
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 26 June 2023
Sec. Genitourinary Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1162644
This article is part of the Research Topic Impact of DNA Damage Repair Alteration on Prostate Cancer Progression and Metastasis View all 5 articles
 Natalia Lukashchuk1*
Natalia Lukashchuk1* Alan Barnicle1Carrie A. Adelman1Joshua Armenia2Jinyu Kang3
Alan Barnicle1Carrie A. Adelman1Joshua Armenia2Jinyu Kang3 J. Carl Barrett4Elizabeth A. Harrington1
J. Carl Barrett4Elizabeth A. Harrington1Prostate cancer is among the most common diseases worldwide. Despite recent progress with treatments, patients with advanced prostate cancer have poor outcomes and there is a high unmet need in this population. Understanding molecular determinants underlying prostate cancer and the aggressive phenotype of disease can help with design of better clinical trials and improve treatments for these patients. One of the pathways often altered in advanced prostate cancer is DNA damage response (DDR), including alterations in BRCA1/2 and other homologous recombination repair (HRR) genes. Alterations in the DDR pathway are particularly prevalent in metastatic prostate cancer. In this review, we summarise the prevalence of DDR alterations in primary and advanced prostate cancer and discuss the impact of alterations in the DDR pathway on aggressive disease phenotype, prognosis and the association of germline pathogenic1 alterations in DDR genes with risk of developing prostate cancer.
Prostate cancer is the third most common cancer worldwide, with 1,414,259 new cases and 375,304 deaths in 2020, and the 5th leading cause of cancer deaths worldwide (1, 2). Approximately 80% of men with prostate cancer are diagnosed with localised prostate cancer, and their 10-year survival is up to 99% if diagnosed early. Approximately 10–20% of men with advanced prostate cancer will develop castration-resistant prostate cancer (CRPC) within five years, and at least 84% of these men will have metastases at the time of CRPC diagnosis. Men with metastatic CRPC have poor outcomes (3–5).
Metastatic prostate cancer is a broad term used to describe a range of advanced disease states that are no longer organ-confined. This group includes de novo metastatic castration-sensitive prostate cancer (mCSPC), as well as cancers that progress during or after androgen deprivation therapy (ADT), termed metastatic castration-resistant prostate cancer (mCRPC) (6). Based on clinical trials, median overall survival (OS) for patients with mCRPC is approximately three years, and is even less in the real-world setting (7). Approximately half of patients with mCRPC may receive only one line of active treatment, with diminishing benefit observed with the use of subsequent therapies (5, 8–11).
A variety of life-prolonging agents are approved for the mCRPC population overall, however the most used medicines for patients with mCRPC remain mainly chemotherapy (docetaxel/cabazitaxel) and new hormonal agents (NHAs; e.g abiraterone and enzalutamide) (8, 12–14). Once patients with mCRPC have failed NHA, the benefit from approved therapeutic options appears substantially diminished (12, 15–22). As such, there is a high unmet medical need for patients who have progressed after NHA treatment, and many efforts to find more effective treatment options have failed in the past decade. Recently, poly(ADP-ribose) polymerase inhibitors (PARPi) treatment as monotherapy demonstrated radiographic progression-free survival (rPFS) and OS improvement in biomarker-selected patient populations with an underlying alteration in the homologous recombination repair (HRR) pathway (HRRm, or BRCA1/2 alterations) (23–29) and have been approved as monotherapy for patients with HRR mutant (HRRm) mCRPC post NHA (25, 30, 31). The mechanism of action of PARP inhibitors as monotherapy is described in Figure 1. The androgen receptor, in addition to its role in binding androgen and stimulating prostate cancer cell growth (32), also contributes towards the general repair of DNA damage, including damage not normally repaired by HRR (33–35). This demonstrates that androgen receptor and PARP are both important for the repair of DNA damage in prostate cancer cells and highlights the importance of crosstalk (interaction) between both mechanisms. This evidence provided the rationale for the clinical development of co-administration of NHAs and PARP inhibitors (36). The clinical benefit of PARPi in combination with NHA has been demonstrated in biomarker-selected and biomarker-unselected populations in first-line mCRPC (Table 1) (36–39) and approvals have been granted in some regions (40).
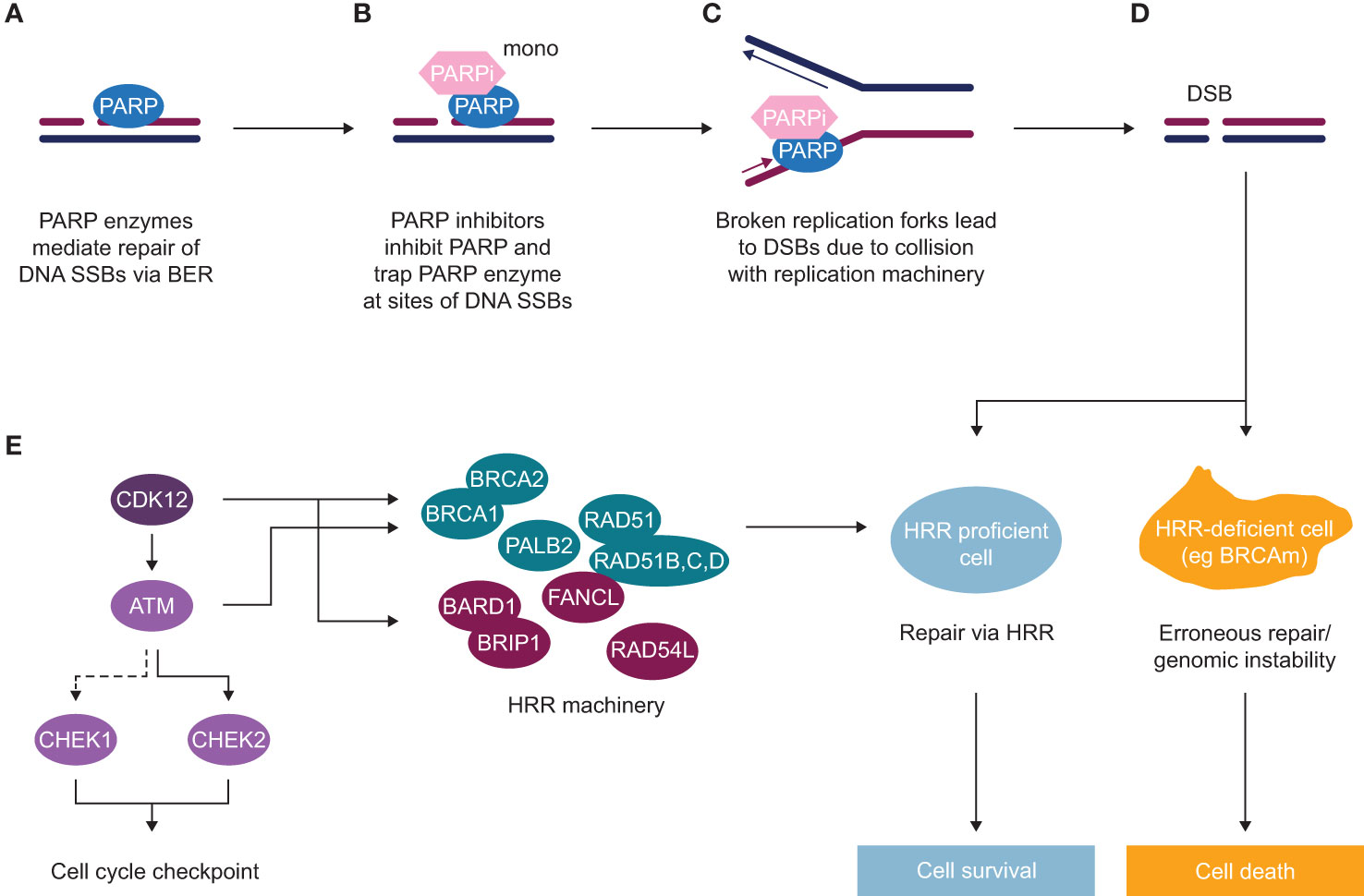
Figure 1 Mechanism of action of PARP inhibitors (as monotherapy) in HRR deficient background (A) Poly(ADP-ribose) polymerase (PARP) enzymes are recruited to sites of DNA single-strand breaks (SSBs) and mediate their repair via base excision repair (BER). (B) PARP inhibitors inhibit PARP and trap PARP enzyme at sites of SSBs. (C) In replicating cells, due to collision of trapped PARP with replication machinery, broken replication forks occur, which lead to double-strand breaks (DSBs). (D) In normal cells or HRR proficient cells, such DSBs get repaired via an accurate DNA repair process of HRR, with some of the key HRR factors depicted in panel (E) In HRR-deficient cells, such as BRCA mutant cells or with alteration in another HRR gene, lack of efficient HRR repair leads to erroneous DNA repair and subsequent genomic instability and cell death. (E) HRR is an accurate DNA repair pathway, which operates in S/G2 cells where the sister chromatid is present. HRR factors include DNA damage sensors and factors involved in direct repair [BRCA1, BRCA2, PALB2, RAD51 and RAD51 paralogues (B–D)], as well as DNA repair by either interacting with key HRR factors (BARD1, BRIP1, FANCL, RAD54L) or regulating HRR gene expression (CDK12), as well as key kinases inducing cell cycle arrest to allow repair to occur (ATM, CHEK1, CHEK2).
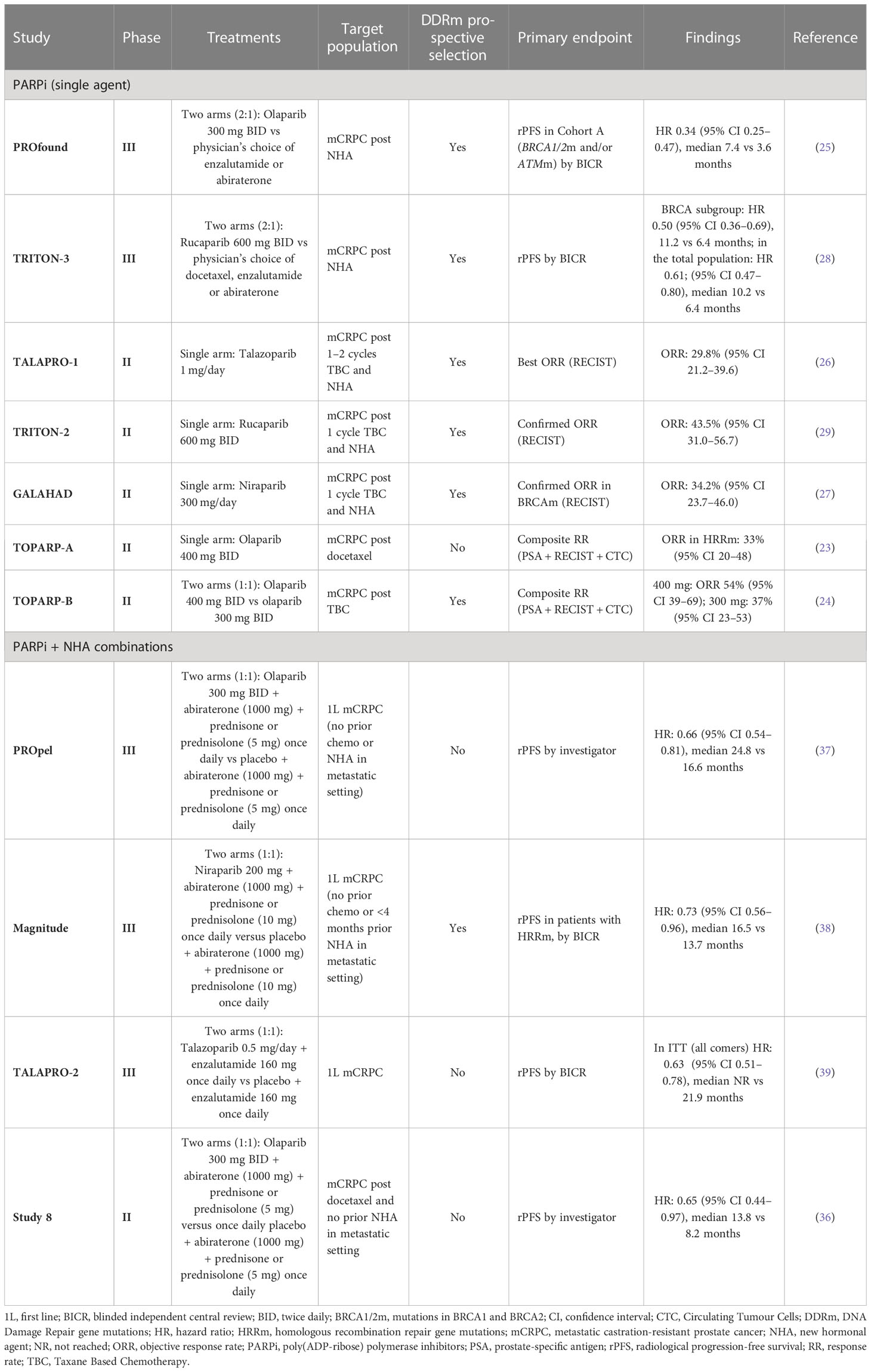
Table 1 Clinical trials involving PARP inhibitors in mCRPC evaluating the predictive value of BRCA mutation/HRR mutation/HRD status.
Despite recent progress in treatments for metastatic disease, there is a high unmet need in this patient population (5, 41). Understanding the biology underlying primary and metastatic prostate cancer, and the differences in prognosis, can help improve prostate cancer treatment and patient outcomes.
Comprehensive molecular characterisation through genetic profiling has revealed a complex and heterogenous genomic landscape of prostate cancer. Multiple landmark genomics studies have identified some of the most recurrent altered genes and pathways in advanced prostate cancer, including genes involved in androgen signalling (50%), PI3K signalling (40%), the cell cycle (24%), WNT/beta-catenin signalling (19%), as well as genes involved in DNA damage response (DDR; 27%), with significant enrichment of all pathways observed in mCRPC (42–46). Alterations in the RAS pathway, including hotspots in BRAF or deleterious alterations in NF1 or RASA1 are detected at lower prevalence than other genes, at around 8% (43). These studies have also highlighted the presence of distinct genomic subtypes defined by rearrangements involving the ERG transcription factor (46%), or hotspot mutations in SPOP (8–11%) and FOXA1 (3%) (44). Due to the highly complex nature of this disease, patients with prostate cancer could greatly benefit from better means of molecular stratification to better select appropriate anti-cancer therapies.
Here we discuss alterations in the DDR pathway, which are frequent in prostate cancer and particularly in advanced stages of disease. DDR is a tightly coordinated pathway that enables cells to control and regulate DNA damage that arises every day. Accumulating damage can lead to mutations and promote genomic instability, which is one of the hallmarks of cancer development (47, 48). Alterations in DDR genes are found to be frequently mutated in many types of cancer, including prostate cancer, where around 23–31% of patients with advanced prostate cancer have been reported to have alterations in DDR genes (43, 45, 46, 49).
As reviewed recently by Morgado & Mateo (50), DDR mutant cancer has been a term used broadly to describe genomic alterations in any gene involved in DDR, including HRR or mismatch repair (MMR) alterations, which have different implications and therapeutic targets in primary and metastatic prostate cancer. Within DDR genes, alterations in genes involved in HRR are most prevalent (23–28%) in mCRPC, with alterations in other pathways of DDR, such as MMR (3–4%) or Fanconi anaemia (FA; around 5%), found at lower prevalence (42, 43, 49, 51, 52).
DNA repair is a complex process that involves sensing DNA damage and downstream signalling cascades to promote DNA repair by recruiting DDR factors and triggering cell cycle checkpoints to allow cells to repair DNA (53, 54). Cancer cells often deregulate the DDR pathway via genomic alterations or epigenetic silencing of DDR genes, which can lead to genomic instability – one of the hallmarks of tumourigenesis (47). Alterations in some of the DDR genes, particularly genes involved in HRR in prostate cancer, are associated with worse prognosis and a higher likelihood of developing metastatic disease (55). The HRR pathway is an accurate pathway that regulates the repair of DNA damage, such as double-strand breaks (DSBs). This pathway relies on the presence of sister chromatid, and therefore only operates in S and G2 stages of the cell cycle when the homologous chromatid is available, whereas the NHEJ pathway takes place in all stages of the cell cycle or in quiescent cells. The HRR pathway is also required for repair of DSBs arising during inter-strand crosslink repair, a process that includes FA factors and other pathways such as nucleotide excision repair and translesion synthesis (47, 48, 56). There are multiple DDR factors that have a direct and indirect role in the HRR pathway: DNA damage sensors (i.e. MRN complex and ATM), signal mediator proteins (i.e. BARD1, BRCA1, PALB2, BRCA2, FANCL, RAD54L), effector proteins directly involved in DNA repair (i.e. RAD51, RAD54L) via strand invasion and replication fork stabilisation, downstream signalling to trigger cell cycle checkpoints (ATM, CHEK2, CHEK1) or regulating transcription of HRR genes (i.e. CDK12); (Figure 1E) (57, 58).
Multiple genes in the HRR pathway are altered in several types of cancers, including in prostate cancer; these include: BRCA1, BRCA2, ATM, CDK12, PALB2, BRIP1, CHEK1, CHEK2, RAD51B, RAD51C, RAD51D, RAD54L, BARD1 and FANCL (Table 2).
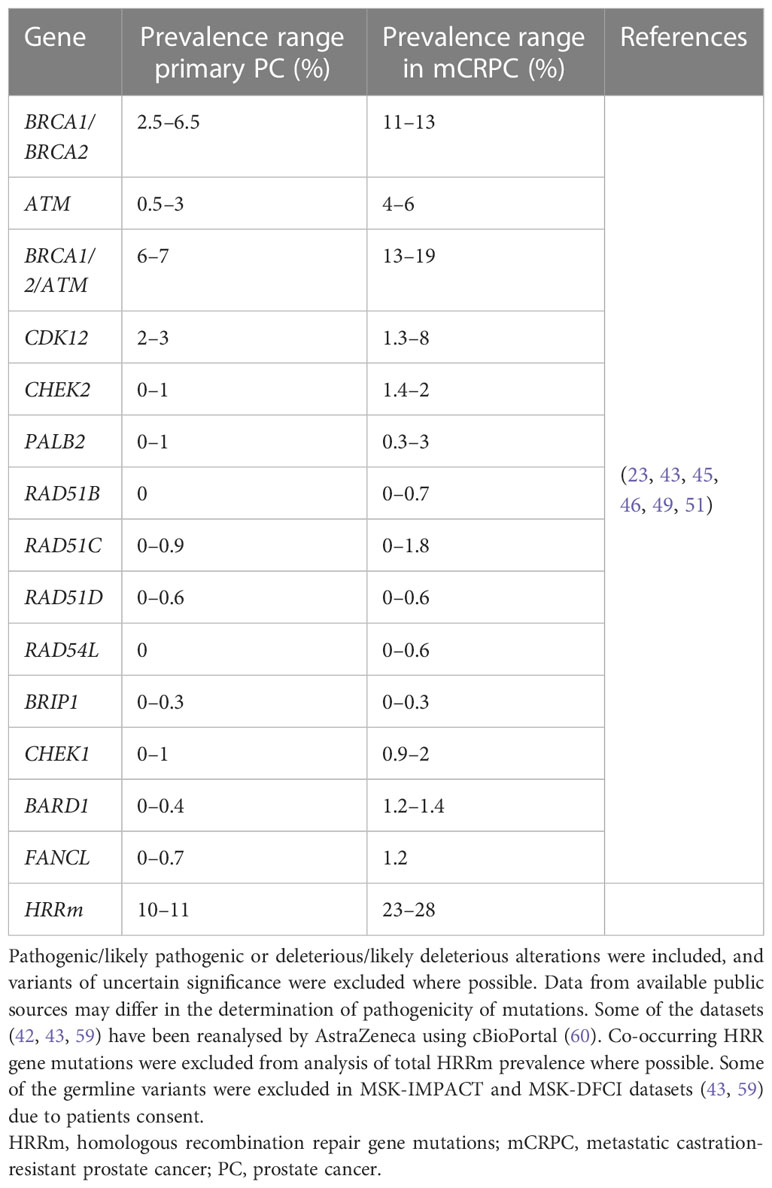
Table 2 Prevalence of HRRm in primary and metastatic prostate cancer.
The most well characterised genes involved in HRR are tumour suppressor genes BRCA1 and BRCA2 (also referred to as ‘BRCA’ genes), alterations in which have been associated with prostate cancer as well as breast, ovarian and pancreatic cancer (61). BRCA1 and BRCA2 proteins play a key role in HRR, where BRCA1 plays a role in the early step of determining DSB repair pathway choice to promote resection and channel it to HRR, for which interaction with BARD1 is important. Later, BRCA1 interacts with PALB2 to bring BRCA2, which promotes RAD51 filament formation and strand invasion, a key step in HRR (62, 63). RAD54L plays a supportive role by promoting RAD51 filament stabilisation (58) BARD1 and BRIP1 are important BRCA1 interacting partners, promoting repair pathway choice, DNA repair and DDR checkpoints (64, 65). There are other important HRR genes that play a direct or indirect role in HRR, such as a kinase, ATM, which is involved in response to DSBs by phosphorylating key DDR proteins to propagate signalling to promote repair or to arrest the cell cycle. ATM directs repair of DSBs associated with replication to HRR by promoting efficient DNA resection (66). One of the key downstream targets of ATM is CHEK2 kinase, the phosphorylation of which leads to activation of the G1 checkpoint (67–70). On the other hand, CHEK1 kinase is phosphorylated by ATR and is involved in triggering G2/M as well as intra-S checkpoints (68, 69, 71–73). CDK12, a kinase with an indirect role in HRR, regulates transcription of HRR genes (74, 75). Alterations in CDK12 lead to a unique tandem duplication genotype (76).
In prostate cancer, alterations in genes involved in HRR are enriched in later/advanced stages of disease compared with primary prostate cancer (46, 55).
Around 10−11% of patients with primary cancer harbour HRR alterations, whereas between 23% and 28% of patients with mCRPCs have loss-of-function mutations in genes involved in the HRR pathway of DDR in the tumour (Table 2).
In the PROfound clinical trial (a randomised, open-label, Phase III trial evaluating the PARP inhibitor olaparib in men with mCRPC who had disease progression while receiving a new hormonal agent, e.g. enzalutamide or abiraterone), an alteration in one or more of 152 prespecified HRR genes was detected in 28% of over 4000 patients screened (25). Very similar prevalence of HRRm was also observed in the Phase III PROpel trial (double-blind, Phase III trial of abiraterone and olaparib versus abiraterone and placebo in patients with mCRPC in the first-line setting), where patients were enrolled regardless of HRR status; 28% of patients enrolled had alterations in HRR genes (37). Comparable prevalence of HRR/DDR genomic alterations were also observed in other clinical trials and datasets involving advanced prostate cancer tumours. In the TOPARP-A clinical trial (Phase II trial in which patients with mCRPC were treated with olaparib), 33% of patients harboured genomic alterations specific to DDR genes (23). Chung et al. used real-world data from routine prospective genomic profiling and showed that 23% of patients had alterations in genes involved in the HRR pathway (49) and a similar prevalence of HRR gene mutations was also demonstrated by Abida et al. in 429 patients with mCRPC (42).
Mutations in the BRCA genes (BRCA1 and/or BRCA2) are the most prevalent HRR gene mutations in mCRPC (with BRCA2 more prevalent than BRCA1), with ATM being the second most frequently mutated (23, 43, 46, 49). In mCRPC, prevalence of BRCAm ranges from around 11% to 13%, and ATMm from 4% to 6% (Table 2). The next most prevalent mutations in HRR genes in mCRPC are CDK12 (1.3–8%), CHEK2 (1.4–2%), PALB2 (0.3–3%), and CHEK1 (0.9–2%) (Table 2). The prevalence of alterations in other HRR genes in prostate cancer is low and ranges between 0% and 1.8% (Table 2).
In summary, between around 23–28% of patients have deleterious alterations in HRR/DDR genes in metastatic prostate cancer, with most studies reporting alterations in the tumour (Table 2) (25, 37, 42, 49)
The origin of alterations in DDR genes found in tumour can be either germline or somatic. The relative ratio of pathogenic germline to somatic mutation events in DDR is highly dependent on the HRR genes interrogated. Lai et al. (52) identified that the ratio of germline to somatic alterations in BRCA1/2 genes was roughly 1:1, whereas other HRR genes such as ATM and CDK12 had a much higher ratio of HRRm that were somatic in origin relative to germline with 70% of ATM alterations and 89% of CDK12 alterations being somatic, based on a validated computational algorithm (77). The prevalence of germline alterations in HRR genes in mCRPC is 12% (78), with 6% in BRCA1/2, ATM 1.6%, CHEK2 1.9% and other HRR genes below 1%. In primary prostate cancer the overall prevalence of germline DDR alterations is lower at 4.6%, with highest prevalence of 1% in ATM, and 0.6% in BRCA1 and 0.2% in BRCA2 (78).
Loss of function of both alleles is needed for inactivation of HRR gene function. Biallelic loss-of-function rate of HRR gene mutations in prostate cancer is high at 73%, with the highest rate in BRCA2 (>90%) and ATM (around 75%); beyond these, the rate is variable (ranging between under 10% for BRIP1 and over 60% for CDK12) (52), suggesting alterations in HRR genes are important drivers of tumourigenesis for prostate cancer. Biallelic inactivation rate was high for both germline and somatic BRCA alterations (52, 79), suggesting both play an important role in prostate cancer tumourigenesis.
Beyond HRR gene alterations described above, epigenetic silencing of another HRR gene – XRCC3 – has been reported in prostate adenocarcinoma (80). XRCC3 is one of the RAD51 paralogues and its role in HRR has been previously reported (81). Interestingly, XRCC3 alterations were mutually exclusive with alterations in BRCA genes in a pan-cancer dataset, similar to other HRR genes, which confirms its functional importance in the HRR pathway. Depletion of this gene sensitised cells to PARP inhibition preclinically (80), which warrants further investigation of loss of XRCC3 expression as a potential biomarker for PARPi sensitivity in clinic.
A consequence of deficiency in the HRR pathway (HRD) is the accumulation of DNA damage leading to genomic instability signatures, or scars, over time. In addition to alterations in HRR genes, HRD/genomic instability can be another way to identify patients who might benefit from PARP inhibitors. HRD has been associated with a clinical benefit for PARP inhibitor treatment in ovarian cancer (82–85) and to platinum-based chemotherapy in breast cancer (86); however, there is lack of evidence of clinical utility of HRD in other tumour types.
In prostate cancer, genomic instability/HRD (as measured by genome-wide loss of heterozygosity [gLOH]) is generally lower than in ovarian, breast or pancreatic cancers (52, 79). Biallelic alterations in BRCA genes or HRR genes are associated with higher gLOH/HRD scores compared with BRCA wild type (wt) or HRR wt prostate cancer tumours, respectively, which is consistent with these alterations leading to deficiency in the HRR pathway (52, 79). However, there are no data on the clinical utility of gLOH/HRD scores in prostate cancer. The distinction between HRD-positive and HRD-negative tumours, based on genomic instability in prostate cancer, is not clear relative to ovarian and breast cancer. The cut-off to identify HRD-positive tumours in prostate cancer would need to be robustly defined, and this is likely to be more challenging than in ovarian cancer due to a lower dynamic range in prostate cancer. Interestingly, Zurita et al. assessed the relationship between genome instability and clinical features and identified that higher genomic instability was associated with higher risk of disease progression to CRPC (87). Recently, a functional biomarker of HRR has been developed based on measuring nuclear foci formed by the key HRR factor, RAD51, at DNA damage sites, which is currently being implemented in clinical trials (88).
Beyond HRR, alterations in other genes in the DDR pathway are relatively low, however some are potentially actionable. Deficiency in MMR is the underlying cause of the microsatellite instability-high (MSI-H) phenotype, which is a biomarker of response to immune checkpoint blockade therapy (89). Prevalence of alterations in the MMR pathway (MLH1, MSH2, MSH6, PMS2) or MSI-H in prostate cancer is much lower than HRRm, at around 3–5% (49). Interestingly, mutual exclusivity between genomic instability/HRD and MSI-H has been reported across tumours (79, 90). In prostate cancer, 12.8% of BRCA1 and 3.4% of BRCA2 alterations co-occurred with MSI-H, and 46.3% of MSI-H had at least one HRR gene mutation; however, most BRCA mutations in the MSI-H segment were monoallelic (90, 91).
Enrichment of BRCA1/2, ATM and CDK12 mutations in advanced prostate cancer has been documented in the literature by several studies (43, 45, 49, 92). Armenia et al. performed a large study that analysed 680 primary tumours and 333 metastatic samples and identified HRR defects in 10% and 27% of the primary and metastatic samples, respectively (43). Similarly, Abida et al. (45) observed an increase in HRR alterations according to disease progression, with 10% of HRR alterations detected in primary tumours, 14% in castration-sensitive prostate cancer and 27% in CRPC. The high representation of BRCA2 mutations in advanced/metastatic prostate cancer is considered to be a consequence of BRCA2 mutations being associated with a particularly aggressive phenotype (49, 93–95) rather than these mutations (e.g. androgen receptor mutations and amplifications) being acquired under treatment with standard therapies (96). An increase in the prevalence of DDR alterations in metastatic compared with primary prostate cancer could either be due to disease progression or therapy exposure, or could be due to a worse prognosis for patients with mutations/alterations in DDR genes (DDRm) prostate cancer. Recent analysis of paired tumour samples from patients with prostate cancer (treatment-naïve and metastatic tumour samples) showed that in most cases, alterations in DDR genes were already present in the primary prostate cancer sample, suggesting that this is an early event in tumourigenesis (97). On the contrary, alterations of AR/TP53/RB1 are enriched at later disease stages (97). These data suggest that genomic instability associated with alterations in HRR genes leads to a more aggressive disease, which is more likely to metastasise, highlighting the need to treat those patients early.
Family history is an important risk factor to be considered for development of prostate cancer. Germline alterations in MMR genes (MLH1, MSH2, MSH6, PMS2) and the HRR pathway (BRCA1/2, ATM, PALB2, CHEK2), particularly BRCA and ATM, are associated with increased risk of hereditary prostate cancer, as reported by National Comprehensive Cancer Network (NCCN) guidelines (98). The proportion of patients with prostate cancer with germline mutations in DDR genes increases from around 5% in primary cancer to 12–16% in mCRPC, indicating a more aggressive nature of disease with germline DDR alterations (46, 78, 99).
Men with germline pathogenic BRCA1/2 mutations have an increased risk of prostate cancer and the relative increase in risk of prostate cancer in men <65 years ranges from 1.8-fold to 3.8-fold for germline BRCA1m carriers (100, 101) and from 2.5-fold to 8.6-fold for germline BRCA2m carriers (102–105) compared with non-carriers. A large meta-analysis of 8 cohort, 7 case control, 4 case series, 28 frequency and 11 survival studies found that being a BRCA mutation carrier (BRCA1 and/or BRCA2) was associated with a significant increase in prostate cancer risk (odds ratio [OR] =1.90; 95% CI 1.58, 2.29), with BRCA2 mutations being associated with a greater risk of prostate cancer than BRCA1 (106) (Table 3). Lifetime risk of cancer ranged between 19–61% for BRCA2 carriers, and 7–26% for BRCA1 carriers (108). A prospective study, IMPACT, which is evaluating targeted screening using prostate-specific antigen (PSA) in men with germline pathogenic BRCA1/2 mutations, has reported a higher incidence of prostate cancer in BRCA2 carriers compared with controls at interim analysis (114). Furthermore, germline pathogenic BRCA2 mutations are associated with a particularly aggressive phenotype and poor outcomes (93, 94). Germline pathogenic mutation status of BRCA and ATM distinguishes risk for lethal and indolent prostate cancer and is associated with earlier age at death and shorter survival time (115). Germline pathogenic variants in ATM lead to an approximate fourfold elevated risk of developing prostate cancer, and in addition, they are more likely to develop the disease earlier (109, 116). Beyond BRCA genes, there is evidence of elevated prostate cancer risk for CHEK2 heterozygotes (110, 111) and increased incidence of germline pathogenic CHEK2, BRIP1 and PALB2 mutations in familial cases of prostate cancer (Table 3) (64, 112, 117).
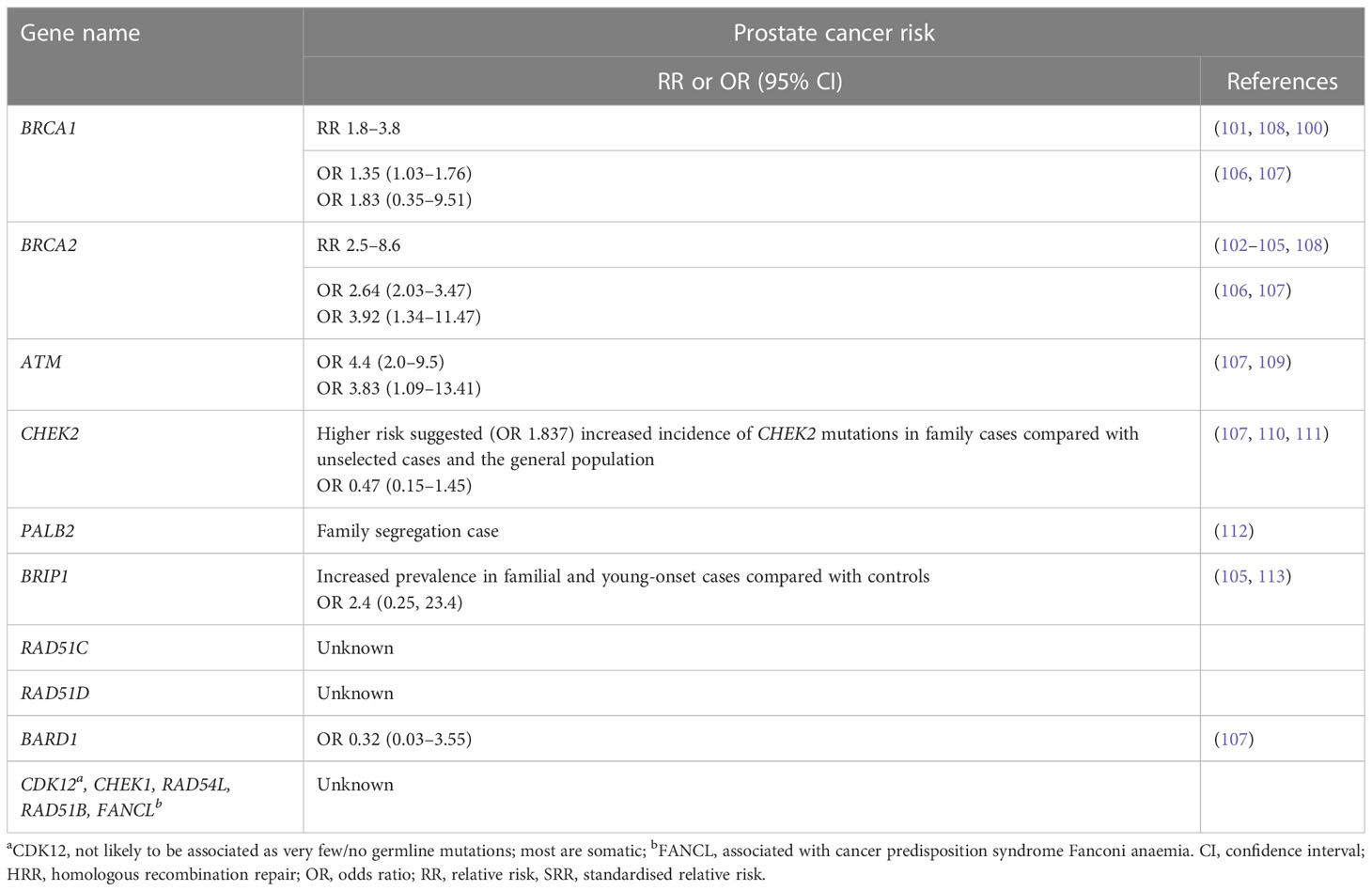
Table 3 Hereditary cancer risk for HRR genes.
In addition to prostate cancer, HRR genes are associated with increased risk of breast, ovarian and pancreatic cancer (BRCA1, BRCA2, ATM, BRIP1, PALB2, BARD1, CHEK2, RAD51C and RAD51D). Alterations in some of the DDR genes are associated with cancer predisposition syndromes (e.g. BRCA1, BRCA2, ATM, FANCL, PALB2, RAD51C, BRIP1, FA genes, MMR genes) (64, 100, 101, 110, 115, 116, 118–127). Germline pathogenic mutations in MMR genes (MLH1, MSH2, MSH6 and PMS2) are the key cause of Lynch syndrome (LS) – an inherited cancer predisposition syndrome leading to increased risk of particularly colorectal cancer and other LS-associated cancers. Patients with LS have a twofold elevated risk of incidence of prostate cancer compared with the general population (128).
Among HRR alterations, germline pathogenic BRCA mutations in particular are associated with a more aggressive prostate cancer and worse outcomes for patients with localised prostate cancer and mCRPC compared to non-carriers. The presence of a germline pathogenic BRCA mutation in prostate cancer is associated with a more aggressive phenotype, such as higher Gleason scores, nodal involvement and presence of metastases at diagnosis, as shown in large retrospective analyses (93, 94). BRCA2 was reported to be an independent prognostic factor in a multivariate analysis, where patients with germline pathogenic BRCA mutations had overall worse outcomes than non-BRCAm patients in a cohort with localised disease as well as in the overall cohort (93, 94). Another study reported that the combined rate of germline BRCA/ATM alterations was significantly higher in patients with lethal prostate cancer than in patients with localised prostate cancer, and patients with germline BRCA/ATM alterations with either localised disease or a diagnosis with metastases, had a shorter prostate-cancer-specific survival compared with non-carriers (129).
Once prostate cancer becomes castration-resistant and progresses to a metastatic stage (mCRPC) the disease is not curable and treatment must focus on extending life, delaying disease progression and improving quality of life (130, 131). Germline mutations in HRRm have been found in around 12% of patients with mCRPC (78). There has been conflicting evidence reported for the association of HRRm and prognosis for patients with mCRPC. Annala et al. reported worse outcomes for germline DDR carriers (17/22 were gBRCAm) compared with non-carriers when treated with first-line androgen receptor signaling inhibitors (132). However, more recently Antonarakis et al. reported that patients with BRCA/ATM mutations (n=9) do better on first-line NHA than those without mutations in these genes (133). These findings are based on a relatively small number of patients with HRRm. A retrospective analysis showed similar OS outcomes for patients with metastatic prostate cancer and with germline mutations in DDR genes compared with those without; however, somatic alterations in DDR genes were not assessed, and a significant proportion of patients were treated with PARPi or platinum-based chemotherapy, which might have contributed to better outcomes for patients with DDRm (50, 96). A prospective study, PRO-REPAIR, evaluated prevalence of germline DDR mutations and their impact on outcomes for patients with mCRPC (99). This trial enrolled unselected patients with mCRPC and screened for germline alterations in 107 DDR genes with the primary objective to assess the impact of germline alterations in ATM/BRCA1/BRCA2/PALB2 on cause-specific survival (CSS) from diagnosis of mCRPC. In PRO-REPAIR, 16% of patients had a germline mutation in a HRR gene (most commonly BRCA and ATM alterations). Although numerically the CSS was shorter in patients with ATM/BRCA1/BRCA2, the difference was not statistically significant; however, patients with germline BRCA2 mutation had considerably shorter CSS than non-carriers and BRCA2 was an independent prognostic factor, where sequence and type of treatment may impact the outcomes (99). Castro et al. reported that treatment sequence is important for prognosis, with patients with mCRPC and DDR mutations having worse outcomes overall but better outcomes following first-line NHA treatment (99). These observations might explain conflicting observations in different studies. In addition to alterations in HRR genes, genomic instability might also be associated with worse outcomes, such as with risk of biochemical recurrence and metastases (134, 135).
High unmet clinical need and poor prognosis for patients with prostate cancer and DDR has triggered an active development of targeted treatment options for these patients (136). As described above, based on the principle of synthetic lethality, cells deficient in the HRR pathway are sensitive to PARP inhibitors (137, 138) (Figure 1). PARP inhibitors as monotherapy have demonstrated efficacy in patients with evidence of deficiency in the HRR pathway in the tumour and have been approved for treatment of HRRm mCRPC (136). The clinical benefit of PARPi in combination with NHA has been demonstrated in biomarker-selected and biomarker-unselected populations in first-line mCRPC (Table 1) (36–39), with approval in some regions, providing additional efficacious treatment options for these patients.
Platinum-based chemotherapy also leads to DNA damage, which is repaired by HRR pathway. Increased platinum sensitivity in tumours deficient in HRR pathway (ie BRCAm) has been reported in other tumour types, such as breast or ovarian cancer (139, 140) Retrospective analyses indicated encouraging anti-tumour activity of platinum-based chemotherapy in advanced prostate cancer patients with BRCA alterations or some DDR alterations with higher PSA response rates in patients with DDR/BRCA alterations compared to patients without (141–143), although these findings need to be validated in a prospective setting.
ATM-deficient cells are dependent on ATR activity, which leads to sensitivity to ATR inhibition preclinically; clinical trials with ATR inhibitors as monotherapy or in combinations are ongoing, including in biomarker-selected patients with ATM- or DDR-deficiency in advanced solid tumours, including prostate cancer, as reviewed recently by Ngoi et al. (144).
Anti-PD1 antibody (Pembrolizumab) is approved by the Food and Drug Administration (FDA) to treat cancers with MMR mutations or MSI-H, including prostate cancer (145, 146). CDK12 inactivation results in tandem duplications in the genome leading to increased fusions and mutations and might lead to increased antigens. Clinical trials with immune checkpoint inhibitors are ongoing for patients with CDK12m and advanced prostate cancer (147–149).
Prostate cancer is the third most common cancer worldwide, and metastatic prostate cancer is associated with poor outcomes and high mortality. There are various genomic alterations commonly associated with prostate cancer, and alterations in the HRR pathway of DDR are prevalent in prostate cancer, ranging from 23–28% (25, 42, 43, 46, 78). Germline alterations in several HRR genes, such as BRCA, ATM and others, are associated with increased prostate cancer risk, and are generally associated with worse prognosis for patients with prostate cancer. Alterations in DDR genes in tumours tend to be an early event in prostate tumourigenesis and are associated with more aggressive disease and likelihood of metastasis (97). With current breakthroughs regarding targeted treatments, PARP inhibitors as monotherapy are an option for patients with HRR alterations in mCRPC, who have progressed on NHA (136) and in some countries, are available in combination with NHA in a broad population of 1L mCRPC (36, 55, 99). Clinical trials are ongoing that are evaluating ATR inhibitors in all solid tumours, including prostate cancer, as reviewed by Ngoi et al. (144). Given the association of some DDR genes with worse prognosis, and DDR alterations being an early event in prostate cancer, future clinical trials for patients with DDRm and an earlier stage of disease are important to improve outcomes for these patients.
NL and AB developed the initial draft of the manuscript. NL, AB, EH, CA and JCB made substantial contributions to the concept and content of review. JA, JK and CA contributed to writing parts of the manuscript. All authors contributed to the article and approved the submitted version.
This manuscript was funded by AstraZeneca.
We thank Mudskipper Business LTD medical writing agency for editing support and review of the manuscript. This support was funded by AstraZeneca
NL, AB, CA, JA, JK, JCB and EH are employees of, and hold stock at AstraZeneca.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin (2018) 68:7–30. doi: 10.3322/caac.21442
4. Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers (2021) 7:9. doi: 10.1038/s41572-020-00243-0
5. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract (2011) 65:1180–92. doi: 10.1111/j.1742-1241.2011.02799.x
6. Ku S-Y, Gleave ME, Beltran H. Towards precision oncology in advanced prostate cancer. Nat Rev Urol (2019) 16:645–54. doi: 10.1038/s41585-019-0237-8
7. Ng K, Smith S, Shamash J. Metastatic hormone-sensitive prostate cancer (mHSPC): advances and treatment strategies in the first-line setting. Oncol Ther (2020) 8:209–30. doi: 10.1007/s40487-020-00119-z
8. Beer TM, Armstrong AJ, Rathkopf D, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol (2017) 71:151–4. doi: 10.1016/j.eururo.2016.07.032
9. Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PFA, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol (2015) 16:152–60. doi: 10.1016/S1470-2045(14)71205-7
10. de Bono JS, Smith MR, Saad F, Rathkopf DE, Mulders PFA, Small EJ, et al. Subsequent chemotherapy and treatment patterns after abiraterone acetate in patients with metastatic castration-resistant prostate cancer: Post hoc analysis of COU-AA-302. Eur Urol (2017) 71:656–64. doi: 10.1016/j.eururo.2016.06.033
11. George DJ, Sartor O, Miller K, Saad F, Tombal B, Kalinovský J, et al. Treatment patterns and outcomes in patients with metastatic castration-resistant prostate cancer in a real-world clinical practice setting in the united states. Clin Genitourin Cancer (2020) 18:284–94. doi: 10.1016/j.clgc.2019.12.019
12. Flaig TW, Potluri RC, Ng Y, Todd MB, Mehra M. Treatment evolution for metastatic castration-resistant prostate cancer with recent introduction of novel agents: retrospective analysis of real-world data. Cancer Med (2016) 5:182–91. doi: 10.1002/cam4.576
13. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. New Engl J Med (2013) 368:138–48. doi: 10.1056/NEJMoa1209096
14. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. New Engl J Med (2014) 371:424–33. doi: 10.1056/NEJMoa1405095
15. Mezynski J, Pezaro C, Bianchini D, Zivi A, Sandhu S, Thompson E, et al. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: clinical evidence for cross-resistance? Ann Oncol (2012) 23:2943–7. doi: 10.1093/annonc/mds119
16. Azad AA, Eigl BJ, Leibowitz-Amit R, Lester R, Kollmannsberger C, Murray N, et al. Outcomes with abiraterone acetate in metastatic castration-resistant prostate cancer patients who have poor performance status. Eur Urol (2015) 67:441–7. doi: 10.1016/j.eururo.2014.01.030
17. Schweizer MT, Zhou XC, Wang H, Bassi S, Carducci MA, Eisenberger MA, et al. The influence of prior abiraterone treatment on the clinical activity of docetaxel in men with metastatic castration-resistant prostate cancer. Eur Urol (2014) 66:646–52. doi: 10.1016/j.eururo.2014.01.018
18. Cheng HH, Gulati R, Azad A, Nadal R, Twardowski P, Vaishampayan UN, et al. Activity of enzalutamide in men with metastatic castration-resistant prostate cancer is affected by prior treatment with abiraterone and/or docetaxel. Prostate Cancer Prostatic Dis (2015) 18:122–7. doi: 10.1038/pcan.2014.53
19. Suzman DL, Luber B, Schweizer MT, Nadal R, Antonarakis ES. Clinical activity of enzalutamide versus docetaxel in men with castration-resistant prostate cancer progressing after abiraterone. Prostate (2014) 74:1278–85. doi: 10.1002/pros.22844
20. Moreira RB, Debiasi M, Francini E, Nuzzo PV, Velasco G, Maluf FC, et al. Differential side effects profile in patients with mCRPC treated with abiraterone or enzalutamide: a meta-analysis of randomized controlled trials. Oncotarget (2017) 8:84572–8. doi: 10.18632/oncotarget.20028
21. Parker C, Heidenreich A, Nilsson S, Shore N. Current approaches to incorporation of radium-223 in clinical practice. Prostate Cancer Prostatic Dis (2018) 21:37–47. doi: 10.1038/s41391-017-0020-y
22. Den RB, George D, Pieczonka C, McNamara M. Ra-223 treatment for bone metastases in castrate-resistant prostate cancer. Am J Clin Oncol (2019) 42:399–406. doi: 10.1097/COC.0000000000000528
23. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. New Engl J Med (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
24. Mateo J, Porta N, McGovern UB, Elliott T, Jones RJ, Syndikus I, et al. TOPARP-b: a phase II randomized trial of the poly(ADP)-ribose polymerase (PARP) inhibitor olaparib for metastatic castration resistant prostate cancers (mCRPC) with DNA damage repair (DDR) alterations. J Clin Oncol (2019) 37:5005–5. doi: 10.1200/JCO.2019.37.15_suppl.5005
25. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. New Engl J Med (2020) 382:2091–102. doi: 10.1056/NEJMoa1911440
26. de Bono JS, Mehra N, Scagliotti GV, Castro E, Dorff T, Stirling A, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol (2021) 22:1250–64. doi: 10.1016/S1470-2045(21)00376-4
27. Smith MR, Scher HI, Sandhu S, Efstathiou E, Lara PN, Yu EY, et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol (2022) 23:362–73. doi: 10.1016/S1470-2045(21)00757-9
28. Fizazi K, Piulats JM, Reaume MN, Ostler P, McDermott R, Gingerich JR, et al. Rucaparib or physician’s choice in metastatic prostate cancer. New Engl J Med (2023) 388:719–32. doi: 10.1056/NEJMoa2214676
29. Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol (2020) 38:3763–72. doi: 10.1200/JCO.20.01035
30. Clovis Oncology, Inc. TRITON3 phase 3 trial of rubraca® (rucaparib) achieves primary endpoint in men with metastatic castration-resistant prostate cancer with BRCA or ATM mutations . Available at: https://ir.clovisoncology.com/investors-and-news/news-releases/press-release-details/2022/TRITON3-Phase-3-Trial-of-Rubraca-rucaparib-Achieves-Primary-Endpoint-in-Men-with-Metastatic-Castration-Resistant-Prostate-Cancer-with-BRCA-or-ATM-Mutations/default.aspx (Accessed December 21, 2022).
31. Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. New Engl J Med (2020) 383:2345–57. doi: 10.1056/NEJMoa2022485
32. Westaby D, Fenor de la Maza M de LD, Paschalis A, Jimenez-Vacas JM, Welti J, de Bono J, et al. A new old target: androgen receptor signaling and advanced prostate cancer. Annu Rev Pharmacol Toxicol (2022) 62:131–53. doi: 10.1146/annurev-pharmtox-052220-015912
33. Tarish FL, Schultz N, Tanoglidi A, Hamberg H, Letocha H, Karaszi K, et al. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci Transl Med (2015) 7:312re11. doi: 10.1126/scitranslmed.aac5671
34. Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone–DNA repair circuit governs the response to genotoxic insult. Cancer Discovery (2013) 3:1254–71. doi: 10.1158/2159-8290.CD-13-0108
35. Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discovery (2013) 3:1245–53. doi: 10.1158/2159-8290.CD-13-0172
36. Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol (2018) 19:975–86. doi: 10.1016/S1470-2045(18)30365-6
37. Clarke NW, Armstrong AJ, Thiery-Vuillemin A, Oya M, Shore N, Loredo E, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evidence (2022) 1(9). doi: 10.1056/EVIDOA2200043
38. Chi KN, Rathkopf DE, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol (2022) 40:12–2. doi: 10.1200/JCO.2022.40.6_suppl.012
39. Agarwal N, Azad AA, Carles J, Fay AP, Matsubara N, Heinrich D, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. Lancet (2023). doi: 10.1016/S0140-6736(23)01055-3
40. Lynparza in combination with abiraterone approved in the EU as 1st-line treatment for patients with metastatic castration-resistant prostate cancer . Available at: https://www.astrazeneca.com/media-centre/press-releases/2022/lynparza-approved-in-eu-for-prostate-cancer.html (Accessed March 21, 2023).
41. Liu JM, Lin CC, Liu KL, Lin CF, Chen BY, Chen TH, et al. Second-line hormonal therapy for the management of metastatic castration-resistant prostate cancer: a real-world data study using a claims database. Sci Rep 2020 10:1 (2020) 10:1–7. doi: 10.1038/s41598-020-61235-4
42. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci (2019) 116:11428–36. doi: 10.1073/pnas.1902651116
43. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
44. Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The molecular taxonomy of primary prostate cancer. Cell (2015) 163:1011–25. doi: 10.1016/j.cell.2015.10.025
45. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol (2017) 1:1–16. doi: 10.1200/PO.17.00029
46. Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
47. Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage repair in cancer: from mechanisms to applications. Ann Transl Med (2020) 8:1685–5. doi: 10.21037/atm-20-2920
48. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature (2012) 481:287–94. doi: 10.1038/nature10760
49. Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ, et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol (2019) 3:1–23. doi: 10.1200/PO.18.00283
50. Cresta Morgado P, Mateo J. Clinical implications of homologous recombination repair mutations in prostate cancer. Prostate (2022) 82:S45–59. doi: 10.1002/pros.24352
51. de Bono JS, Fizazi K, Saad F, Shore N, Sandhu SK, Mehra N, et al. Central, prospective detection of homologous recombination repair gene mutations (HRRm) in tumour tissue from >4000 men with metastatic castration-resistant prostate cancer (mCRPC) screened for the PROfound study. Ann Oncol (2019) 30:v328–9. doi: 10.1093/ANNONC/MDZ248.004
52. Lai Z, Brosnan M, Sokol ES, Xie M, Dry JR, Harrington EA, et al. Landscape of homologous recombination deficiencies in solid tumours: analyses of two independent genomic datasets. BMC Cancer (2022) 22:13. doi: 10.1186/s12885-021-09082-y
53. Blackford A, Jackson SP. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol Cell (2017) 66:801–17. doi: 10.1016/j.molcel.2017.05.015
54. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
55. Lang SH, Swift SL, White H, Misso K, Kleijnen J, Quek RGW. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int J Oncol (2019) 55:597–616. doi: 10.3892/IJO.2019.4842/HTML
56. Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Research/Fundamental Mol Mech Mutagenesis (2001) 477:131–53. doi: 10.1016/S0027-5107(01)00115-4
57. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
58. Mazin AV, Mazina OM, Bugreev DV, Rossi MJ. Rad54, the motor of homologous recombination. DNA Repair (Amst) (2010) 9:286–302. doi: 10.1016/j.dnarep.2009.12.006
59. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol (2017) 1:1–16. doi: 10.1200/PO.17.00029
60. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:l1. doi: 10.1126/scisignal.2004088
61. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, et al. Cancers associated with BRCA 1 and BRCA 2 mutations other than breast and ovarian. Cancer (2015) 121:269–75. doi: 10.1002/cncr.29041
62. Taylor MRG, Špírek M, Chaurasiya KR, Ward JD, Carzaniga R, Yu X, et al. Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell (2015) 162:271–86. doi: 10.1016/j.cell.2015.06.015
63. Bonilla B, Hengel SR, Grundy MK, Bernstein KA. RAD51 gene family structure and function. Annu Rev Genet (2020) 54:25–46. doi: 10.1146/annurev-genet-021920-092410
64. Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. JNCI: J Natl Cancer Institute (2015) 107(11):djv214. doi: 10.1093/jnci/djv214
65. Christou C, Kyriacou K. BRCA1 and its network of interacting partners. Biol (Basel) (2013) 2:40–63. doi: 10.3390/biology2010040
66. Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, et al. ATM Orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun (2019) 10:87. doi: 10.1038/s41467-018-07729-2
67. Bartek J, Falck J, Lukas J. Chk2 kinase [[/amp]]mdash; a busy messenger. Nat Rev Mol Cell Biol (2001) 2:877–86. doi: 10.1038/35103059
68. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol (2009) 21:245–55. doi: 10.1016/j.ceb.2009.01.018
69. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell (2003) 3:421–9. doi: 10.1016/S1535-6108(03)00110-7
70. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol (2004) 24:708–18. doi: 10.1128/MCB.24.2.708-718.2004
71. Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol (2005) 7:195–201. doi: 10.1038/ncb1212
72. Booth L, Roberts J, Poklepovic A, Dent P. The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to kill carcinoma cells. Cancer Biol Ther (2018) 19:786–96. doi: 10.1080/15384047.2018.1472189
73. Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer (2014) 134:1013–23. doi: 10.1002/ijc.28226
74. Dubbury SJ, Boutz PL, Sharp PA. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature (2018) 564:141–5. doi: 10.1038/s41586-018-0758-y
75. Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res (2014) 74:287–97. doi: 10.1158/0008-5472.CAN-13-2541
76. Pan E, Cabal A, Javier-DesLoges J, Patel D, Panian J, Lee S, et al. Analysis of CDK12 alterations in a pan-cancer database. Cancer Med (2022) 11:753–63. doi: 10.1002/cam4.4483
77. Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PloS Comput Biol (2018) 14:e1005965. doi: 10.1371/JOURNAL.PCBI.1005965
78. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. New Engl J Med (2016) 375:443–53. doi: 10.1056/NEJMoa1603144
79. Sokol ES, Pavlick D, Khiabanian H, Frampton GM, Ross JS, Gregg JP, et al. Pan-cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome-wide loss of heterozygosity. JCO Precis Oncol (2020) 4:442–65. doi: 10.1200/PO.19.00345
80. Jamal K, Galbiati A, Armenia J, Illuzzi G, Hall J, Bentouati S, et al. Drug–gene interaction screens coupled to tumor data analyses identify the most clinically relevant cancer vulnerabilities driving sensitivity to PARP inhibition. Cancer Res Commun (2022) 2:1244–54. doi: 10.1158/2767-9764.CRC-22-0119
81. Garcin EB, Gon S, Sullivan MR, Brunette GJ, Cian A, Concordet J-P, et al. Differential requirements for the RAD51 paralogs in genome repair and maintenance in human cells. PloS Genet (2019) 15:e1008355. doi: 10.1371/journal.pgen.1008355
82. Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. New Engl J Med (2019) 381:2416–28. doi: 10.1056/NEJMoa1911361
83. Patel JN, Braicu I, Timms KM, Solimeno C, Tshiaba P, Reid J, et al. Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br J Cancer (2018) 119:1060–6. doi: 10.1038/s41416-018-0268-6
84. Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol (2017) 18:75–87. doi: 10.1016/S1470-2045(16)30559-9
85. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 390:1949–61. doi: 10.1016/S0140-6736(17)32440-6
86. Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res (2016) 22:3764–73. doi: 10.1158/1078-0432.CCR-15-2477
87. Zurita AJ, Graf RP, Villacampa G, Raskina K, Sokol E, Jin D, et al. Genomic biomarkers and genome-wide loss-of-Heterozygosity scores in metastatic prostate cancer following progression on androgen-targeting therapies. JCO Precis Oncol (2022) 6:e2200195. doi: 10.1200/PO.22.00195
88. Carreira S, Porta N, Arce-Gallego S, Seed G, Llop-Guevara A, Bianchini D, et al. Biomarkers associating with PARP inhibitor benefit in prostate cancer in the TOPARP-b trial. Cancer Discovery (2021) 11:2812–27. doi: 10.1158/2159-8290.CD-21-0007
89. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
90. Westphalen CB, Fine AD, André F, Ganesan S, Heinemann V, Rouleau E, et al. Pan-cancer analysis of homologous recombination repair–associated gene alterations and genome-wide loss-of-Heterozygosity score. Clin Cancer Res (2022) 28:1412–21. doi: 10.1158/1078-0432.CCR-21-2096
91. Sokol ES, Jin DX, Fine A, Trabucco SE, Maund S, Frampton G, et al. PARP inhibitor insensitivity to BRCA1/2 monoallelic mutations in microsatellite instability-high cancers. JCO Precis Oncol (2022) 6:e2100531. doi: 10.1200/PO.21.00531
92. Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, Nelson PS, et al. DNA Repair in prostate cancer: biology and clinical implications. Eur Urol (2017) 71:417–25. doi: 10.1016/j.eururo.2016.08.037
93. Castro E, Goh C, Leongamornlert D, Saunders E, Tymrakiewicz M, Dadaev T, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol (2015) 68:186–93. doi: 10.1016/j.eururo.2014.10.022
94. Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol (2013) 31:1748–57. doi: 10.1200/JCO.2012.43.1882
95. Hatano K, Nonomura N. Genomic profiling of prostate cancer: an updated review. World J Mens Health (2022) 40:368. doi: 10.5534/wjmh.210072
96. Mateo J, Carreira S, Seed G, Chandler R, Dolling D, Figueiredo I, et al. Genomic profiling of primary prostate tumors from patients who develop metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol (2018) 36:5013–3. doi: 10.1200/JCO.2018.36.15_suppl.5013
97. Mateo J, Seed G, Bertan C, Rescigno P, Dolling D, Figueiredo I, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest (2020) 130:1743–51. doi: 10.1172/JCI132031
98. Schaeffer E, Srinivas S, Antonarakis ES, Armstrong AJ, Bekelman JE, Cheng H, et al. NCCN guidelines insights: prostate cancer, version 1.2021. J Natl Compr Cancer Network (2021) 19:134–43. doi: 10.6004/jnccn.2021.0008
99. Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-b: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol (2019) 37:490–503. doi: 10.1200/JCO.18.00358
100. Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer (2012) 106:1697–701. doi: 10.1038/bjc.2012.146
101. Thompson D, Easton F. Cancer incidence in BRCA1 mutation carriers. CancerSpectrum Knowledge Environ (2002) 94:1358–65. doi: 10.1093/jnci/94.18.1358
102. van Asperen CJ. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet (2005) 42:711–9. doi: 10.1136/jmg.2004.028829
103. Breast cancer linkage consortium. Cancer risks in BRCA2 mutation carriers. JNCI J Natl Cancer Institute (1999) 91:1310–6. doi: 10.1093/jnci/91.15.1310
104. Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, Vora K, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res (2010) 16:2115–21. doi: 10.1158/1078-0432.CCR-09-2871
105. Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer (2011) 105:1230–4. doi: 10.1038/bjc.2011.383
106. Oh M, Alkhushaym N, Fallatah S, Althagafi A, Aljadeed R, Alsowaida Y, et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: a meta-analysis. Prostate (2019) 79:880–95. doi: 10.1002/pros.23795
107. Matejcic M, Patel Y, Lilyquist J, Hu C, Lee KY, Gnanaolivu RD, et al. Pathogenic variants in cancer predisposition genes and prostate cancer risk in men of African ancestry. JCO Precis Oncol (2020) 4:32–43. doi: 10.1200/PO.19.00179
108. Lecarpentier J, Silvestri V, Kuchenbaecker KB, Barrowdale D, Dennis J, McGuffog L, et al. Prediction of breast and prostate cancer risks in Male BRCA1 and BRCA2 mutation carriers using polygenic risk scores. J Clin Oncol (2017) 35:2240–50. doi: 10.1200/JCO.2016.69.4935
109. Karlsson Q, Brook MN, Dadaev T, Wakerell S, Saunders EJ, Muir K, et al. Rare germline variants in ATM predispose to prostate cancer: a PRACTICAL consortium study. Eur Urol Oncol (2021) 4:570–9. doi: 10.1016/j.euo.2020.12.001
110. Wang Y, Dai B, Ye D. CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis. Int J Clin Exp Med (2015) 8:15708–15.
111. Cybulski C, Wokolorczyk D, Huzarski T, Byrski T, Gronwald J, Gorski B, et al. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J Med Genet (2006) 43:863–6. doi: 10.1136/jmg.2006.044974
112. Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature (2007) 446:316–9. doi: 10.1038/nature05609
113. Leongamornlert D, Saunders E, Dadaev T, Tymrakiewicz M, Goh C, Jugurnauth-Little S, et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br J Cancer (2014) 110:1663–72. doi: 10.1038/bjc.2014.30
114. Page EC, Bancroft EK, Brook MN, Assel M, Hassan Al Battat M, Thomas S, et al. Interim results from the IMPACT study: evidence for prostate-specific antigen screening in BRCA2 mutation carriers. Eur Urol (2019) 76:831–42. doi: 10.1016/j.eururo.2019.08.019
115. Na R, Zheng SL, Han M, Yu H, Jiang D, Shah S, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol (2017) 71:740–7. doi: 10.1016/j.eururo.2016.11.033
116. Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. JNCI: J Natl Cancer Institute (2005) 97:813–22. doi: 10.1093/jnci/dji141
117. Näslund-Koch C, Nordestgaard BG, Bojesen SE. Increased risk for other cancers in addition to breast cancer for CHEK2 *1100delC heterozygotes estimated from the Copenhagen general population study. J Clin Oncol (2016) 34:1208–16. doi: 10.1200/JCO.2015.63.3594
118. Ford D. Risks of cancer in BRCA1-mutation carriers. Lancet (1994) 343:692–5. doi: 10.1016/S0140-6736(94)91578-4
119. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, et al. Compromised BRCA1–PALB2 interaction is associated with breast cancer risk. Oncogene (2017) 36:4161–70. doi: 10.1038/onc.2017.46
120. Risch HA, McLaughlin JR, Cole DEC, Rosen B, Bradley L, Kwan E, et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet (2001) 68:700–10. doi: 10.1086/318787
121. Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet (2003) 72:1117–30. doi: 10.1086/375033
122. Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, et al. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer (2009) 100:426–30. doi: 10.1038/sj.bjc.6604847
123. Nysom K, Leblond P, Frappaz D, Aerts I, Varlet P, Giangaspero F, et al. PALB2 reversion mutations in breast, prostate, and ovarian carcinomas. Ann Oncol (2017) 28:v23–4. doi: 10.1093/ANNONC/MDX363.004
124. CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis - PubMed. Available at: https://ncbi.nlm.nih.gov/26629066/ (Accessed pril 13, 2022).
125. Mavrou A, Tsangaris GTH, Roma E, Kolialexi A. The ATM gene and ataxia telangiectasia. Anticancer Res (2008) 28:401–5.
126. McKinnon PJ. ATM And ataxia telangiectasia. EMBO Rep (2004) 5:772–6. doi: 10.1038/sj.embor.7400210
127. Che R, Zhang J, Nepal M, Han B, Fei P. Multifaceted fanconi anemia signaling. Trends Genet (2018) 34:171–83. doi: 10.1016/j.tig.2017.11.006
128. Raymond VM, Mukherjee B, Wang F, Huang S-C, Stoffel EM, Kastrinos F, et al. Elevated risk of prostate cancer among men with lynch syndrome. J Clin Oncol (2013) 31:1713–8. doi: 10.1200/JCO.2012.44.1238
129. Na R, Zheng SL, Han M, Yu H, Jiang D, Shah S, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death [figure presented]. Eur Urol (2017) 71:740–7. doi: 10.1016/j.eururo.2016.11.033
130. Caffo O, De Giorgi U, Fratino L, Alesini D, Zagonel V, Facchini G, et al. Clinical outcomes of castration-resistant prostate cancer treatments administered as third or fourth line following failure of docetaxel and other second-line treatment: results of an Italian multicentre study. Eur Urol (2015) 68:147–53. doi: 10.1016/j.eururo.2014.10.014
131. Scher HI, Solo K, Valant J, Todd MB, Mehra M. Prevalence of prostate cancer clinical states and mortality in the united states: estimates using a dynamic progression model. PloS One (2015) 10:e0139440. doi: 10.1371/journal.pone.0139440
132. Annala M, Struss WJ, Warner EW, Beja K, Vandekerkhove G, Wong A, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair–deficient prostate cancer. Eur Urol (2017) 72:34–42. doi: 10.1016/j.eururo.2017.02.023
133. Antonarakis ES, Lu C, Luber B, Liang C, Wang H, Chen Y, et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur Urol (2018) 74:218–25. doi: 10.1016/j.eururo.2018.01.035
134. Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol (2014) 15:1521–32. doi: 10.1016/S1470-2045(14)71021-6
135. Lalonde E, Alkallas R, Chua MLK, Fraser M, Haider S, Meng A, et al. Translating a prognostic DNA genomic classifier into the clinic: retrospective validation in 563 localized prostate tumors. Eur Urol (2017) 72:22–31. doi: 10.1016/j.eururo.2016.10.013
136. Herzog TJ, Vergote I, Gomella LG, Milenkova T, French T, Tonikian R, et al. Testing for homologous recombination repair or homologous recombination deficiency for poly (ADP-ribose) polymerase inhibitors: a current perspective. Eur J Cancer (2022) 179:136–46. doi: 10.1016/j.ejca.2022.10.021
137. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434:913–7. doi: 10.1038/nature03443
138. Farmer H, McCabe H, Lord CJ, Tutt AHJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nat 2005 434:7035 (2005) 434:917–21. doi: 10.1038/nature03445
139. Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT trial. Nat Med (2018) 24:628–37. doi: 10.1038/s41591-018-0009-7
140. Tan DSP, Kaye SB. Chemotherapy for patients with BRCA1 and BRCA2 –mutated ovarian cancer: same or different? American Society of clinical oncology educational book (2015) 35:114–21. doi: 10.14694/EdBook_AM.2015.35.114
141. Mota JM, Barnett E, Nauseef JT, Nguyen B, Stopsack KH, Wibmer A, et al. Platinum-based chemotherapy in metastatic prostate cancer with DNA repair gene alterations. JCO Precis Oncol (2020) 4:355–66. doi: 10.1200/PO.19.00346
142. Pomerantz MM, Spisák S, Jia L, Cronin AM, Csabai I, Ledet E, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer (2017) 123:3532–9. doi: 10.1002/cncr.30808
143. Schmid S, Omlin A, Higano C, Sweeney C, Martinez Chanza N, Mehra N, et al. Activity of platinum-based chemotherapy in patients with advanced prostate cancer with and without DNA repair gene aberrations. JAMA Netw Open (2020) 3:e2021692. doi: 10.1001/jamanetworkopen.2020.21692
144. Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer (2021) 7:930–57. doi: 10.1016/j.trecan.2021.06.002
145. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer (2018) 6:35. doi: 10.1186/s40425-018-0342-x
146. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. New Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
147. de Almeida DVP, Fong L, Rettig MB, Autio KA. Immune checkpoint blockade for prostate cancer: niche role or next breakthrough? American Society of clinical oncology educational book (2020) 40:e89–e106. doi: 10.1200/EDBK_278853
148. Alva AS, Li J, Chou J, Reimers MA, McKay RR, Zhang J, et al. Phase 2 trial of immunotherapy in tumors with CDK12 inactivation (IMPACT): results from cohort a of patients (pts) with metastatic castration resistant prostate cancer (mCRPC) receiving dual immune checkpoint inhibition (ICI). J Clin Oncol (2022) 40:103–3. doi: 10.1200/JCO.2022.40.6_suppl.103
Keywords: DNA damage response, homologous recombination repair, alterations, prognosis, prostate cancer
Citation: Lukashchuk N, Barnicle A, Adelman CA, Armenia J, Kang J, Barrett JC and Harrington EA (2023) Impact of DNA damage repair alterations on prostate cancer progression and metastasis. Front. Oncol. 13:1162644. doi: 10.3389/fonc.2023.1162644
Received: 09 February 2023; Accepted: 01 June 2023;
Published: 26 June 2023.
Edited by:
Navonil De Sarkar, Fred Hutchinson Cancer Research Center, United StatesReviewed by:
Takeshi Yuasa, Japanese Foundation For Cancer Research, JapanCopyright © 2023 Lukashchuk, Barnicle, Adelman, Armenia, Kang, Barrett and Harrington. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia Lukashchuk, bmF0YWxpYS5sdWthc2hjaHVrQGFzdHJhemVuZWNhLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.