
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 01 May 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1166238
This article is part of the Research TopicIdentification, Risk Stratification, and Optimized Management for Lynch SyndromeView all 14 articles
 Madeleine H. Williams1†
Madeleine H. Williams1† Andreas V. Hadjinicolaou2,3*†
Andreas V. Hadjinicolaou2,3*† Benjamin C. Norton4†Rawen Kader5Laurence B. Lovat5
Benjamin C. Norton4†Rawen Kader5Laurence B. Lovat5Lynch syndrome (LS) is an inherited cancer predisposition syndrome associated with high lifetime risk of developing tumours, most notably colorectal and endometrial. It arises in the context of pathogenic germline variants in one of the mismatch repair genes, that are necessary to maintain genomic stability. LS remains underdiagnosed in the population despite national recommendations for empirical testing in all new colorectal and endometrial cancer cases. There are now well-established colorectal cancer surveillance programmes, but the high rate of interval cancers identified, coupled with a paucity of high-quality evidence for extra-colonic cancer surveillance, means there is still much that can be achieved in diagnosis, risk-stratification and management. The widespread adoption of preventative pharmacological measures is on the horizon and there are exciting advances in the role of immunotherapy and anti-cancer vaccines for treatment of these highly immunogenic LS-associated tumours. In this review, we explore the current landscape and future perspectives for the identification, risk stratification and optimised management of LS with a focus on the gastrointestinal system. We highlight the current guidelines on diagnosis, surveillance, prevention and treatment and link molecular disease mechanisms to clinical practice recommendations.
Lynch Syndrome (LS) is a hereditary cancer predisposition syndrome characterised by a high lifetime risk of developing cancers, primarily colorectal and endometrial (1). These cancers exhibit microsatellite instability (MSI) due to defects in the cellular mismatch repair (MMR) system (2). LS is associated with other malignancies including gastrointestinal (GI) (e.g. gastric, small intestinal, hepato-biliary and pancreatic) and extra-GI cancers (e.g. prostate, ovaries, skin, central nervous system and upper urinary tract) (3). LS follows an autosomal dominant pattern of inheritance with germline pathogenic variants in one of the MMR genes, which, in health, maintain genomic stability (4). An estimated 1/450 people in the UK have LS (5), and of those, only 5% are diagnosed. The lifetime risk of colorectal cancer (CRC) in LS patients can vary from 10-80% dependent on the MMR mutation and age, and it is thought to be responsible for 3-5% of all CRCs (6, 7). This makes LS one of the most frequently encountered cancer susceptibility syndromes.
A prototypical cancer surveillance programme using colonoscopy exists for CRC in the setting of LS, but quality data on the role of surveillance for other LS-associated tumours is limited. In recognition of the growing need for new approaches to improve survival, this review explores the current landscape and future perspectives for the detection, risk stratification and management of LS.
LS is due to a pathogenic variant within one of the MMR genes: MLH1, MSH2, MSH6 or PMS2 (4). MLH1/MSH2 mutations are responsible for 70-90% of LS cases and carry significantly higher lifetime cancer risk (8). A small proportion of LS cases (1-3%) arise secondary to constitutional epimutations of the MLH1 or MSH2 genes (9). The heterozygous, loss-of-function, germline mutations in MMR genes are phenotypically dominant but may also convey vulnerability to a second, somatic mutation in the wildtype (normal) allele. Tumorigenesis then develops due to deficient mismatch repair (dMMR) and accumulation of further mutations including in small regions of repeated DNA called microsatellites. This gives rise to microsatellite instability (MSI); the genetic signature of LS-associated tumours.
The need to differentiate between sporadic and inherited CRC in patients with dMMR tumours is crucial because of downstream implications for cancer surveillance. Unfortunately, this is not always straightforward and we are increasingly aware of a heterogenous patient group with Lynch-like syndrome (LLS) defined as dMMR tumours where LS is suspected but no pathological germline MMR mutation is identified (10).
The diagnosis of LS is made in symptomatic patients presenting with a LS-associated cancer, or among asymptomatic patients with a confirmed familial pathogenic variant. In symptomatic cases, the tumour is subjected to molecular profiling for evidence of dMMR. MSI is assessed either using polymerase chain reaction (PCR)-based testing or loss of/abnormal protein expression of MLH1, MSH2, MSH6 or PMS2 using immunohistochemistry (IHC) (11). Both methods have high sensitivity (PCR 92.9%, IHC 92.4%), specificity (PCR 86.3%, HCI 87.8%) and negative predictive values (PCR 99.6%, IHC 99.6%) for LS (12).
An abnormal result must be followed by referral for genetic testing and counselling. Younger patients (<40 years old) should be referred directly for germline testing according to the NHS National Genomic Medicine Service (GMS) Lynch Syndrome Project guidelines (13). Among families with a confirmed pathogenic MMR variant, asymptomatic patients can be referred for cascade genetic testing directly without the need for findings consistent with CRC.
Since 2017, the National Institute for Clinical Excellence (NICE) has recommended testing all newly identified CRCs for dMMR by IHC or for MSI to guide the need for LS evaluation (11). This guidance was expanded to IHC testing in all new endometrial cancers in 2020 (14). These recommendations have superseded the previously used Amsterdam Criteria and Bethesda Guidelines (15, 16) which mainly relied on crude measures such as family history and age of cancer onset (17). Looking to the future, NICE have proposed an accelerated review of next generation sequencing (NGS) as a potential index test for paired tumour-germline profiling in all newly diagnosed CRCs (18). NGS enables identification of MSI using computational algorithms such as mSINGS, MSISenory, and MANTIS among others (19). It can simultaneously sequence the whole exome looking for markers of MSI, compared to a normal/baseline sample, which is measured against a threshold value. Concurrently, exome tumour sequencing can be paired with a blood sample to enable differentiation between somatic and germline variants (20). This paired testing is superior to traditional stepwise testing, which would enable earlier, more precise and personalised risk stratification in suspected LS cases (21).
Over the last few decades, there has been great insight into the natural history of LS patients with thousands of unique germline MMR gene variants identified and recorded in international databases such as InSiGHT (22). However, having a pathogenic variant does not result in a uniform diagnosis across all patients, with great genetic variability observed due to penetrance (i.e. the probability of a gene being expressed) and expressivity (i.e. if the gene is penetrant, the variability in that expression). The establishment of the Prospective Lynch Syndrome Database (PLSD), an international, multi-centre, observational prospective study, has improved understanding of the cumulative incidence and survival of LS-associated cancer patients (between 25-75 years) and equipped us with age and cancer-specific risk estimates for each pathogenic MMR variant (Table 1) (24, 25). However, it is important to acknowledge its limitations such as the absence of a control group who did not undergo surveillance and granular data such as cancer-specific survival.
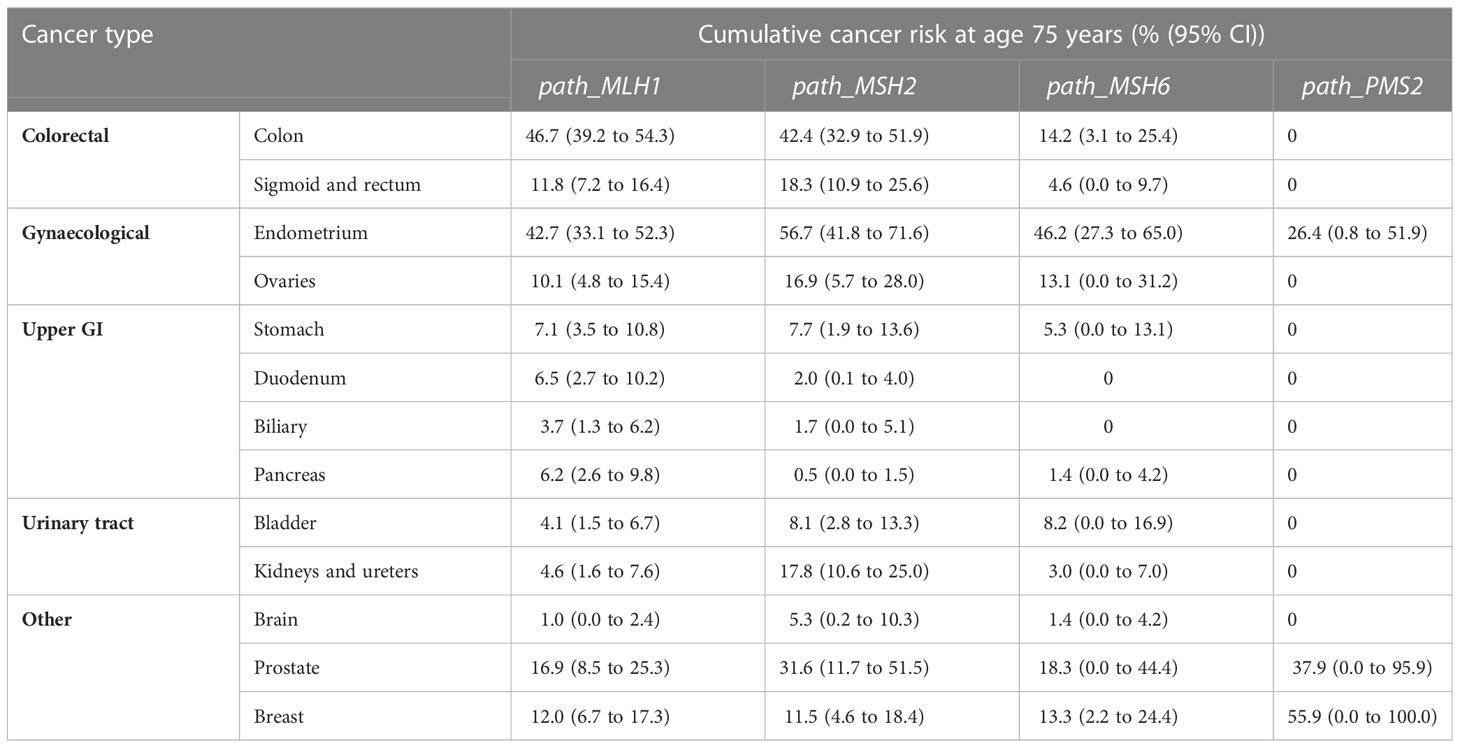
Table 1 Cumulative incidence of individual cancers in patients with pathogenic MMR variants between 25-75 years old (23).
These limitations have somewhat been addressed by the international multi-centre International Mismatch Repair Consortium (IMRC) (26). In contrast to the PLSD, in which all cases have undergone at least one colonscopy, IMRC data derives from retrospective segregation analysis of LS families, including older generations who did not receive comparable colonoscopic surveillance. Contrary to expectations, incidence of CRC in path_MLH1 and path_MSH2 carriers in the PLSD group (who underwent colonoscopy and polypectomy) was significantly higher than in the IMRC series. Differences in data fidelity between the two databases could have influenced these findings (27).
Over the last decade, significant improvements have been made in the personalised risk stratification of patients with LS. However, the optimal timing of surveillance is still to be determined and there is a paucity of data for extra-colonic tumours and surveillance in older age patients (28).
Current consensus favours conoloscopy for CRC surveillance in asymptomatic patients with LS. A landmark prospective study from Finland in 2000 demonstrated that 3-yearly colonoscopy in LS decreased CRC incidence and mortality (29, 30), with other non-randomised studies replicating these findings (31, 32). However, many of these are somewhat limited in their granularity of data. For example, in the aforementioned study, all participants who attended a colonscopy were deemed to be compliant with surveillance regardless of the frequency of their surveillance or whether they had any actual further colonoscopies at all. More recently, a retrospective cohort study (33) used a unique time-based model to explore the effect of surveillance interval in LS (<27 months vs >27 months vs no surveillance), demonstrating that shorter intervals reduced the risk of first CRC diagnosis. These findings could encourage adherence to timely surveillance in at-risk individuals, although an important limation of this study in the context of colonoscopy, was the inclusion of other surveillance techniques such as CT colonography, MRI and barium enema.
The optimal strategy for CRC surveillance in LS remains the subject of ongoing research. Guidelines vary internationally (outlined in Table 2) (3, 10, 34–45), with the European Society of Gastrointestinal Endoscopy (ESGE) recommending 2 yearly (36). Interestingly, 98% of centres favoured colonscopy every 1-2 years when reported to the IMRC (46). The prevalence of CRC is low in patients with LS under the age of 25 regardless of genotype, however data from both PLSD and IMRC support the notion that those with the higher penetrance MHL1 and MSH2 variants typically develop CRC earlier in life than their MSH6 and PMS2 counterparts (26, 47), hence the decision by some to begin surveillance earlier for MSH1/MSH2 carriers (Table 2). In patients with PMS2 variants, carcinogenesis may be more akin to the traditional adenoma-carcinoma sequence (25, 48) leading to low CRC incidence which may justify the suggestion from the European Mallorca guidelines for 5-yearly surveillance (35).
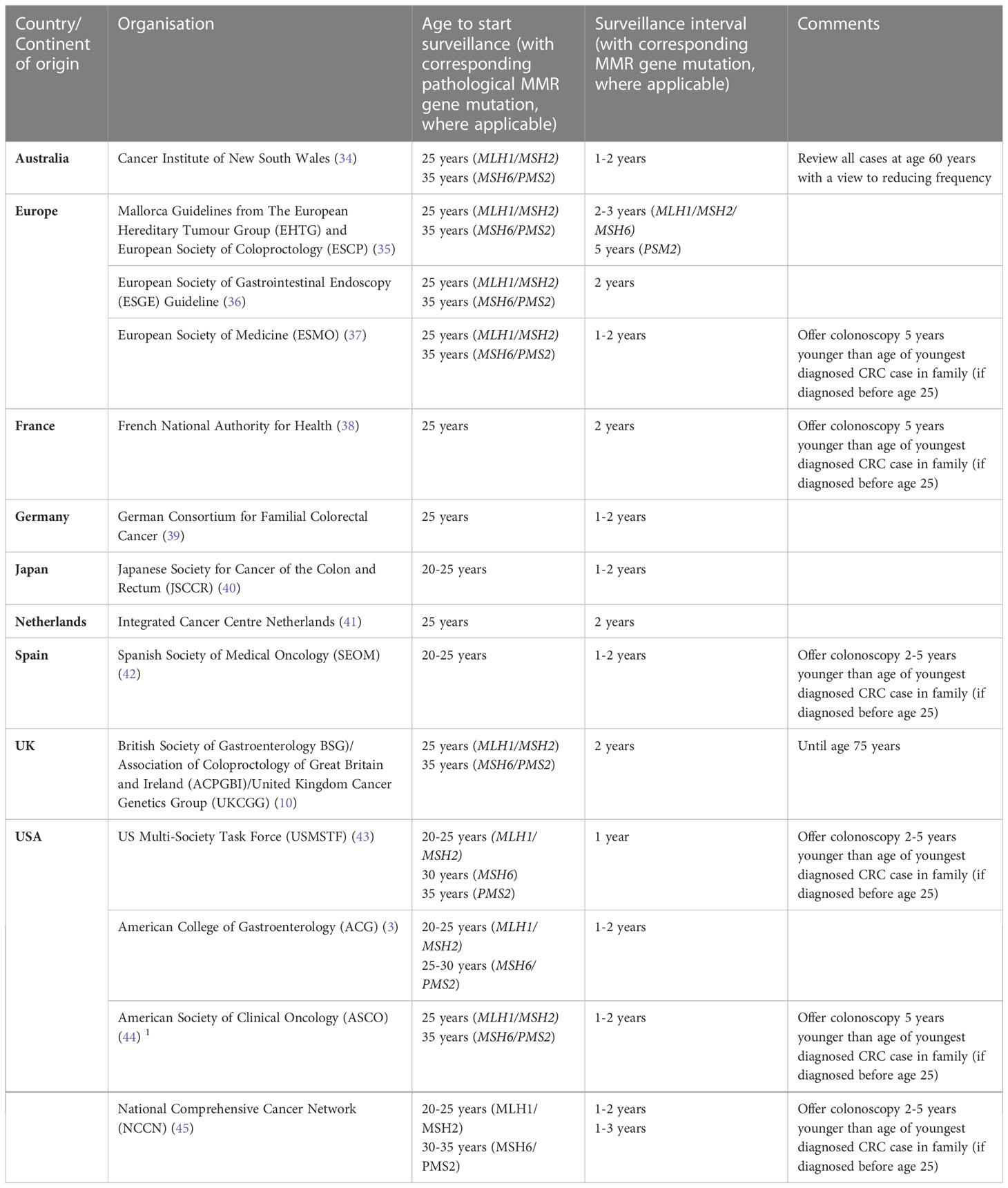
Table 2 Current recommendations for colorectal cancer surveillance from different national and international organisations.
Whilst 1-2 yearly colonoscopy in LS is widely practiced, prospective observational cohorts have demonstrated that lifetime risk of CRC, including metchronous tumours, remains as high as 36% (49, 50) and do not necessarly improve by increasing surveillance frequency (51). Analysis of 2747 LS patients showed no significant difference between incidence and stage of CRC between annual, 1-2 yearly and 3 yearly surveillance (52). It has also been suggested that frequent surveillance could lead to over-diagnosis by detecting tumours that may not have become clinically significant (53). Compliance issues too may be an argument for longer surveillance intervals. In one study, loss to follow-up rates were higher among participants randomised to annual screening than those having 2 or 5-yearly surveillance (54). Considering these findings it is perhaps unsurprising that consensus on surveillance strategy is difficult to establish.
There are various hypotheses as to why the rate of interval CRC is still high despite best efforts in surveillance programmes. First, it has been suggested that CRC in LS develops through accelerated tumorigenesis compared with sporadic CRC (55). This assumes a prior optimally performed colonoscopy. Second, adenomas in LS are often proximal, flat, and harder to detect, which could lead to missed lesions, especially during inadequately performed colonoscopy (56). Finally, LS-associated CRCs may have a unique, non-polypous carcinogenesis pathway that allow them to develop from endoscopically undectable lesions (e.g. colonic crypts) (57). The aforementioned failure to reduce CRC incidence by reducing surveillance intervals suggests that accelerated carcinogenesis is less likely and has led to a switch of focus on optimising the colonoscopic procedure and adherence to key performance indictors for colonoscoy (10, 58–60).
High quality colonoscopy is crucial to the detection of both sporadic and hereditary CRC (61), especially in LS where lesions may be difficult to detect. To achieve this, different advanced imaging modalities including dye-based and virtual chromoendoscopy (VCE) have been assessed in patients with LS. A recent meta-analysis of four prospective studies comparing standard white light endoscopy (WLE) to chromoendoscopy using dye-spray showed that the latter was superior for detection of any adenomatous, flat, or proximal lesion (62). European guidelines suggest chromoendoscopy as an adjunct, whereas BSG guidelines advise that it offers no advantage to high-definition white light endoscopy (HDWLE) (10, 35, 36).
VCE is increasingly popular owing to its ease of use. Back-to-back studies comparing imaging modalities immediately following one another have shown a benefit for both narrow band imaging (NBI; Olympus) and iScan (Pentax) in LS polyp detection (63, 64). However, these comparisons have also shown higher lesion detection with dye-based chromoendoscopy versus NBI (65, 66). A recent multi-centre RCT compared HDWLE to Linked colour imaging (LCI; Fujifilm) among 357 patients with pathogenic LS variants and found no significant difference in polyp detection rate (44.4% vs. 36.0%; p=0.12) (67). Thus,at best, advanced imaging techniques can be an adjunct to HDWLE but cannot replace standard care.
In another growing field, the use of real-time artificial intelligence (AI)-colonoscopy has demonstrated enhanced detection of polyps and adenomas in average risk CRCs (68–71). A recent German RCT demonstrated a higher (albeit not statisticaly significant) rate of lesion detection, including LS-relevant flat lesions, by AI-colonoscopy than HDWLE in a LS cohort (72).
A recent systemic review (73) brought attention to non-invasive biomarkers such as plasma-based methylated SEPTIN9, Big Adenine Tract-26 (a faceal marker of MSI), faecal sulfate-reducing bacteria Desulfovibrio and faecal immunochemical testing (FIT) in the detection of CRC and adenomas in LS, although further evidence is required to support their use in practice. A 2017 meta-analysis reported that FIT had a sensitivity of 85% for CRC and 46% for advanced adenomas in asymptomatic adults with a family history, suggesting that FIT alone would miss advanced neoplasia (74). However, during the COVID-19 pandemic in England, when access to non-urgent colonoscopy services was restricted, a temporary system based on FIT was introduced to risk stratify patients with LS to urgent colonoscopy (75). This formed the basis for an ongoing UK-based multi-centre prospective study examining a potential future role for FIT testing in LS (76).
Recommendations for the surveillance of LS-associated extra-colonic cancers are vary. For gastric cancers, most guidelines support routine testing for, and eradication of, Helicobacter pylori. American, Japanese and certain European guidelines advocate for regular oesophagogastroduodenoscopy (OGD) starting from 30-35 years of age (3, 37, 40, 77).
Beyond careful inspection of the duodenum and terminal ileum at OGD and colonoscopy respectively, routine testing for small bowel cancers is not typically recommended, though capsule endoscopy has been suggested for unexplained iron deficiency anaemia or abdominal pain (78).
LS families have been estimated to have an 8.6-fold increased risk of pancreatic cancer compared to the normal population (79) and surveillance using MRI or endoscopic ultrasound has been proposed for high-risk groups and carriers (80). However, low diagnostic yields and poor outcomes from surgical treatment of suspicious pancreatic lesions largely negate any theoretical benefit (10). Surveillance practices for LS-associated gynaecological cancers lack consensus and have not demonstrated a mortality benefit (81). American and European Oncology guidelines advocate for annual transvaginal ultrasound and endometrial sampling from the ages of 30-35, and prophylactic hysterectomy and bilateral salpingoophrectomy once child bearing completes, although the evidence for this is weak (3, 35, 37, 77). There is currently insufficient evidence to recommend screening for other extra-colonic LS cancers.
Unlike CRCs, for which standarised mortality ratios have been reported to decrease over time in LS cohorts, risk of death from LS-associated extra-colonic tumours is significantly increased compared with the general population (82). In a retrospective Finnish cohort, 7.2% of patients developed urothelial, prostate or gastric cancer, with one in five dying from the disease (83). Extra-colonic surveillance may benefit those with cancer at a young age who have a higher lifetime risk of subsequent cancer, but this needs addressing in well-designed prospective trials.
Most data on modifiable risk factors such as poor diet, high alcohol intake, smoking, lack of exercise and high body mass index (BMI) are extrapolated from sporadic CRC cohorts (84). Weak evidence specific to LS suggests lower CRC risk in patients who consume more fruit and higher risk in smokers (85). Subgroup analyses from the Colorectal Adenoma/Carcinoma Prevention Programme 2 (CAPP2) trial revealed a significant association between obesity and CRC risk (86). Two prospective cohort studies demonstrated a 30% increased risk of CRC for every 5.0 kg/m2 increase in BMI in early adulthood and an association between an overweight BMI and CRC risk in men (87, 88).
Aspirin is the only recommended chemoprophylaxis in LS. Its potential benefit was first highlighted by meta-analyses associating long-term use with lower incidence of all cancers, especially proximal CRC (89, 90). Subsequently the double-blinded RCT CAPP2, of 861 LS patients demonstrated that the use of 600mg/day of aspirin for 2-4 years was linked with a significantly lower risk of all LS-associated cancers after 10 year follow-up (91). A successive ongoing trial, CAPP3, aims to establish optimal dosing, meanwhile international guidelines have varied in their adoption of the CAPP2 findings. In the UK, both the BSG and NICE support the use of 150mg aspirin daily (300 mg if obese) in patients under 70 years old for 2-5 years (10, 92). American guidelines by contrast have refrained from recommending its use given data is currently derived from a single trial (3, 77).
Data on advanced endoscopic techniques to remove early-stage colorectal tumours in LS is lacking, therefore current practice heavily favours surgical resection. Endoscopic management follows guidance for non-LS colorectal polyps (93). As such, it is critical to optimise complete resection rates in LS-associated polypectomies, particularly for flat serrated polyps (94, 95).
The role of surgery in LS-associated CRC is two-fold: to resect the advanced neoplastic lesion and reduce the risk of metachronous disease. Meta-analyses have demonstrated a lower incidence of metachronous CRC in those who underwent extended resection (total/subtotal colectomy with ileorectal/ileosigmoidal anastomosis) versus segmental resection for a first CRC (96, 97) with absolute risk for metachronous tumour of 4.7% and 22.4%, respectively, over 100.7 months follow-up (98).
The risk of metachronous disease applies mainly to MHL1 and MSH2 pathogenic variant carriers and thus, in this context, most guidelines recommend the use of extended colectomy for a first CRC, particularly in younger patients (3, 10, 35, 37). For carriers of MSH6 and PMS2 variants there is insufficient evidence of oncological benefit to support the same approach, thus,for a first CRC, UK guidelines consider the two surgeries equal (10), whereas European guidelines advocate segmental resection unless there is a metachronous CRC (35).
Systemic anti-cancer treatment options for LS-CRCs were previously confined to the four chemotherapeutic agents used in sporadic CRCs (fluorouracil, leucovorin, oxaliplatin and irinotecan) with no consideration given to MSI or MMR status. Studies that explored the efficacy of these treatments in MSI-high CRCs were conflicting, not specific to LS and limited by small sample sizes (99–102). A single LS-CRC-specific retrospective study found no survival benefit associated with adjuvant fluorouracil (103). Nevertheless these agents remain in use as adjuvant treatment for some high-risk or late stage MSI-H/dMMR CRCs, both sporadic and LS-associated (104).
MMR-deficient CRCs demonstrate higher levels of immunogenicity than their MMR-proficient counterparts. MMR deficiency allows accumulation of point mutations in microsatellite sequences which can cause translational frameshifts, generating carboxy-terminal frameshift peptides (FSPs) that serve as “neoantigens” recognised by and stimulating the anti-tumour host immune response. The immunoreactive nature of MSI-high/dMMR CRCs prompted use of checkpoint inhibitors. The phase three KEYNOTE-177 trial demonstrated that pembrolizumab (anti-PD1) doubles the median progression-free survival compared to standard chemotherapy (16.5 vs 8.2 months) (105). As such, pembrolizumab is now approved by the USA Food and Drug Administration and recommended first-line treatment in the UK for metastatic MSI-high/dMMR CRCs. A second PD-1 inhibitor, nivolumab, is also NICE-approved for combination use with ipilimumab following standard combination chemotherapy (106).
It remains unknown whether LS-CRCs and sporadic MSI-high/dMMR CRCs share a common response to checkpoint inhibitor therapy. The higher neoantigen load in LS-CRCs might suggest an even more pronounced response, but available studies of checkpoint inhibitors that include LS patients are largely limited by small subgroup numbers and have not demonstrated a difference in response rates (107–110).
The compelling evidence for interplay between host immune surveillance and LS tumours has provided the conceptual basis for the use of vaccines to augment the adaptive immune response in LS. The high burden of foreign FSPs in LS makes them excellent vaccine targets (111, 112). Although not specifically tested in LS-CRC, FSP-based vaccination induced significant humoral and T-cell responses in a first-in-human, phase I/IIa clinical trial (113) as well as in a mouse model of conditional MSH2 knockout (114). The same principles underpin the use of cancer vaccines to prevent tumour development from premalignant polyps by targeting CRC-associated antigens such as MUC1 and CEA, a theory currently being tested and with promising results in mouse models (115, 116).
Lynch syndrome is encountered by many clinicians at some stage in their practice and yet remains under-diagnosed with historically limited success in risk stratification and management. The PLSD international database continues to expand our knowledge of LS-associated cancer risk. However, we have yet to obtain international consensus on the optimal surveillance strategies, which will be essential among a population of patients who are living beyond their index cancer. The advent of NGS into clinical practice will undoubtably improve detection rates and allow for more effective, precise, and personalised management programmes for patients with LS. Finally, over the next decade it will be exciting to see improvements in the preventative strategies that can be offered to patients in the form of aspirin, or even anti-cancer vaccines, as we continue to attempt to disrupt the natural history of this prevalent cancer predisposition syndrome.
MW, AH, BN, RK and LL contributed to conception and design of the review. MW, AH and BN performed literature review and wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Curtius K, Gupta S, Boland CR. Review article: lynch syndrome-a mechanistic and clinical management update. Aliment Pharmacol Ther (2022) 55(8):960–77. doi: 10.1111/apt.16826
2. Worthley DL, Leggett BA. Colorectal cancer: molecular features and clinical opportunities. Clin Biochem Rev (2010) 31(2):31–8.
3. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol (2015) 110(2):223–62. doi: 10.1038/ajg.2014.435
4. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology (2010) 138(6):2044–58. doi: 10.1053/j.gastro.2010.01.054
5. Patel AP, Wang M, Fahed AC, Mason-Suares H, Brockman D, Pelletier R, et al. Association of rare pathogenic DNA variants for familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and lynch syndrome with disease risk in adults according to family history. JAMA Netw Open (2020) 3(4):e203959. doi: 10.1001/jamanetworkopen.2020.3959
6. Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med (2020) 22(1):15–25. doi: 10.1038/s41436-019-0596-9
7. Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A, . Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med (2014) 16(1):101–16. doi: 10.1038/gim.2013.166
8. Peltomaki P. Update on lynch syndrome genomics. Fam Cancer (2016) 15(3):385–93. doi: 10.1007/s10689-016-9882-8
9. Hitchins MP. Inheritance of epigenetic aberrations (constitutional epimutations) in cancer susceptibility. Adv Genet (2010) 70:201–43. doi: 10.1016/B978-0-12-380866-0.60008-3
10. Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, et al. Guidelines for the management of hereditary colorectal cancer from the British society of gastroenterology (BSG)/Association of coloproctology of great Britain and Ireland (ACPGBI)/United kingdom cancer genetics group (UKCGG). Gut (2020) 69(3):411–44. doi: 10.1136/gutjnl-2019-319915
11. NICE. Molecular testing strategies for lynch syndrome in people with colorectal cancer. NICE diagnostic guidelines. (2017).
12. Pearlman R, Frankel WL, Swanson BJ, Jones D, Zhao W, Yilmaz A, et al. Prospective statewide study of universal screening for hereditary colorectal cancer: the Ohio colorectal cancer prevention initiative. JCO Precis Oncol (2021) 5:779–91. doi: 10.1200/PO.20.00525
13. Burn J, Monahan K, Lalloo F, Peters K, Hardy S, Jessop J, et al Implementing lynch syndrome testing and surveillance pathways. NHS England (2021).
14. NICE. Testing strategies for lynch syndrome in people with endometrial cancer. NICE Diagnostic Guidance (2020). Available at: https://www.nice.org.uk/guidance/dg42
15. Vasen HF, Watson P, Mecklin JP, Lynch HT, International Collaborative Group on HNPCC. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, lynch syndrome) proposed by the international collaborative group on HNPCC. Gastroenterology (1999) 116(6):1453–6. doi: 10.1016/S0016-5085(99)70510-X
16. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst (2004) 96(4):261–8. doi: 10.1093/jnci/djh034
17. Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet (2000) 37(9):641–5. doi: 10.1136/jmg.37.9.641
18. NICE. Molecular testing strategies for lynch syndrome in people with colorectal cancer. NICE diagnostic guidelines. (2022).
19. Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol (2019) 46(3):261–70. doi: 10.1053/j.seminoncol.2019.08.003
20. Gray PN, Tsai P, Chen D, Wu S, Hoo J, Mu W, et al. TumorNext-Lynch-MMR: a comprehensive next generation sequencing assay for the detection of germline and somatic mutations in genes associated with mismatch repair deficiency and lynch syndrome. Oncotarget (2018) 9(29):20304–22. doi: 10.18632/oncotarget.24854
21. Salvador MU, Truelson MRF, Mason C, Souders B, LaDuca H, Dougall B, et al. Comprehensive paired Tumor/Germline testing for lynch syndrome: bringing resolution to the diagnostic process. J Clin Oncol (2019) 37(8):647–57. doi: 10.1200/JCO.18.00696
22. Plazzer JP, Sijmons RH, Woods MO, Peltomäki P, Thompson B, Den Dunnen JT, et al. The InSiGHT database: utilizing 100 years of insights into lynch syndrome. Fam Cancer (2013) 12(2):175–80. doi: 10.1007/s10689-013-9616-0
23. European Hereditary Tumour Group. The prospective lynch syndrome database. (2023). Available at: https://www.ehtg.org/
24. Moller P. The prospective lynch syndrome database reports enable evidence-based personal precision health care. Hered Cancer Clin Pract (2020) 18:6. doi: 10.1186/s13053-020-0138-0
25. Moller P, Seppala TT, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut (2018) 67(7):1306–16. doi: 10.1136/gutjnl-2017-314057
26. International Mismatch Repair Consortium. Variation in the risk of colorectal cancer in families with lynch syndrome: a retrospective cohort study. Lancet Oncol (2021) 22(7):1014–22. doi: 10.1016/S1470-2045(21)00189-3
27. Møller P, Seppälä T, Dowty JG, Haupt S, Dominguez-Valentin M, Sunde L, et al. Colorectal cancer incidences in lynch syndrome: a comparison of results from the prospective lynch syndrome database and the international mismatch repair consortium. Hered Cancer Clin Pract (2022) 20:36. doi: 10.1186/s13053-022-00241-1
28. Seppala TT, Dominguez-Valentin M, Sampson JR, Møller P. Prospective observational data informs understanding and future management of lynch syndrome: insights from the prospective lynch syndrome database (PLSD). Fam Cancer (2021) 20(1):35–9. doi: 10.1007/s10689-020-00193-2
29. Jarvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology (1995) 108(5):1405–11. doi: 10.1016/0016-5085(95)90688-6
30. Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology (2000) 118(5):829–34. doi: 10.1016/S0016-5085(00)70168-5
31. Newton K, Green K, Lalloo F, Evans DG, Hill J. Colonoscopy screening compliance and outcomes in patients with lynch syndrome. Colorectal Dis (2015) 17(1):38–46. doi: 10.1111/codi.12778
32. Stupart DA, Goldberg PA, Algar U, Ramesar R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis (2009) 11(2):126–30. doi: 10.1111/j.1463-1318.2008.01702.x
33. Lindberg LJ, Rasmussen M, Andersen KK, Nilbert M, Therkildsen C. Benefit from extended surveillance interval on colorectal cancer risk in lynch syndrome. Colorectal Dis (2020) 22(5):529–36. doi: 10.1111/codi.14926
34. Wales CINS. Cancer genetics referral guidelines for colorectal cancer or polyposis risk assessment and consideration of genetic testing. (2022).
35. Seppala TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, et al. European Guidelines from the EHTG and ESCP for lynch syndrome: an updated third edition of the mallorca guidelines based on gene and gender. Br J Surg (2021) 108(5):484–98. doi: 10.1002/bjs.11902
36. van Leerdam ME, Roos VH, van Hooft JE, Balaguer F, Dekker E, Kaminski MF, et al. Endoscopic management of lynch syndrome and of familial risk of colorectal cancer: European society of gastrointestinal endoscopy (ESGE) guideline. Endoscopy (2019) 51(11):1082–93. doi: 10.1055/a-1016-4977
37. Stjepanovic N, Moreira L, Carneiro F, Balaguer F, Cervantes A, Balmaña J, et al. Hereditary gastrointestinal cancers: ESMO clinical practice guidelines for diagnosis, treatment and follow-updagger. Ann Oncol (2019) 30(10):1558–71. doi: 10.1093/annonc/mdz233
38. French National Authority for Health. Colorectal Cancer Screening and Prevention: Update of the Periodic Health Examination (PHA) Code of Practice. (2013). Available at: https://www.has-sante.fr/jcms/c_1623732/en/depistage-et-prevention-du-cancer-colorectal
39. Hüneburg R, Aretz S, Büttner R, Daum S, Engel C, Fechner G, et al. Current recommendations for surveillance, risk reduction and therapy in lynch syndrome patients. Z Gastroenterol (2019) 57(11):1309–20. doi: 10.1055/a-1008-9827
40. Ishida H, Yamaguchi T, Tanakaya K, Akagi K, Inoue Y, Kumamoto K, et al. Japanese Society for cancer of the colon and rectum (JSCCR) guidelines 2016 for the clinical practice of hereditary colorectal cancer (Translated version). J Anus Rectum Colon (2018) 2(Suppl I):S1–S51. doi: 10.23922/jarc.2017-028
42. Guillen-Ponce C, Lastra E, Lorenzo-Lorenzo I, Martín Gómez T, Morales Chamorro R, Sánchez-Heras AB, et al. SEOM clinical guideline on hereditary colorectal cancer (2019). Clin Transl Oncol (2020) 22(2):201–12. doi: 10.1007/s12094-019-02272-y
43. Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, et al. Guidelines on genetic evaluation and management of lynch syndrome: a consensus statement by the US multi-society task force on colorectal cancer. Am J Gastroenterol (2014) 109(8):1159–79. doi: 10.1038/ajg.2014.186
44. Stoffel EM, Mangu PB, Gruber SB, Hamilton SR, Kalady MF, Lau MW, et al. Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk–colorectal cancer: European society for medical oncology clinical practice guidelines. J Clin Oncol (2015) 33(2):209–17. doi: 10.1200/JCO.2014.58.1322
45. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J Natl Compr Canc Netw 17(9):1032–41. doi: 10.6004/jnccn.2016.0108
46. Pan JY, Haile RW, Templeton A, Macrae F, Qin F, Sundaram V, et al. Worldwide practice patterns in lynch syndrome diagnosis and management, based on data from the international mismatch repair consortium. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc (2018) 16(12):1901. doi: 10.1016/j.cgh.2018.04.025
47. Moller P, Seppala T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer incidence and survival in lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective lynch syndrome database. Gut (2017) 66(3):464–72. doi: 10.1136/gutjnl-2015-309675
48. Ten Broeke SW, van Bavel TC, Jansen AML, Gómez-García E, Hes FJ, van Hest LP, et al. Molecular background of colorectal tumors from patients with lynch syndrome associated with germline variants in PMS2. Gastroenterology (2018) 155(3):844–51. doi: 10.1053/j.gastro.2018.05.020
49. Ahadova A, Seppälä TT, Engel C, Gallon R, Burn J, Holinski-Feder E, et al. The “unnatural” history of colorectal cancer in lynch syndrome: lessons from colonoscopy surveillance. Int J Cancer (2021) 148(4):800–11. doi: 10.1002/ijc.33224
50. Moller P, Seppala T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective lynch syndrome database. Gut (2017) 66(9):1657–64. doi: 10.1136/gutjnl-2016-311403
51. Seppala T, Pylvanainen K, Evans DG, Järvinen H, Renkonen-Sinisalo L, Bernstein I, et al. Colorectal cancer incidence in path MLH1 carriers subjected to different follow-up protocols: a prospective lynch syndrome database report. Hered Cancer Clin Pract (2017) 15(1):18. doi: 10.1186/s13053-017-0078-5
52. Engel C, Vasen HF, Seppala T, Aretz S, Bigirwamungu-Bargeman M, de Boer SY, et al. No difference in colorectal cancer incidence or stage at detection by colonoscopy among 3 countries with different lynch syndrome surveillance policies. Gastroenterology (2018) 155(5):1400–9 e2. doi: 10.1053/j.gastro.2018.07.030
53. Seppala TT, Ahadova A, Dominguez-Valentin M, Macrae F, Evans DG, Therkildsen, et al. Lack of association between screening interval and cancer stage in lynch syndrome may be accounted for by over-diagnosis; a prospective lynch syndrome database report. Hered Cancer Clin Pract (2019) 17:8. doi: 10.1186/s13053-019-0106-8
54. Lund J, Scholefield J, Grainge M, Smith SJ, Mangham C, Armitage NC, et al. Risks, costs, and compliance limit colorectal adenoma surveillance: lessons from a randomised trial. Gut (2001) 49(1):91. doi: 10.1136/gut.49.1.91
55. Edelstein DL, Axilbund J, Baxter M, Hylind LM, Romans K, Griffin CA, et al. Rapid development of colorectal neoplasia in patients with lynch syndrome. Clin Gastroenterol Hepatol (2011) 9(4):340–3. doi: 10.1016/j.cgh.2010.10.033
56. Iino H, Simms L, Young J, Arnold J, Winship IM, Webb SI, et al. DNA Microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer. Gut (2000) 47(1):37–42. doi: 10.1136/gut.47.1.37
57. Ahadova A, von Knebel Doeberitz M, Bläker H, Kloor M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: a model of interval cancers in lynch syndrome. Fam Cancer (2016) 15(4):579–86. doi: 10.1007/s10689-016-9899-z
58. Sanchez A, Roos VH, Navarro M, Pineda M, Caballol B, Moreno L, et al. Quality of colonoscopy is associated with adenoma detection and postcolonoscopy colorectal cancer prevention in lynch syndrome. Clin Gastroenterol Hepatol (2022) 20(3):611–21.e9. doi: 10.1016/j.cgh.2020.11.002
59. Haanstra JF, Vasen HF, Sanduleanu S, van der Wouden EJ, Koornstra JJ, Kleibeuker JH, et al. Quality colonoscopy and risk of interval cancer in lynch syndrome. Int J Colorectal Dis (2013) 28(12):1643–9. doi: 10.1007/s00384-013-1745-2
60. Rees CJ, Thomas Gibson S, Rutter MD, Baragwanath P, Pullan R, Feeney M, et al. UK Key performance indicators and quality assurance standards for colonoscopy. Gut (2016) 65(12):1923–9. doi: 10.1136/gutjnl-2016-312044
61. Bisschops R, East JE, Hassan C, Hazewinkel Y, Kamiński MF, Neumann H, et al. Advanced imaging for detection and differentiation of colorectal neoplasia: European society of gastrointestinal endoscopy (ESGE) guideline - update 2019. Endoscopy (2019) 51(12):1155–79. doi: 10.1055/a-1031-7657
62. Har-Noy O, Yung DE, Koulaouzidis A, Eliakim R, Kopylov U, Avidan B, et al. Chromoendoscopy or white light endoscopy for neoplasia detection in lynch syndrome, a meta-analysis. Dig Liver Dis (2019) 51(11):1515–21. doi: 10.1016/j.dld.2019.07.018
63. Bisschops R, Tejpar S, Willekens H, De Hertogh G, Van Cutsem E. Virtual chromoendoscopy (I-SCAN) detects more polyps in patients with lynch syndrome: a randomized controlled crossover trial. Endoscopy (2017) 49(4):342–50. doi: 10.1055/s-0042-121005
64. East JE, Suzuki N, Stavrinidis M, Guenther T, Thomas HJ, Saunders BP. Narrow band imaging for colonoscopic surveillance in hereditary non-polyposis colorectal cancer. Gut (2008) 57(1):65–70. doi: 10.1136/gut.2007.128926
65. Huneburg R, Lammert F, Rabe C, Rahner N, Kahl P, Büttner R, et al. Chromocolonoscopy detects more adenomas than white light colonoscopy or narrow band imaging colonoscopy in hereditary nonpolyposis colorectal cancer screening. Endoscopy (2009) 41(4):316–22. doi: 10.1055/s-0028-1119628
66. Cellier C, Perrod G, Colas C, Dhooge M, Saurin JC, Lecomte T, et al. Back-to-Back comparison of colonoscopy with virtual chromoendoscopy using a third-generation narrow-band imaging system to chromoendoscopy with indigo carmine in patients with lynch syndrome. Am J Gastroenterol (2019) 114(10):1665–70. doi: 10.14309/ajg.0000000000000386
67. Houwen B, Hazewinkel Y, Pellise M, Rivero-Sánchez L, Balaguer F, Bisschops R, et al. Linked colour imaging for the detection of polyps in patients with lynch syndrome: a multicentre, parallel randomised controlled trial. Gut (2022) 71(3):553–60. doi: 10.1136/gutjnl-2020-323132
68. Gong D, Wu L, Zhang J, Mu G, Shen L, Liu J, et al. Detection of colorectal adenomas with a real-time computer-aided system (ENDOANGEL): a randomised controlled study. Lancet Gastroenterol Hepatol (2020) 5(4):352–61. doi: 10.1016/S2468-1253(19)30413-3
69. Su JR, Li Z, Shao XJ, Ji CR, Ji R, Zhou RC, et al. Impact of a real-time automatic quality control system on colorectal polyp and adenoma detection: a prospective randomized controlled study (with videos). Gastrointest Endosc (2020) 91(2):415–24.e4. doi: 10.1016/j.gie.2019.08.026
70. Wang P, Berzin TM, Glissen Brown JR, Bharadwaj S, Becq A, Xiao X, et al. Real-time automatic detection system increases colonoscopic polyp and adenoma detection rates: a prospective randomised controlled study. Gut (2019) 68(10):1813–9. doi: 10.1136/gutjnl-2018-317500
71. Repici A, Spadaccini M, Antonelli G, Correale L, Maselli R, Galtieri PA, et al. Artificial intelligence and colonoscopy experience: lessons from two randomised trials. Gut (2022) 71(4):757–65. doi: 10.1136/gutjnl-2021-324471
72. Hüneburg R, Bucksch K, Schmeisser F, Heling D, Marwitz T, Aretz S, et al. Real-time use of artificial intelligence (CADEYE) in colorectal cancer surveillance of patients with lynch syndrome-a randomized controlled pilot trial (CADLY). United Eur Gastroenterol J (2022) 11(1):60–8. doi: 10.1002/ueg2.12354
73. van Liere E, de Boer N, Dekker E, van Leerdam M, de Meij T, Ramsoekh D, et al. Systematic review: non-endoscopic surveillance for colorectal neoplasia in individuals with lynch syndrome. Aliment Pharmacol Ther (2022) 55(7):778–88. doi: 10.1111/apt.16824
74. Katsoula A, Paschos P, Haidich AB, Tsapas A, Giouleme O. Diagnostic accuracy of fecal immunochemical test in patients at increased risk for colorectal cancer: a meta-analysis. JAMA Intern Med (2017) 177(8):1110–8. doi: 10.1001/jamainternmed.2017.2309
75. Anne G Lincoln SCB, Sasieni P, Monahan KJ. Risk-stratified FIT for urgent colonoscopy in lynch syndrome: a clinical service throughout the COVID-19 pandemic. J Clin Oncol (2022) 40(16). doi: 10.1200/JCO.2022.40.16_suppl.10606
76. Lincoln A, Benton S, Piggott C, North BV, Rigney J, Young C, et al. Exploring the utility and acceptability of faecal immunochemical testing (FIT) as a novel intervention for the improvement of colorectal cancer (CRC) surveillance in individuals with lynch syndrome (FIT for lynch study): a single-arm, prospective, multi-centre, non-randomised study. BMC Cancer (2022) 22(1):1144. doi: 10.1186/s12885-022-10217-y
77. Stoffel EM, Mangu PB, Limburg PJ, Hamilton SR, Kalady MF, Lau MW, et al. Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European society for medical oncology clinical practice guidelines. J Oncol Pract (2015) 11(3):e437–41. doi: 10.1200/JOP.2015.003665
78. ten Kate GL, Kleibeuker JH, Nagengast FM, Craanen M, Cats A, Menko FH, et al. Is surveillance of the small bowel indicated for lynch syndrome families? Gut (2007) 56(9):1198–201. doi: 10.1136/gut.2006.118299
79. Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with lynch syndrome. JAMA (2009) 302(16):1790–5. doi: 10.1001/jama.2009.1529
80. Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International cancer of the pancreas screening (CAPS) consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut (2013) 62(3):339–47. doi: 10.1136/gutjnl-2012-303108
81. Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T, Burn J, Cornes JM, et al. The Manchester international consensus group recommendations for the management of gynecological cancers in lynch syndrome. Genet Med (2019) 21(10):2390–400. doi: 10.1038/s41436-019-0489-y
82. Evans DG, Ingham SL. Reduced life expectancy seen in hereditary diseases which predispose to early-onset tumors. Appl Clin Genet (2013) 6:53–61. doi: 10.2147/TACG.S35605
83. Pylvanainen K, Lehtinen T, Kellokumpu I, Järvinen H, Mecklin JP. Causes of death of mutation carriers in Finnish lynch syndrome families. Fam Cancer (2012) 11(3):467–71. doi: 10.1007/s10689-012-9537-3
84. Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the united states. CA Cancer J Clin (2018) 68(1):31–54. doi: 10.3322/caac.21440
85. Diergaarde B, Braam H, Vasen HF, Nagengast FM, van Muijen GN, Kok FJ, et al. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol (2007) 5(6):736–42. doi: 10.1016/j.cgh.2007.02.019
86. Movahedi M, Bishop DT, Macrae F, Mecklin JP, Moeslein G, Olschwang S, et al. Obesity, aspirin, and risk of colorectal cancer in carriers of hereditary colorectal cancer: a prospective investigation in the CAPP2 study. J Clin Oncol (2015) 33(31):3591–7. doi: 10.1200/JCO.2014.58.9952
87. Botma A, Nagengast FM, Braem MG, Hendriks JC, Kleibeuker JH, Vasen HF, et al. Body mass index increases risk of colorectal adenomas in men with lynch syndrome: the GEOLynch cohort study. J Clin Oncol (2010) 28(28):4346–53. doi: 10.1200/JCO.2010.28.0453
88. Win AK, Dowty JG, English DR, Campbell PT, Young JP, Winship I, et al. Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer (2011) 105(1):162–9. doi: 10.1038/bjc.2011.172
89. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet (2011) 377(9759):31–41. doi: 10.1016/S0140-6736(10)62110-1
90. Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet (2010) 376(9754):1741–50. doi: 10.1016/S0140-6736(10)61543-7
91. Burn J, Sheth H, Elliott F, Reed L, Macrae F, Mecklin JP, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet (2020) 395(10240):1855–63. doi: 10.1016/S0140-6736(20)30366-4
93. Ferlitsch M, Moss A, Hassan C, Bhandari P, Dumonceau JM, Paspatis G, et al. Colorectal polypectomy and endoscopic mucosal resection (EMR): European society of gastrointestinal endoscopy (ESGE) clinical guideline. Endoscopy (2017) 49(3):270–97. doi: 10.1055/s-0043-102569
94. Suzuki S, Ikehara H, Gotoda T. Should large sessile serrated lesions be treated with cold snare polypectomy? Dig Endosc (2022) 34(3):485–7. doi: 10.1111/den.14257
95. Takeuchi Y, Shichijo S, Uedo N, Kawakami Y, Okubo Y, Tani Y, et al. Safety and efficacy of cold versus hot snare polypectomy including colorectal polyps >/=1 cm in size. Dig Endosc (2022) 34(2):274–83. doi: 10.1111/den.14096
96. Heneghan HM, Martin ST, Winter DC. Segmental vs extended colectomy in the management of hereditary nonpolyposis colorectal cancer: a systematic review and meta-analysis. Colorectal Dis (2015) 17(5):382–9. doi: 10.1111/codi.12868
97. Anele CC, Adegbola SO, Askari A, Rajendran A, Clark SK, Latchford A, et al. Risk of metachronous colorectal cancer following colectomy in lynch syndrome: a systematic review and meta-analysis. Colorectal Dis (2017) 19(6):528–36. doi: 10.1111/codi.13679
98. Malik SS, Lythgoe MP, McPhail M, Monahan KJ. Metachronous colorectal cancer following segmental or extended colectomy in lynch syndrome: a systematic review and meta-analysis. Fam Cancer (2018) 17(4):557–64. doi: 10.1007/s10689-017-0062-2
99. Fallik D, Borrini F, Boige V, Viguier J, Jacob S, Miquel C, et al. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res (2003) 63(18):5738–44.
100. Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology (2004) 126(2):394–401. doi: 10.1053/j.gastro.2003.12.023
101. Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med (2003) 349(3):247–57. doi: 10.1056/NEJMoa022289
102. Liang JT, Huang KC, Lai HS, Lee PH, Cheng YM, Hsu HC, et al. High-frequency microsatellite instability predicts better chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV sporadic colorectal cancer after palliative bowel resection. Int J Cancer (2002) 101(6):519–25. doi: 10.1002/ijc.10643
103. de Vos tot Nederveen Cappel WH, Meulenbeld HJ, Kleibeuker JH, Nagengast FM, Menko FH, Griffioen G, et al. Survival after adjuvant 5-FU treatment for stage III colon cancer in hereditary nonpolyposis colorectal cancer. Int J Cancer (2004) 109(3):468–71. doi: 10.1002/ijc.11712
104. Argiles G, Tabernero J, Labianca R, Hochhauser D, Salazar R, Iveson T, et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 31(10):1291–305. doi: 10.1016/j.annonc.2020.06.022
105. Diaz LA Jr., Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol (2022) 23(5):659–70. doi: 10.1016/S1470-2045(22)00197-8
106. NICE. Nivolumab with ipilimumab for previously treated metastatic colorectal cancer with high microsatellite instability or mismatch repair deficiency. technology appraisal guidance. (2021).
107. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
108. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
109. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-Deficient/Microsatellite instability-high metastatic colorectal cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
110. Therkildsen C, Jensen LH, Rasmussen M, Bernstein I. An update on immune checkpoint therapy for the treatment of lynch syndrome. Clin Exp Gastroenterol (2021) 14:181–97. doi: 10.2147/CEG.S278054
111. Hernandez-Sanchez A, Grossman M, Yeung K, Sei SS, Lipkin S, Kloor M. Vaccines for immunoprevention of DNA mismatch repair deficient cancers. J Immunother Cancer (2022) 10(6). doi: 10.1136/jitc-2021-004416
112. Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology (2008) 134(4):988–97. doi: 10.1053/j.gastro.2008.01.015
113. Kloor M, Reuschenbach M, Pauligk C, Karbach J, Rafiyan MR, Al-Batran SE, et al. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: a phase I/IIa clinical trial. Clin Cancer Res (2020) 26(17):4503–10. doi: 10.1158/1078-0432.CCR-19-3517
114. Gebert J, Gelincik O, Oezcan-Wahlbrink M, Marshall JD, Hernandez-Sanchez A, Urban K, et al. Recurrent frameshift neoantigen vaccine elicits protective immunity with reduced tumor burden and improved overall survival in a lynch syndrome mouse model. Gastroenterology (2021) 161(4):1288–302.e13. doi: 10.1053/j.gastro.2021.06.073
115. Zeytin HE, Patel AC, Rogers CJ, Canter D, Hursting SD, Schlom J, et al. Combination of a poxvirus-based vaccine with a cyclooxygenase-2 inhibitor (celecoxib) elicits antitumor immunity and long-term survival in CEA.Tg/MIN mice. Cancer Res (2004) 64(10):3668–78. doi: 10.1158/0008-5472.CAN-03-3878
Keywords: lynch syndrome, mismatch repair (MMR) deficiency, colorectal cancer, surveillance, cancer diagnosis, cancer treatment
Citation: Williams MH, Hadjinicolaou AV, Norton BC, Kader R and Lovat LB (2023) Lynch syndrome: from detection to treatment. Front. Oncol. 13:1166238. doi: 10.3389/fonc.2023.1166238
Received: 15 February 2023; Accepted: 11 April 2023;
Published: 01 May 2023.
Edited by:
Christina Therkildsen, Copenhagen University Hospital, DenmarkReviewed by:
Wei Chen, The Ohio State University, United StatesCopyright © 2023 Williams, Hadjinicolaou, Norton, Kader and Lovat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas V. Hadjinicolaou, YWg0OTlAY2FtLmFjLnVr
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.