 Alicia Garcia-Gimenez1
Alicia Garcia-Gimenez1 Simon E. Richardson1,2*
Simon E. Richardson1,2*- 1Department of Haematology, Wellcome Trust—Medical Research Council Cambridge Stem Cell Institute, University of Cambridge, Cambridge, United Kingdom
- 2Cambridge University Hospitals, Cambridge, United Kingdom
B cell precursor acute lymphoblastic leukemia (BCP-ALL) is a malignant disorder of immature B lineage immune progenitors and is the commonest cancer in children. Despite treatment advances it remains a leading cause of death in childhood and response rates in adults remain poor. A preleukemic state predisposing children to BCP-ALL frequently arises in utero, with an incidence far higher than that of transformed leukemia, offering the potential for early intervention to prevent disease. Understanding the natural history of this disease requires an appreciation of how cell-extrinsic pressures, including microenvironment, immune surveillance and chemotherapy direct cell-intrinsic genetic and epigenetic evolution. In this review, we outline how microenvironmental factors interact with BCP-ALL at different stages of tumorigenesis and highlight emerging therapeutic avenues.
Introduction
Cancer is a clonal outgrowth of cells that have adapted to gain a competitive advantage over their physiologically-constrained competitors. Classically this has been viewed as a stepwise evolutionary process, characterized by the sequential acquisition of driver mutations in critical tumor suppressor and oncogenes. However, powerful non-genetic factors also direct and constrain tumor evolution, including cell-intrinsic “epigenetic” states acquired from the developmental origins, stem/progenitor cell programming and epigenetic dysregulation of the cell of origin, and cell-extrinsic factors, notably microenvironmental niches and the selective pressure of immune surveillance and therapy. In this review we will outline how cell-extrinsic factors contribute to the initiation and evolution of B-cell precursor acute lymphoblastic leukemia (BCP-ALL), highlighting opportunities for therapeutic intervention at different stages of disease progression and chemoresistance.
B-Cell precursor acute lymphoblastic leukemia
BCP-ALL is an aggressive malignancy of immature B-lineage immune cell progenitors. Leukemic cells arise in the bone marrow (BM) and infiltrate extramedullary sites, notably the reticuloendothelial system (liver, spleen & lymph nodes) and so-called sanctuary sites (central nervous system (CNS) and testes) (1). BCP-ALL is predominantly a disease of childhood and there is compelling evidence in a number of genetic subgroups that a pre-leukemic state is initiated in utero, with B-cells harboring clonal genetic fusions or immunoglobulin gene rearrangements identifiable at birth on neonatal blood samples from affected children and/or shared in the blood of monochorionic twins (2). Importantly, the incidence of pre-leukemia in children far exceeds that of overt disease, indicating that these cells require further genetic and/or other events to transform to frank leukemia.
The first hit mutations that can initiate pre-leukemia are highly diverse between patients, but recent work has shown that the disease can be classified into 23 distinct subgroups based on underlying transcriptional signatures (3, 4). In contrast, the second-hit mutations implicated in disease transformation and progression are relatively conserved across genetic subtypes, implicating common pathways in disease progression including activation of signaling pathways (e.g. RAS, JAK-STAT), loss of transcriptional master regulators (e.g. PAX5, IKZF1) and perturbation of epigenetic co-regulators (e.g. CREBBP) (3–6).
Modern response-adapted multiagent chemotherapy regimens can cure the majority of children, albeit at the expense of toxicity (7, 8). Treatment of adult B-ALL, however, is more challenging with approximately 50% overall survival even in patients fit enough to undergo allogeneic bone marrow transplantation (9, 10). Introduction of novel targeted therapies (e.g. BCR::ABL1 tyrosine kinase inhibitors) and immunotherapy (e.g. CD19-CD3 bispecific T cell engager (BiTEs) and chimeric antigen receptor (CAR)-T cells) are improving outcomes for certain high-risk and relapsed cases. Nevertheless, some groups of patients continue to fare poorly, including those with high-risk genetic drivers (e.g. E2A::HLF, KMT2A-rearranged BCP-ALL), certain age groups (e.g. infants, elderly), those with poor-risk second hit mutations (e.g. CREBBP-RAS) and those who relapse in the CNS.
Microenvironmental and cell-extrinsic factors are increasingly thought to play a both supportive and constraining roles throughout the pathogenesis of BCP-ALL (Figure 1A). Understanding how they contribute to pre-leukemic initiation, the fate of preleukemic clones and mechanisms of resistance will offer new avenues for understanding disease mechanism and facilitate the development of novel therapeutic interventions.
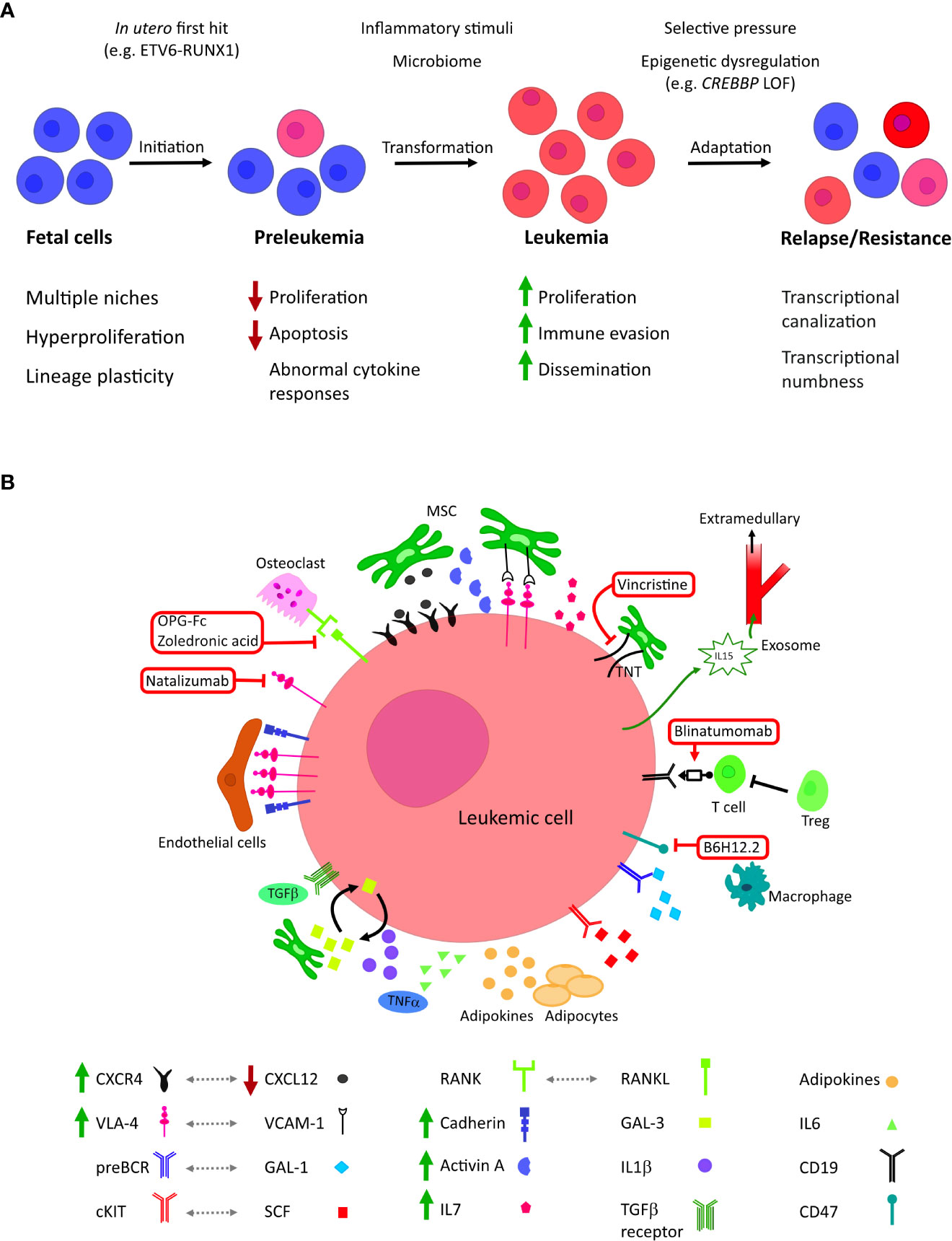
Figure 1 (A) The roles of microenvironmental factors at different stages of tumor evolution in childhood BCP-ALL. BCP-ALL is thought to initiate in utero where B lineage progenitors exhibit unique characteristics, including inhabiting diverse niches. First-hit mutations create a clinically silent preleukemic state that persists into childhood. In a small number of children, this pre leukemic state transforms to acute leukemia; this is thought to be driven by the acquisition of co-operative second hit mutations, possibly in response to inflammatory signaling. Leukemic blasts evolve to colonize further niches and evade treatment, altering their epigenetic state to become less dependent on external stimuli. (B) Cellular and molecular microenvironmental factors associated with BCP-ALL. Current and investigational therapeutic targets are highlighted in red: mitochondrial transfer through tunnelling nanotubules (TNT) can be inhibited by the microtubule inhibitor vincristine; blinatumomab co-associates B-lineage blasts with tumour-targeting cytotoxic T cells; neutralization of CD47 by B6H12.2 antibody restores the macrophage phagocytic response against leukemic blasts; zoledronic acid and recombinant OPG-Fc reduce bone loss, inhibiting leukemic growth and prolonging survival in vivo; integrin 4 inhibitor Natalizumab sensitizes leukemic blasts to chemotherapy.
Niche in normal B cell development
The BM niche is formed by hematopoietic and non-hematopoietic cells from different lineages. Bone forming cells (osteoblasts and osteoclasts), adipocytes, reticular cells, endothelial cells, mesenchymal stromal cells (MSC) and neural cells help create and support BM homeostasis and hematopoiesis, through direct cellular interactions, the production of soluble cytokines and the maintenance of the extracellular matrix (11–13). Interactions between hematopoietic stem/progenitor cells (HSPCs) and the BM microenvironment are essential for both commitment into the B lineage and survival following successful rearrangement of the heavy and light chain immunoglobulin loci, evaluated by pro-survival signaling through the pre-B cell receptor (preBCR) and BCR complexes respectively. A number of soluble factors are known to be essential for the commitment into the B lineage, including FLT3L and SCF (14, 15). Multipotent hematopoietic progenitor cells and early pre-proB cells directly co-associate with CXCL12-expressing stromal cells (16), with osteoblasts appearing particularly important for successful B lineage differentiation (17). CXCL12 seems important in both attracting B progenitors to specific BM niches and by inducing direct interactions through the integrin VLA-4 - VCAM1 axis (18).
Different parts of the BM niche exhibit different metabolic characteristics, in particular oxygenation, which is thought to regulate REDOX-sensitive transcription factors such as the hypoxia inducible factors as well as the B cell master regulator PAX5 (19). Maturing proB cells migrate to areas of high IL7 expression and complete RAG-mediated rearrangement of their IGH locus to become preB cells, which express the preBCR. Successful recombination of the IGH locus results in effective signaling through the preBCR, which is in part activated by stromal Galectin 1 binding (20). Successful light chain recombination completes BM B cell maturation, producing a repertoire of naïve B cells with a unique BCR competent for antigenic stimulation in lymph nodes.
B cell progenitors are particularly abundant during early embryonic development. During ontogeny hematopoiesis arises in multiple waves from diverse sites, including primitive hematopoiesis in the yolk sac (YS) before definitive HSCs are specified in the aorto-gonad-mesonephros (AGM) region of the dorsal aorta. The progeny of these HSPCs establish differentiation hierarchies in multiple niches, notably the YS, fetal liver (FL), placenta and BM (21). The precise lineages of the B cell progenitors produced in these sites remains controversial (22), but it appears that both the ontogenic origins and the niche in which these cells reside are associated with significant differences to adult lymphopoiesis, in particular the enhanced proliferative state of FL HSPCs (23, 24). These differences indicate that fetal hematopoiesis may be structured differently to adult, providing a unique cellular context for the in utero initiation of childhood leukemia.
Subversion of B-cell-microenvironmental interactions promotes pre-leukemic development
There is evidence that pre-leukemic cells are supported by the microenvironment following disease initiation and that the microenvironment becomes remodeled to permit and support disease progression (25) (Figure 1B).
Much work has been undertaken in the paradigmatic pre-leukemia initiated in utero by the ETV6::RUNX1 gene fusion, the commonest single genetic cause of childhood BCP-ALL. Pre-leukemic in vitro model systems expressing the first-hit mutation show aberrant co-expression of myeloid and erythroid cytokine receptors on lymphoid cells, indicating that even at the earliest stages, pre-leukemic cells significantly change the way that they sense and respond to their microenvironment (26–28). Functionally, preleukemic cells are thought to be maintained as a small population, with niche factors contributing to their maintenance in a relatively quiescent (26, 29) and anti-apoptotic state (30).
Prenatally, the major site of pre-leukemia is likely to be the FL, although direct transplacental exposure to mutagens has been implicated in the generation of first-hit mutations, and by the time of birth pre-leukemic cells are readily detectable in the peripheral blood and umbilical cord. HSCs emerging from the AGM are attracted to and retained in the FL niche by factors including SCF, CXCL12 and β1 integrin, factors which are also supportive of B cell development (31, 32). Compared to both adult and fetal BM, FL hematopoiesis is characterized by extremely high levels of proliferation (33), along with other differences in cell surface markers, lineage differentiation and gene expression profiles (23, 34). B-lymphopoiesis is particularly prominent in the FL and exhibits a number of qualitative differences to adult including lack of TDT expression, IL7R independence and that the earliest committed B cells appear to emerge from a developmentally-restricted progenitor that exhibits unique co-expression of myeloid and lymphoid programming and potential (28). These differences might explain some of the unique features of childhood BCP-ALL, including its relatively high incidence, propensity for unique genetic drivers rarely seen in adults (e.g. ETV6::RUNX1, hyperdiploidy) and tendency to lineage promiscuity.
By the end of gestation, it is probable that pre-malignant cells have colonized the BM. There is evidence that multiple first-hit mutations can change B cell adhesion/migration properties; for example, ETV6::RUNX1 is associated with a cell-intrinsic defect in CXCR4-CXCL12 signaling (35) and KMT2A-mutated BCP-ALL up-regulates protocadherin genes (36). An area of active interest is whether pre-leukemic cells can modify the BM niche prior to transformation; given the changes seen at the time of diagnosis (see below) this seems likely and novel experimental co-culture techniques will help in delineating these interactions.
Inflammatory stimuli aid the transformation and maintenance of overt leukemia
The incidence of detectable pre-leukemia in asymptomatic children is many times that of the incidence of overt childhood BCP-ALL; for example, ETV6::RUNX1+ pre-leukemia is approximately 100-500x more common than ETV6::RUNX1+ BCP-ALL (37, 38). This indicates the importance of second-hit events in driving disease transformation, thought mainly to be the acquisition of co-operative mutations. Conversely, it demonstrates that the majority of pre-leukemic clones in children lack sufficient self-renewal potential to persist into adulthood, either in a cell-intrinsic manner, or due to an inability to adapt to changing niches during development (39).
Epidemiological evidence points to delayed exposure to infectious stimuli as a risk factor for BCP-ALL transformation. This has led to the hypothesis that an under-exposed immune system in early life results in subsequent overactivation of cytokine signaling and hyper-mutagenesis in pre-malignant cells, potentially driven by aberrant activation-induced cytidine deaminase (AID) (40) or recombination-activating gene (RAG) activity (41, 42). Furthermore, the neonatal blood of children who develop leukemia exhibits measurable differences in cytokine concentrations, indicating a degree of immune dysregulation prior to environmental exposures to pathogens (43, 44). The specific pathogens involved in this mechanism are unknown, but significant epidemiological associations with outbreaks of swine flu (45), influenza (46) and SARS (47) viruses have been reported. The effects of both exposure to Covid19 and the lack of infectious exposure in children during prolonged lockdowns are an area of active research (48).
Experimental evidence shows that pre-leukemic mouse models housed in clean specified pathogen free facilities develop increased rates of leukemia in response to infectious stimuli (49, 50) and that inhibition of inflammatory stimulation can mitigate transformation to BCP-ALL in a Pax5 pre-leukemic model (51). There is evidence that MSC-derived pro-inflammatory TGFβ family members (including TGFβ and Activin A) can favor the growth of ETV6::RUNX1+ pre-leukemic cells (26, 52) and that the acute-phase response cytokines (IL6, TNFα and IL1β) can co-operate with MSCs to generate a pro-leukemic niche for ETV6::RUNX1+ cells and drive a hyper-mutagenic state (53). Intriguingly, gut commensal microbes are known to affect the immune microenvironment systemically and it has been shown in murine pre-leukemic models that microbiome disruption by antibiotic administration in early life can induce the development of leukemia (54).
Once transformed, it is established that BCP-ALL actively remodels its interaction with the BM niche into a permissive or supportive microenvironment (55). The BCP-ALL-MSC interaction appears key to this, with down-regulation of the CXCL12 chemokine-axis favoring BCP-ALL MSC interactions over those of normal HSCs (56). Subversion of a number soluble and surface ligand axes has been implicated in promoting leukemic survival, including TGFβ, Cadherin-wnt-Catenin, Notch and Integrin pathways. Furthermore, BCP-ALL cells inhibit osteoblast function through the RANK-RANKL axis, remodeling the BM space and contributing to clinically-meaningful trabecular bone destruction (57). Zoledronic acid (58) and the recombinant RANKL antagonist, OPG-Fc (59) have shown efficacy in restoring bone homeostasis and reducing disease burden in in vivo models. MSC-derived Galectin 3 can be internalized by BCP-ALL blasts stimulating autocrine production of Galectin 3, driving disease progression in a cell autonomous manner (60). This serves as an example of how leukemic cells can also adapt to circumvent their own dependency on microenvironmental factors.
Epigenetic adaptation to extracellular cues underlies tumor evolution and resistance
During tumor evolution BCP-ALL subclones increasingly compete with each other, adapting to diversify their signaling, metabolic and niche dependencies, as well as evading external pressures including immune surveillance and chemotherapy.
In experimental models, interactions between BCP-ALL and factors from BM perivascular, endosteal, and hematopoietic niches maintain BCP-ALL survival and quiescence in response to chemotherapy (61). A preclinical in vivo model has shown that targeting the integrin VLA-4 using the therapeutic antibody Natalizumab sensitized BCP-ALL cells to cytotoxic chemotherapy (62). A fascinating mechanism of chemoresistance has been demonstrated by Burt and colleagues, who showed that direct connections between BCP-ALL blasts and MSCs by tunneling nanotubules (TNT) could lead to mitochondrial transfer, protecting BCP-ALL cells from toxicity from chemotherapy-induced reactive oxygen species (63). The microtubule inhibitor vincristine, a key component of current BCP-ALL chemotherapy regimens, was shown to disrupt TNT formation, ameliorating this mechanism of resistance. Adipocytes have been shown to induce chemoresistance by both directly sequestering chemotherapeutic agents and by increasing leukemic blasts fitness through the production of pro- and anti-inflammatory adipokines, fueling the leukemic cells with free fatty acids and promoting oxidative phosphorylation (64, 65). The upregulation of Galectin-9 by adipokines is responsible for several of these effects with preliminary studies implicating Galectin-9 as a therapeutic target (66). Interestingly, the role of adipocytes has been shown to alter dynamically during treatment, with a particular role in promoting a quiescent chemo-resistant state in regenerating bone marrow. Mechanistically this was associated with a global suppression of protein translation, which could be overcome by inhibition of GCN2, restoring chemosensitivity (67).
The established graft-versus-leukemia effects seen after allogeneic transplantation, and more recently the advent of highly-efficacious BiTE and CAR-T immunotherapies in BCP-ALL, highlights the necessity for continuous T cell immune evasion during leukemic evolution. The efficacy of the CD19-CD3 BiTE blinatumomab demonstrates the ongoing presence of anti-BCP-ALL cytotoxic T cells in frank disease. Blinatumomab failure has been associated with higher numbers of inhibitory Treg cells, implicating these negative regulators of the cellular immune response as potential effectors of immune evasion (68). Furthermore, BCP-ALL blasts overexpress the surface CD47, a “don’t eat me” signal that inhibits phagocytosis by macrophages. The anti-CD47 neutralizing antibody B6H12.2 has been shown to relieve this block in in vitro and in vivo models and this strategy has shown promise in other leukemia subtypes (69).
Clinically important sites of relapse include the CNS and testes. These are considered sanctuary sites, with demonstrably lower levels of exposure to chemotherapy drugs in addition to being relatively privileged sites from immune surveillance (70–72). Extramedullary niches share an impaired CXCR4-CXCL12 axis (73, 74), hypoxic conditions (75) and the presence of the cytokine SCF (76). B lymphocytes have a physiological ability to enter the CNS and the ability to cross the blood-cerebrospinal fluid barrier is a generic feature of BCP-ALL blasts (77). A number of surface receptors are thought to contribute to this, including classical B-cell progenitor markers such as CD79a and IL7R (78, 79). Leukemic blasts also produce exosomes with soluble molecules (e.g. IL15) that alter distant niches such as the CNS (80). Once in the CNS, leukemic cells adapt to their new environment, including by adjusting to relative hypoxia (75) and by changing their metabolic requirements towards fatty acid metabolism (81, 82), changes that potentially provide unique therapeutic vulnerabilities.
As BCP-ALL progresses, cells appear to become more autonomous, characterized by reduced dependence on external/niche stimuli and the ability to tolerate more extreme environmental and therapeutic selective pressure. This was initially thought to be due to the positive selection of genetically heterogenous subclones, favoring those harboring mutations that provided selective advantages to particular evolutionary pressures. Clinical and experimental studies, however, have failed to demonstrate recurrent selection of genetic sub-clones during treatment, notwithstanding mutations in a small number of direct drug targets (e.g. BCR::ABL1 tyrosine kinase mutations and mutations in key members of the glucocorticoid or mercaptopurine pathways). Instead, leukemic cells appear to adapt to treatment-related selective pressure by “transcriptional canalization”, characterized by increased quiescence and a reduction in their global transcriptional heterogeneity (83). In a genetically highly diverse disease, this acquired loss of heterogeneity at the gene expression level could provide common therapeutic vulnerabilities.
Epigenetic dysregulation is a hallmark of many cancers and the genes encoding epigenetic co-regulators are commonly mutated in BCP-ALL. An intriguing study has recently shown that perturbation of multiple different epigenetic regulators across diverse cancer models tended to increase the tolerance of cancer cells to environmental stress (84). This “transcriptional numbness” to selective pressure lowers the probability of cell death, providing a phenotypic inertia that facilitates survival and adaptation of mutant cells. In the case of BCP-ALL, a potential exemplar of this are loss of function mutations in the transcriptional co-activator CREBBP. CREBBP deletion, or point mutations affecting its enzymatic acetyltransferase domain, are enriched in relapsed BCP-ALL (6) and high-risk genetic subtypes such as near-haploid BCP-ALL (85). It has also been associated with relapse risk in the otherwise good-risk subgroup of pediatric hyperdiploid BCP-ALL (4, 86). A number of mechanisms could account for this risk, including induction of glucocorticoid resistance (6, 87) and/or potentiation of cytokine signaling though the RAS pathway (88), as well as potentially phenotypic inertia related to loss of transcriptional co-activation. Understanding how cell-intrinsic epigenetic reprograming mediates BCP-ALL adaptation is therefore essential to better appreciating and targeting the cell-extrinsic dependencies seen at different stages of disease progression.
Discussion
BCP-ALL cells are highly dependent on microenvironmental niches at all stages of tumor development. Characterizing these shifting dependencies is an essential component of undertesting the selective pressure that drives tumor evolution through pre-leukemic initiation, leukemic transformation, frank leukemia, tissue infiltration and chemoresistance. New model systems are emerging to examine these factors, including humanized in vivo models and advanced 2D and 3D co-culture systems (89, 90). Perturbation of critical soluble factors, cell-cell and immune interactions are providing promising novel therapeutic avenues, as well as new insights into the mechanism of action of established drugs. Delineating the role that epigenetic reprogramming plays during tumor evolution and treatment resistance will provide opportunities to target the most resistant cases. Conversely, the clear dependencies of early pre-leukemic cells on microenvironmental factors might afford the potential for early intervention and the tantalizing possibility of making childhood BCP-ALL a preventable disease (91).
Author contributions
AG-G and SR drafted the manuscript. A-GG and SR reviewed and approved the final submission. All authors contributed to the article and approved the submitted version.
Funding
SR is supported by a Clinician Scientist Fellowship from Cancer. Research UK (C67279/A27957) and Leukaemia UK John Goldman. Fellowship (2022/JGF/004). Research in the Wellcome - MRC. Cambridge Stem Cell Institute is funded by a grant from the Wellcome Trust (203151/Z/16/Z). Research in the Cancer Research. UK Cambridge Centre is supported by a Cancer Research UK Major CentreAward (C9685/A25117).
Acknowledgments
We apologize to those authors whose work we were unable to cite due to space constraints.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
2. Ford AM, Colman S, Greaves M. Covert pre-leukaemic clones in healthy co-twins of patients with childhood acute lymphoblastic leukaemia. Leuk (2022) 37(1):47–52, 1-6. doi: 10.1038/s41375-022-01756-1
3. Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of b-progenitor acute lymphoblastic leukemia. Nat Genet (2019) 51(2):296–307. doi: 10.1038/s41588-018-0315-5
4. Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet (2022) 54(9):1376–89. doi: 10.1038/s41588-022-01159-z
5. Malinowska-Ozdowy K, Frech C, Schönegger A, Eckert C, Cazzaniga G, Stanulla M, et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. (2015) 29:1656–67. doi: 10.1038/leu.2015.107
6. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nat (2011) 471(7337):235–9. doi: 10.1038/nature09727
7. Demidowicz E, Pogorzała M, Łęcka M, Żołnowska H, Marjańska A, Kubicka M, et al. Outcome of pediatric acute lymphoblastic leukemia: Sixty years of progress. Anticancer Res (2019) 39(9):5203–7. doi: 10.21873/anticanres.13717
8. Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J Clin Oncol (2012) 30(14):1663–9. doi: 10.1200/JCO.2011.37.8018
9. Ssenyonga N, Stiller C, Nakata K, Shalkow J, Redmond S, Bulliard JL, et al. Worldwide trends in population-based survival for children, adolescents, and young adults diagnosed with leukaemia, by subtype, during 2000–14 (CONCORD-3): analysis of individual data from 258 cancer registries in 61 countries. Lancet Child Adolesc Heal (2022) 6(6):409–31. doi: 10.1016/S2352-4642(22)00095-5
10. Marks DI, Clifton-Hadley L, Copland M, Hussain J, Menne TF, McMillan A, et al. In-vivo T-cell depleted reduced-intensity conditioned allogeneic haematopoietic stem-cell transplantation for patients with acute lymphoblastic leukaemia in first remission: results from the prospective, single-arm evaluation of the UKALL14 trial. Lancet Haematol (2022) 9(4):e276–88. doi: 10.1016/S2352-3026(22)00036-9
11. Fröbel J, Landspersky T, Percin G, Schreck C, Rahmig S, Ori A, et al. The hematopoietic bone marrow niche ecosystem. Front Cell Dev Biol (2021) 9:1958. doi: 10.3389/fcell.2021.705410
12. Bianco P. Bone and the hematopoietic niche: A tale of two stem cells. Blood. (2011) 117(20):5281–8. doi: 10.1182/blood-2011-01-315069
13. Man Y, Yao X, Yang T, Wang Y. Hematopoietic stem cell niche during homeostasis, malignancy, and bone marrow transplantation. Front Cell Dev Biol (2021) 9:14. doi: 10.3389/fcell.2021.621214
14. Sitnicka E, Bryder D, Theilgaard-Mönch K, Buza-Vidas N, Adolfsson J, Jacobsen SEW. Key role of flt3 ligand in regulation of the common lymphoid progenitor but not in maintenance of the hematopoietic stem cell pool. Immunity. (2002) 17(4):463–72. doi: 10.1016/S1074-7613(02)00419-3
15. Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-KitW/W mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. (2002) 17(3):277–88. doi: 10.1016/S1074-7613(02)00386-2
16. Tokoyoda K, Egawa T, Sugiyama T, Choi BIL, Nagasawa T. Cellular niches controlling b lymphocyte behavior within bone marrow during development. Immunity. (2004) 20(6):707–18. doi: 10.1016/j.immuni.2004.05.001
17. Galán-Díez M, Kousteni S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr Mol Biol Rep (2017) 3(2):53–62. doi: 10.1007/s40610-017-0055-9
18. Park S-Y, Wolfram P, Canty K, Harley B, Nombela-Arrieta C, Pivarnik G, et al. Focal adhesion kinase regulates the localization and retention of pro-b cells in bone marrow microenvironments. J Immunol (2013) 190(3):1094–102. doi: 10.4049/jimmunol.1202639
19. Borzillo GV, Ashmun RA, Sherr CJ. Macrophage lineage switching of murine early pre-b lymphoid cells expressing transduced fms genes. Mol Cell Biol (1990) 10(6):2703–14. doi: 10.1128/mcb.10.6.2703-2714.1990
20. Gauthier L, Rossi B, Roux F, Termine E, Schiff C. Galectin-1 is a stromal cell ligand of the pre-b cell receptor (BCR) implicated in synapse formation between pre-b and stromal cells and in pre-BCR triggering. Proc Natl Acad Sci U S A. (2002) 99(20):13014–9. doi: 10.1073/pnas.202323999
21. Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. (2011) 138(6):1017–31. doi: 10.1242/dev.040998
22. Böiers C, Carrelha J, Lutteropp M, Luc S, Green JCA, Azzoni E, et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell (2013) 13(5):535–48. doi: 10.1016/j.stem.2013.08.012
23. Popescu DM, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver haematopoiesis. Nat (2019) 574(7778):365–71. doi: 10.1038/s41586-019-1652-y
24. O’Byrne S, Elliott N, Rice S, Buck G, Fordham N, Garnett C, et al. Discovery of a CD10-negative b-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood. (2019) 134(13):1059–71. doi: 10.1182/blood.2019001289
25. Dander E, Palmi C, D’amico G, Cazzaniga G. The bone marrow niche in b-cell acute lymphoblastic leukemia: The role of microenvironment from pre-leukemia to overt leukemia. Int J Mol Sci (2021) 22(9):4426. doi: 10.3390/ijms22094426
26. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-β pathway in early b lineage progenitor cells. J Clin Invest (2009) 119(4):826–36. doi: 10.1172/JCI36428
27. Torrano V, Procter J, Cardus P, Greaves M, Ford AM. ETV6-RUNX1 promotes survival of early b lineage progenitor cells via a dysregulated erythropoietin receptor. Blood. (2011) 118(18):4910–8. doi: 10.1182/blood-2011-05-354266
28. Böiers C, Richardson SE, Laycock E, Zriwil A, Turati VA, Brown J, et al. A human IPS model implicates embryonic b-myeloid fate restriction as developmental susceptibility to b acute lymphoblastic leukemia-associated ETV6-RUNX1. Dev Cell (2018) 44(3):362–377.e7. doi: 10.1016/j.devcel.2017.12.005
29. Wray JP, Deltcheva EM, Boiers C, Richardson S, Chhetri JB, Brown J, et al. Regulome analysis in b-acute lymphoblastic leukemia exposes core binding factor addiction as a therapeutic vulnerability. Nat Commun (2022) 13(1):1–18. doi: 10.1038/s41467-022-34653-3
30. Gefen N, Binder V, Zaliova M, Linka Y, Morrow M, Novosel A, et al. Hsa-mir-125b-2 is highly expressed in childhood ETV6/RUNX1 (TEL/AML1) leukemias and confers survival advantage to growth inhibitory signals independent of p53. Leuk (2010) 24(1):89–96. doi: 10.1038/leu.2009.208
31. Christensen JL, Wright DE, Wagers AJ, Weissman IL. Circulation and chemotaxis of fetal hematopoietic stem cells. PloS Biol (2004) 2(3):e75. doi: 10.1371/journal.pbio.0020075
32. Potocnik AJ, Brakebusch C, Fässler R. Fetal and adult hematopoietic stem cells require β1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity. (2000) 12(6):653–63. doi: 10.1016/S1074-7613(00)80216-2
33. Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest (2006) 116(10):2808–16. doi: 10.1172/JCI28310
34. Lewis K, Yoshimoto M, Takebe T. Fetal liver hematopoiesis: from development to delivery. Stem Cell Res Ther (2021) 12(1):1–8. doi: 10.1186/s13287-021-02189-w
35. Palmi C, Fazio G, Savino AM, Procter J, Howell L, Cazzaniga V, et al. Cytoskeletal regulatory gene expression and migratory properties of b-cell progenitors are affected by the ETV6-RUNX1 rearrangement. Mol Cancer Res (2014) 12(12):1796–806. doi: 10.1158/1541-7786.MCR-14-0056-T
36. Zhang H, Cheng J, Li Z, Xi Y. Identification of hub genes and molecular mechanisms in infant acute lymphoblastic leukemia with MLL gene rearrangement. PeerJ. (2019) 2019(8):e7628. doi: 10.7717/peerj.7628
37. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. (2002) 99(12):8242–7. doi: 10.1073/pnas.112218799
38. Schäfer D, Olsen M, Lähnemann D, Stanulla M, Slany R, Schmiegelow K, et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood. (2018) 131(7):821–6. doi: 10.1182/blood-2017-09-808402
39. Zanetti C, Kumar R, Ender J, Godavarthy PS, Hartmann M, Hey J, et al. The age of the bone marrow microenvironment influences b-cell acute lymphoblastic leukemia progression via CXCR5-CXCL13. Blood. (2021) 138(19):1870–84. doi: 10.1182/blood.2021011557
40. Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol (2015) 16(7):766–74. doi: 10.1038/ni.3160
41. Kirkham CM, Scott JNF, Wang X, Smith AL, Kupinski AP, Ford AM, et al. Cut-and-Run: A distinct mechanism by which V(D)J recombination causes genome instability. Mol Cell (2019) 74(3):584–597.e9. doi: 10.1016/j.molcel.2019.02.025
42. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet (2014) 46(2):116–25. doi: 10.1038/ng.2874
43. Chang JS, Zhou M, Buffler PA, Chokkalingam AP, Metayer C, Wiemels JL. Profound deficit of IL10 at birth in children who develop childhood acute lymphoblastic leukemia. Cancer Epidemiol Biomarkers Prev (2011) 20(8):1736–40. doi: 10.1158/1055-9965.EPI-11-0162
44. Søegaard SH, Rostgaard K, Skogstrand K, Wiemels JL, Schmiegelow K, Hjalgrim H. Neonatal inflammatory markers are associated with childhood b-cell precursor acute lymphoblastic leukemia. Cancer Res (2018) 78(18):5458–63. doi: 10.1158/0008-5472.CAN-18-0831
45. Cazzaniga G, Bisanti L, Randi G, Deandrea S, Bungaro S, Pregliasco F, et al. Possible role of pandemic AH1N1 swine flu virus in a childhood leukemia cluster. Leuk (2017) 31(8):1819–21. doi: 10.1038/leu.2017.127
46. Kroll ME, Draper GJ, Stiller CA, Murphy MFG. Childhood leukemia incidence in Britain, 1974–2000: Time trends and possible relation to influenza epidemics. JNCI J Natl Cancer Inst (2006) 98(6):417–20. doi: 10.1093/jnci/djj095
47. Li CK, Zee B, Lee J, Chik KW, Ha SY, Lee V. Impact of SARS on development of childhood acute lymphoblastic leukaemia. Leukemia. (2007) 21(7):1353–6. doi: 10.1038/sj.leu.2404729
48. Greaves M. COVID-19 and childhood acute lymphoblastic leukemia. Pediatr Blood Cancer (2020) 67(12):e28481. doi: 10.1002/pbc.28481
49. Rodríguez-Hernández G, Opitz FV, Delgado P, Walter C, Álvarez-Prado ÁF, González-Herrero I, et al. Infectious stimuli promote malignant b-cell acute lymphoblastic leukemia in the absence of AID. Nat Commun (2019) 10:5563. doi: 10.1038/s41467-019-13570-y
50. Martín-Lorenzo A, Hauer J, Vicente-Dueñas C, Auer F, González-Herrero I, García-Ramírez I, et al. Infection exposure is a causal factor in b-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer Discovery (2015) 5(12):1328–43. doi: 10.1158/2159-8290.CD-15-0892
51. Isidro-Hernández M, Mayado A, Casado-García A, Martínez-Cano J, Palmi C, Fazio G, et al. Inhibition of inflammatory signaling in Pax5 mutant cells mitigates b-cell leukemogenesis. Sci Rep (2020) 10(1):1–14. doi: 10.1038/s41598-020-76206-y
52. Portale F, Cricrì G, Bresolin S, Lupi M, Gaspari S, Silvestri D, et al. ActivinA: a new leukemia-promoting factor conferring migratory advantage to b-cell precursor-acute lymphoblastic leukemic cells. Haematologica. (2019) 104(3):533–45. doi: 10.3324/haematol.2018.188664
53. Beneforti L, Dander E, Bresolin S, Bueno C, Acunzo D, Bertagna M, et al. Pro-inflammatory cytokines favor the emergence of ETV6-RUNX1-positive pre-leukemic cells in a model of mesenchymal niche. Br J Haematol (2020) 190(2):262–73. doi: 10.1111/bjh.16523
54. Vicente-Duenãs C, Janssen S, Oldenburg M, Auer F, Lez-Herrero ISG, Casado-García A, et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood. (2020) 136(18):2003–17. doi: 10.1182/blood.2019004381
55. Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell (2014) 25(6):778–93. doi: 10.1016/j.ccr.2014.04.015
56. Balandrán JC, Purizaca J, Enciso J, Dozal D, Sandoval A, Jiménez-Hernández E, et al. Pro-inflammatory-related loss of CXCL12 niche promotes acute lymphoblastic leukemic progression at the expense of normal lymphopoiesis. Front Immunol (2017) 7:666. doi: 10.3389/fimmu.2016.00666
57. Rajakumar SA, Danska JS. Bad to the bone: B cell acute lymphoblastic leukemia cells mediate bone destruction. Mol Cell Oncol (2020) 8(1):1835423. doi: 10.1080/2372355620201835423
58. Cheung LC, Tickner J, Hughes AM, Skut P, Howlett M, Foley B, et al. New therapeutic opportunities from dissecting the pre-b leukemia bone marrow microenvironment. Leukemia. (2018) 32:2326–38. doi: 10.1038/s41375-018-0144-7
59. Rajakumar SA, Papp E, Lee KK, Grandal I, Merico D, Liu CC, et al. B cell acute lymphoblastic leukemia cells mediate RANK-RANKL-dependent bone destruction. Sci Transl Med (2020) 12(561):5942. doi: 10.1126/scitranslmed.aba5942
60. Fei F, Joo EJ, Tarighat SS, Schiffer I, Paz H, Fabbri M, et al. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through galectin-3. Oncotarget. (2015) 6(13):11378–94. doi: 10.18632/oncotarget.3409
61. Ma C, Witkowski MT, Harris J, Dolgalev I, Sreeram S, Qian W, et al. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in b cell acute lymphoblastic leukemia bone marrow niche. Sci Adv (2020) 6(44):eaba5536. doi: 10.1126/sciadv.aba5536
62. Hsieh Y, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, et al. Integrin alpha4 blockade sensitizes drug resistant pre-b acute lymphoblastic leukemia to chemotherapy. Blood. (2013) 121(10):1814–8. doi: 10.1182/blood-2012-01-406272
63. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood. (2019) 134(17):1415–29. doi: 10.1182/blood.2019001398
64. Tucci J, Chen T, Margulis K, Orgel E, Paszkiewicz RL, Cohen MD, et al. Adipocytes provide fatty acids to acute lymphoblastic leukemia cells. Front Oncol (2021) 11:665763. doi: 10.3389/fonc.2021.665763
65. Sheng X, Parmentier JH, Tucci J, Pei H, Cortez-Toledo O, Dieli-Conwright CM, et al. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res (2017) 15(12):1704–13. doi: 10.1158/1541-7786.MCR-17-0338
66. Lee M, Hamilton JAG, Talekar GR, Ross AJ, Michael L, Rupji M, et al. Obesity-induced galectin-9 is a therapeutic target in b-cell acute lymphoblastic leukemia. Nat Commun (2022) 13(1):1157. doi: 10.1038/s41467-022-28839-y
67. Heydt Q, Xintaropoulou C, Clear A, Austin M, Pislariu I, Miraki-Moud F, et al. Adipocytes disrupt the translational programme of acute lymphoblastic leukaemia to favour tumour survival and persistence. Nat Commun (2021) 12(1):1–18. doi: 10.1038/s41467-021-25540-4
68. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with b-precursor ALL. Leuk (2017) 31(10):2181–90. doi: 10.1038/leu.2017.41
69. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res (2011) 71(4):1374–84. doi: 10.1158/0008-5472.CAN-10-2238
70. Dave DS, Leppert JT, Rajfer J. Is the testis a chemo-privileged site? is there a blood-testis barrier? Rev Urol (2007) 9(1):28.
71. Akers SM, Rellick SL, Fortney JE, Gibson LF. Cellular elements of the subarachnoid space promote ALL survival during chemotherapy. Leuk Res (2011) 35(6):705–11. doi: 10.1016/j.leukres.2010.12.031
72. Frishman-Levy L, Shemesh A, Bar-Sinai A, Ma C, Ni Z, Frenkel S, et al. Central nervous system acute lymphoblastic leukemia: role of natural killer cells. Blood. (2015) 125(22):3420–31. doi: 10.1182/blood-2014-08-595108
73. Juarez J, Dela Pena A, Baraz R, Hewson J, Khoo M, Cisterne A, et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leuk (2007) 21(6):1249–57. doi: 10.1038/sj.leu.2404684
74. Kato I, Niwa A, Heike T, Fujino H, Saito MK, Umeda K, et al. Identification of hepatic niche harboring human acute lymphoblastic leukemic cells via the SDF-1/CXCR4 axis. PloS One (2011) 6(11):e27042. doi: 10.1371/journal.pone.0027042
75. Kato I, Nishinaka Y, Nakamura M, Akarca AU, Niwa A, Ozawa H, et al. Hypoxic adaptation of leukemic cells infiltrating the CNS affords a therapeutic strategy targeting VEGFA. Blood. (2017) 129(23):3126–9. doi: 10.1182/blood-2016-06-721712
76. Kallmann BA, Wagner S, Hummel V, Buttmann M, Bayas A, Tonn JC, et al. Characteristic gene expression profile of primary human cerebral endothelial cells. FASEB J (2002) 16(6):589–91. doi: 10.1096/fj.01-0594fje
77. Williams MTS, Yousafzai YM, Elder A, Rehe K, Bomken S, Frishman-Levy L, et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood. (2016) 127(16):1998–2006. doi: 10.1182/blood-2015-08-665034
78. Lenk L, Carlet M, Vogiatzi F, Spory L, Winterberg D, Cousins A, et al. CD79a promotes CNS-infiltration and leukemia engraftment in pediatric b-cell precursor acute lymphoblastic leukemia. Commun Biol (2021) 4(1):73. doi: 10.1038/s42003-020-01591-z
79. Alsadeq A, Lenk L, Vadakumchery A, Cousins A, Vokuhl C, Khadour A, et al. IL7R is associated with CNS infiltration and relapse in pediatric b-cell precursor acute lymphoblastic leukemia. Blood. (2018) 132(15):1614. doi: 10.1182/blood-2018-04-844209
80. Kinjyo I, Bragin D, Grattan R, Winter SS, Wilson BS. Leukemia-derived exosomes and cytokines pave the way for entry into the brain. J Leukoc Biol (2019) 105(4):741–53. doi: 10.1002/JLB.3A0218-054R
81. Cousins A, Olivares O, Markert E, Manoharan A, Bubnova X, Bresolin S, et al. Central nervous system involvement in childhood acute lymphoblastic leukemia is linked to upregulation of cholesterol biosynthetic pathways. Leukemia. (2022) 36(12):2903–7. doi: 10.1038/s41375-022-01722-x
82. Savino AM, Fernandes SI, Olivares O, Zemlyansky A, Cousins A, Markert EK, et al. Metabolic adaptation of acute lymphoblastic leukemia to the central nervous system microenvironment is dependent on stearoyl CoA desaturase. Nat cancer (2020) 1(10):998–1009. doi: 10.1038/s43018-020-00115-2
83. Turati VA, Guerra-Assunção JA, Potter NE, Gupta R, Ecker S, Daneviciute A, et al. Chemotherapy induces canalization of cell state in childhood b-cell precursor acute lymphoblastic leukemia. Nat Cancer (2021) 2(8):835–52. doi: 10.1038/s43018-021-00219-3
84. Loukas I, Simeoni F, Milan M, Inglese P, Patel H, Goldstone R, et al. Selective advantage of epigenetically disrupted cancer cells via phenotypic inertia. Cancer Cell (2023) 41(1):70–87.e14. doi: 10.1016/j.ccell.2022.10.002
85. Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet (2013) 45:242–52. doi: 10.1038/ng.2532
86. Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leuk (2012) 26(8):1797–803. doi: 10.1038/leu.2012.60
87. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. (2014) 33(17):2169–78. doi: 10.1038/onc.2013.169
88. Dixon ZA, Nicholson L, Zeppetzauer M, Matheson E, Sinclair P, Harrison CJ, et al. CREBBP knockdown enhances RAS/RAF/MEK/ERK signaling in ras pathway mutated acute lymphoblastic leukemia but does not modulate chemotherapeutic response. Haematologica. (2017) 102(4):736–45. doi: 10.3324/haematol.2016.145177
89. Pal D, Blair H, Parker J, Hockney S, Beckett M, Singh M, et al. hiPSC-derived bone marrow milieu identifies a clinically actionable driver of niche-mediated treatment resistance in leukemia. Cell Rep Med (2022) 3(8):100717. doi: 10.1016/j.xcrm.2022.100717
90. Shen ZH, Zeng DF, Wang XY, Ma YY, Zhang X, Kong PY. Targeting of the leukemia microenvironment by c(RGDfV) overcomes the resistance to chemotherapy in acute myeloid leukemia in biomimetic polystyrene scaffolds. Oncol Lett (2016) 12(5):3278–84. doi: 10.3892/ol.2016.5042
Keywords: microenvironment, tumor evolution, chemoresistance, epigenetics, B- cell precursor acute lymphoblastic leukemia (BCP-ALL)
Citation: Garcia-Gimenez A and Richardson SE (2023) The role of microenvironment in the initiation and evolution of B-cell precursor acute lymphoblastic leukemia. Front. Oncol. 13:1150612. doi: 10.3389/fonc.2023.1150612
Received: 24 January 2023; Accepted: 21 February 2023;
Published: 07 March 2023.
Edited by:
Silvia Deaglio, University of Turin, ItalyReviewed by:
Anthony Ford, Institute of Cancer Research (ICR), United KingdomChristina Halsey, University of Glasgow, United Kingdom
Copyright © 2023 Garcia-Gimenez and Richardson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon E. Richardson, c2VyMzJAY2FtLmFjLnVr