 Hongfeng Zheng
Hongfeng Zheng Jiangui Liu
Jiangui Liu Xiuwu Pan
Xiuwu Pan Xingang Cui
Xingang Cui- Department of Urology, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
Wilms tumor, originating from aberrant fetal nephrogenesis, is the most common renal malignancy in childhood. The overall survival of children is approximately 90%. Although existing risk-stratification systems are helpful in identifying patients with poor prognosis, the recurrence rate of Wilms tumors remains as high as 15%. To resolve this clinical problem, diverse studies on the occurrence and progression of the disease have been conducted, and the results are encouraging. A series of molecular biomarkers have been identified with further studies on the mechanism of tumorigenesis. Some of these show prognostic value and have been introduced into clinical practice. Identification of these biomarkers can supplement the existing risk-stratification systems. In the future, more biomarkers will be discovered, and more studies are required to validate their roles in improving the detection rate of occurrence or recurrence of Wilms tumor and to enhance clinical outcomes.
1 Introduction of Wilms tumor
Wilms tumor (WT), also known as nephroblastoma, is the most common renal malignancy in childhood, accounting for approximately 90% of all renal tumors in children. Approximately 95% of patients with WT are under 10 years of age (1). Current standardized diagnostic and therapeutic procedures have made it possible to cure nearly 90% of children with WT. According to the International Society of Pediatric Oncology (SIOP) report (2), the two-year event-free survival (EFS) and overall survival (OS) were 87% and 93%, respectively in children with WT who received the SIOP-2001 protocol with preoperative chemotherapy, while the Children’s Oncology Group (COG) trials report (3) similar results, in which patients received direct operation.
However, postoperative recurrence and high-risk tumors remain formidable clinical challenges. Recurrence rate in WT is approximately 15% of children and is positively correlated with histological risk (2, 4, 5). The anaplastic subtype is the most common histological type in Wilms tumor, which is associated with poorer outcomes (6, 7). Risk stratification systems have been developed to assess clinical outcomes by stratifying tumors at different risk levels. Both the SIOP and COG protocols recognize tumor stage, histology, and volume as prognostic factors to divide patients into subgroups and formulate postoperative therapeutic strategies. In addition, the significance of genetic aberrations is underlined by COG, knowing that a gain of 1q leads to a high risk of relapse and death. With the identification of more WT-associated genes and proteins (4), the relationship between these biomarkers and clinical outcomes has also been gradually disclosed.
In this review article, we begin with the relationship between nephrogenesis and tumorigenesis. We focused on some WT-related genetic abnormalities, briefly overview their pathophysiological mechanism in tumorigenesis, and identify their potential clinical value in WT (Table 1). We then discuss copy number variations mentioned in the COG stratification system, which are regarded as prognostic factors for assessing tumor recurrence and extra mortality in a particular cohort. We will also introduce some lncRNA-related studies on WT. Finally, as liquid biopsy is a hot topic in cancer research, we summarized relevant studies and discussed how liquid biopsy was applied to improve WT diagnosis. Although many of these biomarkers are limited by additional factors such as tumor histology, tumor stage, and therapeutic regimens, they have potential value in the diagnosis, prognostic prediction, and therapeutic assessment of patients with WT.
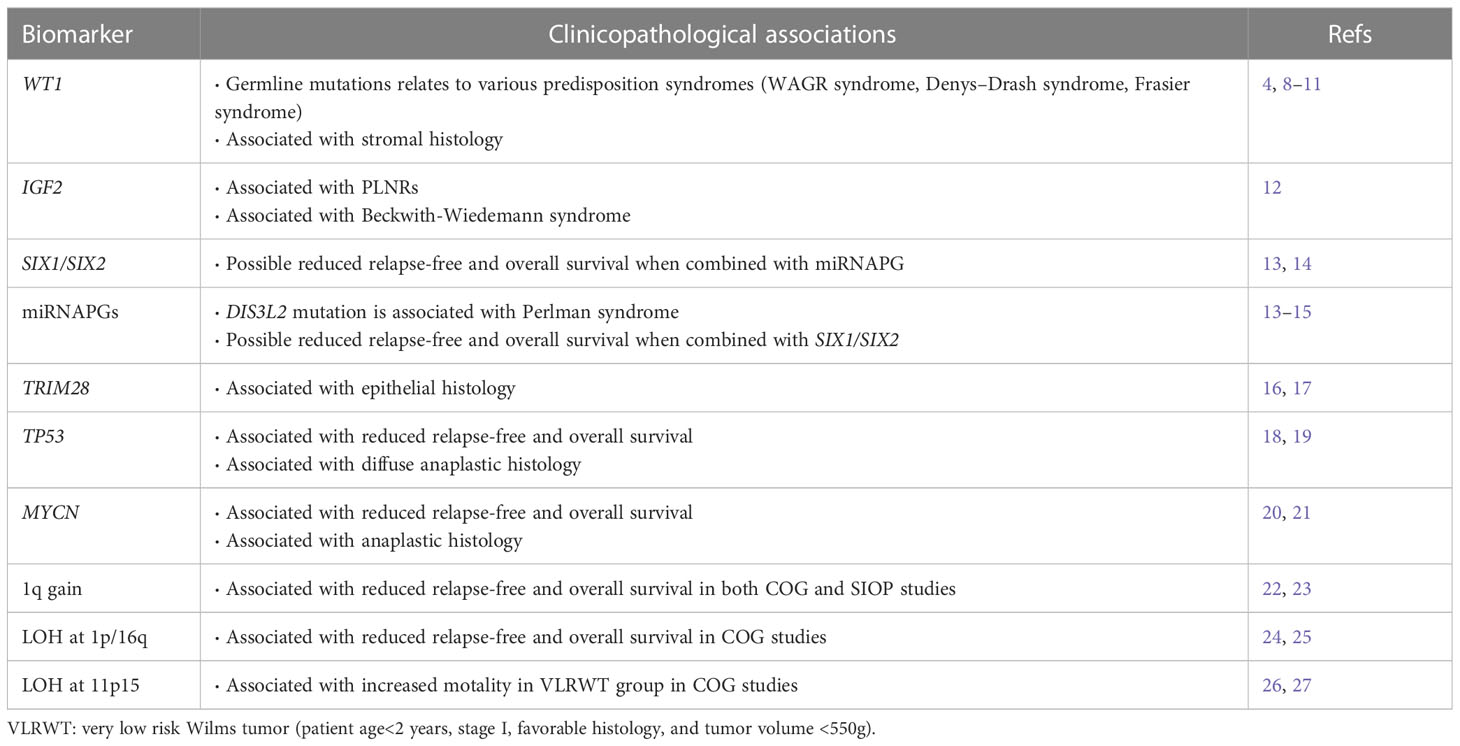
Table 1 Genetic abnormorlities in Wilms tumor.
2 Fetal nephrogenesis and Wilms tumorigenesis
Accumulating evidence suggests that Wilms tumor originates from aberrant fetal renal development, which evolves into the definitive human kidney and originates from the ureteric bud and metanephrogenic tissue during the fifth week of embryonic development (28). The ureteric bud sprouts from the mesonephric duct branch and invades the metanephric mesenchyme. Under ureteric bud induction, mesenchymal cells condense and undergo mesenchymal-to-epithelial transition (MET), leading to renal vesicles. The ureteric bud and its branches eventually form the collecting duct system, while renal vesicle polarization and elongation form the proximal and distal tubules and loops of Henle. In this process, a complex network of genes controls the balance between self-renewal and differentiation (Figure 1).
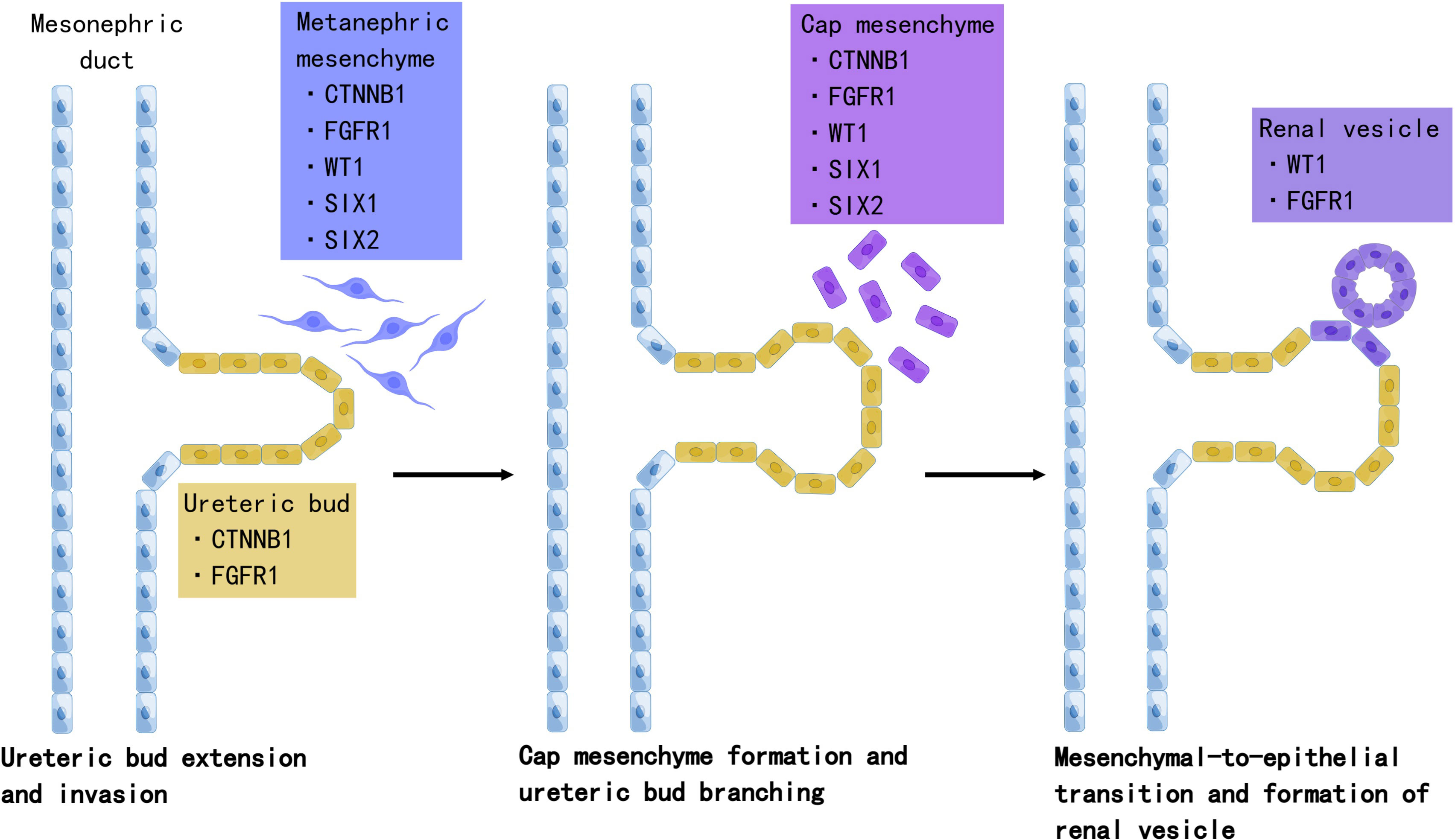
Figure 1 The role of Wilms tumor genes in nephrogenesis. Development of the definitive kidney starts around the fifth week of gestation. Several genes are involved in this process. WT1 is a key regulator of the entire process, including the development of the metanephric mesenchyme to the cap mesenchyme and renal vesicles. SIX2 maintains the population of mesenchymal progenitors in an undifferentiated state. Together, WT1, SIX2, and CTNNB1 function to facilitate the FGFR pathway. FGFR1 plays an important role in nephron progenitor cell survival, branching of ureteric buds, and elongation of primitive renal vesicles into comma- and S-shaped bodies that eventually form mature nephrons. Mutations in these genes have been associated with Wilms tumorigenesis. Created by Figdraw.
In WT mice, the process of nephrogenesis can be disrupted at different levels, leading to incomplete differentiation arrest of renal progenitor cells. Thus, WTs are often called the tri-phasic type, because they comprise blastemal, stromal, and epithelial cells, which correlate with cap mesenchyme, uninduced metanephric mesenchyme, and renal epithelial cells, respectively (29). Single-cell transcriptomes in 2018 revealed the relationship between WTs and fetal developing nephron populations, supporting the hypothesis that Wilms tumor is closely linked to stalled renal organogenesis (30). We selected WT genes and discussed their relationship with nephrogenesis and tumorigenesis.
3 Genetic abnormalities in WT
3.1 Wilms tumor gene 1
WT1 was the first gene implicated in Wilms tumorigenesis (31). WT1 encodes an important transcription factor that regulates over 100 genes and is involved in all stages of fetal kidney development (32, 33). In homozygous WT1 knock-out mice, the development of the metanephric kidney failed (34). Germline WT1 abnormalities contribute to several WT-associated predisposition syndromes. One of the most common syndromes is WAGR syndrome, which is characterized by Wilms tumor, aniridia, genitourinary anomalies, and a range of developmental delays (WAGR). WAGR is caused by microdeletions at 11p13, including WT1 deletion and adjacent PAX6. Denys–Drash Syndrome (DDS) underlay byWT1 missense mutation is characterized by ambiguous genitalia and nephropathy secondary to diffuse mesangial sclerosis (8, 9). Moreover, mutations alter the balance of WT1 splice isoforms, resulting in Frasier Syndrome, which carries the risk of gonadoblastoma and focal glomerulosclerosis (10). The relationship between predisposing genetic conditions and tumor relapse has been reported in previous studies. Both COG and SIOP studies (11, 35) reported that a higher relapse rate was not observed in patients with WAGR than in patients with non-syndromic WT patients, excluding the effect of metachronous tumors. Besides, somatic WT1 mutations were found in 10%–20% WT patients, without showing independent prognostic value (4, 36).
3.2 Insulin-like growth factor 2
Abnormal methylation at 11p15 is the most common genomic change found in the WT, and the IGF2/H19 domain was detected in this chromosomal region (37). IGF2 encodes an embryonal growth factor and is regulated by a non-coding RNA transcribed by H19. IGF pathway is overactivated by the biallelic expression of IGF2, which results from H19 hypermethylation and subsequent loss of imprinting of IGF2 (38, 39). During nephrogenesis, perilobar nephrogenic rests (PLNR) are associated with biallelic expression of IGF2, which is considered an early event in tumorigenesis (12). Multiple germline changes at 11p15, including epimutation of H19 or loss of heterozygosity at IGF2, are responsible for Beckwith–Wiedemann syndrome, which is susceptible to embryonal tumors, including WT (40). Coorens et al. (41) observed that hypermethylation of H19 with subsequent overexpression of IGF2 was directly associated with clonal nephrogenesis and the development of Wilms tumor in a cohort of 23 patients with WT. Although the prognostic value of IGF2/H19 was not explored, the authors suggested that the relationship between clonal nephrogenesis and formation of WT should be emphasized, which could be utilized to guide the surveillance schedule of patients with WT.
3.3 SIX1/SIX2
Several studies (13, 14) have identified SIX1 and SIX2 as WT-specific oncogenes, both of which are associated with the blastemal subtype, another high-risk histology in the SIOP protocols. SIX1 and SIX2 are key regulators of nephrogenesis. Expression of cell cycle genes was found to be upregulated in SIX1- and SIX2-mutant WT mice, and loss of SIX1 resulted in mesenchymal apoptosis in SIX1-knockout mice, while SIX2 activity maintained the number of nephrogenic progenitors in undifferentiated blastemal tissues (42, 43). In addition, SIX2 overactivation in a renal cell line increased the percentage of cells in the S-phase (13, 14). Walz et al. reported that patients with combined SIX1/SIX2 and microRNA processing genes (miRNAPGs) mutations had a significant higher relapse rate (80%, p = 0.001)and a higher mortality (40%), though the SIX1/SIX2 and miRNAPGs variants alone did not show bad outcomes (14).
3.4 microRNA processing genes and microRNA
Whole-genome and whole-exosome sequencing of WT have been used to identify unique mutations in microRNA processing genes (miRNAPGs), including DROSHA, DICER1, DGCR8, XPO5, and TARBP2 (13–15), which lead to impaired miRNA biogenesis (Figure 2). Approximately 33% of the WTs examined carried mutations in miRNAPGs (44). Combined mutations in both SIX1/SIX2 and miRNAPG resulted in poorer outcomes in a COG study (14). As microRNAs (miRNAs) are critical regulators of kidney morphogenesis by modulating diverse biological processes in different renal cell lineages, mutations in miRNAPGs lead to the downregulation of important microRNAs (miRNAs). Global downregulation of mature let7 family miRNAs occurs in DROSHA mutants, resulting in failure of epithelial differentiation (15, 44). The RNA-binding protein Lin28 suppresses the processing of let7 miRNA, and the balance between them controls the timing of nephrogenesis in mice (45). Overexpression of LIN28 inhibits the differentiation of nephrogenic progenitors, thus causing neoplastic transformation, which is similar to the situation in human WT (46). Copy number gain of LIN28B and loss of let7 were observed in 25% and 46% of the human WT, respectively (20). DIS3L2, which encodes an exoribonuclease responsible for degrading preprocessed forms of let7, was found to be mutated in Perlman syndrome, which is characterized by macrosomia, polyhydramnios, facial dysmorphology, renal dysplasia, and predisposition to WT (47). The miR-200 family, which is key to the mesenchymal-to-epithelial transition, was also found to be downregulated as a result of miRNAPG mutations and is associated with an undifferentiated blastemal histology (14). A review by Cerqueira et al. (48) summarized multiple studies and found that aberrant expression of specific miRNAs was correlated with the etiology of WT. These miRNAs not only function as oncogenes but also as tumor suppressors in WT development.
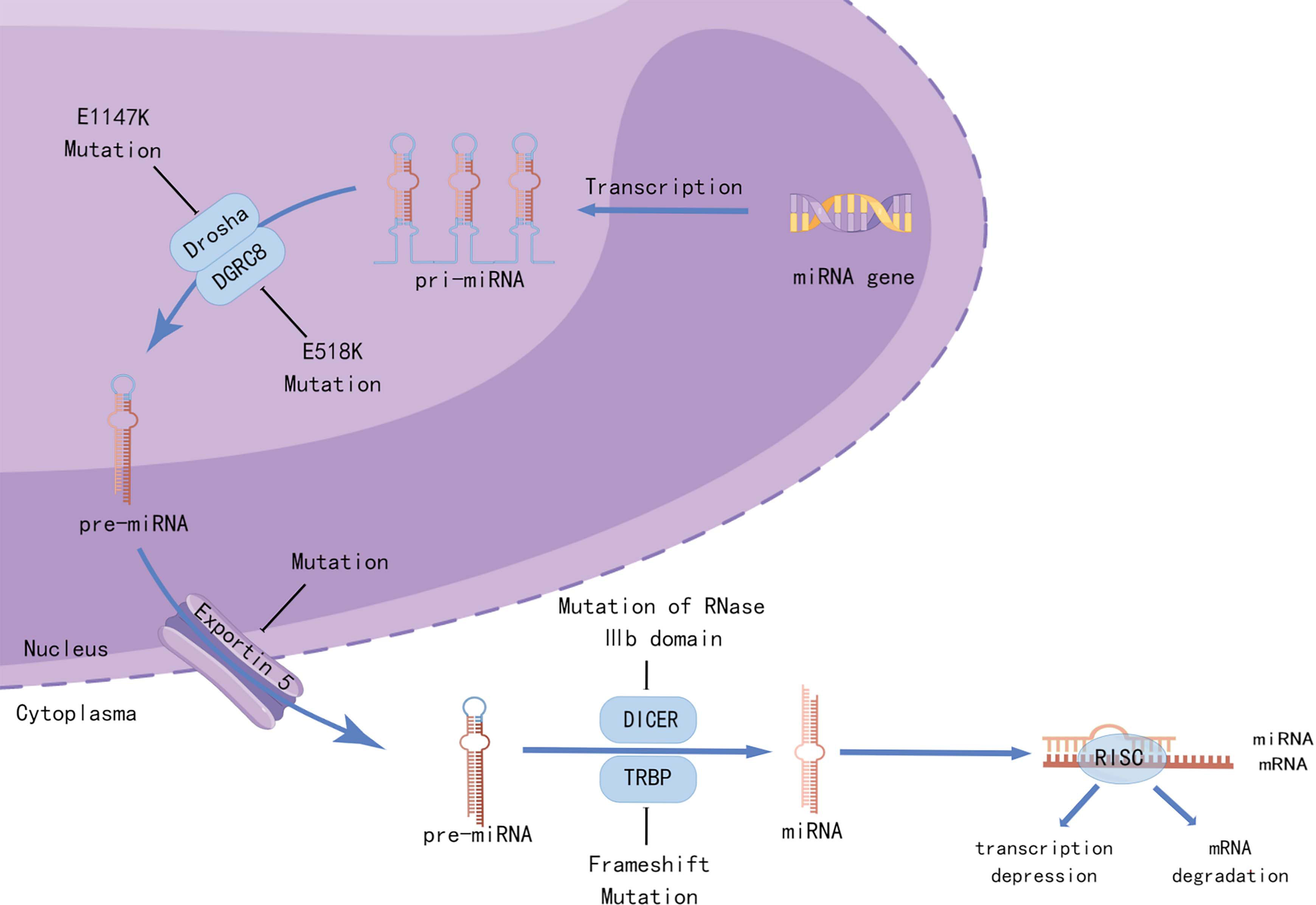
Figure 2 Mutations in miRNA-processing genes lead to aberrant miRNA biogenesis. Recurrent mutations in the metal-binding (Mg2+) residue of the RNase IIIb domain of DROSHA (E1147K) or the doublestranded RNA-binding domain of DGRC8 (E518K) disrupted the cleavage of pri-miRNAs into pre-miRNAs. Mutations in XPO5 (encoding exportin 5) prevent pre-miRNA export, resulting in premiRNA accumulation in the nucleus. Frameshift mutations in TARBP2 (encoding TRBP) and those affecting the RNase IIIb domain of DICER1 can disrupt the processing of pre-miRNAs into mature miRNAs. Created by Figdraw.
Notably, the expression levels of some miRNAs were associated with clinical outcomes. One study (49) reported the upregulation of 14 miRNAs in the serum of patients with WT. They found that the expression levels of miR-110-5p and miR-130-3p could be used to differentiate WT children from healthy children. Apart from their potential predictive value, there are several additional reasons to support miRNAs as detectable biomarkers. MicroRNAs are widely distributed in various organisms. Apart from their intracellular location, their distribution in body fluids makes it non-invasive to capture sufficient samples (50). In addition, circulating miRNAs are conjugated to other macromolecules, thus facilitating their stable storage (51, 52). However, hurdles also exist and should be overcome using standardized methodologies for the purification and analysis of samples. In addition, studies with large sample sizes are required. In conclusion, miRNAs have great potential as biomarkers because of their unique biological features and potential clinicopathological value.
3.5 TRIM28
TRIM28, a classic WT tumor suppressor gene, is predisposed to familial or non-familial WT with germline mutations (16, 17). WT with TRIM28 mutations is associated with epithelial histology, which shows a better prognosis. Hol et al. (16) reviewed all previously reported cases, and follow-up data were available for 13 patients with germline pathogenic variants in TRIM28 and found that no relapse occurred in any of these patients. Although the epithelial histological type has been reported to be associated with good outcomes (53), whether the prognostic value of TRIM28 mutations is independent of epithelial histology remains to be validated. As TRIM28 germline mutation can be simply detected by immunohistochemistry using anti-KAP1 antibody in WT patients (17), it can be used to recognize other young family members predisposed to tumors.
3.6 TP53
Somatic mutations in TP53 are one of the most frequent alternations in human cancers, and germline mutations are the underlying cause of Li–Fraumeni syndrome, which predisposes to a range of cancers (54). In patients with WTs, TP53 mutations is frequently detected in the anaplastic subtype, especially in diffuse anaplastic Wilms tumor (DAWT) (7, 55). Ooms et al. (18) reported TP53 mutations in 57 (48%) of 118 DAWT cases, 13 (11%) cases of copy loss without mutation, and 48 (41%) cases lacking both. In contrast to those with TP53 abnormalities, DAWTs with TP53-wide-type indicate lower relapse and death rates in stage III/IV patients. As diffuse anaplasia correlates with poor outcomes, TP53 status further improves risk stratification in DAWT, meaning that patients with TP53 mutations should receive more intensive treatment (19). In view of the correlation between TP53 mutations and DAWT, early identification of this high-risk histological subtype could be done by detecting TP53 mutations in circulating tumor DNA to determine whether intensive preoperative chemotherapy should be provided (56).
TP53 mutations are not limited to anaplasia. In blastemal and some intermediate-risk histology subtypes, TP53 mutations were also observed to be correlated with a high risk of death (13). Wegert et al. (55) suggested that TP53 might play a driving role in the histological progression of WTs, as partial features of anaplasia were found in some blastemal tumors. TP53-screening should be launched at an early stage, not only to identify anaplasia before surgery, but also to access tumor progression. As intratumoral heterogeneity may cause trouble, multiple sampling is needed by applying liquid biopsies to capture adequate tumor circulating DNA and harbored TP53 mutations. We further discuss circulating tumor DNA in Section 5.
3.7 MYCN
Mutations in MYCN have also been associated with high-risk anaplastic histology. Williams et al. (21) reported that 30.4% (7/23) samples had MYCN gain in the diffuse anaplastic subtype compared to 11.2% (30/269) in other subtypes, indicating a significant association (p = 0.0159). In this study, MYCN gain was found to be correlated with poorer relapse-free survival and OS in cases of all histology and in cases with diffuse anaplasia. Interestingly, MYCN mutations are three times less frequent in DAWTs in the COG cohort (20). Although this skewing did not reach statistical significance, it agrees with the conclusion of most recent studies that MYCN mutations have prognostic value, whether anaplastic or not.
4 Long noncoding RNAs in WT
Long noncoding RNAs (lncRNAs) are a large group of nonprotein-coding RNAs consisting of more than 200 nucleotides. lncRNAs are involved in many biological processes, including gene silencing, gene imprinting, RNA interference, and protein translation and modification (57–59). Disruption of lncRNA expression is intrinsically linked to a variety of diseases, including cancer (60). The role of lncRNAs in WT has not been fully elucidated, although relatively few studies have been conducted in the recent years. For example, WT1, the most prominent WT relative gene, is directly or indirectly regulated by lncRNAs. WT1 antisense RNA (WT1-AS), originating from the intron region of WT1, can bind to WT1 mRNA and regulate WT1 protein expression by RNA–RNA interactions (61). Recent studies have demonstrated that WT1-AS plays a significant role in many tumors; however, its roles vary among different tumors. Dallosso et al. found high expression levels of WT1-AS in WT (62); however, its relationship with clinical outcomes and prognosis has not been clarified. However, the specific mechanisms of action need to be elucidated.
According to the competing endogenous RNA (ceRNA) theory, lncRNAs regulate the expression of target genes by adsorbing miRNAs (63). To further explore the role of lncRNAs in tumorigenesis, several studies have established ceRNA networks to identify the potential lncRNAs as much as possible involved in WT. Wang et al. (64) constructed a lncRNA–miRNA–mRNA ceRNA network consisting of 32 lncRNAs, 14 miRNAs, and 158 mRNAs. Subsequently, three lncRNAs, three miRNAs, and 17 mRNAs were found to be associated with OS. Of the three lncRNAs, MYCN opposite strand (MYCNOS), deleted in lymphocytic 2 (DLEU2), was highly expressed in the late stages of WT and correlated with poorer OS, whereas upregulation of chromosome 8 open reading frame 31 (C9orf31) in the early stage may play a protective role. Similar results regarding MYCNOS and DEUL2 have also been reported in other studies on neuroblastoma, laryngeal carcinoma, and leukemia (65–67). In addition to the prognostic correlation, some studies have established predictive survival models. Liu et al. (68) constructed three models based on survival-associated RNAs (lncRNAs, miRNAs, and mRNAs) from primary solid WT tissue and AUC values of these models were all greater than 0.7, denoting excellent model performance. Although significant results have been obtained, more applicable predictive models must be built based on multicenter data and various pathological tissues.
5 Copy number variations in stratification system
Both the SIOP and COG use stages and histological subtypes were used to stratify risks in postoperative patients. Since 2005, COG has included a molecular marker in risk stratification, recommending that children whose tumors have loss of heterozygosity (LOH) for alleles spanning chromosomes 1p and 16q should receive more intensive chemotherapy (26, 69). In addition, 1q gain and LOH at 11p15 showed clinical value in a particular subgroup of patients. Although the precise mechanism of oncogenesis in tumors with these copy number variations remains unclear, their association with relapse and death is important in clinical practice.
5.1 1q gain
The gain of chromosome arm 1q is a significant factor associated with poorer clinical outcomes in terms of reduced OS and shorter EFS in both COG and SIOP-treated patients (22, 23, 70, 71). In COG studies, gain of 1q was detected in 27% of patients with favorable histology WT (FHWT) and showed significance in OS and EFS as a marker independent of tumor stage (22, 23). The COG is planning to incorporate it into risk stratification in the next series of studies. In addition, the SIOP study has recognized 1q gain as a potential prognostic biomarker in WT, and they aimed to further validate its role in the stratification of patients who have received preoperative chemotherapy (70).
5.2 LOH at 1p and 16q
According to a previous National Wilms Tumor Study Group (NWTSG) study (69), LOH at 1p only (LOH 1p), 16q only (LOH 16q), and combined 1p and 16q (LOH 1p/16q) was associated with an adverse outcomes in patients with stage I/II favorable-histology WT treated with immediate nephrectomy. In patients with stage III/IV disease, only LOH 1p/16q is associated with an increased risk of relapse and death. Another study (24) reported that in patients with non-anaplastic WT, only LOH 1p had prognostic value, while LOH 16q and LOH 1p/16q did not. Messahel et al. (25) found that LOH 16q and LOH 1p/16q were related to increased risk of relapse and death in patients with favorable histology tumor, whether the patients had received initial therapies or not. LOH 1p and/or LOH 16q appeared to have an independent prognostic effect in the 1q-gain-negative group when patients with or without 1q gain were analyzed separately (23). In summary, LOH 1p, LOH 16q, and LOH 1p/16q have limited but not completely independent prognostic values and are applied in a particular subgroup of patients in COG studies.
In the SIOP study, neither LOH 1p nor LOH 16q, nor LOH 1p/16q can be considered as a single biomarker related to poorer EFS or OS at p = 0.05, whether in the univariate or multivariate analysis (70), which conflicts with the COG observations. The prognostic value of LOH 1p and/or 16q should be further validated in SIOP patients (72).
5.3 LOH at 11p15
COG stratification defines a group of patients as a very low-risk subgroup (younger than 2 years, stage I, favorable histology, and tumor volume <550 g), who are at low risk of relapse and only need to undergo direct surgery without adjuvant chemotherapy (26). However, if LOH at 11p15 exists, operation-only treatment is not effective and chemotherapy is indispensable because LOH at 11p15 is associated with a higher rate of relapse (26, 27).
6 Liquid biopsy
Tumor biopsies at diagnosis, resection, or relapse are the gold standards for identifying tumor biology, diagnosis, and therapeutic decision-making. However, the shortcomings are also obvious, such as unavoidable trauma caused by puncture or surgery and over-dependence on imaging examination. Solid pediatric tumors are more likely to shed tumor cells, DNA, RNA, or proteins into the blood or urine. Since blood or urine samples are easily available at any time, identification of these tumor markers in body fluids, also known as liquid biopsy, is a better and potential measure to manage tumor patients. Liquid biopsy has unique advantages in that it can screen primary lesions, monitor recurrence, and assess the treatment effect in patients with WT by identifying tumor markers in a real-time manner (73–76).
A signature of 176 circulating miRNAs was diagnostic of WT and could distinguish healthy children (77). TP53 mutations in circulating tumor DNA (ctDNA) detected by liquid biopsy can help to identify DAWT at an early stage, one of the most invasive subtypes of WT (56). A COG trial (74) reported the detection of ctDNA in the serum of 41/50 (82%) and urine of 13/50 (26%) patients with stage III/IV disease, and the agreement between serum ctDNA and tumor sequencing results was highly significant. Detectable ctDNAs include CNVs (1q gain, LOH at 1p and/or 16q) and single-nucleotide variants (WT1, CTNNB1, MYCN, and TP53). OS and EFS in patients with detectable ctDNA in serum were poorer than those in patients without (positive vs. negative group: 82.79% vs. 100% for OS, 80.41% vs. 100% for 4 years-EFS), whereas the discrimination effect of urine ctDNA was not significant between the two groups (positive group vs. negative group: 76.92% vs. 91.43% for OS, 76.92% vs. 88.57% for 4 years-EFS) (74). Moreover, circulating miRNA detection can be used to differentiate WT from other pediatric tumors (78). In other cancers, ctDNA has been shown to capture the presence of subclonal heterogeneity better than solitary biopsies (79–81), which provides a reference for WT management.
In addition to nucleic acid detection, protein biomarkers can be profiled using high-resolution mass spectrometry (HRMS) proteomics of urinary specimens. Previous studies (82–84) reported that neuron-specific enolase, basic fibroblast growth factor (bFGF), and hyaluronidase have been reported to be enriched in the urine of patients with Wilms tumor. In addition, bFGF overexpression in urine is related to the WT stage and has prognostic value (83). Ortiz et al. (85) reported that prohibitin in FHWTs acted as a prognostic marker in tumor relapse and a cutoff threshold of 998 ng/ml was a predictor of recurrence, especially recurrence in the abdomen (AUC: 0.78 for all recurrence, 0.96 for abdominal recurrence). DACT2 and DAD1 proteins were only mentioned briefly in their study and no further validated. The review by Coppes et al. (86) mentioned “paraneoplastic syndromes” in WT and several associated factors, including neuron-specific enolase (NSE), hyaluronic acid (HA), hyaluronic acid-stimulating activity (HSA), and hyaluronidase, all of which may predict recurrence or evaluate the therapeutic effect. SIOP aims to establish biobanks by collecting serial blood and urine samples, as well as tumor and germline material at diagnosis and specific time points during treatment for international collaborative studies (87). Similarly, the COG study also utilized liquid biopsy to test the potential benefits of diagnostics, monitoring of therapy, and detection of residual disease (88).
7 Conclusion
Remarkable progress has been made in the early detection and management of cancer progression and recurrence owing to advances in risk stratification systems, treatment, and follow-up protocols. As tumor stage and histological subtype have clearly shown relevant prognostic value, the introduction of Wilms tumor biomarkers has further completed the risk stratification systems, the targeting capability of the treatment measures, and follow-up plans. Among various biomarkers, copy number variations, such as 1p/16q LOH have displayed significant prognostic value and have been successfully applied in COG protocols. TP53 and MYCN mutations have confirmed clinicopathological associations, showing promising application potential. Others, especially miRNAs and proteins, also exhibited their potential as novel tumor biomarkers in the future due to their close association with tumorigenesis. In addition to prognostic value, alterations in some biomarkers are early events in WT tumorigenesis, showing promising perspectives in predicting tumorigenesis before routine laboratory tests and imaging examinations. Because blood and urine samples are easily available, all biomarkers can be monitored dynamically. These measures will greatly improve the primary or secondary tumor screening rate and shorten the window period.
With further research on the mechanism of tumor occurrence and progression, the future objectives of research should focus on saving patients with relapsed and refractory Wilms tumor, while, on the other hand, identifying children with excellent prognosis to release their therapeutic burden. Future studies should continue to discover more biomarkers, clarify their underlying biological mechanisms, and define their predictive and prognostic value for the benefit of WT patients.
Author contributions
All authors had full access to all the data in the study and take responsibility for the integrity of the literature. All authors were involved in critical revision of the manuscript for important intellectual content. All authors have read and agreed to the published version of the manuscript.
Funding
This work was sponsored by the National Natural Science Foundation of China (No. 81974391, 82072806, 82173265); Leading health talents of Shanghai Municipal Health Commission (2022LJ002); Shanghai Rising-Star Program (23QC1401400); the Natural Science Foundation of Shanghai (23ZR1441300).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Spreafico F, Fernandez CV, Brok J, Nakata K, Vujanic G, Geller JI, et al. Wilms tumour. Nat Rev Dis Primer (2021) 7(1):75. doi: 10.1038/s41572-021-00308-8
2. Brok J, Treger TD, Gooskens SL, van den Heuvel-Eibrink MM, Pritchard-Jones K. Biology and treatment of renal tumours in childhood. Eur J Cancer Oxf Engl 1990 (2016) 68:179–95. doi: 10.1016/j.ejca.2016.09.005
3. Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, et al. Advances in wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33(27):2999–3007. doi: 10.1200/JCO.2015.62.1888
4. Treger TD, Chowdhury T, Pritchard-Jones K, Behjati S. The genetic changes of wilms tumour. Nat Rev Nephrol (2019) 15(4):240–51. doi: 10.1038/s41581-019-0112-0
5. Brok J, Lopez-Yurda M, Tinteren HV, Treger TD, Furtwängler R, Graf N, et al. Relapse of wilms’ tumour and detection methods: a retrospective analysis of the 2001 renal tumour study group-international society of paediatric oncology wilms’ tumour protocol database. Lancet Oncol (2018) 19(8):1072–81. doi: 10.1016/S1470-2045(18)30293-6
6. Bonadio JF, Storer B, Norkool P, Farewell VT, Beckwith JB, D’Angio GJ. Anaplastic wilms’ tumor: clinical and pathologic studies. J Clin Oncol Off J Am Soc Clin Oncol (1985) 3(4):513–20. doi: 10.1200/JCO.1985.3.4.513
7. Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B, Adam M, et al. Anaplastic wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet (1994) 7(1):91–7. doi: 10.1038/ng0594-91
8. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, et al. Germline mutations in the wilms’ tumor suppressor gene are associated with abnormal urogenital development in denys-drash syndrome. Cell (1991) 67(2):437–47. doi: 10.1016/0092-8674(91)90194-4
10. Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for frasier syndrome. Nat Genet (1997) 17(4):467–70. doi: 10.1038/ng1297-467
11. Hol JA, Jongmans MCJ, Sudour-Bonnange H, Ramírez-Villar GL, Chowdhury T, Rechnitzer C, et al. Clinical characteristics and outcomes of children with WAGR syndrome and wilms tumor and/or nephroblastomatosis: the 30-year SIOP-RTSG experience. Cancer (2021) 127(4):628–38. doi: 10.1002/cncr.33304
12. Satoh Y, Nakadate H, Nakagawachi T, Higashimoto K, Joh K, Masaki Z, et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic wilms’ tumours. Br J Cancer (2006) 95(4):541–7. doi: 10.1038/sj.bjc.6603302
13. Wegert J, Ishaque N, Vardapour R, Geörg C, Gu Z, Bieg M, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type wilms tumors. Cancer Cell (2015) 27(2):298–311. doi: 10.1016/j.ccell.2015.01.002
14. Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology wilms tumors. Cancer Cell (2015) 27(2):286–97. doi: 10.1016/j.ccell.2015.01.003
15. Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in wilms tumours. Nat Commun (2014) 2:4802. doi: 10.1038/ncomms5802
16. Hol JA, Diets IJ, de Krijger RR, van den Heuvel-Eibrink MM, Jongmans MC, Kuiper RP. TRIM28 variants and wilms’ tumour predisposition. J Pathol (2021) 254(4):494–504. doi: 10.1002/path.5639
17. Diets IJ, Hoyer J, Ekici AB, Popp B, Hoogerbrugge N, van Reijmersdal SV, et al. TRIM28 haploinsufficiency predisposes to wilms tumor. Int J Cancer (2019) 145(4):941–51. doi: 10.1002/ijc.32167
18. Ooms AHAG, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, et al. Significance of TP53 mutation in wilms tumors with diffuse anaplasia: a report from the children’s oncology group. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22(22):5582–91. doi: 10.1158/1078-0432.CCR-16-0985
19. Maschietto M, Williams RD, Chagtai T, Popov SD, Sebire NJ, Vujanic G, et al. TP53 mutational status is a potential marker for risk stratification in wilms tumour with diffuse anaplasia. PloS One (2014) 9(10):e109924. doi: 10.1371/journal.pone.0109924
20. Gadd S, Huff V, Walz AL, Ooms AHAG, Armstrong AE, Gerhard DS, et al. A children’s oncology group and TARGET initiative exploring the genetic landscape of wilms tumor. Nat Genet (2017) 49(10):1487–94. doi: 10.1038/ng.3940
21. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, et al. Multiple mechanisms of MYCN dysregulation in wilms tumour. Oncotarget (2015) 6(9):7232–43. doi: 10.18632/oncotarget.3377
22. Gratias EJ, Jennings LJ, Anderson JR, Dome JS, Grundy P, Perlman EJ. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology wilms tumor: a report from the children’s oncology group. Cancer (2013) 119(21):3887–94. doi: 10.1002/cncr.28239
23. Gratias EJ, Dome JS, Jennings LJ, Chi YY, Tian J, Anderson J, et al. Association of chromosome 1q gain with inferior survival in favorable-histology wilms tumor: a report from the children’s oncology group. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(26):3189–94. doi: 10.1200/JCO.2015.66.1140
24. Spreafico F, Gamba B, Mariani L, Collini P, D’Angelo P, Pession A, et al. Loss of heterozygosity analysis at different chromosome regions in wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian association of pediatric hematology and oncology. J Urol (2013) 189(1):260–6. doi: 10.1016/j.juro.2012.09.009
25. Messahel B, Williams R, Ridolfi A, A’hern R, Warren W, Tinworth L, et al. Allele loss at 16q defines poorer prognosis wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a children’s cancer and leukaemia group (CCLG) study. Eur J Cancer Oxf Engl 1990 (2009) 45(5):819–26. doi: 10.1016/j.ejca.2009.01.005
26. Fernandez CV, Perlman EJ, Mullen EA, Chi YY, Hamilton TE, Gow KW, et al. Clinical outcome and biological predictors of relapse after nephrectomy only for very low-risk wilms tumor: a report from children’s oncology group AREN0532. Ann Surg (2017) 265(4):835–40. doi: 10.1097/SLA.0000000000001716
27. Perlman EJ, Grundy PE, Anderson JR, Jennings LJ, Green DM, Dome JS, et al. WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children’s oncology group study. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29(6):698–703. doi: 10.1200/JCO.2010.31.5192
28. Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nat Rev Genet (2002) 3(7):533–43. doi: 10.1038/nrg842
29. Trink A, Kanter I, Pode-Shakked N, Urbach A, Dekel B, Kalisky T. Geometry of gene expression space of wilms’ tumors from human patients. Neoplasia N Y N (2018) 20(8):871–81. doi: 10.1016/j.neo.2018.06.006
30. Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science (2018) 361(6402):594–9. doi: 10.1126/science.aat1699
31. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 wilms’ tumor locus. Cell (1990) 60(3):509–20. doi: 10.1016/0092-8674(90)90601-A
32. Rivera MN, Haber DA. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer (2005) 5(9):699–712. doi: 10.1038/nrc1696
33. Hartwig S, Ho J, Pandey P, Macisaac K, Taglienti M, Xiang M, et al. Genomic characterization of wilms’ tumor suppressor 1 targets in nephron progenitor cells during kidney development. Dev Camb Engl (2010) 137(7):1189–203. doi: 10.1242/dev.045732
34. Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene (2001) 273(2):141–61. doi: 10.1016/S0378-1119(01)00593-5
35. Breslow NE, Norris R, Norkool PA, Kang T, Beckwith JB, Perlman EJ, et al. Characteristics and outcomes of children with the wilms tumor-aniridia syndrome: a report from the national wilms tumor study group. J Clin Oncol Off J Am Soc Clin Oncol (2003) 21(24):4579–85. doi: 10.1200/JCO.2003.06.096
36. Gadd S, Huff V, Huang CC, Ruteshouser EC, Dome JS, Grundy PE, et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of wilms tumor: a children’s oncology group study. Neoplasia N Y N (2012) 14(8):742–56. doi: 10.1593/neo.12714
37. Karnik P, Chen P, Paris M, Yeger H, Williams BR. Loss of heterozygosity at chromosome 11p15 in wilms tumors: identification of two independent regions. Oncogene (1998) 17(2):237–40. doi: 10.1038/sj.onc.1201959
38. Okamoto K, Morison IM, Taniguchi T, Reeve AE. Epigenetic changes at the insulin-like growth factor II/H19 locus in developing kidney is an early event in wilms tumorigenesis. Proc Natl Acad Sci USA (1997) 94(10):5367–71. doi: 10.1073/pnas.94.10.5367
39. Charlton J, Williams RD, Sebire NJ, Popov S, Vujanic G, Chagtai T, et al. Comparative methylome analysis identifies new tumour subtypes and biomarkers for transformation of nephrogenic rests into wilms tumour. Genome Med (2015) 7(1):11. doi: 10.1186/s13073-015-0136-4
40. Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, et al. Molecular subtypes and phenotypic expression of beckwith-wiedemann syndrome. Eur J Hum Genet EJHG (2005) 13(9):1025–32. doi: 10.1038/sj.ejhg.5201463
41. Coorens TH, Treger TD, Al-Saadi R, Moore L, Tran MG, Mitchell TJ, et al. Embryonal precursors of wilms tumor. Science (2019) 366(6470):1247–51. doi: 10.1126/science.aax1323
42. Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Dev Camb Engl (2003) 130(14):3085–94. doi: 10.1242/dev.00536
43. Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J (2006) 25(21):5214–28. doi: 10.1038/sj.emboj.7601381
44. Torrezan GT, Ferreira EN, Nakahata AM, Barros BDF, Castro MTM, Correa BR, et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in wilms tumour. Nat Commun (2014) 5:4039. doi: 10.1038/ncomms5039
45. Yermalovich AV, Osborne JK, Sousa P, Han A, Kinney MA, Chen MJ, et al. Lin28 and let-7 regulate the timing of cessation of murine nephrogenesis. Nat Commun (2019) 10(1):168. doi: 10.1038/s41467-018-08127-4
46. Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, et al. Lin28 sustains early renal progenitors and induces wilms tumor. Genes Dev (2014) 28(9):971–82. doi: 10.1101/gad.237149.113
47. Chang HM, Triboulet R, Thornton JE, Gregory RI. A role for the perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature (2013) 497(7448):244–8. doi: 10.1038/nature12119
48. Cerqueira DM, Tayeb M, Ho J. MicroRNAs in kidney development and disease. JCI Insight (2022) 7(9):e158277. doi: 10.1172/jci.insight.158277
49. Ludwig N, Nourkami-Tutdibi N, Backes C, Lenhof HP, Graf N, Keller A, et al. Circulating serum miRNAs as potential biomarkers for nephroblastoma. Pediatr Blood Cancer (2015) 62(8):1360–7. doi: 10.1002/pbc.25481
50. Turchinovich A, Weiz L, Burwinkel B. Extracellular miRNAs: the mystery of their origin and function. Trends Biochem Sci (2012) 37(11):460–5. doi: 10.1016/j.tibs.2012.08.003
51. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA (2008) 105(30):10513–8. doi: 10.1073/pnas.0804549105
52. Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res (2008) 18(10):997–1006. doi: 10.1038/cr.2008.282
53. Verschuur AC, Vujanic GM, Van Tinteren H, Jones KP, de Kraker J, Sandstedt B. Stromal and epithelial predominant wilms tumours have an excellent outcome: the SIOP 93 01 experience. Pediatr Blood Cancer (2010) 55(2):233–8. doi: 10.1002/pbc.22496
54. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol (2010) 2(1):a001008. doi: 10.1101/cshperspect.a001008
55. Wegert J, Vokuhl C, Ziegler B, Ernestus K, Leuschner I, Furtwängler R, et al. TP53 alterations in wilms tumour represent progression events with strong intratumour heterogeneity that are closely linked but not limited to anaplasia. J Pathol Clin Res (2017) 3(4):234–48. doi: 10.1002/cjp2.77
56. Treger TD, Chagtai T, Butcher R, Cresswell GD, Al-Saadi R, Brok J, et al. Somatic TP53 mutations are detectable in circulating tumor DNA from children with anaplastic wilms tumors. Transl Oncol (2018) 11(6):1301–6. doi: 10.1016/j.tranon.2018.08.006
57. St Laurent G, Wahlestedt C, Kapranov P. The landscape of long non-coding RNA classification. Trends Genet TIG (2015) 31(5):239–51. doi: 10.1016/j.tig.2015.03.007
58. Li CH, Chen Y. Insight into the role of long noncoding RNA in cancer development and progression. Int Rev Cell Mol Biol (2016) 326:36–37. doi: 10.1016/bs.ircmb.2016.04.001
59. Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature (2012) 491(7424):454–7. doi: 10.1038/nature11508
60. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12(12):861–74. doi: 10.1038/nrg3074
61. Moorwood K, Charles AK, Salpekar A, Wallace JI, Brown KW, Malik K. Antisense WT1 transcription parallels sense mRNA and protein expression in fetal kidney and can elevate protein levels in vitro. J Pathol (1998) 185(4):356–358. doi: 10.1002/(SICI)1096-9896(199808)185:4<352::AID-PATH119>3.0.CO;2-#
62. Dallosso AR, Hancock AL, Malik S, Salpekar A, King-Underwood L, Pritchard-Jones K, et al. Alternately spliced WT1 antisense transcripts interact with WT1 sense RNA and show epigenetic and splicing defects in cancer. RNA (2007) 13(12):2287–99. doi: 10.1261/rna.562907
63. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell (2011) 146(3):353–8. doi: 10.1016/j.cell.2011.07.014
64. Wang Z, Cheng H, Qi L, Sui D. Comprehensive analysis of long non−coding RNA using an associated competitive endogenous RNA network in wilms tumor. Mol Med Rep (2020) 22(1):105–16. doi: 10.3892/mmr.2020.11124
65. Zhao X, Li D, Pu J, Mei H, Yang D, Xiang X, et al. CTCF cooperates with noncoding RNA MYCNOS to promote neuroblastoma progression through facilitating MYCN expression. Oncogene (2016) 35(27):3565–76. doi: 10.1038/onc.2015.422
66. Xie ZZ, Xiao ZC, Song YX, Li W, Tan GL. Long non-coding RNA Dleu2 affects proliferation, migration and invasion ability of laryngeal carcinoma cells through triggering miR-16-1 pathway. Eur Rev Med Pharmacol Sci (2018) 22(7):1963–70. doi: 10.26355/eurrev_201804_14723
67. Lerner M, Harada M, Lovén J, Castro J, Davis Z, Oscier D, et al. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp Cell Res (2009) 315(17):2941–52. doi: 10.1016/j.yexcr.2009.07.001
68. Liu H, Zhang M, Shi M, Zhang T, Zhang Z, Cui Q, et al. A survival-related competitive endogenous RNA network of prognostic lncRNAs, miRNAs, and mRNAs in wilms tumor. Front Oncol (2021) 11:608433. doi: 10.3389/fonc.2021.608433
69. Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology wilms tumor: a report from the national wilms tumor study group. J Clin Oncol Off J Am Soc Clin Oncol (2005) 23(29):7312–21. doi: 10.1200/JCO.2005.01.2799
70. Chagtai T, Zill C, Dainese L, Wegert J, Savola S, Popov S, et al. Gain of 1q as a prognostic biomarker in wilms tumors (WTs) treated with preoperative chemotherapy in the international society of paediatric oncology (SIOP) WT 2001 trial: a SIOP renal tumours biology consortium study. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(26):3195–203. doi: 10.1200/JCO.2015.66.0001
71. Segers H, van den Heuvel-Eibrink MM, Williams RD, van Tinteren H, Vujanic G, Pieters R, et al. Gain of 1q is a marker of poor prognosis in wilms’ tumors. Genes Chromosomes Cancer (2013) 52(11):1065–74. doi: 10.1002/gcc.22101
72. van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, van Tinteren H, Furtwängler R, Verschuur AC, et al. Position paper: rationale for the treatment of wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat Rev Urol (2017) 14(12):743–52. doi: 10.1038/nrurol.2017.163
73. Weiser DA, West-Szymanski DC, Fraint E, Weiner S, Rivas MA, Zhao CWT, et al. Progress toward liquid biopsies in pediatric solid tumors. Cancer Metastasis Rev (2019) 38(4):553–71. doi: 10.1007/s10555-019-09825-1
74. Madanat-Harjuoja LM, Renfro LA, Klega K, Tornwall B, Thorner AR, Nag A, et al. Circulating tumor DNA as a biomarker in patients with stage III and IV wilms tumor: analysis from a children’s oncology group trial, AREN0533. J Clin Oncol Off J Am Soc Clin Oncol (2022) 40(26):3047–56. doi: 10.1200/JCO.22.00098
75. Jiménez I, Chicard M, Colmet-Daage L, Clément N, Danzon A, Lapouble E, et al. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int J Cancer (2019) 144(1):68–79. doi: 10.1002/ijc.31620
76. Miguez ACK, Barros BD de F, de Souza JES, da Costa CML, Cunha IW, Barbosa PNVP, et al. Assessment of somatic mutations in urine and plasma of wilms tumor patients. Cancer Med (2020) 9(16):5948–59. doi: 10.1002/cam4.3236
77. Schmitt J, Backes C, Nourkami-Tutdibi N, Leidinger P, Deutscher S, Beier M, et al. Treatment-independent miRNA signature in blood of wilms tumor patients. BMC Genomics (2012) 13:379. doi: 10.1186/1471-2164-13-379
78. Murray MJ, Raby KL, Saini HK, Bailey S, Wool SV, Tunnacliffe JM, et al. Solid tumors of childhood display specific serum microRNA profiles. Cancer Epidemiol biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol (2015) 24(2):350–60. doi: 10.1158/1055-9965.EPI-14-0669
79. Abbou SD, Shulman DS, DuBois SG, Crompton BD. Assessment of circulating tumor DNA in pediatric solid tumors: the promise of liquid biopsies. Pediatr Blood Cancer (2019) 66(5):e27595. doi: 10.1002/pbc.27595
80. Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med (2018) 379(18):1754–65. doi: 10.1056/NEJMra1706174
81. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early stage lung cancer evolution. Nature (2017) 545(7655):446–51. doi: 10.1038/nature22364
82. Stern M, Longaker MT, Adzick NS, Harrison MR, Stern R. Hyaluronidase levels in urine from wilms’ tumor patients. J Natl Cancer Inst (1991) 83(21):1569–74. doi: 10.1093/jnci/83.21.1569
83. Lin RY, Argenta PA, Sullivan KM, Adzick NS. Diagnostic and prognostic role of basic fibroblast growth factor in wilms’ tumor patients. Clin Cancer Res Off J Am Assoc Cancer Res (1995) 1(3):327–31.
84. Lin RY, Argenta PA, Sullivan KM, Stern R, Adzick NS. Urinary hyaluronic acid is a wilms’ tumor marker. J Pediatr Surg (1995) 30(2):304–8. doi: 10.1016/0022-3468(95)90578-2
85. Ortiz MV, Ahmed S, Burns M, Henssen AG, Hollmann TJ, MacArthur I, et al. Prohibitin is a prognostic marker and therapeutic target to block chemotherapy resistance in wilms’ tumor. JCI Insight (2019) 4(15):127098. doi: 10.1172/jci.insight.127098
86. Coppes MJ. Serum biological markers and paraneoplastic syndromes in wilms tumor. Med Pediatr Oncol (1993) 21(3):213–21. doi: 10.1002/mpo.2950210311
87. Vujanić GM, Gessler M, Ooms AHAG, Collini P, Coulomb-l’Hermine A, D’Hooghe E, et al. The UMBRELLA SIOP–RTSG 2016 wilms tumour pathology and molecular biology protocol. Nat Rev Urol (2018) 15(11):693–701. doi: 10.1038/s41585-018-0100-3
88. Children’s Oncology Group. Treatment of newly diagnosed diffuse anaplastic wilms tumors (DAWT) and relapsed favorable histology wilms tumors (FHWT). clinicaltrials.gov (2022). Available at: https://clinicaltrials.gov/ct2/show/NCT04322318.
Keywords: Wilms tumor, genetic abnormalities, biomarkers, stratification system, liquid biopsy
Citation: Zheng H, Liu J, Pan X and Cui X (2023) Biomarkers for patients with Wilms tumor: a review. Front. Oncol. 13:1137346. doi: 10.3389/fonc.2023.1137346
Received: 04 January 2023; Accepted: 27 June 2023;
Published: 24 July 2023.
Edited by:
Andrea Di Cataldo, University of Catania, ItalyReviewed by:
Atsushi Makimoto, Tokyo Metropolitan Children’s Medical Center, JapanHarold “Bo” Newton Lovvorn, III, Vanderbilt University Medical Center, United States
Copyright © 2023 Zheng, Liu, Pan and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuwu Pan, cGFueGl1d3VAMTI2LmNvbQ==; Xingang Cui, Y3VpeGluZ2FuZ0B4aW5odWFtZWQuY29tLmNu