 Laura Patras
Laura Patras Doru Paul
Doru Paul Irina R. Matei
Irina R. Matei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 07 June 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1163786
This article is part of the Research TopicCellular and Molecular Characteristics of the Pre-metastatic NicheView all 6 articles
The discovery that primary tumors condition distant organ sites of future metastasis for seeding by disseminating tumor cells through a process described as the pre-metastatic niche (PMN) formation revolutionized our understanding of cancer progression and opened new avenues for therapeutic interventions. Given the inherent inefficiency of metastasis, PMN generation is crucial to ensure the survival of rare tumor cells in the otherwise hostile environments of metastatic organs. Early on, it was recognized that preparing the “soil” of the distal organ to support the outgrowth of metastatic cells is the initiating event in PMN development, achieved through the remodeling of the organ’s extracellular matrix (ECM). Remote restructuring of ECM at future sites of metastasis under the influence of primary tumor-secreted factors is an iterative process orchestrated through the crosstalk between resident stromal cells, such as fibroblasts, epithelial and endothelial cells, and recruited innate immune cells. In this review, we will explore the ECM changes, cellular effectors, and the mechanisms of ECM remodeling throughout PMN progression, as well as its impact on shaping the PMN and ultimately promoting metastasis. Moreover, we highlight the clinical and translational implications of PMN ECM changes and opportunities for therapeutically targeting the ECM to hinder PMN formation.
The extracellular matrix (ECM) is a complex three-dimensional extracellular network surrounding and supporting all cells and tissues in the body. Although fundamentally composed of water, proteins, and polysaccharides, the ECM is highly heterogeneous, with a tissue-specific physical, topological, and biochemical composition generated during tissue development through dynamic bidirectional interactions between cells (epithelial, fibroblast, adipocyte, and endothelial) and their microenvironment (1). The ECM provides a structural foundation for cells within a tissue and ensures mechanical integrity. The molecular cues provided by the ECM modulate intercellular interactions, cell behavior, vascular development, and immune function, ultimately orchestrating tissue development, phenotype, and homeostasis (1–3).
The core matrisome in mammals comprises around 300 proteins with specific functions, mainly macromolecules such as proteoglycans (PGs) (compressibility), fibrous proteins such as collagens (tensile strength), elastins (elasticity), fibronectins (FNs) (stiffness), glycoproteins and laminins (cell adhesion) (1, 4–6), and ECM-associated proteins known as matricellular proteins (e.g., tenascin C, thrombospondin-1 (TSP-1), periostin (POSTN), osteopontin, and osteonectin, also known as secreted protein acidic and rich in cysteine (SPARC)) (7–10), without a primary structural role (reviewed in 3). ECM constituents, PGs, glycosaminoglycans (GAGs), and fibrous proteins assemble into solid gel scaffolds with distinct biochemical and biomechanical properties, which are dynamically remodeled and tailored depending on the specific physiological structure and functions of the tissue (e.g., bone loose connective tissue and blood plasma) (5, 11, 12). Tissue-specific differences in ECM organization and structure (13, 14) are ensured by variations in ECM composition, isoform expression (15, 16), alternative splicing (17), and post-translational modifications (PTMs) such as glycosylation (13, 14) that shape the functional outcome of ECM–cellular interactions.
ECM architecture consists of two distinct but interconnected structures: the basement membrane (BM), a condensed thin matrix lining the basal side of epithelial and endothelial tissues (18, 19), and the interstitial matrix (IM), an interlaced porous structure characteristic of interstitial connective tissues, which is the preponderant ECM (20, 21) (Table 1). The BM consists primarily of collagen IV and laminins, which confer resistance to tensile force, as well as entactin/nidogen and heparan sulfate (HS) proteoglycans (31, 70–72). Critical for vascular function, the BM regulates endothelial barrier function and cellular transmigration across the endothelium, while modular PGs such as perlecan, agrin, and collagen type XVIII (endostatin precursor) regulate angiogenesis (73–75). Moreover, the BM enforces tissue compartmentalization while conveying extracellular microenvironment messages to epithelial cells or endothelial cells and pericytes in blood vessels (14, 76). The IM is primarily assembled from collagens (e.g., type I and III), which, together with fibronectin, provide a structural foundation for tissue function and mechanical integrity, protecting the tissue by acting as a compression buffer against mechanical stress (77). Tenascin and anionic proteoglycans maintain tissue hydration and the pH of the local microenvironment (1, 5, 11, 17), while physiological crosslinking of the matrix maintains its integrity and provides resistance to proteolytic degradation (13, 54).
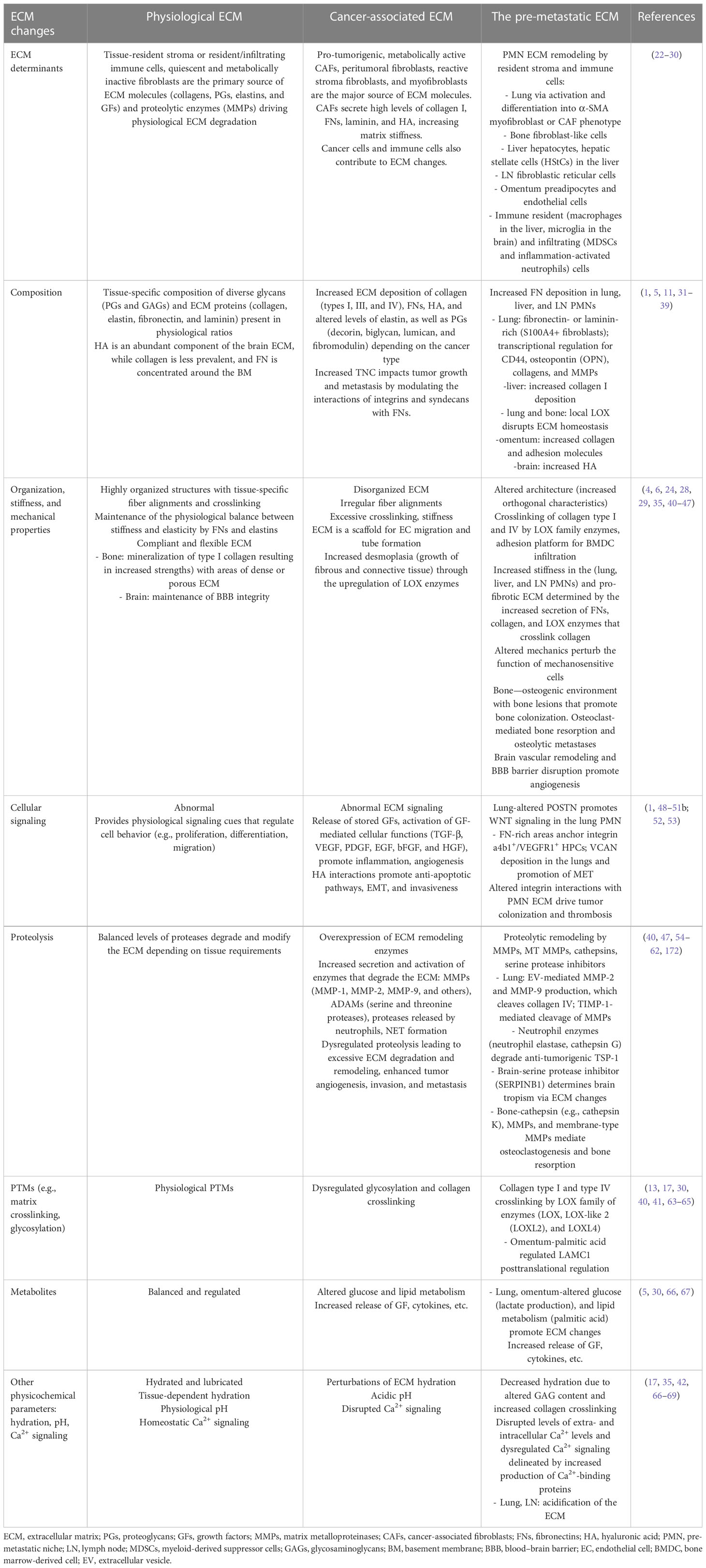
Table 1 ECM evolution during cancer progression.
The ECM stores and binds growth factors (GFs), cytokines, and extracellular vesicles (EVs), regulating their availability to cells depending on the physiological state of the tissue (5). The binding or trafficking of EVs, nanosized (50–150) membranous structures conveying bioactive cargo (proteins, lipids, metabolites, and nucleic acids) (78) throughout the ECM facilitates intercellular communication in the microenvironment (reviewed in 79, 80). A particular subtype of EVs, matrix-bound nanovesicles (MBVs), form an integral functional component of the connective tissue ECM by activating macrophages and supporting differentiation in neuroblastoma (81).
Communication between cells and their surrounding ECM occurs through focal adhesions and is mediated by ECM-binding receptors. Among these, integrins are the most prominent receptor family and are instrumental for the adhesion and migration of immune cells, thus facilitating systemic immune responses. Integrins bind ECM components such as fibronectin, laminin, collagen, and fibrinogen, as well as other integrins on the surface of cells (82–85). Non-integrin ECM receptors include glycoproteins such as syndecans, discoidin domain receptors, and CD44 (86–90). Importantly, by binding GFs and interacting with cell-surface receptors, the ECM elicits signal transduction and regulates gene transcription (1), thereby actively regulating cell growth, survival, differentiation, and migration. Matricellular proteins, such as TNC, TSP-1, and POSTN, alter the interactions between cellular transmembrane receptors and fibrous ECM molecules, thereby modulating cell proliferation and differentiation and playing a central role in tissue homeostasis (3). For example, hyaluronic acid (HA), also known as hyaluronan, is an anionic non-sulfated GAG synthesized by one of the three hyaluronan synthetases (HAS1, HAS2, and HAS3) (91) and a dominant constituent of the connective, epithelial, and neural ECM (32) essential for embryonic development, wound healing, and inflammation (91, 92).
Fibroblasts are the primary cellular components that “build” the connective tissue ECM and the major type of mechanoresponsive cells. In addition to being the primary source of most ECM components including collagens, PGs, elastin, growth factors [e.g., transforming growth factor beta (TGF-β) and platelet-derived growth factor (PDGF)] and cytokines [e.g., tumor necrosis factor-alpha (TNF-α) and interleukin 1 beta (IL-1β)], fibroblasts also secrete matrix metalloproteinases (MMPs), thereby emerging as the primary players in regulating tissue growth, ECM remodeling, wound healing, and fibrosis (22, 23). By producing both fibronectin and elastin, fibroblasts control the balance between stiffness and elasticity of the ECM, which in turn orchestrates cell migration, gene expression, and differentiation, as cells can sense ECM rigidity.
In cancer, quiescent and metabolically inactive fibroblasts are distinct from cancer-associated fibroblasts (CAFs), ubiquitous cells present in all solid tumors that become the major source of ECM molecules. CAFs develop from various cell types (e.g., stellate cells, bone marrow-derived cells (BMDCs), adipose tissue mesenchymal stromal cells (MSCs), and resident quiescent fibroblasts) and secrete higher levels of collagen I, FNs, laminin, and HA than normal fibroblasts, increasing ECM stiffness (24–26). FNs, laminin, and HA are also critical extracellular drivers of malignancy (6, 11, 63, 93) and reviewed in Frantz et al. (1) and LeBleu et al. (19).
Dysregulation of ECM homeostasis consists of alterations (e.g., ECM degradation and ECM deposition) in the dynamic ECM composition, organization, and mechanical properties that contribute to neoplastic progression and is a prerequisite for metastatic disease (3, 94). Typical changes in cancer-associated ECM were comprehensively reviewed by Frantz et al. (1). These include i) alterations in BM function with a loss of apical–basal polarity, along with cellular proliferation and metastatic cell migration; ii) contributions of differentiated fibroblasts (peritumoral fibroblasts, reactive stroma fibroblasts, CAFs, and myofibroblasts) to increased ECM deposition of collagen (types I, III, and IV), FNs, and elastin, as well as PGs (decorin, biglycan, lumican, and fibromodulin); iii) increased levels of TGF-β, VEGF, PDGF, EGF, bFGF, and HGF, promoting vessel growth and inflammation (1) (Figure 1).
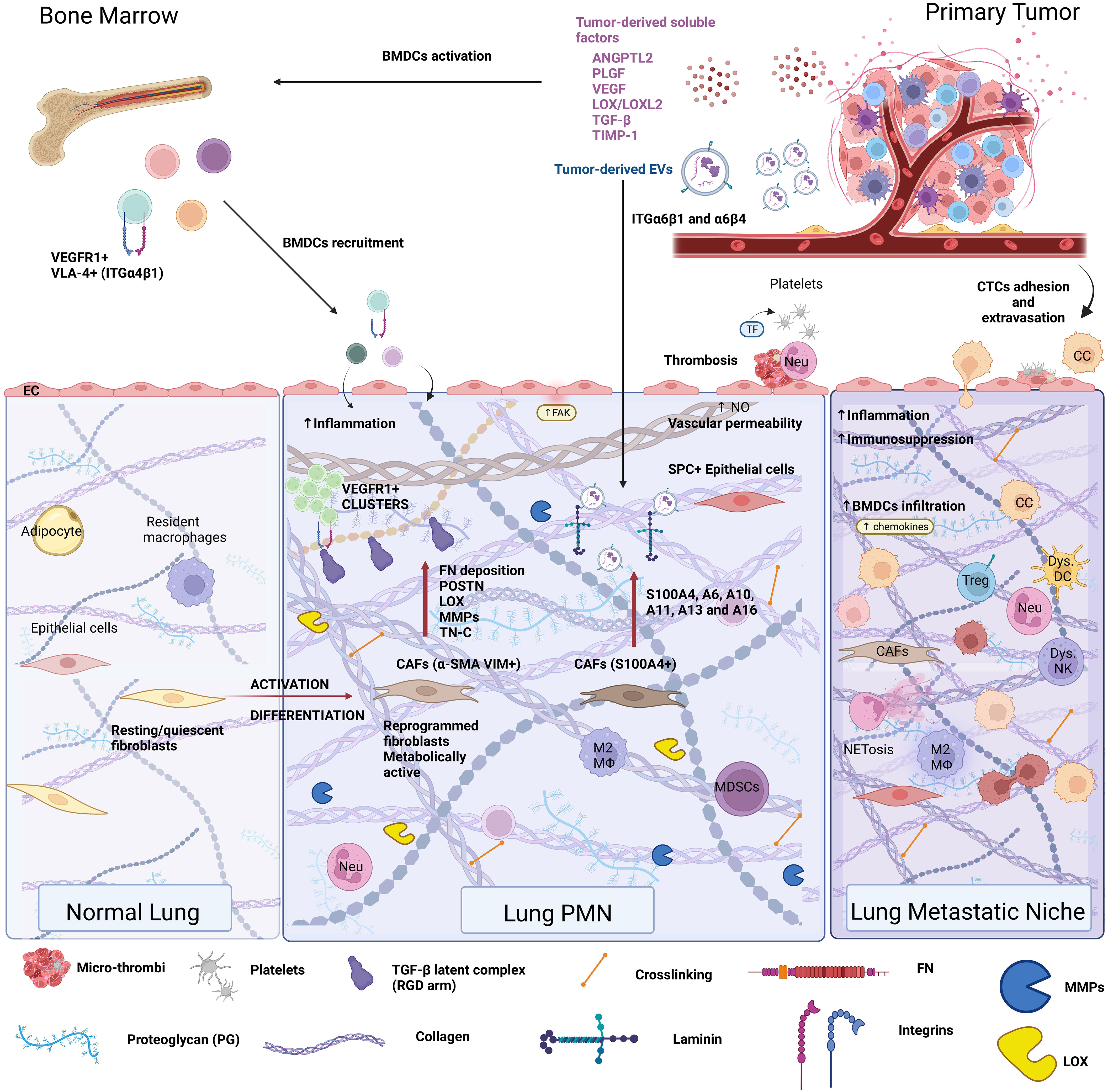
Figure 1 Extracellular matrix composition in the pre-metastatic niche. The remote conditioning of future organ sites of metastasis by the primary tumor is termed pre-metastatic niche formation. The lung pre-metastatic niche is depicted here, as it has been the most extensively characterized. Primary tumors secrete soluble factors, such as cytokines, chemokines, hormones, metabolites, and extracellular vesicles (EVs) that condition the future site of metastasis as well as the bone marrow. Bone marrow conditioning results in egress of VEGFR1+VLA-4+ bone marrow-derived progenitor cells, which are recruited to the pre-metastatic niche. Conditioning of the pre-metastatic niche induces vascular permeability, thrombosis, inflammation, and stromal cell activation, all of which cooperate in extracellular matrix remodeling. Pre-metastatic niche development generates a favorable milieu for circulating tumor cell (CTC) seeding and outgrowth, resulting in the formation of the metastatic niche.
Cancer-associated ECM alterations also include the loss of a differentiated phenotype. Interestingly, embryonic forms of FNs (i.e., oncofetal FNs) are re-expressed in cancer along with glycosylation dysregulation (95). Therefore, changes that perturb ECM homeostasis such as inflammation associated with wound healing, aging, obesity, or cancer include increased secretion and activation of enzymes that degrade the ECM (MMPs, serine, and threonine proteases), release stored GFs, and activate GF-mediated cellular functions. To this end, MMPs are essential in regulating the complex interplay between cells and the ECM (13).
Vascular BM components are also involved in the regulation of tumor angiogenesis (14, 74, 96). Matrix degradation promotes malignant progression and metastasis (97), as cancer cells strictly depend on ECM degradation for migration and intravasation and, thus, to escape their original environment. Moreover, the metastatic potential of most tumors correlates with the enzymatic degradation of BM collagen (98). Tumor microenvironment (TME) hypoxia drives ECM remodeling and collagen crosslinking to stiffen the tissue stroma during metastatic progression, promoting transformation, cancer cell survival, and outgrowth, motility, and invasion, ultimately enabling metastatic dissemination and facilitating the establishment of tumor cells at distant sites (40). Hypoxic tumors secrete LOX family enzymes that initiate ECM degradation and epithelial-to-mesenchymal transition (EMT), altering tumor cell behavior (40). Reducing LOX-mediated collagen crosslinking was sufficient to prevent tissue fibrosis and to increase ECM stiffness, focal adhesions, GF signaling, and cancer invasiveness in murine models of cancer (64). Changes associated with desmoplasia (i.e., growth of fibrous and connective tissue) are a common occurrence in tumors (e.g., pancreatic cancer (99)) and confer a fibrotic state due to increased ECM deposition and enhanced PTMs of ECM proteins (100). As a result, the tumor ECM has altered biomechanical properties due to crosslinking and orientation and has increased stiffness. Importantly, cancer cell interactions with a stiffer ECM provide them with a proliferative advantage (2) and instigate cancer progression (101).
Many matricellular proteins are increased in adult ECM when normal physiology is altered. Increased HA levels are observed in many tumor types and correlate with malignancy and metastasis (91, 102). HA specifically interacts with cell surface receptors CD44 and CD168 (also known as Receptor for Hyaluronan Mediated Motility) and can be retained on the cell surface after its synthesis. HA presence on the cell surface generates a voluminous pericellular matrix, which can further incorporate other HA-binding molecules; however, these interactions are not completely understood (91). In cancer, tumor cell–HA interactions influence intracellular signaling such as anti-apoptotic pathways and promote EMT and cell invasiveness (52, 91). POSTN also has a multifaceted role in tumor-associated ECM remodeling (103). In addition, tenascin C promotes glioma cell migration on fibronectin substrates by modulating interactions between αvβ5 integrin and FNs (3, 104). Tenascin C levels are elevated in the cancer-associated stroma, and they can inhibit the interaction between syndecan 4 and FNs, thus promoting tumor growth and metastasis (33).
Altogether, cancer-induced changes in tissue homeostasis alter ECM composition and physicochemical properties through ECM remodeling, ultimately leading to changes in ECM function.
Of the ECM–stroma interactions, perhaps the most critical is the crosstalk with the vasculature, as ECM-mediated regulation of angiogenesis and vasculogenesis is essential for tumor growth, progression, and metastasis. Cancer neoangiogenesis is a complex process regulated through the intricate crosstalk between cancer cells, endothelial cells (ECs), and the ECM that dictates the potential of the tumor to metastasize (105). Since tumor growth is accompanied by hypoxia and limitations in nutrient availability, tumor vascularization, via angiogenesis and vasculogenesis, is a prerequisite for further tumor growth, escape of tumor cells from the primary site, and ultimately metastasis. Primary tumors depend on the VEGF-dependent recruitment of bone marrow-derived precursors (e.g., VEGFR1+ hematopoietic and VEGFR2+ circulating endothelial progenitor cells (EPCs) as a source of ECs) that initiate neovascularization (106) in response to specific TME ECM changes and the chemo-cytokine tumor- and tumor-associated stroma repertoire. For example, CAFs in the tumor stroma secrete stromal cell-derived factor (SDF-1)/CXCL12 and recruit CXCR4+ EPCs, thereby promoting new vessel formation (107). Importantly, the balance between positive and negative regulators of blood vessel formation directs a switch toward an angiogenic milieu in the TME (i.e., “angiogenic switch”), further supporting primary tumor growth and metastasis.
ECM-driven angiogenesis in cancer occurs through several mechanisms: i) the ECM provides a scaffold for EC migration and tube formation; ii) MMP-mediated ECM and BM degradation are required for tumor cell migration and as sources of pro-angiogenic growth factors (e.g., VEGF and FGF) (108); iii) ECM molecules such as FNs (109), HA (93, 108), TSP, angiostatin, and endostatin (73, 108, 110) directly regulate tumor angiogenesis. These mechanisms act in concert.
Matrix-degrading enzymes (MMPs), secreted by cancer cells or TME stroma (CAFs, adipocytes, and immune cells), are critical for neovascularization. MMP-1 secreted by breast cancer cells promotes angiogenesis and bone metastasis (111), while MMP-9 released by tumor-infiltrating neutrophils and macrophages triggers the angiogenic switch by releasing VEGF from tumor matrix to sustain angiogenesis-driven intravasation and metastasis (104, 112, 113). Moreover, vascular cell-derived MMP-9 induces pericyte detachment from vessels undergoing angiogenesis, destabilizing them and promoting vascular leakiness (114, 115). MMP-2, in turn, promotes angiogenesis by cleaving collagen IV and exposing a cryptic αVβ3 integrin binding site (55). MMP-2 and A disintegrin and metalloprotease 10 (ADAM10) also regulate vascular integrity and angiogenesis by degrading adhesion proteins at endothelial junctions, including VE-cadherin (56, 116). Interestingly, MMPs can also inhibit angiogenesis by generating endogenous angiogenesis inhibitors (e.g., cleavage of collagen chains and plasminogen) or by disrupting ligand–receptor interactions (117–120).
Judah Folkman pioneered the research into ECM components that inhibit angiogenesis and tumor growth, angiostatin, and endostatin (73, 110). Endostatin, a C-terminal cleavage product of collagen XVIII, a BM component, generated by MMP-3, MMP-7, MMP-9, MMP-13, MMP-20, or cathepsins (73, 118, 119, 121), interacts with ECs via integrins, glypicans, or VEGFR-2 on their surface (122), inhibiting VEGF and bFGF-induced EC migration and inducing apoptosis (123). Angiostatin, the most potent anti-angiogenic factor, results from proteolytic cleavage by extracellular urokinase, plasminogen activators, MMP-3, MMP-7, MMP-9, and MMP-12 (117, 124–126), and is systemically released by primary tumors, suppressing primary tumor growth and metastasis, explaining why primary tumor removal can lead to outgrowth of occult, pre-existing micro-metastases (73, 127).
Another endogenous matricellular inhibitor of angiogenesis is TSP-1, a calcium-binding glycoprotein belonging to the thrombospondin family that exerts its anti-tumorigenic activity on both tumor and endothelial cells (128, 129), reducing VEGF expression (130, 131) and disrupting VEGF signaling (132). However, TSP-1 also mediates the invasiveness and migratory capacity of several cancers such as melanoma, thyroid, and prostate (133–136) by binding both ECM (FNs, fibrinogen, laminin, and collagen IV) as well as integrins. Indeed, loss of TSP-1 is regarded as an “angiogenic switch” that could drive tumor progression (137). Thus, the TSP-1 role in mediating ECM-dependent tumor progression and angiogenesis is likely highly context and microenvironment dependent.
Integrins, receptors for ECM such as collagen on smooth muscle (α1β1) or epithelial cells and platelets (α2β1), also modulate angiogenesis. For example, integrin α1β1, the major collagen IV receptor, activates endothelial cell signaling pathways leading to proliferation, adhesion, migration, and tubulogenesis (138), while also promoting fibroblast proliferation and inhibiting the synthesis of collagen and MMPs. Integrin α1-deficiency markedly decreased tumor vascularization and increased angiostatin levels in plasma via MMP-7 and MMP-9 (139). FNs, the α5β1 integrin ligand on ECs, are also involved in promoting angiogenesis in a VEGF-independent manner and blocking α5β1 integrin with antibodies, peptides, or antagonists promoting tumor regression (109).
Integrin-mediated signaling is augmented by HA, which is highly produced by tumor or stromal cells (102, 140). HA critically regulates endothelial barrier function, and its processing profoundly impacts cancer-associated vascularization (141, 142). Moreover, HA stimulates EC tube formation via CD44 and CD168/TGFβ Receptor I signaling, which promote plasminogen activator-inhibitor-1 (PAI-1) expression and subsequently ICAM-1, VCAM-1, and MMP-2 expression, thereby promoting tumor angiogenesis (93, 143, 144).
Taken together, these findings support the idea that tumor-associated ECM alterations provide pivotal biochemical and biomechanical cues to cancer and stromal cells in the tumor microenvironment, emerging as critical modulators of cancer hallmarks and virtually supporting every malignant characteristic of tumor progression, such as sustained proliferation, growth, angiogenesis and evasion from apoptosis or antitumor immunity, chronic inflammation, dysregulation of cellular energetics, and acquisition of invasiveness potential (2).
Ever since the discovery of the pre-metastatic niche (PMN) (40, 48, 145), ECM changes have been recognized as a hallmark of PMN formation. ECM remodeling is a prerequisite for PMN initiation by preconditioning the niche, “weaving the nest”, to create a favorable environment for metastatic cell seeding (146, 147). The primary tumor induces ECM deposition at distant sites through its secretome (soluble factors such as cytokines, chemokines, growth factors, and EVs) with angiogenic, inflammatory, chemoattractant, and enzymatic roles that initiate PMN formation (Table 1 and Figure 1).
ECM changes at the pre-metastatic site resemble alterations associated with aging, or primary tumor progression, such as increased fibronectin secretion (27, 48, 148) and stiffness and result from activation and differentiation of stromal cell phenotypes. Specifically, senescent fibroblasts in aging, or pro-tumor fibroblast differentiation in cancer, deposit more fibronectin. PMN-specific changes consist of ECM deposition (mainly mediated by resident-activated stroma/immune cells or infiltrating immune cells) and ECM degradation that altogether remodel the architecture of the distant tissue microenvironment (Figure 1).
Upregulation of FNs and increased FN deposition by activated resident fibroblasts in the lung and resident macrophages in the liver PMN have been described in murine and human models of melanoma (34, 48) and pancreatic cancer (27, 148), as well as in uninvolved lymph nodes of breast cancer patients (149). Fibronectin-rich pre-metastatic organs (lung, liver, testis, spleen, and kidney) are hospitable niches for the recruitment and adhesion of VEGFR1+VLA-4+ BMDCs in murine melanoma and lung cancer models and also in human breast and gastrointestinal cancers, highlighting the clinical relevance of this finding (48). Pancreatic CAFs (α-SMA+) support liver ECM remodeling via pro-inflammatory IL-6 secretion and induction of IL-6/JAK1/STAT3 signaling in hepatocytes that in turn promote the production of FNs and collagen I in the liver (65).
Another matricellular protein known for its various pro-tumorigenic roles in solid cancers (150) and with an emerging role in pre-conditioning the PMN is POSTN. For example, primary tumor-derived TGF-β induces the expression and secretion of POSTN by stromal fibroblasts (α-SMA+VIM+) in the pre-metastatic lung of the MMTV-PyMT breast cancer mouse model, sustaining infiltrating metastasis-initiating cells by promoting WNT signaling (151). POSTN also interacts with type I collagen, fibronectin, NOTCH1, TNC, and BMP1 and engages with αvβ3 and αvβ5 integrins, promoting cell motility throughout the tissue parenchyma (152). Importantly, POSTN is involved in tissue repair and upregulated in damaged tissues, where it also contributes to creating a PMN-like microenvironment during tissue regeneration (152).
Among PGs, versican (VCAN) is key to ECM remodeling at the distant future sites of metastasis, with various functions depending on the cellular source (49, 153). For example, in lung cancer PMN, tumor-derived VCAN activated macrophages, generating an inflammatory environment (153), whereas, in the MMTV-PyMT model, the BMDC-derived VCAN was pivotal for lung PMN development (49).
Primary tumor angiogenesis is closely related to PMN formation, as it entails the chronic release of PMN-inducing factors, such as VEGF, SDF-1, TGF-β, and TNF-α (154), that ultimately reconfigure future metastatic sites to create a permissive niche for cancer cell extravasation and proliferation. Primary tumor-secreted factors, such as VEGF, then induce MMP-9 in lung ECs and macrophages (145). The primary tumor secretome also remotely mobilizes BMDCs to the early PMN through mechanisms that rely on interdependent vascular and ECM remodeling. The seminal paper describing this effect showed that VEGFR1+ hematopoietic progenitor cells (HPCs) are anchored in FN-rich areas of the lung via integrin α4β1 (VLA-4) (48) initiating a feed-forward loop that amplifies ECM remodeling and compromises vascular integrity (e.g., FNs, LOX, and S100A8) (155–157). Also recruited to the lung PMN are CD11b+Gr1+ myeloid progenitor cells, which are major contributors to VCAN (an ECM proteoglycan) deposition in the pre- and metastatic lungs (49). VCAN then promotes mesenchymal-to-epithelial transition (MET) of metastatic tumor cells by interfering with Smad2 activation downstream of TGF-β signaling, leading to metastatic outgrowth (49).
Importantly, circulating anti-angiogenic factors such as TSP-1, angiostatin, and endostatin can counteract the effects of pro-angiogenic tumor-secreted factors, thereby preventing PMN formation. However, TSP-1 can also exert pro-metastatic effects as evidenced in breast cancer cells in vitro (158) and in osteosarcoma patients, in which both lung metastatic primary tumors and lung metastases expressed higher levels of TSP-1 in response to TGF-β, inducing further ECM remodeling by promoting MMP-2, MMP-9, and FN1 (159). However, the pro-metastatic functions of TSP-1 are overshadowed by its anti-metastatic roles (57, 160). For example, in response to tumor-secreted prosaposin (PSAP), Gr1+ BMDC secretes TSP-1 at the PMN site, hindering metastasis, and PSAP mimetic peptides could recapitulate this effect (160). The effects in the distant organ could be ascribed to TSP-1 anti-angiogenic function (130, 131) and blocking of the migration and recruitment of VEGFR1+, VEGF-responsive BMDCs critical for PMN initiation (48, 106, 161). Future studies may unveil the potential of the systemic PSAP/TSP-1 axis in modulating PMN formation (161) and thus could provide insight into the role of circulating anti-angiogenic molecules and circulating ECM in systemic regulation of the “pre-metastatic switch”.
ECM degradation and remodeling are achieved via enzymatic breakage of the ECM by proteases, such as MMPs, serine, and threonine proteases, to generate bioactive molecules as well as reconfigure collagen fibers via enzymatic crosslinking by LOX family enzymes (40, 145, 162). Enzymatic modulation of the PMN ECM is also intricately linked to vascular integrity, as vascular leakiness is another hallmark of PMN formation (163). Major sources of ECM remodeling MMPs are BMDCs (macrophages, CD11b+ myeloid cells, and VEGFR1+ HPCs), stromal fibroblasts, and endothelial cells (40, 48, 145, 147, 164). Importantly, collagen IV peptides resulting from MMP-2 and MMP-9 mediated cleavage chemoattract BMDCs and circulating tumor cells (CTCs) to PMNs and promote metastasis (40). Another ECM remodeling molecule, TIMP-1, specifically cleaves MMPs and controls their enzymatic activities locally (162). High levels of TIMP-1 enhance cancer cell propensity for liver metastasis in murine and human pancreatic, lung, gastric, and ovarian cancers, lymphoma, and fibrosarcoma models (165). In osteosarcoma models, tumor-secreted pro-inflammatory glycoprotein angiopoietin-like protein 2 (ANGPTL2) binding to integrin α5β1 mediates tumor cell intravasation via MMPs, enhancing lung metastasis (166).
LOX family enzymes are critical modulators of PMN ECM remodeling in many cancers, particularly breast cancer. Hypoxic tumors, along with HIF1 activation (e.g., breast cancer and head and neck cancer) are the main source of LOX, which is released systemically and disrupts ECM homeostasis in the pre-metastatic lung or bone (167) by crosslinking ECM molecules and primarily collagen. Collagen type I and type IV crosslinking by LOX, LOX-like 2 (LOXL2), and LOXL4 that accumulate in PMNs of various organs create an adhesion platform for CD11b+ BMDCs (40), which further contributes to PMN formation through a positive feedback loop of MMP production that facilitates the colonization of the distant tissue by CTCs (40, 58, 168). In addition to tumor cells, activated fibroblasts also represent a source of LOX in the pre-metastatic tissue (40, 58).
Small EVs or exosomes are nanosized (50–150) membranous structures released by cells into the extracellular space and systemically mediate intercellular communication, modulating the behavior of the target cells and their microenvironment (169). Tumor-derived EVs exert systemic effects and convey malignant traits from primary tumors to distant organs via functional interactions and/or specific transfer of cargo [proteins, lipids, metabolites, and RNA such as microRNA (miRNA)] that contributes to organotropic metastasis, ECM remodeling, and reprogramming resident cells in the distant tissue (e.g., Kupffer cells, microglia, fibroblasts, and ECs). EVs promote ECM remodeling by altering the physiological architecture of the niche (27, 28, 35, 170, 171). Several studies highlighted the role of primary tumor-derived EVs in promoting ECM remodeling in the pre-metastatic lungs in melanoma (34), pancreatic cancer (41, 59), osteosarcoma (170), nasopharyngeal carcinoma (171), metastatic adenocarcinoma (172), liver (27), pancreatic (27) and gastric (173) cancer models and lymph nodes (LNs) in melanoma (174) and metastatic adenocarcinoma (172) models.
The main physicochemical alterations of the PMN ECM, involving changes in tension, pH, ion composition, and hydration, are driven by BM dysfunction, vascular leakiness, thrombus formation, and changes in the ECM composition such as increased production of ECM proteins and their deposition and increased production of ECM-degrading enzymes that disrupt ECM physiology. Thus, physicochemical ECM changes contribute to the fertile “soil” of the PMN tissue, characterized by a pro-invasive ECM. Specifically, the major physicochemical changes of the ECM encompass changes that impart mechanical properties (ECM mechanics) in relation to molecular weight, size of the ECM, and assembly (structural), and chemical changes such as ionization state, pH, and hydration, which have not been yet studied extensively in the context of the PMN.
Increased fibronectin secretion and deposition (assembling into fibronectin fibrils) (27, 29, 48, 148) increase collagen production and subsequent crosslinking by LOX enzymes (40, 65), imparting major structural changes on the ECM characteristic of the liver and lung PMNs. The pathological matrix deposition and crosslinking disrupt ECM mechano-homeostasis by increasing stiffness (i.e., mechanical force and tensile strength), reducing elasticity and the swelling ratio, characteristic for a fibrotic environment that is biomechanically less compliant and more rigid (175; 29). Therefore, the increase in mechanical force further facilitates BMDC infiltration, creating a highly inflammatory environment (40), and a dense matrix that prevents T-cell recruitment and function, as reported in tumors (176). Concomitantly, the crosslinking of collagen is associated with fiber dehydration and a fibrotic ECM (42). Thus, future studies assessing the PMN hydration state, along with the proteoglycan dysregulation, are warranted.
The inflammation occurring during early PMN formation is accompanied by higher ionization, as described by Kurpińska et al., who showed that calcium (Ca2+) signaling was dominant in the lung PMN as a pre-requisite for cytoskeleton and ECM reorganization (66). Among the most prominent mediators of the PMN inflammatory circuitry are the S100 proteins (e.g., S100A4, S100A9, and S100A8/A9), small Ca2+ signaling molecules that are hallmarks of lung and liver PMN formation (35, 68, 69, 177). The S100 family proteins bind to many proteins including the receptor for advanced glycation end products (RAGE) and extracellular matrix metalloproteinase inducer (EMMPRIN) (69, 178, 179). These receptors are known to promote ECM deposition and enhance pro-invasive ECM properties via MMP induction. Importantly, extracellular Ca2+ can regulate post-translational MMP dynamics (180), thereby showing that the activity of Zn2+-dependent MMP endopeptidases depends not only on the intracellular Zn2+ but also on increased Ca2+ availability and inflammation. Other Ca2+-dependent interactions with ECM molecules include the binding of endostatin to transglutaminase 2 (122) and the dependence of ADAM10-mediated proteolysis on Ca2+ influx (56).
Interestingly, in breast cancer, extravesicular CEMIP promoted brain PMN formation (28) potentially by depolymerizing HA (181), as well as intracellular Ca2+ regulation by releasing Ca2+ from the endoplasmic reticulum deposits (182). Therefore, intracellular calcium release in brain ECs may mediate calcium-dependent cytoskeletal re-arrangements and EC migration (28, 183). Collectively, these studies suggest that disruption of Ca2+ homeostasis may be critical for pre-metastatic ECM reorganization and PMN formation.
Although no exhaustive studies reported data on pH levels in the PMN, several findings suggest that lung PMN could foster an acidic microenvironment. Interstitial macrophages in the lung metabolically reprogrammed by tumor-derived EVs displayed a glycolytic-dominant metabolic profile, which fueled the conversion of pyruvate to lactate and lactate secretion, thereby contributing to the extracellular milieu acidification (67). Moreover, early lung PMN changes in a breast cancer model were associated with altered glucose metabolism and a decrease in lactate dehydrogenase chain b (Ldhb) gene expression (66), which catalyzes the conversion of lactate and NAD+, to pyruvate with and NADH and H+, ultimately contributing to intracellular or extracellular acidification depending on monocarboxylate transporter expression (184). Tumor-derived lactic acid activated the fibroblastic reticular cells in the draining LNs, indicated by increased alpha-SMA, and induced collagen I production along with increased expression of genes involved in ECM remodeling. These changes supported the formation of a pro-fibrotic environment favorable for PMN formation (43). Thus, PMN acidification may play a pro-metastatic role.
In the context of cancer, CAF-secreted EVs mediated a decrease of intracellular lactate in breast cancer cells with a concomitant extracellular environment acidification (185). Whether PMN CAF-derived exosomes can exert similar effects locally remains to be determined. However, it is important to stress that a complete metabolic landscape of the PMN has not yet been established. Thus, it would be important to explore the relationship between lactate production, pH level increases in the PMN and ECM remodeling, and the mechanisms through which they cooperate to promote metastasis.
Several seminal studies (48, 145) demonstrated the propensity of cancer cells to metastasize to specific organs—defined as organotropism—reinforcing Paget’s “seed and soil” theory (186). In this review, we explore the role of the ECM in orchestrating organotropic metastases and emphasize the importance of the “soil” and the specific local interactions that render the soil amenable for successful metastatic seeding. Tumor-derived small EVs orchestrate tumor metastasis to specific organs with a prominent role ascribed to integrins that dictate specific interactions with target cells (27, 28, 35). Along with ECM structure garnering greater interest in cancer metastasis, several studies demonstrated that EVs actively contribute to ECM remodeling at distant sites by reprogramming stromal cells (e.g., fibroblasts, epithelial cells, resident macrophages, and recruited immune cells) to alter the architecture of the microenvironment (27, 35, 41, 170, 171).
The lung was the first metastatic site where pioneering PMN research demonstrated that MMP-9 expression in endothelial cells and macrophages was induced by melanoma-secreted VEGF (145) and that FN (α4β1 ligand) was upregulated by VEGFR1+ integrin α4β1+ (VLA-4+) bone marrow-derived HPCs mobilized to the lung via tumor-secreted VEGF-A and placental growth factor (PIGF) (48) (Figure 1). In breast cancer, essential ECM components dictate the propensity for lung metastases at both cellular and extravesicular levels. Expression of integrin α3β1 on murine mammary carcinoma (4T1) cells was associated with lung colonization via binding laminin α5 (LAMA5) (187), which is widely expressed on the endothelial cell BM in lung capillaries (188). Human lung-tropic breast cancer-derived EVs package laminin-binding ITGα6β4 and ITGα6β1 (35). Moreover, specific uptake of these EVs by S100A4+ fibroblasts in laminin-rich lung microenvironments activated Src and pro-inflammatory S100 gene expression in these cells (35).
Through their pleiotropic functions and prominent role in ECM deposition and remodeling, lung fibroblasts are instrumental players in pre-conditioning the distant organ to create a permissive environment for metastatic colonization by shaping the PMN immune microenvironment (34, 40, 48, 151). Early changes induced at the distant site by the primary tumor include the differentiation of fibroblasts, which are sensitive to ECM mechanics (44). Importantly, lung fibroblasts are also a common target for tumor-derived EVs, with a large number of studies demonstrating that EV-specific functional cargo could induce fibroblast activation and differentiation into myofibroblast or CAF phenotypes, promoting ECM remodeling at the pre-metastatic site (35, 41, 170, 171).
Reprogrammed fibroblasts deposit FN, which becomes an adhesion platform for incoming BMDCs via their integrins (48, 82, 147, 148) and secretes POSTN, which promotes myeloid-derived suppressor cell (MDSC) accumulation and an immune-suppressive environment (189), and CTC colonization (151). The primary tumor-derived or local production of LOX enzymes (LOX and LOXL2) along with the increase in FNs and collagen creates a profibrotic lung microenvironment that is conducive to PMN formation and further supports inflammation and immunosuppression (40, 58, 63, 190). Lastly, a recent study showed that cytotoxic treatment of primary breast cancer (e.g., doxorubicin) induced the functional differentiation of lung PMN-promoting CAFs toward a pro-inflammatory phenotype, albeit with reduced cell migration and contractility and downregulation of fibrosis-related gene signature (191). These results support the idea that therapeutic intervention aimed at re-educating CAFs could hinder ECM remodeling and subsequent PMN formation.
ECM remodeling is intricately linked to vascular permeability and thrombosis, established hallmarks of PMN formation. Due to its structural features, lung tissue enhances the propensity of circulating tumor cells arrest in the dense capillaries and extravasate into the lung parenchyma (192). Cancer also induces systemic thrombosis, which consists of the adhesion and aggregation of platelets (163). Thrombosis is a bona fide example of cell–matrix interactions that occur when vascular integrity is compromised, exposing the BM and IM. Platelet-derived thrombin converts fibrinogen to fibrin that further crosslinks to fibronectin, resulting in platelet aggregation and thrombus formation. Fibronectin deposited at PMNs consequently acts as a scaffold for microthrombi. Fibrin and its precursor fibrinogen are ECM components that drive thrombosis at the distant site. Fibrinogen release from blood vessels and its deposition as fibrin in the pre-metastatic lung of murine melanoma models alters the vasculature integrity and favors microthrombus formation (163). Additionally, platelet ADP receptor P2Y12 activation by ITGαIIbβ3 [a ligand for RGD and AGD-containing ligands, such as fibrinogen, fibrin, and von Willebrand factor (83)] induced platelet aggregation in the lung PMN, promoting VEGFR1+ BMDC recruitment and increasing fibronectin deposition (193).
Innate immune cells that infiltrate the lung and participate in PMN formation, namely, neutrophils, have been shown to promote the release of elastase and cathepsin G proteases, leading to the degradation of TSP-1, which in turn enhanced lung metastasis. Neutrophils contribute to the proteolytic degradation of anti-tumorigenic TSP-1 via neutrophil-mediated degranulation of azurophilic granules to release serine proteases, such as neutrophil elastase (NE) and cathepsin G (57). Thus, the upregulation of neutrophil proteolytic activity and subsequent loss of TSP-1 underlie the PMN lung inflammatory microenvironment and metastatic outgrowth (57). Moreover, neutrophil extracellular trap (NET) formation activated tumor growth by degrading TSP-1 (57, 60), and inflammation-activated neutrophils proteolytically disrupt TSP-1 to enable cancer cell colonization (60) by an unknown mechanism.
Neutrophils play an undisputed critical role in cancer metastasis and their pro-metastatic role as well as functional effects in the PMNs, which is drawing increasing attention. Primary tumor-secreted Cathepsin C (CTSC), a protease that activates other proteases driving the proteolytic remodeling of the ECM, regulates neutrophil infiltration and NET formation in vivo, being critical for the early stage of metastatic colonization of the lung microenvironment by breast cancer cells (60). CTSC levels are highly increased in metastases compared to the primary tumors of breast cancer patients (60). Under inflammatory conditions, the proteolytic ECM remodeling activity of NETs awakened cancer cells from dormancy (194), while NET-associated DNA induced cancer cell chemotaxis (195).
Interestingly, a recent study showed that systemic communication and cooperation with a third organ were involved in regulating the development of PMNs and metastases. For example, liver-entrained leukocytes (B220+CD11b+NK1.1+) expressing coagulation factor X (FX) and vitronectin (VTN)—an adhesive glycoprotein that connects cells and ECM via integrins—traveled to fibrinogen‐rich hyperpermeable regions in pre‐metastatic lungs, where upon induction of TSP expression, they eliminated fibrinogen deposition, thus interfering with thrombotic and fibrotic events in the lung (196).
A variety of EV cargo modulates ECM deposition and function in the PMN. Melanoma EVs conveying onco-fetal insulin-like growth factor 2 mRNA-binding protein 1, involved in post-transcriptional regulation of transcripts encoding proteins for cell adhesion, migration, and ECM remodeling [e.g., CD44, a ligand for HA, osteopontin (OPN), collagens, and MMPs (168)], promoted ECM remodeling and FN deposition (34).
EVs secreted by murine primary pancreatic adenocarcinomas generate a pro-invasive lung niche by evoking invasive alterations to ECM deposition and architecture. Mechanistically, controlled EV loading of podocalyxin—a glycocalyx component and adhesion receptor with negative charge imparted by glycosylation—influenced Rab-coupling protein-dependent ITGα5β1 trafficking in normal fibroblasts in the pre-metastatic lung and altered ECM architecture (i.e., increased orthogonal characteristics), rendering the microenvironment conducive for metastatic cell seeding (41). EV-packaged latent membrane protein 1 (LMP-1) also induced lung fibroblast differentiation into CAFs (expressing α-SMA and fibroblast activation protein (FAP)) in vivo in a nasopharyngeal carcinoma model (171). Meanwhile, osteosarcoma-derived EVs enriched in TGFβ1 activated lung myofibroblast/CAFs as demonstrated by increased α-SMA expression and FN production (170). Moreover, EVs enriched in TNF1α, TGFβ, and IL-6, isolated from hypoxic human prostate (PC3) cancer cells, increased MMP-2, MMP-9, fibronectin, and collagen deposition and stimulated the recruitment of CD11b myeloid cells at the PMN (59). Lastly, EVs from metastatic rat adenocarcinoma (ASML) pre-conditioned the lung for metastasis via CD44v-dependent transcriptional and posttranscriptional regulation of miRNA repertoires. Some of these miRNAs, such as miR-494 and miR-542-3p, target cadherin-17 and can upregulate MMP transcription in vitro, thereby promoting ECM remodeling via functionally modulating target stroma cells in the lung and LNs (172).
Pancreatic cancer-derived EVs enriched in RGD-motif binding integrin αvβ5 exert liver tropism and specifically bind to Kupffer cells, liver-resident macrophages (F4/80+) with phagocytic function, reprogramming them through Src activation and the expression of pro-inflammatory S100 gene expression (35). Kupffer cells are intrinsically susceptible to EV-mediated reprogramming and can modulate ECM changes associated with PMN formation and induction of a pro-metastatic inflammatory milieu that supports tumor colonization. Pancreatic cancer-derived EVs enriched in macrophage inhibitory factor (MIF) were internalized by Kupffer cells (F4/80+) activating signaling pathways (e.g., TGF-β secretion) that reprogrammed hepatic stellate cells (HStCs) (α-SMA+desmin+) toward a myofibroblastic phenotype with increased FN production and deposition that creates a fibrotic environment. ECM remodeling further stimulated BMDC myeloid and neutrophil recruitment to the liver, facilitating PMN formation (27). Another study recently showed that extravesicular ANGPTL1 suppressed MMP-9 production in Kupffer cells, hindering liver vascular leakiness in colorectal cancer (CRC) murine models (197). However, the exosomal miR-135a-5p released from hypoxic colon cancer cells promoted MMP-7 production in Kupffer cells (198).
In CRC-associated liver metastasis, CCL15 mobilized immature myeloid cells to the pre-metastatic liver, producing MMP-2 and MMP-9 that degraded the liver ECM (199). EV-packaged CCL2 induced M2 pro-tumor macrophage differentiation and liver fibrosis that rendered the liver environment more conducive for cancer cell invasion (29). Another ECM modeling molecule, TIMP1, packaged into CRC EVs induced a positive feedback loop by upregulating TIMP1 in recipient fibroblasts in the liver, also resulting in ECM remodeling, as shown in human HCT116, HT29, and SW620 CRC models (44). Moreover, EV-mediated transfer of miR-122-5 upregulated N-cadherin and Vimentin and downregulated E-cadherin in hepatocytes in non-small cell lung cancer (NSCLC), thereby regulating PMN-promoting cell–ECM adhesions (36). Finally, gastric cancer-derived EV EGFR regulated the propensity of gastric cancer cells to colonize the liver by educating stromal cells in the microenvironment. Thus, EV-mediated translocation of EGFR to the plasma membrane of liver stromal cells activated HGF/c-Met signaling, facilitating gastric cancer cell invasion and liver seeding (173).
Altogether, these studies demonstrate that soluble factors or EVs released by primary tumors establish a fibrotic PMN-promoting liver environment via reprogramming local stroma or immune cells (e.g., Kupffer cells and HStCs) and highlight the therapeutic potential of targeting circulating EVs and/or the fibrotic niche for early prevention and therapeutic intervention against cancers with a propensity for liver metastasis.
The brain ECM has unique features: the most abundant ECM component is HA, while collagen is less prevalent, and FN is concentrated around the BM (37), as reviewed in Deasy and Erez (38).
Integrin β3 expression promotes brain tropism and brain PMN formation (200). Another molecule that shows specificity for brain metastatic tropism is a serine protease inhibitor, SERPINB1, highlighting the fact that ECM changes occur in the brain PMN (61). The main cellular players in promoting brain PMN formation are microglia and brain-resident macrophages. Microglia specifically uptake EVs released by brain-tropic metastatic cells, promoting neuroinflammation (i.e., perivascular inflammation) and increased cell adhesion, diapedesis, in the brain microenvironment due to the specific enrichment of hyaluronan-binding protein, also known as hyaluronidase 1 (CEMIP) in these vesicles (28). CEMIP breaks HA, and the resulting by-products are known activators of angiogenesis (32), consistent with the local vascular remodeling (i.e., disruption of the blood–brain barrier) in the pre-metastatic brain (28, 35). Thus, endothelial vasculature is susceptible to alterations mediated by tumor-derived EVs. For example, breast cancer EV-mediated transfer of miR-181c to brain endothelial cells disrupts the blood–brain barrier (45), whereas glioblastoma stem-like cell-derived EV transfer of VEGF-A to endothelial cells stimulated angiogenesis (201).
Interestingly, pro-metastatic EVs secreted by aggressive brain-tropic breast cancer models (MDA-MB-231-BrM2-831) interact systemically with low-density lipoprotein (LDL), which increases their propensity to be internalized by monocytes in the brain microenvironment (202). In contrast, their junctional interactions (e.g., including zona occludens 1, N-cadherin, VE-cadherin, claudin-5, and occludin) with human brain microvascular endothelial cells were similar to those of EVs isolated from non-tropic breast cancer (MDA-MB-231) (202). This study highlights the fact that systemic interactions with other circulating factors synergize with EV-intrinsic properties to determine their functional properties.
Bone has a unique collagen makeup, providing this tissue with increased strength through the mineralization of type I collagen and contains areas with dense or porous ECM matrix (203). The bone marrow is a complex microenvironment that contains multiple cell types of bone stromal cells that contribute to ECM remodeling in the PMN; osteoblasts, myeloid lineage osteoclasts, and fibroblasts are prominent. However, osteoblasts are the specialized cells primarily responsible for bone ECM matrix production.
Various ECM interactions with cellular or extravesicular integrins determine bone tropic metastasis. For example, human and murine breast cancer-secreted EVs enriched in cadherin 11 (CDH11) and α5 integrin delivered runt-related transcription factor 2 (RUNX2) to the bone, inducing an osteogenic environment and bone lesions that promoted bone colonization by cancer cells (46). Increased α5 integrin in breast cancers or bone was associated with osteoclast-mediated bone resorption and osteolytic metastases (50a). Additional integrins on the surface of prostate and breast cancer cells that mediate bone niche colonization are integrins β1 and β3 (51, 53). Integrin interactions and signaling are also pivotal for pancreatic cancer cell migration to the bone. Specifically, pancreatic cancer cell adhesion to collagen I is mediated via ITGα2β1 and stimulates their migration and subsequent bone colonization (51), whereas integrin-mediated cancer–stroma interactions in the bone have various outcomes: the expression of αIIbβ3 promotes platelet aggregation, similarly to the lung, and integrin signaling via αvβ3 and VLA-4 on tumor cells promotes tumor metastasis in the bone niche (53).
Among the cell types shaping the bone ECM, circulating platelets also play an important role in bone remodeling by mediating the crosstalk between the tumor and the bone microenvironment. Sequestration of functional molecules by platelets (platelet TSP-1, TGF-β1, MMP-1, and receptor activator of NF-κB (RANK)) mediates bone-tropic prostate cancer-induced osteoclast differentiation and BMDC infiltration (62, 204). Similarly, the receptor activator of NF-κB ligand (RANKL) regulated osteoclastogenesis and induced bone remodeling and PMN formation (205). Fibroblasts or fibroblast-like cells directly enhance bone resorption by inducing osteoclastogenesis by producing cathepsin (e.g., cathepsin K), MMPs, and MT MMPs (47). However, the crosstalk between these stromal cells in PMN ECM remodeling is not completely understood.
The majority of cancers involve LN metastasis, which is an important clinical indicator of patient outcome (149, 174, 206, 207). LNs are also susceptible to reprogramming or remodeling mediated by EVs, as lymphatics are a major trafficking route for tumor-derived EVs. A recent study described that EV integrin mediated homing to LN in murine melanoma, as well as their target in the pre-metastatic tissue. Specifically, tumor-derived EVs enriched in α4β1 (VLA‐4) or α9β1 were internalized only by lymphatic endothelial cells (VCAM-1+) from the subcapsular and medullary sinuses of B16-F10 melanoma-bearing mouse draining lymph nodes. This resulted in EV-shuttled antigens that mediated immune escape and killing of cytotoxic CD8+ T cells (207). Melanoma EV homed to sentinel LNs and promoted LN remodeling into a hospitable niche before tumor cell colonization by inducing the expression of factors responsible for cell recruitment (GM-CSF in endothelial cells), ECM remodeling, and angiogenesis (174).
In the metastatic rat adenocarcinoma model BSp73ASML (ASML), which has increased susceptibility for PMN formation in LNs and lung tissue, exosomal miR-494 and miR-542-3p transfer to lymph node stromal cells and lung fibroblasts, resulting in the transcriptional upregulation of MMPs and downregulation of cadherin-17, thereby mainly affecting proteases and adhesion molecules, highlighting the myriad of mechanisms through which EVs mediate ECM changes in pre-metastatic tissues (172).
The omentum, a common site for ovarian metastasis, has received great interest in the past years due to its aggressive nature and late diagnosis of ovarian cancer. A recent study revealed collagen-rich omentum as a homing site for ITGα2-mediated peritoneal metastases (39). Omental ECM proteins contributed to early metastasis to the peritoneum by triggering cancer cell adhesion to collagen, cell migration, and activation of focal adhesion kinase. Importantly, both ITGα2 and altered collagen expression in the pre-metastatic omentum were indicative of poor clinical outcomes in ovarian cancer patients (39). This suggests that the omentum is not only a bystander in peritoneal metastases but that it actively participates in ECM dysregulation.
Pancreatic cancer also has the tendency to metastasize to the peritoneum (39) or omentum (208), and Laminin gamma 1 (LAMC1) on pancreatic cancer cells promotes their homing to the peritoneum (30). To gain mechanistic insight into pancreatic cancer propensity to metastasize to the omentum, Feygenzon et al. analyzed the secretome and mRNA expression of tissue explants from human omental fat. These studies revealed higher levels of cancer-related proteins involved in ECM remodeling and ECM-mediated interactions (e.g., TIMP-1, MMP-8, and TSP-1) compared to subcutaneous fat, suggesting that omental fat could reprogram pancreatic cancer cells toward a more aggressive and invasive phenotype, marked by increased levels of ECM proteins and adhesion molecules (208).
LAMC1 is also secreted by gastric cancers, and it accumulated in the pre-metastatic peritoneal microenvironment, where it induced preadipocyte maturation (i.e., differentiation) and altered their metabolism and function, resulting in the release of free fatty acids. Of these fatty acids, palmitic acid regulated LAMC1 in a posttranslational manner and promoted PMN formation and gastric cancer cell colonization of the peritoneum (30).
Finally, the roles of EVs in peritoneal metastasis are only beginning to be explored. Small EVs from malignant ascites of patients with advanced ovarian cancer undergoing adjuvant chemotherapy displayed preferential homing to the peritoneum and were enriched in ECM components and regulatory proteins. These EVs promoted an increase in transforming growth factor-β-I and FNs, which are functionally linked proteins, plasminogen activator inhibitor 1, and FNs, suggesting a role of these EVs in reprogramming peritoneal ECMs toward a pro-metastatic function (209).
The remote control of PMN formation by the primary tumor secretome presents the opportunity for circulating PMN-inducing biomarker detection and monitoring (MMPs, TIMPs, LOX, and EVs) in liquid biopsies from patients. Indeed, several clinical studies correlated the levels of these circulating biomarkers with clinical outcomes and metastasis, which could be further exploited for the development of therapeutic strategies to impair ECM remodeling that supports PMN formation.
For example, the levels of plasma TIMP-1 can be used as a biomarker, as this soluble tumor-derived secreted factor appears to be the main player in determining the propensity for liver PMN formation in various cancers. Indeed, high levels of plasma TIMP-1 are a poor prognostic indicator for liver metastases (210) and for the progression of gastric (211), colorectal (165), and pancreatic (212) cancers. A recent study identified extravesicular TIMP as a critical mediator of liver PMN reprogramming and ECM remodeling, as well as an independent prognostic factor, associated with poor progression-free survival in patients with colorectal liver metastases (44).
In patients with osteosarcoma, the levels of ANGPTL2, which promotes lung PMN formation, were systemically elevated relative to healthy subjects, thus offering the possibility for intervention to hinder ANGPTL2-mediated signaling and subsequent pro-metastatic effects (213). Importantly, high levels of serum MMPs are also reliable biomarkers indicative of metastatic progression and are associated with poor survival in gastric (MMP-14) (214) and breast (MMP-2 and MMP-9) (215) cancer patients.
The potential use of EVs as biomarkers associated with PMN formation was recently highlighted by Hoshino et al. (216), where unbiased proteomic profiling of EVs from human cancer samples (tumor and adjacent tissue explants, plasma, and other biological fluids) allowed the discovery of pan-cancer EVP markers, with high diagnostic and therapeutic potential (216). Therefore, VCAN, TN-C, and thrombospondin are pan-cancer EVP markers and also ECM components that can discriminate tumors from adjacent tissues with high sensitivity and specificity. Moreover, specific EVP adhesion proteins (e.g., CD36, TN-C, THBS2, and VCAN) may also be pan-cancer markers. Additionally, pancreatic cancer-derived EVs were mainly enriched in ECM-associated proteins, thereby reinforcing the possibility to employ EVs for monitoring the stiffness and changes in ECM deposition in early-stage primary tumors, which may allow early PMN detection (216).
The ECM is a key player in therapeutic resistance (217–219). Structural and physical ECM changes negatively modulate responses to therapies. A dense and rigid ECM protects tumor cells from therapeutic agents, regulating their permeability throughout the microenvironment. Likewise, functional modifications of the ECM can actively interfere with cell cycle arrest, apoptosis, and anti-tumor immunity by sustaining a favorable environment for tumor cells.
ECM remodeling mediated by primary tumors represents one of the first, albeit critical, steps in PMN formation, which offers the advantage to target soluble factors secreted by tumors in systemic circulation to hinder PMN formation or to target PMN-resident or PMN-infiltrating immune and stromal cells that are pivotal for altering ECM homeostasis in distant tissues. However, a word of caution is necessary, as sustained inhibition of ECM may adversely affect normal programs of wound healing, and the timing and duration of ECM-directed treatments in cancer are key (220).
Based on the potential of anti-angiogenic therapies to hinder metastasis and interfere with PMN formation, the use of recombinant circulating anti-angiogenic factors should be further evaluated to determine the therapeutic outcome and PMN formation. A recent meta-analysis showed that recombinant endostatin, Endostar (rh‐endostatin), approved for clinical use in China for almost 20 years, improved the prognosis of non‐small cell lung cancer patients with bone metastases when used in combination with other chemotherapeutic agents (221–223).
For example, pharmaceutical inhibition of ANGPTL2 signaling using a non-RGD-based integrin-binding peptide (ATN-161) could be a potential strategy for interfering with lung PMN formation in osteosarcoma, as suggested by preclinical studies (213). Other preclinical data also show that suppressing the interaction of TIMP-1 with the CD63 receptor on hepatic stellate cells effectively prevented liver PMN formation in pancreatic cancer models (212). Moreover, drugs targeting MMPs (MMP-2 and MMP-9) to interfere with ECM degradation are also promising options for preventing ECM disruption in the PMN (reviewed in 224), especially as there are ongoing clinical trials that target MMPs systemically.
The relevance of targeting LOX proteins systemically or at the PMN is based on their pivotal contribution in regulating ECM remodeling, BMDC docking in the distant organ, and the encouraging preclinical data showing that LOX inhibition reduced hypoxia-induced metastasis (140). Previous therapeutic strategies employing LOX inhibitors that underwent clinical trials, such as LOXL2-blocking monoclonal antibody simtuzumab and small molecule LOXL2 inhibitors (e.g., PAT1251 and PXS-2SA), did not appear to be very effective. Currently, tetrathiomolybdate and pemetrexed AB0024 (anti-LOXL2 monoclonal antibody) are being clinically tested for moderate-to-advanced breast cancer and metastatic non-small cell carcinoma (224). However, these trials are tailored for cancers with established metastases and are not designed to evaluate effects on the PMN. Interestingly, an EGFR inhibitor, WZ4002, in combination with silibinin, was effective in decreasing metastasis in preclinical models by blocking EGFR/LOX pathway and may represent a promising alternative strategy to interfere with LOX-mediated PMN formation (225).
Given the critical role of integrins in mediating organotropic metastases and their central role in ECM-based interactions in the PMN, targeting specific heterodimeric partners either systemically or locally is both a challenging and a promising opportunity. For example, Cilengitide, an RGD pentapeptide targeting both αvβ3 and αvβ5 integrins, has been tested in clinical trials on metastatic and non-metastatic tumors (226). Similar to integrin-targeting strategies, disintegrins, such as vicrostatin, have been tailored to target multiple integrins (i.e., αvβ3, αvβ5, and α5β1), and studies in preclinical cancer models highlighted their translational potential (227). Lastly, small molecules interfering with β1-integrin signaling (e.g., ATN-161, and JSM6427) also showed promise in targeting dormant cancer cells and preventing metastatic recurrence in preclinical models (228).
EVs play a key role in PMN formation, and various therapeutic approaches targeting EVs are currently in development (229). One potential therapeutic strategy would be to interfere with EV production. In preclinical studies, reserpine, an anti-hypertensive drug, could suppress tumor-derived EV uptake and disrupt the EV-induced PMN formation and melanoma lung metastasis, suggesting a potential benefit of reserpine in the adjuvant setting for melanoma treatment (230). Ideally, blocking the delivery of specific EV cargo crucial for ECM remodeling at distant sites could improve patient outcomes. Recently, an interesting therapeutic strategy revealed that the inhibition of HSP90AA on human serum-derived EVs packaging TIMP-1 (which binds HSP90AA) attenuates the ECM remodeling effects of these EVs (44), and this intervention could have a high clinical impact in preventing liver PMN formation. Alternatively, blocking TIMP-1 with monoclonal antibodies should allow the specific systemic targeting of this molecule.
As a large number of studies demonstrated that specific EV integrins selectively adhere to ECM components and ECM-enriched areas in distant pre-metastatic organs, a potential therapeutic strategy would involve active targeting or sequestration of specific circulating EVs with the homing capacity to various PMNs using monoclonal antibodies tailored against integrins (α6β4 and αvβ5) or other proteins (S100A4, VCAN, TNC, and TSP), thereby preventing their intrinsic PMN-promoting roles (35, 216).
In addition to interfering with specific components of the ECM matrisome that promote PMN formation, other potential strategies could aim to target or re-educate stromal or immune cells that are critical for ECM remodeling in specific organs (e.g., CAFs, Kupffer cells, and BMDCs). Thus, recent innovative nano-therapies show translational potential in preclinical models. For example, an anti-thrombotic agent based on calcium phosphate nanoparticles coated with heparin strongly reduced ICAM-1, VCAM-1, and MMP-9 in pre-metastatic lungs of murine breast cancers (231). Drug-free peptide nano-blankets assembled in situ interfered with fibroblast activation and subsequent LOX, FN, and periostin accumulation in the early PMN (232).
As opposed to the attention paid to cancer cells, the “soma”, or non-cellular material, has been generally overlooked in the past by oncology researchers. Despite the recent progress in our understanding of the composition and function of the PMN ECM, many outstanding questions remain. For instance, are ECM changes in the PMN reversible, or should therapeutic targeting focus on blocking the already existent PMN-associated ECM alterations? Given the crucial roles of ECM in homeostasis and wound healing, are there cancer-specific/PMN-specific ECM characteristics that can be specifically targeted with minimum side effects, such as particular ECM protein isoforms or post-translational modifications? Since ECM alterations provide a platform for the recruitment of immunosuppressive myeloid subsets, thereby precluding T-cell recruitment or function in the PMN, could disrupting this axis be a promising therapy that could be combined with checkpoint blockade in cancers otherwise refractory to immunotherapy?
To unveil the complete potential of the early detection of ECM pro-metastatic changes (e.g., in patients without metastatic disease) and to identify novel or critical therapeutic targets that are highly specific for PMN formation, a better understanding of ECM molecular and cellular changes in the PMN in cancer patients is needed. Precedent exists in preclinical models, illustrated, for example, by the work of Hebert et al., who conducted a comprehensive, unbiased mass spectrometry analysis of the ECM in metastatic niches such as the brain, lungs, liver, and bone marrow for MDA-MB-231 TNBC (61). This study highlighted the fact that tumor cells and stroma cooperate during metastatic niche formation, with stromal cells building the core, structural matrisome, and tumor cells producing ECM-associated proteins, secreted factors, and ECM regulators. A similar analysis dissecting the PMN matrisome in conjunction with the tumor secretome in patients may unveil novel biomarkers, leading to the discovery of therapies designed to “demolish the nest”, hindering ECM remodeling and preventing PMN formation in patients with early-stage primary tumors. Moreover, using novel innovative techniques such as MicroMap in PMN preclinical models and patient tissue samples may enable the discovery of novel, PMN-specific ECM–receptor interactions that could be targeted in the clinic (233).
From a therapeutic perspective, preclinical studies often highlight the possibility of using both novel therapies currently under clinical investigation as well as repurposing already Food and Drug Administration (FDA)-approved therapies that could interfere with all aspects of ECM-associated changes in distant organs. However, clinical trials that include investigation of PMN or ECM-associated changes at future metastatic sites are relatively rare, despite the availability of a number of non-invasive clinical tools that could reveal PMN formation. This impedes the identification and clinical validation of PMN/ECM-associated markers that could be detected early on during the cancer progression timeline, before metastatic onset. As an example, the use of new technologies or existing imaging techniques, such as the ultrasound-based FibroScan or artificial intelligence-assisted computer tomography, may help detect structural or quantitative ECM changes such as fibronectin deposition, tissue stiffness, and fibrotic environments, which occur in the lung, liver, and LNs, and therefore guide therapeutic decisions. Our overview of the current knowledge of ECM biology, often taken for granted or dismissed as peripheral, highlights its pleiotropic roles at each step of the metastatic cascade and its theranostic as well as therapeutic potential.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the National Cancer Institute (No. CA270998) and the STARR Cancer Consortium (No. SCC I16-0057) to IM, and the Association of Transdisciplinary Society of Personalized Oncology for Combating Cancer to LP.
The authors thank Dr. David Lyden for feedback and mentorship. The figure was created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci (2010) 123:4195–200. doi: 10.1242/jcs.023820
2. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep (2014) 15:1243–53. doi: 10.15252/embr.201439246
3. Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell (2019) 49:332–46. doi: 10.1016/j.devcel.2019.03.026
4. Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Elastic proteins: biological roles and mechanical properties. Philos Trans R Soc Lond B Biol Sci (2002) 357:121–32. doi: 10.1098/rstb.2001.1022
5. Hynes RO. The extracellular matrix: not just pretty fibrils. Science (2009) 326:1216–9. doi: 10.1126/science.1176009
6. Iwasaki A, Sakai K, Moriya K, Sasaki T, Keene DR, Akhtar R, et al. Molecular mechanism responsible for fibronectin-controlled alterations in matrix stiffness in advanced chronic liver fibrogenesis. J Biol Chem (2016) 291:72–88. doi: 10.1074/jbc.M115.691519
7. Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA (1990) 87:6624–8. doi: 10.1073/pnas.87.17.6624
8. Li G, Oparil S, Sanders JM, Zhang L, Dai M, Chen LB, et al. Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis (2006) 188:292–300. doi: 10.1016/j.atherosclerosis.2005.11.002
9. Manabe R, Tsutsui K, Yamada T, Kimura M, Nakano I, Shimono C, et al. Transcriptome-based systematic identification of extracellular matrix proteins. Proc Natl Acad Sci USA (2008) 105:12849–54. doi: 10.1073/pnas.0803640105
10. Tichet M, Prod’Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun (2015) 6:6993. doi: 10.1038/ncomms7993
11. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol (2010) 22:697–706. doi: 10.1016/j.ceb.2010.08.015
12. Zimmermann R, Werner C, Sterling J. Exploring structure–property relationships of GAGs to tailor ECM-mimicking hydrogels. Polymers (2018) 10:1376. doi: 10.3390/polym10121376
13. Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix: structure and composition of the extracellular matrix. J Pathol (2003) 200:423–8. doi: 10.1002/path.1437
14. Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer (2003) 3:422–33. doi: 10.1038/nrc1094
15. Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem (1997) 272:31525–32. doi: 10.1074/jbc.272.50.31525
16. Sugawara K, Tsuruta D, Ishii M, Jones JCR, Kobayashi H. Laminin-332 and -511 in skin. Exp Dermatol (2008) 17:473–80. doi: 10.1111/j.1600-0625.2008.00721.x
17. Borsi L, Balza E, Gaggero B, Allemanni G, Zardi L. The alternative splicing pattern of the tenascin-c pre-mRNA is controlled by the extracellular pH. J Biol Chem (1995) 270:6243–5. doi: 10.1074/jbc.270.11.6243
18. Kleinman HK, McGarvey ML, Hassell JR, Star VL, Cannon FB, Laurie GW, et al. Basement membrane complexes with biological activity. Biochemistry (1986) 25:312–8. doi: 10.1021/bi00350a005
19. LeBleu VS, MacDonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (2007) 232:1121–9. doi: 10.3181/0703-MR-72
20. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol (2014) 15:786–801. doi: 10.1038/nrm3904
21. Tien J, Ghani U, Dance YW, Seibel AJ, Karakan M.Ç., Ekinci KL, et al. Matrix pore size governs escape of human breast cancer cells from a microtumor to an empty cavity. iScience (2020) 23:101673. doi: 10.1016/j.isci.2020.101673
22. Camelliti P, Borg T, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res (2005) 65:40–51. doi: 10.1016/j.cardiores.2004.08.020
23. Wang JH-C, Thampatty BP, Lin J-S, Im H-J. Mechanoregulation of gene expression in fibroblasts. Gene (2007) 391:1–15. doi: 10.1016/j.gene.2007.01.014
24. Bauer J, Emon MAB, Staudacher JJ, Thomas AL, Zessner-Spitzenberg J, Mancinelli G, et al. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin a signaling. Sci Rep (2020) 10:50. doi: 10.1038/s41598-019-55687-6
25. Manoukian P, Bijlsma M, van Laarhoven H. The cellular origins of cancer-associated fibroblasts and their opposing contributions to pancreatic cancer growth. Front Cell Dev Biol (2021) 9:743907. doi: 10.3389/fcell.2021.743907
26. Han L, Wu Y, Fang K, Sweeney S, Roesner UK, Parrish M, et al. The splanchnic mesenchyme is the tissue of origin for pancreatic fibroblasts during homeostasis and tumorigenesis. Nat Commun (2023) 14:1. doi: 10.1038/s41467-022-34464-6
27. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol (2015) 17:816–26. doi: 10.1038/ncb3169
28. Rodrigues G, Hoshino A, Kenific CM, Matei IR, Steiner L, Freitas D, et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol (2019) 21:1403–12. doi: 10.1038/s41556-019-0404-4
29. Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka-Kondoh S, et al. Inactivation of chemokine (C-c motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci USA (2010) 107:13063–8. doi: 10.1073/pnas.1002372107
30. Fang Y, Dou R, Huang S, Han L, Fu H, Yang C, et al. LAMC1-mediated preadipocytes differentiation promoted peritoneum pre-metastatic niche formation and gastric cancer metastasis. Int J Biol Sci (2022) 18:3082–101. doi: 10.7150/ijbs.70524
31. Aumailley M, Timpl R. Attachment of cells to basement membrane collagen type IV. J Cell Biol (1986) 103:1569–75. doi: 10.1083/jcb.103.4.1569
32. Fraser JRE, Laurent TC, Laurent UBG. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med (1997) 242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x
33. Tucker RP, Chiquet-Ehrismann R. The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta BBA - Mol Cell Res (2009) 1793:888–92. doi: 10.1016/j.bbamcr.2008.12.012
34. Ghoshal A, Rodrigues LC, Gowda CP, Elcheva IA, Liu Z, Abraham T, et al. Extracellular vesicle-dependent effect of RNA-binding protein IGF2BP1 on melanoma metastasis. Oncogene (2019) 38:4182–96. doi: 10.1038/s41388-019-0797-3
35. Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature (2015) 527:329–35. doi: 10.1038/nature15756
36. Li C, Qin F, Wang W, Ni Y, Gao M, Guo M, et al. hnRNPA2B1-mediated extracellular vesicles sorting of miR-122-5p potentially promotes lung cancer progression. Int J Mol Sci (2021) 22:12866. doi: 10.3390/ijms222312866
37. George N, Geller HM. Extracellular matrix and traumatic brain injury. J Neurosci Res (2018) 96:573–88. doi: 10.1002/jnr.24151
38. Deasy SK, Erez N. A glitch in the matrix: organ-specific matrisomes in metastatic niches. Trends Cell Biol (2022) 32:110–23. doi: 10.1016/j.tcb.2021.08.001
39. Huang Y-L, Liang C-Y, Ritz D, Coelho R, Septiadi D, Estermann M, et al. Collagen-rich omentum is a premetastatic niche for integrin α2-mediated peritoneal metastasis. eLife (2020) 9:e59442. doi: 10.7554/eLife.59442
40. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell (2009) 15:35–44. doi: 10.1016/j.ccr.2008.11.012
41. Novo D, Heath N, Mitchell L, Caligiuri G, MacFarlane A, Reijmer D, et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat Commun (2018) 9:5069. doi: 10.1038/s41467-018-07339-y
42. Miles CA, Avery NC, Rodin VV, Bailey AJ. The increase in denaturation temperature following cross-linking of collagen is caused by dehydration of the fibres. J Mol Biol (2005) 346:551–6. doi: 10.1016/j.jmb.2004.12.001
43. Riedel A, Helal M, Pedro L, Swietlik JJ, Shorthouse D, Schmitz W, et al. Tumor-derived lactic acid modulates activation and metabolic status of draining lymph node stroma. Cancer Immunol Res (2022) 10:482–97. doi: 10.1158/2326-6066.CIR-21-0778
44. Rao VS, Gu Q, Tzschentke S, Lin K, Ganig N, Thepkaysone M-L, et al. Extravesicular TIMP-1 is a non-invasive independent prognostic marker and potential therapeutic target in colorectal liver metastases. Oncogene (2022) 41:1809–20. doi: 10.1038/s41388-022-02218-9
45. Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, et al. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun (2015) 6:6716. doi: 10.1038/ncomms7716
46. Li X-Q, Zhang R, Lu H, Yue X-M, Huang Y-F. Extracellular vesicle-packaged CDH11 and ITGA5 induce the premetastatic niche for bone colonization of breast cancer cells. Cancer Res (2022) 82:1560–74. doi: 10.1158/0008-5472.CAN-21-1331
47. Pap T, Claus A, Ohtsu S, Hummel KM, Schwartz P, Drynda S, et al. Osteoclast-independent bone resorption by fibroblast-like cells. Arthritis Res Ther (2003) 5:R163–173. doi: 10.1186/ar752
48. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438:820–7. doi: 10.1038/nature04186
49. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72:1384–94. doi: 10.1158/0008-5472.CAN-11-2905
50. Pantano F, Croset M, Driouch K, Bednarz-Knoll N, Iuliani M, Ribelli G, et al. Integrin alpha5 in human breast cancer is a mediator of bone metastasis and a therapeutic target for the treatment of osteolytic lesions. Oncogene (2021) 40:1284–99. doi: 10.1038/s41388-020-01603-6
51. Hall CL, Dubyk CW, Riesenberger TA, Shein D, Keller ET, van Golen KL. Type I collagen receptor (alpha2beta1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia N Y N (2008) 10:797–803. doi: 10.1593/neo.08380
52. Preca B-T, Bajdak K, Mock K, Lehmann W, Sundararajan V, Bronsert P, et al. A novel ZEB1/HAS2 positive feedback loop promotes EMT in breast cancer. Oncotarget (2017) 8:11530–43. doi: 10.18632/oncotarget.14563
53. Schneider JG, Amend SR, Weilbaecher KN. Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone (2011) 48:54–65. doi: 10.1016/j.bone.2010.09.016
54. Yang Y-F, Sun YY, Acott TS, Keller KE. Effects of induction and inhibition of matrix cross-linking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells. Sci Rep (2016) 6:30505. doi: 10.1038/srep30505
55. Xu J, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, Moon YS, et al. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol (2001) 154:1069–79. doi: 10.1083/jcb.200103111
56. Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, et al. ADAM10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res (2008) 102:1192–201. doi: 10.1161/CIRCRESAHA.107.169805
57. El Rayes T, Catena R, Lee S, Stawowczyk M, Joshi N, Fischbach C, et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of tsp-1. Proc Natl Acad Sci USA (2015) 112:16000–5. doi: 10.1073/pnas.1507294112
58. Wong CC-L, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA (2011) 108:16369–74. doi: 10.1073/pnas.1113483108
59. Deep G, Jain A, Kumar A, Agarwal C, Kim S, Leevy WM, et al. Exosomes secreted by prostate cancer cells under hypoxia promote matrix metalloproteinases activity at pre-metastatic niches. Mol Carcinog (2020) 59:323–32. doi: 10.1002/mc.23157
60. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin c promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell (2021) 39:423–37.e7. doi: 10.1016/j.ccell.2020.12.012
61. Hebert JD, Myers SA, Naba A, Abbruzzese G, Lamar JM, Carr SA, et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res (2020) 80:1475–85. doi: 10.1158/0008-5472.CAN-19-2961
62. Kerr BA, McCabe NP, Feng W, Byzova TV. Platelets govern pre-metastatic tumor communication to bone. Oncogene (2013) 32:4319–24. doi: 10.1038/onc.2012.447
63. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y, et al. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res CR (2018) 37:99. doi: 10.1186/s13046-018-0761-z
64. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
65. Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature (2019) 567:249–52. doi: 10.1038/s41586-019-1004-y
66. Kurpińska A, Suraj J, Bonar E, Zakrzewska A, Stojak M, Sternak M, et al. Proteomic characterization of early lung response to breast cancer metastasis in mice. Exp Mol Pathol (2019) 107:129–40. doi: 10.1016/j.yexmp.2019.02.001
67. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab (2021) 33:2040–2058.e10. doi: 10.1016/j.cmet.2021.09.002
68. Mathisen B, Lindstad RI, Hansen J, El-Gewely SA, Maelandsmo GM, Hovig E, et al. S100A4 regulates membrane induced activation of matrix metalloproteinase-2 in osteosarcoma cells. Clin Exp Metastasis (2003) 20:701–11. doi: 10.1023/b:clin.0000006819.21361.03
69. Grum-Schwensen B, Klingelhöfer J, Beck M, Bonefeld CM, Hamerlik P, Guldberg P, et al. S100A4-neutralizing antibody suppresses spontaneous tumor progression, pre-metastatic niche formation and alters T-cell polarization balance. BMC Cancer (2015) 15:44. doi: 10.1186/s12885-015-1034-2
70. Sinkus R, Lorenzen J, Schrader D, Lorenzen M, Dargatz M, Holz D. High-resolution tensor MR elastography for breast tumour detection. Phys Med Biol (2000) 45:1649–64. doi: 10.1088/0031-9155/45/6/317
71. Myllyharju J. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet (2004) 20:33–43. doi: 10.1016/j.tig.2003.11.004
72. Kass L, Erler JT, Dembo M, Weaver VM. Mammary epithelial cell: influence of extracellular matrix composition and organization during development and tumorigenesis. Int J Biochem Cell Biol (2007) 39:1987–94. doi: 10.1016/j.biocel.2007.06.025
73. O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell (1997) 88:277–85. doi: 10.1016/s0092-8674(00)81848-6
74. Iozzo RV, Zoeller JJ, Nyström A. Basement membrane proteoglycans: modulators par excellence of cancer growth and angiogenesis. Mol Cells (2009) 27:503–13. doi: 10.1007/s10059-009-0069-0
75. Kawahara R, Granato DC, Carnielli CM, Cervigne NK, Oliveria CE, Martinez CAR, et al. Agrin and perlecan mediate tumorigenic processes in oral squamous cell carcinoma. PloS One (2014) 9:e115004. doi: 10.1371/journal.pone.0115004
76. Birbrair A, Zhang T, Wang Z-M, Messi ML, Mintz A, Delbono O. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol (2013) 305:C1098–1113. doi: 10.1152/ajpcell.00171.2013
77. Vuorio E, de Crombrugghe B. The family of collagen genes. Annu Rev Biochem (1990) 59:837–72. doi: 10.1146/annurev.bi.59.070190.004201
78. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9:654–9. doi: 10.1038/ncb1596
79. Yáñez-Mó M, Siljander PR-M, Andreu Z, Bedina Zavec A, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles (2015) 4:27066. doi: 10.3402/jev.v4.27066
80. Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. BioScience (2015) 65:783–97. doi: 10.1093/biosci/biv084
81. Huleihel L, Hussey GS, Naranjo JD, Zhang L, Dziki JL, Turner NJ, et al. Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv (2016) 2:e1600502. doi: 10.1126/sciadv.1600502
82. Hynes RO. Integrins: a family of cell surface receptors. Cell (1987) 48:549–54. doi: 10.1016/0092-8674(87)90233-9
83. Sánchez-Cortés J, Mrksich M. The platelet integrin alphaIIbbeta3 binds to the RGD and AGD motifs in fibrinogen. Chem Biol (2009) 16:990–1000. doi: 10.1016/j.chembiol.2009.08.012
84. Roca-Cusachs P, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc Natl Acad Sci USA (2013) 110:E1361–1370. doi: 10.1073/pnas.1220723110
85. Høye AM, Erler JT. Structural ECM components in the premetastatic and metastatic niche. Am J Physiol Cell Physiol (2016) 310:C955–967. doi: 10.1152/ajpcell.00326.2015
86. Bass MD, Humphries MJ. Cytoplasmic interactions of syndecan-4 orchestrate adhesion receptor and growth factor receptor signalling. Biochem J (2002) 368:1–15. doi: 10.1042/bj20021228
87. Cichy J, Puré E. The liberation of CD44. J Cell Biol (2003) 161:839–43. doi: 10.1083/jcb.200302098
88. Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol (2007) 26:146–55. doi: 10.1016/j.matbio.2006.10.007
89. Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci (2009) 122:1472–2. doi: 10.1242/jcs.052910
90. Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res (2010) 339:31–46. doi: 10.1007/s00441-009-0829-3
91. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer (2004) 4:528–39. doi: 10.1038/nrc1391
92. Tempel C, Gilead A, Neeman M. Hyaluronic acid as an anti-angiogenic shield in the preovulatory rat Follicle1. Biol Reprod (2000) 63:134–40. doi: 10.1095/biolreprod63.1.134
93. Park D, Kim Y, Kim H, Kim K, Lee Y-S, Choe J, et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFβ receptor interaction via CD44-PKCδ. Mol Cells (2012) 33:563–74. doi: 10.1007/s10059-012-2294-1
94. Paolillo, Schinelli. Extracellular matrix alterations in metastatic processes. Int J Mol Sci (2019) 20:4947. doi: 10.3390/ijms20194947
95. Patten J, Wang K. Fibronectin in development and wound healing. Adv Drug Deliv Rev (2021) 170:353–68. doi: 10.1016/j.addr.2020.09.005
96. Kuo HJ, Maslen CL, Keene DR, Glanville RW. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem (1997) 272:26522–9. doi: 10.1074/jbc.272.42.26522
97. Erler JT, Weaver VM. Three-dimensional context regulation of metastasis. Clin Exp Metastasis (2009) 26:35–49. doi: 10.1007/s10585-008-9209-8
98. Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature (1980) 284:67–8. doi: 10.1038/284067a0
99. Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol (2009) 7:S44–7. doi: 10.1016/j.cgh.2009.07.039
100. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol (2012) 196:395–406. doi: 10.1083/jcb.201102147
101. Ishihara S, Haga H. Matrix stiffness contributes to cancer progression by regulating transcription factors. Cancers (2022) 14:1049. doi: 10.3390/cancers14041049
102. Knudson W, Biswas C, Toole BP. Interactions between human tumor cells and fibroblasts stimulate hyaluronate synthesis. Proc Natl Acad Sci USA (1984) 81:6767–71. doi: 10.1073/pnas.81.21.6767
103. Cui D, Huang Z, Liu Y, Ouyang G. The multifaceted role of periostin in priming the tumor microenvironments for tumor progression. Cell Mol Life Sci (2017) 74:4287–91. doi: 10.1007/s00018-017-2646-2
104. Deryugina EI, Bourdon MA. Tenascin mediates human glioma cell migration and modulates cell migration on fibronectin. J Cell Sci (1996) 109(Pt 3):643–52. doi: 10.1242/jcs.109.3.643
105. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med (1995) 1:27–31. doi: 10.1038/nm0195-27
106. Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow–derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med (2001) 7:1194–201. doi: 10.1038/nm1101-1194
107. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
109. Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol (2000) 156:1345–62. doi: 10.1016/s0002-9440(10)65005-5
110. O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell (1994) 79:315–28. doi: 10.1016/0092-8674(94)90200-3
111. Eck SM, Hoopes PJ, Petrella BL, Coon CI, Brinckerhoff CE. Matrix metalloproteinase-1 promotes breast cancer angiogenesis and osteolysis in a novel in vivo model. Breast Cancer Res Treat (2009) 116:79–90. doi: 10.1007/s10549-008-0085-3
112. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2:737–44. doi: 10.1038/35036374
113. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA (2007) 104:20262–7. doi: 10.1073/pnas.0706438104
114. Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, Shalinsky DR, et al. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res (2004) 64:1675–86. doi: 10.1158/0008-5472.can-03-0160
115. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med (2005) 9:267–85. doi: 10.1111/j.1582-4934.2005.tb00355.x
116. Silletti S, Kessler T, Goldberg J, Boger DL, Cheresh DA. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. proc. Natl Acad Sci USA (2001) 98:119–24. doi: 10.1073/pnas.98.1.119
117. Dong Z, Kumar R, Yang X, Fidler IJ. Macrophage-derived metalloelastase is responsible for the generation of angiostatin in Lewis lung carcinoma. Cell (1997) 88:801–10. doi: 10.1016/S0092-8674(00)81926-1
118. Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA (2001) 98:1024–9. doi: 10.1073/pnas.98.3.1024
119. Heljasvaara R, Nyberg P, Luostarinen J, Parikka M, Heikkilä P, Rehn M, et al. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp Cell Res (2005) 307:292–304. doi: 10.1016/j.yexcr.2005.03.021
120. Brauer R, Beck IM, Roderfeld M, Roeb E, Sedlacek R. Matrix metalloproteinase-19 inhibits growth of endothelial cells by generating angiostatin-like fragments from plasminogen. BMC Biochem (2011) 12:38. doi: 10.1186/1471-2091-12-38
121. Felbor U, Dreier L, Bryant RA, Ploegh HL, Olsen BR, Mothes W. Secreted cathepsin l generates endostatin from collagen XVIII. EMBO J (2000) 19:1187–94. doi: 10.1093/emboj/19.6.1187
122. Faye C, Inforzato A, Bignon M, Hartmann DJ, Muller L, Ballut L, et al. Transglutaminase-2: a new endostatin partner in the extracellular matrix of endothelial cells. Biochem J (2010) 427:467–75. doi: 10.1042/BJ20091594
123. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA (2003) 100:4766–71. doi: 10.1073/pnas.0730882100
124. Gately S, Twardowski P, Stack MS, Cundiff DL, Grella D, Castellino FJ, et al. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc Natl Acad Sci USA (1997) 94:10868–72. doi: 10.1073/pnas.94.20.10868
125. Patterson BC, Sang QA. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase b/type IV collagenase (MMP-9). J Biol Chem (1997) 272:28823–5. doi: 10.1074/jbc.272.46.28823
126. Lijnen HR, Ugwu F, Bini A, Collen D. Generation of an angiostatin-like fragment from plasminogen by stromelysin-1 (MMP-3). Biochemistry (1998) 37:4699–702. doi: 10.1021/bi9731798
127. Sim BK, O’Reilly MS, Liang H, Fortier AH, He W, Madsen JW, et al. A recombinant human angiostatin protein inhibits experimental primary and metastatic cancer. Cancer Res (1997) 57:1329–34.
128. Sund M, Hamano Y, Sugimoto H, Sudhakar A, Soubasakos M, Yerramalla U, et al. Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. Proc Natl Acad Sci USA (2005) 102:2934–9. doi: 10.1073/pnas.0500180102
129. Xie L, Duncan MB, Pahler J, Sugimoto H, Martino M, Lively J, et al. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc Natl Acad Sci USA (2011) 108:9939–44. doi: 10.1073/pnas.1105041108
130. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb. Perspect Biol (2011) 3:a009712. doi: 10.1101/cshperspect.a009712
131. Tsuchida R, Osawa T, Wang F, Nishii R, Das B, Tsuchida S, et al. BMP4/Thrombospondin-1 loop paracrinically inhibits tumor angiogenesis and suppresses the growth of solid tumors. Oncogene (2014) 33:3803–11. doi: 10.1038/onc.2013.358
132. Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci USA (2001) 98:12485–90. doi: 10.1073/pnas.171460498
133. Sid B, Langlois B, Sartelet H, Bellon G, Dedieu S, Martiny L. Thrombospondin-1 enhances human thyroid carcinoma cell invasion through urokinase activity. Int J Biochem Cell Biol (2008) 40:1890–900. doi: 10.1016/j.biocel.2008.01.023
134. Firlej V, Mathieu JRR, Gilbert C, Lemonnier L, Nakhlé J, Gallou-Kabani C, et al. Thrombospondin-1 triggers cell migration and development of advanced prostate tumors. Cancer Res (2011) 71:7649–58. doi: 10.1158/0008-5472.CAN-11-0833
135. Borsotti P, Ghilardi C, Ostano P, Silini A, Dossi R, Pinessi D, et al. Thrombospondin-1 is part of a slug-independent motility and metastatic program in cutaneous melanoma, in association with VEGFR-1 and FGF-2. Pigment Cell Melanoma Res (2015) 28:73–81. doi: 10.1111/pcmr.12319
136. Huang T, Sun L, Yuan X, Qiu H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget (2017) 8:84546–58. doi: 10.18632/oncotarget.19165
137. Naumov GN, Bender E, Zurakowski D, Kang S-Y, Sampson D, Flynn E, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst (2006) 98:316–25. doi: 10.1093/jnci/djj068
138. Abair TD, Bulus N, Borza C, Sundaramoorthy M, Zent R, Pozzi A. Functional analysis of the cytoplasmic domain of the integrin {alpha}1 subunit in endothelial cells. Blood (2008) 112:3242–54. doi: 10.1182/blood-2007-12-126433
139. Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci USA (2000) 97:2202–7. doi: 10.1073/pnas.040378497
140. Chopra V, Sangarappillai RM, Romero-Canelón I, Jones AM. Lysyl oxidase like-2 (LOXL2): an emerging oncology target. Adv Ther (2020) 3:1900119. doi: 10.1002/adtp.201900119
141. West DC, Kumar S. Hyaluronan and angiogenesis. Ciba Found Symp (1989) 143:187–201. doi: 10.1002/9780470513774.ch12
142. Slevin M, Kumar S, Gaffney J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J Biol Chem (2002) 277:41046–59. doi: 10.1074/jbc.M109443200
143. Koyama H, Hibi T, Isogai Z, Yoneda M, Fujimori M, Amano J, et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: possible involvement of versican/PG-m. Am J Pathol (2007) 170:1086–99. doi: 10.2353/ajpath.2007.060793
144. Golshani R, Lopez L, Estrella V, Kramer M, Iida N, Lokeshwar VB. Hyaluronic acid synthase-1 expression regulates bladder cancer growth, invasion, and angiogenesis through CD44. Cancer Res (2008) 68:483–91. doi: 10.1158/0008-5472.CAN-07-2140
145. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell (2002) 2:289–300. doi: 10.1016/S1535-6108(02)00153-8
146. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
147. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer (2017) 17:302–17. doi: 10.1038/nrc.2017.6
148. Lu X, Kang Y. Organotropism of breast cancer metastasis. J Mammary Gland Biol Neoplasia (2007) 12:153–62. doi: 10.1007/s10911-007-9047-3
149. Chen XW, Yu TJ, Zhang J, Li Y, Chen HL, Yang GF, et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene (2017) 36:5045–57. doi: 10.1038/onc.2017.118
150. González-González L, Alonso J. Periostin: a matricellular protein with multiple functions in cancer development and progression. Front Oncol (2018) 8:225. doi: 10.3389/fonc.2018.00225
151. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H-A, Delaloye J-F, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2012) 481:85–9. doi: 10.1038/nature10694
152. Kudo A. Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell. Cell Mol Life Sci (2011) 68:3201–7. doi: 10.1007/s00018-011-0784-5
153. Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. doi: 10.1038/nature07623
154. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9:285–93. doi: 10.1038/nrc2621
155. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol (2006) 8:1369–75. doi: 10.1038/ncb1507
156. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the “pre-metastatic niche”: within bone and beyond. Cancer Metastasis Rev (2006) 25:521–9. doi: 10.1007/s10555-006-9036-9
157. Carlini MJ, De Lorenzo MS, Puricelli L. Cross-talk between tumor cells and the microenvironment at the metastatic niche. Curr Pharm Biotechnol (2011) 12:1900–8. doi: 10.2174/138920111798377058
158. Wang TN, Qian X, Granick MS, Solomon MP, Rothman VL, Berger DH, et al. Thrombospondin-1 (TSP-1) promotes the invasive properties of human breast cancer. J Surg Res (1996) 63:39–43. doi: 10.1006/jsre.1996.0219
159. Hu C, Wen J, Gong L, Chen X, Wang J, Hu F, et al. Thrombospondin-1 promotes cell migration, invasion and lung metastasis of osteosarcoma through FAK dependent pathway. Oncotarget (2017) 8:75881–92. doi: 10.18632/oncotarget.17427
160. Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov (2013) 3:578–89. doi: 10.1158/2159-8290.CD-12-0476
161. Kang S-Y, Halvorsen OJ, Gravdal K, Bhattacharya N, Lee JM, Liu NW, et al. Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and tsp-1. Proc Natl Acad Sci USA (2009) 106:12115–20. doi: 10.1073/pnas.0903120106
162. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res (2003) 92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D
163. Huang Y, Song N, Ding Y, Yuan S, Li X, Cai H, et al. Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis. Cancer Res (2009) 69:7529–37. doi: 10.1158/0008-5472.CAN-08-4382
164. van Deventer HW, Wu QP, Bergstralh DT, Davis BK, O’Connor BP, Ting JP-Y, et al. C-c chemokine receptor 5 on pulmonary fibrocytes facilitates migration and promotes metastasis via matrix metalloproteinase 9. Am J Pathol (2008) 173:253–64. doi: 10.2353/ajpath.2008.070732
165. Seubert B, Grünwald B, Kobuch J, Cui H, Schelter F, Schaten S, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatol Baltim Md (2015) 61:238–48. doi: 10.1002/hep.27378
166. Odagiri H, Kadomatsu T, Endo M, Masuda T, Morioka MS, Fukuhara S, et al. The secreted protein ANGPTL2 promotes metastasis of osteosarcoma cells through integrin α5β1, p38 MAPK, and matrix metalloproteinases. Sci Signal (2014) 7:ra7. doi: 10.1126/scisignal.2004612
167. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522:106–10. doi: 10.1038/nature14492
168. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
169. Lucotti S, Kenific CM, Zhang H, Lyden D. Extracellular vesicles and particles impact the systemic landscape of cancer. EMBO J (2022) 41:e109288. doi: 10.15252/embj.2021109288
170. Mazumdar A, Urdinez J, Boro A, Migliavacca J, Arlt MJE, Muff R, et al. Osteosarcoma-derived extracellular vesicles induce lung fibroblast reprogramming. Int J Mol Sci (2020) 21:5451. doi: 10.3390/ijms21155451
171. Wu X, Zhou Z, Xu S, Liao C, Chen X, Li B, et al. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett (2020) 478:93–106. doi: 10.1016/j.canlet.2020.03.004
172. Rana S, Malinowska K, Zöller M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia N Y N (2013) 15:281–95. doi: 10.1593/neo.122010
173. Zhang H, Deng T, Liu R, Bai M, Zhou L, Li S, et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun (2017) 8:15016. doi: 10.1038/ncomms15016
174. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res (2011) 71:3792–801. doi: 10.1158/0008-5472.CAN-10-4455
175. Jones MG, Andriotis OG, Roberts JJ, Lunn K, Tear VJ, Cao L, et al. Nanoscale dysregulation of collagen structure-function disrupts mechano-homeostasis and mediates pulmonary fibrosis. eLife (2018) 7:e36354. doi: 10.7554/eLife.36354
176. Nicolas-Boluda A, Vaquero J, Vimeux L, Guilbert T, Barrin S, Kantari-Mimoun C, et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. eLife (2021) 10:e58688. doi: 10.7554/eLife.58688
177. Lukanidin E, Sleeman JP. Building the niche: the role of the S100 proteins in metastatic growth. Semin Cancer Biol (2012) 22:216–25. doi: 10.1016/j.semcancer.2012.02.006
178. Boye K, Nesland JM, Sandstad B, Haugland Haugen M, Mælandsmo GM, Flatmark K. EMMPRIN is associated with S100A4 and predicts patient outcome in colorectal cancer. Br J Cancer (2012) 107:667–74. doi: 10.1038/bjc.2012.293
179. Hibino T, Sakaguchi M, Miyamoto S, Yamamoto M, Motoyama A, Hosoi J, et al. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res (2013) 73:172–83. doi: 10.1158/0008-5472.CAN-11-3843
180. Munshi HG, Wu YI, Ariztia EV, Stack MS. Calcium regulation of matrix metalloproteinase-mediated migration in oral squamous cell carcinoma cells. J Biol Chem (2002) 277:41480–8. doi: 10.1074/jbc.M207695200
181. Yoshida H, Nagaoka A, Kusaka-Kikushima A, Tobiishi M, Kawabata K, Sayo T, et al. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci USA (2013) 110:5612–7. doi: 10.1073/pnas.1215432110
182. Evensen NA, Kuscu C, Nguyen H-L, Zarrabi K, Dufour A, Kadam P, et al. Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein, in cancer cell migration. J Natl Cancer Inst (2013) 105:1402–16. doi: 10.1093/jnci/djt224
183. Tran QK, Ohashi K, Watanabe H. Calcium signalling in endothelial cells. Cardiovasc Res (2000) 48:13–22. doi: 10.1016/s0008-6363(00)00172-3
184. Brisson L, Bański P, Sboarina M, Dethier C, Danhier P, Fontenille M-J, et al. Lactate dehydrogenase b controls lysosome activity and autophagy in cancer. Cancer Cell (2016) 30:418–31. doi: 10.1016/j.ccell.2016.08.005
185. Liu Y, Hua F, Zhan Y, Yang Y, Xie J, Cheng Y, et al. Carcinoma associated fibroblasts small extracellular vesicles with low miR-7641 promotes breast cancer stemness and glycolysis by HIF-1α. Cell Death Discov (2021) 7:176. doi: 10.1038/s41420-021-00524-x
186. Paget S. The distribution of secondary growths in cancer of the breast. 1889 Cancer Metastasis Rev (1989) 8:98–101.
187. Zhou B, Gibson-Corley KN, Herndon ME, Sun Y, Gustafson-Wagner E, Teoh-Fitzgerald M, et al. Integrin α3β1 can function to promote spontaneous metastasis and lung colonization of invasive breast carcinoma. Mol Cancer Res MCR (2014) 12:143–54. doi: 10.1158/1541-7786.MCR-13-0184
188. Sorokin LM, Pausch F, Frieser M, Kröger S, Ohage E, Deutzmann R. Developmental regulation of the laminin alpha5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol (1997) 189:285–300. doi: 10.1006/dbio.1997.8668
189. Wang Z, Xiong S, Mao Y, Chen M, Ma X, Zhou X, et al. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J Pathol (2016) 239:484–95. doi: 10.1002/path.4747
190. Cox TR, Bird D, Baker A-M, Barker HE, Ho MW-Y, Lang G, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res (2013) 73:1721–32. doi: 10.1158/0008-5472.CAN-12-2233
191. Monteran L, Ershaid N, Doron H, Zait Y, Scharff Y, Ben-Yosef S, et al. Chemotherapy-induced complement signaling modulates immunosuppression and metastatic relapse in breast cancer. Nat Commun (2022) 13:5797. doi: 10.1038/s41467-022-33598-x
192. Cucanic O, Farnsworth RH, Stacker SA. The cellular and molecular mediators of metastasis to the lung. Growth Factors Chur Switz (2022) 40:119–52. doi: 10.1080/08977194.2022.2087520
193. Wang Y, Sun Y, Li D, Zhang L, Wang K, Zuo Y, et al. Platelet P2Y12 is involved in murine pulmonary metastasis. PloS One (2013) 8:e80780. doi: 10.1371/journal.pone.0080780
194. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361:eaao4227. doi: 10.1126/science.aao4227
195. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA Of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
196. Hiratsuka S, Tomita T, Mishima T, Matsunaga Y, Omori T, Ishibashi S, et al. Hepato-entrained B220+CD11c+NK1.1+ cells regulate pre-metastatic niche formation in the lung. EMBO Mol Med (2018) 10:e8643. doi: 10.15252/emmm.201708643
197. Jiang K, Chen H, Fang Y, Chen L, Zhong C, Bu T, et al. Exosomal ANGPTL1 attenuates colorectal cancer liver metastasis by regulating kupffer cell secretion pattern and impeding MMP9 induced vascular leakiness. J Exp Clin Cancer Res CR (2021) 40:21. doi: 10.1186/s13046-020-01816-3
198. Sun H, Meng Q, Shi C, Yang H, Li X, Wu S, et al. Hypoxia-inducible exosomes facilitate liver-tropic premetastatic niche in colorectal cancer. Hepatol Baltim Md (2021) 74:2633–51. doi: 10.1002/hep.32009
199. Chen C, Yao X, Xu Y, Zhang Q, Wang H, Zhao L, et al. Dahuang zhechong pill suppresses colorectal cancer liver metastasis via ameliorating exosomal CCL2 primed pre-metastatic niche. J Ethnopharmacol (2019) 238:111878. doi: 10.1016/j.jep.2019.111878
200. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol (1996) 12:697–715. doi: 10.1146/annurev.cellbio.12.1.697
201. Treps L, Edmond S, Harford-Wright E, Galan-Moya EM, Schmitt A, Azzi S, et al. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene (2016) 35:2615–23. doi: 10.1038/onc.2015.317
202. Busatto S, Yang Y, Walker SA, Davidovich I, Lin W-H, Lewis-Tuffin L, et al. Brain metastases-derived extracellular vesicles induce binding and aggregation of low-density lipoprotein. J Nanobiotechnology (2020) 18:162. doi: 10.1186/s12951-020-00722-2
203. Kolb AD, Bussard KM. The bone extracellular matrix as an ideal milieu for cancer cell metastases. Cancers (2019) 11:1020. doi: 10.3390/cancers11071020
204. Kerr BA, Harris KS, Shi L, Willey JS, Soto-Pantoja DR, Byzova TV. Platelet TSP-1 controls prostate cancer-induced osteoclast differentiation and bone marrow-derived cell mobilization through TGFβ-1. Am J Clin Exp Urol (2021) 9:18–31.
205. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante ACT, Kestelman F, Chaves SB, et al. T Cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS One (2013) 8:e68171. doi: 10.1371/journal.pone.0068171
206. Bekkhus T, Martikainen T, Olofsson A, Franzén Boger M, Vasiliu Bacovia D, Wärnberg F, et al. Remodeling of the lymph node high endothelial venules reflects tumor invasiveness in breast cancer and is associated with dysregulation of perivascular stromal cells. Cancers (2021) 13:211. doi: 10.3390/cancers13020211
207. Leary N, Walser S, He Y, Cousin N, Pereira P, Gallo A, et al. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J Extracell Vesicles (2022) 11:e12197. doi: 10.1002/jev2.12197
208. Feygenzon V, Loewenstein S, Lubezky N, Pasmanic-Chor M, Sher O, Klausner JM, et al. Unique cellular interactions between pancreatic cancer cells and the omentum. PloS One (2017) 12:e0179862. doi: 10.1371/journal.pone.0179862
209. Bortot B, Apollonio M, Rampazzo E, Valle F, Brucale M, Ridolfi A, et al. Small extracellular vesicles from malignant ascites of patients with advanced ovarian cancer provide insights into the dynamics of the extracellular matrix. Mol Oncol (2021) 15:3596–614. doi: 10.1002/1878-0261.13110
210. Bunatova K, Pesta M, Kulda V, Topolcan O, Vrzalova J, Sutnar A, et al. Plasma TIMP1 level is a prognostic factor in patients with liver metastases. Anticancer Res (2012) 32:4601–6.
211. Qin L, Wang Y, Yang N, Zhang Y, Zhao T, Wu Y, et al. Tissue inhibitor of metalloproteinase-1 (TIMP-1) as a prognostic biomarker in gastrointestinal cancer: a meta-analysis. PeerJ (2021) 9:e10859. doi: 10.7717/peerj.10859
212. Grünwald B, Harant V, Schaten S, Frühschütz M, Spallek R, Höchst B, et al. Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via CD63 signaling to create a premetastatic niche in the liver. Gastroenterology (2016) 151:1011–1024.e7. doi: 10.1053/j.gastro.2016.07.043
213. Charan M, Dravid P, Cam M, Setty B, Roberts RD, Houghton PJ, et al. Tumor secreted ANGPTL2 facilitates recruitment of neutrophils to the lung to promote lung pre-metastatic niche formation and targeting ANGPTL2 signaling affects metastatic disease. Oncotarget (2020) 11:510–22. doi: 10.18632/oncotarget.27433
214. Kasurinen A, Tervahartiala T, Laitinen A, Kokkola A, Sorsa T, Böckelman C, et al. High serum MMP-14 predicts worse survival in gastric cancer. PloS One (2018) 13:e0208800. doi: 10.1371/journal.pone.0208800
215. Ren F, Tang R, Zhang X, Madushi WM, Luo D, Dang Y, et al. Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis. PloS One (2015) 10:e0135544. doi: 10.1371/journal.pone.0135544
216. Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi M, Hernandez J, et al. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell (2020) 182:1044–1061.e18. doi: 10.1016/j.cell.2020.07.009
217. Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA, et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8+ T cell exhaustion. Nat Commun (2020) 11:4520. doi: 10.1038/s41467-020-18298-8
218. Jurj A, Ionescu C, Berindan-Neagoe I, Braicu C. The extracellular matrix alteration, implication in modulation of drug resistance mechanism: friends or foes? J Exp Clin Cancer Res CR (2022) 41:276. doi: 10.1186/s13046-022-02484-1
219. He X, Yang Y, Han Y, Cao C, Zhang Z, Li L, et al. Extracellular matrix physical properties govern the diffusion of nanoparticles in tumor microenvironment. Proc Natl Acad Sci USA (2023) 120:e2209260120. doi: 10.1073/pnas.2209260120
220. Cox TR, Erler JT. Fibrosis, cancer and the premetastatic niche. Breast Cancer Manage (2014) 3:453–5. doi: 10.2217/bmt.14.36
221. Zhang R, Wang Z-Y, Li Y-H, Lu Y-H, Wang S, Yu W-X, et al. Usefulness of dynamic contrast-enhanced magnetic resonance imaging for predicting treatment response to vinorelbine-cisplatin with or without recombinant human endostatin in bone metastasis of non-small cell lung cancer. Am J Cancer Res (2016) 6:2890–900.
222. Li K, Shi M, Qin S. Current status and study progress of recombinant human endostatin in cancer treatment. Oncol Ther (2018) 6:21–43. doi: 10.1007/s40487-017-0055-1
223. Pu X, Wang Q, Liu L, Chen B, Li K, Zhou Y, et al. Rh-Endostatin plus camrelizumab and chemotherapy in first-line treatment of advanced non-small cell lung cancer: a multicenter retrospective study. Cancer Med (2022) 12(7):7724–33. doi: 10.1002/cam4.5526
224. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
225. Hou X, Du H, Quan X, Shi L, Zhang Q, Wu Y, et al. Silibinin inhibits NSCLC metastasis by targeting the EGFR/LOX pathway. Front Pharmacol (2018) 9:21. doi: 10.3389/fphar.2018.00021
226. Mas-Moruno C, Rechenmacher F, Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med Chem (2010) 10:753–68. doi: 10.2174/187152010794728639
227. Minea RO, Helchowski CM, Zidovetzki SJ, Costa FK, Swenson SD, Markland FS. Vicrostatin - an anti-invasive multi-integrin targeting chimeric disintegrin with tumor anti-angiogenic and pro-apoptotic activities. PloS One (2010) 5:e10929. doi: 10.1371/journal.pone.0010929
228. Barkan D, Chambers AF. β1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res Off J Am Assoc Cancer Res (2011) 17:7219–23. doi: 10.1158/1078-0432.CCR-11-0642
229. Wang H, Pan J, Barsky L, Jacob JC, Zheng Y, Gao C, et al. Characteristics of pre-metastatic niche: the landscape of molecular and cellular pathways. Mol Biomed (2021) 2:3. doi: 10.1186/s43556-020-00022-z
230. Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell (2019) 35:33–45.e6. doi: 10.1016/j.ccell.2018.12.001
231. Xu M, Hu K, Liu Y, Huang Y, Liu S, Chen Y, et al. Systemic metastasis-targeted nanotherapeutic reinforces tumor surgical resection and chemotherapy. Nat Commun (2021) 12:3187. doi: 10.1038/s41467-021-23466-5
232. Zhou Y, Ke P, Bao X, Wu H, Xia Y, Zhang Z, et al. Peptide nano-blanket impedes fibroblasts activation and subsequent formation of pre-metastatic niche. Nat Commun (2022) 13:2906. doi: 10.1038/s41467-022-30634-8
Keywords: pre-metastatic niche, extracellular matrix, systemic disease, extracellular vesicles, cancer metastasis
Citation: Patras L, Paul D and Matei IR (2023) Weaving the nest: extracellular matrix roles in pre-metastatic niche formation. Front. Oncol. 13:1163786. doi: 10.3389/fonc.2023.1163786
Received: 11 February 2023; Accepted: 15 May 2023;
Published: 07 June 2023.
Edited by:
Bruce Zetter, Harvard Medical School, United StatesReviewed by:
Randolph Watnick, Boston Children’s Hospital and Harvard Medical School, United StatesCopyright © 2023 Patras, Paul and Matei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Doru Paul, ZG9wOTA1NEBtZWQuY29ybmVsbC5lZHU=; Irina R. Matei, aXJtMjIyNEBtZWQuY29ybmVsbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.