 Eduardo R. M. Cabral1
Eduardo R. M. Cabral1 Marilia F. Pacanhella2
Marilia F. Pacanhella2 Andre V. H. Lengert1
Andre V. H. Lengert1 Mariana B. dos Reis1
Mariana B. dos Reis1 Leticia F. Leal1,2
Leticia F. Leal1,2 Marcos A. de Lima1
Marcos A. de Lima1 Aline L. V. da Silva1
Aline L. V. da Silva1 Icaro A. Pinto1
Icaro A. Pinto1 Rui M. Reis1,3,4Mariana T. Pinto1
Rui M. Reis1,3,4Mariana T. Pinto1 Flavio M. Cárcano1,5*
Flavio M. Cárcano1,5*- 1Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos, Brazil
- 2Barretos School of Health Sciences Dr. Paulo Prata – FACISB, Barretos, Brazil
- 3Life and Health Sciences Research Institute (ICVS), Medical School, University of Minho, Braga, Portugal
- 43ICVS/3B’s-PT Government Associate Laboratory, Braga, Portugal
- 5Division of Genitourinary Medical Oncology, Oncoclínicas, Belo Horizonte, Brazil
Background: Testicular Germ Cell Tumors (TGCT) are the most common cancer among young adult men. The TGCT histopathology is diverse, and the frequency of genomic alterations, along with their prognostic role, remains largely unexplored. Herein, we evaluate the mutation profile of a 15-driver gene panel and copy number variation of KRAS in a large series of TGCT from a single reference cancer center.
Materials and methods: A cohort of 97 patients with TGCT, diagnosed at the Barretos Cancer Hospital, was evaluated. Real-time PCR was used to assess copy number variation (CNV) of the KRAS gene in 51 cases, and the mutation analysis was performed using the TruSight Tumor 15 (Illumina) panel (TST15) in 65 patients. Univariate analysis was used to compare sample categories in relation to mutational frequencies. Survival analysis was conducted by the Kaplan–Meier method and log-rank test.
Results: KRAS copy number gain was a very frequent event (80.4%) in TGCT and presented a worse prognosis compared with the group with no KRAS copy gain (10y-OS, 90% vs. 81.5%, p = 0.048). Among the 65 TGCT cases, different variants were identified in 11 of 15 genes of the panel, and the TP53 gene was the most recurrently mutated driver gene (27.7%). Variants were also detected in genes such as KIT, KRAS, PDGFRA, EGFR, BRAF, RET, NRAS, PIK3CA, MET, and ERBB2, with some of them potentially targetable.
Conclusion: Although larger studies incorporating collaborative networks may shed the light on the molecular landscape of TGCT, our findings unveal the potential of actionable variants in clinical management for applying targeted therapies.
1 Introduction
Testicular germ cell tumors (TGCTs) are relatively rare tumors, accounting for about 1.7% of all cancers (1). However, they are the most common malignant tumors among men aged between 15 and 44 years worldwide (1). TGCTs are highly curable, and since the employment of platinum-based combination chemotherapy regimens began (2, 3), the overall cure rates have dramatically increased to over 95% across all disease stages, however cure rates for IGCCCG poor risk disease remain less than 80% (4–8). Ten to twenty per cent of patients with advanced disease are resistant to cisplatin and present with a less favorable prognosis, with relapses requiring second-line treatment (9, 10).
The process of gonocyte maturation and cell differentiation driven by mutations, associated with the microenvironment interaction, leads to the formation of in situ germ cell neoplasms (GCNIS) (11, 12). These precursor cells remain senescent until puberty and develop into TGCT under the effect of sex hormones and additional genetic changes (11). The TGCTs are subclassified into two major histological groups: seminomas and non-seminomas. Seminomas are formed by cells similar to GCNIS, with suppressed differentiation, high sensitivity to chemotherapy and radiotherapy, and better prognosis, whereas non-seminomas have different histologies due to greater differentiation (embryonic carcinoma, teratoma, yolk sac tumors, and choriocarcinoma), resistance to radiotherapy and more aggressive behavior (13).
The genetic composition of TGCTs portrays the embryonic characteristics of their precursor, the primordial germ cells (PGCs). The molecular biology of TGCTs is not still fully elucidated, despite intensive research on the molecular mechanisms involved in their development (14, 15). Copy number alterations are the most consistent genomic alterations in all histological subtypes. Aneuploidy is considered an important biomarker for TGCT, with the number of chromosomes varying between 50 and 70, and gains in the short arm of chromosome 12 (isochromosome 12p) (16–20).
Recurrent somatic mutations in TGCTs are low, with the exception of variants in KIT and KRAS oncogenes that are frequently described (21, 22). KIT and KRAS mutations are reported more frequently in seminomas when compared with non-seminomas (23–25). Moreover, high levels of expression and KRAS gene amplification have been described as mutually exclusive events in TGCTs (26, 27). The molecular profile of Brazilian TGCT is poorly explored, and the mutational landscape remains unknown (15, 28–30).
Importantly, a molecular understanding of TGCTs might unveil signatures that will guide a more specific and effective treatment, as well as medical decision-making. Therefore, the present study aims to evaluate the mutational profile of driver genes and KRAS copy number variation in Brazilian TGCT patients.
2 Materials and methods
2.1 Study population material
We performed a retrospective cohort study of all TGCT Brazilian patients who attended Barretos Cancer Hospital. This study was approved by the Barretos Cancer Hospital IRB under Protocol No. 784/2014. Clinicopathologic data were retrieved from the electronic medical records, including data on age, date of initial diagnosis, histological type, pathologic diagnosis, overall survival, and treatment history with chemotherapy. All information that could identify the patients was collected and managed using REDCap electronic data capture tools hosted at Barretos Cancer Hospital (31, 32), to ensure the confidentiality of the data and the anonymity of the patients. REDCap (Research Electronic Data Capture) is a secure, web-based software platform designed to support data capture for research studies, providing 1) an intuitive interface for validated data capture; 2) audit trails for tracking data manipulation and export procedures; 3) automated export procedures for seamless data downloads to common statistical packages; and 4) procedures for data integration and interoperability with external sources.
Formalin-fixed paraffin-embedded (FFPE) tumoral tissues were retrieved by macrodissection guided by hematoxylin and eosin-stained slides. The macrodissection of mixed tumor cases was performed combining all histology subtypes in the sample. All the samples are from primary tumor before any systemic treatment exposure and were reviewed by an independent pathologist. Biological samples of fresh frozen tumoral tissue were retrieved at the Biobank of the Barretos Cancer Hospital (BB-BCH), ensuring the quality of processes and the suitability of the biospecimens for research (33).
For mutational analysis, among the 183 cases diagnosed with TCGT feasible for inclusion in this study, 75 did not meet the quality control criteria for DNA extraction (minimum 70% tumor; maximum 30% necrosis); so 108 cases had their genetic material extracted. After the multiplex PCR for the GAPDH gene, used as a quality control for FFPE samples, 12 cases did not present intact and good quality DNA and, for this reason, could not proceed to the preparation of libraries for sequencing. Of the 96 samples that went on to prepare libraries, 32 did not have their targets amplified. Ultimately, only 65 cases were included for the Next Generation Sequencing (NGS) variant identification analyses. For Copy Number Variation (CNV) analysis for KRAS, among the 183 cases diagnosed with TCGT and feasible for inclusion in this study, 66 cases did not have fresh frozen tissue samples available in the Biobank. Out of 117 cases with available samples, seven post-chemotherapy cases were excluded. Among the 110 cases feasible for inclusion in this study, 59 did not meet the quality control criteria for DNA extraction (minimum 70% tumor; maximum 30% necrosis). Ultimately, only 51 cases were included for the CNV analysis for KRAS gene. The cases selection flowchart is presented in the Figure S1.
2.2 DNA isolation and integration analysis
FFPE samples were kept in the stove for 20 min at 80°C. The rehydration of the sample was then performed by incubation with decreasing alcohol percentages (100, 70, and 50% ethanol) for 1 min at room temperature (RT). After that, samples were left in milli-Q H2O until the extraction step. Tumor DNA from FFPE was extracted using QIAamp DNA FFPE Tissue Kit (Qiagen), following the manufacturer’s protocols as previously reported (28). Tumor DNA samples were quantified using Qubit® 2.0 Fluorometer technology (Invitrogen). A multiplex PCR for GAPDH gene was used as quality control for FFPE samples, and those with amplifications of longer fragments (200bp, 300bp, and 400bp) (34) were considered eligible for NGS workflow. Amplifications were performed with a final volume of 30 µL, containing 1.5 mM MgCl2 (Invitrogen), 0.2 mM dNTPs (Invitrogen), 0.133 µM of each primer, 1 unit of Taq DNA Polymerase (Invitrogen), and 50 ng of tumor DNA. The reactions were performed in a ProFlex thermocycler (Thermo Fisher Scientific) using the following amplification parameters: 94°C for 4 minutes, 35 cycles of 94°C for 1 minute, 56°C for 1 minute, 72°C for 1 minute, and a final extension of 72°C for 7 minutes. The amplified DNA was assessed on a 2% agarose gel and only samples with 200 bp or more were accepted for further preparation of sequencing libraries for mutation analysis.
Tumor DNA from fresh frozen tissue samples was extracted using the DNeasy Blood and Tissue kit (Qiagen® Hilden-Germany), following the manufacturer’s instructions, and by Biobank Barretos procedures (NEUBER et al., 2021). DNA was quantified using NanoDrop™ Spectrophotometer (Thermo Scientific).
2.3 Copy number variation analysis of KRAS gene
For CNV analysis (n=51), a specific TaqMan assay for the KRAS gene (Hs02739788_cn—Thermo Fisher Scientific) was used. The reference gene Ribonuclease P (RNAse P 4403326 —Thermo Fisher Scientific), located on chromosome 14, is known to contain two copies in the human genome and was used to calculate the number of copies of the target gene. The TaqMan® KRAS Copy Number Assay contained two primers and an FAM™ dye-labeled MGB probe to detect the target genomic DNA sequence; the Ribonuclease P reference assay contained two primers and a VIC® and TAMRA™ dye-labeled probe to detect the genomic DNA reference sequence. As controls for KRAS copy number, DNA from the GCT cell line (NTERA-2), which contains five copies of KRAS and a normal blood sample, which contains two copies, were used. The qPCR reactions were performed using 20 ng of DNA, 10 μL TaqMan Genotyping Master Mix (Thermo Fisher Scientific), 1 μL of probes and region-specific primers and controls, and nuclease-free ultrapure water in a total volume of 20 μL. The copy number variation assays were performed simultaneously with the reference assay in a qPCR duplex using QuantStudio 6 equipment (Thermo Fisher Scientific) and cycling with an initial temperature of 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. The Ct values of each reference sample and gene were exported to the CopyCaller™ software (Applied BioSystems, USA), in which a comparative Ct quantification (ΔΔCt) analysis of the real-time data was performed. The comparative Ct (ΔΔCt) method calculated the difference (ΔCt) between the threshold cycles of the target and reference assay sequences and then compared the ΔCt values of the test samples to a calibrator sample (normal blood), which contained a known number of copies of the target sequence (two copies). All qPCR reactions were performed in technical triplicates. A variation in the number of copies of the KRAS gene was considered when the cases presented a copy number different from two for this gene. Via the method used, it was not possible to differentiate gain from amplification, only to define a variation in the number of copies different from normal (two copies of the KRAS gene).
2.4 Mutational analysis
To evaluate the driver mutations (n=65), the TruSight Tumor 15 panel (TST15 - Illumina) was used, as reported by the manufacturer’s instructions (35). The panel provides a comprehensive assessment of 15 driver genes (TP53, KRAS, KIT, NRAS, BRAF, EGFR, MET, PIK3CA, PDGFRA, AKT1, ERBB2, RET, GNA11, GNAQ, and FOXL2). For library preparation, targets were enriched in a multiplex PCR, with an initial input of 20 ng/μL of DNA, according to the manufacturer’s instructions. After library preparation, the DNA amplification was verified by electrophoresis. The libraries were paired-end-sequenced using MiSeq Reagent Kit v3, 600 Cycles on the Illumina MiSeq instrument (Illumina). Demultiplexed data and FASTQ files were generated using BaseSpace software (Illumina).
The reads were aligned with the reference genome (GRCh37/hg19) using the algorithm BWA-MEM-PAIRED, and Sequence Alignment Map (SAM) output format files were converted to the Binary SAM (BAM) format using SAMtools. BAM files were processed using the Genome Analysis ToolKIT (GATK). Realignment and variant calling were performed using GATK HaplotypeCaller and the Freebayes genetic variant detector using BED–Capture. The annotation of variants was performed using the Varstation® (36) analysis platform, via the Varstation Annotation Somatic software.
A customized analysis filter was used, which retained pathogenic and likely pathogenic variants of uncertain/unknown significance (VUS) and drug response, as well as those with population frequency less than 1% (gnomAD, 1000 genomes, ABRaOM); allelic frequency (VAF) greater than or equal to 3%; vertical coverage (depth) greater than or equal to 500x in the exonic and splicing regions, and frameshift, non-synonymous, or stopgain type. Variants leading to loss of function, “high‐impact” variants (frameshift, nonsense, and canonical splice site variants), and missense variants were selected. In addition, benign and likely benign variants were excluded from the analyses.
The manual curation of the variants was carried out using public databases (Clinvar, IARC TP53, Varsome, COSMIC, gnomAD, ABraOM, and CGI) for the better classification of the detected variants according to their effect, pathogenicity, and frequency in the general population. CGI was also employed to verify the status of driver variants, and only known variants or those predicted as drivers in cancer-related genes were retained. The variants were classified according to public databases as pathogenic, likely pathogenic, uncertain significance (VUS), and drug response.
2.5 Statistical analysis
Univariate analysis was used to compare sample categories in relation to mutation frequencies. For this, the χ2 test or Fisher’s exact test was used, according to the characteristics of the sample. Survival curves were plotted using the Kaplan–Meier method and events of interest (death record) were considered for the outcome of overall survival (OS). Alive patients and those lost to follow-up were discounted. Univariate comparisons of survival times were performed using the log-rank test. Statistical analyses were performed using IBM SPSS Statistics for Windows, Version 2.0 (IBM), and alpha=0.05 with 80% power in a two-tailed hypothesis test.
3 Results
3.1 Clinicopathologic characterization of TCGT patients
Out of the 97 cases analyzed, the average age at diagnosis was 32.5 years (range, 18-50 years), and most cases (57.7%) were non-seminomas (Table 1). Among the histological group of non-seminomatous tumors, 41.2% presented a histology of mixed tumors. In total, 67% of cases were in the advanced stages of the disease (stage II and III—AJCC), of which 39.2% were low risk, according to the IGCCCG risk.

Table 1 Characteristics of patients with TGCT at the Barretos Cancer Hospital.
The most used treatment strategy was the bleomycin, etoposide, and cisplatin (PEB) combination (57.7%). Considering all of the chemotherapy regimens, 63.9% were responsive and 6.2% were refractory. The remaining 29.9% corresponds to those who did not undergo chemotherapy (n = 25) and/or to those who died (n = 4) before the date set for the first cycle.
The overall survival probability of the analyzed cohort was 91.5% in the 5-year follow-up period and 83.2% in the 10-year follow-up period; the overall survival according to AJCC staging, histological group, IGCCCG risk, and chemosensitivity status were evaluated and did not differ from the results in the literature (Figure S2).
3.2 CNV analysis for KRAS
In total, 51 cases were analyzed for KRAS CNV, and it was observed that 41 cases (80.4%) showed at least one copy number gain, with a variation of three to seven KRAS copies (Figure 1). Non-seminoma cases showed a non significant trend of greater variation in the number of copies of the KRAS gene, when compared to seminoma cases (47.1% vs 33.3%; p = 0.160), and none of the cases showed loss of copy number.
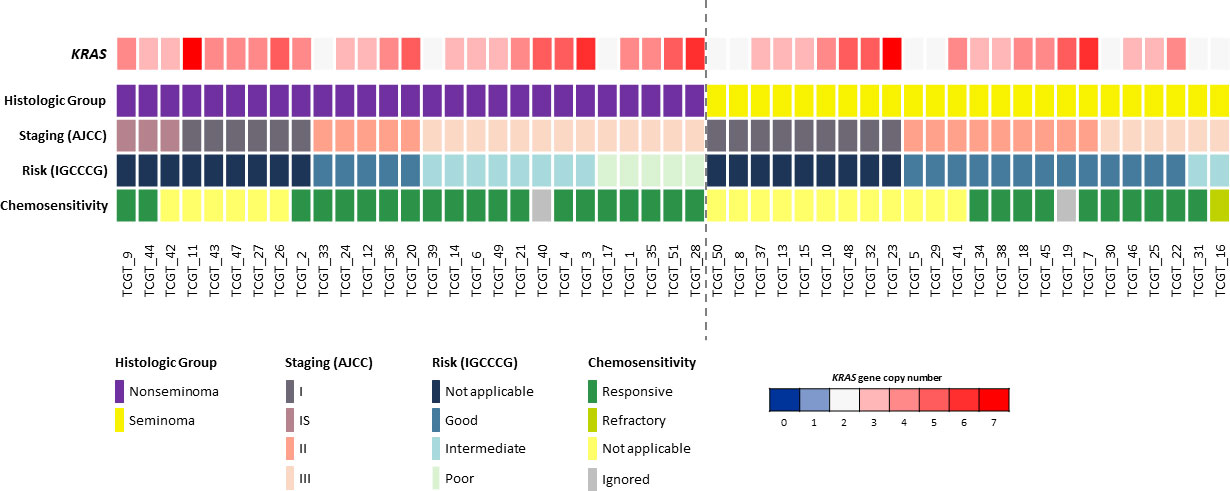
Figure 1 Copy number variations of the KRAS gene compared to clinicopathologic data (histological group, staging, risk and chemosensitivity). Only TGCTs evaluated for copy number variation are presented (n = 51). Each column represents an individual patient. The dotted line in the center of the figure separates seminoma and non-seminoma cases. A color scale (blue to red) is used to represent the variation in the number of copies of the KRAS gene, in which the dark blue color represents the deletion of two copies of this gene (0 copies); light blue the deletion of only one copy; white represents that there is no loss or gain of copies; and light red to dark red colors represent a gain of copies of this gene (1 to 7 copies).
KRAS copy gain was compared to the clinicopathologicdata of the TGCT patients, including age, histological group, stage (AJCC), risk (IGCCCG), chemosensitivity to cisplatin-based treatment, and overall survival; however, no significant association between KRAS CNV status and patient’s features was observed (Table S1). Regarding overall survival (10-year period), the KRAS copy gain group presented a worse prognosis compared with the group with no KRAS copy gain (10y-OS, 90% vs. 81.5%, p = 0.048) (Figure 2).

Figure 2 Overall Survival stratified by KRAS copy number in TGCT.
3.3 Called variants by next generation sequencing
Sixty-five cases were analyzed for the presence of variants and a total of 82,975 variants were initially detected; the variants were filtered out according to the pipeline, and we identified 73 different variants (amongst them, pathogenic, likely pathogenic, VUS, and drug response) (Table S2). Furthermore, after CGI was employed to verify the variant prediction status, only known variants and/or predicted drivers in cancer-related genes were maintained. So, among the 65 cases analyzed for the presence of variants in this study, 35 cases (53.8%) showed a total of 51 different variants (known and/or predicted as drivers) that were identified in 11 of 15 genes of the panel.
TP53 alterations were the most common in our cases (27.7%, n=18), followed by KIT (18.5%, n=12), KRAS (7.7%, n=5), PDGFRA (7.7%, n= 5), EGFR (6.2%, n=4), BRAF (4.6%, n= 3), RET (4.6%, n=3), NRAS (3.1%, n= 2), PIK3CA (3.1%, n= 2), MET (3.1%, n=2) and ERBB2 (1,5%, n=1). No variants were identified on AKT1, FOXL2, GNAQ, or GNA11 genes in any of the cases (Figure 3).
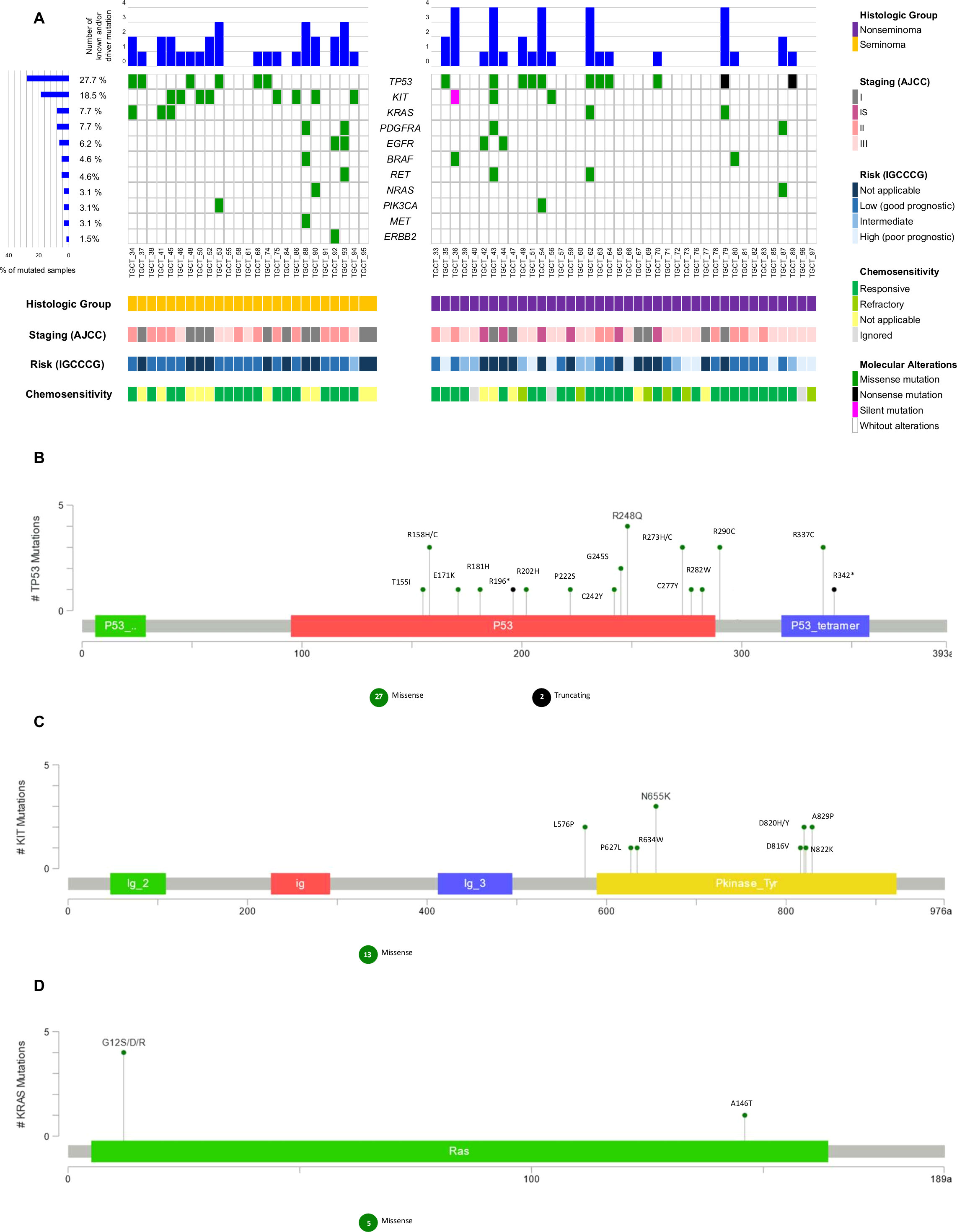
Figure 3 Waterfall plot of the known and/or predicted driver mutation spectrum of Testicular Germ Cell Tumor (TGCT). (A) Plots show the frequency of samples mutated for seminomas and non-seminomas. The upper panel demonstrates the frequency of mutation for each sample. The left panel shows the frequency of samples harboring mutations according to the gene. The lower panel indicates clinicopathologic data. (B) Variant distribution in TP53 gene. (C) Variant distribution in KIT gene. (D) Variant distribution in KRAS gene. All graphs depict a lollipop plot showing identified variants relative to a schematic representation of the gene. Any position with a mutation is shown with a circle, and the length of the line depends on the number of mutations detected at that codon. The grey bar represents the entire protein with different amino acid positions (aa). The colored boxes are specific functional domains. On top of the lollipops, the most frequent variants are annotated as the amino acid change at that specific site.
We further analyzed the variants according to the histologic group, staging (AJCC), risk (IGCCCG), and chemosensitivity (Figure 3). The frequency of known and/or predicted driver mutations in seminomas was higher (72% of cases with at least one variant) when compared to non-seminomas (42.5% of cases with at least one variant) (p = 0.024) (Table S3).
TP53, KIT, and KRAS genes were the three most mutated in our case series; TP53/KIT and KIT/KRAS genes were mutually exclusively mutated, except for a non-seminoma case and a seminoma case, respectively.
Mutations in the TP53 gene were identified more frequently among non-seminomas when compared to seminomas, and more than one variant was observed for the same case. Three non-seminoma cases and one seminoma case, all of which were chemotherapy-responsive, carried the R248Q variant. The R337C and R290C variants were observed in more than two cases. A case of poor prognostic non-seminoma presented three distinct variants, and another four cases also presented two or more variants concomitantly (Figures 3A, B; Table S2).
Only one chemotherapy-refractory non-seminoma case (staging III and intermediate risk) presented two different variants in the TP53 gene (R158C and C277Y) (Table S2).
Mutations in the KIT gene were identified more frequently among seminomas when compared to non-seminomas, and more than one variant was observed in only one case. L576P and A829P variants were observed in more than one case. One non-seminoma case with a poor prognosis, a carrier of the N655K variant, died before the date set for the first cycle of adjuvant chemotherapy, at 20 years old. No variant in the KIT gene was found in chemotherapy-refractory cases (Figures 3A–C; Table S2).
Mutations in the KRAS gene were identified more frequently among seminomas when compared to non-seminomas, and four different variants were observed in five TGCT cases. One non-seminoma case and one seminoma case, both chemotherapy-responsive, carried the G12S variant. Two other variants, also on the KRAS gene codon 12 (G12R and G12D), were observed in seminoma cases. The A146T variant was identified in non-seminoma case (staging III and intermediate risk) (Figures 3A–D; Table S2).
Analysis comparing the mutational status of genes with clinicopathologic data revealed that KIT (p = 0.007) was significantly mutated in seminomas, and EGFR (p = 0.010) in stage IS. All clinical and molecular comparisons of the other genes are shown in Table S4.
We further compared the mutational status with overall survival, but no significant association was found (Tables S5, 6; Figure S3).
4 Discussion
The analysis of tumor mutation and KRAS copy number variation revealed expressive KRAS copy number gain, well-known variants and potentially targetable variants never described before in TGCT.
The findings of KRAS copy number gain might be explained due to the gain of the 12p isochromosome, commonly found in almost all TGCTs, especially in non-seminomas. KRAS is located in the 12p11.2–p12.1 region of chromosome 12, and its overexpression seems to be associated with the development of TGCTs (17, 37). In seminomas, Loveday et al. (17) demonstrated that KRAS copy number was relatively high, but Shen et al. (18) identified a subset of seminomas with decreased KRAS copy number. We were not able to identify statistical differences between seminomas and non-seminomas regarding KRAS copy number gain, and none of our cases had loss of copy number. Therefore, the role of the KRAS copy number in TGCT needs to be explored further. Worse cancer-specific and overall survival have been associated to KRAS mutation and copy number gain in lung cancer (38, 39). Using cell free DNA somatic mutation analysis, KRAS mutation and copy number gain were associated to worse outcomes in pancreatic cancer (40). We demonstrated a worse prognosis associated to KRAS copy gain when compared to the group with no gain, and this finding has not been addressed before in TGCT.
The gain of 12-p occurs in utero at the gonocyte stage and the development of GCNIS, a TGCT precursor, after birth (41). This pathogenesis involves the blood–testis barrier, which harbors a specialized microenvironment and an immunological barrier that prevents the production of antibodies against meiotic and postmeiotic germ cells (42). This complex and overprotected microenvironment might, at least in part, explain why point mutations are rare events in TGCTs. Although rare, mutation might occur mainly in the TP53, KIT, KRAS, BRAF, and NRAS genes (18, 41, 43).
TP53 is one of the most frequently mutated genes in different types of cancer (44, 45) and variants in TP53 have been identified in non-seminomas with no changes in seminomas (46). However, the COSMIC database shows that mutations in TP53 are rare in TGCTs, with a frequency of 4% considering all histological subtypes. Here, among the known and predicted driver mutations, 26% were found in the TP53 gene, which was the most commonly mutated in our series of cases, mainly in non-seminomas. Fifteen variants were detected in TP53, and ten of them (R290C, R158H, G245S, C242Y, C277Y, E171K, R158C, R181H, R196* and R342*) have not been described in TGCT previously. Although there were no cases of Li–Fraumeni syndrome in our dataset, some of the variants found were associated with that syndrome (R282W, R342*, R196*) in some aspects (47–50). TP53 has been associated to the platinum-resistance mechanism (51), but we did not find any association, and there is no evident cause for the high frequency of TP53 in our study.
KIT mutation is more prevalent in seminomas, and it was the second most frequently mutated gene among our cases, with almost all cases being seminomas (23, 25, 52, 53). KIT variants have been identified in TGCT precursor cells, suggesting that the KIT gene may be largely responsible for the development of TGCTs, and the identification of these variants might assist in the diagnosis of the disease at an early stage (54). We found eight variants of KIT, and it is noteworthy that the D816V, D820H, and D820Y variants, located in the same region as other KIT mutations (exon 17), presumably also encode a constitutively activated protein that confers resistance to imatinib (55, 56). Addionally, KRAS mutations have been associated to cisplatin-resistant TGCTs (57), but we did not find any association in our results. We found four KRAS variants (A146T, G12D, G12R, G12S) and two NRAS variants (G12A, G60R), and this is the first time the NRAS G60R variant has been described. None of the PDGFRA variants identified have been previously described in TGCT either.
Interestingly, targetable variants were found in genes such as BRAF, EGFR, and RET. BRAF mutations are rare in TGCT (28, 52) and the majority of anti-BRAF drugs target V600E variants. However, we found three cases of BRAF T599I variants, and it has been suggested that vemurafenib might control cancer, such as melanoma, even in patients with that variant (58). Four genomic alterations were detected in EGFR (A822T, R776H, S768N, T790M), and the T790M variant has been described in advanced non-small cell lung cancer (NSCLC) as an acquired variant associated with Tyrosine Kinase Inhibitors (TKIs) resistance (59, 60). A third generation of anti-EGFR drugs has been developed to overcome this resistance in lung cancer with the T790M variant, including osimertinib (61). RET inhibitors such as pralsetinib and selpercatinib have been developed and approved by the FDA to treat patients with RET alterations (62). We found two different variants in RET (R912W, T930M), and none of them have been reported in TGCT before. Although rare, targetable mutations in TGCT increase the opportunities for precision medicine within the scenario of the accelerated approval of new drugs, mainly in cases of refractory disease.
There are limitations to our study, such as the low number of TGCT cases, the retrospective design, the single institution setting, the low number of events to analyze survival, and the large number of mixed tumors. All these limitations increase bias and might lead to misinterpretation of the results. Additionally, the sequencing analysis of the presence study was limited tumor tissue, since it was a retrospective nature of the study and we did not have availability of blood or normal tissue of the cases. Therefore, we could not exclude the germline or somatic nature of the variants identified. The analysis based on important and consolidated databases might minimize the presence of polymorphisms. However, we found new variants never described before in TGCT, and we were able to show targetable variants using a reliable mutational panel. Moreover, patients with admixed American ancestry, like Brazilian people, are commonly underrepresented in genomic profile studies, although it has been increasing (63). The populations previously studied are mostly European, so the cases in our Brazilian cohort are expressive, and might bring new insights and generate new hypotheses in precision oncology.
In conclusion, the increase in the number of copies of the KRAS gene is a very frequent event in TGCT, and non-seminomas are most highly associated with a higher rate of this amplification. Although point mutations are rare events in TGCTs, relevant gene variants are identified mainly in TP53, KIT, KRAS, and NRAS genes, and 40% of the evaluable genes have at least one known and/or predicted driver variant in one of the 15 genes tested, with TP53 being the most commonly mutated gene. Although larger studies enrolling collaborative networks might shed light on the molecular landscape of TGCTas well as limited evidence available for use of targeted approaches in TGCT, our findings unveil the potential of actionable variants in clinical management when applying targeted therapies, and further evaluation of these approaches is necessary.
Data availability statement
The data presented in the study are deposited in the Sequence Read Archive (SRA) repository, accession number PRJNA942937 (https://www.ncbi.nlm.nih.gov/sra/PRJNA942937)
Ethics statement
The studies involving human participants were reviewed andapproved by Barretos Cancer Hospital IRB under Protocol No. 784/2014. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
FC, MTP, and RR designed the study and critically revised the results. EC and MFP performed the study. AL, MR, and LL supervised the experiments. EC, ML, AS and IP analyzed the data. EC wrote the manuscript draft. FC, MTP, RR, LL, and MR revised the manuscript. All authors contributed to the article and approved the submitted version
Funding
This study was partially supported by the Public Ministry of Labor Campinas (Research, Prevention, and Education of Occupational Cancer—15ª zone, Campinas, Brazil). RMR was supported by the National Council for Scientific and Technological Development (CNPq, Brazil) as Research Productivity Scholarship—Level 1B; FMC was supported by PAIP (Researcher Support Program) from Barretos Cancer Hospital. MBR is sponsored by Coordination for the Improvement of Higher Education Personnel—Brazil (CAPES). MTP is supported by FAPESP, Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil (2019/07502-8). MFP was supported by FAPESP, Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil (2019/14512-0).
Acknowledgments
We would like to thank all our patients who contributed to this study. The authors are very grateful to Ana Carolina Laus for all the support at CPOM/Barretos; Daniel Preto for the collection of clinicopathologic data of patients; and Varsomics for the Varstation solution (www.varsomics.com/en) used in the analysis of the gene mutation variants.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1133363/full#supplementary-material
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Einhorn LH. Treatment of testicular cancer: a new and improved model. J Clin Oncol (1990) 8(11):1777–81. doi: 10.1200/JCO.1990.8.11.1777
3. Shibley L, Brown M, Schuttinga J, Rothen-berg M, Whalen J. Cisplatin-based combination chemotherapy in the treatment of advanced-stage testicular cancer: cost-benefit analysis. JNCI: J Natl Cancer Institute (1990) 82(3):186–92. doi: 10.1093/jnci/82.3.186
4. Nakamura T, Miki T. Recent strategy for the management of advanced testicular cancer. Int J Urol (2010) 17(2):148–57. doi: 10.1111/j.1442-2042.2009.02431.x
5. Shanmugalingam T, Soultati A, Chowdhury S, Rudman S, Van Hemelrijck M. Global incidence and outcome of testicular cancer. Clin Epidemiol (2013) 5:417. doi: 10.2147/CLEP.S34430
6. Bakardjieva-Mihaylova V, Skvarova Kramarzova K, Slamova M, Svaton M, Rejlova K, Zaliova M. Molecular basis of cisplatin resistance in testicular germ cell tumors. Cancers (2019) 11(9):1316. doi: 10.3390/cancers11091316
7. Beyer J, Collette L, Sauvé N, Daugaard G, Feldman DR, Tandstad T, et al. Survival and new prognosticators in metastatic seminoma: results from the IGCCCG-update consortium. J Clin Oncol (2021) 39(14):1553–62. doi: 10.1200/JCO.20.03292
8. Gillessen S, Sauvé N, Collette L, Daugaard G, de Wit R, Albany C, et al. Predicting outcomes in men with metastatic nonseminomatous germ cell tumors (NSGCT): results from the IGCCCG update consortium. J Clin Oncol (2021) 39(14):1563–74. doi: 10.1200/JCO.20.03296
9. Beyer J, Albers P, Altena R, Aparicio J, Bokemeyer C, Busch J. Maintaining success, reducing treatment burden, focusing on survivorship: highlights from the third European consensus conference on diagnosis and treatment of germ-cell cancer. Ann Oncol (2013) 24(4):878–88. doi: 10.1093/annonc/mds579
10. Vries G, Rosas-Plaza X, Van Vugt MA, Gietema JA, Jong S. Testicular cancer: determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat Rev (2020) 88:102054. doi: 10.1016/j.ctrv.2020.102054
11. Boublikova L, Buchler T, Stary J, Abrahamova J, Trka J. Molecular biology of testicular germ cell tumors: Unique features awaiting clinical application. Crit Rev Oncol Hematol (2014) 89(3):366–85. doi: 10.1016/j.critrevonc.2013.10.001
12. Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs–part a: renal, penile, and testicular tumours. Eur Urol (2016) 70:93–105. doi: 10.1016/j.eururo.2016.02.029
13. Bray F, Ferlay J, Devesa SS, McGlynn KA, Møller H. Interpreting the international trends in testicular seminoma and nonseminoma incidence. Nat Clin Pract Urol (2006) 3(10):532–43. doi: 10.1038/ncpuro0606
14. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol (2018) 2018:1–8. doi: 10.1155/2018/9059382
15. Pinto MT, Cárcano FM, Vieira AGS, Cabral ERM, Lopes LF. Molecular biology of pediatric and adult Male germ cell tumors. Cancers (2021) 13(10):2349. doi: 10.3390/cancers13102349
16. Frigyesi A, Gisselsson D, Hansen GB, Soller M, Mitelman F, Höglund M. A model for karyotypic evolution in testicular germ cell tumors. Genes Chromosomes Cancer (2004) 40(3):172–8. doi: 10.1002/gcc.20032
17. Loveday C, Litchfield K, Proszek PZ, Cornish AJ, Santo F, Levy M, et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat Commun (2020) 11(1):1–12. doi: 10.1038/s41467-020-15768-x
18. Shen H, Shih J, Hollern DP, Wang L, Bowlby R, Tickoo SK, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep (2018) 23(11):3392–406. doi: 10.1016/j.celrep.2018.05.039
19. Fichtner A, Richter A, Filmar S, Gaisa NT, Schweyer S, Reis H, et al. The detection of isochromosome i (12p) in malignant germ cell tumours and tumours with somatic malignant transformation by the use of quantitative real-time polymerase chain reaction. Histopathology (2021) 78(4):593–606. doi: 10.1111/his.14258
20. Cheng L, Davidson DD, Montironi R, Wang M, Lopez-Beltran A, Masterson TA, et al. Fluorescence in situ hybridization (FISH) detection of chromosomal 12p anomalies in testicular germ cell tumors. In: Bagrodia A, Amatudra JF, editors. Testicular germ cell tumors. New York: Humana (2021). p. 49–63.
21. Cutcutache I, Suzuki Y, Tan IB, Ramgopal S, Zhang S, Ramnarayanan K, et al. Exome-wide sequencing shows low mutation rates and identifies novel mutated genes in seminomas. Eur Urol (2015) 68(1):77–83. doi: 10.1016/j.eururo.2014.12.040
22. Taylor-Weiner A, Zack T, O’Donnell E, Guerriero JL, Bernard B, Reddy A, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature (2016) 540(7631):114–8. doi: 10.1038/nature20596
23. Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, et al. KIT mutations are common in testicular seminomas. Am J Pathol (2004) 164(1):305–13. doi: 10.1016/S0002-9440(10)63120-3
24. McIntyre A, Summersgill B, Grygalewicz B, Gillis AJ, Stoop J, van Gurp RJ, et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res (2005) 65(18):8085–9. doi: 10.1158/0008-5472.CAN-05-0471
25. Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun (2015) 6(1):1–8. doi: 10.1038/ncomms6973
26. McIntyre A, Summersgill B, Spendlove HE, Huddart R, Houlston R, Shipley J. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia (2005) 7(12):1047–52. doi: 10.1593/neo.05514
27. Goddard NC, McIntyre A, Summersgill B, Gilbert D, Kitazawa S, Shipley J. KIT and RAS signalling pathways in testicular germ cell tumours: new data and a review of the literature. Int J andrology (2007) 30(4):337–49. doi: 10.1111/j.1365-2605.2007.00769.x
28. Cárcano FM, Lengert AH, Vidal DO, Scapulatempo Neto C, Queiroz L, Marques H. Absence of microsatellite instability and BRAF (V600E) mutation in testicular germ cell tumors. Andrology (2016) 4(5):866–72. doi: 10.1111/andr.12200
29. da Silva Martinelli CM, Lengert AH, Cárcano FM, Silva ECA, Brait M, Lopes LF, et al. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget (2017) 8(31):50608. doi: 10.18632/oncotarget.11167
30. Pinto F, Cárcano FM, da Silva ECA, Vidal DO, Scapulatempo-Neto C, Lopes LF, et al. Brachyury oncogene is a prognostic factor in high-risk testicular germ cell tumors. Andrology (2018) 6(4):597–604. doi: 10.1111/andr.12495
31. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap) – a metadata-driven methodology and workflow process for providing translational research informatics support. J BioMed Inform (2009) 42(2):377–81. doi: 10.1016/j.jbi.2008.08.010
32. Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O’Neal L, et al. REDCap consortium - the REDCap consortium: Building an international community of software partners. J BioMed Inform (2019) 9:1–10. doi: 10.1016/j.jbi.2019.103208
33. Neuber AC, Tostes CH, Ribeiro AG, Marczynski GT, Komoto TT, Rogeri CD, et al. Quality assessment of cryopreserved human biological samples from biobank of barretos cancer hospital. Biopreservation Biobanking (2022) 23(2):271–84. doi: 10.1089/bio.2021.0131
34. van Beers EH, Joosse SA, Ligtenberg MJ, Fles R, Hogervorst FB, Verhoef S, et al. A multiplex PCR predictor for aCGH success of FFPE samples. Br J Cancer (2006) 94(2):333–7. doi: 10.1038/sj.bjc.6602889
35. Campanella NC, Silva EC, Dix G, de Lima Vazquez F, Escremim de Paula F, Berardinelli GN, et al. Mutational profiling of driver tumor suppressor and oncogenic genes in Brazilian malignant pleural mesotheliomas. Pathobiology (2020) 87(3):208–16. doi: 10.1159/000507373
36. Faria ACO, Caraciolo MP, Minillo RM, Almeida TF, Pereira SM, Cervato MC, Filho JBO. Varstation: a complete and efficient tool to support NGS data analysis. bioRxiv (2019) 833582. doi: 10.1101/833582
37. Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene (2003) 22(12):1880–91. doi: 10.1038/sj.onc.1206302
38. Sasaki H, Hikosaka Y, Kawano O, Moriyama S, Yano M, Fujii Y. Evaluation of kras gene mutation and copy number gain in non-small cell lung cancer. J Thorac Oncol (2011) 6(1):15–20. doi: 10.1097/JTO.0b013e31820594f0
39. Fung AS, Karimi M, Michiels S, Seymour L, Brambilla E, Le-Chevalier T, et al. Prognostic and predictive effect of KRAS gene copy number and mutation status in early stage non-small cell lung cancer patients. Trans Lung Cancer Res (2021) 10(2):826. doi: 10.21037/tlcr-20-927
40. Mohan S, Ayub M, Rothwell DG, Gulati S, Kilerci B, Hollebecque A, et al. Analysis of circulating cell-free DNA identifies KRAS copy number gain and mutation as a novel prognostic marker in pancreatic cancer. Sci Rep (2019) 9(1):1–6. doi: 10.1038/s41598-019-47489-7
41. Sheikine Y, Genega E, Melamed J, Lee P, Reuter VE, Ye H. Molecular genetics of testicular germ cell tumors. Am J Cancer Res (2012) 2(2):153.
42. Cheng CY, Mruk DD. The blood-testis barrier and its implications for male contraception. Pharmacol Rev (2012) 64(1):16–64. doi: 10.1124/pr.110.002790
43. Gonzalez-Exposito R, Merino M, Aguayo C. Molecular biology of testicular germ cell tumors. Clin Trans Oncol (2016) 18(6):550–6. doi: 10.1007/s12094-015-1423-7
44. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat (2007) 28(6):622–9. doi: 10.1002/humu.20495
45. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep (2019) 28(5):1370–84. doi: 10.1016/j.celrep.2019.07.001
46. Necchi A, Bratslavsky G, Corona RJ, Chung JH, Millis SZ, Elvin JA, et al. Genomic characterization of testicular germ cell tumors relapsing after chemotherapy. Eur Urol Focus (2020) 6(1):122–30. doi: 10.1016/j.euf.2018.07.013
47. Zhang Y, Coillie SV, Fang JY, Xu J. Gain of function of mutant p53: R282W on the peak? Oncogenesis (2016) 5(2):e196–6. doi: 10.1038/oncsis.2016.8
48. Mannan AU, Singh J, Lakshmikeshava R, Thota N, Singh S, Sowmya TS, et al. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: implications of embracing a multi-gene panel in molecular diagnosis in India. J Hum Genet (2016) 61(6):515–22. doi: 10.1038/jhg.2016.4
49. Hettmer S, Archer NM, Somers GR, Novokmet A, Wagers AJ, Diller L, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer (2014) 120(7):1068–75. doi: 10.1002/cncr.28507
50. Fiszer-Maliszewska L, Kazanowska B, Padzik J. p53 tetramerization domain mutations: germline R342X and R342P, and somatic R337G identified in pediatric patients with Li–fraumeni syndrome and a child with adrenocortical carcinoma. Familial Cancer (2009) 8(4):541–6. doi: 10.1007/s10689-009-9284-2
51. Ottaviano M, Giunta EF, Rescigno P, Pereira Mestre R, Marandino L, Tortora M, et al. The enigmatic role of TP53 in germ cell tumours: Are we missing something? Int J Mol Sci (2021) 22(13):7160. doi: 10.3390/ijms22137160
52. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res (2019) 47(D1):D941–7. doi: 10.1093/nar/gky1015
53. Coffey J, Linger R, Pugh J, Dudakia D, Sokal M, Easton DF, et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer (2008) 47(1):34–42. doi: 10.1002/gcc.20503
54. Biermann K, Göke F, Nettersheim D, Eckert D, Zhou H, Kahl P, et al. C-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathology: A J Pathological Soc Great Britain Ireland (2007) 213(3):311–8. doi: 10.1002/path.2225
55. Willmore-Payne C, Holden JA, Chadwick BE, Layfield LJ. Detection of c-kit exons 11-and 17-activating mutations in testicular seminomas by high-resolution melting amplicon analysis. Modern Pathol (2006) 19(9):1164–9. doi: 10.1038/modpathol.3800623
56. Ichimura K, Fukushima S, Totoki Y, Matsushita Y, Otsuka A, Tomiyama A, et al. Recurrent neomorphic mutations of MTOR in central nervous system and testicular germ cell tumors may be targeted for therapy. Acta neuropathologica (2016) 131(6):889–901. doi: 10.1007/s00401-016-1557-x
57. Feldman DR, Iyer G, Van Alstine L, Patil S, Al-Ahmadie H, Reuter VE, et al. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin Cancer Res (2014) 20(14):3712–20. doi: 10.1158/1078-0432.CCR-13-2868
58. Gallo S, Coha V, Caravelli D, Becco P, Venesio T, Zaccagna A, et al. BRAF-inhibitors can exert control of disease in BRAF T599I mutated melanoma: A case report. Melanoma Res (2018) 28(2):143–6. doi: 10.1097/CMR.0000000000000417
59. Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol (2016) 9(1):1–7. doi: 10.1186/s13045-016-0268-z
60. Jenkins S, Yang JC, Ramalingam SS, Yu K, Patel S, Weston S, et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non–small cell lung cancer. J Thorac Oncol (2017) 12(7):1061–70. doi: 10.1016/j.jtho.2017.04.003
61. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. New Engl J Med (2020) 382(1):41–50. doi: 10.1056/NEJMoa1913662
62. Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann Oncol (2021) 32(2):261–8. doi: 10.1016/j.annonc.2020.10.599
Keywords: testicular neoplasms, mutation, testicular germ cell tumor, K-ras Gene, TP53 Gene, next-generation sequencing
Citation: Cabral ERM, Pacanhella MF, Lengert AH, Reis MB, Leal LF, Lima MA, Silva ALV, Pinto IA, Reis RM, Pinto MT and Cárcano FM (2023) Somatic mutation detection and KRAS amplification in testicular germ cell tumors. Front. Oncol. 13:1133363. doi: 10.3389/fonc.2023.1133363
Received: 28 December 2022; Accepted: 27 February 2023;
Published: 16 March 2023.
Edited by:
Arun Azad, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Anis Hamid, Eastern Health, AustraliaCiara Conduit, Peter MacCallum Cancer Centre, Australia
Copyright © 2023 Cabral, Pacanhella, Lengert, Reis, Leal, Lima, Silva, Pinto, Reis, Pinto and Cárcano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flavio M. Cárcano, Y2FyY2Fuby5mbUBnbWFpbC5jb20=