytokines (IL-10 and TGF-β), metabolic activities consisting of depletion of L-arginine, ROS production and immune checkpoint engagement. TAMs secrete TNF-α and IL-10 to promote the expression of PD-L1, thereby inhibiting the function of antitumor T cells (59). TGF-β from TAMs promote TAMs to secrete CCL22 and then recruit Treg into TME; Moreover, TGF-β also increase the expression of CTLA-4 and PD-1 on the surface of CD8+ T cells, and reduce the production of granzyme and IFN. TAMs secrete chemokines such as CCL to recruit Tregs into TME, and produce IL-10 and PGE2 to induce Treg differentiation in TME (60). Moreover, Zhou et al. reported that exosomes played a critical role in the interaction between TAMs and T cells. Exosomes including miR-29a-3p and miR-21-5p derived from TAMs directly suppressed STAT3 and induced an imbalance Treg/Th17 cells, generating an immune suppressive microenvironment (61). TAMs upregulated the expression of B7-H4 on the surface of cancer cells through EGFR/mitogen activated protein kinase (MAPK) pathway, and attenuated the cytolytic effect of T cells (62). TAMs in pancreatic TME can serve as a potential target for immunotherapy. IFN-γ possesses immunoregulatory and anti-tumor properties. IFN-γ directly converts TAMs to the M1 subtype. IFN-γ enhances the efficacy of PD1 blockade therapy by inhibiting CXCR2-expressing M2 TAMs tumor trafficking and infiltration (63). A deeper understanding of roles of TAMs in immunotherapy will help explore the targeting TAMs as an adjuvant therapy in tumor immunotherapies.
As one of the important sources of CAFs in pancreatic cancer, PSCs is a fibroblast like cell containing vitamin A lipid droplets in the cytoplasm which secrete extracellular matrix (ECM) components and various cytokines (64). Quiescent PSCs can be activated by cytokines such as interleukin and TGF-β, and the activated PSCs secrete soluble cytokines, including IL-10, MCP-1 and VEGF which interact with immune cells in TME, resulting in the increasing the number of immunosuppressive cells and decreasing the infiltration of anti-tumor immune cells in TME. Moreover, activated PSCs produce large amounts of ECM components such as fibronectin and type I collagen, which form a barrier to immune cell infiltration and facilitate pancreatic cancer cells immune escape (65). Inhibiting the secretion of matrix by PSCs can increase the transport efficiency of chemotherapeutic drugs in TME and improve the response of pancreatic cancer to immunotherapy. Dynamic and bidirectional crosstalk exists between PSCs and other stromal cells in the pancreatic TME. PSCs and macrophages interact with cytokines reciprocally. PSCs induces macrophages to differentiate into M2 type with elevated expression of IL-10, TGF-β, PDGF-β, CD206, and CD301 and decreased expression of M1 macrophage marker iNOS. M2 macrophages increase the expression of TGF-β and PDGF which promote the proliferation and activation of PSCs (66). PSCs promote the differentiation and migration of MDSCs into TME by activating IL-6 and CXCL12. In addition, IL-6 and CXCL12 also promote the infiltration of Treg into TME. PSCs promote the apoptosis of CD4+ and CD8+T cells in pancreatic cancer by secreting galectin 1 (67). PSCs reduce the secretion of Th1 cytokines such as IL -2 and IFN-γ, and enhance the secretion of Th2 cytokines including IL-4 and IL-5, resulting an imbalance of Th1/Th2 cytokines. Further elucidation of the role of PSCs in immunosuppression could lead to exploring novel therapeutic targets in pancreatic cancer (68).
A large amount of TANs infiltrate in the pancreatic cancer. The crosstalk between tumor cells and TANs drive tumor growth and progression. TANs have tumor promoting effects and is associated with poor prognosis of pancreatic cancer via immunosuppression and regulation of other immune cell populations. TANs are classified as N1 (antitumor) or N2 (protumor) according to their activation, cytokine status and effects on tumor cell growing (107). In pancreatic TME, stromal cells and cytokines can regulate the recruitment, activation and differentiation of neutrophils. CAFs participate in the recruitment of neutrophils to TME, especially by secreting CXC1, CXCL2, CXCL5, CXCL6, CXCL2 and chemokine ligand 2. Tumor-derived cytokines, such as TGF-β and GM-CSF extend survival of TANs in TME. TGF-β in TME secreted by CAFs induces neutrophils to differentiate into N2 TANs, which in turn recruit Treg cells through CCL17 secretion (69, 70). When IFN-β or TGF-β in TME is inhibited, TANs show the N1 phenotype of tumor inhibition. When TGF-β is increased at high levels, TANs showed the N2 phenotype of immunosuppression and cancer promotion, and their gene expression patterns also changed, such as up regulation of CCL17, ARG1 and MMP9. N2 neutrophils leads to somatic DNA instability and promotes tumorigenesis by producing ROS, NO and PEG2. Zhou et al. reported that TANs recruited macrophages and Treg cells to infiltrate the tumor site by secreting CCL2 and CCL17, and contributed to sorafenib resistance. The number of infiltrated TANs could be used as a biomarker for predicting responsiveness to sorafenib (71). TANs also accumulate in tumor tissues and secrete elastase, which promotes EMT and secretion of MMP-9 in pancreatic cancer, and promotes tumor invasion and angiogenesis. Moreover, TANs inhibit antitumor immunity by up regulating ARG1 and PD-1. There exists mutual influence between TANs and TAMs and they coordinate effects within the TME. N2 TANs promotes TAMs polarization via secretion of IL-13 (72). TANs and TAMs secrete CCL17 to induce the recruitment of Treg cells to the TME. The interaction between TANs and TAMs requires targeting common TAM and TAN signal pathway as a means to enhance the efficacy of current therapies.
Dendritic cells (DCs), as one of the most powerful APCs in the immune system, are central regulators of the adaptive immune response and essential for T cell-mediated cancer immunity (108). DCs express co-stimulatory molecules (CD80, CD40, CD70 and CD86) and antigen-presenting molecules such as MHC-I and MHC-II, and DCs deliver the co-stimulatory signals to activate T cells. DCs also enhance T cell expansion and polarization to secret of IFN-γ via cytokine signals including IL-12 and IL-15 (109). Therefore, DCs play a critical role in anti-tumor and immunoregulatory activities. The study of DCs infiltrated in pancreatic TME is of great significance to understand the mechanism of tumor immune evasion. CD103+ DCs in tumors are necessary to recruit effector T cells into the TME and priming tumor-specific CD8+ T cells. It is reported that the recruitment of effector T cell subsets into TME depended on the expression of CXCL9 and CXCL10 which mainly released from CD103+ DCs in tumors (110). Abound infiltration of CD103+ DCs in tumor improve responses to therapeutic PD-L1 and BRAF blockade (111). However, A network of immunosuppressive factors in TME inhibit DCs infiltration and their anti-tumor activity. Tumor decrease DCs infiltration by reducing CCL4 expression and producing PGE2. DCs infiltrated in tumor enhance expression of T cell immunoglobulin mucin receptor 3 (TIM3) which sequesters high mobility group protein B1 (HMGB1), resulting in impairment of DC activation and antigen presentation (112). Metabolites in the TME such as lactic acid which is a major metabolic product of tumor cells dampen DCs differentiation and activation. DCs interact with Treg cells to induce immune tolerance. Treg cells prevent the expression of CD80 and CD86 to inhibit the maturation of DCs (113). In addition, Treg cells regulate the cytokines produced by DCs. For example, Treg cells decrease IL-6 and increase IL-10 expression in DCs. In metastatic pancreatic cancer, DCs induce the proliferation of Treg cells and inhibit CD8+ T cell-mediated tumor immunity via MGL2 and PD-L2, establishing an immunosuppressive microenvironment favor for metastasis formation (114). DCs also regulate the balance between Th17 and Treg cells. Some cytokines in TME, such as macrophage colony stimulating factor, IL-4, IL-6, IL-10, TGF-β and VEGF can alter the differentiation of DCs and inhibit the activation and maturation of DCs, resulting in a lack of mature and functional DCs in tumor patients. These immature DCs do not express or down regulate costimulatory molecules, MHCII molecules and antigen presentation related transporters, but express indoleamine 2,3-dioxygenase, which degrade tryptophan and inhibit T cell immunity (115). Surgical resection, chemoradiotherapy or immunochemotherapy can increase the number of DCs in the circulation and restore its function to some extent. Increased DCs not only prolong the survival, but also significantly reduce the risk of postoperative infection complications in patients undergoing pancreatectomy. These findings suggest that immunotherapy to increase the number of DCs and restore their function is beneficial to improve the prognosis of patients (116).
DC vaccine is the focus of tumor immunotherapy. The treatment strategy based on DCs is to use patients’ own DCs to produce therapeutic vaccine. First, DCs are collected from the patient, matured in vitro, loaded with tumor antigen and injected back into the patient. After injection, DCs present tumor antigen to T cells, leading to T cell activation and initiating T cell response. DC vaccine has been proved to be safe for the treatment of pancreatic cancer (117). It is confirmed that the combination of gemcitabine and DC vaccine can promote the recruitment of CD8+ T cells and enhance CTLs mediated tumor cell death in a mouse model of pancreatic cancer (118). Recent multicenter clinical studies have also confirmed that DC vaccine and chemotherapy drugs induced to produce tumor antigen-specific CTLs, having a synergistic effect on pancreatic cancer patients (119). In order to restore the function of DCs in TME, many attempts have been made. It is reported that impaired function of DCs as well as the immunosuppressive effect of Treg can be reversed by activating TLR-7/8 and TLR-9 signal pathways (120). Because the suppressor of cytokine signaling 1 (SOCS1) is a negative regulator that inhibit the maturation of DCs, some studies have designed a siRNA to interfere with SOCS1, aiming to promote the antigen presentation of DCs, activate TLRs signal pathway and promote the maturation of DCs (121).
Natural killer (NK) cells are an important part of tumor immunosurveillance with ability of directly killing cancer cells or indirectly promoting the immune responses. NK cells secrete a large number of cytokines including GM-CSF, IFN-γ, TNF-α and IL-3 which affect the activity of other immune cells such dendritic cells, neutrophils and macrophages, thus influencing the subsequent T and B cell responses (73). Chemo-attractants/receptors (including CCL5/CCR5, CCL27/CCR10 and CX3CL1/CX3CR1) and immunomodulation of chemokine axes (including HLA-G and CD47) are involved in controlling NK cell recruitment to the tumor. Extracellular matrix (ECM) barriers also play a role in regulating NK cells infiltration into TME (74). The activation and functional status of NK cells are depended on interacting signals between activating co- stimulatory and inhibitory signals. Activating signals mainly comprise integrins, killing-receptors (CD16, NKp30, NKp40 and NKp44), and other receptors such as NKp80, SLAMs, CD18, CD2 and TLR3/9. Inhibitory signals include Ly49s, NKG2A and LLT1 (Summarized in (75)). NK cells kill or ignore target cells based on the balance between ligands expressed on tumor cells that mediated inhibitory and activated signal. NK cells induce apoptosis of cells through producing a huge amount of IFN-γ, perforin and granzyme family molecules after re-stimulation. The sensitivity of target cells to NK cells mediated cell lysis mainly depends on the expression of MHC I. NK cells selectively lyse cells with low expression of MHC type I molecules or cells that lose the expression of MHC I. Virus infected cells or tumor cells down regulate the expression of MHC molecules, and become possible targets for NK cells (76). The cytotoxic ability of NK cells can be enhanced by IL-2, IL-12, IL-15 and IFN-α/β) while their cytotoxic functions are inhibited by immunosuppressive factors such as TGF-β, IL-10, PGE2, and IDO1/2 (75). Close interaction and a positive feedback loop exist between macrophages and NK cells in the TME. NK cells activate macrophages and promote M1-like macrophage polarization to secrete a large number of cytokines such as IL-12/18 by expressing IFN-γ, TNF-α and GM-CSF. Meanwhile, IL-12/-18 secreted by activated macrophages increase of NK cell cytotoxicity and IFN-γ production, and enhance the expression of CD80, CD86, HLA-DR and HLA-DQ (77). The interaction between NK cells and DCs can lead to the activation of NK cells, cytokine production, cytotoxicity and the maturation of DCs (78). NK cell toxicity, IFN-γ production and NKG2D expression are blocked by MDSCs and Treg cells in a TGF-β-dependent mechanism in pancreatic TME. TGF-β is also able to decrease NKp30- and NKG2D- expression on NK cells, thus interfering possibly the crosstalk of NK cells and DCs (79, 80). In the TME of pancreatic cancer, the receptor NKG2D, NKp30 and NKp46 related to NK cell activity are significantly downregulated. Therefore, the number of NK cells in pancreatic cancer tissue is decreased significantly. However, due to heterogeneity of NK cells and complexity of tumor intrinsic signal pathways, the specific role of NK cells in cancer immune response is depends on distinct cancer types and need further elucidation.
Mast cells derived from CD34+/CD117+ pluripotent hematopoietic stem cells release chemokines and cytokines after activation. MCs activation is triggered by a variety of receptors such as high-affinity IgE receptor (FcepsilonRI), TLRs, complement receptors and adenosine receptor (81). MCs are recruited to the TME by pancreatic cancer cells, and the infiltrated MCs promoted pancreatic cancer progression in a matrix metalloproteinase (MMPs) -dependent manner (82). Moreover, there is complex interaction between mast cells and stellate cells in pancreatic cancer. Stellate cells activate mast cells to secrete IL-13 and tryptase which promote the proliferation of CAFs and PSCs, establishing a feed-forward loop between MCs and PSCs (83). MCs activated CAFs and TGF-β signal pathway to increase the resistance to Gemcitabine/Nabpaclitaxel and promote tumor invasion in pancreatic cancer via PAR-2, ERK1/2 and Akt activation (84). Inhibiting MCs with AMD3100 suppresses tumor growth. Activated MCs inhibit the anti-tumor immune response. MCs promote M2-TAMs polarization and proliferation by PI3K and free adenosine (85). MCs mobilize MDSCs infiltration into tumors and induce MDSCs to secrete IL-17 or histamine, then recruiting Tregs into pancreatic TME and enhancing their suppressive function (86). Histamine reduce the ligands of NK cell receptor expressed on tumor cells and interfere with the recognition and cytotoxicity of tumor cells mediated by NK cells. New possibilities for therapeutic intervention based on MCs need further exploration of the crosstalk between MCs and stromal cell in pancreatic TME.
Stromal cells and cytokines in pancreatic TME promote tumor growth and metastasis by inhibiting the anti-tumor immunity and enhancing the activity of immunosuppressive cells. It is a problem to be solved in tumor immunotherapy that how to reprogram this environment conducive to tumor growth so that the anti-tumor immunity can be restored. New immunotherapy for cancer is expected to be a breakthrough in the treatment of pancreatic cancer. Immunotherapy is rated as the first of the top ten scientific and technological breakthroughs in 2013 because of its remarkable effect on tumor which strongly proves the future potential of tumor immunotherapy (122) Investigations into an increasing variety of immunotherapies and combing with other traditional chemoradiotherapy are urgent need for more effective therapeutic strategies. Combination therapy approaches, such as combining immune checkpoint inhibitors with chemotherapy or radiation therapy, may be necessary to achieve optimal outcomes in this disease. Immunotherapy strategy for pancreatic cancer based on tumor microenvironment is summarized in Table 2. From the perspective of clinical application, it is important to find a biomarkers to identify patients who are likely to respond to immunotherapy. Currently, there is no established biomarker for predicting response to immunotherapy in pancreatic cancer, and this has limited the success of immunotherapy. More research is needed to identify biomarkers that can predict response to immunotherapy, such as tumor mutational burden, microsatellite instability, and immune cell infiltration, which could help guide patient selection and treatment decisions.
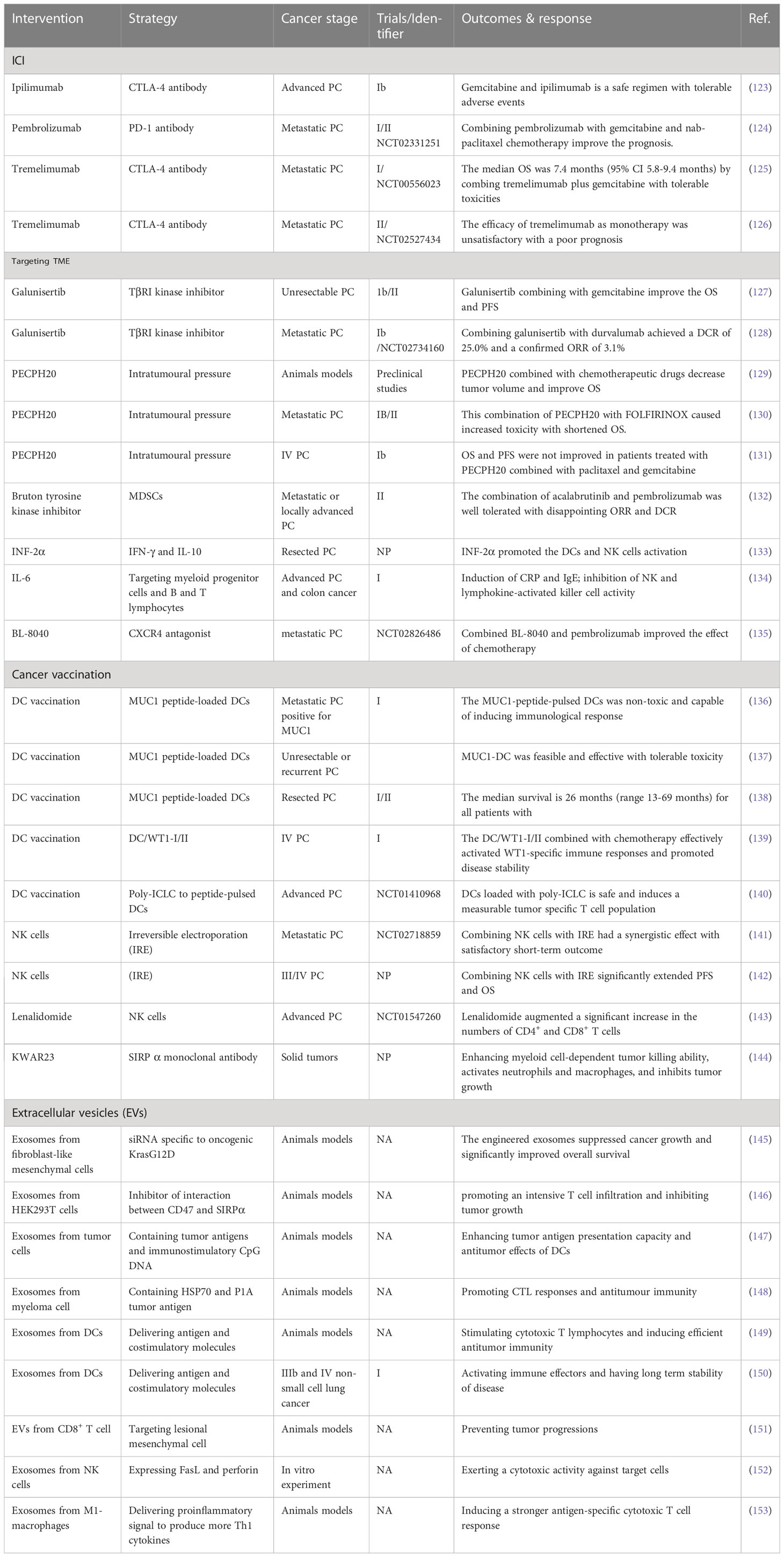
Table 2 Immunotherapy strategy for pancreatic cancer based on tumor microenvironment.
Immune checkpoint proteins are molecules that produce costimulatory or inhibitory signals in the immune response and regulate the host immune response under normal conditions. Recent studies have mainly focused on the immune checkpoint PD-1, its ligand PD-L1 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) which negatively regulate T cell function (154). PD⁃L1 highly expressed in tumors binds to PD-1 on the surface of T cells and limits T cell activation, resulting in tumor immune escape. Therefore, the application of PD-1 or PD-L1 antibody may restore the state of T cells and rectify the immunosuppression of TME for PD-L1 positive tumors. CTLA-4 is an immune checkpoint molecule expressed on Treg. Clearing Treg cells by anti CTLA-4 monoclonal antibodies relieves the inhibition of CTLs and activate T cell immune response. Combinations of immune checkpoint inhibitors with radiotherapy and/or chemotherapy have shown efficacy in pancreatic cancer. Ipilimumab, an anti-CTLA-4 IgG1 monoclonal antibody (mAb), is approved for cancer therapy in USA and Europe in 2011. A phase Ib clinical trial showed that gemcitabine and ipilimumab is a safe regimen with tolerable adverse events for pancreatic cancer, with a median progression-free survival (PFS) of 2.78 months (95% CI 1.61–4.83) and median overall survival (OS) of 6.90 months (95% CI 2.63–9.57) (123). In 2014, Japan and the United States approved pembrolizumab, the first anti-PD-1 monoclonal antibody, for metastatic melanoma. A phase I/II study (NCT02331251) confirmed the safety of pembrolizumab. Moreover, the study also demonstrated that the median PFS and OS with 9.1 and 15.0 months were achieved respectively by combining pembrolizumab with gemcitabine and nab-paclitaxel chemotherapy. The prognosis was slightly improved over previously reported results for standard regiments (124). The efficacy of tremelimumab, another anti-CTLA-4 inhibitor, was evaluated in a phase I trial (NCT00556023). The median OS was 7.4 months (95% CI 5.8-9.4 months) by combing tremelimumab with gemcitabine. Two patients achieved partial response and 7 showed stable disease for more than 10 weeks at the end of treatment (125). However, the efficacy of tremelimumab as monotherapy assessed by a phase II open label study (NCT02527434) was unsatisfactory with 18 out of 20 pancreatic cancer patients demonstrating progressive disease and a poor median OS of 4 months (95% CI 2.83–5.42) (126). The indications of immune checkpoint inhibitors are gradually extending, and there are also survival benefits in pancreatic cancer, renal cell carcinoma, gastric cancer, and non-small cell lung cancer. Blocking of the above two immune checkpoint proteins (PD-1/CTLA-4) may produces a synergistic effect on tumor. However, related complications such as autoimmune hepatitis, myocarditis, nervous system inflammation and pneumonia have been reported with the wide use of immune checkpoint inhibitors (155). It is reported that the mortality of autoimmune myocarditis caused by anti PD-1 therapy is as high as 46% (156). Therefore, effective intervention or method should be taken to avoid possible serious complications before the application of immune checkpoint drugs in clinical practice.
Although the ECM in TME is not as important as infiltrating immune cells in tumor immunotherapy, this component has a critical impact on tumor immunotherapy. Methods have been adopted to reduce extracellular matrix hardness and fibrosis, thus improving immune cell infiltration and drug delivery (157). Inhibition of protein tyrosine kinase that is involved in matrix fiber formation can prolong the survival of pancreatic cancer and make them more sensitive to T cell adoptive reinfusion and PD-1 blocking therapy. The infiltration of immune cell into TME is enhanced via digesting the fibrous stroma through applying of IL-15 activated NK cells, whole-cell vaccines secreting GM-CSF and CD40 specific monoclonal antibody. TGF-β secreted by CAFs in pancreatic TME accelerates formation of fibrous matrix by transforming the phenotype of fibroblasts. Furthermore, TGF-β increases production of matrix proteins, including collagen, fibronectin, proteoglycans and tenascin via enhancing the expression of ECM-associated genes in epithelial cells, including collagen type 1 α1 (COL1A1), lysyl oxidase homologue 4 (LOXL4) and MMP (158). TGF-β can be used as a target for enhanced cancer chemotherapy and immunotherapy. The TβRI kinase inhibitor galunisertib (LY2157299) combining with gemcitabine improve the OS and PFS for patients with unresectable pancreatic cancer in a clinical trial (127). A multinational, phase Ib study (NCT02734160) evaluated the safety and efficacy of the galunisertib co-administered with the anti-programmed death-ligand 1 (PD-L1) antibody durvalumab in metastatic pancreatic cancer. Galunisertib in combination with durvalumab achieved a disease control rate of 25.0% and a confirmed objective response rate of 3.1%. Median OS and PFS were 5.72 months (95% CI: 4.01 to 8.38) and 1.87 months (95% CI: 1.58 to 3.09), respectively (128). The effect of nanomedicine characterized with high permeability, tumor-targeting and substantial retention function on extracellular matrix of pancreatic cancer has been confirmed. Pegylated recombinant human hyaluronidase (PECPH20) decrease intratumoural interstitial fluid pressure and increase in vessel diameter in the tumor-bearing mouse models. Preclinical studies have shown that PECPH20 combined with chemotherapeutic drugs substantially decrease tumor volume and improve the overall survival time (129). A clinical trial evaluated the efficacy of PECPH20 combined with modified FOLFIRINOX in patients with untreated metastatic pancreatic cancer. Compared with the modified FOLFIRINOX alone, the OS of patients in the combination group was shortened and the incidence of related adverse events was significantly increased. This combination resulted in decreased treatment duration due to increased toxicity (130). OS and PFS were not improved in patients with metastatic pancreatic cancer treated with PECPH20 combined with paclitaxel and gemcitabine (131). The disappointing results indicate that it is not enough to only target fibrous tissue proliferation. In addition, although ECM in pancreatic TME is a physical barrier that hinders drug delivery, it also plays a protective role in inhibiting tumor growth and progression. The strategy selectively targeting ECM barrier of pancreatic cancer is worth further exploring.
In view of the inhibitory effect of MDSCs in tumor immunity, many studies focus on finding ways to block the signal transduction of MDSCs. Animal model studies have confirmed that gemcitabine significantly reduce MDSCs in the spleen without altering the number of T and B lymphocytes. Bruton tyrosine kinase (BTK), a non-receptor enzyme in the Tec kinase family, is critical to maintain the desmoplastic microenvironment surrounded by an abundance of Tregs, MDSCs, TAMs and MCs in pancreatic cancer. BTK inhibition converts M2-like macrophages to an M1-like phenotype and promotes the cytotoxicity of CD8+ T cells. Inhibition of BTK with ibrutinib enhances T cell-dependent antitumor immune responses and improves responsiveness of pancreas cancer to gemcitabine (159). A randomized phase II clinical trial (NCT02362048) reported that the combination of BTK inhibitor acalabrutinib and pembrolizumab led to reduction of reductions in granulocytic (CD15+) MDSCs in peripheral blood of patient with pancreatic cancer, but the overall response rate and disease control rate were disappointing (132). Many studies have confirmed the importance of CAFs in promoting the occurrence and development of pancreatic cancer. However, some problems exist in targeting CAFs. One is that CAFs are highly heterogeneous. Inflammatory CAFs with low expression of a-SMA and high expression of IL-6 promote tumor growth, while myofibroblastic CAFs with high expression of a-SMA inhibit tumor growth (160). In addition, the phenotype CAFs of different subtypes can be transformed intercellularly. Meflin-positive CAFs have tumor suppressive properties by remodeling the extracellular matrix. However, meflin-positive CAFs differentiate into cancer-promoting phenotypes with the tumor progresses (34). Therefore, how to identify and remodel various properties of CAFs is the key to target CAFs for pancreatic cancer treatment.
The safety and efficacy of DC-based immunotherapy for pancreatic cancer have been evaluated. Studies show that patients with pancreatic cancer receiving DC vaccination-based immunotherapy achieves a significantly better survival period (161, 162). Peptide-loaded DC vaccines selectively targeting tumor-associated antigens (TAAs) such as mucin 1 (MUC1), Wilms’ tumor gene 1 (WT1) and mutant K-RAS also enhance more cytotoxic lymphocyte response. Clinical trials showed that MUC1 peptide-loaded DCs were safe and effective for unresectable, recurrent or refractory pancreatic cancer (136–138). A phase I trial to investigated the safety and efficacy following treatment with DCs pulsed with a mixture of three types of WT1 peptides in combination with chemotherapy in pancreatic cancer and intrahepatic cholangiocarcinoma. The DC/WT1-I/II combined with chemotherapy effectively activated WT1-specific immune responses and promoted disease stability in advanced pancreatic cancer (139). DCs enhance more effectively the anti-tumor immune response when cultured in presence of toll-like receptor (TLR)-3 agonist poly-ICLC. Mehrotra, et al. (NCT01410968) showed that the addition of poly-ICLC to peptide-pulsed DCs was safe and produced increased numbers of tumor specific T cell population in patients with advanced pancreatic cancer (140). Even though DC vaccines-based immunotherapy have effective therapeutic effects on pancreatic cancer, the effect is limited because decreased expression of MHC molecules and costimulatory molecules on cancer cells mediates the tumor immune evasion. A series of clinical studies have shown that adoptive NK cell transfer has anti-tumor effects on pancreatic cancer. A clinical trial (NCT02718859) showed combining allogeneic NK cells with irreversible electroporation (IRE) had a synergistic effect on pancreatic cancer, demonstrating satisfactory short-term outcome and the quality of life of the patients (141). In addition, percutaneous IRE in combining with allogeneic NK cells also significantly extended the PFS and OS in advanced pancreatic cancer (142). Lenalidomide, initially approved by the U. S. Food and Drug Administration (FDA) for multiple myeloma (MM), has anti-tumor effects by augmenting the cytotoxicity of NK cells. A clinical trial (NCT01547260) found lenalidomide augmented a significant increase in the numbers of CD4+ and CD8+ T cells in patients with advanced pancreatic cancer. However, combination of lenalidomide with gemcitabine have no therapeutic impact compared to gemcitabine alone (143).
The tryptophan catabolic enzyme IDO produced by tumor cells in TME promote the activation of Treg cells and increase the infiltration of MDSCs. Therefore, depletion of IDO inhibits tumor stem cells proliferation pathways and improves immunotherapeutic vaccines susceptibility by reducing TLRs 2 to 9, NF-κβ1-2, Wnt/β-catenin and TGF-β (163). CD47 is expressed in a variety of tumor cells, and mediates a “don’t eat me” signal after binding the immune checkpoint protein SIRP-α expressed on myeloid cells (monocytes, macrophages, and DCs), which contributes to the resistance of tumors to phagocyte-dependent clearance. KWAR23, a SIRP α monoclonal antibody which blocks its binding to CD47, enhances myeloid cell-dependent tumor killing ability, activates neutrophils and macrophages, and inhibits tumor growth (144). IFN enhances function of DCs and NK cells, and improve the survival of T cells, which is already used in cancer therapy. Karakhanova et al. reported that INF-2α increased amount of IFN-γ and IL-10 in the serum, and promoted the DCs and NK cells activation following the IFN-2α therapy in pancreatic cancer patients. However, activation of anti-tumor immunity as well as immunosuppressive response are induced in the unspecific immunotherapy with INF-2α (133). IL-6, a glycoprotein of 184 amino acids, acts on myeloid progenitor cells and B and T lymphocytes. A phase I clinical study showed that IL-6 induced C-reactive protein and IgE levels, and had a suppressive effect on NK and lymphokine-activated killer activity in patients with advanced pancreatic cancer and colon cancer (134). Combined CXCR4 and PD-1 blockade may expand the benefit of chemotherapy. A phase IIa clinical trial (NCT02826486) conformed the synergistic effects of combing CXCR4 antagonist BL-8040 (motixafortide) with pembrolizumab and chemotherapy on metastatic pancreatic cancer (135).
Accumulating evidence indicates that extracellular vesicles (EVs) including microvesicles (MVs) and exosomes mediate reciprocal communication between pancreatic cancer and stromal cells in TME, and ultimately exerts influence on the biologic features of recipient cells, thus promoting cancer progression and evasion of immune surveillance (164, 165). Exosomes derived from pancreatic cancer cause an imbalance of immune cells via increasing MDSCs, while reducing DCs. Tumor-derived EVs inhibit NK cell cytotoxicity by decreasing the expression of NKG2D, INF-γ and TNF-α (166). TGF-β1 contained in pancreatic cancer-derived EVs suppresses NK cell cytotoxic activity against tumor cells via SMAD2/3-dependent signal pathway, resulting in immune tolerance and tumor immunosuppression (167). The EVs are modified for clinical applications through artificially integration of specific loadings such as drugs or tumor targeting molecules. Exosomes from normal fibroblast-like mesenchymal cells carry siRNA specific to oncogenic KrasG12D which is commonly mutated in pancreatic cancer. The engineered exosomes suppressed cancer growth and significantly improved overall survival (145). Exosomes secreted from immune cells including DCs, macrophages and CD8+ T cells express SIRPα, PD1, or tumor antigen peptides, which can be used as immune modulators. SIRPa-exosomes, acting as immune checkpoint blockade, antagonizes the crosstalk between CD47 and SIRPa, and induce tumor phagocytosis. SIRPa-exosomes enhance effective anti-tumor T cell response and increase T cell infiltration in cancer (146). Engineered exosomes from tumor cells containing immunostimulatory CpG DNA or tumor antigens (TRP2, gp100, endogenous P1A tumor antigen and HSP70) stimulate DCs maturation and T cell immune responses (147, 148). EVs released from immune cells also can be used as a vaccine adjuvant in cancer immunotherapy. Clinical trials showed that exosomes from DCs loaded with tumor antigen activated the strong cytotoxicity of CD8+ T cells and promoted antitumor immune responses (149, 150). EVs derived from activated CD8+ T cell inhibit tumor progression by depletion of mesenchymal tumor stromal cells (151). Exosomes from NK cells and M1-like macrophages exert a stronger cytotoxic T cell response against tumor cells (152, 153). However, some challenges in EVs-based immunotherapy still remain including lack of methods for efficient isolation, purification, and identification of specific EV populations. Additionally, the antigen loading efficiency of EVs should be improved (168).
Oncogenic K-Ras is involved in tumorigenesis in pancreatic cancer and contribute to immunosuppressive microenvironment. More than 85% patients with pancreatic cancers harbor the G12 mutation in K-Ras. K-Ras mutation increases robustly MEK/ERK activity by upregulating the EGFR and RAS levels (169). Due to the multiple alternative pathways of K-Ras, there is none of specific K-Ras inhibitors that have been applied in clinical practice. Moreover, inhibitors of downstream molecules of K-Ras such as RAF/MEK/ERK pathway or the PI3K/PDK1/AKT/mTOR pathway show little efficacy (170). Multidrug combinations of MEK inhibitors and PI3K inhibitors contribute to apoptosis in pancreatic cancer and achieve a longer median survival (171). Ulixertinib, an ERK inhibitor, combined with MEK inhibitors produces potent synergistic effects on tumor (172). More studies are needed in the evaluation of combinatorial effect of these therapies altogether to improve survival rate.
Immunotherapy has a positive therapeutic effect on patients with advanced pancreatic cancer. Tumors in TME dynamically evolves by blocking the effect of immunotherapy and increasing drug resistance. Because of the complexity of tumor immunosuppressive microenvironment, immunotherapy including immune checkpoint inhibitors, interfering with stromal cells or cytokines in pancreatic TME and cancer vaccines still need to be improved. The efficacy of immunotherapy combinations with chemoradiotherapy or other molecularly targeted agents is also disappointing for the majority of clinical trials. The main reasons hindering the effect of immunotherapy include: (1) the low response of host immune system to tumor antigen; (2) low infiltration of immune cells in pancreatic TME; (3) formation of immunosuppressive TME which is characterized by a profoundly desmoplastic stroma. A large number of immunosuppressive cells including MDSCs, TAMs, CAFs, and Tregs in pancreatic TME decrease the efficacy of immunotherapy (173–175). A variety of immunotherapeutic drugs targeting TME have been developed. However, it is evident that single-agent immunotherapies are unlikely to be successful in pancreatic cancer due to the diversified immunosuppressive signals, and the heterogeneity of TME at different stages of cancer progression is complex, which limits the accuracy and effectiveness of immunotherapy. Considering the limitations of current immunotherapy strategy, it may be improved from the following aspects: (1) screening markers sensitive to the immunotherapy strategies. Sensitive biomarkers predict the effectiveness of immunotherapy, dynamically monitor the treatment process, and adjust the therapeutic schedule accordingly; (2) combine multiple immunotherapy strategies. It is necessary to explore the combined treatment strategies that strengthen the effect of cytotoxicity and improve the sensitivity of immunotherapy through the combination of radiotherapy, chemotherapy and immunotherapy, or the application of multiple immunotherapy strategies; (3) develop new immune activation strategies. By developing a new immune activation vector will affect the immune state of pancreatic TME, and enhance immune response against tumor (176). Therefore, it is still necessary to explore the underlying mechanism of immunosuppressive TME, which contribute to the success of combination therapy for pancreatic cancer.
SL and YL designed the review structure and wrote the manuscript. SX performed the scientific literature search, collected and analyzed data. WP, JW and HZ prepared the tables and figures. All authors contributed to the article and approved the submitted version.
The study was supported by the National Natural Science Foundation of China (Grant No. 81802888), the Key Research and Development Program of Shandong (No. 2018GSF118088), and the General Financial Grant from the China Postdoctoral Science Foundation (No. 2016M592143).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
APCs, antigen presenting cells; ARG, arginase 1; CAFs, cancer‐associated fibroblasts; CAR-T, chimeric antigen receptor; CTLs, cytotoxic T lymphocytes; CTLA‐4, CTL‐associated antigen 4; CSF-1R, colony stimulating factor 1 receptor; CXCR2, CXC chemokine receptor 2; DCs, dendritic cells; ECM, extracellular matrix; EVs, extracellular vesicles; EGF, epithelial growth factor; EGFR, epithelial growth ligands of the factor receptor; HMGB1, high mobility group protein B1; HIF1-α, hypoxia inducible factor 1-α; IDO, indoleamine 2,3-dioxygenase; iNOS, inducible nitric oxide synthase; MCP-1, monocyte chemotactic protein-1; MDSCs, myeloid derived suppressor cells; MHC, major histocompatibility antigens; M-CSF, macrophage colony-stimulating factor; MUC1, mucin 1; NK, natural killer; OS, overall survival; PD-1, programmed death‐1 receptor, PD-L1, programmed death‐1 receptor–ligand; PSCs, pancreatic stellate cells; PGE2, prostaglandin E2; PFS, progression-free survival; RAGE, receptor for advanced glycation end products; SOCS1, suppressor of cytokine signaling 1; TAMs, tumor associated macrophages; TDO, tryptophan-2,3-dioxygenase; Tregs, regulatory T cells; TEM, tumor microenvironment; TANs, Tumor associated neutrophils; TLR4, Toll-like receptor 4; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J Clin (2020) 70(5):375–403. doi: 10.3322/caac.21626
3. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
4. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol (2020) 17(9):527–40. doi: 10.1038/s41571-020-0363-5
5. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20(1):131. doi: 10.1186/s12943-021-01428-1
6. Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer (2019) 7(1):224. doi: 10.1186/s40425-019-0667-0
7. Huber M, Brehm CU, Gress TM, Buchholz M, Alashkar Alhamwe B, von Strandmann EP, et al. The immune microenvironment in pancreatic cancer. Int J Mol Sci (2020) 21(19):7307. doi: 10.3390/ijms21197307
8. Dougan SK. The pancreatic cancer microenvironment. Cancer J (2017) 23(6):321–5. doi: 10.1097/PPO.0000000000000288
9. Peran I, Dakshanamurthy S, McCoy MD, Mavropoulos A, Allo B, Sebastian A, et al. Cadherin 11 promotes immunosuppression and extracellular matrix deposition to support growth of pancreatic tumors and resistance to gemcitabine in mice. Gastroenterology (2021) 160(4):1359–72.e13. doi: 10.1053/j.gastro.2020.11.044
10. Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer (2018) 17(1):108. doi: 10.1186/s12943-018-0858-1
11. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):1–12. doi: 10.1038/s41423-019-0306-1
12. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98(6):913–22. doi: 10.1189/jlb.4RI0515-204R
13. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
14. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
15. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol (2018) 200(2):422–31. doi: 10.4049/jimmunol.1701019
16. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
17. Lunardi S, Muschel RJ, Brunner TB. The stromal compartments in pancreatic cancer: are there any therapeutic targets? Cancer Lett (2014) 343(2):147–55. doi: 10.1016/j.canlet.2013.09.039
18. Yan X, Takahara M, Xie L, Gondo C, Setsu N, Oda Y, et al. Arginine metabolism in soft tissue sarcoma. J Dermatol Sci (2011) 61(3):211–5. doi: 10.1016/j.jdermsci.2010.12.009
19. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. L-arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol (2004) 232(1-2):21–31. doi: 10.1016/j.cellimm.2005.01.004
20. Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
21. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701. doi: 10.4049/jimmunol.0900092
22. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res (2001) 61(12):4756–60.
23. Wei J, Zhang M, Zhou J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res (2015) 228(3):695–701. doi: 10.1016/j.psychres.2015.06.002
24. Lelis FJN, Jaufmann J, Singh A, Fromm K, Teschner AC, Pöschel S, et al. Myeloid-derived suppressor cells modulate b-cell responses. Immunol Lett (2017) 188:108–15. doi: 10.1016/j.imlet.2017.07.003
25. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol (2020) 11:938. doi: 10.3389/fimmu.2020.00938
26. Malek E, de Lima M, Letterio JJ, Kim BG, Finke JH, Driscoll JJ, et al. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev (2016) 30(5):341–8. doi: 10.1016/j.blre.2016.04.002
27. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882
28. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
29. Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, et al. Targeting MDSC for immune-checkpoint blockade in cancer immunotherapy: Current progress and new prospects. Clin Med Insights Oncol (2021) 15:11795549211035540. doi: 10.1177/11795549211035540
30. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
31. Li Y, Wang J, Wang H, Zhang S, Wei Y, Liu S. The interplay between inflammation and stromal components in pancreatic cancer. Front Immunol (2022) 13:850093. doi: 10.3389/fimmu.2022.850093
32. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
33. Chen J, Ye Y, Liu P, Yu W, Wei F, Li H, et al. Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol (2017) 78(2):113–9. doi: 10.1016/j.humimm.2016.12.001
34. Geng X, Chen H, Zhao L, Hu J, Yang W, Li G, et al. Cancer-associated fibroblast (CAF) heterogeneity and targeting therapy of CAFs in pancreatic cancer. Front Cell Dev Biol (2021) 9:655152. doi: 10.3389/fcell.2021.655152
35. Mhaidly R, Mechta-Grigoriou F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin Immunol (2020) 48:101417. doi: 10.1016/j.smim.2020.101417
36. De Jaeghere EA, Denys HG, De Wever O. Fibroblasts fuel immune escape in the tumor microenvironment. Trends Cancer (2019) 5(11):704–23. doi: 10.1016/j.trecan.2019.09.009
37. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, et al. Cancer-stimulated CAFs enhance monocyte differentiation and protumoral TAM activation via IL6 and GM-CSF secretion. Clin Cancer Res (2018) 24(21):5407–21. doi: 10.1158/1078-0432.CCR-18-0125
38. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36(8):1090–101. doi: 10.1038/onc.2016.273
39. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res (2020) 8(4):436–50. doi: 10.1158/2326-6066.CIR-19-0507
40. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-associated fibroblasts affect intratumoral CD8+ and FoxP3+ T cells Via IL6 in the tumor microenvironment. Clin Cancer Res (2018) 24(19):4820–33. doi: 10.1158/1078-0432.CCR-18-0205
41. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci USA (2014) 111(11):4203–8. doi: 10.1073/pnas.1319269111
42. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci USA (2009) 106(49):20847–52. doi: 10.1073/pnas.0906481106
43. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
44. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther (2021) 6(1):218. doi: 10.1038/s41392-021-00641-0
45. Seifert AM, Eymer A, Heiduk M, Wehner R, Tunger A, von Renesse J, et al. PD-1 expression by lymph node and intratumoral regulatory T cells is associated with lymph node metastasis in pancreatic cancer. Cancers (Basel) (2020) 12(10):2756. doi: 10.3390/cancers12102756
46. Whiteside TL. FOXP3+ treg as a therapeutic target for promoting anti-tumor immunity. Expert Opin Ther Targets (2018) 22(4):353–63. doi: 10.1080/14728222.2018.1451514
47. Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 attracts regulatory T cells: Implication for pancreatic cancer. Oncoimmunology (2015) 4(9):e1027473. doi: 10.1080/2162402X.2015.1027473
48. Wang X, Wang L, Mo Q, Dong Y, Wang G, Ji A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int J Clin Exp Pathol (2015) 8(5):5702–8.
49. Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst (2007) 99(15):1188–99. doi: 10.1093/jnci/djm064
50. Bommireddy R, Doetschman T. TGFbeta1 and treg cells: alliance for tolerance. Trends Mol Med (2007) 13(11):492–501. doi: 10.1016/j.molmed.2007.08.005
51. Kryczek I, Wei S, Zou L, Zhu G, Mottram P, Xu H, et al. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J Immunol (2006) 177(1):40–4. doi: 10.4049/jimmunol.177.1.40
52. Nakamura T, Shima T, Saeki A, Hidaka T, Nakashima A, Takikawa O, et al. Expression of indoleamine 2, 3-dioxygenase and the recruitment of Foxp3-expressing regulatory T cells in the development and progression of uterine cervical cancer. Cancer Sci (2007) 98(6):874–81. doi: 10.1111/j.1349-7006.2007.00470.x
53. Cassetta L, Pollard JW. Tumor-associated macrophages. Curr Biol (2020) 30(6):R246–8. doi: 10.1016/j.cub.2020.01.031
54. Medvedeva GF, Kuzmina DO, Nuzhina J, Shtil AA, Dukhinova MS. How macrophages become transcriptionally dysregulated: A hidden impact of antitumor therapy. Int J Mol Sci (2021) 22(5):2662. doi: 10.3390/ijms22052662
55. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
56. Piao C, Zhang WM, Li TT, Zhang CC, Qiu S, Liu Y, et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res (2018) 366(2):127–38. doi: 10.1016/j.yexcr.2018.03.009
57. Dong L, Chen C, Zhang Y, Guo P, Wang Z, Li J, et al. The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8+ T cell dysfunction and tumor growth. Cancer Cell (2021) 39(7):945–57.e10. doi: 10.1016/j.ccell.2021.04.016
58. Mazzone M, Menga A, Castegna A. Metabolism and TAM functions-it takes two to tango. FEBS J (2018) 285(4):700–16. doi: 10.1111/febs.14295
59. Yan S, Wan G. Tumor-associated macrophages in immunotherapy. FEBS J (2021) 288(21):6174–86. doi: 10.1111/febs.15726
60. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
61. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol Res (2018) 6(12):1578–92. doi: 10.1158/2326-6066.CIR-17-0479
62. Podojil JR, Miller SD. Potential targeting of B7-H4 for the treatment of cancer. Immunol Rev (2017) 276(1):40–51. doi: 10.1111/imr.12530
63. Zhang M, Huang L, Ding G, Huang H, Cao G, Sun X, et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J Immunother Cancer (2020) 8(1):e000308. doi: 10.1136/jitc-2019-000308
64. Schnittert J, Bansal R, Prakash J. Targeting pancreatic stellate cells in cancer. Trends Cancer (2019) 5(2):128–42. doi: 10.1016/j.trecan.2019.01.001
65. Farran B, Nagaraju GP. The dynamic interactions between the stroma, pancreatic stellate cells and pancreatic tumor development: Novel therapeutic targets. Cytokine Growth Factor Rev (2019) 48:11–23. doi: 10.1016/j.cytogfr.2019.07.001
66. Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. BioMed Pharmacother (2020) 131:110693. doi: 10.1016/j.biopha.2020.110693
67. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer (2012) 130(10):2337–48. doi: 10.1002/ijc.26290
68. Masamune A, Shimosegawa T. Pancreatic stellate cells: A dynamic player of the intercellular communication in pancreatic cancer. Clin Res Hepatol Gastroenterol (2015) 39 Suppl 1:S98–103. doi: 10.1016/j.clinre.2015.05.018
69. Wu L, Zhang XH. Tumor-associated neutrophils and macrophages-heterogenous but not chaotic. Front Immunol (2020) 11:553967. doi: 10.3389/fimmu.2020.553967
70. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
71. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology (2016) 150(7):1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
72. Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many faces of tumor-associated neutrophils and macrophages: Considerations for targeting innate immune cells in cancer. Trends Cancer (2019) 5(12):789–98. doi: 10.1016/j.trecan.2019.10.013
73. Sivori S, Pende D, Quatrini L, Pietra G, Della Chiesa M, Vacca P, et al. NK cells and ILCs in tumor immunotherapy. Mol Aspects Med (2021) 80:100870. doi: 10.1016/j.mam.2020.100870
74. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol (2020) 21(8):835–47. doi: 10.1038/s41590-020-0728-z
75. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19(1):120. doi: 10.1186/s12943-020-01238-x
76. Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res (2003) 90:127–56. doi: 10.1016/s0065-230x(03)90004-2
77. Zhou J, Zhang S, Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharmacol (2021) 101(Pt B):108374. doi: 10.1016/j.intimp.2021.108374
78. Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. Trends Immunol (2004) 25(1):47–52. doi: 10.1016/j.it.2003.10.012
79. Jacobs B, Ullrich E. The interaction of NK cells and dendritic cells in the tumor environment: how to enforce NK cell & DC action under immunosuppressive conditions? Curr Med Chem (2012) 19(12):1771–9. doi: 10.2174/092986712800099857
80. Mahmood S, Upreti D, Sow I, Amari A, Nandagopal S, Kung SK. Bidirectional interactions of NK cells and dendritic cells in immunotherapy: current and future perspective. Immunotherapy (2015) 7(3):301–8. doi: 10.2217/imt.14.122
81. Aponte-López A, Muñoz-Cruz S. Mast cells in the tumor microenvironment. Adv Exp Med Biol (2020) 1273:159–73. doi: 10.1007/978-3-030-49270-0_9
82. Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res (2010) 16(8):2257–65. doi: 10.1158/1078-0432.CCR-09-1230
83. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res (2013) 73(13):3927–37. doi: 10.1158/0008-5472.CAN-12-4479
84. Porcelli L, Iacobazzi RM, Di Fonte R, Serratì S, Intini A, Solimando AG, et al. CAFs and TGF-β signaling activation by mast cells contribute to resistance to Gemcitabine/Nabpaclitaxel in pancreatic cancer. Cancers (Basel) (2019) 11(3):330. doi: 10.3390/cancers11030330
85. Khan MW, Keshavarzian A, Gounaris E, Melson JE, Cheon EC, Blatner NR, et al. PI3K/AKT signaling is essential for communication between tissue-infiltrating mast cells, macrophages, and epithelial cells in colitis-induced cancer. Clin Cancer Res (2013) 19(9):2342–54. doi: 10.1158/1078-0432.CCR-12-2623
86. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, et al. Mast cells mobilize myeloid-derived suppressor cells and treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PloS One (2010) 5(1):e8922. doi: 10.1371/journal.pone.0008922
87. Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity (2021) 54(5):875–84. doi: 10.1016/j.immuni.2021.04.004
88. Bruger AM, Dorhoi A, Esendagli G, Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, et al. How to measure the immunosuppressive activity of MDSC: assays, problems and potential solutions. Cancer Immunol Immunother (2019) 68(4):631–44. doi: 10.1007/s00262-018-2170-8
89. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
90. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun (2020) 11(1):404. doi: 10.1038/s41467-019-14134-w
91. Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun (2018) 9(1):1056. doi: 10.1038/s41467-018-03348-z
92. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T cells to protect tumour cells. Nat Commun (2018) 9(1):948. doi: 10.1038/s41467-018-03347-0
93. Takahashi H, Sakakura K, Kawabata-Iwakawa R, Rokudai S, Toyoda M, Nishiyama M, et al. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol Immunother (2015) 64(11):1407–17. doi: 10.1007/s00262-015-1742-0
94. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discovery (2020) 10(9):1330–51. doi: 10.1158/2159-8290.CD-19-1384
95. Li Z, Zhou J, Zhang J, Li S, Wang H, Du J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int J Cancer (2019) 145(7):1946–57. doi: 10.1002/ijc.32278
96. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res (2016) 76(14):4124–35. doi: 10.1158/0008-5472.CAN-15-2973
97. Bughda R, Dimou P, D'Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: Launching an attack on tumor stroma. Immunotargets Ther (2021) 10:313–23. doi: 10.2147/ITT.S291767
98. Duperret EK, Trautz A, Ammons D, Perales-Puchalt A, Wise MC, Yan J, et al. Alteration of the tumor stroma using a consensus DNA vaccine targeting fibroblast activation protein (FAP) synergizes with antitumor vaccine therapy in mice. Clin Cancer Res (2018) 24(5):1190–201. doi: 10.1158/1078-0432.CCR-17-2033
99. Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci USA (2019) 116(22):10674–80. doi: 10.1073/pnas.1819889116
100. Gorchs L, Ahmed S, Mayer C, Knauf A, Fernández Moro C, Svensson M, et al. The vitamin d analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci Rep (2020) 10(1):17444. doi: 10.1038/s41598-020-74368-3
101. Kamali Zonouzi S, Pezeshki PS, Razi S, Rezaei N. Cancer-associated fibroblasts in colorectal cancer. Clin Transl Oncol (2022) 24(5):757–69. doi: 10.1007/s12094-021-02734-2
102. Wu X, Hu W, Lu L, Zhao Y, Zhou Y, Xiao Z, et al. Repurposing vitamin d for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm Sin B (2019) 9(2):203–19. doi: 10.1016/j.apsb.2018.09.002
103. Morita M, Okuyama M, Akutsu T, Ohdaira H, Suzuki Y, Urashima M. Vitamin d supplementation regulates postoperative serum levels of PD-L1 in patients with digestive tract cancer and improves survivals in the highest quintile of PD-L1: A Post hoc analysis of the AMATERASU randomized controlled trial. Nutrients (2021) 13(6):1987. doi: 10.3390/nu13061987
104. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
105. Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery (2020) 10(3):422–39. doi: 10.1158/2159-8290.CD-19-0958
106. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
107. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol (2019) 9:1146. doi: 10.3389/fonc.2019.01146
108. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
109. Martinek J, Wu TC, Cadena D, Banchereau J, Palucka K. Interplay between dendritic cells and cancer cells. Int Rev Cell Mol Biol (2019) 348:179–215. doi: 10.1016/bs.ircmb.2019.07.008
110. Zhou Y, Slone N, Chrisikos TT, Kyrysyuk O, Babcock RL, Medik YB, et al. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103+ conventional dendritic cells. J Immunother Cancer (2020) 8(1):e000474. doi: 10.1136/jitc-2019-000474
111. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
112. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
113. Yin X, Chen S, Eisenbarth SC. Dendritic cell regulation of T helper cells. Annu Rev Immunol (2021) 39:759–90. doi: 10.1146/annurev-immunol-101819-025146
114. Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, et al. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res (2017) 77(15):4158–70. doi: 10.1158/0008-5472.CAN-16-2212
115. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
116. Nava S, Lisini D, Frigerio S, Bersano A. Dendritic cells and cancer immunotherapy: The adjuvant effect. Int J Mol Sci (2021) 22(22):12339. doi: 10.3390/ijms222212339
117. Yang J, Eresen A, Shangguan J, Ma Q, Yaghmai V, Zhang Z. Irreversible electroporation ablation overcomes tumor-associated immunosuppression to improve the efficacy of DC vaccination in a mice model of pancreatic cancer. Oncoimmunology (2021) 10(1):1875638. doi: 10.1080/2162402X.2021.1875638
118. Bauer C, Sterzik A, Bauernfeind F, Duewell P, Conrad C, Kiefl R, et al. Concomitant gemcitabine therapy negatively affects DC vaccine-induced CD8(+) T-cell and b-cell responses but improves clinical efficacy in a murine pancreatic carcinoma model. Cancer Immunol Immunother (2014) 63(4):321–33. doi: 10.1007/s00262-013-1510-y
119. Mayanagi S, Kitago M, Sakurai T, Matsuda T, Fujita T, Higuchi H, et al. Phase I pilot study of wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci (2015) 106(4):397–406. doi: 10.1111/cas.12621
120. Boullart AC, Aarntzen EH, Verdijk P, Jacobs JF, Schuurhuis DH, Benitez-Ribas D, et al. Maturation of monocyte-derived dendritic cells with toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol Immunother (2008) 57(11):1589–97. doi: 10.1007/s00262-008-0489-2
121. Zhang J, Yu J, Yang L, Li H, Wei F, Zhao H, et al. Enhanced activation of human dendritic cells by silencing SOCS1 and activating TLRs simultaneously. Cancer Immunol Immunother (2012) 61(10):1653–61. doi: 10.1007/s00262-012-1218-4
123. Kamath SD, Kalyan A, Kircher S, Nimeiri H, Fought AJ, Benson A, et al. Ipilimumab and gemcitabine for advanced pancreatic cancer: A phase ib study. Oncologist (2020) 25(5):e808–15. doi: 10.1634/theoncologist.2019-0473
124. Weiss GJ, Blaydorn L, Beck J, Bornemann-Kolatzki K, Urnovitz H, Schütz E, et al. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Invest New Drugs (2018) 36(1):96–102. doi: 10.1007/s10637-017-0525-1
125. Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH Jr, Bagalà C, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol (2014) 25(9):1750–5. doi: 10.1093/annonc/mdu205
126. Sharma P, Sohn J, Shin SJ, Oh DY, Keam B, Lee HJ, et al. Efficacy and tolerability of tremelimumab in locally advanced or metastatic urothelial carcinoma patients who have failed first-line platinum-based chemotherapy. Clin Cancer Res (2020) 26(1):61–70. doi: 10.1158/1078-0432.CCR-19-1635
127. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer (2018) 119(10):1208–14. doi: 10.1038/s41416-018-0246-z
128. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer (2021) 9(3):e002068. doi: 10.1136/jitc-2020-002068
129. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi: 10.1016/j.ccr.2012.01.007
130. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol (2019) 37(13):1062–9. doi: 10.1200/JCO.18.01295
131. Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, et al. Phase ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res (2016) 22(12):2848–54. doi: 10.1158/1078-0432.CCR-15-2010
132. Overman M, Javle M, Davis RE, Vats P, Kumar-Sinha C, Xiao L, et al. Randomized phase II study of the bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J Immunother Cancer (2020) 8(1):e000587. doi: 10.1136/jitc-2020-000587
133. Karakhanova S, Mosl B, Harig S, von Ahn K, Fritz J, Schmidt J, et al. Influence of interferon-alpha combined with chemo (radio) therapy on immunological parameters in pancreatic adenocarcinoma. Int J Mol Sci (2014) 15(3):4104–25. doi: 10.3390/ijms15034104
134. Scheid C, Young R, McDermott R, Fitzsimmons L, Scarffe JH, Stern PL. Immune function of patients receiving recombinant human interleukin-6 (IL-6) in a phase I clinical study: induction of c-reactive protein and IgE and inhibition of natural killer and lymphokine-activated killer cell activity. Cancer Immunol Immunother (1994) 38(2):119–26. doi: 10.1007/BF01526207
135. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med (2020) 26(6):878–85. doi: 10.1038/s41591-020-0880-x
136. Rong Y, Qin X, Jin D, Lou W, Wu L, Wang D, et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exp Med (2012) 12(3):173–80. doi: 10.1007/s10238-011-0159-0
137. Kondo H, Hazama S, Kawaoka T, Yoshino S, Yoshida S, Tokuno K, et al. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res (2008) 28(1B):379–87.
138. Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, McKolanis JR, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther (2008) 6(B):955–64.
139. Koido S, Homma S, Okamoto M, Takakura K, Mori M, Yoshizaki S, et al. Treatment with chemotherapy and dendritic cells pulsed with multiple wilms' tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin Cancer Res (2014) 20(16):4228–39. doi: 10.1158/1078-0432.CCR-14-0314
140. Mehrotra S, Britten CD, Chin S, Garrett-Mayer E, Cloud CA, Li M, et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J Hematol Oncol (2017) 10(1):82. doi: 10.1186/s13045-017-0459-2
141. Lin M, Liang S, Wang X, Liang Y, Zhang M, Chen J, et al. Short-term clinical efficacy of percutaneous irreversible electroporation combined with allogeneic natural killer cell for treating metastatic pancreatic cancer. Immunol Lett (2017) 186:20–7. doi: 10.1016/j.imlet.2017.03.018
142. Lin M, Liang S, Wang X, Liang Y, Zhang M, Chen J, et al. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: a promising treatment. J Cancer Res Clin Oncol (2017) 143(12):2607–18. doi: 10.1007/s00432-017-2513-4
143. Ullenhag GJ, Mozaffari F, Broberg M, Mellstedt H, Liljefors M. Clinical and immune effects of lenalidomide in combination with gemcitabine in patients with advanced pancreatic cancer. PloS One (2017) 12(1):e0169736. doi: 10.1371/journal.pone.0169736
144. McCracken MN, Cha AC, Weissman IL. Molecular pathways: Activating T cells after cancer cell phagocytosis from blockade of CD47 "Don't eat me" signals. Clin Cancer Res (2015) 21(16):3597–601. doi: 10.1158/1078-0432.CCR-14-2520
145. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature (2017) 546(7659):498–503. doi: 10.1038/nature22341
146. Koh E, Lee EJ, Nam GH, Hong Y, Cho E, Yang Y, et al. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials (2017) 121:121–9. doi: 10.1016/j.biomaterials.2017.01.004
147. Morishita M, Takahashi Y, Matsumoto A, Nishikawa M, Takakura Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials (2016) 111:55–65. doi: 10.1016/j.biomaterials.2016.09.031
148. Xie Y, Bai O, Zhang H, Yuan J, Zong S, Chibbar R, et al. Membrane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) CTL- and NK-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J Cell Mol Med (2010) 14(11):2655–66. doi: 10.1111/j.1582-4934.2009.00851.x
149. Hao S, Bai O, Yuan J, Qureshi M, Xiang J. Dendritic cell-derived exosomes stimulate stronger CD8+ CTL responses and antitumor immunity than tumor cell-derived exosomes. Cell Mol Immunol (2006) 3(3):205–11.
150. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med (2005) 3(1):9. doi: 10.1186/1479-5876-3-9
151. Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H, et al. Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun (2018) 9(1):435. doi: 10.1038/s41467-018-02865-1
152. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol (2012) 189(6):2833–42. doi: 10.4049/jimmunol.1101988
153. Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther (2017) 25(7):1665–75. doi: 10.1016/j.ymthe.2017.02.007
154. Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer (2018) 4(6):418–28. doi: 10.1016/j.trecan.2018.04.001
155. AstraZeneca. In: Study of tremelimumab in patients with advanced solid tumors. Available at: https://clinicaltrials.gov/show/NCT02527434.
156. Varricchi G, Galdiero MR, Marone G, Criscuolo G, Triassi M, Bonaduce D, et al. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open (2017) 2(4):e000247. doi: 10.1136/esmoopen-2017-000247
157. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol (2020) 17(8):487–505. doi: 10.1038/s41575-020-0300-1
158. Chen J, Ding ZY, Li S, Liu S, Xiao C, Li Z, et al. Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics (2021) 11(3):1345–63. doi: 10.7150/thno.51383
159. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discovery (2016) 6(3):270–85. doi: 10.1158/2159-8290.CD-15-0827
160. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
161. Stift A, Friedl J, Dubsky P, Bachleitner-Hofmann T, Schueller G, Zontsich T, et al. Dendritic cell-based vaccination in solid cancer. J Clin Oncol (2003) 21(1):135–42. doi: 10.1200/JCO.2003.02.135
162. Nakamura M, Wada J, Suzuki H, Tanaka M, Katano M, Morisaki T. Long-term outcome of immunotherapy for patients with refractory pancreatic cancer. Anticancer Res (2009) 29(3):831–6.
163. Alahdal M, Xing Y, Tang T, Liang J. 1-Methyl-D-tryptophan reduces tumor CD133+ cells, wnt/β-catenin and NF-κβp65 while enhances lymphocytes NF-κβ2, STAT3, and STAT4 pathways in murine pancreatic adenocarcinoma. Sci Rep (2018) 8(1):9869. doi: 10.1038/s41598-018-28238-8
164. Li Y, Zhao W, Wang Y, Wang H, Liu S. Extracellular vesicle-mediated crosstalk between pancreatic cancer and stromal cells in the tumor microenvironment. J Nanobiotechnol (2022) 20(1):208. doi: 10.1186/s12951-022-01382-0
165. Moeng S, Son SW, Lee JS, Lee HY, Kim TH, Choi SY, et al. Extracellular vesicles (EVs) and pancreatic cancer: From the role of EVs to the interference with EV-mediated reciprocal communication. Biomedicines (2020) 8(8):267. doi: 10.3390/biomedicines8080267
166. Nannan L, Oudart JB, Monboisse JC, Ramont L, Brassart-Pasco S, Brassart B. Extracellular vesicle-dependent cross-talk in cancer-focus on pancreatic cancer. Front Oncol (2020) 10:1456. doi: 10.3389/fonc.2020.01456
167. Zhao J, Schlößer HA, Wang Z, Qin J, Li J, Popp F, et al. Tumor-derived extracellular vesicles inhibit natural killer cell function in pancreatic cancer. Cancers (Basel) (2019) 11(6):874. doi: 10.3390/cancers11060874
168. Xie F, Zhou X, Fang M, Li H, Su P, Tu Y, et al. Extracellular vesicles in cancer immune microenvironment and cancer immunotherapy. Adv Sci (Weinh) (2019) 6(24):1901779. doi: 10.1002/advs.201901779
169. Javadrashid D, Baghbanzadeh A, Derakhshani A, Leone P, Silvestris N, Racanelli V, et al. Pancreatic cancer signaling pathways, genetic alterations, and tumor microenvironment: The barriers affecting the method of treatment. Biomedicines (2021) 9(4):373. doi: 10.3390/biomedicines9040373
170. Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S, et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol (2020) 13(1):130. doi: 10.1186/s13045-020-00958-3
171. Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res (2015) 21(2):396–404. doi: 10.1158/1078-0432.CCR-14-1591
172. Smalley I, Smalley KSM. ERK inhibition: A new front in the war against MAPK pathway-driven cancers? Cancer Discovery (2018) 8(2):140–2. doi: 10.1158/2159-8290.CD-17-1355
173. Bear AS, Vonderheide RH, O'Hara MH. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell (2020) 38(6):788–802. doi: 10.1016/j.ccell.2020.08.004
174. Rocha FG. Landmark series: Immunotherapy and targeted therapy for pancreatic cancer. Ann Surg Oncol (2021) 28(3):1400–6. doi: 10.1245/s10434-020-09367-9
175. Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: Challenges and opportunities. Gastroenterology (2019) 156(7):2056–72. doi: 10.1053/j.gastro.2018.12.038
Keywords: pancreatic cancer, immunosuppression, tumor microenvironment, immunotherapy, clinical trial
Citation: Li Y, Xiang S, Pan W, Wang J, Zhan H and Liu S (2023) Targeting tumor immunosuppressive microenvironment for pancreatic cancer immunotherapy: Current research and future perspective. Front. Oncol. 13:1166860. doi: 10.3389/fonc.2023.1166860
Received: 15 February 2023; Accepted: 20 March 2023;
Published: 29 March 2023.
Edited by:
Andrey Zamyatnin, I.M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Antonella Argentiero, National Cancer Institute Foundation (IRCCS), ItalyCopyright © 2023 Li, Xiang, Pan, Wang, Zhan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanglong Liu, bGl1c2hhbmdsb25nQHFkdS5lZHUuY24=; Hanxiang Zhan, emhhbmhhbnhpYW5nQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.