 Emanuela Guerra
Emanuela Guerra Roberta Di Pietro
Roberta Di Pietro Gianmarco Stati
Gianmarco Stati Saverio Alberti
Saverio Alberti- 1Laboratory of Cancer Pathology, Center for Advanced Studies and Technology (CAST), “G. d’Annunzio” University of Chieti-Pescara, Chieti, Italy
- 2Department of Medical, Oral and Biotechnological Sciences, “G. d’Annunzio” University of Chieti-Pescara, Chieti, Italy
- 3Department of Medicine and Aging Sciences, Section of Biomorphology, G. d’Annunzio University of Chieti-Pescara, Chieti, Italy
- 4Sbarro Institute for Cancer Research and Molecular Medicine, Center for Biotechnology, Department of Biology, College of Science and Technology, Temple University, Philadelphia, PA, United States
- 5Unit of Medical Genetics, Department of Biomedical Sciences - Biomedical Sciences (BIOMORF), University of Messina, Messina, Italy
The advent of high throughput DNA sequencing is providing massive amounts of tumor-associated mutation data. Implicit in these analyses is the assumption that, by acquiring a series of hallmark changes, normal cells evolve along a neoplastic path. However, the lack of correlation between cancer risk and global exposure to mutagenic factors provides arguments against this model. This suggested that additional, non-mutagenic factors are at work in cancer development. A candidate determinant is TROP2, that stands out for its expression in the majority of solid tumors in human, for its impact on the prognosis of most solid cancers and for its role as driver of cancer growth and metastatic diffusion, through overexpression as a wild-type form. The Trop-2 signaling network encompasses CREB1, Jun, NF-κB, Rb, STAT1 and STAT3, through induction of cyclin D1 and MAPK/ERK. Notably, Trop-2-driven pathways vastly overlap with those activated by most functionally relevant/most frequently mutated RAS and TP53, and are co-expressed in a large fraction of individual tumor cases, suggesting functional overlap. Mutated Ras was shown to synergize with the TROP2-CYCLIND1 mRNA chimera in transforming primary cells into tumorigenic ones. Genomic loss of TROP2 was found to promote carcinogenesis in squamous cell carcinomas through modulation of Src and mutated Ras pathways. DNA methylation and TP53 status were shown to cause genome instability and TROP gene amplification, together with Trop-2 protein overexpression. These findings suggest that mutagenic and the TROP2 non-mutagenic pathways deeply intertwine in driving transformed cell growth and malignant progression of solid cancers.
1 Introduction
The advent of high throughput DNA sequencing is providing massive amounts of data on tumor-associated mutations (1) and on the landscape of genetic alterations in cancer (2). Oncogene/tumor suppressor gene mutation models have been adopted as a conceptual framework for multi-stage tumor progression. Underlying these analyses there is the assumption that normal cells progressively evolve along the tumor progression path (3). In contrast with this model, no correlation can be found between cancer risk and body size (i.e. number of cell replications) or longevity (i.e. duration of exposure to mutagenic factors), which is known as the Peto’s paradox (4).
This suggested that non-mutagenic mechanisms (5) cooperate with mutagenic oncogenic events (6) to drive tumor progression. Trop-2 is a key candidate for such mechanisms (7). Upregulation of Trop-2 has been associated to poor prognosis of lung (8), breast (9), pancreas (10, 11), stomach (12), head and neck (13), ovary (14) and colon-rectum cancers (11), suggesting a pivotal role of this molecule in tumor progression (15) and metastatic diffusion (16). Notably, the Trop-2 non-mutagenic signature of cancer-driving signaling networks (17) appears to vastly overlap with signatures of mutated oncogenes (18). Experimental evidence for this has been obtained in mTROP2 knockout mouse models (19) and in human tumors, whereby interaction between Cyclin D1, TROP2 and mutated RAS leads to the transformation of primary, naïve cells into tumorigenic ones (20). DNA methylation and TP53 status were additionally shown to cause genome instability, TROP gene amplification and Trop protein overexpression (21). Thus, mutagenic and TROP2 non-mutagenic pathways may deeply intertwine in driving cancer cell growth, and play a convergent role in tumor progression in solid cancers.
2 Impact of Trop-2 in cancer and genetic diseases
2.1 Trop-2 in cancer
TROP2 is a candidate non-mutated cancer driver, that stands out for its expression in the majority of solid tumors in human (7, 22, 23). Trop-2 (AC: P09758) is a type-I transmembrane protein, encoded by the tumor-associated calcium signal transducer 2 (TROP2/TACSTD2/M1S1/GA733-1) gene (7, 24, 25), a retrotransposon of the TROP1/TACSTD1/EPCAM gene (24, 26). The extracellular domain of Trop-2 (residues 27–274 in human) encompasses a cysteine-rich N-terminal region, which hosts a GA733 type 1 motif (residues 27-69) (27), and a thyroglobulin type-1 domain (residues 70-148), followed by a C-terminal domain devoid of cysteines (residues 149-274) (7). The 26-amino acid intracellular domain contains a HIKE motif (28), and two PKC phosphorylation sites, at Ser303 and Ser322 (29).
Trop-2 induces tumor (23) and cancer stem cell growth (30). Our findings showed that Trop-2 expression is upregulated in tumors, regardless of baseline expression in normal tissues (23). Representative portraits of Trop-2 protein in 30 neoplasia types are shown in Figure 1. TROP2 mRNA expression levels in normal tissues are shown in Figure 2. These findings first suggested that Trop-2 expression in cancer cells provided a selective advantage. Consistent with this, upregulation of wtTrop-2 was shown to stimulate tumor growth in proportion to expression levels in vivo (23). This correspondingly raised interest in Trop-2 as a target for immunotherapy in solid cancer (32, 33), and in the development of next-generation anti-Trop-2 antibodies (34).
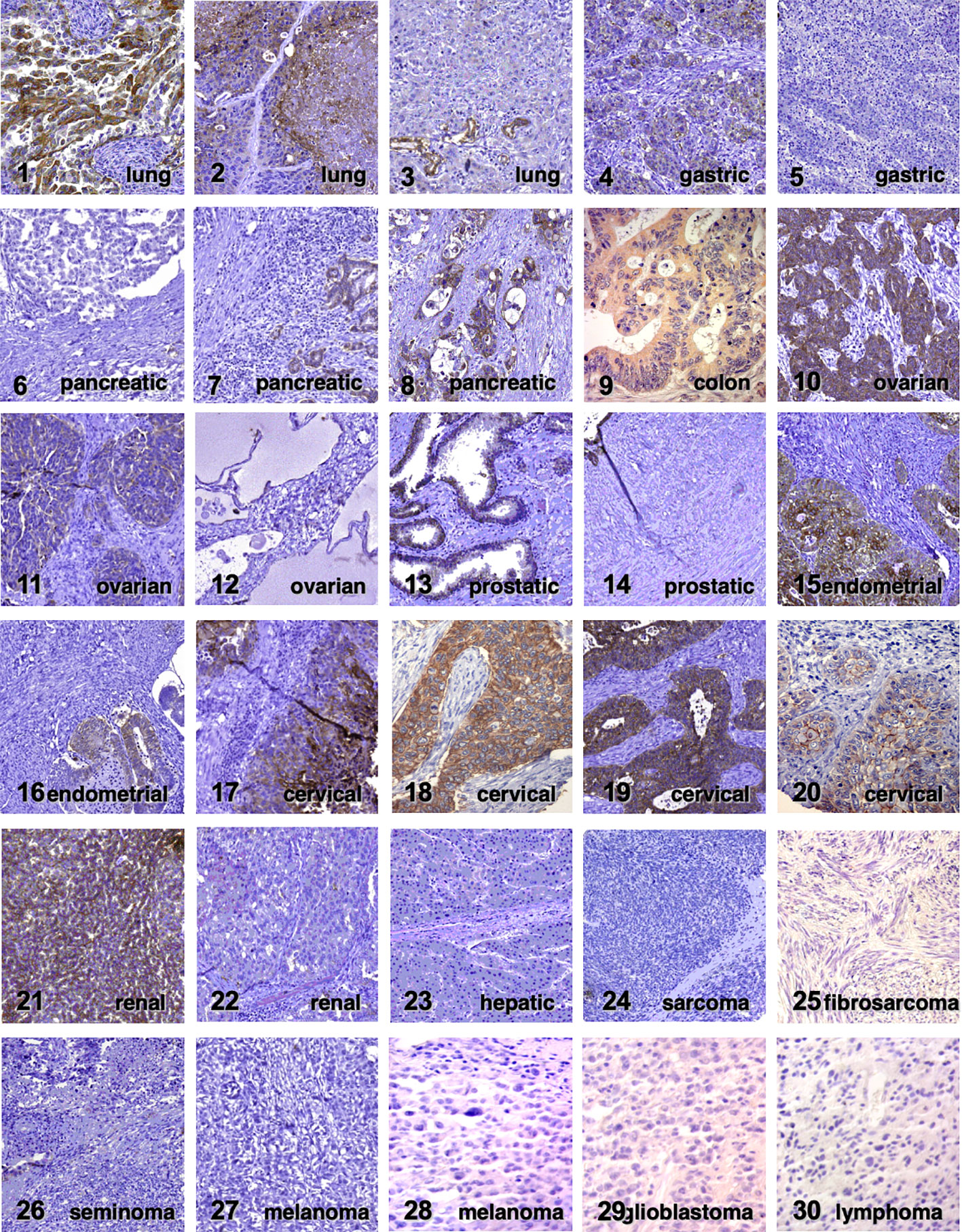
Figure 1 Trop-2 expression in cancer. Immunohistochemical analysis of Trop-2 protein expression in representative tumor cases (23). Tumor histotypes are indicated. Four expression subgroups (absent, weak, moderate and intense) were defined according to Spizzo et al. (31). Negative tumors are cases 5, 6, 12, 14, 22-30. Weak expression was detected in cases 7, 11, 20. Moderate expression was found in cases 3, 4, 8, 13, 16, 21. Intense expression was observed in cases 1, 2, 9, 10, 15, 17-19.
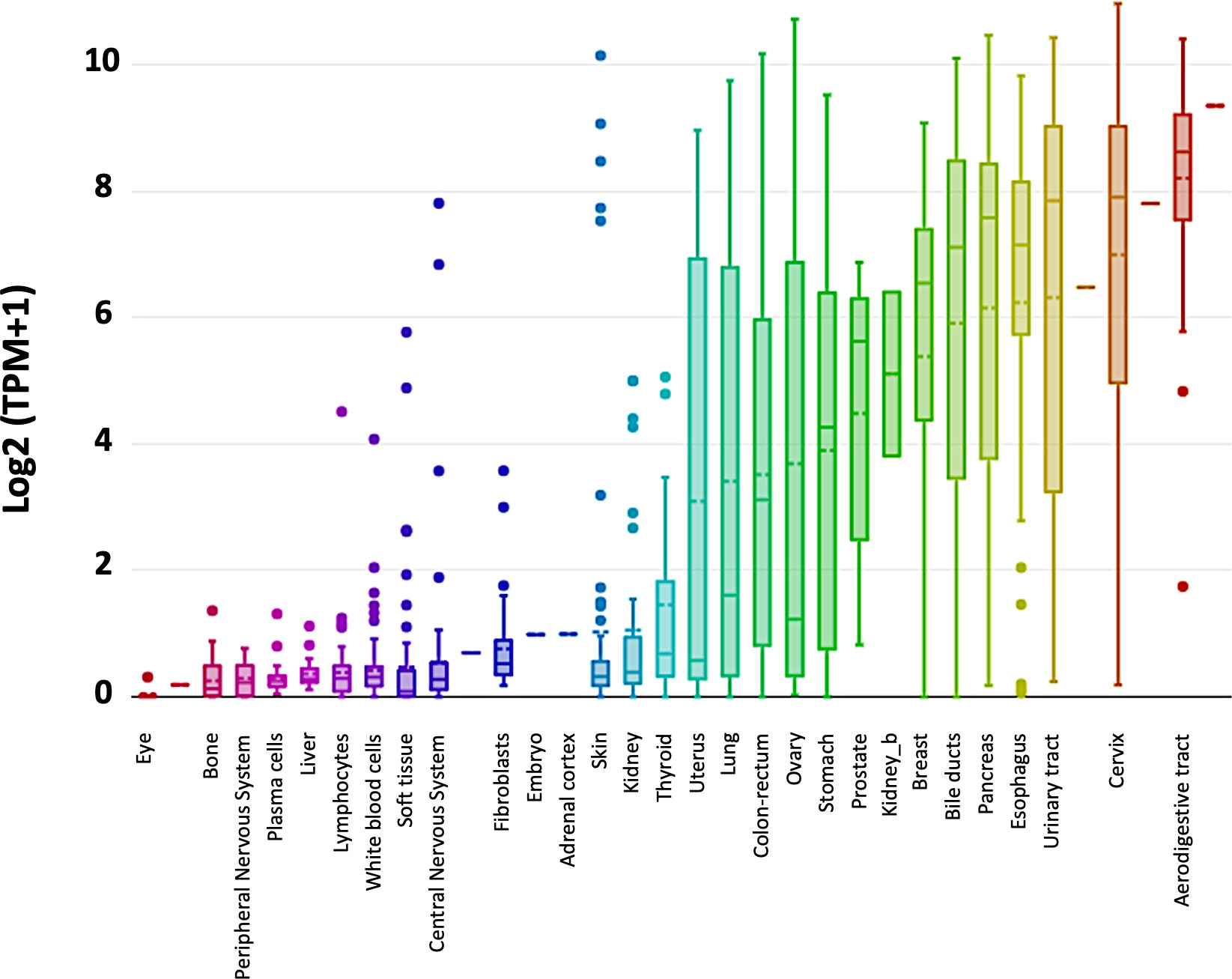
Figure 2 TROP2 gene expression in normal tissues. TROP2 mRNA expression levels were obtained from the DepMap portal (https://depmap.org/portal/) (Expression public 20Q2) and expressed as Log2 (TPM+1). Whisker plots show value distributions of TROP2 mRNA levels across different normal tissues. Boxes corresponding to individual tissues are color coded according to their median intensity (horizontal bars within each box). Dots indicate outlier measurements.
Using antibody targeting and N-terminal Edman degradation, we showed that Trop-2 undergoes cleavage in the first loop of the thyroglobulin domain in the extracellular region, between residues R87 and T88. ADAM10 was shown to be effector protease at this site (16). Molecular modeling indicated that this cleavage induces a profound rearrangement of the Trop-2 structure, which suggested impact on its biological function. No Trop-2 cleavage was detected in normal human tissues, whereas most tumors, including skin, ovary, colon and breast cancers, showed Trop-2 proteolysis. Proteolysis of Trop-2 at R87-T88 was shown to trigger cancer cell growth and metastatic spreading (11).
2.2 Trop-2 alterations cause a rare genetic disease
Given the role of Trop-2 in cancer, a role in cancer inheritance was explored. Genomic loss of the murine TROP2 (mTrop2) gene was shown to promote carcinogenesis in squamous cell carcinomas through modulation of Arf, Src and mutated Ras pathways in mTrop2 mouse knockouts (19).
Genomic alterations of the human TROP2 were shown to cause a rare genetic disease, whereby TACSTD2 mutations induce an amyloid corneal dystrophy (Gelatinous Drop-like Corneal Dystrophy, GDLD) (35), while other organs that host Trop-2-expressing epithelia remain devoid of amyloid deposits. A common GDLD mutation is the substitution of the codon for glutamine at position 118 with a stop codon, in Japan (36) and worldwide (37). However, a broad spectrum of mutations has been subsequently detected (38), and novel, more rare mutations are still being added to this list (39). No clear association of GDLD mutations to altered cancer incidence has been reported (23).
2.3 Epigenetic TROP2 signatures in cancer
The expression of the TROP2 gene was found to be under epigenetic control in choriocarcinomas, i.e. trophoblast-derived malignancies (40, 41). This was shown by associating the expression of TROP1, TROP2 and control genes to their native genomic configuration and DNA methylation status in choriocarcinoma cells. In other words, intact gene coding capacity was shown by transfection and expression of the encoded protein, as modulated by their respective DNA methylation status (40). Epigenetic control was shown for HLA class I genes, for the T-cell differentiation antigens CD5 and CD8, for TROP1 and TROP2. Treating choriocarcinoma cells with the lowest expression ability with 5-azacytidine led to DNA demethylation, and to differential reexpression of cell surface antigen genes. Further cultivation of choriocarcinoma cells in the absence of 5-azacytidine resulted in renewed methylation of their DNA and reversion to baseline low expression capacity (40).
Cases with epigenetic reduction of the expression of the TROP2 gene were identified in prostate cancer (42). TACSTD2 was unmethylated in prostatic intraepithelial neoplasia and hypermethylated/down-regulated in 17% of prostate cancers (42). Low TROP2 expression was observed in lung adenocarcinomas, as compared with normal lung tissues. Bisulphite DNA sequencing and methylation-specific polymerase chain reaction showed that loss of expression was due to hypermethylation of the TROP2 promoter region. Consistent with this, DNA demethylation with 5-Aza-deoxycytidine led to activation of TROP2 expression (43). TROP2 was found highly expressed in normal bile duct epithelia, but down-regulated in cholangiocarcinoma cells. Sixty percent of cholangiocarcinomas revealed TROP2 promoter hypermethylation and TROP2 knockdown significantly enhanced the proliferation and migration of cholangiocarcinoma cell lines (44).
2.4 Post-transcriptional and post-translational modulation of TROP2 expression in cancer
The expression of TROP2 in cancer was shown to be modulated by both post-transcriptional and post-translational mechanisms. A bi-cistronic CYCLIN D1-TROP2 mRNA chimera was isolated from human ovarian and mammary cancer cells (20). The CYCLIN D1-TROP2 mRNA was shown to transform naïve, primary cells and to induce aggressive tumor growth. Silencing of the chimeric mRNA inhibited breast cancer growth. The CYCLIN D1-TROP2 mRNA was found expressed by a large fraction of human gastro-intestinal, ovarian and endometrial tumors. The chimeric mRNA was shown to be of post-transcriptional origin, and independently translated the Cyclin D1 and Trop-2 proteins. Truncation of the 3’ UTR of the CYCLIN D1 mRNA led to higher mRNA stability, for inappropriate expression during the cell cycle. There was a quantitative correlation between the chimeric mRNA and the transcriptional levels of CYCLIN D1 and TROP2. Hence, an oncogenic determinant appears linked to levels of expression of the parental moieties, in the absence of both epigenetic changes and of mutagenic alterations. This mechanism of cell transformation appears widespread in human cancers (20).
A distinct post-transcriptional TROP2 modulation mechanism was shown to involve the Trop-2/miR-125b axis in the progression from normal urothelium to non-invasive and invasive urothelial cancer (45). miR-125b inhibits Trop-2 expression. Experimental findings showed that a progressive increase in Trop-2 protein levels along urothelial cancer progression is induced by miR-125b downregulation, and this correlates with the severity of the disease (45).
Post-transcriptional modulation of Trop-2 expression was also found to be dependent on environmental factors. In SW480 colon cancer cells (–),-epigallocatechin-3-gallate (EGCG) affected the post-transcriptional processing of the TROP2 mRNA, which was quickly and specifically degraded in the presence of EGCG. Furthermore, EGCG was found to suppress Trop-2 expression at a post-translational level in HCT-116 cells, by affecting the stability of the Trop-2 protein (46).
2.5 The intertwining of mutagenic and Trop-2 non-mutagenic signatures in cancer
DNA methylation was shown to mediate TROP gene copy number variation, with corresponding additional alteration of protein expression levels (40, 41), suggesting a corresponding stronger drive for cancer cell proliferation (23). This was shown by selection for high protein expressors at a clonal level, which was found to lead to progressively higher expression of CD5, CD8α, TROP2 through gene amplification (41). A lack of TROP1 gene amplification was shown to depend on a unique pattern of gene methylation. This regulatory mode was lost upon treatment with the DNA demethylating agent 5-azacytidine (41) and demethylated TROP1 genes were found to amplify efficiently and progressively. Thus, DNA methylation not only regulates the expression of the TROP genes (40), but also is a determinant of TROP gene amplification in tumor cells (41). Mutations of TP53 were subsequently shown to induce loss of DNA methylation in the TROP1 gene, which then led to gene amplification (21). This was reverted by transduction of a wtTP53 gene or by inducing methylation of the genomic DNA with the Sss I DNA methylase (21), demonstrating a mechanistic link between mutations of the TP53 tumor suppressor gene, genomic instability/gene copy number variation (47) and overexpression of the TROP genes.
The expression of the TROP2 gene was shown to depend on a large network of transcription factors, that includes p63/p53L, ERG, GRHL1/Get-1, HNF1A/TCF-1, HNF4A, SPI1/PU.1, WT1, GLIS2, AIRE, FOXM1 and FOXP3 (17). TROP2 upregulation was found to then drive the expression and activation of CREB1, Jun, NF-κB, Rb, STAT1 and STAT3 through induction of the cyclin D1 and MAPK/ERK kinase pathways. High-throughput proteomic analysis led us to identify AKT as a central hub of the Trop-2 activation network in human cancer cells. AKT inhibitors only blocked the growth of Trop-2–expressing tumors, but were ineffective on Trop-2–null cells, indicating Trop-2 as a pivotal AKT activator for tumor growth (48). AKT was also shown to be central to Trop-2 signaling in lung adenocarcinomas, as inhibition of TROP2 expression, via DNA methylation, enacted IGF-1R signaling and AKT/β-catenin (43). These signaling pathways were shown to be triggered by a Trop-2, Na+/K+ ATPase, CD9, PKCα, cofilin membrane signaling super-complex. This super-complex was found to be ubiquitous, but essentially dormant in normal cells, and was shown to be activated by Trop-2 to trigger colorectal cancer growth and invasion (29).
These findings suggested functional interaction and potential synergy of mutagenic and non-mutagenic cancer drivers. Trop-2-triggered PKCα, tetraspanins, AKT, Jun, NF-κB, Rb, STAT1, STAT3, cyclin D1, MAPK/ERK kinases fall within main hallmarks of tumor progression (6, 18, 49), and comprise signaling networks triggered by mutated oncogenes (1). As indicated above, DNA methylation and TP53 status are instrumental to TROP gene copy number variation and to consequent aberrant overexpression of wtTrops (21, 41).
Consistent with this, genomic loss of mTrop2 rendered Arf-null mice susceptible to the formation of biphasic sarcomatoid carcinomas upon carcinogen exposure in skin cancer-resistant mouse strains (C57BL/6). Ras-transformed keratinocytes derived from mTrop2(-/-)Arf(-/-) mice exhibit enhanced proliferative and migratory capacity as well as increased activation of MAPK and Src (19). Ras-transformed mTrop2(-/-) keratinocytes were shown to undergo epithelial to mesenchymal transition (EMT) and formed tumors with spindle cell histology. Consistent with this, TROP2 mRNA levels were found to be down-regulated in head and neck squamous cell carcinomas undergoing EMT (19). Mutated Ras was shown to synergize with the TROP2-CYCLIND1 mRNA chimera in transforming primary cells in vitro and in inducing tumor growth in vivo (20). The vast overexpression of Trop-2 in main cancer types, e.g. breast (76%), stomach (70%), prostate (80%), pancreas (83%), cervix (91%) cancers (23), indicates that these mutagenic and non-mutagenic signatures are candidate to frequently interact in cancer cells (1).
2.6 Mutagenic and Trop-2 non-mutagenic signatures – the pancreatic cancer case
A prognostic impact of Trop-2 and of activation-inducing ADAM10 was found in major clinically-relevant cancer types (8, 11, 16). Among them, Trop-2 was shown to impact on the progression and metastatic relapse of pancreatic cancer (PC) (Figure 3) (10, 11). PC is a very aggressive disease with a poor prognosis (5-year survival: ∼ 6%). Recent evidence on mutagenic and non-mutagenic signatures in PC has started shedding light on its pathogenic determinants. KRAS mutations have been detected in about 90% of pre-neoplastic non-invasive lesions (low-grade pancreatic intraepithelial neoplasia) (51), though more rarely at more advanced stages of PC, suggesting a ‘common ground’ for early PC development. Data on TROP2 prevalence added to this scenario, as overexpression of Trop-2 was found in the majority of PC patients (10). TROP2 mRNA and protein levels were subsequently shown to be sharp prognostic biomarkers for PC (16) (Figure 3). These findings suggested a driving role of Trop-2 in PC (16) and a convergent impact of TROP2 and KRAS signaling pathways (17) at early stages of cancer development (23). Along PC grade progression, additional mutations were shown to occur in cell growth/death regulatory genes including CDKN2A and TP53. Invasive PC carry mutations in SMAD4 in about 55% of the cases (51), suggesting again a broad, common ground for PC development.
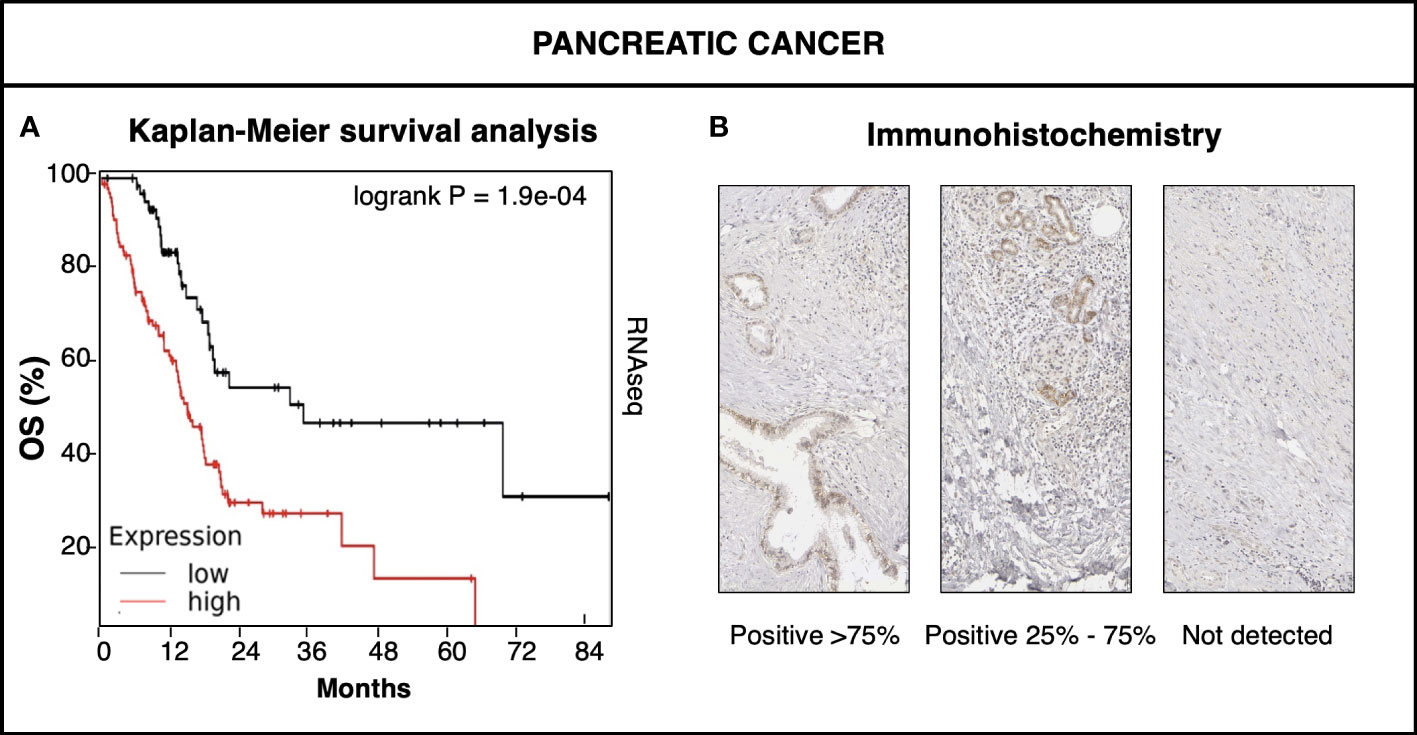
Figure 3 Trop-2 impact on pancreatic cancer. (A) KMPlot database (www.kmplot.com) data (50) of expression of TROP2 mRNA in pancreatic cancer versus patient overall survival (OS). RNAseq high (red) versus low (black) TROP2 mRNA levels were compared by Kaplan-Meier survival analysis. Logrank P value of survival curve comparison is shown. (B) Immunohistochemical analysis of Trop-2 protein expression in representative cases of pancreatic cancer. Percent expression of Trop-2 by cancer cells is indicated. Expression classes were categorized as high (>75% of cells) medium (25-75% of cells) and low/nil (<25% of cells) (23).
Aberrant DNA methylation of gene promoter CpG islands was detected in CACNA1G, CDH1, CDKN2A, DAPK1, MGMT, MINT1-2-31-32, MLH1, RARB, THBS1 and TIMP3 (52, 53). Hypomethylation of the CLDN4, LCN2, MSLN, PSCA, S100A4, TFF2 and YWHAS genes, with corresponding high levels of transcription in PC, was found in parallel (54–56). Consistent with findings that methylation of BRCA1 is associated to metastatic relapse (57), promoter methylation of BRCA1 was found in 46% of PC (58), suggesting a driving role in PC occurrence and progression (59, 60). Parallel impact was shown for BRCA-1 and BRCA-2 mutations (59, 60), suggesting a convergent mechanism of inactivation of shared target genes in PC. DNA methylation interacts with modified histones and plays a role in chromatin remodeling and heterochromatin formation (61). Unsurprisingly, mutations of the SWI/SNF chromatin remodeling genes were found in 15% of PC cases (51), suggesting impact on downstream epigenetic changes.
These findings suggested a mechanistic overlap between the Trop-2 signature and mutagenic/epigenetic PC driving changes, and supports a model of PC progression, through accumulation of genetic and epigenetic changes during tumor initiation, promotion and progression (62). Corresponding impact of genetic and epigenetic changes was demonstrated for PC response to therapy (63, 64), extending the value of Trop-2 as a therapy target in PC (33), through the targeting of the Trop-2 activated form by next-generation monoclonal antibodies (34).
3 Conclusions
Modelling of cancer genetic signatures, according to a multi-stage, somatic mutation-based carcinogenesis process, has generated key insight on cancer development, and has led to major advances of anticancer therapy (6, 65). However, mutation-only tumor progression models may risk missing major regulatory paths and networks, that are driven by epigenetic components (6), together with non-mutagenic (23), cancer-prone, acquired phenotypes (16, 66).
Upregulation of Trop-2 was shown to quantitatively stimulate human cancer growth (23) and metastasis (16) in the absence of detectable TROP2 gene mutations. Acquisition of stable, potentially heritable modes of gene expression, as driven by gene expression regulatory loops/miRNA (45, 46) and by post-transcriptional/post-translational events, like oncogenic mRNA chimeras (20, 67, 68) were shown to contribute to key steps of oncogenic transformation. Taken together, these findings suggest that mutagenic and the TROP2 non-mutagenic pathways deeply intertwine in driving cancer cell growth, for a pivotal role in tumor progression in solid cancers. Next-generation identification of oncogenic mutagenic/non-mutagenic signatures in cancer development may provide additional insight and generate novel models of cancer development.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
SA designed the strategy of the article. EG, RDP, GS, SA contributed to data collection, writing of the article and revision of the final text. All authors contributed to the article and approved the submitted version.
Funding
We thank the Italian Ministry of Development (FESR 2016-2018. SSI000651, art. 69 Reg. CE n. 1083/2006 and Reg. CE n. 1828/2006), the Italian Ministry of University and Research - Smart Cities and Communities and Social Innovation - “Health @ home” SCN_00558, the Marie Curie Transfer of Knowledge Fellowship “Advanced structure prediction methods” – EC VI Framework Programme (Contract 014541), the Horizon 2020-SMEINST-2015-1, PIC 944224288, Region Abruzzo (POR FESR 2007-2013: Activity 1.1.1 line B, C78C14000100005) for support. The sponsors had no role in the design and conduct of this study, nor in the collection, analysis and interpretation of the data, nor in the preparation, review or approval of the manuscript.
Acknowledgments
We thank the students and colleagues who supported the authors during the course of this work, through discussion, advice and technical assistance. The support of Oncoxx Biotech srl, of Mediterranea Theranostic srl and of FIRA – Finanziaria Regionale Abruzzese is gratefully acknowledged.
Conflict of interest
EG is an inventor in patents WO201687651 and WO201784763. SA is an inventor in patents WO201089782, WO201687651 and WO201784763 and founder of Oncoxx Biotech Srl and Mediterranea Theranostic Srl.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CGI, CpG islands; CP, Chronic pancreatitis; GDLD, Gelatinous Drop-like Corneal Dystrophy; PC, Pancreatic cancer.
References
1. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature (2020) 578:94–101. doi: 10.1038/s41586-020-1943-3
2. Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature (2020) 578:82–93. doi: 10.1038/s41586-020-1969-6
3. Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer (1953) 7:68–72. doi: 10.1038/bjc.1953.8
4. Peto R. Epidemiology, multistage models, and short-term mutagenicity tests. Int J Epidemiol (2016) 45:621–37. doi: 10.1093/ije/dyv199
5. Huang S. Tumor progression: chance and necessity in Darwinian and lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol (2012) 110:69–86. doi: 10.1016/j.pbiomolbio.2012.05.001
6. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
7. Fornaro M, Dell'Arciprete R, Stella M, Bucci C, Nutini M, Capri MG, et al. Cloning of the gene encoding TROP-2, a cell-surface glycoprotein expressed by human carcinomas. Int J Cancer (1995) 62:610–8. doi: 10.1002/ijc.2910620520
8. Relli V, Trerotola M, Guerra E, Alberti S. Distinct lung cancer subtypes associate to distinct drivers of tumor progression. Oncotarget (2018) 9:35528–40. doi: 10.18632/oncotarget.26217
9. Ambrogi F, Fornili M, Boracchi P, Trerotola M, Relli V, Simeone P, et al. Trop-2 is a determinant of breast cancer survival. PloS One (2014) 9:e96993. doi: 10.1371/journal.pone.0096993
10. Fong D, Moser P, Krammel C, Gostner JM, Margreiter R, Mitterer M, et al. High expression of TROP2 correlates with poor prognosis in pancreatic cancer. Br J Cancer (2008) 99:1290–5. doi: 10.1038/sj.bjc.6604677
11. Guerra E, Trerotola M, Relli V, Lattanzio R, Tripaldi R, Vacca G, et al. Trop-2 induces ADAM10-mediated cleavage of e-cadherin and drives EMT-less metastasis in colon cancer. Neoplasia (2021) 23:898–911. doi: 10.1016/j.neo.2021.07.002
12. Muhlmann G, Spizzo G, Gostner J, Zitt M, Maier H, Moser P, et al. TROP2 expression as prognostic marker for gastric carcinoma. J Clin Pathol (2009) 62:152–8. doi: 10.1136/jcp.2008.060590
13. Fong D, Spizzo G, Gostner JM, Gastl G, Moser P, Krammel C, et al. TROP2: a novel prognostic marker in squamous cell carcinoma of the oral cavity. Mod Pathol (2008) 21:186–91. doi: 10.1038/modpathol.3801001
14. Bignotti E, Todeschini P, Calza S, Falchetti M, Ravanini M, Tassi RA, et al. Trop-2 overexpression as an independent marker for poor overall survival in ovarian carcinoma patients. Eur J Cancer (2010) 46:944–53. doi: 10.1016/j.ejca.2009.12.019
15. Hsu E-C, Rice MA, Bermudez A, Marques FJG, Aslan M, Liu S, et al. Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proc Natl Acad Sci (2020) 117:2032. doi: 10.1073/pnas.1905384117
16. Trerotola M, Guerra E, Ali Z, Aloisi AL, Ceci M, Simeone P, et al. Trop-2 cleavage by ADAM10 is an activator switch for cancer growth and metastasis. Neoplasia (2021) 23:415–28. doi: 10.1016/j.neo.2021.03.006
17. Guerra E, Trerotola M, Aloisi AL, Tripaldi R, Vacca G, La Sorda R, et al. The trop-2 signalling network in cancer growth. Oncogene (2013) 32:1594–600. doi: 10.1038/onc.2012.151
18. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
19. Wang J, Zhang K, Grabowska D, Li A, Dong Y, Day R, et al. Loss of Trop2 promotes carcinogenesis and features of epithelial to mesenchymal transition in squamous cell carcinoma. Mol Cancer Res (2011) 9:1686–95. doi: 10.1158/1541-7786.MCR-11-0241
20. Guerra E, Trerotola M, Dell’ Arciprete R, Bonasera V, Palombo B, El-Sewedy T, et al. A bi-cistronic CYCLIN D1-TROP2 mRNA chimera demonstrates a novel oncogenic mechanism in human cancer. Cancer Res (2008) 68:8113–21. doi: 10.1158/0008-5472.CAN-07-6135
21. Nasr AF, Nutini M, Palombo B, Guerra E, Alberti S. Mutations ofTP53 induce loss of DNA methylation and amplification of the TROP1 gene. Oncogene (2003) 22:1668–77. doi: 10.1038/sj.onc.1206248
22. Alberti S, Miotti S, Stella M, Klein CE, Fornaro M, Ménard S, et al. Biochemical characterization of trop-2, a cell surface molecule expressed by human carcinomas: formal proof that the monoclonal antibodies T16 and MOv-16 recognize trop-2. Hybridoma (1992) 11:539–5. doi: 10.1089/hyb.1992.11.539
23. Trerotola M, Cantanelli P, Guerra E, Tripaldi R, Aloisi AL, Bonasera V, et al. Up-regulation of trop-2 quantitatively stimulates human cancer growth. Oncogene (2013) 32:222–33. doi: 10.1038/onc.2012.36
24. Linnenbach AJ, Seng BA, Wu S, Robbins S, Scollon M, Pyrc JJ, et al. Retroposition in a family of carcinoma-associated antigen genes. Mol Cell Biol (1993) 13:1507–15. doi: 10.1128/mcb.13.3.1507-1515.1993
25. Calabrese G, Crescenzi C, Morizio E, Palka G, Guerra E, Alberti S. Assignment of TACSTD1 (alias TROP1, M4S1) to human chromosome 2p21 and refinement of mapping of TACSTD2 (alias TROP2, M1S1) to human chromosome 1p32 by in situ hybridization. Cytogenet Cell Genet (2001) 92:164–5. doi: 10.1159/000056891
26. Zanna P, Trerotola M, Vacca G, Bonasera V, Palombo B, Guerra E, et al. Trop-1 are conserved growth stimulatory molecules that mark early stages of tumor progression. Cancer (2007) 110:452–64. doi: 10.1002/cncr.22785
27. Chong JM, Speicher DW. Determination of disulfide bond assignments and n-glycosylation sites of the human gastrointestinal carcinoma antigen GA733-2 (CO17-1A, EGP, KS1-4, KSA, and ep-CAM). J Biol Chem (2001) 276:5804–13. doi: 10.1074/jbc.M008839200
28. Ciccarelli F, Acciarito A, Alberti S. Large And diverse numbers of human diseases with HIKE mutations. Hum Mol Genet (2000) 9:1001–7. doi: 10.1093/hmg/9.6.1001
29. Guerra E, Relli V, Ceci M, Tripaldi R, Simeone P, Aloisi AL, et al. Trop-2, Na+/K+ ATPase, CD9, PKCα, cofilin assemble a membrane signaling super-complex that drives colorectal cancer growth and invasion. Oncogene (2022) 41:1795–808. doi: 10.1038/s41388-022-02220-1
30. Stoyanova T, Goldstein AS, Cai H, Drake JM, Huang J, Witte ON. Regulated proteolysis of Trop2 drives epithelial hyperplasia and stem cell self-renewal via beta-catenin signaling. Genes Dev (2012) 26:2271–85. doi: 10.1101/gad.196451.112
31. Spizzo G, Fong D, Wurm M, Ensinger C, Obrist P, Hofer C, et al. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. J Clin Pathol (2011) 64:415–20. doi: 10.1136/jcp.2011.090274
32. Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. New Engl J Med (2021) 384:1529–41. doi: 10.1056/NEJMoa2028485
33. Guerra E, Alberti S. The anti-Trop-2 antibody-drug conjugate sacituzumab govitecan–effectiveness, pitfalls and promises. Ann Trans Med (2022) 10:501–5. doi: 10.21037/atm-22-621
34. Guerra E, Trerotola M, Relli V, Lattanzio R, Tripaldi R, Ceci M, et al. 3D-informed targeting of the trop-2 signal-activation site drives selective cancer vulnerability. Mol Cancer Ther (2023) 22:790–804. doi: 10.1158/1535-7163.MCT-22-0352
35. Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet (1999) 21:420–3. doi: 10.1038/7759
36. Fujiki K, Nakayasu K, Kanai A. Corneal dystrophies in Japan. J Hum Genet (2001) 46:431–5. doi: 10.1007/s100380170041
37. Ren Z, Lin PY, Klintworth GK, Iwata F, Munier FL, Schorderet DF, et al. Allelic and locus heterogeneity in autosomal recessive gelatinous drop-like corneal dystrophy. Hum Genet (2002) 110:568–77. doi: 10.1007/s00439-002-0729-z
38. Alavi A, Elahi E, Tehrani MH, Amoli FA, Javadi MA, Rafati N, et al. Four mutations (three novel, one founder) in TACSTD2 among Iranian GDLD patients. Invest Ophthalmol Vis Sci (2007) 48:4490–7. doi: 10.1167/iovs.07-0264
39. Cabral-Macias J, Zenteno JC, Ramirez-Miranda A, Navas A, Bermudez-Magner JA, Boullosa-Grana VM, et al. Familial gelatinous drop-like corneal dystrophy caused by a novel nonsense TACSTD2 mutation. Cornea (2016) 35(7):987–90. doi: 10.1097/ICO.0000000000000863
40. Alberti S, Herzenberg LA. DNA Methylation prevents transfection of genes for specific surface antigens. Proc Natl Acad Sci USA (1988) 85:8391–4. doi: 10.1073/pnas.85.22.8391
41. Alberti S, Nutini M, Herzenberg LA. DNA Methylation prevents the amplification of TROP1, a tumor associated cell surface antigen gene. Proc Natl Acad Sci USA (1994) 91:5833–7. doi: 10.1073/pnas.91.13.5833
42. Ibragimova I, Ibanez de Caceres I, Hoffman AM, Potapova A, Dulaimi E, Al-Saleem T, et al. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila) (2011) 3:1084–92. doi: 10.1158/1940-6207.CAPR-10-0039
43. Lin J-C, Wu Y-Y, Wu J-Y, Lin T-C, Wu C-T, Chang Y-L, et al. TROP2 is epigenetically inactivated and modulates IGF-1R signalling in lung adenocarcinoma. EMBO Mol Med (2012) 4:472–85. doi: 10.1002/emmm.201200222
44. Sawanyawisuth K, Tantapotinan N, Kraiklang R, Puapairoj A, Wongkham C, Riggins GJ, et al. Suppression of trophoblast cell surface antigen 2 enhances proliferation and migration in liver fluke-associated cholangiocarcinoma. Ann Hepatol (2016) 15:71–81. doi: 10.5604/16652681.1184223
45. Avellini C, Licini C, Lazzarini R, Gesuita R, Guerra E, Tossetta G, et al. The trophoblast cell surface antigen 2 and miR-125b axis in urothelial bladder cancer. Oncotarget (2017) 8:58642–53. doi: 10.18632/oncotarget.17407
46. Sukhthankar M, Alberti S, Baek SJ. (-)-Epigallocatechin-3-Gallate (EGCG) post-transcriptionally and post-translationally suppresses the cell proliferative protein TROP2 in human colorectal cancer cells. Anticancer Res (2010) 30:2497–503.
47. Funnell T, O’Flanagan CH, Williams MJ, McPherson A, McKinney S, Kabeer F, et al. Single-cell genomic variation induced by mutational processes in cancer. Nature (2022) 6121(7938):106–115. doi: 10.1038/s41586-022-05249-0
48. Guerra E, Trerotola M, Tripaldi R, Aloisi AL, Simeone P, Sacchetti A, et al. Trop-2 induces tumor growth through akt and determines sensitivity to akt inhibitors. Clin Cancer Res (2016) 22:4197–205. doi: 10.1158/1078-0432.CCR-15-1701
49. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
50. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PloS One (2013) 8:e82241. doi: 10.1371/journal.pone.0082241
51. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med (2014) 371:1039–49. doi: 10.1056/NEJMra1404198
52. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer (2011) 11:726–34. doi: 10.1038/nrc3130
53. Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res (2000) 60:1835–9.
54. Zhao Y, Sun J, Zhang H, Guo S, Gu J, Wang W, et al. High-frequency aberrantly methylated targets in pancreatic adenocarcinoma identified via global DNA methylation analysis using methylCap-seq. Clin Epigenet (2014) 6:18. doi: 10.1186/1868-7083-6-18
55. Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res (2003) 63:4158–66.
56. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (2008) 321:1801–6. doi: 10.1126/science.1164368
57. Li Y, Melnikov AA, Levenson V, Guerra E, Simeone P, Alberti S, et al. A seven-gene CpG-island methylation panel predicts breast cancer progression. BMC Cancer (2015) 15:417. doi: 10.1186/s12885-015-1412-9
58. Guo M, Jia Y, Yu Z, House MG, Esteller M, Brock MV, et al. Epigenetic changes associated with neoplasms of the exocrine and endocrine pancreas. Discov Med (2014) 17:67–73.
59. Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst (2018) 110:1067–74. doi: 10.1093/jnci/djy024
60. Abe T, Blackford AL, Tamura K, Ford M, McCormick P, Chuidian M, et al. Deleterious germline mutations are a risk factor for neoplastic progression among high-risk individuals undergoing pancreatic surveillance. J Clin Oncol (2019) 37:1070–80. doi: 10.1200/JCO.18.01512
61. Ren W, Fan H, Grimm SA, Guo Y, Kim JJ, Yin J, et al. Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc Natl Acad Sci USA (2020) 117:18439–47. doi: 10.1073/pnas.2009316117
62. Maiuri AR, O'Hagan HM. Interplay between inflammation and epigenetic changes in cancer. Prog Mol Biol Transl Sci (2016) 144:69–117. doi: 10.1016/bs.pmbts.2016.09.002
63. O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol (2020) 38:1378–88. doi: 10.1200/JCO.19.02931
64. Janysek DC, Kim J, Duijf PHG, Dray E. Clinical use and mechanisms of resistance for PARP inhibitors in homologous recombination-deficient cancers. Trans Oncol (2021) 14:101012. doi: 10.1016/j.tranon.2021.101012
65. De Vita VT, Lawrence TS, Rosenberg SA. De vita, hellman & rosenberg's cancer: principles & practice of oncology. 10th edn. Philadelphia: Lippincott Williams & Wilkins (2015).
66. Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet (2021) 22:3–18. doi: 10.1038/s41576-020-0265-5
67. Romani A, Guerra M, Trerotola M, Alberti S. Detection and analysis of spliced chimeric mRNAs in sequence databanks. Nucleic Acids Res (2003) 31:1–8. doi: 10.1093/nar/gng017
Keywords: gelatinous drop-like corneal dystrophy (GDLD), pancreatic cancer (PC), Trop-2, genomic mutations, gene expression profiles, tumor progression
Citation: Guerra E, Di Pietro R, Stati G and Alberti S (2023) A non-mutated TROP2 fingerprint in cancer genetics. Front. Oncol. 13:1151090. doi: 10.3389/fonc.2023.1151090
Received: 25 January 2023; Accepted: 12 June 2023;
Published: 29 June 2023.
Edited by:
Hua Tan, National Human Genome Research Institute (NIH), United StatesReviewed by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesWilliam Garrow Kerr, Upstate Medical University, United States
Yuriy L. Orlov, I.M. Sechenov First Moscow State Medical University, Russia
Copyright © 2023 Guerra, Di Pietro, Stati and Alberti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saverio Alberti, YWxiZXJ0aS5zYXZlcmlvQGdtYWlsLmNvbQ==