
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 26 June 2023
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1153082
This article is part of the Research Topic Implementation of Genomic and Epigenomic Innovation in Clinical Cancer Diagnostics View all 8 articles
 Anne Tierens1
Anne Tierens1 Elizabeth Kagotho2
Elizabeth Kagotho2 Satoru Shinriki3Andrew Seto4
Satoru Shinriki3Andrew Seto4 Adam C. Smith1,4Melanie Care4Dawn Maze5
Adam C. Smith1,4Melanie Care4Dawn Maze5 Hassan Sibai5Karen W. Yee5Andre C. Schuh5Dennis Dong Hwan Kim5Vikas Gupta5
Hassan Sibai5Karen W. Yee5Andre C. Schuh5Dennis Dong Hwan Kim5Vikas Gupta5 Mark D. Minden5
Mark D. Minden5 Hirotaka Matsui3
Hirotaka Matsui3 José-Mario Capo-Chichi1,4*
José-Mario Capo-Chichi1,4*Introduction: Inherited DDX41 mutations cause familial predisposition to hematologic malignancies including acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), with the majority of DDX41 mutated MDS/AMLs described to date harboring germline DDX41 and co-occurring somatic DDX41 variants. DDX41-AMLs were shown to share distinguishing clinical features such as a late AML onset and an indolent disease associated with a favorable outcome. However, genotype-phenotype correlation in DDX41-MDS/AMLs remain poorly understood.
Methods: Here, we studied the genetic profile, bone marrow morphology and immunophenotype of 51 patients with DDX41 mutations. We further assessed the functional impact of ten previously uncharacterized DDX41 variants of uncertain significance.
Results: Our results demonstrate that MDS/AML cases harboring two DDX41 variants share specific clinicopathologic hallmarks that are not seen in other patients with monoallelic DDX41 related hematologic malignancies. We further showed that the features seen in these individuals with two DDX41 variants were concordant with biallelic DDX41 disruption.
Discussion: Here, we expand on previous clinicopathologic findings on DDX41 mutated hematologic malignancies. Functional analyses conducted in this study unraveled previously uncharacterized DDX41 alleles and further illustrate the implication of biallelic disruption in the pathophysiology of this distinct AML entity.
Germline mutations in DDX41 cause familial predisposition to myeloid malignancies including acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) (1–9). Single acquired DDX41 variants on their own were not identified as major contributors to the pathophysiology of AML, MDS and myeloproliferative neoplasms (MPN). However, not uncommon is the co-occurrence of additional somatic DDX41 variants in individuals with a germline DDX41 mutation (1–9). Such AML presenting with multiple DDX41 variants share distinguishing features, suggesting that the presence of germline DDX41 and acquired DDX41 mutation(s) defines a specific AML subgroup. For example, DDX41-AML is a late onset disease with an indolent presentation and a favorable outcome; features that contrast with other types of inherited or acquired AMLs (1–9). Several somatic and germline DDX41 mutations have been reported to date. The deleterious potential of loss of function variants (i.e frameshift, nonsense and splicing) can be easily inferred. However, the functional impact of numerous missense, in-frame and start loss variants identified in DDX41 has not been assayed. Functional characterization of such DDX41 variants of uncertain significance (VUS) is of great clinical value. For instance, elucidating the role of a previously uncharacterized germline DDX41 variant is of crucial importance to genetic counselling of an affected carrier as well as family risk relatives, given considerations of familial predisposition to myeloid malignancies.
Here, we sought to analyze the genomic architecture, bone marrow morphology as well as immunophenotypes of patients with DDX41 mutated AML, MDS and MPN to explore possible genotype–phenotype correlations between individuals with these DDX41 related hematologic malignancies. To this effect, we further investigated the impact of ten previously reported, yet uncharacterized DDX41 VUS. Our results expand on previous reports on DDX41 related malignancies. Unique to this study, we provide supportive in-vivo and in vitro evidence that biallelic disruption of DDX41 is associated with unique clinicopathologic features observed in this distinct AML entity.
The study cohort constituted of 1930 patients with myeloid malignancies diagnosed at the Princess Margaret Centre. DNA was extracted from peripheral blood (PB) or bone marrow (BM) samples of myeloid malignancies cases studied herein. The study was approved by the University Health Network Research Ethics Board.
G-banding analyses were conducted on all cases with analyzable metaphases. Where G-banding analyses were unsuccessful, reflex FISH testing was performed to rule out possible deletions on chromosome 5 where DDX41 is located.
Genomic DNA was sheared and library capture by hybridization was performed by using a pool of custom probes targeting hotspot sequences or exonic coding regions and flanking intronic sequences of 49 genes considered clinically relevant in hematologic malignancies (Table S1). The gene targets include the entire consensus coding DNA regions of DDX41. Libraries were visualized (TapeStation, Agilent Technologies), and sequencing was performed by using the Illumina Next-Generation Sequencing platform (paired-end sequencing, MiSeq version 2). Bioinformatics analyses included alignment to the human genome reference build GRCh37/hg19 using the Burrows-Wheeler Alignment, processing and quality metrics using the Genome Analysis Toolkit and Picard, and variant calling using VarScan2. Variants not meeting laboratory-defined quality metrics (read depth <100, population frequency >1% in the gnomAD database, or VAF <2%) were removed from further analysis.
Variant filtering was performed by using the Alissa Clinical Informatics Platform (Agilent Technologies). Variants lying outside of coding sequences or canonical splice sites were filtered out, along with synonymous alterations. The remaining variants were classified as pathogenic/likely pathogenic or variants of uncertain significance following the CAP/AMP/ASCO somatic variants classification guidelines (10). Variant annotation was conducted as previously described (11–13). Briefly, evidence were gathered from published literature (eg, PubMed: ncbi.nlm.nih.gov/pubmed/), as well as cancer-wide (eg, COSMIC: cancer.sanger.ac.uk/cosmic) and gene-specific (eg. TP53 IARC: http://p53.iarc.fr/) repositories. Pathogenic (Tier-1) or likely pathogenic (Tier-2) variants included the following: (1) variants affecting hotspot codons (eg. p.R525H in DDX41); (2) variants occurring at critical gene regions or protein functional domains in which other deleterious variants have been reported in myeloid neoplasms (eg. truncating variants in the DEAD box of DDX41); and (3) variants predicted to disrupt the normal activity of the encoded protein (eg. start loss variant in DDX41). Variants not meeting any of the aforementioned criteria were classified as variants of uncertain significance (Tier-3).
For immunphenotyping, heparin anticoagulated bone marrow aspirates were processed according to the Euroflow standard procedures for sample preparation, staining and acquisition as previously described (14). Bone marrow smears were stained with Wright Giemsa. Bone marrow trephine were fixed in B+ decalcified and stained with Hematoxylin and Eosin. Acquisition was performed on Lyric Instruments (Becton Dickenson (BD) Biosciences, San Jose, CA, USA). The files were analyzed using Infinicyt v.2.1 (Cytognos TM, Salamanca, Spain).
DDX41 variant expression vectors (p.Met1?, p.Ala11Thr, p.Met155Ile p.Pro213Arg, p.Gly218Asp, p.Thr320Ile, p.Glu345Lys, p.Ala346Pro, p.Ser363del, p.Arg479Trp, p.Arg525His and p.Asp570Gly) were constructed using the KOD-Plus-Mutagenesis Kit (Toyobo Co., Ltd., Tokyo, Japan) based on the pcDNA4 myc-His plasmid inserted with wild-type (WT) DDX41 (15). The primers used for the construction are shown in Table S2.
The pLKO.1 puro vector inserted with an shRNA targeting the 3’UTR region of the DDX41 gene (shDDX41-UTR) was transfected into HEK293FT cells together with the psPAX2/pMD2G packaging plasmids to produce lentivirus for DDX41 knockdown. The target sequence for DDX41 knockdown is CCTGTGCTCTTCAGAATTA. pLKO.1-scramble shRNA (shScr), a gift of David Sabatini (Addgene plasmid #1864), was used for control (16). The virus in the culture supernatant was infected to HEK293T cells, and the knockdown was confirmed by western blot analysis.
DDX41-expressing plasmid (WT, p.R525H and other 11 variants) or an empty plasmid for a negative control were transfected into HEK293T cells using Polyethylenimine MAX (Polysciences, Warrington, PA, USA) according to the manufacturer’s instructions. The culture medium was replaced with fresh medium 8 hours after the transfection, after which the cells were infected with the lentivirus for DDX41 knockdown as described above. The number of viable cells was counted by trypan blue staining on day 6 after the infection. A schematic illustration of the experimental procedure was shown in Figure S1.
The cells were washed with PBS and fixed overnight in 70% ethanol chilled at -20°C. Fixed cells were washed with PBS and incubated with propidium iodide (50 μg/mL) and RNase A (10 μg/mL) for 20 min at room temperature. After filtration, the cell suspensions were analyzed using FACSVerse (BD Biosciences, San Jose, CA, USA).
For cell viability assay and cell cycle analysis, the experiments were performed in triplicate. Due to the limited sample numbers in each assay, all individual data points for these experiments were plotted alongside error bars.
We have sequenced the tumor of 1930 patients with myeloid malignancies including AML, MDS and MPN using a targeted myeloid next-generation sequencing (NGS) panel (Table S1). In total, we have identified 51 cases (2.6%) with at least one mutation in DDX41 (Table 1). Our study cohort consist of 13 DDX41-MPNs and 38 DDX41-AMLs. Cytogenetic analyses conducted in parallel to NGS revealed a deletion chromosome 5q35 encompassing DDX41 in one patient (case M22) that also harbored a DDX41 point mutation (Table 1).
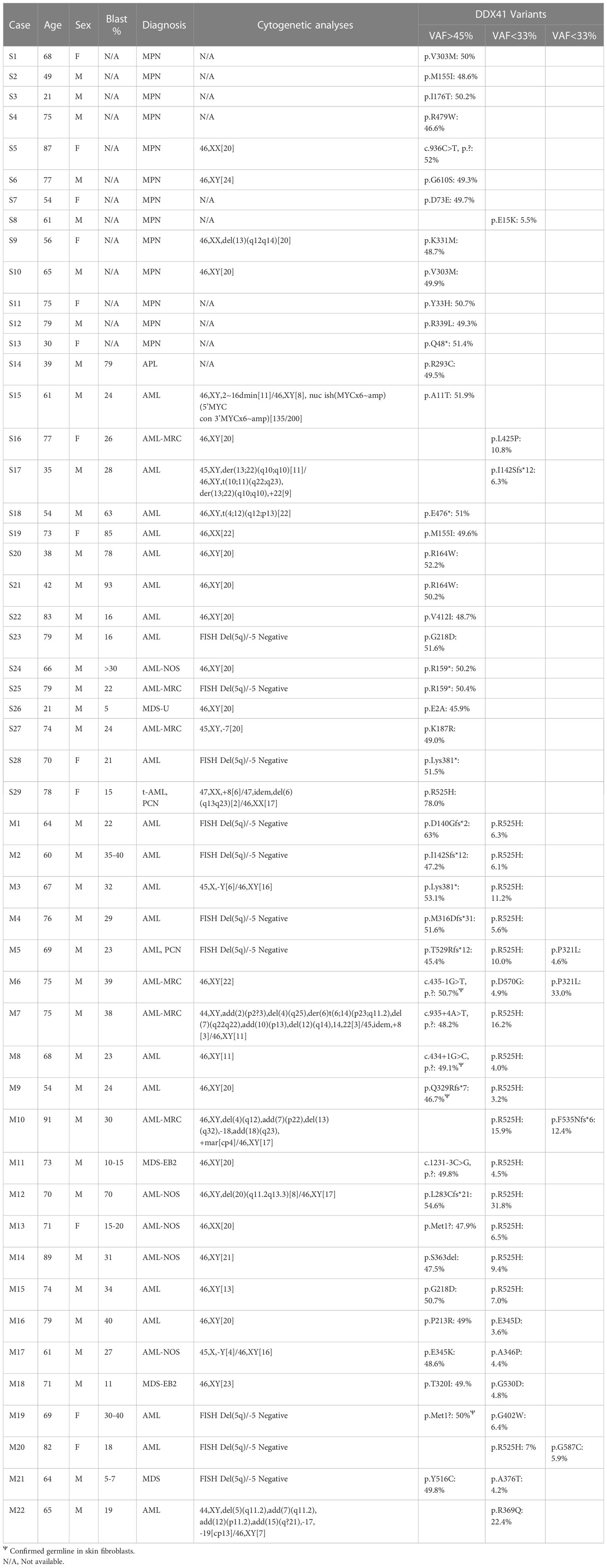
Table 1 Genomics findings in myeloid cases with mutations in DDX41.
All the DDX41-MPNs (cases S1-S13) harbored a single DDX41 variant (DDX41-s). The DDX41-AMLs constituted of 16 DDX41-s (cases S14-S29), as well as 22 cases (cases M1-M22) with multiple alterations in DDX41 (DDX41-m). The study cohort composed of 13 females and 38 males (Table 1). DDX41-AMLs were predominantly males (DDX41-s AMLs = 12 or 75%, DDX41-m AMLs = 18 or 82%) compared to DDX41-MPNs (7 or 54%). DDX41-m presented with AML at older age (average = 71, minimum = 54, maximum = 91) compared to the DDX41-s AMLs (average = 61, minimum = 21, maximum = 83) and the DDX41-s MPNs (average = 61, minimum = 21, maximum = 87).
We identified patients with one (n=29), two (n=20) or three (n=2) genetic lesions (i.e DNA point mutations and chromosome deletion) in DDX41. Variants detected in DDX41 were distributed in two distinct clusters of variant allele frequency (VAF) with germline-like variants having a VAF >45% (average: 50%, maximum: 63%) and somatic variants VAF <33% (average: 9.4%, minimum: 3.2%). Repartition of variants within these VAF clusters applied to all the DDX41 cases analyzed herein, except for an outlier (case S29) harboring the somatic hotspot p.R252H variant at 78%. Germline variants were confirmed by sequencing skin fibroblasts, where possible (Table 1).
In total we identified 74 DDX41 variants including 44 germline-like and 30 somatic. More than half (74%) of these variants (germline = 33, somatic = 22) have been previously reported in a DDX41-mutated hematologic malignancy or were seen in more than one DDX41 mutated patient from this study cohort (Table 1, Figure 1). The most common DDX41 variants was the hotspot p.R525H (n=16).
Suspicious germline-like variants consisted of 24 missenses, 2 start-losses, 1 in-frame, 6 nonsenses, 6 frameshifts and 5 splicing variants (Figure 1). Somatic variants included 28 missenses and 2 frameshift variants. The 19 loss-of function (start loss, nonsense, frameshift and splicing) germline DDX41 variants were classified as likely pathogenic variants using the ACMG germline variant classification criteria (17). However, the remaining 25 germline variants (missense and in-frame) were considered as variants of uncertain significance (VUS) due to the lack of evidence of their impact on the encoded protein. Previously, we showed that the somatic hotspot p.R525H variant cannot rescue the growth of hematopoietic cells in which endogenous DDX41 had been eliminated (15). Based on this functional evidence, the p.R525H hotspot was determined to be a Tier-1 (pathogenic) variant using the AMP/CAP/ASCO somatic variant classification scheme (10). When applying the same scheme to other somatic DDX41 variants, the 2 frameshifts were interpreted as Tier-2 (likely pathogenic) variants and 12 previously uncharacterized missense somatic variants were determined to be Tier-3 (VUS). In summary, of the 74 DDX41 variants identified herein, half (germline and somatic) were classified as pathogenic or likely pathogenic variants and the other half including 25 germline and 12 somatic were VUS.
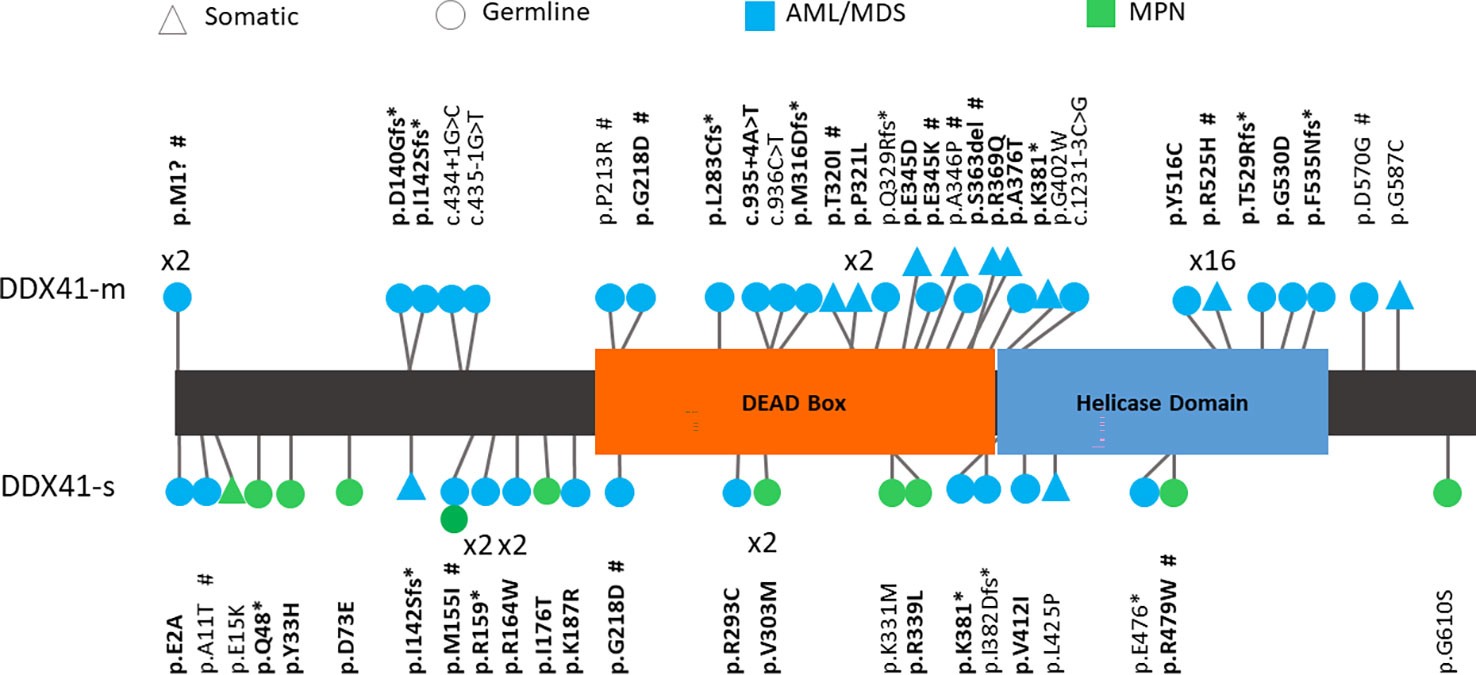
Figure 1 Schematic representation of variants detected in DDX41. Bold – Variants previously reported. # - Variant functionally assessed in this study.
The majority of variants identified in the DDX41-s were of different nature and were located elsewhere in the protein than those seen in the DDX41-m. For instance, 12 out of 16 (75%) of the DDX41-s harbored a single missense VUS in non-catalytic regions (N- and C-terminal ends) of DDX41. In contrast, such missense VUS where only identified in 2 out of 8 (25%) of the DDX41-m. Instead 25 out of the 39 variants (64%) detected in the DDX41-m, were disruptive variants affecting the catalytic core DEAD box and (Figure 1). These deleterious DDX41-m variants include the hotspot p.R525H (n=16) and 9 additional loss-of-function (nonsense, frameshift, splicing variants).
NGS detected additional variants in myeloid genes in the DDX41-MPNs and DDX41-AMLs (Figure 2). These co-occurring variants were predominantly seen in ASXL1 (n=15), TET2 (n=12), CUX1 (n=10) and TP53 (n=9). In aggregate, DNA transcription (n=54) and DNA methylation (n=43) genes accumulated the most variants. Overall, DDX41-s MPNs (minimum: 1, maximum: 10, average: 4), DDX41-s AMLs (minimum: 1, maximum: 8, average: 4) and DDX41-m AMLs (minimum: 2, maximum: 9, average: 5) shared similar numbers of co-occurring myeloid variants. However, variants affecting myeloid defining biomarkers were more frequently seen in the DDX41s-MPNs (JAK2 = 6, CALR = 3, MPL =1) and the DDX41-s AMLs (CEBPA biallelic =2, NPM1 = 3, RUNX1 = 2) compared to the DDX41-m AMLs (RUNX1 = 1, Figure 2) (18). Furthermore, molecular and cytogenetics tests conducted in parallel to NGS identified other abnormalities (PML-RARA fusion =1, double minute chromosomes with MYC amplification = 1, KMT2A rearrangement =1, ETV6 rearrangement =1) that could explain the origin of AML in 4 additional DDX41-s AMLs (19–21). In summary, the origin of MPN could be associated to the presence of a co-occurring variant and not a DDX41 mutation, in 10 out of 13 (77%) of the DDX41-MPNs. Similarly, the etiology of AML could be linked to a genetic lesion not impacting DDX41 in 11 out of 18 (61%) of the DDX41-s. However, such myeloid defining alterations were not unraveled in the 22 DDX41-m studied herein (Figure 2).
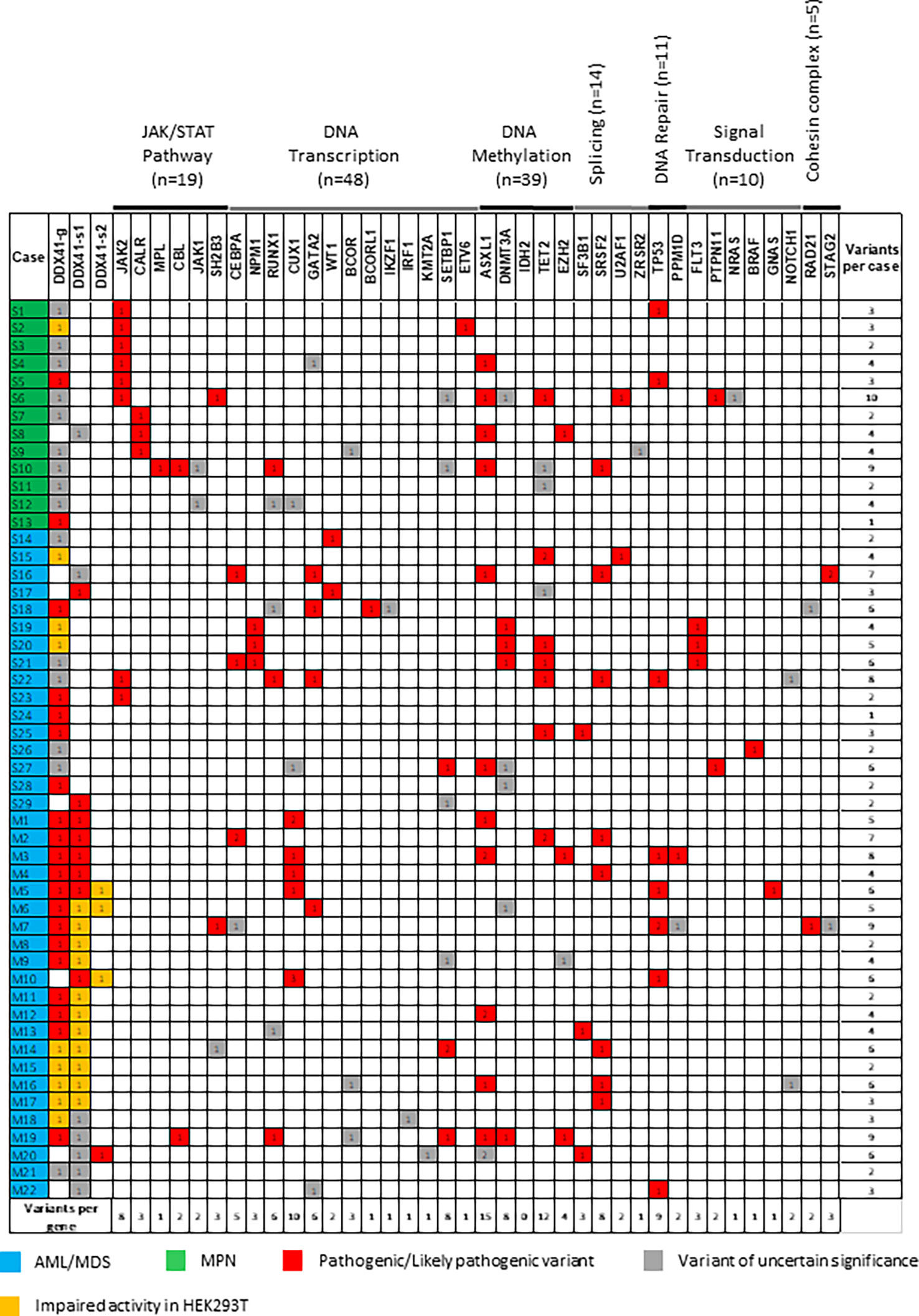
Figure 2 Co-occurring variants in DDX41 mutated patients. DDX41-g. DDX41 germline. DDX41-s. DDX41 somatic.
NGS identified 37 variants (25 germline and 12 somatic) variants that were determined to be VUS following ACMG, AMP, CAP, ASCO variants classifications schemes. More than half of these VUS (n =28) have been reported in association to DDX41 mutated hematologic malignancies, but were never characterized. Here we sought to investigate the impact of 10 (8 germline and 2 somatic) of these VUS in HEK293T cells (Table 1, Figure 1). In selecting those 10 variants for functional assessment, priority was given to variants meeting the following criteria: (a) frequency of variant in DDX41 mutated malignancies and (b) location of variant within the encoded DDX41 protein. As such, we retained 1 in-frame and 9 missenses variants for functional analyses conducted in HEK293T cells.
We have previously demonstrated that the hotspot p.R525H variant is a deleterious variant as this variant failed to rescue the cellular growth of hematopoietic cells in which endogenous DDX41 had been eliminated (15). Here, we investigated the impact of DDX41 mutants on the proliferation of HEK293T cells, within which the endogenous DDX41 expression was repressed using a 3’UTR shRNA. First, we confirmed that consistent with previous results, p.R525H also failed to restore cellular growth in which endogenous DDX41 was suppressed as elicited by 61% cells recovered compared to HEK293T cells transduced with wild-type DDX41 (Figure 3A). We have previously showed that DDX41 deficient hematopoietic cells can progress to the S phase of the cell cycle, albeit with mild DNA replication. However, these cells are stalled in the G2/M phase just before entering mitosis and arrest in the G2/M phase correlates with impaired cellular proliferation in these DDX41 deficient cells (22). Here, we observed that the p.R525H mutant also distorted normal cell cycle of HEK293T cells in which endogenous DDX41 was suppressed with fewer cells observed in G0/G1 (33.9 versus 43.8) and more cells detected in the S (25.3 versus 22.5) and G2/M (38.8 versus 31.8) phases compared to HEK293T cells transduced with wild-type DDX41 (Figures 3B, C). Accumulation of cells in the G2/M phase is reminiscent of previous observations made in DDX41 deficient hematopoietic cells (22).
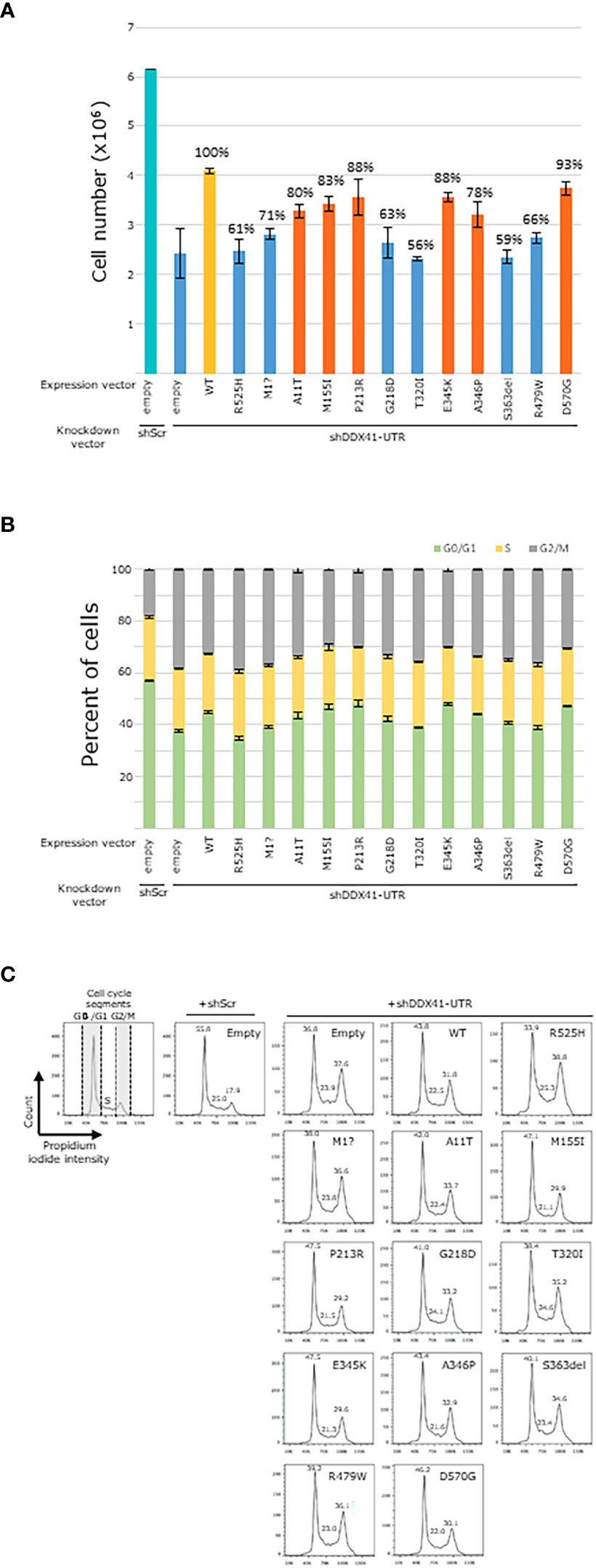
Figure 3 Functional characterization of DDX41 variants in HEK293T cells. (A). Representative images of HEK293T cell count measurements performed in triplicate are shown. Percentage of cellular growth compared to HEK293T cells transduced with wild type DDX41 is included on top of histograms. Blue. Variants showing 75% or fewer HEK293T cells when compared to HEK293T cells transduced with wild type DDX41. Orange. Variants showing greater than 75% of HEK293T cells when compared to HEK293T cells transduced with wild type DDX41. (B). Plots indicate G0/G1 (green), S (yellow) and G2/M (gray) phases of all individual data points. (C). Impact of selected DDX41 variants on cell cycle kinetics in HEK293T DDX41 knockdown model. Values of each cell cycle phase are indicated in the histogram.
To further validate the use of the HEK293T paradigm for our functional analyses, we also showed that another common germline DDX41 allele, the start loss variant p.Met1?, elicited similar deficits on HEK293T proliferation (71% cells recovered compared to wild-type DDX41) and cell cycle (number of HEK293T cells in G0/G1: 38, S: 23.8 and G2/M: 36.6; Figure 3). Thus, the p.R525H and p.Met1? alleles show dysfunction in the HEK293T paradigm. Findings of disruption of DDX41 by the p.Met1? allele is corroborated by previous studies showing that p.Met1? abrogates normal protein translation in HEK293T cells resulting in a shortened protein product with altered cellular localization (2).
Based on these observations, we assessed the ability of the ten DDX41 VUS to impact the proliferation as well as cell cycle of HEK293T cells (Figure 3). Four variants (p.G218D, p.T320I, p.S363del and p.R479W) also disrupted cellular growth in HEK293T to comparable levels (between 56% and 66% of cells recovered compared to wild-type DDX41) to those observed with p.R525H and p.Met1? (blue bars, Figure 3A). Six other variants (p.A11T, p.M155I, p.P213R, p.E345K, p.A346P and p.D570G) also failed to restore cellular growth of HEK293T cells; although between 78% and 93% of cells were recovered compared to wild-type DDX41 (orange bars, Figure 3A). Our results showed that the same DDX41 mutants also impaired normal cell cycle in HEK293T cells. Although, cell cycle kinetics varied between the DDX41 mutants investigated herein; overall, all the mutant alleles showed a distortion of the cell cycle with an accumulation of cells in G2/M which are reminiscent of previous observations made in hematopoietic cells (22). In summary, we showed that the ten DDX41 variants analyzed herein impaired cellular proliferation and normal cell cycle in HEK293T cells suggesting that these are deleterious DDX41 alleles.
We reviewed the bone marrow (BM) features of the DDX41 mutated AML, MDS and MPN patients. Morphologic features seen in the DDX41-s (AMLs and MPNs) were heterogeneous and did not have any distinctive hallmarks differentiating them from non-mutated DDX41 AMLs or MPNs seen at our institution. Unlike the DDX41-s, the DDX41-m presented with variable degrees of pancytopenia at presentation and no or rare circulating blasts. This is in contrast to DDX41-s AMLs where circulating blasts were seen, and occasionally at high levels. The majority of DDX41-m AMLs were classified as AML NOS and AML MRC, the latter often secondary to MDS (Table 1). BM of DDX41-m AMLs were hypo- to normocellular often with relative erythroid hyperplasia and granulocytic and megakaryocytic hypoplasia. Blasts were small, without distinctive morphologic characteristics and at the lower limit for the diagnosis of AML (i.e. in the 20-30% range). The presence of dysplastic changes varied but were frequently minimal, not always meeting the morphologic criteria for MDS (Figure 4).
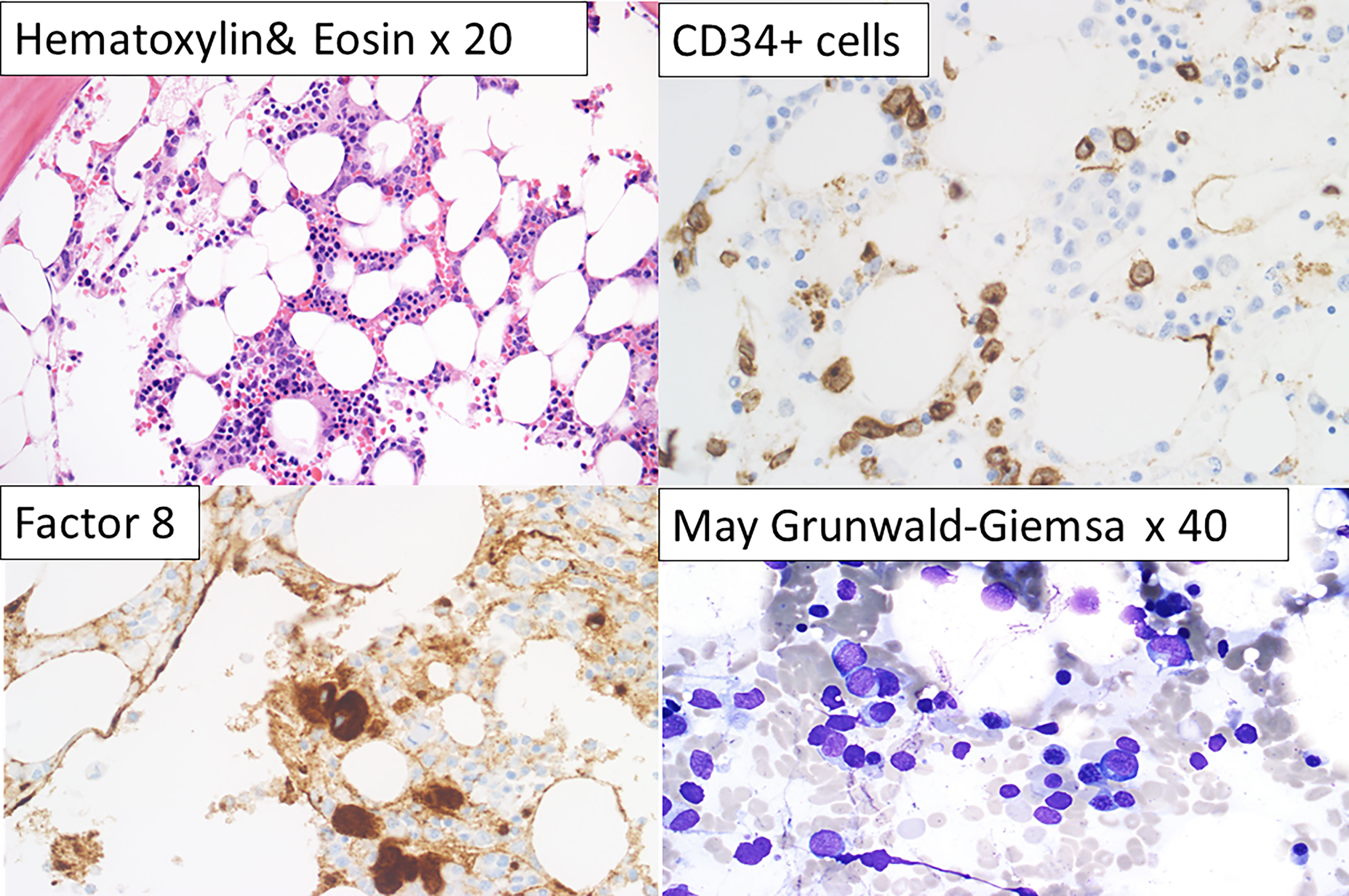
Figure 4 Characteristic bone marrow features of DDX41-m AML. Slightly hypo cellular bone marrow for the patient’s age with relative erythroid hyperplasia, granulocytic and megakaryocytic hypoplasia (Hematoxylin and Eosin, x 20). Small clusters of CD34+ cells (x63). Factor 8 positive normal and small megakaryocytes (x63). Bone marrow particle showing erythroid cells and blasts (May-Grunwald x40).
Given that DDX41-m AML present with clinicopathologic characteristics reminiscent of MDS/AML, we sought to explore whether the immunophenotype of the DDX41-m AML has common features with MDS with excess blasts. We performed an immunophneotpic analysis to look for changes associated with MDS and compared the antigen expression profiles with those of patients with MDS with excess blasts. BM samples of patients with non-hematologic malignancies served as normal controls. We analyzed 12 DDX41-m (cases M5-M6, M8-M11, M15-M20) on which suitable BM sample could be obtained our investigations, 10 normal controls and 10 MDS samples and predominantly focused our analysis on CD34+ progenitor cells and nucleated red blood cells often showing aberrant immunophenotypes in MDS (Figures 5, 6). CD34+ cells were characterized by decreased expression of CD117 and HLA-DR compared to the expression on normal CD34+ cells but similar to that of CD34+ cells of MDS (Figure 5A). We did not find consistent changes in the expression of the myeloid markers CD13, CD33 and CD45 (Figure 5B). Of interest, the CD34+ cells did not express mature myeloid markers including CD11b CD36 and CD64 (not shown). CD34+ B cells precursors were often <5% of total CD34+ cells. Next, we examined the expression of CD36 and CD71 on nucleated red cells because of their frequent aberrant expression in MDS. Unlike nucleated red cells of MDS, the expression of CD36 and CD71 was normal with similar expression patterns as observed in normal BM samples (Figure 5C). Maturing granulocytes and monocytes showed normal maturation patterns.
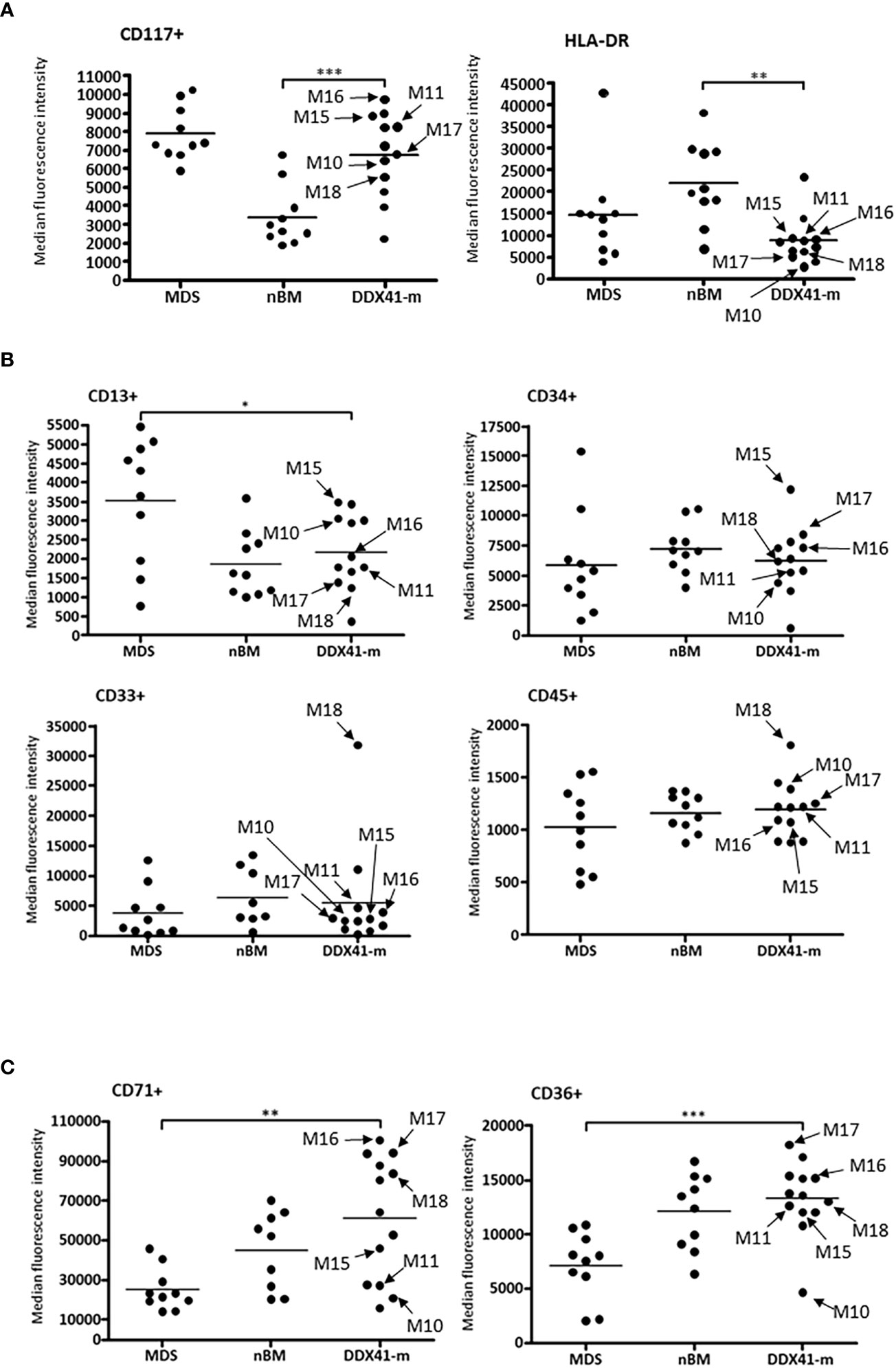
Figure 5 Antigen expression profiles of nucleated red cells and CD34 + cells. Arrows identifying specific DDX41-m discussed in the manuscript. (A). Median expression patterns of CD117 and HLA-DR on CD34 + cells of DDX41-m AML, MDS and nBM. Highest to lowest CD117 expression values for DDX41-m: M16, M9, M6, M15, M8, M11, M20, M17, M10, M18 and M5. Highest to lowest HLA-DR expression values for DDX41-m: M20, M6, M15, M11, M16, M9, M8, M18, M17, M10 and M5. (B). Median expression patterns of CD13, CD33, CD34 and CD45 on CD34 + cells of DDX41-m AML, MDS and nBM. Highest to lowest CD13 expression values for DDX41-m: M15, M19, M10, M20, M6, M16, M8, M11, M17, M18 and M5. Highest to lowest CD33 expression values for DDX41-m: M18, M20, M11, M16, M15, M17, M6, M8, M10, M9 and M5. Highest to lowest CD34 expression values for DDX41-m: M15, M17, M8, M16, M20, M6, M18, M11, M10, M9 and M5. Highest to lowest CD45 expression values for DDX41-m: M18, M6, M10, M17, M8, M11, M20, M16, M15, M5 and M9. (C). Median expression patterns of CD36 and CD71 on nucleated red cells of DDX41-m AML, Myelodysplastic syndrome (MDS) and normal BM (nBM). Highest to lowest CD71 expression values for DDX41-m: M16, M6, M17, M20, M18, M8, M5, M15, M11, M10, M9 and M19. Highest to lowest CD36 expression values for DDX41-m: M17, M20, M16, M19, M8, M5, M18, M11, M15, M9, M6 and M10. *p-value ≤0.05; **p-value ≤0.01; ***p-value ≤0.001.
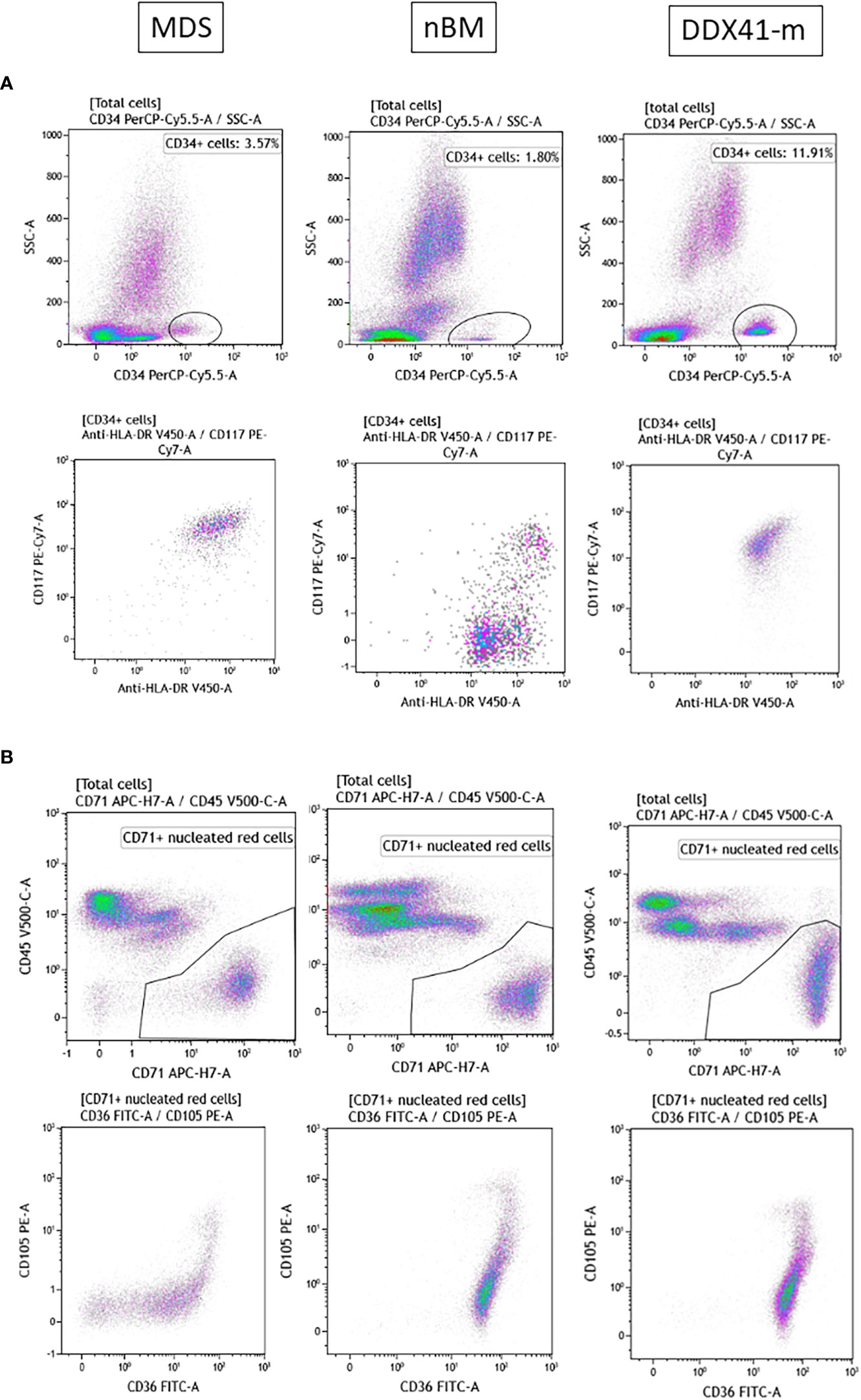
Figure 6 Representative FACS plot of the antigen expression profiles of nucleated red cells and CD34 + cells. MDS. Myelodysplastic syndrome. nBM. Normal BM. DDX41-m AML/MDS. MDS/AML with multiple DDX41 mutations. (A).CD34+ cells are gated on a CD34+ and SSC dot-plot. Expression of CD117 and HLA-DR on CD34+ cells. (B). CD71+ Nucleated red cells are gated on a CD45 and CD71 dot-plot. Expression of CD36, CD71 and CD105.
We applied a targeted myeloid NGS panel to 1930 patients with hematologic malignancies seen at our institution and identified 51 cases (13 MPNs and 38 MDS/AMLs) carrying at least one mutation in DDX41. The 2.6% incidence for DDX41 AML/MDS and MPN patients identified in this study is comparable to the pooled incidence of 3.3% reported in a systematic review of 20 studies on DDX41 mutated myeloid neoplasms (7). As shown by others (1–9), the majority of DDX41-MDS/AMLs (22 out of 38) harbored multiple (somatic combined to germline) DDX41 variants (DDX41-m, cases M1-M22); but of note, we also identified 16 MDS/AMLs (cases S14-S29) with a single DDX41 variant (DDX41-s). In contrast to the MDS/AMLs, all the MPNs (S1-S13) in our study carried a single DDX41 variant, consistent with previous reports of solitary DDX41 mutation in DDX41-MPNs (3, 20). Of interest, most of the DDX41-MPNs (10 out 13), also harbor co-occurring MPN disease-causing mutations (JAK2 = 6, CALR = 3, MPL =1), which correlates with findings of JAK2-mutated MPNs in other DDX41-MPNs previously reported (3). Furthermore, the morphologic features seen in these DDX41-MPNs were in line with those typically encountered in corresponding non DDX41 mutated (eg. JAK2, CALR and MPL) MPNs. Similar to the DDX41-s MPNs, the origin of AML could be attributed to other myeloid defining alterations than DDX41 mutations in the majority (9 out of 16) of the DDX41-s AMLs (Table 1, Figure 2). In keeping with a different AML etiology (eg. mutations in CEBPA, NPM1), the clinicopathologic features of the DDX41-s AMLs were heterogeneous and differed from what was seen in DDX41-m AMLs.
In our case series, the underlying cause of AML and MPN could be attributed to well-characterized oncogenic drivers other than a single DDX41 variant, suggesting that monoallelic DDX41 variants do not drive the pathophysiology of these myeloid malignancies. For instance, we have identified several DDX41-s that shared the same single DDX41 variant (eg. p.V303M in S1 and S10, p.M155I in S2 and S19, p.R164W in S20 and S21, p.R159* in S24 and S25) but yet had diverse AML or MPN presentations implying a limited contribution of such solitary DDX41 variants to the biology of these myeloid malignancies. In keeping with a lesser deleterious potential of monoallelic DDX41 mutations, the majority of variants identified in the DDX41-s were missense variants located outside of the DEAD box and helicase domain of DDX41 (Figure 1, Bottom). This is in contrast to the DDX41-m that have a predominance of loss-of-function variants clustering within the catalytic regions of the encoded protein (Figure 1, Top). Of note, even the presence of a single truncating DDX41 variant, was not sufficient to reproduce hallmarks of MDS/AML seen in the DDX41-m. For example, cases S28 and M3 shared the same truncating p.K381* DDX41 variant, but have distinct AML presentations at diagnosis (Table 1). Similar observations were made in cases S17 versus M2 (same p.I142Sfs*12 variant) or cases S23 versus M15 (same p.G218D variant). In all three scenarios, the difference of AML biology in the DDX41-s versus DDX41-m was attributed to the co-occurrence of a secondary disruptive DDX41 variant (i.e p.R525H). These findings are in keeping with the preserved normal hematopoiesis demonstrated in mice with monoallelic DDX41 mutations. In fact, functional analyses showed that hematopoietic defects in these mice were only elicited in the context of biallelic DDX41 dysfunction (23).
As described by others, the DDX41-m MDS/AMLs from this study were predominantly males of older age (1–9). Contrasting with the DDX41-s (MPNs and AMLs), the origin of AML in the DDX41-AMLs could not be associated to mutations in other myeloid defining biomarkers. Instead, we have encountered several DDX41-m AMLs (eg. M8, M11, M15 and M21) with a normal karyotype and no other myeloid genes variants than the two DDX41 variants identified, suggesting that biallelic DDX41 disruption is implicated in the pathophysiology of AML in these individuals. In keeping with a DDX41 driven AML biology, the DDX41-m AMLs shared common features that are not observed in the DDX41-s AMLs or other non DDX41 mutated AMLs (Figure 5). As reported by others, DDX41-AMLs have variable degrees of pancytopenia at presentation and no or rare circulating blasts and variable degrees of uni- and multi-lineage dysplasia, often not meeting the morphologic criteria of MDS (1–9) (Figure 4). In keeping with the morphologic findings, the major myeloid lineages including nucleated red cells, maturing granulocytes and monocytes do not show unequivocal changes in their antigen expression (Figures 5, 6). In contrast, the CD34+ myeloid cells have immunophenotypic characteristics akin to MDS CD34+ myeloid cells. The lack of CD36 and CD64 expression and decreased expression of CD117 and HLA-DR suggest a maturation/differentiation block in DDX41-m AML similar to high-grade MDS.
Having found that DDX41-m shared distinct BM immunophenotypic hallmarks that are not observed in DDX41-s, we sought to explore possible correlations with biallelic DDX41 dysfunction. We thus studied the impact of ten previously uncharacterized missense and in-frame VUSs on DDX41 function. Previously, we demonstrated that the somatic p.R525H hotspot variant is unable to rescue cellular growth and normal cell cycle in DDX41 deficient hematopoietic cells (15). Here using a similar experimental approach, we showed that p.R525H also fail to restore growth and cell cycle in HEK293T cells, within which the endogenous DDX41 expression was repressed using a 3’UTR shRNA (Figure S1). Similar to p.R252H, DDX41 impairment was also seen for another common loss-of-function DDX41 variant (p.Met1)? (2), further reinforcing the validity of the HEK293T cellular paradigm for our functional analyses.
Using the HEK293T model, we have identified four variants (p.G218D, p.T320I, p.S363del and p.R479W) that recovered between 56% and 66% of HEK293T cells, compared to HEK293T cells transfected with wild-type DDX41 (blue bars, Figure 3A). For reference, similar experiments using the common disruptive DDX41 p.R525H allele in hematopoietic cells (15, 22) yielded 61% of HEK293T cells. We have also encountered six additional variants (p.A11T, p.M155I, p.P213R, p.E345K, p.A346P and p.D570G) that recouped between 78% and 93% of HEK293T cells compared to HEK293T cells transfected with wild-type DDX41 (orange bars, Figure 3A). Overall, the same DDX41 mutants also impaired normal cell cycle with a trend towards an accumulation of cells in the G2/M phase (Figures 3B, C). We have previously demonstrated cell cycle arrest in G2/M to be a hallmark of DDX41 dysfunction in hematopoietic cells and to correlate with reduced cellular proliferation. In keeping with these observations, our results suggest that defective cell cycle and ineffective cellular proliferation in HEK293T cells are at least partly due to loss of DDX41 activity caused by the mutant alleles investigated herein (22). Features of DDX41-m persisted, even in patients showing lesser deficits on HEK293T proliferation. For instance, in case M17 harboring the p.E345K and p.A346P variants (HEK2963T cells count: p.E345K = 88% and p.A346P = 78%, respectively), the expression profile of early myeloid progenitors and/or markers of dyserythropoiesis (Figure 5) were comparable to that seen in patient M15 with the p.G218D and p.R525H alleles (HEK2963T cells count: p.G218D = 63% and p.R525H = 61%, respectively).
In this study, we have unraveled the functional role of ten previously uncharacterized DDX41 variants. Together, evidence gathered on these variants are in favor of a deleterious impact on DDX41 activity. Notable examples include p.G218D, p.T320I, p.E345K, p.A346D, p.S363del and p.R479W.
The p.G218D variant was detected in two DDX41-AMLs (one DDX41-s and one DDX41-m) in our study cohort (Table 1, Figure 2). This variant was also previously identified in another DDX41-s AML (5) and two additional patients in the context of familial predisposition to hematologic malignancies (Clinvar). The p.G218D variant is very rarely seen in control individuals from the genome aggregation database (gnomAD) as shown by the minor allele frequency (MAF) of 0.0017% (5/282742 alleles). This variant is located within the catalytic DEAD box of DDX41 and is predicted to affect protein function by in sillico prediction algorithms (MutationTaster, LRT, Polyphen-2, PROVEAN, SIFT). The p.G218D variant was previously reported as VUS (one submitter) and likely pathogenic (one submitter), in the context of familial predisposition to hematologic malignancies (Clinvar). Functional analyses conducted herein show that this variant impair cellular proliferation and cell cycle in HEK293T cells (Figure 3). Furthermore, BM evaluation of patient M15 harboring p.G218D combined to p.R525H showed that this patient shared similar hallmarks to other DDX41-m and suggests biallelic DDX41 dysfunction (Figure 5).
The p.T320I variant was detected in one DDX41-m AML in our study cohort (Table 1, Figure 1). This variant was also previously identified in another patient in the context of familial predisposition to hematologic malignancies (Clinvar), where it has been classified as a VUS (Clinvar). The p.T320I variant was not previously seen in control individuals from gnomAD. This variant is located within the catalytic DEAD box of DDX41 and is predicted to affect protein function by in sillico prediction algorithms (MutationTaster, LRT, Polyphen-2, PROVEAN, SIFT). Functional analyses conducted herein show that this variant impair cellular proliferation and cell cycle in HEK293T cells (Figure 3). BM features of patient M18 harboring p.T320I combined to p.G530D showed similar hallmarks to patient M15 and other DDX41-m, in keeping with biallelic DDX41 dysfunction (Figure 5).
The p.E345K variant was detected in one DDX41-m AML in our study cohort (Table 1, Figure 1). This variant was also identified in another patient in the context of familial predisposition to hematologic malignancies (Clinvar), where it has been classified as a VUS (Clinvar). Furthermore, one DDX41-m AML from this study (Figure 1) as well as and two other patients (one AML and one MDS with excess blast) harboring a different missense variant (p.E345D) affecting the same E345 residue have been identified. In both of these external cases, E345D was detected in combination to a germline frameshift DDX41 variant (3, 4). The p.E345K was not previously seen in control individuals from gnomAD. This variant is located within the catalytic DEAD box of DDX41 and is predicted to affect protein function by in sillico prediction algorithms (MutationTaster, LRT, Polyphen-2, PROVEAN, SIFT). Functional analyses conducted herein show that this variant impair cellular proliferation and cell cycle in HEK293T cells (Figure 3).
The p.A346P variant was detected in one DDX41-m AML in our study cohort (Table 1, Figure 1). Two other patients (one AML and one MDS/AML) harboring a different missense variant (p.A346T) affecting the same A346 residue have been identified. In both cases, A346T was detected in combination to another deleterious DDX41 alleles (germline p.Met1? variant and p.R525H) (3, 5). The p.A346P variant was not previously seen in control individuals from gnomAD. This variant is located within the catalytic DEAD box of DDX41 and is predicted to affect protein function by in sillico prediction algorithms (MutationTaster, LRT, Polyphen-2, PROVEAN, SIFT). Functional analyses conducted herein show that this variant results in impairment of DDX41 activity on cellular proliferation and cell cycle in HEK293T cells (Figure 3). Of note, the BM features of patient M17 harboring p.A346P combined to p.E345K (see above) were suggestive of biallelic DDX41 dysfunction as seen with other DDX41-m investigated herein (Figure 5).
The p.S363del variant was detected in one DDX41-m AML in our study cohort (Table 1, Figure 2) as well as another DDX41-m AML (4). The p.S363del variant is very rarely seen in control individuals from gnomAD as shown by with a minor allele frequency (MAF) of 0.00039% (1/251030 alleles). This variant affects the catalytic DEAD box of DDX41. Functional analyses conducted herein show that this variant impair cellular proliferation and cell cycle in HEK293T cells (Figure 3).
The p.R479W variant was detected in one DDX41-s MPN in our study cohort (Table 1, Figure 2). This variant was also previously identified in another DDX41-s that presented with MDS (4). Three other patients (one AML, one MDS and one MDS/MPN) harboring a different missense variant (p.R479Q) affecting the same R479 residue have been reported (4). The p.R479W is very rarely seen in control individuals from the genome aggregation database (gnomAD) as shown by with a minor allele frequency (MAF) of 0.0016% (4/251358 alleles). This variant is located within the catalytic helicase domain of DDX41 and is predicted to affect protein function by in sillico prediction algorithms (MutationTaster, LRT, Polyphen-2, PROVEAN, SIFT). Functional analyses conducted herein show that this variant impair cellular proliferation and cell cycle in HEK293T cells (Figure 3).
In this study, we have identified loss-of-function (eg. nonsense, frameshift, splicing) DDX41 variants as well as disruptive DDX41 alleles from the functional analyses conducted. The nature of individual variants (eg. frameshift versus missense) did not seem to determine patient outcome. Rather, the combination of two disruptive DDX41 variants was associated with similar clinical pathologic findings and an overall favorable AML outcome compared to other AML entities. Our study showed evidence of biallelic DDX41 disruption in 17 out of the 22 DDX41-m (cases M1-M17) as highlighted by the combination of at least two deleterious variants (Figure 2). These include cases M1-M5 (carrying two loss-of-function DDX41 variants), cases M6-M13 (carrying a loss-of-function variant combined to a disruptive DDX41 allele in HEK293T cells) and cases M14 and M17 (carrying two disruptive DDX41 alleles in HEK293T cells). Of note, from short reads NGS data, we were able to confirm that the deleterious DDX41 variants in cases M5 (p.R525H and p.T529Rfs*12), M10 (p.R525H and p.F535Nfs*6) and M17 (p.E345K and p.A346T) were in trans configuration, further corroborating biallelic DDX41 disruption. Furthermore, BM morphology as well as immunophenotype of the DDX41-m investigated herein were similar and distinct from pathophysiologic findings in DDX41-s, MDS or normal bone marrows (Figures 5, 6). Unique hallmarks seen in these DDX41-m and not the DDX41-s are in keeping with findings that defective hematopoiesis only occur in the context of biallelic DDX41dysfunction (23).
As shown by others, DDX41-m typically involve a germline and a somatic DDX41 variant. However, in this study, we have also identified two individuals harboring two somatic deleterious DDX41 alleles (eg. M10: p.R525H at 15.9% and p.F535Nfs*6 at 12.4%; M20: p.R252H at 7% and p.G587C at 5.9%) indicating that biallelic DDX41 disruption can also occur in the context of two somatic variants. Although rare (4 out of 277), biallelic somatic variants (based on VAF< 10%) were previously reported in individuals having MDS with excess blasts (6). Implications of such biallelic somatic variants are multifold. First, testing methodologies should be adapted to improve the detection of the somatic DDX41 variants that are commonly encountered at low levels (eg. in this study average VAF: 9.5%, minimum VAF: 3.2%). Second, cases with two somatic DDX41 variants share similar clinicopathologic findings to other DDX41-m AMLs with a germline and a somatic DDX41 allele and as such should be managed accordingly. Third, unlike other DDX41-m AMLs, germline testing is not warranted in the context of two somatic DDX41 alleles are detected.
In summary, we have studied the genomic and BM features of 51 patients with DDX41 mutations. Of these, only individuals with two DDX41 variants shared similar clinicopathologic findings as described by others (1–9). Here, we also observed that unlike individuals with monoallelic DDX41 alterations, patients harboring two DDX41 alleles present with common BM morphology and immunophenotypic hallmarks. As highlighted by others, DDX41-m AMLs have a more favorable outcome than seen in other AML entities; thus, accurate identification and interpretation of DDX41-m variants, particularly germline mutations, are of crucial importance to clinical management of affected individuals and genetic counselling of at risk family members. In this study, we have determined the deleterious potential of ten previously uncharacterized DDX41 variants. Here, we have corroborated genomic and clinicopathologic findings on DDX41 mutated MDS/AML (1–9). Our work contributes to expanding knowledge on these DDX41 related malignancies with a particular emphasis on biallelic DDX41 disruption and deleterious DDX41 alleles unraveled herein. Furthermore, pathologic hallmarks of DDX41-m evaluated in this study can serve as a basis for molecular testing in patients with suspicion of biallelic DDX41 dysfunction. More extensive studies including such as single cell RNA sequencing will help shed more light on the biological implication of DDX41 in the pathophysiology of AML at a cellular level.
The data presented in the study is deposited in the ClinVar repository, accession number SUB13174259.
The studies involving human participants were reviewed and approved by UHN research and ethics board. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
J-MC-C, AT and HM designed and supervised the experiments conducted in this study. DM, HS, KY, ASc, DK and MM were involved in the clinical management of patients and contributed to samples collection. J-MC-C, MC and AS conducted molecular analyses on the patients. ASm conducted cytogenetic analyses. AT and EK performed tumor assessment on the patients. AT and MM acquired clinicopathological findings on the patients. SS and HM performed functional validation experiments. J-MC-C, MM, AT and HM wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the Ontario Molecular Pathology Research Network - CPTRG.024.
This study was supported by grants from the Department of Laboratory Medicine and Pathobiology, University of Toronto and the Ontario Molecular Pathology Research Network. We also acknowledge the contribution of patients studied herein as well as geneticists and hematopathologist staffs from the Department of Laboratory Medicine and Pathobiology, University of Toronto.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1153082/full#supplementary-material
1. Polprasert C, Schulze I, Sekeres Mikkael A, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell (2015) 27(5):658–70. doi: 10.1016/j.ccell.2015.03.017
2. Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood (2016) 127(8):1017–23. doi: 10.1182/blood-2015-10-676098
3. Quesada AE, Routbort MJ, DiNardo CD, Bueso-Ramos CE, Kanagal-Shamanna R, Khoury JD, et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am J Hematol (2019) 94(7):757–66. doi: 10.1002/ajh.25486
4. Sébert M, Passet M, Raimbault A, Rahmé R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood (2019) 134(17):1441–4. doi: 10.1182/blood.2019000909
5. Bannon SA, Routbort MJ, Montalban-Bravo G, Mehta RS, Jelloul FZ, Takahashi K, et al. Next-generation sequencing of DDX41 in myeloid neoplasms leads to increased detection of germline alterations. Front Oncol (2021) 10:582213. doi: 10.3389/fonc.2020.582213
6. Qu S, Li B, Qin T, Xu Z, Pan L, Hu N, et al. Molecular and clinical features of myeloid neoplasms with somatic DDX41 mutations. Br J Haematol (2021) 192(6):1006–10. doi: 10.1111/bjh.16668
7. Wan Z, Han B. Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data. Ther Adv Hematol (2021) 12. doi: 10.1177/20406207211032433
8. Li P, White T, Xie W, Cui W, Peker D, Zeng G, et al. AML with germline DDX41 variants is a clinicopathologically distinct entity with an indolent clinical course and favorable outcome. Leukemia (2021). doi: 10.1038/s41375-021-01404-0
9. Alkhateeb HB, Nanaa A, Viswanatha DS, Foran JM, Badar T, Sproat L, et al. Genetic features and clinical outcomes of patients with isolated and comutated DDX41-mutated myeloid neoplasms. Blood Adv (2021). doi: 10.1182/bloodadvances.2021005738
10. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, American society of clinical oncology, and college of American pathologists. J Mol Diagn (2017) 19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002
11. McNamara CJ, Panzarella T, Kennedy JA, Arruda A, Claudio JO, Daher-Reyes G, et al. The mutational landscape of accelerated- and blast-phase myeloproliferative neoplasms impacts patient outcomes. Blood Adv (2018) 2(20):2658–71. doi: 10.1182/bloodadvances.2018021469
12. Spiegel JY, McNamara C, Kennedy JA, Panzarella T, Arruda A, Stockley T, et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv (2017) 1(20):1729–38. doi: 10.1182/bloodadvances.2017009530
13. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med (2017) 376(6):536–47. doi: 10.1056/NEJMoa1611604
14. Kalina T, Flores-Montero J, van der Velden VH, Martin-Ayuso M, Böttcher S, Ritgen M, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia (2012) 26(9):1986–2010. doi: 10.1038/leu.2012.122
15. Kadono M, Kanai A, Nagamachi A, Shinriki S, Kawata J, Iwato K, et al. Biological implications of somatic DDX41 p.R525H mutation in acute myeloid leukemia. Exp Hematol (2016) 44:745–754.e4. doi: 10.1016/j.exphem.2016.04.017
16. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi: 10.1126/science.1106148
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
18. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
19. Thomas L, Stamberg J, Gojo I, Ning Y, Rapoport AP. Double minute chromosomes in monoblastic (M5) and myeloblastic (M2) acute myeloid leukemia: Two case reports and a review of literature. Am J Hematol (2004) 77(1):55–61. doi: 10.1002/ajh.20151
20. Huh YO, Tang G, Talwalkar SS, Khoury JD, Ohanian M, Bueso-Ramos CE, et al. Double minute chromosomes in acute myeloid leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia are associated with micronuclei, MYC or MLL amplification, and complex karyotype. Cancer Genet (2016) 209(7):313–20. doi: 10.1016/j.cancergen.2016.05.072
21. Kim B, Yun W, Lee ST, Choi JR, Yoo KH, Koo HH, et al. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci Rep (2020) 10(1):14297. doi: 10.1038/s41598-020-71386-z
22. Shinriki S, Hirayama M, Nagamachi A, Yokoyama A, Kawamura T, Kanai A, et al. DDX41 coordinates RNA splicing and transcriptional elongation to prevent DNA replication stress in hematopoietic cells. Leukemia (2022) 36(11):2605–20. doi: 10.1038/s41375-022-01708-9
Keywords: acute leukemia, myelodysplastic syndrome, DDX41 mutations, biallelic, clinical pathology
Citation: Tierens A, Kagotho E, Shinriki S, Seto A, Smith AC, Care M, Maze D, Sibai H, Yee KW, Schuh AC, Kim DDH, Gupta V, Minden MD, Matsui H and Capo-Chichi JM (2023) Biallelic disruption of DDX41 activity is associated with distinct genomic and immunophenotypic hallmarks in acute leukemia. Front. Oncol. 13:1153082. doi: 10.3389/fonc.2023.1153082
Received: 28 January 2023; Accepted: 20 March 2023;
Published: 26 June 2023.
Edited by:
Laila Schenkel, London Health Sciences Centre, CanadaReviewed by:
Daniela Damiani, Hematology and Stem Cell Transplant, ItalyCopyright © 2023 Tierens, Kagotho, Shinriki, Seto, Smith, Care, Maze, Sibai, Yee, Schuh, Kim, Gupta, Minden, Matsui and Capo-Chichi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José-Mario Capo-Chichi, am9zZS1tYXJpby5jYXBvLWNoaWNoaUB1aG4uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.