
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 05 May 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1155511
This article is part of the Research Topic Clinical implications of targeting lipid metabolism and associated pathways for cancer therapy View all 6 articles
 Zhengzhen Xia1
Zhengzhen Xia1 Yi Quan1,2*
Yi Quan1,2*Ferroptosis is a type of lipid peroxidation-induced, iron-dependent programmed cell death. Emerging evidence suggests that ferroptosis is intimately connected to tumorigenesis, development, treatment and plays a major role in tumor immune regulation. This study focused on the connection between ferroptosis and immune regulation, which may offer a theoretical basis for targeting ferroptosis and tumor immunotherapy.
Unlike apoptosis, different types of necrosis, and autophagy, ferroptosis is a recently identified type of programmed cell death that is regulated by iron-dependent lethal lipid peroxidation (1). Ferroptosis mainly occurs as a result of reduced biological activity of glutathione peroxidase 4 (GPX4), lipid peroxidation, and rise of reactive oxygen species (ROS). A crucial factor in the occurrence of ferroptosis is GPX4. GPX4 inhibits ferroptosis by reducing ROS to glutathione (GSH) (2). The cystine/glutamate antibody (system xc-) made up of light chain subunit SLC7A11 and heavy chain subunit SLC3A2, which exchange cystine and glutamate in equal amounts. The ingested cystine undergoes a series of reactions to synthesize glutathione and maintain GPX4 activity. Tumor immunotherapy is a prospective method to address the treatment of malignancies in clinical practice, but only a small proportion of patients are benefited from this approach (3). The tumor microenvironment (TME) also has a significant role in treatment outcome. TME is a hypoxic, acidified, inflammatory, and immunosuppressive state. TME contains a considerable number of immune cells, such as effector CD8 T cells with antitumor effects, natural killer (NK) cells, dendritic cells (DCs), and regulatory T cells (Tregs) with immunosuppressive functions, as well as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and cancer-associated fibroblasts cells (CAFs). The combination of immunotherapy with other therapeutic modalities reverses the immunosuppressive state and has a synergistic antitumor effect (4). In this case, targeted ferroptosis combined with immunotherapy is a viable therapeutic tool. This review describes the interaction between ferroptosis and tumor immunotherapy in order to provide a theoretical reference for the combination of targeted ferroptosis with immunotherapy.
Hypoxia is a common phenomenon in solid tumors, wherein chronic hypoxia promotes tumor growth, metastasis (5), and therapy resistance (6) by remodeling the microenvironment. The stabilization of hypoxia-induced hypoxia-inducible factors (including HIF1α, HIF2α, and HIF-3α) activate hypoxia target genes thereby regulating angiogenesis, cell growth, glycolysis, DNA damage repair, and even apoptosis (7). Under hypoxia, HIF regulates CD8 T cell infiltration and activity via von Hippel-Lindau (VHL) (8). HIF-1α is the key promoter of immunosuppressive TME (9). Hypoxic TME inhibits the activation of cytotoxic T lymphocytes (CTLs), reduces interferon-gamma (IFNγ) production and release, promotes Treg activity (10, 11), promotes the differentiation of MDSCs to immunosuppressive TAMs to enhance immunosuppression (12), impairs DC (13) and NK cell (14, 15) activation and function, and induces epithelial to mesenchymal transition (EMT) (16, 17). In addition, hypoxia regulates the levels of various microRNAs (miRNAs) and long non-coding RNAs (IncRNAs), and these ncRNAs influence the activation of HIF-1α and consequently create positive or negative feedbacks to regulate hypoxic TME (18–21).
The weak acidity of TME is caused by the excessive consumption of glucose by cancer cells, resulting in lactate accumulation. The Warburg effect states that the energy of cancer cells is dominated by glycolysis even under oxygen-sufficient conditions, producing large amounts of lactate (22). The creation of lactate impairs the CTLs’ capacity to multiply and produce cytokines, which reduces their cytotoxic activity (23), suppresses the production of IFNγ by NK and NKT cells, and reduces their antitumor function (24, 25), mediates the differentiation of DCs (26), increases Treg activity (27), and recruits macrophages and induces their functional polarization into TAMs (28), leading to immunosuppression and promoting tumor cell growth and metastasis.
Strikingly, cancer-related inflammation is caused by proinflammatory cytokines and chemokines produced by tumor cells, resulting from cancer gene-mediated malignant transformation (29). Chronic inflammation has been proven to be among the primary causes of tumor occurrence and growth (30). Macrophages and lymphocytes are the primary immune cells that infiltrate sites of persistent inflammation, which can indirectly or directly promote tumor development by releasing various cytokines. These cytokines are categorized into proinflammatory cytokines [interleukin (IL)-1, IL-6, IL-15, IL-17, IL-23, tumor necrosis factor-alpha (TNF-α), and IFN-γ] and anti-inflammatory cytokines [IL-4, IL-10, IL-13, and tumor growth factor-beta (TGF-β)] (31). Inflammation is an effective inducer of EMT in tumors, TGF- β, TNF-α, IL‐1β, IL-6, IL-8, and other proinflammatory cytokines play a role in the initiation and maintenance of tumor EMT, increasing the invasiveness of cancer cells and promoting tumor metastasis (32–35). Another mechanism of cancer-related inflammation involves the emergence of damage-associated molecular patterns (DAMPs). Various endogenous ligands of DAMPs are produced in reacting to hypoxia, cell stress, and tissue damage and then detected by pattern recognition receptors (PRRs) generated by innate immune cells. These DAMPs include high-mobility group box-1 (HMGB1), heat shock protein, S100 protein, and ATP (36, 37).
Hypoxia upregulates iron regulatory protein 2 (IRP2) through a post-translational mechanism to increase iron transport (TFRC) and SLC11A2 while decreasing iron storage (FTH) (38). A vital cellular regulator of tumor hypoxia, carbonic anhydrase IX (CAIX), controls intracellular and extracellular pH and acidosis (39). CAIX prevents cancer cells from undergoing iron death via the cystine/glutamate antiporter xCT (40). A previous study has shown that hypoxia protects cancer cells from iron death through multiple pathways. Hypoxia-induced HIF-1α/lncRNA-PMAN prevents ferroptosis in gastric cancer by enhancing SLC7A11 mRNA stability and increasing the expression of SLC7A11 at the posttranscriptional level (41). The CBSLR/CBS signal axis regulates the methylation of ACSL4 to prevent ferroptosis in gastric cancer by polyubiquitination (42). Blocking ferroptosis of hepatocellular carcinoma by inhibiting METTL14 triggered YTHDF2-dependent silencing of SLC7A11 (43). In addition, hypoxia reduces NCOA4 in macrophages, thereby increasing FTMT and FTH and preventing ferroptosis (44).
Recently, Zhao et al. designed a dual hypoxia-sensitive polymeric nanocarrier containing azobenzene and nitroimidazole, encapsulating the small molecule ferroptosis inducer RSL3, and validated it in a nude mouse model of ovarian cancer. The study also demonstrated that the polymeric carriers depleted NADPH in tumor cells under hypoxic conditions, leading to depletion of GSH and Trx and further inhibition of GPX4 activity, thereby sensitizing tumor cells to ferroptosis. Furthermore, it functions like a selective ferroptosis sensitizer solely in hypoxic tumors, without damaging healthy organs (45). This study provides a novel idea for efficacy enhancement and toxicity reduction of ferroptosis antitumor therapy.
The rapid proliferation of cancer cells relies on glycolysis as an energy source, producing a large amount of lactic acid. The lactic acid in the microenvironment provides energy for cell growth and development, affects the biochemical function of proteins in cells as a vital signaling molecule, regulates the biological functions of various cells, and promotes the malignant proliferation and progress of tumors (46). Luo et al. confirmed that lactic acid inhibits the ferroptosis of hepatoma cells through in vivo and in vitro experiments. Excessive lactic acid upregulated the expression of hydroxycarboxylic acid receptor 1 (HCAR1) and monocarboxylate transporter 1 (MCT1) and inhibited ferroptosis of liver cancer cells by reducing the lipid peroxidation of liver cancer. Strikingly, the inhibition of HCAR1/MCT1 increases the proportion of AMP/ATP in hepatoma cells, enhances the AMP-activated protein kinase’s phosphorylation, downregulates the level of mature of sterol regulatory element-binding protein 1 and its target gene stearoyl-coenzyme A (CoA) desaturase-1, and enhances the ferroptosis sensitivity of hepatoma cells (47). This finding suggested that additional studies on the mechanism of lactic acid and ferroptosis would identify the drug targets for tumor treatment and improve the prognosis of patients.
Proinflammatory factors in the TME may promote or inhibit tumor initiation and progression by affecting tumor ferroptosis. In head and neck squamous cell cancer, IL-6 inhibits ferroptosis by transcriptionally increasing xCT expression through the JAK2/STAT3 pathway (48). miR-539 directly triggers the SAPK/JNK protein kinase through targeted TNF-α-induced protein 8 to promote colorectal cancer ferroptosis (49). CircABCB10 silencing promotes rectum cancer cell ferroptosis and apoptosis through targeting the miR-326/CCL5 axis (50). Circ-IL-4 receptor controls the miR-541-3p/GPX4 axis to inhibit ferroptosis and promote carcinogenesis in hepatocellular carcinoma (51). HMGB1 regulates acute myeloid leukemia cell ferroptosis through the RAS-JNK/p38 pathway (52).
The main agents of antitumor immunity, CD8 T cells secrete IL-2, IL-12, and (IFNγ) in TME, which increases the ability of CD8 T cells to target and destroy tumor cells. Interestingly, IFNγ produced by immunotherapy-activated CD8 T cells suppresses the expression of SLC3A2 and SLC7A11, two components of the glutamate-cystine anti-transport protein system xc-, and prevents tumor cells from utilizing cystine, promoting lipid peroxidation and ferroptosis in tumor cells (53, 54). Another study found that IFNγ generated in the TME by activated T cells paired with arachidonic acid affected spontaneous antitumor immunity in vivo by inducing tumor cell ferroptosis through acyl-CoA synthetase long-chain family member 4 (ACSL4) (55). These studies highlighted that targeting the metabolism associated with tumor ferroptosis can improve the efficacy of cancer immunotherapy.
Recently, Ma et al. found that oxLDL in the TME induced iron death and p38 phosphorylation by lipid peroxidation in CD8+ T cells in a CD36-dependent manner. Also, the activation of P38 induced CD8 T cell death, suppressed IFNγ and TNFα generation in T cells, depleted CD8 T cells, and reduced antitumor effects (56, 57) (Figure 1). In addition, the research team also discovered that anti-PD-1 antibodies and CD36 deletion in CD8 T cells together showed superior antitumor effects to either treatment alone (57). This research suggested that targeting CD36 and ferroptosis might be a viable option for improving the antitumor efficacy of T-cell based immunotherapy.
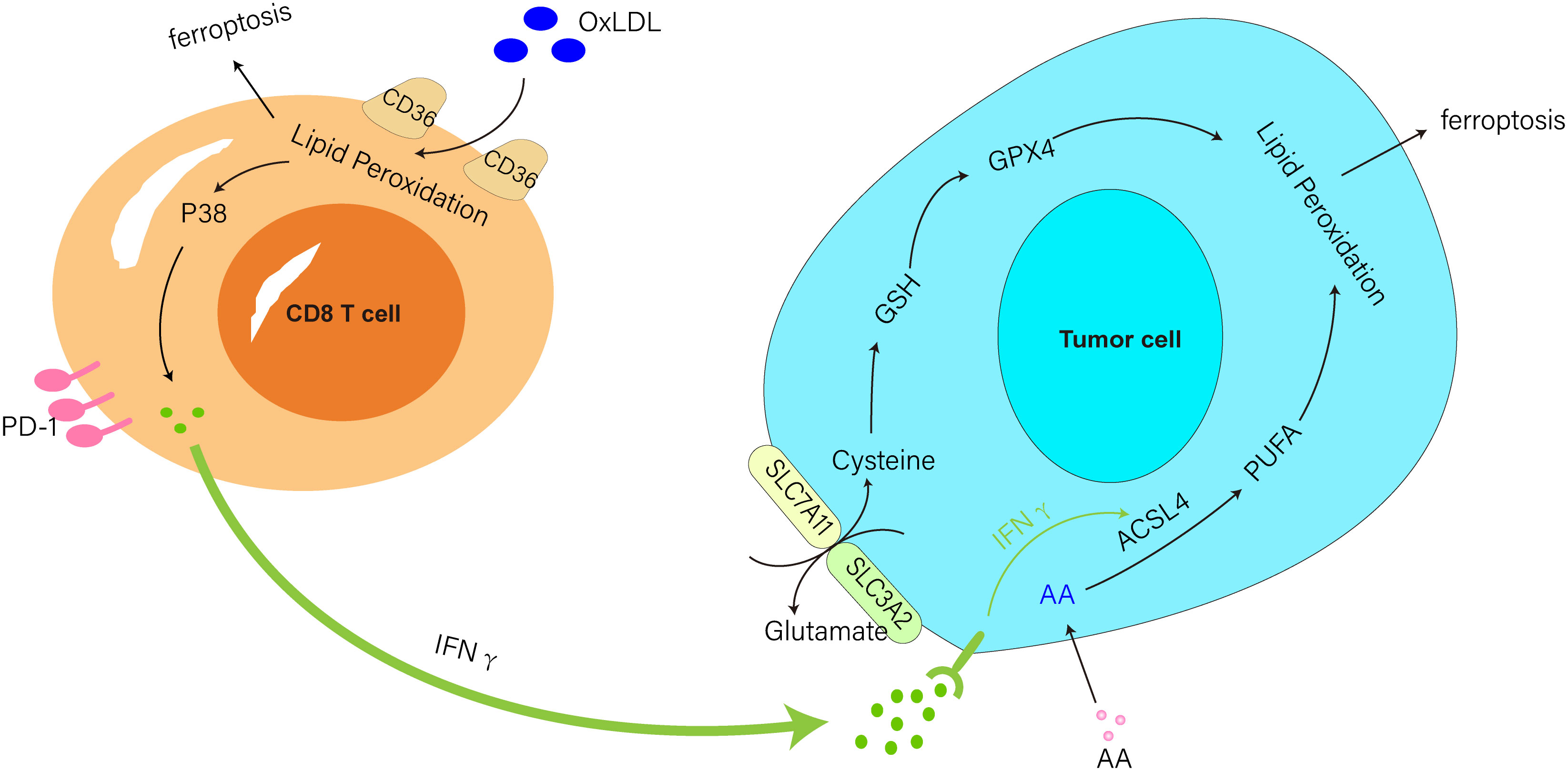
Figure 1 IFNγ produced from CD8 T cells triggered by immunotherapy downregulated SLC3A2 and SLC7A11 activity and aided ferroptosis in tumor cells. IFNγ produced by activated T cells paired with AA-induced tumor cell ferroptosis via ACSL4. CD36 mediated the uptake of oxLDL by CD8 T cells to induce ferroptosis and P38 phosphorylation and inhibited IFNγ production in T cells.
NK cells are nature leukomonocytes that are essential for tumor surveillance and control and in tumor immunotherapy due to their ability to kill abnormal cells without any antigenic stimulation and persistent immune memory. NK cells trigger stressed cell death primarily by the release of perforin, granzyme, and other lytic granule molecules (58). In addition, NK cells have a cytotoxic capacity similar to that of CD8 T cells, regulating immunity and killing tumor cells by releasing IFN-γ (59). Another study demonstrated that activating mitochondrial apoptosis in cancer cells enhances NK cell death. Thus, initiating mitochondrial apoptosis via BH3 mimics can synergize with NK cells to kill cancer cells (60). Moreover, the synergistic effect of BH3 mimetic drugs in combination with ferroptosis induces cancer cell death (61). Then, targeted ferroptosis combined with NK cell therapy may also be a viable strategy. Kim et al. found that the use of clinical-grade iron oxide nanoparticles (ferumoxytol) to mediate ferroptosis in prostate cancer activated NK cells and increased the cytotoxic function of NK cells. Furthermore, in the therapy of ferroptosis+NK cells, tumor cells expressed HMGB1 and PD-L1, and a clear regression of tumor volume after ferumol-mediated ferroptosis and NK cell treatment was observed in a prostate cancer mouse model (62). These findings suggested that ferroptosis can improve NK cell activity.
DCs are robust antigen-presenting cells (APCs) in the organism that promote immunization or tolerance by sampling and presenting antigens to T cells as well as transmitting immunomodulatory signals via cell-cell interaction and cytokines. promoting immunity or tolerance by sampling and presenting antigens to T cells and delivering immunomodulatory signals through cell-cell contact and cytokines (63). DCs consist of exogenous antigens on major histocompatibility class I (MHC-I) molecules and initiate CD8 T-cell antitumor immunity via antigen cross-presentation (64). The activation of DCs can be achieved by PRRs on DCs that recognize pathogen-associated molecular patterns (PAMPs) or DAMPs (65). However, a new research study found that although ferroptosis is capable of releasing cytokines and DAMPs, it does not activate antitumor immune responses, inhibits cross-presentation of soluble antigens to DCs, impairs DC maturation, and inhibits phagocytosis of tumor cells by DCs (66). According to this phenomenon, cancer cells’ ferroptosis may not represent an immunological type of cell death.
Tregs are essential for maintaining self-tolerance and immune cell homeostasis, and their mediated immunosuppression is a major obstacle to tumor immunotherapy. Tregs exhibit their immunosuppressive function through various mechanisms, including inhibition of APCs via cytotoxic T lymphocyte antigen-4 (CTLA-4), suppression of cytokine secretion (IL-10, TGF-β, and IL-35), and competitive depletion of IL-2. Oxidative stress-induced apoptosis of Treg cells in tumors amplifies their immunosuppressive effects and abolishes the efficacy of PD-L1 blockade therapy (67). A current research revealed that GPX4 prevents lipid peroxidation and ferroptosis in Tregs and that GPX4-deficient Tregs promote mitochondrial superoxide production and IL-1β production, thereby promoting T helper 17 (Th17) responses. Furthermore, GPX4 deficiency, rather than intra-tumor Treg cell apoptosis, enhances antitumor immune function and inhibits tumor growth by inducing ferroptosis (68). The current results indicated that induction of iron death in Treg cells could be an effective strategy to improve cancer therapy.
MDSCs constitute a group of immunosuppressive immature bone marrow cells that promotes tumor progression through various non-immune mechanisms to suppress antitumor immunity and support tumor growth and are essential for encouraging tumor immune escape (69). N-Acyl sphingomyelin hydrolase (ASAH2) is overexpressed in MDSCs of colon cancer and protects MDSCs from ferroptosis by regulating p53 stabilization and inhibiting Hmox1 expression from reducing lipid ROS. The novel MDSC-targeting medication NC06 induces MDSC ferroptosis by activating the p53-Hmox1 pathway and decreases MDSC accumulation to stimulate T-cell tumor infiltration and inhibit tumor growth in vivo (70). Thus, induction of MDSC ferroptosis is a potentially effective therapy for tumor immunotherapy.
Macrophages can be divided into two subtypes based on their function and activation. M1 macrophages (driven by bacterial products and interferons produced during type 1 immune responses activated by type 1 T helper and innate lymphoid cells) kill tumor cells and M2 macrophages (driven by type 2 T helper and innate lymphoid cells, such as IL-4 and IL-13) promote cancer progression and immunosuppression (71). TAMs are widely considered to be M2 macrophages, and the M2-like TAMs prevent CD8 T-cell antitumor effect and DC development by secreting transforming growth factor-beta (TGF-β) and IL-10 (72). Several studies have focused on targeting macrophages, and Wang et al. showed that ZVI-NP selectively triggers ferroptosis in lung cancer cells by inhibiting the NRF2-mediated cytoprotective program, shifts pro-tumor M2 macrophages to antitumor M1, enhances antitumor immunity by increasing the cytotoxic function of CD8 T cells and decreasing the Tregs ratio (73). Gu et al. found that MIL88B/RSL3 nano preparations inhibited metastatic tumor activity by polarizing macrophages from pro-tumor M2 to antitumor M1 phenotypes via a ferroptosis-enhanced metabolic program (from mitochondrial oxidative phosphorylation to glycolysis), activated of numerous proinflammatory signals, and greatly inhibited tumor angiogenesis (74). Xu et al. demonstrated that downregulation of matrix remodeling-associated protein 8 (MXRA8) induced ferroptosis in gliomas by elevating Fe levels and attenuating M2 macrophage infiltration to inhibit tumor progression (75). Li et al. found that dihydroartemisinin produced ferroptosis in TAM-induced ROS/LPO, resulting in DNA damage, which in turn activated downstream NF-κB to remodel TAM to M1 phenotype, and that the remodeled M1 macrophages exerted antitumor effects (76). Tang et al. discovered that macrophages with xCT deletion dramatically reduced TAM infiltration, blocked M2-like phenotypic changes in HCC tumor tissue, activated and elevated ferroptosis activity, and hampered tumor growth and metastasis. Moreover, PD-L1 expression in macrophages was markedly elevated by xCT-mediated macrophage ferroptosis, which also enhanced the anti-tumor effectiveness of anti-PD-L1 therapy (77). According to research by Hao et al., blocking APOC1 can encourage M2 macrophages to become M1 macrophages through the iron-sparing pathway, modifying the TME and enhancing the effectiveness of anti-PD1 immunotherapy for hepatocellular carcinoma (78). However, some studies have shown drawbacks in ferroptosis-mediated cancer treatment. The study also demonstrated that, although ferroptosis increases immune cell activation and infiltration, it attenuates the antitumor cytotoxic killing effect. Conversely, TAMs infiltration are reduced and converted TAM to M1-like phenotype when ferrostatin-1 and anti-PD1/L1 antibodies are combined (79) (Figure 2). The above results suggested that ferroptosis plays a major role in TAM infiltration and activation in TME.
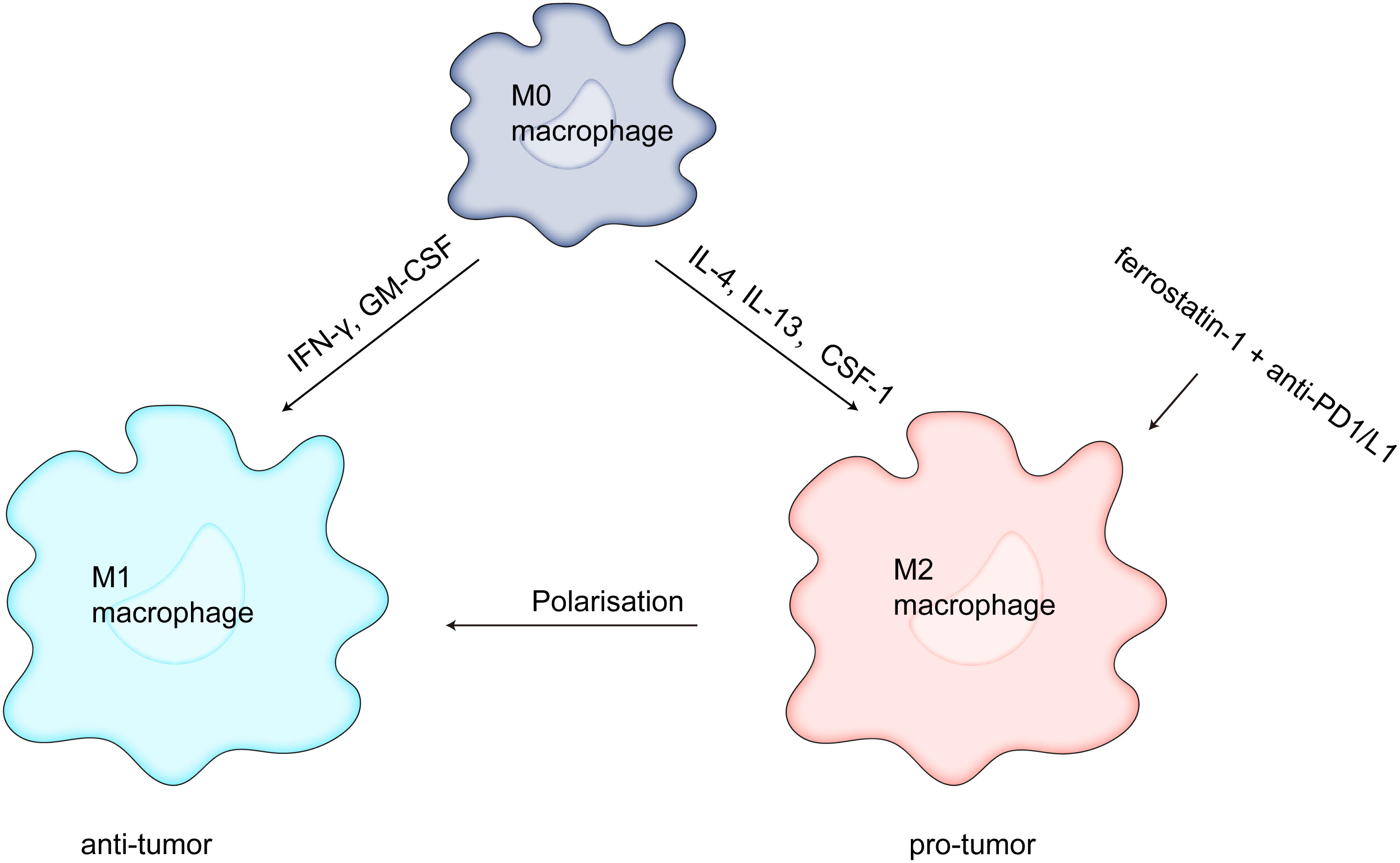
Figure 2 Ferrostatin-1 and anti-PD1/L1 together diminish TAM infiltration and change the M2-like phenotype into the M1-like phenotype, which strengthens the anticancer impact.
As the main component of TME, TAN promotes tumor progression and metastasis in communication through a variety of growth factors, chemokines, inflammatory factors, and other immune cells (80). Similar to macrophages, neutrophils can polarize into an antineoplastic (N1) or tumor-promoting (N2) phenotype. N1 TAN exerts antitumor activity through direct or indirect cytotoxicity, while N2 TAN stimulates immunosuppression, tumor growth, angiogenesis, and metastasis through DNA instability or the release of cytokines and chemokines (81). Li et al. demonstrated that tumor lesions occurring during early tumor progression (i.e. ischemia) draw neutrophils to the areas of tissue injury, where neutrophils destroy tumor cells by ferroptosis, resulting in necrosis. In addition, a cycle of positive feedback between necrosis and neutrophil infiltration increases intratumoral necrosis and speeds up glioma mortality (82).
CAFs are critical components of TME and sources of cytokines, growth factors, and exosomes (83). CAF-secreted exosomes influence tumor phenotypes, whereas exosomes released by tumor cells activate CAFs (84). CAFs derived exosomes transmit biological information via miRNAs, lncRNAs, circRNAs, lipids, and proteins and promote tumor growth (85), invasion (86, 87), metastasis (88), and angiogenesis (89), induce EMT (90, 91), endow tumor with chemical (92, 93) and radiation resistance (94), impede the action of tumor-infiltrating immune cells (95), and regulate TME’s antitumor immunological state. A recent study found that CAFs suppress ALOX15 expression by secreting exosomal miR-522, affecting lipid-ROS production in tumors to suppress ferroptosis in gastric cancer cells. In addition, chemotoxicity affects tumor growth and attenuates cisplatin chemotherapeutic efficacy by boosting miR-522 release from CAFs through triggering the USP7/hnRNPA1 pathway, whereas preventing miR-522 secretion could inhibit tumor growth and enhance sensitivity to cisplatin (96). The present study illustrated that CAFs-derived exosomes regulate tumor ferroptosis by transporting special signaling information and triggering cell ferroptosis in tumors by blocking the packaging of specific miRNAs into exosomes could be a viable novel therapeutic approach.
Immunotherapy is a potential method for treating cancer that achieves tumor clearance through reshaping the TME and activating antitumor immune responses. Current studies with immunotherapy alone have failed to demonstrate significant survival benefits, possibly because of TME’s intricate immunosuppressive nature.The tumor microenvironment inhibits the anti-tumor effects of CTL, NK, and DC due to its hypoxia, acidification, and inflammatory characteristics. It also increases the activity of immunosuppressive cells, puts the tumor microenvironment in an immunosuppressive state, and prevents the ferroptosis of tumor cells(Figure 3). Therefore, reversing the immunosuppressive state of the TME is critical to the success of tumor therapy. Ferroptosis can be promoted or inhibited by immune cells that attack the tumor microenvironment, and the identification of ferroptosis offers fresh hope for halting tumor growth. The combination of targeting the TME and ferroptosis enhances the antitumor effect by promoting the activation and presentation of effector T cells and dendritic cells, inhibiting the activity of immunosuppressive cells and relieving the immunosuppressive state of the tumor microenvironment. Although the combination of targeted ferroptosis therapy with immunotherapy may improve efficacy, the potential drug resistance of ferroptosis cannot be ignored. Consequently, a thorough investigation of ferroptosis’ function in the TME is necessary to build a creative strategy for the creation of fresh cancer treatment options
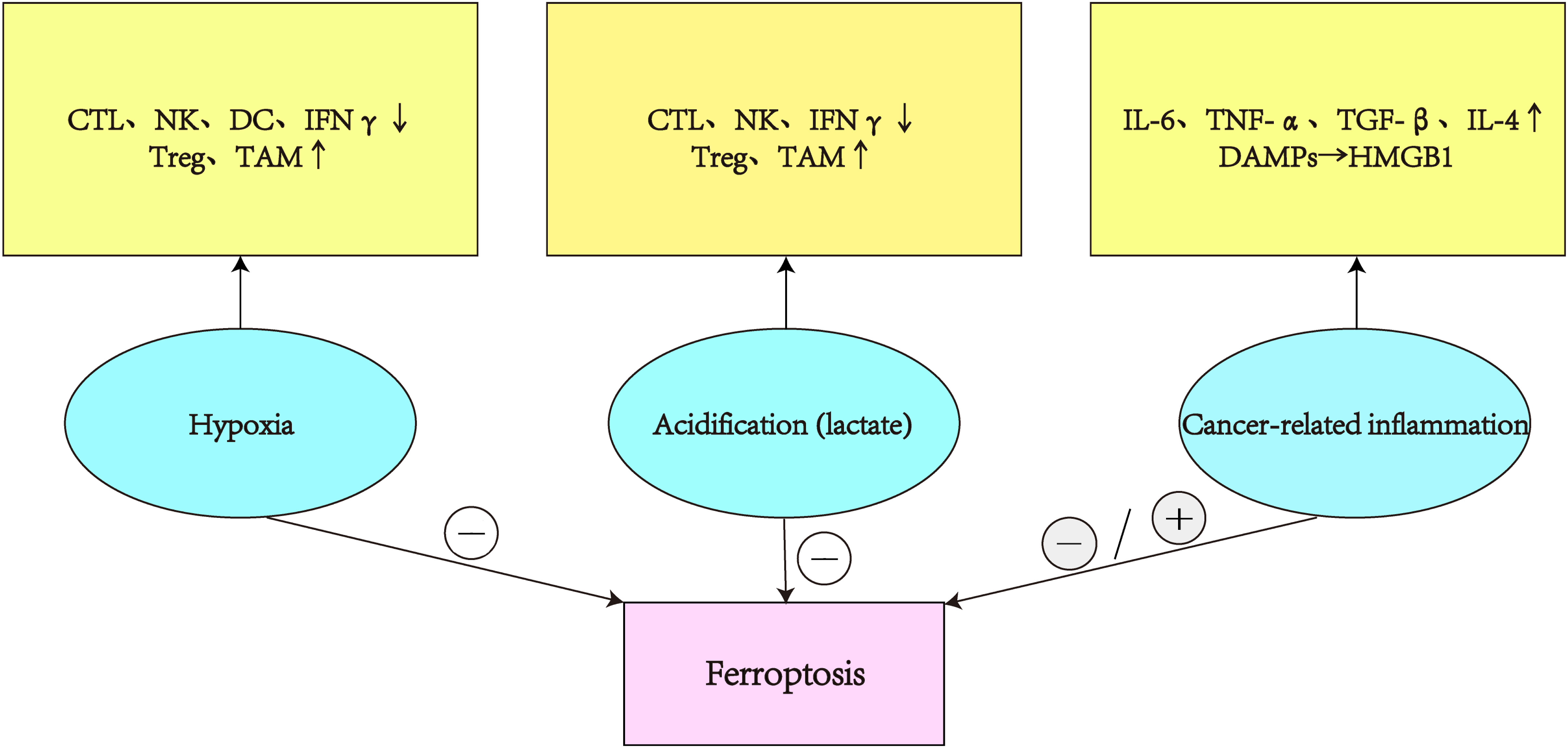
Figure 3 In the TME, hypoxia and acidity reduce the activity of CTL, NK, DC, and INF, boost the immunosuppressive effects of Treg and TAM, and prevent tumor ferroptosis. Chronic inflammation of tumors secretes various cytokines that promote or inhibit tumor ferroptosis.
ZX and YQ provided direction and guidance throughout the preparation of this manuscript. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
2. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
3. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
4. Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med (2021) 27(9):856–67. doi: 10.1016/j.molmed.2021.06.014
5. Rankin EB, Nam JM, Giaccia AJ. Hypoxia: signaling the metastatic cascade. Trends Cancer (2016) 2(6):295–304. doi: 10.1016/j.trecan.2016.05.006
6. Ikeda S, Tagawa H. Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci (2021) 112(10):3995–4004. doi: 10.1111/cas.15087
7. Han J, Li J, Ho JC, Chia GS, Kato H, Jha S, et al. Hypoxia is a key driver of alternative splicing in human breast cancer cells. Sci Rep (2017) 7(1):4108. doi: 10.1038/s41598-017-04333-0
8. Liikanen I, Lauhan C, Quon S, Omilusik K, Phan AT, Bartroli LB, et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J Clin Invest (2021) 131(7):e143729. doi: 10.1172/JCI143729
9. Mortezaee K, Majidpoor J. The impact of hypoxia on immune state in cancer. Life Sci (2021) 286:120057. doi: 10.1016/j.lfs.2021.120057
10. Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, et al. Hypoxia enhances immunosuppression by inhibiting CD4+ effector T cell function and promoting treg activity. Cell Physiol Biochem (2017) 41(4):1271–84. doi: 10.1159/000464429
11. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting edge: hypoxia-inducible factor 1 negatively regulates Th1 function. J Immunol (2015) 195(4):1372–6. doi: 10.4049/jimmunol.1402552
12. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
13. Tran CW, Gold MJ, Garcia-Batres C, Tai K, Elford AR, Himmel ME, et al. Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity. PloS One (2020) 15(12):e0244366. doi: 10.1371/journal.pone.0244366
14. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PloS One (2013) 8(5):e64835. doi: 10.1371/journal.pone.0064835
15. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol (2013) 43(10):2756–64. doi: 10.1002/eji.201343448
16. Hapke RY, Haake SM. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett (2020) 487:10–20. doi: 10.1016/j.canlet.2020.05.012
17. Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB, et al. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology (2017) 6(2):e1271858. doi: 10.1080/2162402X.2016.1271858
18. Dang K, Myers KA. The role of hypoxia-induced miR-210 in cancer progression. Int J Mol Sci (2015) 16(3):6353–72. doi: 10.3390/ijms16036353
19. To KKW, Cho WCS. Exosome secretion from hypoxic cancer cells reshapes the tumor microenvironment and mediates drug resistance. Cancer Drug Resist (2022) 5(3):577–94. doi: 10.20517/cdr.2022.38
20. Zheng F, Chen J, Zhang X, Wang Z, Chen J, Lin X, et al. The HIF-1alpha antisense long non-coding RNA drives a positive feedback loop of HIF-1alpha mediated transactivation and glycolysis. Nat Commun (2021) 12(1):1341. doi: 10.1038/s41467-021-21535-3
21. Wang X, Li L, Zhao K, Lin Q, Li H, Xue X, et al. A novel LncRNA HITT forms a regulatory loop with HIF-1alpha to modulate angiogenesis and tumor growth. Cell Death Differ (2020) 27(4):1431–46. doi: 10.1038/s41418-019-0449-8
22. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
23. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
24. Xie D, Zhu S, Bai L. Lactic acid in tumor microenvironments causes dysfunction of NKT cells by interfering with mTOR signaling. Sci China Life Sci (2016) 59(12):1290–6. doi: 10.1007/s11427-016-0348-7
25. Potzl J, Roser D, Bankel L, Homberg N, Geishauser A, Brenner CD, et al. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-gamma and induces NK cell-dependent lymphoma control without other immunotherapies. Int J Cancer (2017) 140(9):2125–33. doi: 10.1002/ijc.30646
26. Nasi A, Fekete T, Krishnamurthy A, Snowden S, Rajnavolgyi E, Catrina AI, et al. Dendritic cell reprogramming by endogenously produced lactic acid. J Immunol (2013) 191(6):3090–9. doi: 10.4049/jimmunol.1300772
27. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
28. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
29. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
30. Balkwill F, Mantovani A. Inflammation and cancer: back to virchow? Lancet (9255) 2001:539–45:357. doi: 10.1016/S0140-6736(00)04046-0
31. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther (2021) 6(1):263. doi: 10.1038/s41392-021-00658-5
32. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol (2017) 11(7):805–23. doi: 10.1002/1878-0261.12095
33. Dominguez C, David JM, Palena C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin Cancer Biol (2017) 47:177–84. doi: 10.1016/j.semcancer.2017.08.002
34. Liu J, Lin PC, Zhou BP. Inflammation fuels tumor progress and metastasis. Curr Pharm Des (2015) 21(21):3032–40. doi: 10.2174/1381612821666150514105741
35. Baram T, Rubinstein-Achiasaf L, Ben-Yaakov H, Ben-Baruch A. Inflammation-driven breast tumor cell plasticity: Stemness/EMT, therapy resistance and dormancy. Front Oncol (2020) 10:614468. doi: 10.3389/fonc.2020.614468
36. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):1–12. doi: 10.1038/s41423-019-0306-1
37. Castellani P, Balza E, Rubartelli A. Inflammation, DAMPs, tumor development, and progression: a vicious circle orchestrated by redox signaling. Antioxid Redox Signal (2014) 20(7):1086–97. doi: 10.1089/ars.2012.5164
38. Li Z, Jiang L, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol (2019) 26:101297. doi: 10.1016/j.redox.2019.101297
39. McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers (Basel) (2022) 14(14):3297. doi: 10.3390/cancers14143297
40. Chafe SC, Vizeacoumar FS, Venkateswaran G, Nemirovsky O, Awrey S, Brown WS, et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci Adv (2021) 7(35):eabj0364. doi: 10.1126/sciadv.abj0364
41. Lin Z, Song J, Gao Y, Huang S, Dou R, Zhong P, et al. Hypoxia-induced HIF-1alpha/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol (2022) 52:102312. doi: 10.1016/j.redox.2022.102312
42. Yang H, Hu Y, Weng M, Liu X, Wan P, Hu Y, et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J Adv Res (2022) 37:91–106. doi: 10.1016/j.jare.2021.10.001
43. Fan Z, Yang G, Zhang W, Liu Q, Liu G, Liu P, et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J Cell Mol Med (2021) 25(21):10197–212. doi: 10.1111/jcmm.16957
44. Fuhrmann DC, Mondorf A, Beifuss J, Jung M, Brune B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol (2020) 36:101670. doi: 10.1016/j.redox.2020.101670
45. Guo X, Liu F, Deng J, Dai P, Qin Y, Li Z, et al. Electron-accepting micelles deplete reduced nicotinamide adenine dinucleotide phosphate and impair two antioxidant cascades for ferroptosis-induced tumor eradication. ACS Nano (2020) 14(11):14715–30. doi: 10.1021/acsnano.0c00764
46. Zhang Y, Zhai Z, Duan J, Wang X, Zhong J, Wu L, et al. Lactate: the mediator of metabolism and immunosuppression. Front Endocrinol (Lausanne) (2022) 13:901495. doi: 10.3389/fendo.2022.901495
47. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep (2020) 33(10):108487. doi: 10.1016/j.celrep.2020.108487
48. Li M, Jin S, Zhang Z, Ma H, Yang X. Interleukin-6 facilitates tumor progression by inducing ferroptosis resistance in head and neck squamous cell carcinoma. Cancer Lett (2022) 527:28–40. doi: 10.1016/j.canlet.2021.12.011
49. Yang Y, Lin Z, Han Z, Wu Z, Hua J, Zhong R, et al. miR-539 activates the SAPK/JNK signaling pathway to promote ferropotosis in colorectal cancer by directly targeting TIPE. Cell Death Discovery (2021) 7(1):272. doi: 10.1038/s41420-021-00659-x
50. Xian ZY, Hu B, Wang T, Cai JL, Zeng JY, Zou Q, et al. CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer. Neoplasma (2020) 67(5):1063–73. doi: 10.4149/neo_2020_191024N1084
51. Xu Q, Zhou L, Yang G, Meng F, Wan Y, Wang L, et al. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int (2020) 44(11):2344–56. doi: 10.1002/cbin.11444
52. Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am J Cancer Res (2019) 9(4):730–9.
53. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
54. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discovery (2019) 9(12):1673–85. doi: 10.1158/2159-8290.CD-19-0338
55. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell (2022) 40(4):365–78.e6. doi: 10.1016/j.ccell.2022.02.003
56. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity (2021) 54(7):1561–77.e7. doi: 10.1016/j.immuni.2021.05.003
57. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
58. Zhang M, Shao W, Yang T, Liu H, Guo S, Zhao D, et al. Conscription of immune cells by light-activatable silencing NK-derived exosome (LASNEO) for synergetic tumor eradication. Adv Sci (Weinh) (2022) 9(22):e2201135. doi: 10.1002/advs.202201135
59. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19(1):120. doi: 10.1158/1557-3125.HIPPO19-IA12
60. Pan R, Ryan J, Pan D, Wucherpfennig KW, Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell (2022) 185(9):1521–38.e18. doi: 10.1016/j.cell.2022.03.030
61. Moujalled D, Southon AG, Saleh E, Brinkmann K, Ke F, Iliopoulos M, et al. BH3 mimetic drugs cooperate with temozolomide, JQ1 and inducers of ferroptosis in killing glioblastoma multiforme cells. Cell Death Differ (2022) 29(7):1335–48. doi: 10.1038/s41418-022-00977-2
62. Kim KS, Choi B, Choi H, Ko MJ, Kim DH, Kim DH. Enhanced natural killer cell anti-tumor activity with nanoparticles mediated ferroptosis and potential therapeutic application in prostate cancer. J Nanobiotechnology (2022) 20(1):428. doi: 10.1186/s12951-022-01635-y
63. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
64. MacNabb BW, Tumuluru S, Chen X, Godfrey J, Kasal DN, Yu J, et al. Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity (2022) 55(6):982–97.e8. doi: 10.1016/j.immuni.2022.04.016
65. Zelenay S, Reis e Sousa C. Adaptive immunity after cell death. Trends Immunol (2013) 34(7):329–35. doi: 10.1016/j.it.2013.03.005
66. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun (2022) 13(1):3676. doi: 10.1038/s41467-022-31218-2
67. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
68. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35(11):109235. doi: 10.1016/j.celrep.2021.109235
69. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
70. Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, et al. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol (2021) 206(6):1395–404. doi: 10.4049/jimmunol.2000500
71. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
72. Shen L, Zhou Y, He H, Chen W, Lenahan C, Li X, et al. Crosstalk between macrophages, T cells, and iron metabolism in tumor microenvironment. Oxid Med Cell Longev (2021) 2021:8865791. doi: 10.1155/2021/8865791
73. Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics (2021) 11(14):7072–91. doi: 10.7150/thno.57803
74. Gu Z, Liu T, Liu C, Yang Y, Tang J, Song H, et al. Ferroptosis-strengthened metabolic and inflammatory regulation of tumor-associated macrophages provokes potent tumoricidal activities. Nano Lett (2021) 21(15):6471–9. doi: 10.1021/acs.nanolett.1c01401
75. Xu Z, Chen X, Song L, Yuan F, Yan Y. Matrix remodeling-associated protein 8 as a novel indicator contributing to glioma immune response by regulating ferroptosis. Front Immunol (2022) 13:834595. doi: 10.3389/fimmu.2022.834595
76. Li LG, Peng XC, Yu TT, Xu HZ, Han N, Yang XX, et al. Dihydroartemisinin remodels macrophage into an M1 phenotype via ferroptosis-mediated DNA damage. Front Pharmacol (2022) 13:949835. doi: 10.3389/fphar.2022.949835
77. Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the anti-PD-1/L1 response. Adv Sci (Weinh) (2023) 10(2):e2203973. doi: 10.1002/advs.202203973
78. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol (2022) 56:102463. doi: 10.1016/j.redox.2022.102463
79. Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol (2022) 24(7):1113–25. doi: 10.1093/neuonc/noac033
80. Yan M, Zheng M, Niu R, Yang X, Tian S, Fan L, et al. Roles of tumor-associated neutrophils in tumor metastasis and its clinical applications. Front Cell Dev Biol (2022) 10:938289. doi: 10.3389/fcell.2022.938289
81. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol (2019) 9:1146. doi: 10.3389/fonc.2019.01146
82. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun (2020) 11(1):5424. doi: 10.1038/s41467-020-19193-y
83. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
84. Yang X, Li Y, Zou L, Zhu Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front Oncol (2019) 9:356. doi: 10.3389/fonc.2019.00356
85. Fan JT, Zhou ZY, Luo YL, Luo Q, Chen SB, Zhao JC, et al. Exosomal lncRNA NEAT1 from cancer-associated fibroblasts facilitates endometrial cancer progression via miR-26a/b-5p-mediated STAT3/YKL-40 signaling pathway. Neoplasia (2021) 23(7):692–703. doi: 10.1016/j.neo.2021.05.004
86. Shi H, Huang S, Qin M, Xue X, Guo X, Jiang L, et al. Exosomal circ_0088300 derived from cancer-associated fibroblasts acts as a miR-1305 sponge and promotes gastric carcinoma cell tumorigenesis. Front Cell Dev Biol (2021) 9:676319. doi: 10.3389/fcell.2021.676319
87. Zhou L, Li J, Tang Y, Yang M. Exosomal LncRNA LINC00659 transferred from cancer-associated fibroblasts promotes colorectal cancer cell progression via miR-342-3p/ANXA2 axis. J Transl Med (2021) 19(1):8. doi: 10.1186/s12967-020-02648-7
88. Chen B, Sang Y, Song X, Zhang D, Wang L, Zhao W, et al. Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics (2021) 11(8):3932–47. doi: 10.7150/thno.53412
89. Fan J, Xu G, Chang Z, Zhu L, Yao J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin Sci (Lond) (2020) 134(7):807–25. doi: 10.1042/CS20200039
90. Shelton M, Anene CA, Nsengimana J, Roberts W, Newton-Bishop J, Boyne JR. The role of CAF derived exosomal microRNAs in the tumour microenvironment of melanoma. Biochim Biophys Acta Rev Cancer (2021) 1875(1):188456. doi: 10.1016/j.bbcan.2020.188456
91. Shi Z, Jiang T, Cao B, Sun X, Liu J. CAF-derived exosomes deliver LINC01410 to promote epithelial-mesenchymal transition of esophageal squamous cell carcinoma. Exp Cell Res (2022) 412(2):113033. doi: 10.1016/j.yexcr.2022.113033
92. Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol (2019) 20(1):12. doi: 10.1186/s13059-018-1604-0
93. Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics (2018) 8(14):3932–48. doi: 10.7150/thno.25541
94. Chen X, Liu Y, Zhang Q, Liu B, Cheng Y, Zhang Y, et al. Exosomal miR-590-3p derived from cancer-associated fibroblasts confers radioresistance in colorectal cancer. Mol Ther Nucleic Acids (2021) 24:113–26. doi: 10.1016/j.omtn.2020.11.003
95. Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer-associated fibroblasts-derived exosomes suppress immune cell function in breast cancer via the miR-92/PD-L1 pathway. Front Immunol (2020) 11:2026. doi: 10.3389/fimmu.2020.02026
Keywords: ferroptosis, tumor microenvironment, tumor immunotherapy, lipid peroxidation, micro RNA
Citation: Xia Z and Quan Y (2023) Effect of tumor microenvironment on ferroptosis: inhibition or promotion. Front. Oncol. 13:1155511. doi: 10.3389/fonc.2023.1155511
Received: 31 January 2023; Accepted: 24 April 2023;
Published: 05 May 2023.
Edited by:
Jianxun J. Song, Texas A&M Health Science Center, United StatesReviewed by:
Jugal Das, Texas A&M Health Science Center, United StatesCopyright © 2023 Xia and Quan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Quan, cXVhbnlfaUBzaW5hLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.