 Donatella Talotta
Donatella Talotta Mohammad Almasri
Mohammad Almasri Chiara Cosentino
Chiara Cosentino Gianluca Gaidano
Gianluca Gaidano Riccardo Moia
Riccardo Moia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 20 April 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1164517
This article is part of the Research TopicThe Emerging Role of Liquid Biopsies in Hematologic DisordersView all 7 articles
The assessment of the cancer mutational profile is crucial for patient management, stratification, and therapeutic decisions. At present, in hematological malignancies with a solid mass, such as lymphomas, tumor genomic profiling is generally performed on the tissue biopsy, but the tumor may harbor genetic lesions that are unique to other anatomical compartments. The analysis of circulating tumor DNA (ctDNA) on the liquid biopsy is an emerging approach that allows genotyping and monitoring of the disease during therapy and follow-up. This review presents the different methods for ctDNA analysis and describes the application of liquid biopsy in different hematological malignancies. In diffuse large B-cell lymphoma (DLBCL) and Hodgkin lymphoma (HL), ctDNA analysis on the liquid biopsy recapitulates the mutational profile of the tissue biopsy and can identify mutations otherwise absent on the tissue biopsy. In addition, changes in the ctDNA amount after one or two courses of chemotherapy significantly predict patient outcomes. ctDNA analysis has also been tested in myeloid neoplasms with promising results. In addition to mutational analysis, liquid biopsy also carries potential future applications of ctDNA, including the analysis of ctDNA fragmentation and epigenetic patterns. On these grounds, several clinical trials aiming at incorporating ctDNA analysis for treatment tailoring are currently ongoing in hematological malignancies.
The assessment of the cancer mutational profile is nowadays crucial for patient management, stratification, and therapeutic decisions. At present, in hematological malignancies with solid masses, such as lymphomas, tumor genomic profiling is generally performed on the tissue biopsy. Different genomic and molecular analyses on tissue biopsy complement histological diagnosis for the detection of molecular biomarkers with prognostic and therapeutic implications (1–3). However, the current approach for accessing tumor material consists of invasive procedures with limitations with respect to feasibility and the collection of serial samples for real-time monitoring. Moreover, tissue biopsy is usually confined to one single tumor site, thus limiting a comprehensive characterization of the tumor genome, which might vary among different anatomical sites (4).
Liquid biopsy is an emerging approach for characterizing tumors through the isolation and analysis of different cancer-derived components released in any body fluids (i.e., blood, urine, liquor, or others) (Figure 1). These “clues” released from tumors may be represented by circulating tumor cells (CTCs), circulating cell-free nucleic acids, exosomes, or tumor-educated platelets (TEPs) (5). In the context of hematological malignancies, liquid biopsy is a minimally invasive and real-time procedure that can potentially overcome the intrinsic limitations of tissue biopsies, which expose patients to procedural risks and cannot account for spatial intratumor heterogeneity. Different synchronous sources of tumor DNA complement each other in informing on driver gene mutations harboring potential prognostic and/or predictive value (6). The analysis of circulating tumor DNA (ctDNA) from plasma represents the most used application of liquid biopsy in hematological malignances for disease genotyping and for dynamic monitoring of the disease response during therapy (7, 8). The role of liquid biopsy for minimal residual disease (MRD) monitoring has been extensively reviewed elsewhere (9). In the present review, we will discuss the current application of liquid biopsy in lymphoid and myeloid neoplasms and the potential future applications that may allow a more personalized treatment approach in every single patient (Figure 2).
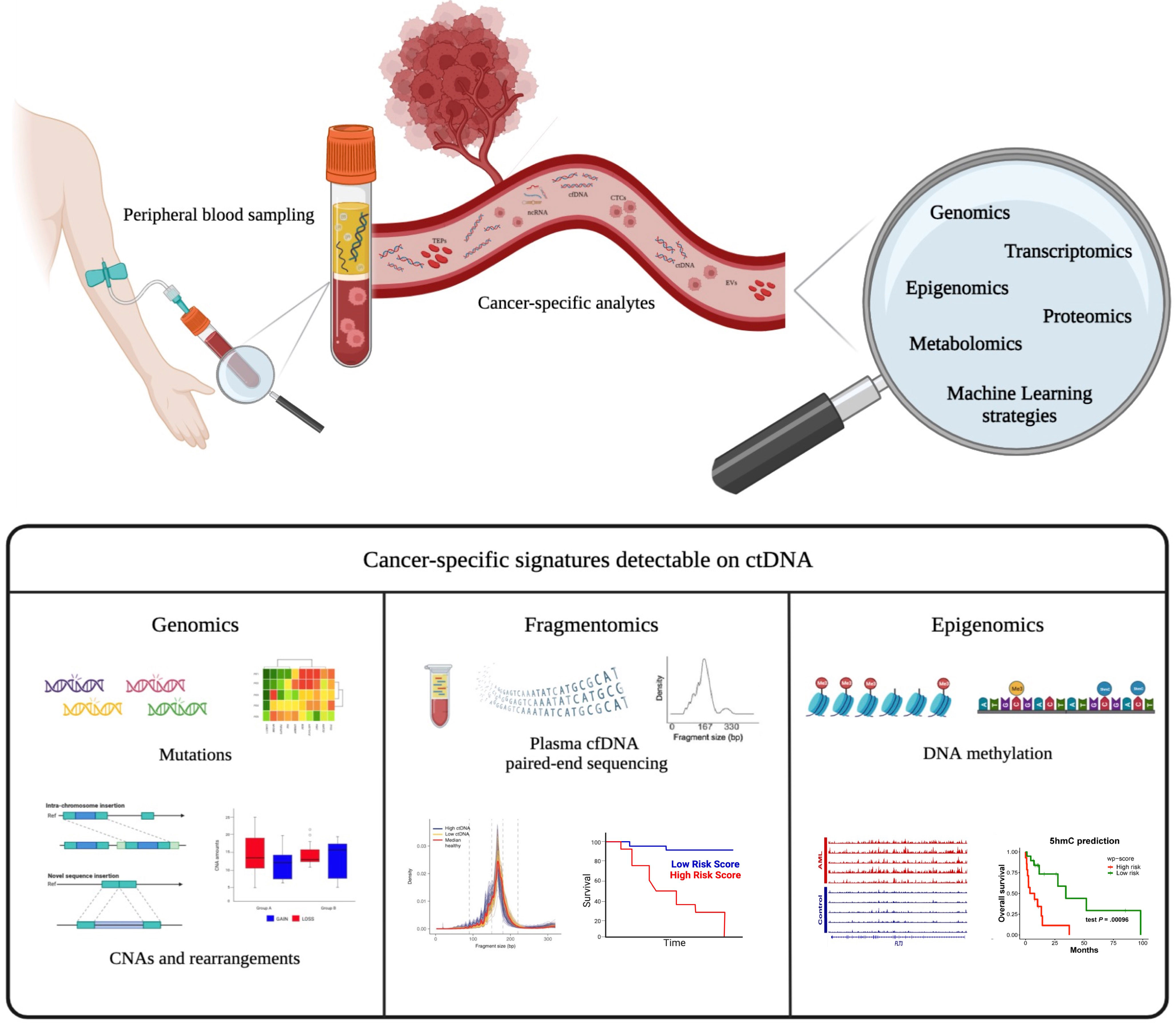
Figure 1 Liquid biopsy as a tool for identifying various cancer-specific analytes. Tumor-specific circulating analytes potentially detected by liquid biopsy include cell-free DNA (cfDNA) and/or circulating tumor DNA (ctDNA), circulating cells (CTCs), extracellular vesicles (EVs), and tumor-educated platelets (TEPs). Different cancer-specific genomic signatures can be revealed from ctDNA, including mutations, copy number alterations (CNAs), fragmentomics, and DNA methylation patterns. These biological data could be integrated using innovative bioinformatic pipelines to improve precision medicine.
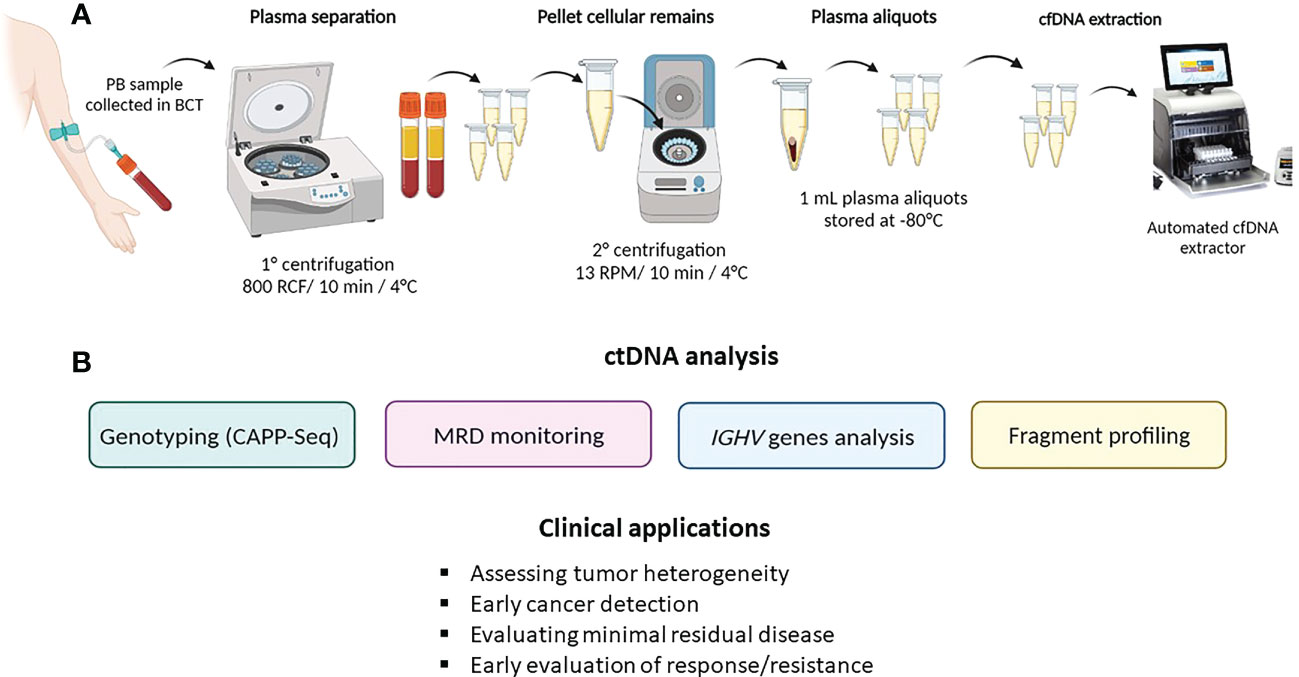
Figure 2 ctDNA collection method and analysis. (A) describes ctDNA isolation and extraction from peripheral blood (PB); (B) summarizes the main ctDNA applications in hematological malignancies.
The definition of circulating cell-free DNA (cfDNA) refers to short DNA fragments (180-200 base pairs) shed by apoptotic or necrotic cells in the bloodstream (10). In healthy subjects, the concentration of cfDNA in plasma ranges from 1 to 16.8 ng/ml, has a short half-life (approximately 2 h), and can increase following conditions such as physical exercise, acute trauma, stroke, and infection (11). In cancer patients, cfDNA levels are significantly higher than in healthy individuals, since apoptotic and necrotic remains within the tumoral mass are not cleared efficiently by phagocytes, leading to the accumulation of cellular debris, including cfDNA, that is subsequently released into the circulation (7). In these patients, cfDNA comprises molecules that are derived both from healthy cells and tumor cells. To differentiate the origin, the fraction of cfDNA derived from tumors cells by multiple shedding mechanisms (apoptosis, necrosis, and secretion) is known as circulating tumor DNA (ctDNA) (7, 12). In lymphoma patients, ctDNA levels vary greatly across different lymphoma subtypes and reflect tumor type, clinical stage, and tumor burden. Overall, the mean concentration of ctDNA in lymphoma patients is 30 ng/ml (13).
As previously stated, ctDNA can be isolated and analyzed from any fluid of the body. Given the high accessibility of peripheral blood, the watery fluid portion, namely, the serum or plasma, represents the most frequent source of ctDNA. Despite the fact that the amount of ctDNA fragments is similar in both serum and plasma (14), the latter represents the optimal specimen type for ctDNA analysis because the amount of cfDNA released by normal leukocytes that dilutes the ctDNA is lower in plasma than in serum. Blood samples should be collected in tubes containing K2EDTA and processed for plasma separation within 6 h in order to avoid contamination derived from genomic DNA released by leukocytes (15, 16). Alternatively, specific blood collection tubes (i.e., Streck BCT) allow the preservation and stabilization of ctDNA for up to 14 days, thus allowing the safe shipment of samples (16, 17).
To preserve cfDNA and minimize leukocyte lysis, it is recommended to perform peripheral blood processing through a sequential pair of centrifugations prior to storage and/or cfDNA extraction (15). Studies indicate that purified plasma can be frozen and safely preserved in single-use aliquots (18, 19).
In hematological malignancies, ctDNA can be analyzed for the evaluation of gene mutations or the analysis of the unique immunoglobulin heavy chain (IGHV) rearrangement in cases of B-cell neoplasms (20–23).
Disease genotyping on ctDNA allows the identification of somatic cancer gene mutations without the need of previous analyses of the tumor biopsy, therefore working in a tissue biopsy-free manner. As previously stated, this approach has the ability to potentially identify mutations derived from different tumor sites thus allowing the characterization of the entire tumor heterogeneity (7). Ultra-deep next-generation sequencing (NGS) methods allow massive parallel sequencing of DNA molecules in a single flow cell (24) and can detect a large spectrum of genetic alterations, including point mutations, insertions/deletions, rearrangements, and copy number changes (25). The CAncer Personalized Profiling by deep sequencing (CAPP-seq) is a highly sensitive and specific targeted capture NGS method to detect tumor-specific mutations in ctDNA in molecularly heterogeneous tumors (25). CAPP-seq utilizes a disease-specific “selector,” which is a set of exonic and intronic targets that cover regions of known recurrent mutations in a particular cancer type. The subsequent amplification and sequencing of the target regions allow the quantification of ctDNA based on the detection of cancer-specific mutations (13).
Almost all B-cell tumors harbor a unique IGHV rearrangement that can be identified on ctDNA using polymerase chain reaction (PCR) or NGS-based techniques (20, 26, 27). In cases of unproductive IGHV rearrangement, the light chain IGK or IGL sequence could be used as a surrogate marker. However, to ensure the tumor origin of the IGHV rearrangement identified on the liquid biopsy, the IGHV rearrangement should be initially identified on the tissue biopsy. NGS-based methods, such as clonoSEQ (28), rely on universal primer sets targeting the immunoglobulin heavy or light chains. This assay is FDA-approved for MRD detection in patients with chronic lymphocytic leukemia (CLL), multiple myeloma (MM), and B-cell acute lymphoblastic leukemia (B-ALL) (29) and has been extensively used for MRD monitoring of ctDNA in lymphoma patients (20–22, 29, 30). However, these methods for immunoglobulin analysis capture only one single molecular marker and fail to detect clonal V(D)J rearrangements in ~20% of lymphoma due to the high rates of somatic hypermutation (SHM) (20, 22, 24).
Diffuse large B-cell lymphoma (DLBCL) is a highly heterogeneous disease both from a molecular and clinical standpoint. Molecular heterogeneity is present also within the same patient, and different anatomical sites may harbor different genetic lesions. On these grounds, liquid biopsy has been demonstrated to be able to detect mutations otherwise absent in the single tissue biopsy. Different studies have demonstrated that lymphoma-related genetic alterations can be detected from ctDNA using CAPP-seq even without a baseline tumor tissue biopsy (4, 31, 32). The sensitivity and specificity of targeted gene mutation analysis in ctDNA versus tissue biopsy in DLBCL patients have been assessed in several independent CAPP-seq studies (4, 31). The percentage of mutations (i.e., true-positive rate) identified both in the tissue biopsy and in ctDNA ranges between 95% and 99%. Moreover, liquid biopsy is able to detect a fraction of mutations (approximately 15%-20%) that are not otherwise identified in the lymph node biopsy. Conversely, only a small fraction of mutations (1% to 5%) characterized by low allelic abundance was identified in the lymph node biopsy only and was not detected in ctDNA (i.e., false-negative rate) (4, 31).
Although the majority of DLBCL patients are cured with the first-line chemoimmunotherapy R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone), novel emerging therapies have been recently approved as additional treatment options (33, 34). Despite these advancements, a considerable fraction of DLBCL patients (~40%) still relapse or are refractory (35, 36). The identification of clinical and molecular biomarkers that better identify high-risk patients may improve the outcome and the cure rates of DLBCL patients. At present, the therapy response criteria in lymphomas do not include MRD markers at the end of the therapy (37). The quantification of ctDNA during and after therapy represents a potential opportunity to evaluate MRD in DLBCL.
Different techniques have been used to evaluate MRD in DLBCL, using both IGHV rearrangement and gene mutations using CAPP-Seq (21, 30, 38, 39). By using a quantitative high-throughput method to analyze IGHV rearrangements, ctDNA measurement has been shown to detect disease progression months before conventional imaging in DLBCL (21). The other approach based on gene mutations relies on the CAPP-seq as a tool for the quantification of ctDNA at baseline and during the course of R-CHOP therapy. The 2-log drop from baseline ctDNA after one cycle [early molecular response (EMR)] and the 2.5-log decrease after two cycles [major molecular response (MMR)] were identified as the best cutoff to split patient outcome (21, 30, 38, 39). In addition, the molecular response by ctDNA remained an independent prognostic parameter for both event-free survival (EFS) and overall survival (OS) in multivariate analysis including the international prognostic index (IPI) score, cell of origin, and interim positron emission tomography combined with computed tomography (PET/CT) scan (40).
Recent advances in molecular biology and in silico strategies that suppress technical errors can substantially improve the sensitivity of targeted NGS-based approaches. A series of error-suppression methods have been applied to lower the background rate, resulting in a sensitivity of ~2.5:100,000 (41, 42). For example, CAPP-seq can be combined with a unique barcoding strategy with a downstream bioinformatic algorithm that largely eliminates sequencing errors and stereotypic background, facilitating ctDNA detection down to an allele frequency (AF) of ~0.002% (31, 39).
To further maximize the analytical sensitivity and to reduce background error rates, a recent innovative approach named phased variant enrichment and detection sequencing (PhasED-seq) can track two or more variants (“phased variants”) on the same strand of one single DNA molecule (43). This method lowers both the technical and the biological background signals and maintains high genome recovery, thus facilitating ctDNA monitoring down to an analytical detection limit of ~0.00005% (39, 42, 44). PhasED-seq seems particularly useful in B-cell malignancies since multiple-phased variants occur in stereotyped portions of the tumor genome mostly due to on-target and aberrant SHM driven by activation-induced deaminase (AID) (45). PhasED-seq can significantly improve sensitivity for ctDNA detection in lymphoma patients, but additional studies of PhasED-seq will be required to confirm its superiority to CAPP-seq for the identification of early relapsing patients and to identify the best clinical settings in which this technology should be used (42).
DLBCLs that involve the central nervous system (CNS), namely, CNS lymphomas (CNSLs), are distinguished into primary and secondary DLBCLs of the CNS. Primary CNS lymphoma (PCNSL) refers to a rare subtype of DLBCL involving the brain, eyes, leptomeninges, or spinal cord without extracranial involvement. Secondary DLBCL CNS lymphoma (SCNSL) denotes an isolated relapse of DLBCL within the CNS or synchronous CNS with systemic involvement (46–48). The diagnosis of CNSL requires invasive neurosurgical procedures that often cannot be safely performed or are delayed because of concurrent therapies (49–51). Consequently, the clinical outcomes of patients with CNSL are extremely heterogeneous, and many patients suffer recurrence or experience early mortality after first-line therapy (50). Novel diagnostic strategies and biomarkers are necessary to non-invasively detect CNSL, to better stratify patients into risk groups, and to predict therapy responses. Initially, liquid biopsy in PCNSL has been mainly explored in cerebrospinal fluid (CSF) since the low abundance of ctDNA in plasma hampered the detection of the disease (52–55). Recently, thanks to the improved sensitivity of liquid biopsy combining CAPP-seq and PhasED-seq approaches, ctDNA has been detected with a high concordant rate in both plasma and CSF in patients with CNSL (56, 57). Plasma ctDNA analysis allowed the identification of recurrent mutations within genes coding for components of the B-cell receptor signaling pathway, such as MYD88, PIM1, and CD79b (58, 59).
After demonstrating that ctDNA analysis can genotype CNSL, the potential prognostic value of ctDNA before and after therapy was evaluated. Patients with positive ctDNA in pretreatment plasma also had significantly shorter PFS and OS when adjusted in a multivariate manner for established clinical and radiographic risk factors. Moreover, similar to DLBCL, measurable residual disease detection by plasma ctDNA during treatment identified patients with a particularly poor prognosis following curative-intent immunochemotherapy (56).
The low abundance of malignant Hodgkin/Reed-Sternberg (HRS) cells in biopsy samples (0.1%-3%) represents the main obstacle to the comprehensive genomic characterization of Hodgkin lymphoma (HL) (60). To overcome the limitations of tissue-based genotyping, plasma ctDNA was identified as a reliable source of tumor DNA for HL mutational profiling (32, 61, 62). The levels of cfDNA in HL patients are two-fold higher than in healthy subjects (~3,400 vs. ~1,700 hGE/ml of plasma), with a median ctDNA level in HL of ~200 hGE/ml (63). Even though the tumor cell volume in cHL is 10-fold smaller than that of other aggressive lymphomas, the correlation between ctDNA levels and radiologic tumor volume in HL is remarkably similar to DLBCL (42, 61). Thus, cHL seems to release a greater amount of ctDNA than DLBCL, a fact that may be due to the high apoptotic HRS cell rate (64).
On these grounds, efforts were initially focused on HL genotyping on the liquid biopsy. Using a CAPP-seq panel made up of frequently mutated genes in HL, ctDNA successfully detected 87% of mutations identified in the microdissected HRS cells. This study also allowed us to refine the current knowledge of cHL genetics. In addition, the authors also analyzed ctDNA of patients at the time of relapse. Longitudinal ctDNA profiling allowed us to identify treatment-dependent patterns of clonal evolution in patients relapsing after chemotherapy and in patients maintaining partial remission under immunotherapy (32). Immune evasion represents one of the pathogenetic hallmarks of HL and efforts have also been made to identify genetic lesions of immune checkpoint genes on ctDNA. Genomic profiling of cfDNA can precisely detect and type ~80% of copy number abnormalities (CNAs) of chromosome 9p24.1, which leads to PD-1 ligand overexpression and is associated with superior outcome in cHL (65, 66).
PET/CT is considered the gold standard for staging and response assessment in cHL (67, 68). At the time of diagnosis, the total metabolic tumor volume (TMTV) represents a functional and quantitative parameter that strongly predicts the outcome in cHL (69–71). Interestingly, the amount of ctDNA in plasma correlates with TMTV, indicating that ctDNA quantification may be coupled to TMTV to better predict HL outcome at the time of diagnosis (42). In addition to baseline assessment, interim PET/CT scan after two cycles of ABVD in advanced HL represents a pivotal timepoint in treatment tailoring. Negative PET/CT patients continue the remaining ABVD cycles; conversely, positive PET/CT patients are switched to a more intensified regimen. However, a meta-analysis demonstrated a certain degree of inaccuracy of this application (72), and the combination of PET/CT scan with ctDNA analysis may fill this gap. Spina et al. observed that a >2-log reduction (DLBCL threshold) in ctDNA load after two chemotherapy courses is associated with complete response and cure in cHL. Vice versa, a drop of <2-log in ctDNA after two chemotherapy courses is associated with progression and shorter survival (32). Therefore, quantification of ctDNA complements interim PET/CT in determining residual disease in cHL. Indeed, cured patients who were inconsistently judged as interim PET/CT-positive had a >2-log drop in ctDNA, whereas relapsing patients who were inconsistently judged as interim PET/CT-negative had a less than 2-log drop in ctDNA.
The diagnosis of most myeloid neoplasms is based on morphological, immunophenotypic, and molecular characterization of bone marrow (BM) aspirates and biopsies. In addition, MRD monitoring in these neoplasms, with the exception of chronic myeloid leukemia, is currently based on sequential results obtained from BM (73). Initial studies demonstrated that cfDNA in patients with myeloid neoplasms is higher than in healthy controls, and it can be used as a reliable tool for the identification of genomic abnormalities specific to myeloid neoplasm (74, 75).
The first evidence of cfDNA value in myeloid neoplasms is derived from acute myeloid leukemia (AML) (76). More recently, targeted NGS of ctDNA has been demonstrated to detect clinically relevant mutations missed by conventional BM analysis, thus providing a complementary tool for the assessment and monitoring of AML patients (77). MRD monitoring in AML on ctDNA has also been evaluated in AML patients after allogeneic hematopoietic stem cell transplantation (alloSCT). After identifying driver mutations in 51 patients using NGS, at least one personalized digital polymerase chain reaction assay per case was designed to evaluate MRD. By analyzing multiple timepoints after alloSCT, the persistence of mutations in ctDNA over time was associated with shorter outcomes (78).
Similar approaches have also been used in myelodysplastic syndrome (MDS) patients. Serial ctDNA monitoring using digital PCR allows the detection and tracking of both driver mutations and karyotypic abnormalities during treatment and can anticipate therapy failure in MDS (79, 80). A more recent study evaluated the molecular and cytogenetic profile of MDS by NGS on ctDNA and compared the results to paired bone BM DNA samples. The mutational profile on ctDNA showed 92.1% of concordance with BM, and also the variant allele frequency of ctDNA correlated with that identified in BM. NGS on ctDNA and microarrays were highly concordant in detecting chromosomal alterations, and all cytogenetic aberrations detected by NGS in BM DNA were also detected on ctDNA. These results suggest that the analysis of ctDNA is a promising strategy for performing molecular characterization and monitoring of patients with MDS (81).
Fewer data have been reported about the use of liquid biopsy in Philadelphia-negative myeloproliferative neoplasms (MPNs). One study showed that ctDNA levels in these diseases are higher than those in healthy controls. Additionally, higher ctDNA levels have been observed in primary myelofibrosis patients than in patients with essential thrombocythemia or polycythemia vera. Similar to other hematological tumors, ctDNA reflects the mutational profile identified in genomic DNA from peripheral blood granulocytes or from BM (82).
MM is characterized by the uncontrolled proliferation of atypical plasma cells (83–85). Although MM is still considered a single condition, the clinical presentation, treatment response, and survival outcomes of patients are heterogeneous and rely on specific genetic lesions, including translocations involving chromosome 14q, deletion of chromosome 17p, and amplification of chromosome 1q (86).
MM is characterized by the coexistence of heterogeneous clones in the same patient that evolve during the disease course. As in other hematological malignancies, ctDNA can detect MM-specific mutations (87–89). Similar to DLBCL, the molecular analysis on BM aspirates may not reflect the different molecular profiles of the various MM subclones (90). On these grounds, the analysis of plasma-derived ctDNA can comprehensively describe the spatial mutational heterogeneity of MM (91).
MRD is also an important prognostic marker in MM and is currently investigated by multiparametric flow cytometry (MFC) or by allele-specific oligonucleotide PCR (ASO-PCR) sequencing or by NGS to detect IgH gene rearrangements in BM aspirates (92–97). Efforts are ongoing to evaluate the potential role of ctDNA as a marker of MRD and MM (9, 98).
The study of cfDNA fragmentation patterns in liquid biopsy, also known as “fragmentomics,” and its correlation with clinical outcome has become an active area of research in recent years (99). The concept of cfDNA fragmentomics was first introduced in 2015 (100), followed by the development of several computational and experimental approaches to measure the cfDNA fragmentation patterns in plasma (101). The fragment length of cfDNA typically shows peaks of ~166 bp or multiples, which supports the evidence that apoptosis can be the main mechanism of cfDNA release. Patients with cancer present ctDNA molecules with a shorter size distribution than the background cfDNA mainly derived from hematopoietic cells (102–105). It was recently demonstrated that ctDNA fragment lengths of lymphoma patients may also vary in each individual with a correlation with disease stage. Moreover, the fragmentation patterns predict outcomes in DLBCL, therefore pointing at fragmentomics as a novel disease prognostic biomarker (44) (Figure 1).
Tumor-specific DNA methylation changes are an important regulatory mechanism of gene expression that occur early in neoplastic development (106). These changes can be potentially detected in plasma even before a clinical diagnosis of cancer (107). Epigenetic alteration-based cfDNA sequencing exploits the entire pool of cfDNA rather than limiting the analysis to somatically mutated ctDNA only (108). Furthermore, epigenetic sequencing has been considered a promising alternative considering that methylation sites are scattered and ubiquitous across the human genome (109).
In the context of hematological malignancies, abnormal methylation patterns detected in cfDNA are associated with poor outcomes in DLBCL (110, 111). Recently, aberrant changes in 5-hydroxymethylcytosine (5hmC), a unique epigenetic feature in many cancers, have been identified in plasma cfDNA as an emerging and more specific biomarker for AML diagnosis and prognosis (112–114). In addition, specific DNA methylation patterns can accurately determine the tumor type from cfDNA, thus allowing a non-invasive cancer classification (112, 115–117). Conceivably, the integration of epigenetic and mutational analyses of ctDNA molecules is a promising approach to better characterize tumors (Figure 1).
Since ctDNA load represents a powerful prognostic biomarker for treatment tailoring, the next step essential for the introduction of ctDNA analysis in clinical practice is its evaluation in prospective clinical trials. Several ongoing clinical trials incorporate ctDNA analysis to guide treatment choices (Table 1) (https://clinicaltrials.gov/).
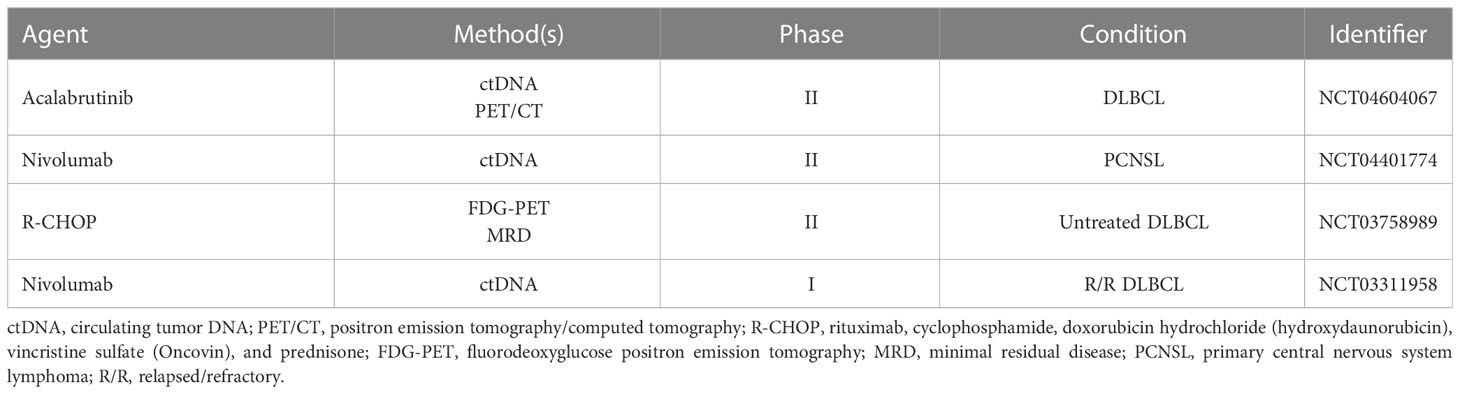
Table 1 Clinical trials using ctDNA for treatment tailoring.
The phase II SAKK (NCT04604067) trial aims to evaluate a PET/CT and ctDNA-oriented therapy in DLBCL in order to test whether the addition of acalabrutinib to R-CHOP may improve the PFS in DLBCL patients harboring the MYD88 L265P and/or CD79A/B mutations or in patients who have positive PET/CT and no molecular response (<2log10 reduction of ctDNA) after two courses of R-CHOP.
The phase II NCT04401774 trial is an open-label trial for patients who have completed first-line high-dose methotrexate-based chemotherapy for primary central nervous system lymphoma (PCNSL) but who have persistent ctDNA in their CSF after treatment despite radiologic response. The aim of this trial is to test whether nivolumab maintenance is safe and prevents or postpones overt clinical relapses.
The phase II NCT03758989 trial aims to measure the levels of ctDNA in patients with early-stage DLBCL to assess the change in ctDNA during treatment in order to prospectively identify biomarkers of treatment failure and to use ctDNA as a future tool for response-adapted therapy. Another objective of this study is to determine the correlation between FDG-PET and MRD, measured by ctDNA.
Patients with relapsed or refractory (R/R) DLBCL usually experience a dismal outcome, and the persistence of ctDNA after first-line chemoimmunotherapy represents a powerful biomarker of early relapse. In this context, the phase I NCT03311958 trial will explore the role of nivolumab. Patients positive for ctDNA after first-line chemoimmunotherapy will be treated with nivolumab for a period of 2 years to avoid complete relapse.
The analysis of ctDNA from the liquid biopsy is being increasingly investigated in hematological malignancies as a reliable approach for tumor genotyping, outcome prediction, and disease monitoring during the course of therapy. Modern technologies have allowed the integration of standard molecular profiling with liquid biopsy for biomarker detection and analysis. The analysis of ctDNA by CAPP-seq and by the highly sensitive PhasED-seq in DLBCL and HL is a consolidated technique that allows to predict outcomes at baseline and to evaluate minimal residual disease after chemoimmunotherapy, thus anticipating clinical relapses. Different interventional clinical trials that use ctDNA analysis for treatment tailoring are ongoing. However, these high-throughput technologies often provide data not easily reproducible across laboratories, and the standardization and validation of these assays will be essential before their introduction into clinical practice.
Most efforts of liquid biopsy in hematological malignancies have been carried out by the analysis of gene mutations using the CAPP-seq and PhasED-seq methods. Liquid biopsy, however, could be also exploited to analyze other clues of the disease, including copy number abnormalities, fragmentomics, and epigenetic patterns of ctDNA that, coupled to novel statistical methods and innovative machine learning approaches, may further improve the molecular characterization of hematological diseases.
Overall, ctDNA analysis on the liquid biopsy represents a step forward toward precision medicine in patients with hematological malignancies, especially in patients with DLBCL and cHL. The combination of ctDNA dynamics with PET/CT scans at interim timepoints may further improve outcome prediction and treatment tailoring during the course of therapy. Patients with no evidence of residual disease may reduce treatment intensity; conversely, patients with persistent disease may benefit from treatment intensification. Ongoing clinical trials will answer these important questions in the near future.
All authors contributed to the article and approved the submitted version.
This work was supported by the Molecular Bases of Disease Dissemination in Lymphoid Malignancies to Optimize Curative Therapeutic Strategies (5 x 1000 No. 21198), the Associazione Italiana per la Ricerca sul Cancro Foundation Milan, Italy; Progetti di Rilevante Interesse Nazionale (PRIN; 2015ZMRFEA), Rome, Italy; the AGING Project – Department of Excellence – DIMET, Università del Piemonte Orientale, Novara, Italy; and Ricerca Finalizzata 2018 (project RF-2018-12365790), MoH, Rome, Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cirillo M, Craig AFM, Borchmann S, Kurtz DM. Liquid biopsy in lymphoma: molecular methods and clinical applications. Cancer Treat Rev (2020) 91:102106. doi: 10.1016/j.ctrv.2020.102106
2. Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med (2020) 12:8. doi: 10.1186/s13073-019-0703-1
3. Chan HT, Chin YM, Low SK. Circulating tumor dna-based genomic profiling assays in adult solid tumors for precision oncology: recent advancements and future challenges. Cancers (Basel) (2022) 14(13):3275. doi: 10.3390/cancers14133275
4. Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse Large b-cell lymphoma genotyping on the liquid biopsy. Blood (2017) 129:1947–57. doi: 10.1182/blood-2016-05-719641
5. Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet (2019) 20:71–88. doi: 10.1038/s41576-018-0071-5
6. Moia R, Favini C, Ferri V, Forestieri G, Terzi Di Bergamo L, Schipani M, et al. Multiregional sequencing and circulating tumour dna analysis provide complementary approaches for comprehensive disease profiling of small lymphocytic lymphoma. Br J Haematol (2021) 195:108–12. doi: 10.1111/bjh.17718
7. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor dna. J Clin Oncol (2014) 32:579–86. doi: 10.1200/JCO.2012.45.2011
8. Li S, Noor ZS, Zeng W, Stackpole ML, Ni X, Zhou Y, et al. Sensitive detection of tumor mutations from blood and its application to immunotherapy prognosis. Nat Commun (2021) 12(1):4172. doi: 10.1038/s41467-021-24457-2
9. Colmenares R, Alvarez N, Barrio S, Martinez-Lopez J, Ayala R. The minimal residual disease using liquid biopsies in hematological malignancies. Cancers (Basel) (2022) 14(5):1310. doi: 10.3390/cancers14051310
10. Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating dna apoptosis and active dna release. Clin Chim Acta (2001) 313:139–42. doi: 10.1016/S0009-8981(01)00665-9
11. Ogawa M, Yokoyama K, Imoto S, Tojo A. Role of circulating tumor dna in hematological malignancy. Cancers (Basel) (2021) 13(9):2078. doi: 10.3390/cancers13092078
12. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant dna to assess tumor dynamics. Nat Med (2008) 14:985–90. doi: 10.1038/nm.1789
13. Rossi D, Spina V, Bruscaggin A, Gaidano G. Liquid biopsy in lymphoma. Haematologica (2019) 104:648–52. doi: 10.3324/haematol.2018.206177
14. Lee JS, Kim M, Seong MW, Kim HS, Lee YK, Kang HJ. Plasma vs. serum in circulating tumor dna measurement: characterization by dna fragment sizing and digital droplet polymerase chain reaction. Clin Chem Lab Med (2020) 58:527–32. doi: 10.1515/cclm-2019-0896
15. Page K, Powles T, Slade MJ, Mt DEB, Walker RA, Coombes RC, et al. The importance of careful blood processing in isolation of cell-free dna. Ann N Y Acad Sci (2006) 1075:313–7. doi: 10.1196/annals.1368.042
16. Kang Q, Henry NL, Paoletti C, Jiang H, Vats P, Chinnaiyan AM, et al. Comparative analysis of circulating tumor dna stability in K3edta, streck, and cellsave blood collection tubes. Clin Biochem (2016) 49:1354–60. doi: 10.1016/j.clinbiochem.2016.03.012
17. Medina Diaz I, Nocon A, Mehnert DH, Fredebohm J, Diehl F, Holtrup F. Performance of streck cfdna blood collection tubes for liquid biopsy testing. PloS One (2016) 11:E0166354. doi: 10.1371/journal.pone.0166354
18. El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free dna: preanalytical considerations. Clin Chim Acta (2013) 424:222–30. doi: 10.1016/j.cca.2013.05.022
19. Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor dna analysis in patients with cancer: American society of clinical oncology and college of American pathologists joint review. J Clin Oncol (2018) 36:1631–41. doi: 10.1200/JCO.2017.76.8671
20. Kurtz DM, Green MR, Bratman SV, Scherer F, Liu CL, Kunder CA, et al. Noninvasive monitoring of diffuse Large b-cell lymphoma by immunoglobulin high-throughput sequencing. Blood (2015) 125:3679–87. doi: 10.1182/blood-2015-03-635169
21. Roschewski M, Dunleavy K, Pittaluga S, Moorhead M, Pepin F, Kong K, et al. Circulating tumour dna and ct monitoring in patients with untreated diffuse Large b-cell lymphoma: a correlative biomarker study. Lancet Oncol (2015) 16:541–9. doi: 10.1016/S1470-2045(15)70106-3
22. Sarkozy C, Huet S, Carlton VE, Fabiani B, Delmer A, Jardin F, et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal ig-vdj sequences At diagnosis in patients with follicular lymphoma. Oncotarget (2017) 8:8765–74. doi: 10.18632/oncotarget.14448
23. Zhang W, Wang W, Han X, Gan Y, Qian L, Zhang Y, et al. Circulating tumor dna by high-throughput sequencing of T cell receptor monitored treatment response and predicted treatment failure in T cell lymphomas. Int J Lab Hematol (2021) 43:1041–9. doi: 10.1111/ijlh.13498
24. Scherer F, Kurtz DM, Diehn M, Alizadeh AA. High-throughput sequencing for noninvasive disease detection in hematologic malignancies. Blood (2017) 130:440–52. doi: 10.1182/blood-2017-03-735639
25. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor dna with broad patient coverage. Nat Med (2014) 20:548–54. doi: 10.1038/nm.3519
26. Frickhofen N, Muller E, Sandherr M, Binder T, Bangerter M, Wiest C, et al. Rearranged ig heavy chain dna is detectable in cell-free blood samples of patients with b-cell neoplasia. Blood (1997) 90:4953–60. doi: 10.1182/blood.V90.12.4953
27. Scheijen B, Meijers RWJ, Rijntjes J, Van Der Klift MY, Mobs M, Steinhilber J, et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by euroclonality-ngs. Leukemia (2019) 33:2227–40. doi: 10.1038/s41375-019-0508-7
28. Monter A, Nomdedeu JF. Clonoseq assay for the detection of lymphoid malignancies. Expert Rev Mol Diagn (2019) 19:571–8. doi: 10.1080/14737159.2019.1627877
29. Ching T, Duncan ME, Newman-Eerkes T, Mcwhorter MME, Tracy JM, Steen MS, et al. Analytical evaluation of the clonoseq assay for establishing measurable (Minimal) residual disease in acute lymphoblastic leukemia, chronic lymphocytic leukemia, and multiple myeloma. BMC Cancer (2020) 20:612. doi: 10.1186/s12885-020-07077-9
30. Frank MJ, Hossain NM, Bukhari A, Dean E, Spiegel JY, Claire GK, et al. Monitoring of circulating tumor dna improves early relapse detection after axicabtagene ciloleucel infusion in Large b-cell lymphoma: results of a prospective multi-institutional trial. J Clin Oncol (2021) 39:3034–43. doi: 10.1200/JCO.21.00377
31. Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor dna. Sci Transl Med (2016) 8:364ra155. doi: 10.1126/scitranslmed.aai8545
32. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor dna reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood (2018) 131:2413–25. doi: 10.1182/blood-2017-11-812073
33. Tilly H, Morschhauser F, Sehn LH, Friedberg JW, Trneny M, Sharman JP, et al. Polatuzumab vedotin in previously untreated diffuse Large b-cell lymphoma. N Engl J Med (2022) 386:351–63. doi: 10.1056/NEJMoa2115304
34. Turot M, Aspas Requena G. New European approval: polatuzumab vedotin associated to rituximab, cyclophosphamide, doxorubicin and prednisone in previously untreated diffuse Large b-cell lymphoma. Bull Cancer (2022) 109:1234–5. doi: 10.1016/j.bulcan.2022.08.011
35. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse Large b-cell lymphoma: results from the international scholar-1 study. Blood (2017) 130:1800–8. doi: 10.1182/blood-2017-03-769620
36. Sawalha Y. Relapsed/Refractory diffuse Large b-cell lymphoma: a look At the approved and emerging therapies. J Pers Med (2021) 11(12):1345. doi: 10.3390/jpm11121345
37. Younes A, Hilden P, Coiffier B, Hagenbeek A, Salles G, Wilson W, et al. International working group consensus response evaluation criteria in lymphoma (Recil 2017). Ann Oncol (2017) 28:1436–47. doi: 10.1093/annonc/mdx097
38. Kurtz DM, Scherer F, Jin MC, Soo J, Craig AFM, Esfahani MS, et al. Circulating tumor dna measurements as early outcome predictors in diffuse Large b-cell lymphoma. J Clin Oncol (2018) 36:2845–53. doi: 10.1200/JCO.2018.78.5246
39. Lauer EM, Mutter J, Scherer F. Circulating tumor dna in b-cell lymphoma: technical advances, clinical applications, and perspectives for translational research. Leukemia (2022) 36:2151–64. doi: 10.1038/s41375-022-01618-w
40. Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, et al. Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell (2019) 178:699–713 E19. doi: 10.1016/j.cell.2019.06.011
41. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor dna. Nat Biotechnol (2016) 34:547–55. doi: 10.1038/nbt.3520
42. Roschewski M, Rossi D, Kurtz DM, Alizadeh AA, Wilson WH. Circulating tumor dna in lymphoma: principles and future directions. Blood Cancer Discov (2022) 3:5–15. doi: 10.1158/2643-3230.BCD-21-0029
43. Kurtz DM, Soo J, Co Ting Keh L, Alig S, Chabon JJ, Sworder BJ, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor dna. Nat Biotechnol (2021) 39:1537–47. doi: 10.1038/s41587-021-00981-w
44. Meriranta L, Alkodsi A, Pasanen A, Lepisto M, Mapar P, Blaker YN, et al. Molecular features encoded in the ctdna reveal heterogeneity and predict outcome in high-risk aggressive b-cell lymphoma. Blood (2022) 139:1863–77. doi: 10.1182/blood.2021012852
45. Scherer F, Navarrete MA, Bertinetti-Lapatki C, Boehm J, Schmitt-Graeff A, Veelken H. Isotype-switched follicular lymphoma displays dissociation between activation-induced cytidine deaminase expression and somatic hypermutation. Leuk Lymphoma (2016) 57:151–60. doi: 10.3109/10428194.2015.1037758
46. Grommes C, DeAngelis LM. Primary cns lymphoma. J Clin Oncol (2017) 35:2410–8. doi: 10.1200/JCO.2017.72.7602
47. Holdhoff M, Wagner-Johnston N, Roschewski M. Systemic approach to recurrent primary cns lymphoma: perspective on current and emerging treatment strategies. Onco Targets Ther (2020) 13:8323–35. doi: 10.2147/OTT.S192379
48. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia (2022) 36:1720–48. doi: 10.1038/s41375-022-01620-2
49. Malone H, Yang J, Hershman DL, Wright JD, Bruce JN, Neugut AI. Complications following stereotactic needle biopsy of intracranial tumors. World Neurosurg (2015) 84:1084–9. doi: 10.1016/j.wneu.2015.05.025
50. Han CH, Batchelor TT. Diagnosis and management of primary central nervous system lymphoma. Cancer (2017) 123:4314–24. doi: 10.1002/cncr.30965
51. Kellermann SG, Hamisch CA, Ruess D, Blau T, Goldbrunner R, Treuer H, et al. Stereotactic biopsy in elderly patients: risk assessment and impact on treatment decision. J Neurooncol (2017) 134:303–7. doi: 10.1007/s11060-017-2522-9
52. Hiemcke-Jiwa LS, Leguit RJ, Snijders TJ, Jiwa NM, Kuiper JJW, De Weger RA, et al. Molecular analysis in liquid biopsies for diagnostics of primary central nervous system lymphoma: review of literature and future opportunities. Crit Rev Oncol Hematol (2018) 127:56–65. doi: 10.1016/j.critrevonc.2018.05.010
53. Rimelen V, Ahle G, Pencreach E, Zinniger N, Debliquis A, Zalmai L, et al. Tumor cell-free dna detection in csf for primary cns lymphoma diagnosis. Acta Neuropathol Commun (2019) 7:43. doi: 10.1186/s40478-019-0692-8
54. Bobillo S, Crespo M, Escudero L, Mayor R, Raheja P, Carpio C, et al. Cell free circulating tumor dna in cerebrospinal fluid detects and monitors central nervous system involvement of b-cell lymphomas. Haematologica (2021) 106:513–21. doi: 10.3324/haematol.2019.241208
55. Ho KG, Bale T, Grommes C, Bhatia A, Malani R. Use of circulating tumor dna to guide treatment of primary central nervous system lymphoma: a case report. Neurooncol Adv (2021) 3:Vdab143. doi: 10.1093/noajnl/vdab143
56. Mutter JA, Alig SK, Esfahani MS, Lauer EM, Mitschke J, Kurtz DM, et al. Circulating tumor dna profiling for detection, risk stratification, and classification of brain lymphomas. J Clin Oncol (2023) 41(9):1684–94. doi: 10.1200/JCO.22.00826
57. Grommes C. Circulating tumor dna in the blood: a new frontier in primary cns lymphoma? J Clin Oncol (2023) 41(9):1649–51. doi: 10.1200/JCO.22.02605
58. Fukumura K, Kawazu M, Kojima S, Ueno T, Sai E, Soda M, et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol (2016) 131:865–75. doi: 10.1007/s00401-016-1536-2
59. Nayyar N, White MD, Gill CM, Lastrapes M, Bertalan M, Kaplan A, et al. Myd88 L265p mutation and Cdkn2a loss are early mutational events in primary central nervous system diffuse Large b-cell lymphomas. Blood Adv (2019) 3:375–83. doi: 10.1182/bloodadvances.2018027672
60. Schmitz R, Stanelle J, Hansmann ML, Kuppers R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu Rev Pathol (2009) 4:151–74. doi: 10.1146/annurev.pathol.4.110807.092209
61. Desch AK, Hartung K, Botzen A, Brobeil A, Rummel M, Kurch L, et al. Genotyping circulating tumor dna of pediatric Hodgkin lymphoma. Leukemia (2020) 34:151–66. doi: 10.1038/s41375-019-0541-6
62. Camus V, Viennot M, Lequesne J, Viailly PJ, Bohers E, Bessi L, et al. Targeted genotyping of circulating tumor dna for classical Hodgkin lymphoma monitoring: a prospective study. Haematologica (2021) 106:154–62. doi: 10.3324/haematol.2019.237719
63. Oki Y, Neelapu SS, Fanale M, Kwak LW, Fayad L, Rodriguez MA, et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br J Haematol (2015) 169:689–93. doi: 10.1111/bjh.13349
64. Georgiadi EC, Sachinis N, Dimtsas G, Vassilakopoulos TP, Kittas C, Doussis-Anagnostopoulou IA. Evaluation of apoptosis in classical hodgkin’s lymphoma comparing different methods. J Buon (2012) 17:746–52.
65. Vandenberghe P, Wlodarska I, Tousseyn T, Dehaspe L, Dierickx D, Verheecke M, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-sternberg cells in early and advanced stage hodgkin’s lymphoma by sequencing of circulating cell-free dna: a technical proof-Of-Principle study. Lancet Haematol (2015) 2:E55–65. doi: 10.1016/S2352-3026(14)00039-8
66. Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major histocompatibility complex class ii and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol (2018) 36:942–50. doi: 10.1200/JCO.2017.77.3994
67. Gallamini A, Hutchings M, Rigacci L, Specht L, Merli F, Hansen M, et al. Early interim 2-[18f]Fluoro-2-Deoxy-D-Glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol (2007) 25:3746–52. doi: 10.1200/JCO.2007.11.6525
68. Zaucha JM, Chauvie S, Zaucha R, Biggii A, Gallamini A. The role of Pet/Ct in the modern treatment of Hodgkin lymphoma. Cancer Treat Rev (2019) 77:44–56. doi: 10.1016/j.ctrv.2019.06.002
69. Cottereau AS, Versari A, Loft A, Casasnovas O, Bellei M, Ricci R, et al. Prognostic value of baseline metabolic tumor volume in early-stage Hodgkin lymphoma in the standard arm of the H10 trial. Blood (2018) 131:1456–63. doi: 10.1182/blood-2017-07-795476
70. Guo B, Tan X, Ke Q, Cen H. Prognostic value of baseline metabolic tumor volume and total lesion glycolysis in patients with lymphoma: a meta-analysis. PloS One (2019) 14:E0210224. doi: 10.1371/journal.pone.0210224
71. Pinochet P, Texte E, Stamatoullas-Bastard A, Vera P, Mihailescu SD, Becker S. Prognostic value of baseline metabolic tumour volume in advanced-stage hodgkin’s lymphoma. Sci Rep (2021) 11:23195. doi: 10.1038/s41598-021-02734-w
72. Terasawa T, Lau J, Bardet S, Couturier O, Hotta T, Hutchings M, et al. Fluorine-18-Fluorodeoxyglucose positron emission tomography for interim response assessment of advanced-stage hodgkin’s lymphoma and diffuse Large b-cell lymphoma: a systematic review. J Clin Oncol (2009) 27:1906–14. doi: 10.1200/JCO.2008.16.0861
73. Abdulmawjood B, Roma-Rodrigues C, Fernandes AR, Baptista PV. Liquid biopsies in myeloid malignancies. Cancer Drug Resist (2019) 2:1044–61. doi: 10.20517/cdr.2019.88
74. Rogers A, Joe Y, Manshouri T, Dey A, Jilani I, Giles F, et al. Relative increase in leukemia-specific dna in peripheral blood plasma from patients with acute myeloid leukemia and myelodysplasia. Blood (2004) 103:2799–801. doi: 10.1182/blood-2003-06-1840
75. Quan J, Gao YJ, Yang ZL, Chen H, Xian JR, Zhang SS, et al. Quantitative detection of circulating nucleophosmin mutations dna in the plasma of patients with acute myeloid leukemia. Int J Med Sci (2015) 12:17–22. doi: 10.7150/ijms.10144
76. Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C, Stroun M. Point mutations of the n-ras gene in the blood plasma dna of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol (1994) 86:774–9. doi: 10.1111/j.1365-2141.1994.tb04828.x
77. Short NJ, Patel KP, Albitar M, Franquiz M, Luthra R, Kanagal-Shamanna R, et al. Targeted next-generation sequencing of circulating cell-free dna vs bone marrow in patients with acute myeloid leukemia. Blood Adv (2020) 4:1670–7. doi: 10.1182/bloodadvances.2019001156
78. Nakamura S, Yokoyama K, Shimizu E, Yusa N, Kondoh K, Ogawa M, et al. Prognostic impact of circulating tumor dna status post-allogeneic hematopoietic stem cell transplantation in aml and mds. Blood (2019) 133:2682–95. doi: 10.1182/blood-2018-10-880690
79. Suzuki Y, Tomita A, Nakamura F, Iriyama C, Shirahata-Adachi M, Shimada K, et al. Peripheral blood cell-free dna is an alternative tumor dna source reflecting disease status in myelodysplastic syndromes. Cancer Sci (2016) 107:1329–37. doi: 10.1111/cas.12994
80. Yeh P, Dickinson M, Ftouni S, Hunter T, Sinha D, Wong SQ, et al. Molecular disease monitoring using circulating tumor dna in myelodysplastic syndromes. Blood (2017) 129:1685–90. doi: 10.1182/blood-2016-09-740308
81. Garcia-Gisbert N, Garcia-Avila S, Merchan B, Salido M, Fernandez-Rodriguez C, Gibert J, et al. Molecular and cytogenetic characterization of myelodysplastic syndromes in cell-free dna. Blood Adv (2022) 6:3178–88. doi: 10.1182/bloodadvances.2021006565
82. Garcia-Gisbert N, Fernandez-Ibarrondo L, Fernandez-Rodriguez C, Gibert J, Andrade-Campos M, Arenillas L, et al. Circulating cell-free dna improves the molecular characterisation of ph-negative myeloproliferative neoplasms. Br J Haematol (2021) 192:300–9. doi: 10.1111/bjh.17087
83. Gupta N, Sharma A, Sharma A. Emerging biomarkers in multiple myeloma: a review. Clin Chim Acta (2020) 503:45–53. doi: 10.1016/j.cca.2019.12.026
84. Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, et al. Diagnosis and management of multiple myeloma: a review. Jama (2022) 327:464–77. doi: 10.1001/jama.2022.0003
85. Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol (2022) 97:1086–107. doi: 10.1002/ajh.26590
86. Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol (2018) 15:409–21. doi: 10.1038/s41571-018-0018-y
87. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun (2014) 5:2997. doi: 10.1038/ncomms3997
88. Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia (2014) 28:384–90. doi: 10.1038/leu.2013.199
89. Rustad EH, Coward E, Skytoen ER, Misund K, Holien T, Standal T, et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica (2017) 102:1266–72. doi: 10.3324/haematol.2016.160564
90. Bahlis NJ. Darwinian Evolution and tiding clones in multiple myeloma. Blood (2012) 120:927–8. doi: 10.1182/blood-2012-06-430645
91. Mithraprabhu S, Khong T, Ramachandran M, Chow A, Klarica D, Mai L, et al. Circulating tumour dna analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia (2017) 31:1695–705. doi: 10.1038/leu.2016.366
92. Sata H, Shibayama H, Maeda I, Habuchi Y, Nakatani E, Fukushima K, et al. Quantitative polymerase chain reaction analysis with allele-specific oligonucleotide primers for individual igh vdj regions to evaluate tumor burden in myeloma patients. Exp Hematol (2015) 43:374–381 E2. doi: 10.1016/j.exphem.2015.01.002
93. Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International myeloma working group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol (2016) 17:E328–46. doi: 10.1016/S1470-2045(16)30206-6
94. Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, Garcia-Sanchez O, Bottcher S, et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia (2017) 31:2094–103. doi: 10.1038/leu.2017.29
95. Kubaczkova V, Vrabel D, Sedlarikova L, Besse L, Sevcikova S. Cell-free dna - minimally invasive marker of hematological malignancies. Eur J Haematol (2017) 99:291–9. doi: 10.1111/ejh.12925
96. Oberle A, Brandt A, Voigtlaender M, Thiele B, Radloff J, Schulenkorf A, et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma dna. Haematologica (2017) 102:1105–11. doi: 10.3324/haematol.2016.161414
97. Wong SW, Shah N, Ledergor G, Martin T, Wolf J, Shui AM, et al. Early dynamics and depth of response in multiple myeloma patients treated with bcma car-T cells. Front Oncol (2021) 11:783703. doi: 10.3389/fonc.2021.783703
98. Pugh TJ. Circulating tumour dna for detecting minimal residual disease in multiple myeloma. Semin Hematol (2018) 55:38–40. doi: 10.1053/j.seminhematol.2018.03.002
99. Ding SC, Lo YMD. Cell-free dna fragmentomics in liquid biopsy. Diagn (Basel) (2022) 12(4):978. doi: 10.3390/diagnostics12040978
100. Ivanov M, Baranova A, Butler T, Spellman P, Mileyko V. Non-random fragmentation patterns in circulating cell-free dna reflect epigenetic regulation. BMC Genomics (2015) 16 Suppl 13:S1. doi: 10.1186/1471-2164-16-S13-S1
101. Liu Y. At The dawn: cell-free dna fragmentomics and gene regulation. Br J Cancer (2022) 126:379–90. doi: 10.1038/s41416-021-01635-z
102. Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma dna in hepatocellular carcinoma patients. Proc Natl Acad Sci USA (2015) 112:E1317–25. doi: 10.1073/pnas.1500076112
103. Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. Fragment length of circulating tumor dna. PloS Genet (2016) 12:E1006162. doi: 10.1371/journal.pgen.1006162
104. Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor dna by fragment size analysis. Sci Transl Med (2018) 10(466):eaat4921. doi: 10.1126/scitranslmed.aat4921
105. Underhill HR. Leveraging the fragment length of circulating tumour dna to improve molecular profiling of solid tumour malignancies with next-generation sequencing: a pathway to advanced non-invasive diagnostics in precision oncology? Mol Diagn Ther (2021) 25:389–408. doi: 10.1007/s40291-021-00534-6
106. Dor Y, Cedar H. Principles of dna methylation and their implications for biology and medicine. Lancet (2018) 392:777–86. doi: 10.1016/S0140-6736(18)31268-6
107. Chen X, Gole J, Gore A, He Q, Lu M, Min J, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun (2020) 11:3475. doi: 10.1038/s41467-020-17316-z
108. van der Pol Y, Mouliere F. Toward the early detection of cancer by decoding the epigenetic and environmental fingerprints of cell-free dna. Cancer Cell (2019) 36:350–68. doi: 10.1016/j.ccell.2019.09.003
109. Gao Q, Zeng Q, Wang Z, Li C, Xu Y, Cui P, et al. Circulating cell-free dna for cancer early detection. Innovation (Camb) (2022) 3:100259. doi: 10.1016/j.xinn.2022.100259
110. Kristensen LS, Hansen JW, Kristensen SS, Tholstrup D, Harslof LB, Pedersen OB, et al. Aberrant methylation of cell-free circulating dna in plasma predicts poor outcome in diffuse Large b cell lymphoma. Clin Epigenet (2016) 8:95. doi: 10.1186/s13148-016-0261-y
111. Wedge E, Hansen JW, Garde C, Asmar F, Tholstrup D, Kristensen SS, et al. Global hypomethylation is an independent prognostic factor in diffuse Large b cell lymphoma. Am J Hematol (2017) 92:689–94. doi: 10.1002/ajh.24751
112. Li W, Zhang X, Lu X, You L, Song Y, Luo Z, et al. 5-hydroxymethylcytosine signatures in circulating cell-free dna as diagnostic biomarkers for human cancers. Cell Res (2017) 27:1243–57. doi: 10.1038/cr.2017.121
113. Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, et al. 5-hydroxymethylcytosine signatures in cell-free dna provide information about tumor types and stages. Cell Res (2017) 27:1231–42. doi: 10.1038/cr.2017.106
114. Shao J, Wang S, West-Szymanski D, Karpus J, Shah S, Ganguly S, et al. Cell-free dna 5-hydroxymethylcytosine is an emerging marker of acute myeloid leukemia. Sci Rep (2022) 12:12410. doi: 10.1038/s41598-022-16685-3
115. Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma dna bisulfite sequencing. Proc Natl Acad Sci USA (2013) 110:18761–8. doi: 10.1073/pnas.1313995110
116. Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma dna tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA (2015) 112:E5503–12. doi: 10.1073/pnas.1508736112
Keywords: liquid biopsy, DLBCL, Hodgkin lymphoma, myeloid neoplasia, precision medicine
Citation: Talotta D, Almasri M, Cosentino C, Gaidano G and Moia R (2023) Liquid biopsy in hematological malignancies: current and future applications. Front. Oncol. 13:1164517. doi: 10.3389/fonc.2023.1164517
Received: 12 February 2023; Accepted: 03 April 2023;
Published: 20 April 2023.
Edited by:
Ioannis Ntanasis-Stathopoulos, National and Kapodistrian University of Athens Medical School, GreeceReviewed by:
Panagiotis Malandrakis, National and Kapodistrian University of Athens, GreeceCopyright © 2023 Talotta, Almasri, Cosentino, Gaidano and Moia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Moia, cmljY2FyZG8ubW9pYUB1bml1cG8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.